 Namrita Kaur
Namrita Kaur Rida Raja
Rida Raja Andrea Ruiz-Velasco
Andrea Ruiz-Velasco Wei Liu*
Wei Liu*
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med. , 15 October 2020
Sec. Cardiovascular Biologics and Regenerative Medicine
Volume 7 - 2020 | https://doi.org/10.3389/fcvm.2020.585309
This article is part of the Research Topic Understanding Molecular Mechanisms in Diabetic Cardiomyopathy (DCM) View all 13 articles
Heart failure is a serious comorbidity and the most common cause of mortality in diabetes patients. Diabetic cardiomyopathy (DCM) features impaired cellular structure and function, culminating in heart failure; however, there is a dearth of specific clinical therapy for treating DCM. Protein homeostasis is pivotal for the maintenance of cellular viability under physiological and pathological conditions, particularly in the irreplaceable cardiomyocytes; therefore, it is tightly regulated by a protein quality control (PQC) system. Three evolutionarily conserved molecular processes, the unfolded protein response (UPR), the ubiquitin-proteasome system (UPS), and autophagy, enhance protein turnover and preserve protein homeostasis by suppressing protein translation, degrading misfolded or unfolded proteins in cytosol or organelles, disposing of damaged and toxic proteins, recycling essential amino acids, and eliminating insoluble protein aggregates. In response to increased cellular protein demand under pathological insults, including the diabetic condition, a coordinated PQC system retains cardiac protein homeostasis and heart performance, on the contrary, inappropriate PQC function exaggerates cardiac proteotoxicity with subsequent heart dysfunction. Further investigation of the PQC mechanisms in diabetes propels a more comprehensive understanding of the molecular pathogenesis of DCM and opens new prospective treatment strategies for heart disease and heart failure in diabetes patients. In this review, the function and regulation of cardiac PQC machinery in diabetes mellitus, and the therapeutic potential for the diabetic heart are discussed.
Diabetes mellitus is one of the fastest-growing health issues worldwide, and it is a major threat to cardiovascular health. In 2019, it was estimated that 463 million people had diabetes, a number predicted to reach 700 million by 2045 (1), and diabetes patients have a 2–5-fold increased risk of developing heart failure (2, 3). Diabetic cardiomyopathy (DCM) refers to the cardiac dysfunction and structural abnormalities subsequent to diabetes, and independent of coronary artery disease, hypertension, and valve malfunctions (4, 5). The systemic metabolic alterations caused by reduced insulin secretion, in type 1 diabetes mellitus (T1DM), or progressive insulin resistance, in type 2 diabetes mellitus (T2DM), constitute continuous cardiac stress that leads to the activation of numerous cellular responses. DCM is characterized by impaired cellular homeostasis, the progressive accumulation of reactive oxygen species (ROS), reactive nitrogen species (RNS) and advanced-end glycation products, organelle dysfunction, and chronic inflammation. Eventually, DCM promotes pathological myocardial remodeling, resulting in cardiac dysfunction. Clinically, diastolic dysfunction is the first manifestation of DCM, followed by systolic dysfunction in later stages, and, ultimately, heart failure (6, 7). However, a single anti-diabetes agent (e.g., metformin or fibrate) is unable to ameliorate multiple comorbid conditions. The combination of individual therapies is indispensable for T2DM patients with other complications, including DCM.
Proteins are the primary managers of cellular homeostasis; therefore, regulation of their synthesis, maturation, and degradation in cardiomyocytes is essential for cardiac performance. To cope with the imbalance in the cardiac protein cycle in response to pathological stress, crucial protein quality control (PQC) systems participate in maintaining cellular protein homeostasis (8, 9) (Figure 1). Endoplasmic reticulum (ER) stress activates the unfolded protein response (UPRER) to reduce protein synthesis, increase the expression of folding chaperones, and degrade non-functional proteins through the ER-associated protein degradation (ERAD) pathway. ERAD recognizes and translocates non-functional proteins into the cytosol for degradation. The ubiquitin-proteasome system (UPS) breaks down most proteins secreted by ERAD and those that have reached the end of their lifespan. Proteins that cannot be processed by the proteasome or protein aggregates are broken down via the autophagy-lysosome system. Similar to ER, mitochondria have a specific UPR (UPRmt) signaling to manage their unfolded protein load and can be selectively marked for autophagic degradation when the damage surpasses their coping capabilities (11). Coordination of PQC systems is adaptive and protective, while impaired PQC contributes to cardiac aging and diseases (12), including DCM (9). Therefore, it is crucial to comprehensively understand the function and regulation of PQC pathways to identify potential therapeutic targets and strategies for DCM.
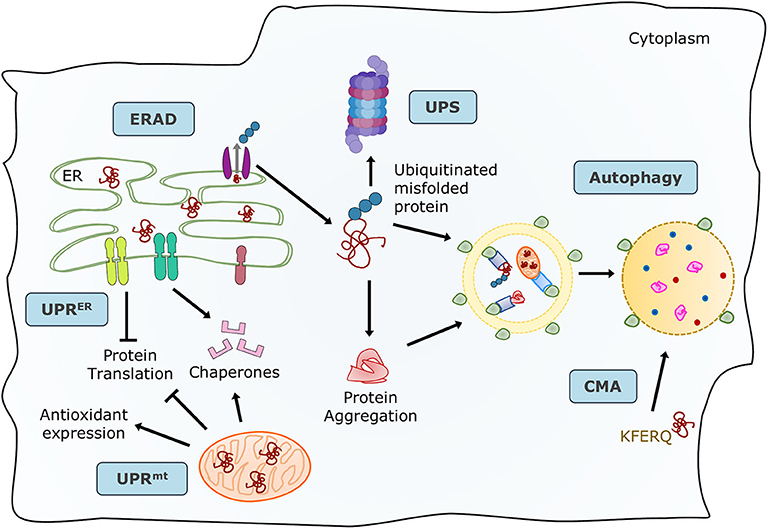
Figure 1. Maintenance of protein homeostasis by the principal PQC systems. Accummulation of misfolded proteins in the ER trigger the UPR to increase its folding capacity by upregulating chaperone expression, and to decrease protein load by inhibiting translation. Misfolded proteins are exported by ERAD complexes that label them and direct them to degradation through the UPS and autophagy. Misfolded or damaged proteins are also sequestered into protein aggregates to reduce their toxicity, these aggregates are processed by the autophagy-lysosome system. Proteins with the KFERQ motif are translocated into the lysosomes via CMA. Additionally, unfolded proteins in mitochondria induce the mtUPR to increase mitocondrial proteases and chaperones. Adapted from Ciechanover et al. (10) and used under CC BY 3.0.
The ER is a central organelle for cellular PQC, operating as the keeper of the multistep maturation process of nascent polypeptides into functional proteins. The ER serves several cellular functions, comprising protein folding, posttranslational modifications, trafficking, calcium homeostasis, and lipid biosynthesis (13–16). Any intracellular and extracellular perturbations to its protein folding capacity result in ER stress and trigger the multi-faceted UPRER necessary for cellular PQC (17–20). Recently, mitochondria have been found to possess their own stress response to manage the unfolded proteins contained within them, also contributing to protein homeostasis (21).
The primary intent of the UPRER is to adapt to any cellular changes by restoring protein homeostasis. The chaperones in the ER-lumen assist protein folding by binding to the hydrophobic regions of the nascent proteins (GRP78, GRP94), promoting glycoprotein interactions (Calreticulin, Calnexin), and facilitating the formation of disulfide bridges (ERP57, ERP78) (22). The master chaperone glucose regulatory protein 78 (GRP78) also binds to calcium, assists in ER permeability during protein translocation, and guides the misfolded proteins for degradation. The initial step of UPRER is the release of the transmembrane sensors, primarily bound to the master chaperone under non-stressed conditions (23). These UPRER sensors, including protein kinase RNA-like endoplasmic reticulum kinase (PERK), inositol-requiring enzyme 1 (IRE1), and activating transcription factor 6 (ATF6), are distinctively activated by stress stimuli and elicit varied adaptive downstream responses. Upon ER stress, PERK is majorly involved in attenuation of translation for lessening ER protein load via eukaryotic translation initiation factor 2α (eIF2α). PERK-eIF2α increases the expression of key genes facilitating UPRER via activating transcription factor (ATF4). On the other hand, the endoribonuclease activity of IRE1 causes splicing of the transcription factor, X-box binding protein 1 (XBP1). The spliced XBP1 (sXBP1) upregulates the expression of genes involved in UPRER signaling (9) and the ERAD pathway (24). IRE1-dependent decay (RIDD) is known to regulate essential ER-localized messenger RNAs (mRNAs) to reduce the inflow of newly synthesized proteins into the ER (25). IRE1 also enhances the degradation of terminally misfolded proteins via ERAD (UPS section) (18). Finally, upon activation, ATF6 translocates from the ER luminal domain to the Golgi apparatus, where site-1 and site-2 proteases cleave it to form an active segment, p50ATF6. The activated ATF6 transcriptionally regulates essential genes responsible for UPRER (26). All three UPRER branches are required to upregulate chaperones expression for assisting protein proper folding (27).
