 Simonetta Ausoni
Simonetta Ausoni Giuseppe Azzarello
Giuseppe Azzarello
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Cardiovasc. Med. , 26 October 2020
Sec. Cardio-Oncology
Volume 7 - 2020 | https://doi.org/10.3389/fcvm.2020.598384
In the last decade, cardiologists and oncologists have provided clinical and experimental evidence that cancer, and not only chemotherapeutic agents, can cause detrimental effects on heart structure and function, a consequence that has serious clinical implications for patient management. In parallel, the intriguing idea that heart failure (HF) may be an oncogenic condition has also received growing attention. A number of epidemiological and clinical studies have reported that patients with HF have a higher risk of developing cancer. Chronic low-grade systemic inflammation has been proposed as a major pathophysiological process linking the failing heart to the multi-step process of carcinogenesis. According to this view, pro-inflammatory mediators secreted by the damaged heart generate a favorable milieu that promotes tumor development and accelerates malignant transformation. HF-associated inflammation synergizes with tumor-associated inflammation, so that over time it is no longer possible to distinguish the effects of one or the other. Experimental studies have just begun to search for the molecular effectors of this process, with the ultimate goal that of identifying mechanisms suitable for anti-cancer target therapy to reduce the risk of incident cancer in patients already affected by HF. In this review we critically discuss strengths and limitations of clinical and experimental studies that support a causal relationship between HF and cancer, and focus on HF-associated inflammation, cardiokines and their endocrine functions linking one and the other disease.
Cancer and cardiovascular diseases are the leading causes of death worldwide, raising the question of whether these diseases have intersecting pathogenetic mechanisms. Clinical and experimental studies have demonstrated that cancer patients develop cardiac disease not only as a consequence of cardiotoxicity induced by chemotherapies, but also because tumor cells release soluble factors that affect various organs at a distance, including the heart (1, 2). Cancer-derived pro-inflammatory molecules cause cardiomyocyte atrophy and tissue remodeling, which can degenerate to cachexia and HF (3–5).
Over the past 10 years, attention has also been moving in an opposite direction, intriguingly posing the question as to whether injured myocardium might of itself trigger cancer development. Thus, cardio-oncology, the discipline that traditionally used to investigate radio and chemotherapy-induced cardiotoxicity, has crossed the orthodox boundaries with expansion of interest in the possibility of bidirectional cancer-heart communication (2, 6–10).
The causal relationship between HF and cancer has been a matter of investigation, and also controversial results, in epidemiologic and clinical studies. An elevated risk of incident cancer has been reported in some studies (11–14), raising the hypothesis that HF may predispose to cancer and that a common milieu characterized by a low-grade chronic inflammation could mediate both conditions. In search of proof of the principle of HF causing cancer, an experimental model of HF and cancer has been also developed (15).
In this review, we provide a critical reappraisal of clinical and experimental studies linking HF and cancer moving from an “oncological point of view” rather than from a “cardiological point of view,” where the latter has been the case in most previous reviews. We discuss the complex HF-associated inflammatory phenotype, the role of secreted cardiokines in generating a systemic inflammatory condition and the potential role and timing of HF-associated inflammation in the multistep process of carcinogenesis. Finally, we identify challenges for future investigation that will be aimed at unraveling possible causal effectors connecting HF and incident cancer, and possibly offer target therapies to reduce the risk of cancer in HF patients.
HF is a life-threatening clinical syndrome characterized by “inherited or acquired abnormality of cardiac structure and function, associated with a constellation of clinical symptoms (dyspnea and fatigue) and signs (edema and rales)” that often severely compromise the quality and duration of life (16). Guidelines from the North American and European cardiology societies classify HF based on the proportion of blood pumped from the ventricles, i.e., the ejection fraction (EF). Accordingly, they distinguish HF with reduced EF (HFrEF) and HF with preserved EF (HFpEF). HFrEF is mainly caused by coronary artery disease followed by myocardial infarction and loss of cardiomyocytes. In this disease the ventricular myocardium can no longer contract properly and EF is severely reduced. Conversely, HFpEF is promoted by multiple comorbidities, such as obesity, diabetes, atherosclerosis, and arterial hypertension. In HFpEF ventricular contractility is preserved, but the ventricular myocardium fails to relax during diastole and consequently filling pressure is increased (17).
In 2013, Hasin et al. (11) published a retrospective analysis with the first strong epidemiological evidence that patients with HFpEF and HFrEF had a higher likelihood of diagnosis of/or of dying from cancer compared with community controls. The study showed a 70% increase in cancer risk over a prolonged median follow-up of more than 5 years, a value that was maintained after correction for comorbidities and risk factors. In a second study on 5,000 patients with chronic HF cancer incidence was 4-fold higher than in control patients. Values positively correlated with circulating levels of brain natriuretic peptide (BNP), a marker of cardiac stress response and tissue damage (18). A large community study based on the Danish National Registries also reported that patients with HF, mainly as HFpEF, had a greater risk of incident cancer (significant incidence rate ratio of 1.24 for all major types of cancer except for prostate cancer) and a worse prognosis (13). While limited by its observational nature, leaving potential only for the generation of hypotheses, this study's strengths were the number of patients examined and the appropriate follow-up. In a prospective cohort-study, Hasin et al. (14) supported previous findings showing that patients who developed HF after myocardial infarction had an increased risk of incident cancer, with a hazard ratio for the association of 2.16, adjusted for age, sex, and comorbidities. An association between HF and incident cancer risk has been also confirmed in a recent pooled analysis (19).
Other clinical studies have questioned the causal relationship between HF and incident cancer providing opposite evidences. In a first post-hoc analysis (Physicians' Health Studies I and II) conducted on a large male population included in two controlled trials on the preventive effect of aspirin and vitamin supplement on incidence of cancer and HF, no correlation was found between the two diseases (20). Though supported by adequate sample size, follow-up and statistics, there were elements of weakness in the study concerned, such as HF diagnosis based on self-reported questionnaires rather than on established criteria, as well as its restriction to a male population. Two additional studies reported no causal relationship between HF and increased cancer risk. In the former study conducted on a large cohort of patients previously affected by myocardial infarction, the authors concluded that occult cancer and shared risk factors were likely responsible for the higher cancer incidence (21). In the second study, the authors attributed the slightly increased cancer risk to baseline comorbidities. Finally, a recent review and meta-analysis focusing on the association between myocardial infarction and cancer reported a 9.5% estimated cancer incidence after myocardial infarction, a value that attained statistical significance only in female patients, and that was substantially restricted to breast cancer (22).
On the basis of the aforementioned results it is clear that the impact of HF on cancer incidence is a complicated issue still open to investigation (23), hopefully through prospective clinical trials with large samples and long-term follow-up. The following biases need to be carefully considered before designing new clinical studies.
1. COMMON RISK FACTORS. The association between HF and cancer could depend on common risk factors, since many patients enrolled in the clinical studies had several comorbidities, such as obesity, diabetes, or hypertension. In addition, smoking and alcohol consumption may have contributed to a confounding of the results. Even though previous epidemiological studies suggest that the association is maintained after data correction for the main risk factors, this key point needs to be duly weighed in well-designed prospective studies. These studies could, at the same time, clarify whether cancer risk in HF patients is generalized or is restricted to specific cancer subtypes.