The ER stress response (ERSR) initially induces an adaptive UPRER to a certain threshold. In the face of chronic pathological stresses, oversaturated ER ensues apoptotic ERSR (28). Overexpression of ATF4 upregulates C/EBP homologous protein (CHOP), growth arrest, and DNA damage-inducible 34 (GADD34) and other pro-apoptotic genes (25). CHOP induces cell death by dysregulating the balance between pro- and anti-apoptotic genes from B-cell lymphoma 2 (BCL2) family. Also, oligomerization of the pro-apoptotic proteins BAX and BAK on the ER membrane causes calcium release into the cytosol, eventually promoting mitochondria-dependent apoptotic pathways (29). IRE1 induces ER-mediated apoptotic mechanisms via recruitment of TNF receptor-associated factor (TRAF) 2 and apoptosis signal-regulating kinase 1 (ASK1), leading to activation of c-Jun N-terminal kinase (JNK), and caspase-12 signaling pathways. In addition to IRE1-regulated caspase 12 cleavage, m-calpain, a cysteine protease, directly cleaves caspase-12 upon stimulation, resulting in its activation (30). The three UPRER branches exist to facilitate both cytoprotective and apoptotic responses depending on the nature of the stimulus (25, 27); therefore, it is not surprising that temporal dynamics of the UPRER has an important role in determining cellular fate.
ER-resident genes have been deemed essential in the heart. GRP78 or XBP1 deficiency is implicated in impaired cardiac development (31, 32) and cardiac dysfunction in response to pathological stresses (33, 34). The increase in protein disulfide isomerase (PDI) (35), an ER chaperone, and sXBP1 expression (36) in ischemic human hearts suggest UPRER is an adaptive component of the cardiac stress response. However, a maladaptive stress response is evident in dilated and failing human hearts marked by an increase in CHOP expression and cell death (37, 38). In light of the clinical evidence, it is apparent that the ERSR has both adaptive and maladaptive roles in cardiac pathology.
ER chaperones promote cell survival under pathological stress in the heart; nevertheless, overexpression is damaging. Cardiac GRP78 knockout in adult mice induced increased cell death, reduced cardiac performance, and caused early mortality (31). Moreover, the pre-induction of GRP78 and GRP94 had a cardioprotective role under oxidative damage in ischemia/reperfusion (39). On the contrary, increased protein synthesis under cardiac hypertrophy upregulated GRP78 expression, simultaneously, under pressure overload, GRP78 overexpression further potentiated hypertrophy by stimulating expression of hypertrophic factors resulting in cardiac dysfunction (40). Additionally, the overexpression of calreticulin, an ER chaperone, resulted in cardiac remodeling, dysfunction, and heart failure due to prolonged UPRER activation. This damaging effect of calreticulin overexpression in vivo was abated by inhibition of IRE1 (41, 42), overall suggesting the importance of balanced UPRER to tackle pathological stress in the heart.
Several animal studies targeting the individual UPRER branches emphasized the importance of UPRER in the pathological hearts of different etiologies. Cardiac PERK deficiency aggravated heart function in response to pressure overload in mice (43), indicating the cytoprotective role of the PERK branch. Moreover, transient IRE1-XBP1 response following pressure overload in mice (44) limited myocardial injury by reducing ER-associated cell death and inflammation (45) and promoting adaptive hypertrophy, in turn preserving contractility in hypertrophic failing hearts (25, 46). Similarly, cardiac XBP1 deficiency enhanced pathological remodeling and dysfunction (47). Lastly, ATF6 deletion in mouse hearts resulted in increased oxidative stress and decreased function after ischemia/reperfusion. The equivalent in vitro ATF6 knockdown model in cardiomyocytes showed similar results, which were obliterated by ATF6 overexpression (48). As noted, transient activation of all three UPRER branches has an adaptive function succeeding acute cardiac ER stress, while sustained activation of UPRER results in irreversible damage to the myocardium. This persistent stress signaling induces cardiomyocyte death via activation of ER-mediated apoptosis following myocardial infarction, ischemia/reperfusion, and pressure overload (23, 49). Also, Miyazaki et al. (50) demonstrated that cardiac CHOP deficiency inhibits ER-mediated myocardial apoptosis and inflammation following reperfusion injury, highlighting the role of maladaptive ERSR.
The role of ER stress in the development of DCM was first observed in failing diabetic human hearts with swollen ERs (51), indicating protein imbalance. They also presented ER-mediated apoptosis, evidenced by increased CHOP and cell death (52). These clinical findings imply that the impaired UPRER predisposes the diabetic heart to failure; however, the precise nexus is elusive. The cardiac fate following ERSR has since been ascertained in several animal models of DCM. In the diabetic models, the elevation of cardiac ER stress-related markers (53) and UPRER genes (54–57) is associated with cardiac abnormalities (58) and apoptosis (59). Although the canonical UPRER signaling is an adaptive response, chronic ER stress is deleterious in the diabetic heart. In T1DM, prolonged ATF6 activation-induced cell death (60), extracellular matrix gene expression, cardiac fibrosis (61), and reduced cardiac compliance in rat models. Moreover, oxidative stress resulted in cardiac dysfunction in type 1 diabetic hearts via persistent PERK signaling (62). The role of over-activated PERK-CHOP and ATF6 ensuing apoptotic signaling via BCL2 associated agonist of cell death (BAD) and contributing to ER-mediated cardiac dysfunction was also recapitulated in T2DM rodent hearts (63). Apoptotic ERSR is associated with pathogenesis of DCM due to irreplaceable cardiomyocyte loss associated with the upregulation of cleaved-caspase 12, CHOP, and JNK in type 1 and type 2 diabetic hearts (51, 54). Altogether, the maladaptive ERSR in DCM prompts organelle dysfunction, cell death, and subsequent myocardial remodeling (64), suggesting that hyperactivated PERK and ATF6 are detrimental in DCM.
Pathological remodeling and cardiac dysfunction in DCM are accompanied by alterations in cellular protein synthesis, which can facilitate ER stress and UPRER. ER stress is an early event in DCM, and the major triggers include hyperglycemia, hyperlipidemia, insulin deficiency/resistance, and inflammation (65, 66) (Figure 2). High glucose and lipid overload induce oxidative stress interceding dysregulated protein homeostasis, prolonged UPRER, and cardiomyocyte death (67). Glucose and lipids upregulated adaptive IRE1-XBP1 signaling (51), and prolonged stress triggered apoptotic CHOP (63), IRE1-JNK (68), and caspase 12 activation in human cardiac cells (69), type 1 (55), and type 2 (30) diabetic rodent hearts. Inflammation and hyperinsulinemia are other factors that instigate ER stress and can be further potentiated by ER stress in a detrimental loop. As a coping mechanism, hyperinsulinemia-induced ER stress has emerged as a new player in the onset of insulin resistance (70, 71), possibly via IRE1/JNK signaling (58), contributing to reduced cardiac function in T2DM (72). On the other hand, pro-inflammatory cytokine interleukin-1β via interleukin 1 receptor-associated kinase 2 (IRAK2) promoted CHOP expression and cell death in T1DM, thereby impairing cardiac function (73). IRAK2 is known to be elevated in the condition of ER stress (74), suggesting a feedback loop mechanism accountable for unalleviated ER stress. However, the direct mechanism of ER-mediated inflammation and cardiac dysfunction in DCM is yet to be determined. Further mechanistic study of the intrinsic details of preferential UPRER under the numerous drivers of ER stress in DCM is essential.
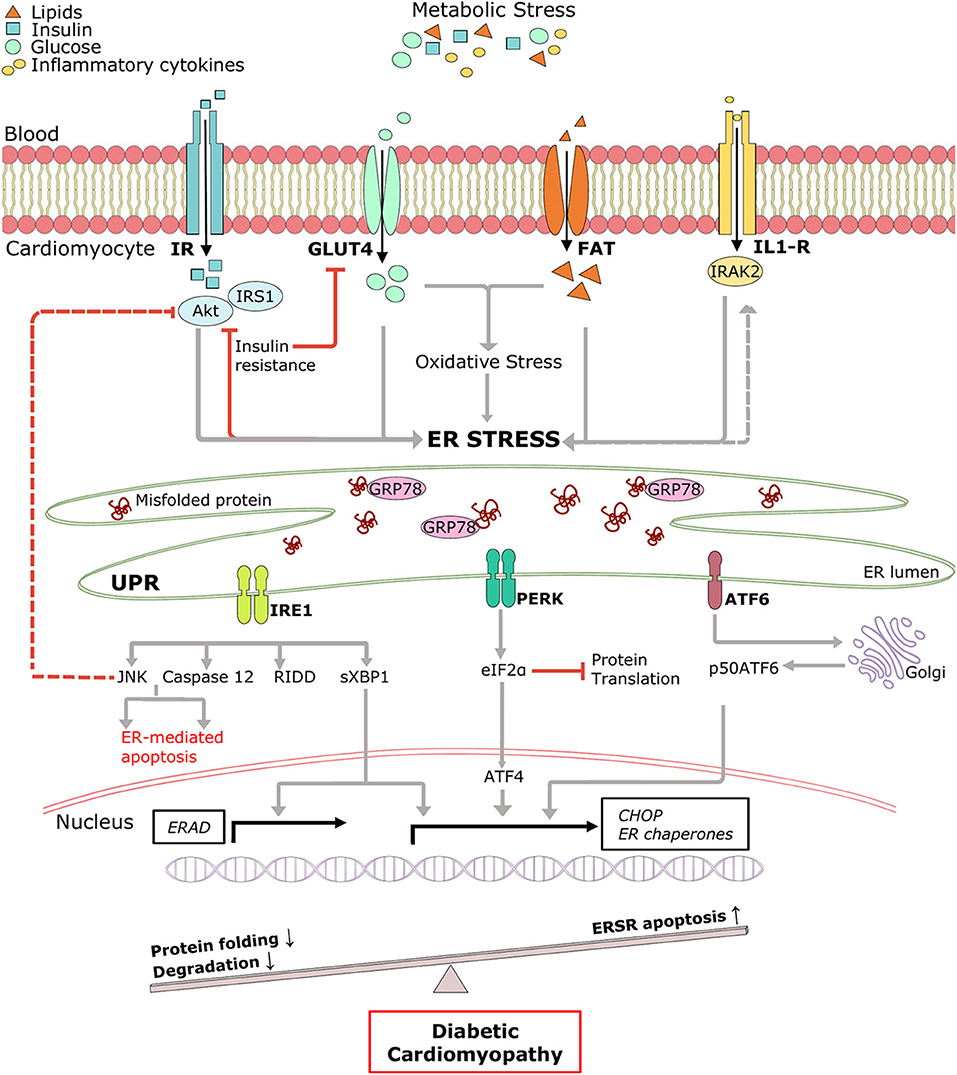
Figure 2. Pictorial representation of triggers of ER stress and role of ERSR activated UPR in the progression of DCM. Under systemic metabolic stress, lipid accumulation, and high glucose directly triggers ER stress and indirectly via oxidative stress. Hyperinsulinemia and inflammatory cytokines also induce ER stress, and in a feedback loop mechanism, ER stress triggers insulin resistance and inflammation (dotted lines). Upon accumulation of misfolded proteins, UPR signaling is activated following dissociation from GRP78. UPR sensors, IRE1, PERK, and ATF6 and its downstream network drives multiple signal outputs such as inhibition of protein translation and increased gene upregulation. Balanced UPR restores protein homeostasis by increasing ER chaperones and ERAD genes. Prolonged stress induces apoptotic ERSR via upregulation of CHOP, contributing to the pathogenesis of DCM. IR, Insulin receptor; IRS1, insulin receptor substrate 1; Akt, protein kinase B; GLUT4, glucose transporter type 4; FAT, fatty acid transporter; IL-R, Interleukin 1β receptor.
The involvement of ERSR in DCM progression is well-accepted; nonetheless, only a few regulatory mechanisms of UPRER in diabetes are documented, where the ER machinery coordinates with several cellular molecules and signaling pathways (Figure 3). For instance, downregulated NAD-dependent protein deacetylase sirtuin 1 (SIRT1) promoted stress signaling pathways such as IRE1-JNK in T1DM (75), and PERK-CHOP and IRE1-caspase 12 signaling in T2DM (76), resulting in ER-mediated apoptosis and cardiac dysfunction. A protein kinase, general control nonderepressible (GCN2), triggered cell death, and cardiac dysfunction directly via the eIF2α-ATF4-CHOP pathway in T1DM and T2DM (77). In addition, increased EGFR tyrosine kinase receptor activation instigated ER stress in T1DM (78) and in T2DM following myocardial infarction (79) by increasing CHOP associated cell death. The ERSR is also regulated via transcription factors. Forkhead box O1 (FOXO1) activation leads to direct and indirect induction of ER stress in DCM via PERK signaling (80, 81), and peroxisome-proliferator activator receptor (PPAR) β/γ activity promotes XBP1 splicing restoring ER balance and providing cryoprotection under diabetic stress in human cardiac cells (69). Additionally, microRNAs (miRNAs) have been observed to regulate UPRER in the diabetic heart. mir455 and mir22 are cardioprotective in T1DM (61) and T2DM (82), respectively. Mir455 reduces cardiac fibrosis via calreticulin suppression, and mir22 alleviates ER-mediated apoptosis via SIRT1 upregulation.
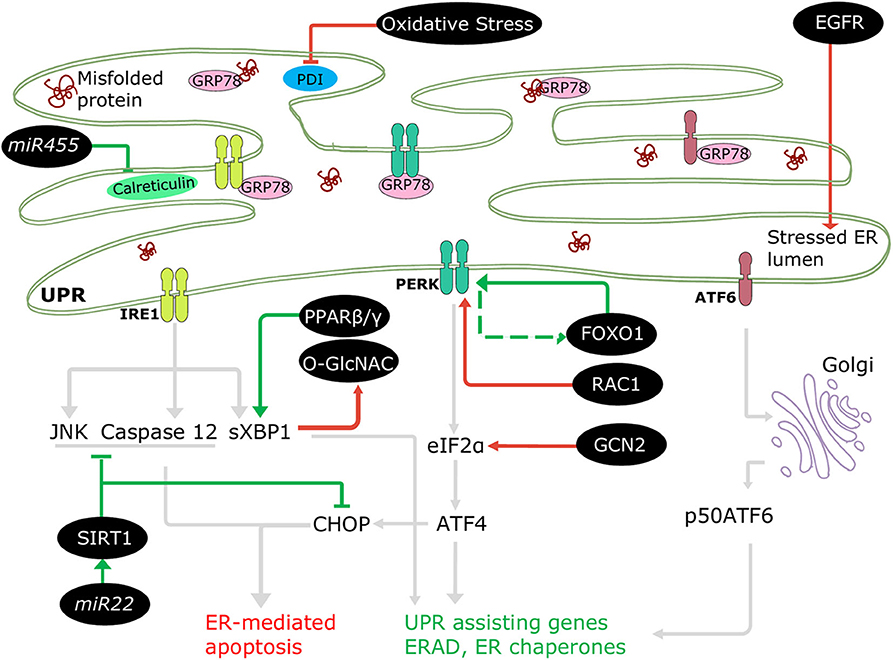
Figure 3. Regulation of ERSR in the diabetic heart. Some cellular molecules (FOXO1, PPARβ/γ, SIRT1, mir22, mir455) participate in regulation of adaptive ERSR by activating UPR and inhibiting ER-mediated apoptosis (green lines). On the other hand, some molecules and stresses (oxidative stress, O-GlcNAC, EGFR, RAC1, GCN2) stimulate maladaptive ERSR (red lines).