2. DETECTION BIAS. The association between HF and cancer may be due to the so-called detection bias, i.e., the closest clinical-instrumental follow-up applied to HF patients. A preliminary answer to this question emerges from studies showing that the highest incidence of cancer was recorded well-beyond the first year after diagnosis of HF, which is generally the period of greatest intensity of cardiology controls (11, 14, 20).
3. CARDIOLOGIC THERAPIES. The potential carcinogenic effect of some cardiologic therapies for HF has also been called into question. Treatment of hypertensive patients with Telmisartan, an angiotensin-receptor blocking drug, was found to be associated with a modestly raised risk of new cancer, and in particular lung carcinomas (24–26). Regarding statins, the inhibitors of 3-hydroxy-3-methyl-glutaryl-coenzyme A (HMG-CoA) and cholesterol synthesis, their carcinogenic risk remains controversial (27). Initial studies in the late 1990s reported an increased cancer-related mortality in patients treated with statins (28). However, further studies did not confirm these data (29), indeed preclinical research demonstrated that statins inhibit tumor growth, induce apoptosis in some cancer types and have a general anti-inflammatory and anti-oxidant protective effect (30).
4. DIAGNOSTIC PROCEDURES of HF and CANCER RISK. HF patients undergo several diagnostic procedures with exposure to ionizing radiations and potential risk of cancer. For instance, the risk associated with coronary angiography is not negligible and justifies adoption of alternative diagnostic procedures (31). Electrocardiogram (ECG)-gated computed tomography angiography (CTA), in the form of prospective analysis, represents an alternative option for radiation safety. Prospective gating only acquires images during the cardiac diastole, instead of throughout the entire cardiac cycle like retrospective gating. This procedure allows high image quality and reduction of the effective radiation dose by up to 90%, thus lowering the cancer induction risk in sensitive organs to 0.13% (32).
Extensive evidence has shown that HF is associated with a chronic inflammatory state and activation of immune response. Inflammation arises locally as a consequence of myocardial injury in HFrEF, and systemically as a consequence of various comorbidities in HFpEF (33, 34) (Figure 1A). In HFrEF, dying or stressed cells release damage-associated molecular patterns (DAMPS), which elicit a robust inflammatory and immune response. Inflammatory cells and also cardiomyocytes, endothelial cells, and cardiac fibroblasts start producing a variety of cytokines—including IL-1β, IL-6, TNF-α, IL-8, IL-18, IL-33—chemokines, and proteins of the pro-inflammatory signaling pathways. Inflammatory mediators blunt responsiveness of cardiomyocytes at multiple levels, decreasing sarcoplasmic reticulum calcium load, and depressing contractility, reducing ATP synthesis, increasing production of oxygen (ROS) and nitrogen (RNS) reactive species (35) and reorienting metabolism from oxidative to glycolytic (so-called metabolic remodeling) (36). Over the weeks, cardiac inflammation exits in repair and tissue remodeling, but inflammation may inexplicably become chronic in some patients, and exacerbate myocardial damage. Given the detrimental effects of an excessive and persistent pro-inflammatory response in HFrEF, therapeutic strategies for attenuating the initial pro-inflammatory response are underway.
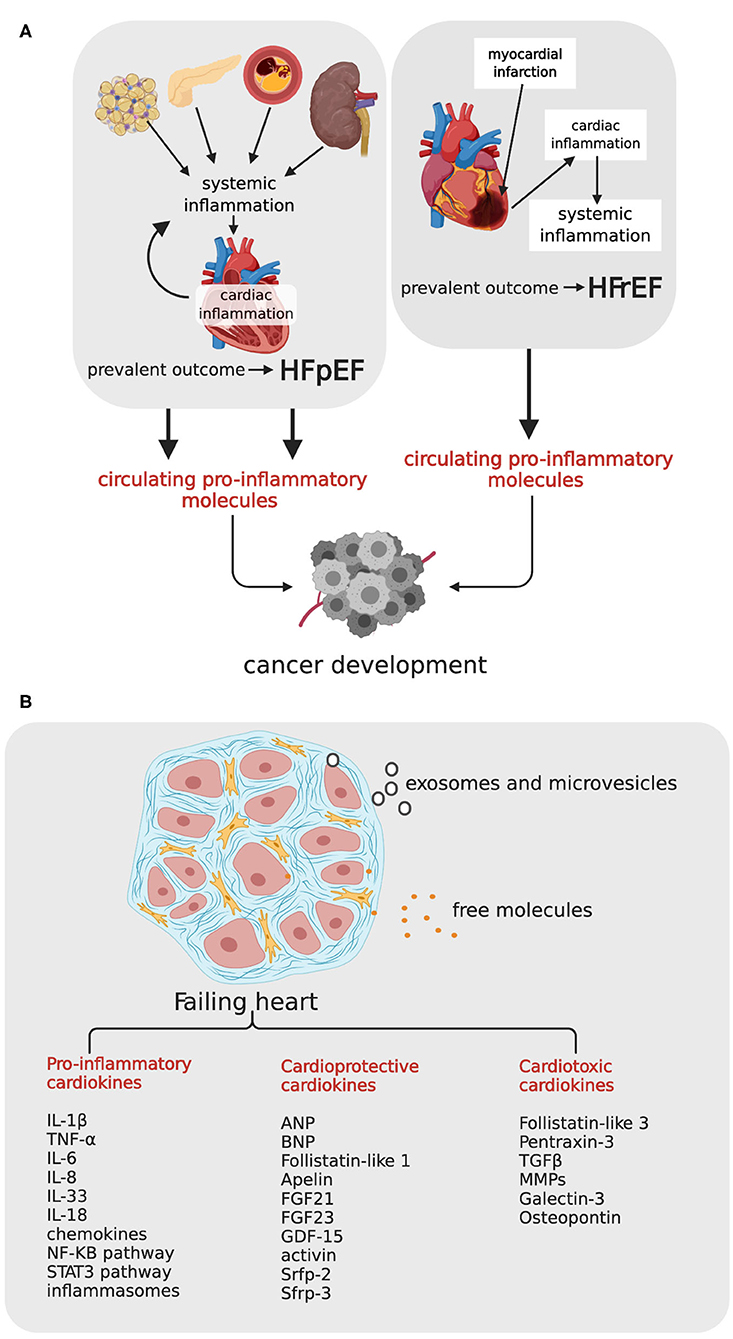
Figure 1. Heart failure, cardiokines, and relationship with incident cancer. (A) HFrEF and HFpEF can potentially promote carcinogenesis by means of circulating pro-inflammatory molecules. In HFrEF consequent to myocardial infarction (right side), heart damage activates cardiac inflammation, which occasionally induces a chronic systemic inflammation and a persistent tissue damage; in HFpEF (left side), comorbidities like obesity, diabetes, atherosclerosis and hypertension, induce a systemic inflammatory state, with consequent cardiac inflammation, and functional impairment. HFpEF, Hear Failure with Preserved Ejection Fraction; HFrEF, Heart Failure with reduced Ejection Fraction. (B) The failing heart, schematically represented as a myocardial tissue section with cardiomyocytes surrounded by fibrotic matrix, secretes a variety of cardiokines, either as free molecules or as small microvesicles and exosomes. TNF-α, tumor necrosis factor-α; IL-1β, interleuchin-1β; IL-6, interleuchin-6; IL-33, interleuchin-33; IL-17, interleuchin-17; MMPs, matrix metalloproteases; TGF-β, transforming growth factor-β; ANP, Atrial Natriuretic Peptide; BNP, Brain Natriuretic Peptide; FGF21, fibroblasts growth factor 21, FGF23, fibroblasts growth factor 23; GDF-15, TGF-β related family members growth differentiation factor-15, Srfp-2 and Srfp-3, secreted frizzled related proteins 2 and 3. Created with Biorender.com.