Apart from the regulated framework of UPRER, diabetic condition also impairs UPRER capacity by directly regulating ER chaperones. Elevated PDI, despite the cardioprotective action under ischemic cardiomyopathy (35), was associated with increased cell death in hearts from diabetes patients (56). The lack of protective effect was attributable to the altered redox state of PDI under type 1 diabetes. Moreover, dual-color fluorescence imaging indicated an abnormal ER oxidative state and altered polarity in diabetic myocardial tissue (83), suggesting ER oxidative state may contribute to impaired UPRER under diabetes. Therefore, the maladaptive ERSR can be promoted by factors independent of canonical UPRER pathways in the diabetic heart. Also, post-translational modifications such as O-GlcNACylation are essential for protein stability and function. The protective effect of XBP1 on O-GlcNACylation (8, 46, 84) is absent in T2DM, leading to cardiac dysfunction (85), which is likely due to delayed UPRER action, as the timely UPRER lacks in the diabetic heart. Despite the growing knowledge about ERSR following various stresses faced by the diabetic heart, it is still unclear about how or when the switch between adaptive UPRER and apoptotic ERSR supervenes.
The mitochondrial proteome contains more than 1,300 proteins and the majority of the nuclear-encoded proteins are imported into the organelle in an unfolded state (86). Mitochondrial PQC entails protein import and folding via chaperones (HSP60, HSP70, and TRAP1) and degradation of misfolded proteins by proteases (ClpP, YME1L1, LonP1, HTRA2/Omi, and Oma1) (87). In response to stresses, the UPRmt initiates a retrograde response to the nucleus to ensure proteome integrity via induction of the UPRmt-related chaperones (88). Akin to UPRER, UPRmt transiently inhibits protein translation and aims to mitigate proteotoxic stress inside the mitochondrion (89, 90). Under physiological conditions, activating transcription factor 5 (ATF5) is imported into the mitochondrion and degraded by LonP1; however, stress targets ATF5 to the nucleus as the transcription factor for the induction of UPRmt (91). Owing to the cross-over among certain stress response proteins (PERK, ATF4, and CHOP) and the physical linkage between ER and mitochondria, both UPRER and UPRmt participate in an integrated stress response to maintain cellular proteostasis (21). For instance, consequent to eIF2α activation, translation of ATF4, CHOP and ATF5 regulate the UPRmt. UPRmt−associated expression of CHOP is identified by binding of c-Jun to the AP-1 promoter region in the CHOP gene (92). CHOP binding, along with MURE1/2 elements in the promoter region, increases transcription of HSP60, ClpP, ATF5, and LonP1. Additionally, misfolded proteins are ubiquitinated in the inner mitochondrial space and degraded by the UPS in the cytosol in a process called mitochondrial associated degradation (93).
Mitochondrial PQC is essential for cardiac structure and function (94). In clinic, patients with ventricular pressure overload due to aortic stenosis had elevated ATF5 and reduced apoptosis (95), suggesting its protective role under cardiac stress. Analogous to clinical observation, silencing ATF5 in cardiomyocytes abated UPRmt and its protection against pressure overload (96). Similarly, pharmacological stimulation of UPRmt ameliorated cardiac dysfunction following ischemic injury via ATF5 induction (97). Moreover, HSP70 overexpression increased the import of antioxidant proteins, reduced cell death, and improved cardiac function against ischemic stress (98). As such, UPRmt is protective under cardiac stress; nonetheless, the role of mitochondrial proteases is still unclear. For instance, under hypoxia, mitochondrial protease LonP1 contributed to ROS accumulation and cell death in cultured cardiomyocytes (99). On the other hand, LonP1 overexpression was found to be protective following ischemic/reperfusion injury in mouse hearts (100) while reduced LonP1 activity in mitochondria contributed to contractile dysfunction after pressure overload (101). Interestingly, the same study demonstrated that LonP1 activation induces UPRER; however, UPRER is activated before UPRmt, suggesting a fine-tuning role of LonP1 in the integrated stress response. More importantly, LonP1 deficiency was compensated via ATF4-dependent fibroblast growth factor (FGF21) activation (92, 101), a marker for mitochondrial stress signaling involved in mediating metabolic changes and ameliorating cardiac dysfunction under several cardiac etiologies, including diabetes (21, 102). Also, the mitochondrial protease, Oma1, is upregulated under cardiac ischemic stress; however, its ablation is protective against heart failure in mice (103).
UPRmt chaperones are likely protective under diabetic stress in the heart. There is reduced expression of the mitochondrial chaperone, HSP70, in T2DM human hearts (104), indicating decreased protein import and UPRmt induction. In the hearts of pre-diabetic rats with hyperinsulinemia, there was an increase in HSP60 expression; however, after prolonged diabetic stress HSP60-mediated myocardial protection decreased due to abated expression (105). Moreover, hyperglycemia reduced TRAP1 expression and activity, ultimately reducing cardiomyocyte viability (106). Furthermore, in T2DM rodent hearts, UPRmt is responsible for the dysregulation of the mitochondrial permeability transition pore, associated with elevated cell death and ischemic injury (107). Overall, adaptive UPRmt is critical for cardiac structure and function under diabetic stress; however, the detailed mechanistic role of the mitochondrial PQC and its therapeutic applications is yet to be cemented in DCM.
The UPS is a major quality control pathway in eukaryotic cells, which plays a fundamental role in maintaining cellular proteostasis and, as such, ensures cell viability and function. The UPS is the primary proteolytic path for ~80% of cellular proteins, most of which are short-lived, misfolded, or damaged (108, 109). Mechanistically, the ubiquitin proteolytic pathway involves two distinct steps: ubiquitylation of protein substrates and degradation of the ubiquitylated proteins by the proteasome (110).
Ubiquitin is a 76-amino acid globular protein that is highly conserved in eukaryotes, and its transfer to target proteins is mediated by a carefully choreographed enzymatic cascade. Initially, ubiquitin is activated to a high-energy thiol ester state by the ubiquitin-activating enzyme E1 in an ATP-dependent manner. Following activation, the ubiquitin moiety is transferred to ubiquitin-conjugating proteins E2 by transesterification. Finally, an E3 ubiquitin ligase catalyzes ubiquitin transfer from the E2-ubiquitin thioester intermediate to a lysine residue on the substrate protein (111–113) (Figure 4A). The human genome encodes ~1,000 E3 ubiquitin ligases, which are subdivided into three major groups, depending on which of the following three domains they possess, namely, really interesting new gene (RING), RING-in-between-RING (RBR), and homologous to the E6-AP carboxyl terminus (HECT). It has been widely reported that the E3 ubiquitin ligases confer specificity to the ubiquitylation process (116). The proteins are targeted by either a single ubiquitin molecule (monoubiquitylation) or ubiquitin chains (polyubiquitylation). To date, eight structurally and functionally distinct ubiquitin linkages (Lys6, Lys11, Lys27, Lys29, Lys33, Lys48, Lys63, and Met1) have been identified, among of them, Lys48 and Lys63 are the most prominent linkage types (117–119). Lys48-ubiquitylated proteins are typically subjected to proteasomal degradation, while Lys63-linked ubiquitin chains mediate autophagic protein quality control (120, 121) (Figure 4B).