In HFpEF, inflammation drives the development of HF through another sequence of events (37). Comorbidities induce a systemic pro-inflammatory state. Myocardial disease begins with coronary endothelial dysfunction and expression of adhesion molecules VCAMs and selectins, and attraction of infiltrating leukocytes. Endothelial cells start producing ROS, and ROS trigger a negative cascade of events with downregulation of nitric oxide (NO) production, decreased NO-stimulated cyclic guanosine monophosphate (cGMP) levels, and decreased protein kinase G (PKG) activity. This sequence culminates in PKG-dependent hypophosphorylation of titin, the protein that regulates cardiomyocyte stiffness, depending on its phosphorylation state. Stiff cardiomyocytes, in association with interstitial fibrosis and capillary rarefaction, contribute to impaired diastolic relaxation, a hallmark of HFpEF (38). Thus, oxidative stress and inflammation, whether the consequence or the cause of HF, generate a chronic systemic condition with adverse clinical outcome. Although low-grade chronic systemic inflammation is often clinically silent, its consequences may also increase the risk of different cancers. In line with these observation, genetic polymorphisms in genes encoding IL-1β, IL-6, IL-8, and anti-inflammatory IL-10, have been shown to predispose affected individuals to cancer (39).
Like other organs, the heart secretes a variety of molecules in response to metabolic and hemodynamic stress. The “cardiac secretome” includes peptides, proteins and coding, and non-coding RNAs released either as free molecules or as microvesicles and exosomes. The expression profile of secreted peptides and proteins—called cardiokines—changes considerably under pathological conditions (40–43). Cardiokines produced by the failing heart include primary citokines and chemokines of inflammation (mostly with detrimental effects for cardiomyocytes), and also proteins involved in stress response, regulation of apoptosis, tissue repair, cardiac remodeling, and angiogenesis (Figure 1B). A large number of cardiokines are functionally intended to maintain cardiac homeostasis and are therefore cardioprotective. This group includes, among others, atrial natriuretic peptide (ANP) and BNP (18), fibroblasts growth factor 21 (FGF21) (44) and 23 (FGF23) (45), TGF-β related family members growth differentiation factor (GDF)-15 (46, 47), activin (48), secreted frizzled related proteins 2 (Srfp-2) (49, 50) and 3 (Srfp-3) (51, 52), and follistatin-like protein-1 (53). Other cardiokines, such as pro-inflammatory follistatin-like protein-3 (54), pentraxin 3 (55), TGF-β matrix metalloproteases (MMPs) (56), as well as other proteins involved in extracellular matrix degradation and remodeling, are cardiotoxic. Up-regulation of these proteins in HF patients is predictive of an adverse outcome.
Since the discovery of natriuretic peptides, it has become evident that cardiokines have a great influence in altering the homeostasis of distant tissues. To test the hypothesis that cardiokines released by the failing heart could induce carcinogenesis Meijers et al. induced myocardial infarction in APCmin mice, a murine model genetically predisposed to develop intestinal polyposis (15). Mice developed more than twice the tumor load as compared to sham-operated controls. To rule out hemodynamic impairment as a causative mechanism for increased tumors, the investigators heterotopically transplanted an additional heart, with previously induced acute myocardial infarction, in the same APCmin mouse strain. Tumor load was increased in the presence of an infarcted transplanted heart, but not with an intact heart, indicating that the pro-carcinogenic effect is due to the failing heart. Using in silico analysis, Meijers et al. identified five potential circulating factors responsible for polyposis, namely Fibronectin, SerpinA3, SerpinA1, Ceruloplasmin, and Paraonase1. The same factors were also up-regulated in HF patients enrolled in the PREVEND study (Prevention of Renal and Vascular End-Stage Disease), together with inflammatory molecules (C-reactive protein, pro-endothelin, pro-calcitonin, and pro-adrenomedullin) and cardioprotective ANP and BNP. Of the five markers identified in APCminmice, only SerpinA3 and SerpinA1 were able to stimulate proliferation of colon cancer cell line HT-29 in vitro and no test in vivo was performed to confirm the results. Thus, Maijers study did not provide conclusive evidence that the identified cardiokines were the true effectors of tumorigenesis.
Other cardiokines with high expression in HF patients have shown tumorigenic potential in previous studies, and are therefore candidates for further investigation. Apelin, a ligand of the G protein-coupled receptor APJ (57), which reduces oxidative stress and prevents cardiac hypertrophy in HF (58), stimulates proliferation of cancer cells in cholangiocarcinoma (CAA) (59), non-small cell lung cancer (NSCLC) (60), gastric cancer (61), prostate cancer (62), ovarian cancer (63), and oral squamous cell carcinoma (64). Similarly, Galectin-3, a carbohydrate-binding protein with pro-inflammatory and pro-fibrotic function in the heart (65), stimulates tumor progression and metastatization (66), and osteopontin, a glycoprotein highly expressed in the post-ischemic heart (67) stimulates primary tumor cell proliferation, angiogenesis, and epithelial- mesenchymal transition (EMT) (68).
More than 100 years ago, Virchow emphasized the association between chronic inflammation and the onset of cancer, starting from the histopathological observation that tumors arise from sites with dense “lymphoreticular infiltration” (69). Association of infection, inflammation and cancer has been reported for a variety of clinical conditions, including inflammatory bowel disease for colorectal cancer, Hepatitis B and C for hepatocarcinoma, schistosoma–induced bladder and colorectal cancer, and Helicobacter pylori infection for gastric mucosa-associated lymphoid tissue lymphoma. In addition, environmental and lifestyle factors that predispose to local inflammation, such as inhaled fine particles, asbestos, tobacco smoke, and alcohol consumption, have been shown to induce cancer (70).
The concept of inflammation as a hallmark of cancer (71, 72) has been proposed for local inflammation and more recently also for systemic inflammation associated with a variety of chronic diseases (73, 74). For instance, metabolic syndrome, a cluster of disorders including obesity, hypertension, dyslipidemia and insulin resistance, predisposes to cancer development, mainly through activation of a chronic inflammatory state sustained by adipose cell-released cytokines (adipokines). Adipokines and locally generated ROS prime both premalignant cells and the microenvironment to facilitate oncogenic transformation (75, 76). In addition to acting locally, adipokines, and the chronic inflammatory microenvironment also mediate crosstalk with distant tissues, thus reflecting the epidemiological evidence that obesity is significantly associated with specific tumor subtypes, mainly breast and pancreas cancer (77).