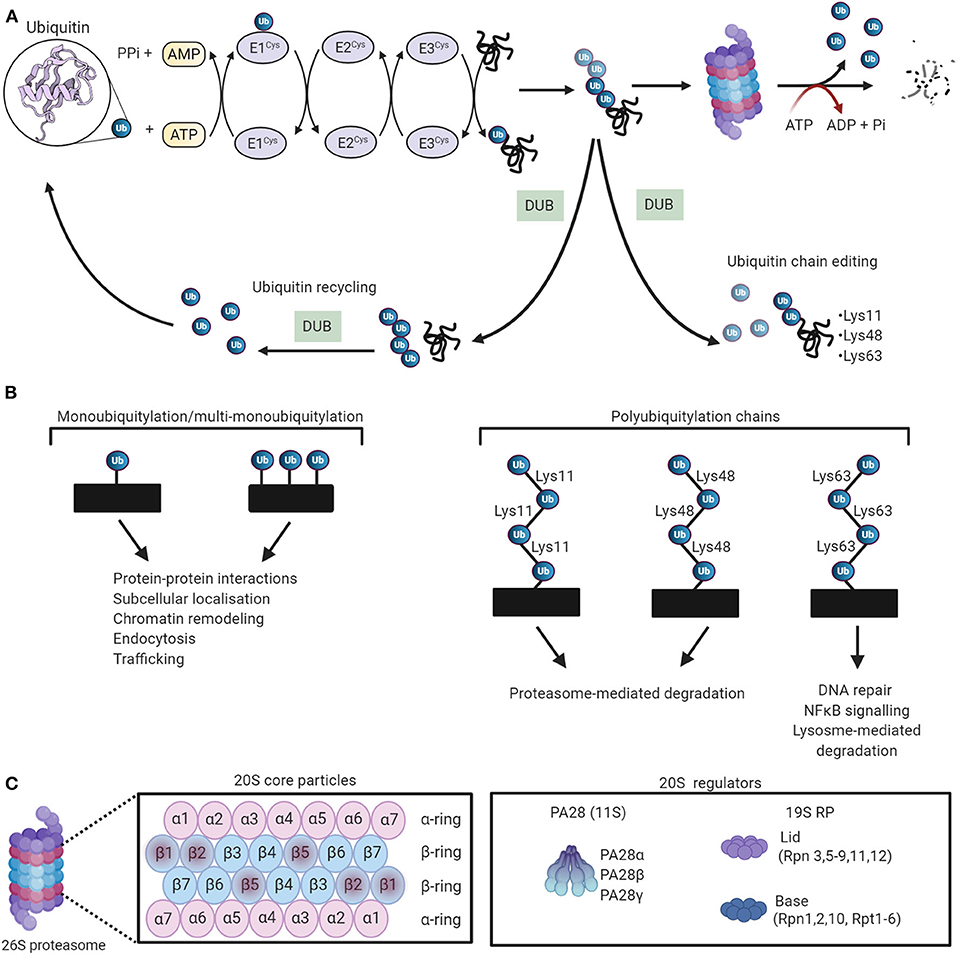
Figure 4. The ubiquitin-proteasome system (UPS) working theory. (A) The UPS marks substrate proteins for degradation via a ubiquitylation system where internal lysine residues of substrate proteins are tagged with ubiquitin (PDB ID: 1UBQ) (114). These ubiquitylated proteins are then degraded by the proteasome. Deubiquitylating enzymes (DUBs) edit ubiquitin chains and mediate ubiquitin recycling. Adapted from Zheng et al. (115) and used under CC BY. (B) The different ubiquitin linkages and their unique biological functions. (C) The proteosomal 20S core particle (CP) consists of four stacked rings, two outer rings composed of seven α subunits, and two inner rings composed of seven β subunits. The proteolytically active sites are localized in the β1, β2, and β5 subunits. The majority of 20S proteasomes are capped with 19S regulatory particles. The 20S can also be activated by PA28. Created with Biorender.com.
Akin to other posttranslational modifications, the ubiquitylation process is reversible; removal of ubiquitin molecules from substrate proteins is mediated by deubiquitylating enzymes (DUBs) (122). DUBs perform critical roles in the ubiquitylation pathway (123). First, de novo ubiquitin is translated as either linear polyubiquitylated chains, or ubiquitin fused to small ribosomal proteins, and DUBs are required to free mono ubiquitin from these precursors. Second, in consort with the E3 ligases, DUBs mediate ubiquitin chain editing, which can alter the ubiquitin signal or protein stability. Finally, DUBs maintain ubiquitin homeostasis by recycling ubiquitin molecules (122, 123).
The degradation of polyubiquitylated proteins is catalyzed by the 26S proteasome, a large ATP-dependent multicatalytic complex composed of a barrel-shaped 20S core protease (CP) capped at one or both ends by the 19S regulatory particle (RP) (124) (Figure 4C). The 20S CP is composed of 28 subunits that are arranged as a cylindrical stack containing four hetroheptameric rings, two peripheral α-rings (α1−7), and two inner β-rings (β1−7). The two β-rings form the central proteolytic chamber, whereas the α subunits guard substrate entry into the chamber, impeding access when the proteasome is in an inactivated state (125, 126). The proteolytic activity of the 20S CP is activated by binding to the 19S RP to establish the proteasome holoenzyme (127). Protein components of the 19S RP recognize ubiquitylated substrates and transport them to the proteolytic core in an ATP-dependent manner (128–131). The peptidase activity of the 20 CP is also activated by other regulatory particles such as the 11S RP, which mediates protein degradation via a ubiquitin- and ATP-independent manner (132). The output of proteasomal degradation is small peptides, which, upon proteasomal exit, are further degraded by a plethora of cytosolic peptidases to generate amino acids to be recycled.
Molecular chaperones are essential for the folding fidelity and conformational integrity of proteins (133, 134), by participating in nascent polypeptide folding, protein transport, assembly of oligomeric complexes, and repair of misfolded proteins (133). In addition, chaperones can also facilitate the degradation of folding-incompetent proteins, thereby preventing their aggregation (133, 135). The C-terminus of Hsc70-interacting protein (CHIP) is a central player in chaperone-mediated degradation (136, 137). CHIP binds with the constitutively expressed HSP70/HSC70 chaperones and members of the ubiquitin conjugating enzyme, such as the Ubc4/5 family, to initiate chaperone substrate sorting to the proteasome or lysosome (135, 138). BAG family molecular chaperone regulator 1 (BAG1) is a co-chaperone which functions as a nucleotide exchange factor triggering ADP dissociation from HSP70/HSC70 proteins and thereafter promoting chaperone substrate release (139). Interestingly, BAG1 can also simultaneously bind to the proteasome via its Ub-like (UBL) domain thereby providing a functional link between chaperones and the proteolytic machinery (140). Conversely, the co-chaperone HSPBP1 attenuates CHIP ubiquitin ligase activity when it is complexed with HSP70/HSC70 and thus inhibits CHIP-mediated degradation (141). Notably, both CHIP and BAG1 exert cytoprotective effects in the heart following ischemia-reperfusion injury (142, 143).
In addition to CHIP and HSP70, chaperones such as HSP20, HSP90, and αB-crystallin (CryAB) are also induced in cardiomyocytes in an effort to buffer misfolded proteins during cardiac stress (137). Numerous studies have highlighted the protective role of these proteins in the heart (137). For instance, HSP90 appears to be cardioprotective in both doxorubicin-induced heart failure and high-glucose induced cell injury (144, 145). Moreover, cardiac specific over-expression of HSP20 attenuates apoptosis, reduces infarct size, and improves cardiac function in mice following ischemia-reperfusion injury (146). Mutations which impair the function of chaperones have been implicated in numerous diseases including cardiomyopathies (147). Pre-clinical studies have demonstrated that transgenic mice expressing an R120G-missense mutation in CRYAB develop restrictive cardiomyopathies and manifest pathological characteristics similar to those observed in clinical desmin-related myopathy (DRC); aberrant protein aggregation in cardiomyocytes and cardiac dysfunction (148, 149).
ERAD is an integral facet of the UPS pathway (150). It is an evolutionarily conserved PQC mechanism in mammalian cells that orchestrates the function of numerous proteins to maintain ER homeostasis (151, 152). Through ERAD, aberrant ER luminal and transmembrane proteins are recognized and retrotranslocated to the cytosolic face where they are modified by the ubiquitylation machinery. The E3 ligases implicated in ERAD include soluble proteins, such as PRKN, ubiquitin conjugation factor E4 A (UBE4A), and CHIP, and ER transmembrane proteins, such as synoviolin (also known as HRD1), TEB4, GP78, and RMA1 (150). ERAD substrates are commonly conjugated to Lys48- and Lys11-linked polyubiquitin chains (153). Once ERAD substrates are adequately ubiquitylated, they are extracted from the ER membrane into the cytosol by the p97-UFD1-NPL4 complex to facilitate their proteasomal degradation (154). As such, if this adaptive ERAD function is defective or insufficient, the UPRER activates destructive cell pathways by transforming into an alternative signaling platform known as the terminal UPRER (155–158).
UPS activity is imperative in the heart as cardiomyocytes are highly susceptible to protein damage due to their constant exposure to metabolic and mechanical stress (159). Additionally, as terminally differentiated cells, cardiomyocytes possess minimal replicative potential; thus, failure to eliminate damaged proteins triggers excessive apoptosis, which is detrimental to the heart. Over the past decade, numerous clinical and experimental studies have documented impaired proteasome function, accumulation of ubiquitylated proteins, and alterations in the expression of UPS components in diseased hearts (159–164). Highlighting the importance of proteasomal integrity, cardiac proteasome inhibition induces heart dysfunction, and pathological hypertrophy in a preclinical mouse model (165). The pharmacological impediment of proteasome activity also leads to maladaptive structural and functional changes in pig hearts, which are consistent with a hypertrophic cardiomyopathy phenotype (166). Similarly, genetic inhibition of cardiac 20S proteasome promotes cardiac dysfunction in mice during systolic overload (138). Moreover, use of proteasome inhibitors (bortezomib, carfilzomib, and ixazomib), as targeted chemotherapeutics, is related to cardiovascular adverse events, including congestive heart failure (167). Of note, perturbations in UPS function have also been documented in doxorubicin-induced cardiotoxicity (168–170).
Inhibition of UBE2V1, a member of the E2 protein family, reduces protein aggregation in a CryABR120G-desmin related myopathy mouse model, improves cardiac function, and enhances survival in vivo (171). Likewise, it has been firmly established that E3 ligases play a significant role in the pathogenesis of heart diseases (Table 1). In a preclinical model of pressure-overload, MURF1 knockout mice displayed exacerbated cardiac hypertrophy in response to mechanical stress (176). Similarly, transgenic mice expressing mutations in Trim63, the gene encoding MURF1, develop cardiac hypertrophy (204). Moreover, Chip–/– mice challenged with ischemia-reperfusion injury were more prone to arrhythmias and had decreased survival rates (143). However, research on the pathological implications of DUBs in the heart is limited (Table 1). A recent study revealed that the expression of ubiquitin carboxyl-terminal hydrolase isozyme L1 (UCHL1) was increased in the cardiomyocytes of hypertrophic and failing hearts (199). Overexpression of UCHL1 exacerbates pressure-overload induced cardiac hypertrophy and dysfunction, which can be reversed by systemic administration of the UCHL1 inhibitor LDN-57444 in mice (199). These studies demonstrate the detrimental effects of UPS malfunction in the myocardium.
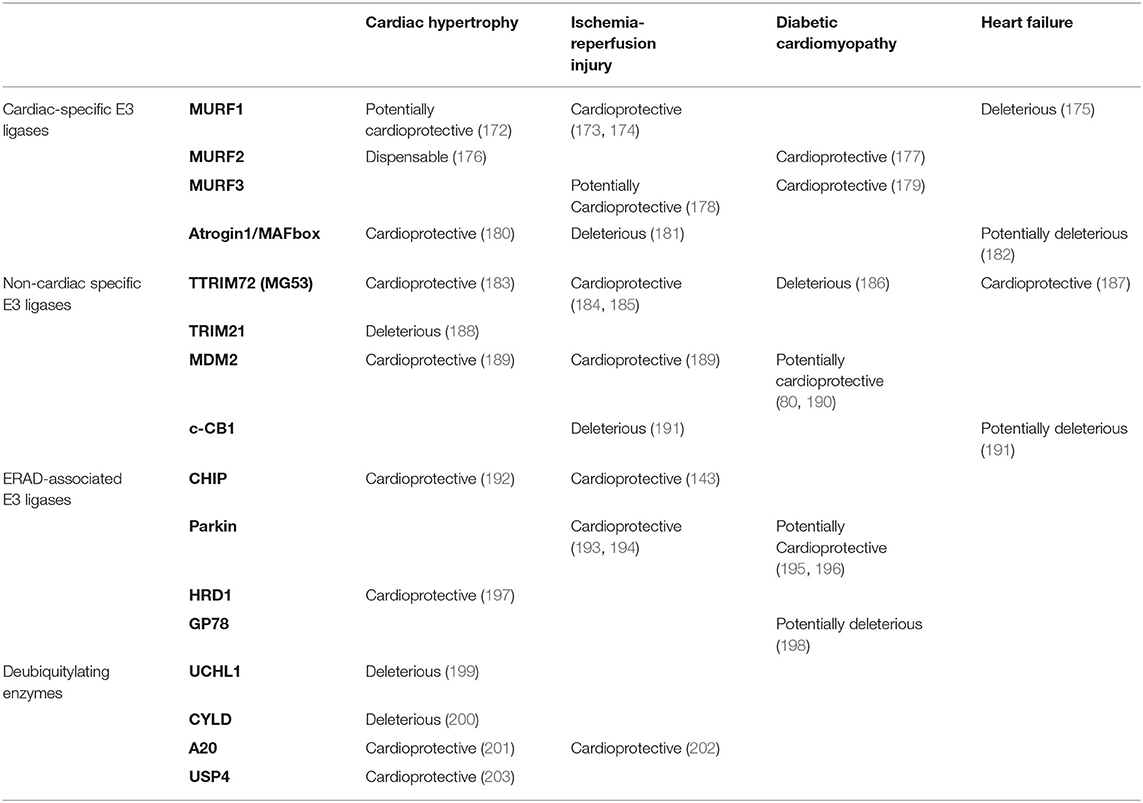
Table 1. The role of the ubiquitylation pathway in cardiovascular disease.
Furthermore, Doroudgar et al. demonstrated that HRD1 plays an essential role in the adaptive ERSR in cardiomyocytes and that cardiac-specific overexpression of HRD1 preserves cardiac structure and function in a mouse model of pathological cardiac hypertrophy (197). Moreover, overexpression of Derlin3, a component of the ERAD retrotranslocation channel, enhances ERAD-dependent disposal of misfolded proteins, attenuates exorbitant ERSR, and reduces caspase activity in response to ischemia/reperfusion injury (205). Conversely, knockdown of Derlin3 impairs the clearance of misfolded ER proteins and augments ischemia-mediated cell death in cardiomyocytes (205). Collectively, these findings suggest that ERAD-associated UPS plays a crucial role in myocardial viability and underscore the importance of PQC mechanisms in the setting of cardiac injury.
The E3 ubiquitin ligases participate in cardiac metabolic regulation, by regulating numerous transcription factors involved in DCM (206) (Figure 5). FOXO1 has emerged as an influential player in the pathogenesis of DCM, which is overactivated in the hearts of murine models of T2DM. This aberrant activation is associated with the development of cardiomyopathy, evidenced by the cardiac-specific deletion of FOXO1 ameliorating high fat diet-induced cardiac dysfunction and preserved insulin responsiveness (80). At the molecular level, several E3 ubiquitin ligases, including CHIP, MDM, and COP1, regulate FOXO (190, 207, 208), as a consequence, the functions of FOXOs are subdued by virtue of their ubiquitin-mediated proteasomal degradation.
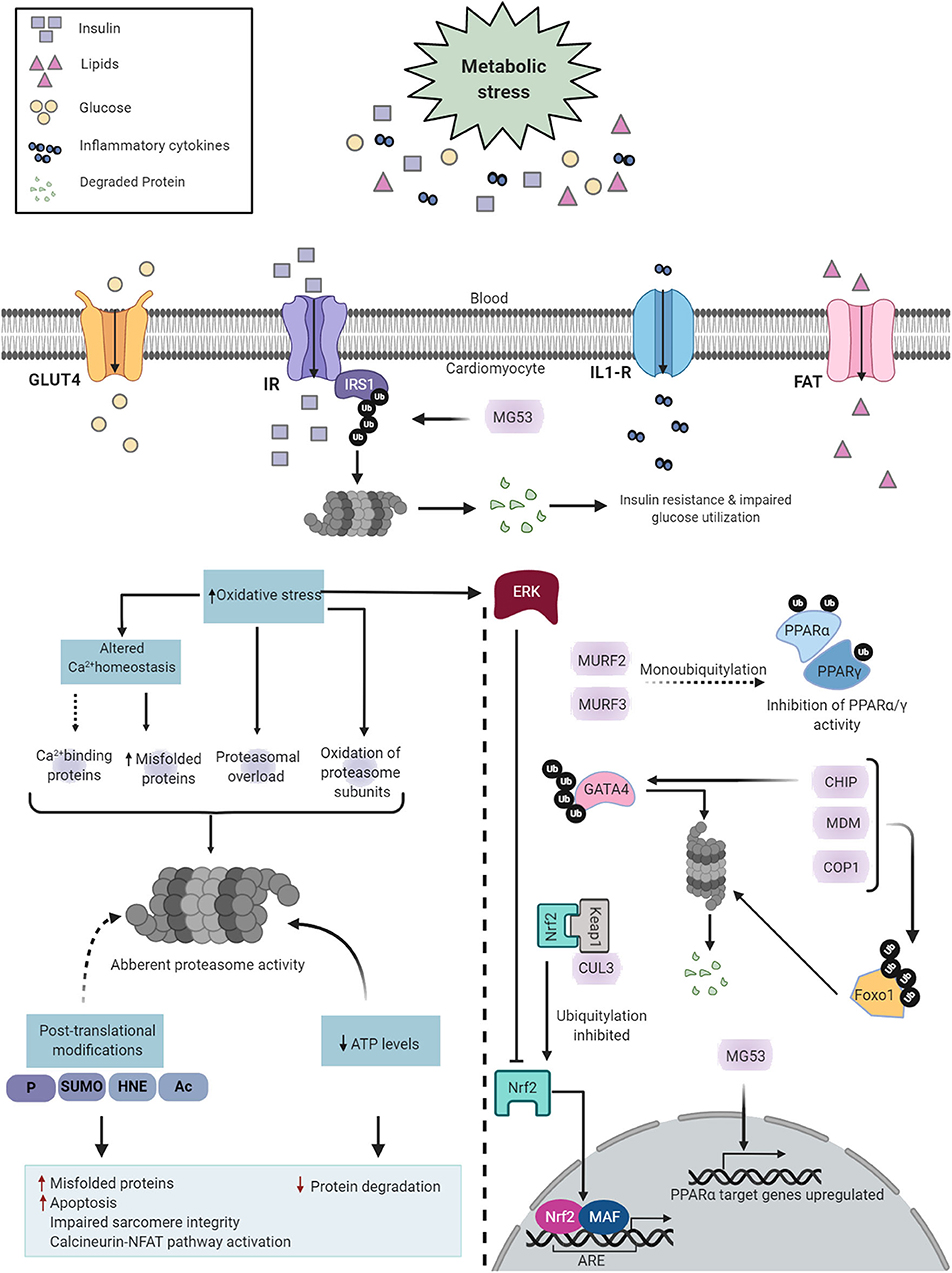
Figure 5. Diabetes-induced cardiac UPS dysfunction. Diabetes induces metabolic alterations in the heart that cause proteasome dysfunction in cardiomyocytes. Proteasome impairment may induce cardiomyopathy through multiple distinct mechanisms such as accumulation of misfolded proteins, enhanced apoptotic activity, contractile dysfunction and activation of calcineurin-NFAT pathway (Left). The E3 ligases regulate key transcription factors involved in DCM (Right). Created with Biorender.com.