The hypothesis that a low-grade chronic inflammation has carcinogenic potential has raised the question of whether anti-inflammatory therapies may reduce the risk of cancer. In the CANTOS trial (Canakinumab Anti-inflammatory Thrombosis Outcome Study), administration of IL-1β-targeting antibody Canakinumab reduced HF-related hospitalization and mortality in patients with previous myocardial infarction (78). In a retrospective analysis, the same authors found a marked reduction in the hazard ratio of lung cancer incidence in patients treated with 150 mg and 300 mg of Canakinumab, compared with placebo controls (79). This finding opened a new research path aimed at investigating the therapeutic potential of anti-inflammatory drugs in cancer patients. Conventional non-steroid anti-inflammatory drugs (NSAIDs), including aspirin, have been also tested. Numerous studies observed that people who regularly take low doses of aspirin may have reduced risks of being diagnosed with or dying from cancer (80). Controlled clinical trials with patients treated with aspirin vs. placebo suggested a long-term risk reduction in the onset of any neoplasm, particularly of gastric, esophageal, colorectal, pancreatic, ovarian, endometrial, breast and prostate cancer, and small intestine neuroendocrine tumors (81, 82). However, the very recent ASPREE study demonstrated that in older adults aspirin had an adverse effect on later stages of cancer evolution, with a higher incidence of metastatic cancers and a higher risk of death (83). These data indicate that it is necessary to remain cautious in defining the preventive anti-cancer effect of aspirin for wide application.
Although results obtained in the CANTOS study and in the experimental model described by Meijers et al. (15) will require further validation, they undoubtedly have the merit of raising the crucial question of whether cardiokines released by the failing heart promote carcinogenesis and if they do what are the mechanisms. The experimental APCmin mouse model is already predisposed to polyposis because it carries a truncating mutation at codon 850 of the Adenomatous polyposis coli (Apc) gene. Mice with heterozygous mutation develop ~30 small intestinal polyps that progressively cause colon obstruction and death in mice within 3 months. Occasionally, polyps progress to adenocarcinoma, and never to metastatic disease (84). One might therefore argue that myocardial infarction and the consequent inflammatory state contribute to speed up a process that would take place in any case. This apparent limitation could nonetheless shed light on what is the most plausible pathogenetic mechanism linking HF and incident cancer (Figure 2). In a low-grade chronic inflammatory state, a large number of molecular pro-inflammatory mediators and inflammatory cells circulate in the bloodstream. Inflammation produce ROS and RNS, which are powerful mutagens and can cause DNA double strand breaks and genomic instability (85, 86). When tumor initiation is nor preceded by systemic inflammation, as it is in APCmin mice, systemic inflammation can nonetheless drive cell proliferation and accelerate carcinogenesis (87). Once generated, the tumor bulk recruits its own inflammatory milieu, which is in continuous cross-talk with HF-associated inflammation, so that over time it is no longer possible to distinguish the effects of one or the other. Inflammation contributes to tumor promotion by editing the tumor microenvironment. Low levels of IL-1β, TNF-α, and IL-6 stimulate angiogenesis, and IL-6 modulates polarization of tumor-associated macrophages (TAM) from M1 (a tumor-eliminating phenotype) to M2 (an anti-inflammatory and tumor-promoting phenotype) (88, 89). Inflammation also regulates cancer stem cells (CSCs) cell cycle and epithelial-mesenchymal transition (EMT), two major events in tumor progression. CSCs, namely those rare immortal cells of the tumor bulk with the capacity for self-renewal, multipotency, tumorigenicity, and quiescence (90), proliferate in response to activation of the NF-kB and STAT3 pathways (91), and EMT, a prerequisite for invasiveness, is favored by exposure of tumor cells to TNF-α, IL-6, and TGF-β (92, 93). Systemic inflammation also reactivates quiescent cancer cells. Experimental studies have proved that sustained lung inflammation and the accompanying formation of neutrophil extracellular traps (NETs) convert dormant cancer cells to aggressive lung metastases through NET-mediated remodeling of the extracellular matrix (94). Finally, intravasation and extravasation of cancer cells during metastatic spread is favored by endothelial dysfunction and expression of adhesion molecules for leucocytes, a process sustained by pro-inflammatory cytokines. HF-associated inflammation thus can play an instrumental role in the control of all steps of carcinogenesis and support the causal relationship between HF and cancer.
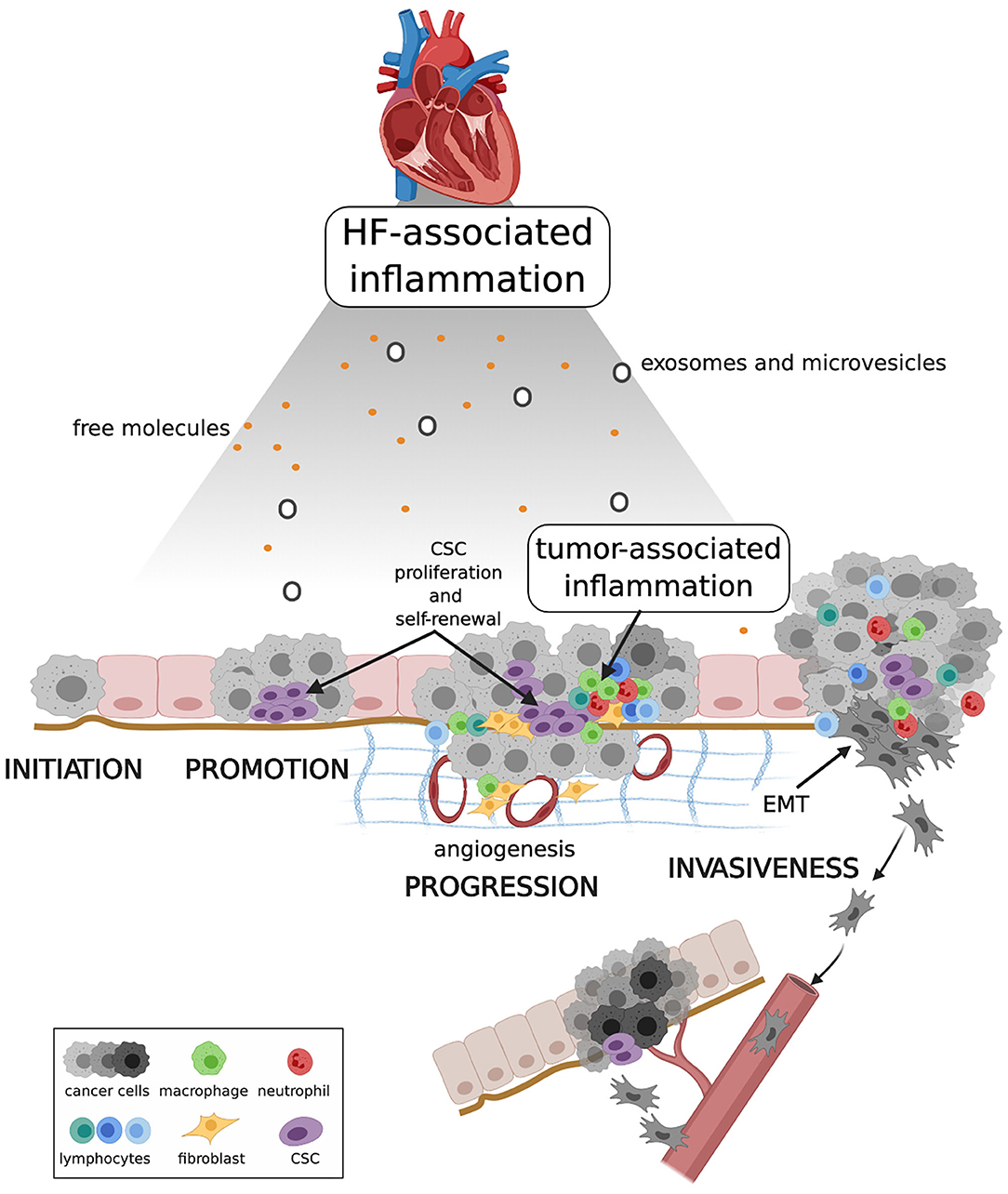
Figure 2. Proposed mechanism linking HF, systemic inflammation and carcinogenesis. HF-associated inflammation can have a direct effect on cancer initiation, and on pre-malignant cell proliferation and resistance to death during cancer promotion. HF-associated inflammation, together with tumor-associated inflammation, shapes the tumor microenvironment, enhancing CSC proliferation and self-renewal, and stimulating angiogenesis and EMT. Collectively these events contribute to tumor progression and invasiveness. CSC, cancer stem cell; EMT, epithelial-mesenchymal transition. Created with Biorender.com.