In addition, GATA4, a member of the GATA zinc-finger transcription factor family, is abundantly expressed in the heart. GATA4 regulates the transcription of numerous cardiac genes, including those involved in myocyte growth and survival (209). In both STZ-induced type 1 diabetic mice and db/db type 2 diabetic mice, GATA4 protein levels are significantly diminished in the heart (210), which is likely associated with the E3 ubiquitin ligase, CHIP (210).
Furthermore, PPARα is a member of the PPAR subfamily of nuclear receptors and acts as a prominent regulator of myocardial fatty acid utilization (211). Transgenic mice with cardiac-specific PPARα overexpression showed cardiac insulin resistance, reduced glucose utilization, lipid accumulation, and cardiomyopathy (212). MG53, an E3 ubiquitin ligase, and MURF1 are both regulators of PPARα (186, 213). Increased protein levels of MG53 results in a DCM-like phenotype (186). Mechanistically, not only does MG53 deteriorate insulin sensitivity, it also positively regulates PPARα, thereby inducing an energy source shift of glucose to fatty acid oxidation (186). Of interest, both MG53 and PPARα were also elevated in the hearts of db/db mice, HFD-induced obese mice, and rhesus monkeys with a spontaneous metabolic syndrome characterized by obesity, hyperlipidemia, and hyperglycemia (186). Moreover, MURF2 and MURF3 attenuate cardiac PPAR isoform activities and protect against DCM in HFD-challenged mice (177, 179).
Finally, the nuclear factor erythroid 2-related factor 2 (NRF2) is the master regulator of the cellular antioxidant response. NRF2 exerts transcriptional action on antioxidant genes through binding to the antioxidant response element (ARE), such as quinone oxidoreductase 1 (NQO1) (214), heme oxygenase-1 (HO1) (215), and superoxide dismutase 1 (SOD1). In addition to its antioxidant capabilities, NRF2 also enhances the clearance of toxic ubiquitylated proteins in the heart (216, 217). KEAP1, as an adaptor of the CUL3-RBX1 E3 ubiquitin ligase, binds NRF2, leading to its ubiquitylation and subsequent proteasomal degradation (218, 219). Human diabetic hearts show a significant reduction in NRF2 protein expression (220), associated with early-onset maladaptive cardiac remodeling and heart failure (220, 221). Both oxidative stress and misfolded proteins synergistically contribute to DCM; therefore, KEAP1 and the CUL3-RBX1 E3 ubiquitin ligase complex represent promising therapeutic targets for diabetic heart disease.
Diabetes induces both structural and functional alterations in the proteasome (Figure 5). In a recent study, Li et al. reported that STZ-induced diabetic mice exhibit a severe and progressive decline in cardiac proteasome activity, evidenced by a cumulative increase in GFPdgn (UPS function reporter) and Lys48-linked ubiquitylated protein levels in the heart (222). These alterations in proteasome activity precede the onset of cardiac dysfunction and thus could potentially play a pathogenic role in diabetic heart disease. In line with this, proteasome functional insufficiency was also reported in the myocardium of Sprague–Dawley rats subjected to T1DM, accompanied by higher levels of ubiquitylated and oxidized proteins (223). Taken together, these studies suggest that diabetes induces dissonance in proteasome activity and thereby distorts myocardial proteostasis.
Although the regulatory events underpinning these observations remain largely elusive, there are multiple mechanisms by which diabetes could lead to proteasome dysfunction, such as ATP depletion, oxidative stress, calcium imbalance, and diabetes-induced posttranslational modifications. ATP is required for both ubiquitin conjugation and the activation of the proteasome (224). Cardiomyocytes subjected to severe ATP depletion manifested profound proteotoxicity and stress-induced cell death (225). As such, reduced ATP levels, as observed in diabetic hearts (226–228), likely contribute to proteasome dysfunction.
Likewise, oxidative damage to proteasome subunits affects proteasome activity (229). Oxidation of the 19S regulatory particles Rpt3 and Rpt5 impairs the degradative capacity of the 26S proteasome (230). Bulteua et al. demonstrated that oxidation of the 20S proteasome also blocks proteasomal peptidase activity (231). Consistently, treatment with the NSAID meclofenamate sodium resulted in increased oxidative stress and concomitant oxidation of proteasome subunits, reducing proteasome activity (159, 232). Moreover, mitochondrial dysfunction-associated accumulation of 4-HNE, a secondary product of lipid peroxidation, directly inhibits the proteasome activity in failing rat heart (233). On the other hand, hyperglycemia-induced oxidative stress promotes aberrant cellular Ca2+ homeostasis (234, 235), subsequently leading to the accumulation of misfolded proteins and proteasomal overload. Altered cellular Ca2+ concentrations may influence the activity of the proteasome more directly by modulating the activity of Ca2+-binding proteins that interact with the proteasome. For instance, calmodulin binds to several non-ATPase subunits of the 26S proteasome and could alter proteasome activity (236).
Cardiac proteasome activity is influenced by posttranslational modifications, such as SUMOylation, glycosylation nitrosylation, and phosphorylation (237), which could be modified by the diabetic myocardial environment (222). Accordingly, protein kinase CβII (PKCβII), a classical PKC isoform, phosphorylates, and inhibits the proteasome activity in failing rat hearts (238). Treatment with a PKCβII inhibitor improves cardiac PQC, function, and survival (238). Abnormal proteasome activity compromises cardiac function through numerous mechanisms (239). Primarily, proteasomal derailment induces cardiac contractile inefficiency by impairing sarcomeres (239). Two parallel processes, assembly, and degradation, are necessary to maintain sarcomere integrity (175). The degradation of sarcomeric proteins is regulated almost exclusively by the UPS (240). Also, proteasome inhibition activates the calcineurin-NFAT pathway in the heart (241), which induces pathological hypertrophic growth (242). Finally, proteasome inhibition has been shown to induce apoptosis in cultured cardiac myocytes (243, 244).
Autophagy is the homeostatic process through which cellular components are delivered to the lysosomes for degradation into their basic units. The cargo managed by the autophagic process includes insoluble and large misfolded proteins that cannot be degraded by the UPS (245), protein aggregates (246), and the proteasome itself (247). Autophagy can be triggered in the heart by various stress signals, such as nutrient deprivation, the absence of growth factors, and UPS malfunction (248).