A number of epidemiological and clinical studies have reported that patients with HF have a higher risk of developing cancer and a chronic low-grade systemic inflammation has been proposed as the major pathophysiological process linking HF and carcinogenesis. The relationship between HF, systemic inflammation, and cancer is complicated and a unifying picture is still lacking (23). In particular, there are outstanding questions that cardiologists and oncologists have yet to answer through cooperative action. Are all HF variants, including the majority of HFpEF, prone to develop incident cancer? Most clinical studies describe HF consequent to myocardial infarction, but the contribution of other HF variants remains largely unexplored. Another key question concerns HF-associated inflammation. Do we deal with an organ-specific and disease-specific inflammation or is it on a par with any other inflammation in its carcinogenic potential? Which cardiokines secreted by the failing heart truly promote carcinogenesis and are therefore worse prognostic? To address these issues a more detailed characterization of the inflammatory profile of cardiokines in various HFrEF and HFpEF is eagerly awaited. In addition, mouse models establishing the carcinogenic potential of specific cardiokines in vivo are essential. A large number of transgenic mice with genetically-induced cardiomyopathies have been characterized, some of which develop progressive HF in a reasonably large window of time that permits cancer development, either genetically-induced or consequent to tumor cell injection (95). These models should be considered when designing future experiments.
For the perspective of preventing cancer in HF patients, cardio-oncologists should consider the possibility of identifying a targetable “cardio-inflammatory” phenotype in HF patients. This approach has important clinical implications in terms of patient stratification, risk assessment, and the determination of specific therapeutic algorithms. So far, therapeutic agents used to manipulate inflammation in patients with HF and cancer have achieved modest results, possibly because these agents may be more effective in preventing cancer or in treating initial cancer, rather than advanced cancer. Targeting IL-1β with Canakinumab opened a new era in the use of selective anti-inflammatory therapies in cardiovascular diseases, but its potential in cancer prevention needs to be further explored. Anti-inflammatory therapies affect immunosuppression and modulation of immunosuppression is one of the most promising therapeutic strategies for cancer (96). Thus, the use of anti-inflammatory or anti-immune agents, alone or in combination, represent a logical focus of therapeutic intervention to establish whether combating inflammation truly reduces the risk of cancer in HF patients.
SA contributed to the conceptualization, writing and editing of the manuscript, and to illustration design. GA contributed to the conceptualization and writing of the manuscript. All authors approved the submitted version.
The paper was funded by the Italian Ministry of Education, University and Research (DOR 2019-2020 to SA) and AVAPO (Volunteers Association for Cancer Patients Assistance-ONLUS Dolo-Mirano (Venice) to GA.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors thank Linguexecutive (VR, Italy) for English language editing, and Marco Ardina for technical assistance.
1. Zamorano JL, Lancellotti P, Rodriguez Munoz D, Aboyans V, Asteggiano R, Galderisi M, et al. 2016 ESC Position Paper on cancer treatments cardiovascular toxicity developed under the auspices of the ESC Committee for Practice Guidelines: the task force for cancer treatments cardiovascular toxicity of the European Society of Cardiology (ESC. Eur J Hear Fail. (2017) 19:9–42. doi: 10.1002/ejhf.654
2. Bertero E, Canepa M, Maack C, Ameri P. Linking heart failure to cancer. Circulation. (2018) 138:735–42. doi: 10.1161/CIRCULATIONAHA.118.033603
3. Anker SD, Sharma R. The syndrome of cardiac cachexia. Int J Cardiol. (2002) 85:51–66. doi: 10.1016/S0167-5273(02)00233-4
4. Anker MS, von Haehling S, Landmesser U, Coats AJS, Anker SD. Cancer and heart failure-more than meets the eye: common risk factors and co-morbidities. Eur J Hear Fail. (2018) 20:1382–4. doi: 10.1002/ejhf.1252
5. Ausoni S, Calamelli S, Saccà S, Azzarello G. How progressive cancer endangers the heart: an intriguing and underestimated problem. Cancer Metastasis Rev. (2020) 39:535–52. doi: 10.1007/s10555-020-09869-8
6. Kazemi-Bajestani SM, Becher H, Fassbender K, Chu Q, Baracos VE. Concurrent evolution of cancer cachexia and heart failure: bilateral effects exist. J Cachexia Sarcopenia Muscle. (2014) 5:95–104. doi: 10.1007/s13539-014-0137-y
7. Bertero E, Ameri P, Maack C. Bidirectional relationship between cancer and heart failure: old and new issues in cardio-oncology. Card Fail Rev. (2019) 5:106–11. doi: 10.15420/cfr.2019.1.2
8. de Boer RA, Meijers WC, van der Meer P, van Veldhuisen DJ. Cancer and heart disease: associations and relations. Eur J Heart Fail. (2019) 21:1515–25. doi: 10.1002/ejhf.1539
9. Kitsis RN, Riquelme JA, Lavandero S. Heart disease and cancer. Circulation. (2018) 138:692–95. doi: 10.1161/CIRCULATIONAHA.118.033907
10. Richards AM. Can heart failure cause cancer? Nat Rev Cardiol. (2019) 3:66–70. doi: 10.1038/s41569-018-0105-x
11. Hasin T, Gerber Y, McNallan SM, Weston SA, Kushwaha SS, Nelson TJ, et al. Patients with heart failure have an increased risk of incident cancer. J Am Coll Cardiol. (2013) 62:881–6. doi: 10.1016/j.jacc.2013.04.088
12. Sakamoto M, Hasegawa T, Asakura M, Kanzaki H, Takahama H, Amaki M, et al. Does the pathophysiology of heart failure prime the incidence of cancer? Hypertens Res. (2017) 40:831–6. doi: 10.1038/hr.2017.45
13. Banke A, Schou M, Videbæk L, Møller JE, Torp-Pedersen C, Gustafsson F, et al. Incidence of cancer in patients with chronic heart failure: a long-term follow-up study. Eur J Heart Fail. (2016) 18:260–6. doi: 10.1002/ejhf.472
14. Hasin T, Gerber Y, Weston SA, Jiang R, Killian JM, Manemann SM, et al. Heart failure after myocardial infarction is associated with increased risk of cancer. J Am Coll Cardiol. (2016) 68:265–71. doi: 10.1016/j.jacc.2016.04.053
15. Meijers WC, Maglione M, Bakker SJL, Oberhuber R, Kieneker LM, De Jong S, et al. Heart failure stimulates tumor growth by circulating factors. Circulation. (2018) 138:678–91. doi: 10.1161/CIRCULATIONAHA.117.030816
17. Ponikowski P, Anker SD, AlHabib KF, Cowie MR, Force TL, Hu S, et al. Heart failure: preventing disease and death worldwide. ESC Hear Fail. (2014) 1:4–25. doi: 10.1002/ehf2.12005
18. Fu S, Ping P, Wang F, Luo L. Synthesis, secretion, function, metabolism and application of natriuretic peptides in heart failure. J Biol Eng. (2018) 12:2. doi: 10.1186/s13036-017-0093-0
19. Zhang H, Gao Y, Wang L, Tian L, An N, Yang X, et al. Does heart failure increase the risk of incident cancer? A meta-analysis and systematic review. Heart Fail Rev. (2019) doi: 10.1007/s10741-019-09876-0
20. Selvaraj S, Bhatt DL, Claggett B, Djoussé L, Shah SJ, Chen J, et al. Lack of association between heart failure and incident cancer. J Am Coll Cardiol. (2018) 71:1501–10. doi: 10.1016/j.jacc.2018.01.069
21. Rinde LB, Småbrekke B, Hald EM, Brodin EE, Njølstad I, Mathiesen EB, et al. Myocardial infarction and future risk of cancer in the general population—the tromsø study. Eur J Epidemiol. (2017) 32:193–201. doi: 10.1007/s10654-017-0231-5
22. Li N, Huang Z, Zhang Y, Sun H, Wang J, Zhao J. Increased cancer risk after myocardial infarction: factor fiction? A systemic review and meta-analysis. Cancer Manag Res. (2019) 11:1959–68. doi: 10.2147/CMAR.S193658
23. Boffetta P, Malhotra J. Impact of heart failure on cancer incidence: a complicated question. J Am Coll Cardiol. (2018) 71:1511–12. doi: 10.1016/j.jacc.2018.02.015
24. Sipahi I, Debanne SM, Rowland DY, Simon DI, Fang JC. Angiotensin-receptor blockade and risk of cancer: meta-analysis of randomised controlled trials. Lancet Oncol. (2010) 11:627–36. doi: 10.1016/S1470-2045(10)70106-6
25. Teo KK. Effects of telmisartan, irbesartan, valsartan, candesartan, and losartan on cancers in 15 trials enrolling 138 769 individuals. J Hypertens. (2011) 29:623–35. doi: 10.1097/HJH.0b013e328344a7de
26. Bangalore S, Kumar S, Kjeldsen SE, Makani H, Grossman E, Wetterslev J, et al. Antihypertensive drugs and risk of cancer: network meta-analyses and trial sequential analyses of 324 168 participants from randomised trials. Lancet Oncol. (2011) 12:65–82. doi: 10.1016/S1470-2045(10)70260-6
27. Zaleska M, Mozenska O, Bil J. Statins use and cancer: an update. Futur Oncol. (2018) 14:1497–509. doi: 10.2217/fon-2017-0543
28. Jacobs DR, Blackburn H, Higgins M, Reed D, Iso H, McMillan G, et al. Report of the conference on low blood cholesterol: mortality associations. Circulation. (1992) 86:1046–60. doi: 10.1161/01.cir.86.3.1046
29. Bonovas S, Filioussi K, Tsavaris N, Sitaras NM. Statins and cancer risk: a literature-based meta-analysis and meta-regression analysis of 35 randomized controlled trials. J Clin Oncol. (2006) 24:4808–17. doi: 10.1200/JCO.2006.06.3560
30. Altwairgi AK. Statins are potential anticancerous agents (Review). Oncol Rep. (2015) 33:1019–39. doi: 10.3892/or.2015.3741
31. Huang B, Li J, Law MWM, Zhang J, Shen Y, Khong PL. Radiation dose and cancer risk in retrospectively and prospectively ECG-gated coronary angiography using 64-slice multidetector CT. Br J Radiol. (2010) 83:152–8. doi: 10.1259/bjr/29879495
32. Mahmoodi M, Chaparian A. Organ doses, effective dose, and cancer risk from coronary CT angiography examinations. Am J Roentgenol. (2020) 214:1131–6. doi: 10.2214/AJR.19.21749
33. Schiattarella GG, Sequeira V, Ameri P. Distinctive patterns of inflammation across the heart failure syndrome. Heart Fail Rev. (2020) doi: 10.1007/s10741-020-09949-5. [Epub ahead of print].
34. Adamo L, Rocha-Resende C, Prabhu SD, Mann DL. Reappraising the role of inflammation in heart failure. Nat Rev Cardiol. (2020) 17:269–85. doi: 10.1038/s41569-019-0315-x
35. Münzel T, Camici GG, Maack C, Bonetti NR, Fuster V, Kovacic JC. Impact of oxidative stress on the heart and vasculature. J Am Coll Cardiol. (2017) 70:212–29. doi: 10.1016/j.jacc.2017.05.035
36. Bertero E, Maack C. Metabolic remodelling in heart failure. Nat Rev Cardiol. (2018) 15:457–70. doi: 10.1038/s41569-018-0116-7
37. Paulus WJ, Tschöpe C. A novel paradigm for heart failure with preserved ejection fraction. J Am Coll Cardiol. (2013) 23:263–71. doi: 10.1016/j.jacc.2013.02.092
38. Sharma K, Kass DA. Heart failure with preserved ejection fraction: mechanisms, clinical features, and therapies. Circ Res. (2014) 115:79–86. doi: 10.1161/CIRCRESAHA.115.302922
39. Michaud DS, Daugherty SE, Berndt SI, Platz EA, Yeager M, Crawford ED, et al. Genetic polymorphisms of interleukin-1B (IL-1B), IL-6, IL-8, and IL-10 and risk of prostate cancer. Cancer Res. (2006) 66:4525–30. doi: 10.1158/0008-5472.CAN-05-3987
40. Shimano M, Ouchi N, Walsh K. Cardiokines: recent progress in elucidating the cardiac secretome. Circulation. (2012) 126:e327–32. doi: 10.1161/CIRCULATIONAHA.112.150656
41. Wu YS, Zhu B, Luo AL, Yang L, Yang C. The role of cardiokines in heart diseases: beneficial or detrimental? Biomed Res Int. (2018) 2018:8207058. doi: 10.1155/2018/8207058
42. Doroudgar S, Glembotski CC. The cardiokine story unfolds: ischemic stress-induced protein secretion in the heart. Trends Mol Med. (2011) 17:207–14. doi: 10.1016/j.molmed.2010.12.003
43. Yu H, Wang Z. Cardiomyocyte-derived exosomes: biological functions and potential therapeutic implications. Front Physiol. (2019) 10:1049. doi: 10.3389/fphys.2019.01049
44. Patel V, Adya R, Chen J, Ramanjaneya M, Bari MF, Bhudia SK, et al. Novel insights into the cardio-protective effects of FGF21 in lean and obese rat hearts. PLoS ONE. (2014) 9:e87102. doi: 10.1371/journal.pone.0087102
45. Faul C, Amaral AP, Oskouei B, Hu MC, Sloan A, Isakova T, et al. FGF23 induces left ventricular hypertrophy. J Clin Invest. (2011) 121:4393–408. doi: 10.1172/JCI46122
46. Kempf T, Eden M, Strelau J, Naguib M, Willenbockel C, Tongers J, et al. The transforming growth factor-β superfamily member growth-differentiation factor-15 protects the heart from ischemia/reperfusion injury. Circ Res. (2006) 98:351–60. doi: 10.1161/01.RES.0000202805.73038.48
47. Kempf T, Zarbock A, Widera C, Butz S, Stadtmann A, Rossaint J, et al. GDF-15 is an inhibitor of leukocyte integrin activation required for survival after myocardial infarction in mice. Nat Med. (2011) 17:581–8. doi: 10.1038/nm.2354
48. Oshima Y, Ouchi N, Shimano M, Pimentel DR, Papanicolaou KN, Panse KD, et al. Activin a and follistatin-like 3 determine the susceptibility of heart to ischemic injury. Circulation. (2009) 120:1606–15. doi: 10.1161/CIRCULATIONAHA.109.872200
49. Kobayashi K, Luo M, Zhang Y, Wilkes DC, Ge G, Grieskamp T, et al. Secreted Frizzled-related protein 2 is a procollagen C proteinase enhancer with a role in fibrosis associated with myocardial infarction. Nat Cell Biol. (2009) 11:46–55. doi: 10.1038/ncb1811
50. He W, Zhang L, Ni A, Zhang Z, Mirotsou M, Mao L, et al. Exogenously administered secreted frizzled related protein 2 (Sfrp2) reduces fibrosis and improves cardiac function in a rat model of myocardial infarction. Proc Natl Acad Sci USA. (2010) 107:21110–5. doi: 10.1073/pnas.1004708107
51. Askevold ET, Aukrust P, Nymo SH, Lunde IG, Kaasbøll OJ, Aakhus S, et al. The cardiokine secreted Frizzled-related protein 3, a modulator of Wnt signalling, in clinical and experimental heart failure. J Intern Med. (2014) 275:621–30. doi: 10.1111/joim.12175
52. Ueland T, Caidahl K, Askevold ET, Karlsson T, Hartford M, Aukrust P. Secreted frizzled-related protein 3 (sFRP3) in acute coronary syndromes. Int J Cardiol. (2015) 190:217–9. doi: 10.1016/j.ijcard.2015.03.401
53. Uematsu M, Nakamura K, Nakamura T, Watanabe Y, Yoshizaki T, Deyama J, et al. Persistent myocardial production of Follistatin-like 1 is associated with left ventricular adverse remodeling in patients with myocardial infarction. J Card Fail. (2020) 26:733–8. doi: 10.1016/j.cardfail.2020.05.015
54. Chaly Y, Hostager B, Smith S, Hirsch R. Follistatin-like protein 1 and its role in inflammation and inflammatory diseases. Immunol Res. (2014) 59:266–72. doi: 10.1007/s12026-014-8526-z
55. Matsubara J, Sugiyama S, Nozaki T, Sugamura K, Konishi M, Ohba K, et al. Pentraxin 3 is a new inflammatory marker correlated with left ventricular diastolic dysfunction and heart failure with normal ejection fraction. J Am Coll Cardiol. (2011) 57:861–9. doi: 10.1016/j.jacc.2010.10.018
56. Lindsey ML. Assigning matrix metalloproteinase roles in ischaemic cardiac remodelling. Nat Rev Cardiol. (2018) 15:471–9. doi: 10.1038/s41569-018-0022-z
57. Tatemoto K, Hosoya M, Habata Y, Fujii R, Kakegawa T, Zou MX, et al. Isolation and characterization of a novel endogenous peptide ligand for the human APJ receptor. Biochem Biophys Res Commun. (1998) 251:471–6. doi: 10.1006/bbrc.1998.9489
58. Foussal C, Lairez O, Calise D, Pathak A, Guilbeau-Frugier C, Valet P, et al. Activation of catalase by apelin prevents oxidative stress-linked cardiac hypertrophy. FEBS Lett. (2010) 584:2363–70. doi: 10.1016/j.febslet.2010.04.025
59. Hall C, Ehrlich L, Venter J, O'Brien A, White T, Zhou T, et al. Inhibition of the apelin/apelin receptor axis decreases cholangiocarcinoma growth. Cancer Lett. (2017) 386:179–88. doi: 10.1016/j.canlet.2016.11.025
60. Berta J, Kenessey I, Dobos J, Tovari J, Klepetko W, Jan Ankersmit H, et al. Apelin expression in human non-small cell lung cancer: role in angiogenesis and prognosis. J Thorac Oncol. (2010) 5:1120–9. doi: 10.1097/JTO.0b013e3181e2c1ff
61. Feng M, Yao G, Yu H, Qing Y, Wang K. Tumor apelin, not serum apelin, is associated with the clinical features and prognosis of gastric cancer. BMC Cancer. (2016) 16:794. doi: 10.1186/s12885-016-2815-y
62. Tekin S, Sandal S, Colak C. Effects of apelin-13 on human prostate cancer lines [insan prostat kanseri hucre serilerinde apelin-13'un etkileri]. Med Sci | Int Med J. (2014) 3:1427–41. doi: 10.5455/medscience.2014.03.8143
63. Hoffmann M, Fiedor E, Ptak A. Bisphenol A and its derivatives tetrabromobisphenol A and tetrachlorobisphenol A induce apelin expression and secretion in ovarian cancer cells through a peroxisome proliferator-activated receptor gamma-dependent mechanism. Toxicol Lett. (2017) 269:15–22. doi: 10.1016/j.toxlet.2017.01.006
64. Heo K, Kim YH, Sung HJ, Li HY, Yoo CW, Kim JY, et al. Hypoxia-induced up-regulation of apelin is associated with a poor prognosis in oral squamous cell carcinoma patients. Oral Oncol. (2012) 48:500–6. doi: 10.1016/j.oraloncology.2011.12.015
65. Besler C, Lang D, Urban D, Rommel KP, Von Roeder M, Fengler K, et al. Plasma and cardiac galectin-3 in patients with heart failure reflects both inflammation and fibrosis: Implications for its use as a biomarker. Circ Hear Fail. (2017) 10:e003804. doi: 10.1161/CIRCHEARTFAILURE.116.003804
66. Liu FT, Rabinovich GA. Galectins as modulators of tumour progression. Nat Rev Cancer. (2005) 5:29–41. doi: 10.1038/nrc1527
67. Scatena M, Liaw L, Giachelli CM. Osteopontin: a multifunctional molecule regulating chronic inflammation and vascular disease. Arterioscler Thromb Vasc Biol. (2007) 27:2302–9. doi: 10.1161/ATVBAHA.107.144824
68. Zhao H, Chen Q, Alam A, Cui J, Suen KC, Soo AP, et al. The role of osteopontin in the progression of solid organ tumour. Cell Death Dis. (2018) 9:356. doi: 10.1038/s41419-018-0391-6
69. Ribbert H. Rudolf Virchow †. Dtsch Medizinische Wochenschrift. (1902) 28:657–8. doi: 10.1055/s-0028-1138926
70. Balkwill F, Mantovani A. Inflammation and cancer: back to virchow? Lancet. (2001) 357:539–45. doi: 10.1016/S0140-6736(00)04046-0
71. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
72. Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. (2008) 454:436–44. doi: 10.1038/nature07205
73. Diakos CI, Charles KA, McMillan DC, Clarke SJ. Cancer-related inflammation and treatment effectiveness. Lancet Oncol. (2014) 15:e493–503. doi: 10.1016/S1470-2045(14)70263-3
74. Greten FR, Grivennikov SI. Inflammation and cancer: triggers, mechanisms, and consequences. Immunity. (2019) 51:27–41. doi: 10.1016/j.immuni.2019.06.025
75. Quail DF, Dannenberg AJ. The obese adipose tissue microenvironment in cancer development and progression. Nat Rev Endocrinol. (2019) 15:139–54. doi: 10.1038/s41574-018-0126-x
76. Mercurio V, Cuomo A, Dessalvi CC, Deidda M, Di Lisi D, Novo G, et al. Redox imbalances in ageing and metabolic alterations: implications in cancer and cardiac diseases. an overview from the working group of cardiotoxicity and cardioprotection of the Italian society of cardiology (SIC). Antioxidants. (2020) 9:641. doi: 10.3390/antiox9070641
77. Bordonaro M, Lazarova D. Hypothesis: Obesity is associated with a lower mutation threshold in colon cancer. J Cancer. (2015) 6:825–31. doi: 10.7150/jca.12352
78. Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. (2017) 377:1119–31. doi: 10.1056/NEJMoa1707914
79. Ridker PM, MacFadyen JG, Thuren T, Everett B, Libby P, Glynn RJ, et al. Effect of interleukin-1β inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet. (2017) 390:1833–42. doi: 10.1016/s0140-6736(17)32247-x
80. Rothwell PM, Fowkes FGR, Belch JF, Ogawa H, Warlow CP, Meade TW. Effect of daily aspirin on long-term risk of death due to cancer: analysis of individual patient data from randomised trials. Lancet. (2011) 377:31–41. doi: 10.1016/S0140-6736(10)62110-1
81. Qiao Y, Yang T, Gan Y, Li W, Wang C, Gong Y, et al. Associations between aspirin use and the risk of cancers: a meta-analysis of observational studies. BMC Cancer. (2018) 18:288. doi: 10.1186/s12885-018-4156-5
82. Rothwell PM, Price JF, Fowkes FGR, Zanchetti A, Roncaglioni MC, Tognoni G, et al. Short-term effects of daily aspirin on cancer incidence, mortality, and non-vascular death: analysis of the time course of risks and benefits in 51 randomised controlled trials. Lancet. (2012) 379:1602–12. doi: 10.1016/S0140-6736(11)61720-0
83. McNeil JJ, Gibbs P, Orchard SG, Lockery JE, Bernstein WB, Cao Y, et al. Effect of aspirin on cancer incidence and mortality in older adults. JNCI J Natl Cancer Inst. (2020) doi: 10.1093/jnci/djaa114
84. Moser AR, Luongo C, Gould KA, McNeley MK, Shoemaker AR, Dove WF. ApcMin: a mouse model for intestinal and mammary tumorigenesis. Eur J Cancer. (1995) 31A:1061–4. doi: 10.1016/0959-8049(95)00181-H
85. Weinberg F, Chandel NS. Reactive oxygen species-dependent signaling regulates cancer. Cell Mol Life Sci. (2009) 66:3663–73. doi: 10.1007/s00018-009-0099-y
86. Weinberg F, Ramnath N, Nagrath D. Reactive oxygen species in the tumor microenvironment: an overview. Cancers. (2019) 11:1191. doi: 10.3390/cancers11081191
87. Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. (2009) 9:798–809. doi: 10.1038/nrc2734
88. Genard G, Lucas S, Michiels C. Reprogramming of tumor-associated macrophages with anticancer therapies: radiotherapy versus chemo- and immunotherapies. Front Immunol. (2017) 8:828. doi: 10.3389/fimmu.2017.00828
89. Aras S, Raza Zaidi M. TAMeless traitors: macrophages in cancer progression and metastasis. Br J Cancer. (2017) 117:1583–91. doi: 10.1038/bjc.2017.356
90. Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. (1997) 3:730–7. doi: 10.1038/nm0797-730
91. Pastò A, Consonni FM, Sica A. Influence of innate immunity on cancer cell stemness. Int J Mol Sci. (2020) 21:3352. doi: 10.3390/ijms21093352
92. Dongre A, Weinberg RA. New insights into the mechanisms of epithelial–mesenchymal transition and implications for cancer. Nat Rev Mol Cell Biol. (2019) 20:69–84. doi: 10.1038/s41580-018-0080-4
93. Sistigu A, Di Modugno F, Manic G, Nisticò P. Deciphering the loop of epithelial-mesenchymal transition, inflammatory cytokines and cancer immunoediting. Cytokine Growth Factor Rev. (2017) 36:67–77. doi: 10.1016/j.cytogfr.2017.05.008
94. Albrengues J, Shields MA, Ng D, Park CG, Ambrico A, Poindexter ME, et al. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science. (2018) 361:eaao4227. doi: 10.1126/science.aao4227
95. Riehle C, Bauersachs J. Small animal models of heart failure. Cardiovasc Res. (2019) 115:1838–49. doi: 10.1093/cvr/cvz161
Keywords: cancer, heart failure, systemic inflammation, cardiokines, carcinogenesis
Citation: Ausoni S and Azzarello G (2020) Development of Cancer in Patients With Heart Failure: How Systemic Inflammation Can Lay the Groundwork. Front. Cardiovasc. Med. 7:598384. doi: 10.3389/fcvm.2020.598384
Received: 26 August 2020; Accepted: 30 September 2020;
Published: 26 October 2020.
Edited by:
Massimo Imazio, University Hospital of the City of Health and Science of Turin, ItalyReviewed by:
George Lazaros, Hippokration General Hospital, GreeceCopyright © 2020 Ausoni and Azzarello. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Simonetta Ausoni, c2ltb25ldHRhLmF1c29uaUB1bmlwZC5pdA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.