Autophagy comprises three types of processes (249). Macroautophagy (hereafter referred to as autophagy) requires the formation of double-membrane vesicles, named autophagosomes, to sequester cytoplasmic components and organelles. Fully developed autophagosomes are fused with the lysosomes, where lysosomal hydrolases break down all the elements contained, including the inner membrane. The second type, microautophagy, is when cytoplasmic components are engulfed and degraded through the invagination of the lysosomal membrane. The third type, chaperone-mediated autophagy (CMA), is the process through which proteins exposing a KFERQ motif are translocated into the lysosomes. Approximately 75% of the human proteome has potential KFERQ motifs (250). Even though autophagy and microautophagy are bulk processes engulfing everything in a section of the cytoplasm, they also function selectively. Organelle (251) and protein aggregate (252) labeling consists of ubiquitination, a process shared with the UPS (253). This label is recognized by autophagy receptors such as p62/sequestosome 1 (SQSTM1), BCL2/adenovirus E1B 19 kDa protein-interacting protein 3 (BNIP3), and BNIP3-like (NIX) (254). Selective autophagy is vital to prevent proteotoxicity and promote cellular survival.
The autophagic molecular machinery consists of numerous autophagy-related proteins (ATG), directing all the stages of autophagy: initiation, elongation, maturation, and degradation (Figure 6A). Initiation begins with ATG1, also known as ULK1, forming the serine/threonine-kinase ULK complex. This complex phosphorylates the class III phosphatidylinositol 3-kinase complex I (PI3KC3-C1) containing beclin 1 (BECN1). The latter complex produces phosphatidylinositol 3,4,5-trisphosphate (PIP3), initiating the formation of the phagophore. Elongation is driven by the lipidation of microtubule-associated proteins 1A/1B light chain 3 (LC3) by ATG7 and the ATG5/ATG12 complex, during which LC3-I is conjugated with phosphatidylethanolamine to form LC3-II. This process allows the incorporation of LC3-II into the membrane and stabilization of the phagophore. At this stage, the autophagy receptors recognize labeled components and bind to LC3-II. When the autophagosome is closed, small GTPases of the Ras-related protein in brain (RAB) family recruit tethering proteins to anchor the autophagosome and the lysosome together, while snap receptor (SNARE) proteins and lysosome-associated membrane glycoprotein 2 (LAMP2) regulate their fusion (255), this is the maturation to autolysosome. A variety of enzymes in the autolysosome carry out the degradation process, after which the macromolecules are released into the cytosol for anabolic reactions.
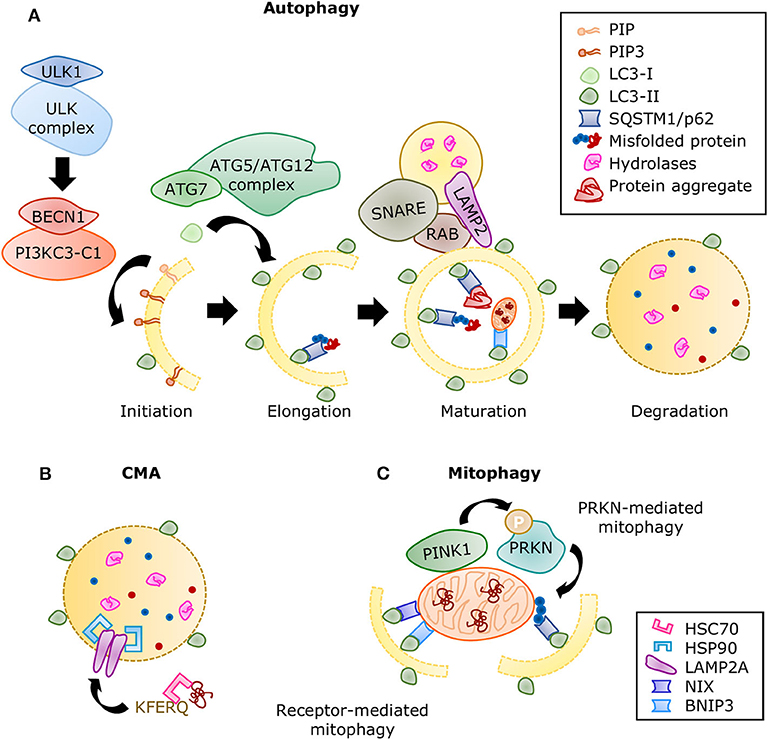
Figure 6. Summary of the autophagic processes. (A) A series of protein complexes drive autophagosome formation and fusion with the lysosome. The autolysosome breaks down misfolded proteins, protein aggregates, and organelles using a variety of hydrolases. Misfolded proteins are directed to selective autophagy through ubiquitination and detection by SQSTM1/p62. Adapted from Ciechanover et al. (10) and used under CC BY 3.0. (B) During CMA, proteins containing the KFERQ motif are recognized by the chaperone HSC70 and translocated directly into the lysosome by a complex of LAMP2A proteins stabilized with HSP90 chaperones. (C) Mitophagy is the selective degradation of nonfunctional mitochondria that have been labeled by the PINK1/PRKN system. Adaptor proteins such as SQSTM1 recognize the ubiquitinated proteins and linking it to the growing autophagosome. The receptor-mediated mitophagy is independent of PINK1/PRKN labeling, but directed by receptors, such as BNIP3 and NIX, which also contain LC3-binding domains.
Molecular chaperones also have a significant role in all three types of autophagy mediating selectivity and stability of the processes. If protein refolding fails, they can also direct cargo for degradation. The function of chaperone heat shock cognate 71kDa protein (HSC70) was first described in CMA. HSC70 recognizes the KFERQ motif in proteins and facilitates their translocation into the lysosome through LAMP2A (249). A second chaperone, heat shock protein (HSP90) enhances binding of the substrates and LAMP2A stability (256) (Figure 6B). Furthermore, HSC70 was later associated with the targeting of cytosolic proteins toward endosomal microautophagy (257) and chaperone-assisted selective autophagy (CASA), both of which can manage the degradation of protein aggregates (258). The substrate and process specificity of chaperones participating in different types of degradation is believed to come from the formation of complexes with co-chaperones, whose availability depends on cell type and stress or stimulus conditions. For example, BAG3 is a co-chaperone that interacts with HSP70 and HSPB8 to trigger selective autophagy of aggregated proteins (259). Its counterpart, BAG1, guides proteins toward proteasomal degradation. Several other co-chaperones have been described (260); however, their specific part in cardiac function is still being explored.
Basal autophagic activity in the myocardium is required to prevent the accumulation of misfolded proteins and the recycling of essential components from defective organelles to sustain cardiac function. Mutations in autophagy-related genes are the cause of dilated cardiomyopathies. The most common is Danon disease (261), a mutation of the LAMP2 gene, characterized by the weakening of the heart, protein aggregation, accumulation of autophagic vesicles in the muscle, and cardiac hypertrophy. The multisystemic disorders rising from loss-of-function mutations in the EPG5(262), PLEKHM2 (263), and BAG3 (264–266) genes have significant cardiovascular manifestations and are the result of defective autophagy. On the contrary, inducing autophagy ameliorates desmin-related cardiomyopathy by clearing the protein aggregates that originate from the mutation of the CRYAB gene (267). Additionally, cardiomyocyte-specific ATG5 deletion in mice, since birth (268) and in the adult stage (269), caused systolic dysfunction and sarcomeric structure disarray without any further stress. Conversely, augmenting basal autophagy by ATG5 overexpression (270) or BECN1 mutation to prevent its association with BCL2 (271) increased longevity in mice. A certain capacity for autophagic processing is elementary for normal cardiac function, and its sustenance counteracts proteotoxicity.
Cardiac autophagic flux is strongly induced by fasting (272), caloric restriction (273), exercise (274), and in neonatal mouse hearts after the placental supply is interrupted at birth (275), which is pivotal for cardiac contractility and survival (276, 277). Fasting and caloric restriction stimulate autophagy in the heart through 5′-AMP-activated protein kinase (AMPK) phosphorylation (272, 273) and increased SIRT1-mediated FOXO1 activity (278). Upon nutrient restoration after birth, insulin and amino acids moderate autophagy. Insulin signaling inhibits autophagy through AKT serine/threonine kinase 1 (AKT)-mediated activation of the mechanistic target of rapamycin kinase complex 1 (MTORC1), an autophagy inhibitor. When postnatal autophagy inhibition was disrupted by genetic deletion of the insulin receptors, cardiomyocyte death, and heart failure occurred due to excessive autophagy (279). Unrestrained autophagy was diminished by the supplementation of amino acids, which suppressed autophagy (279) through RAG protein family regulation of MTORC1 (280). On the other hand, in exercised mouse hearts, AMPK activation promoted the expression of autophagic genes (274) and dissociated the BCL2/BECN1 complex to increase autophagy level (281). However, in the long-term, exercise increased the autophagic capacity by augmenting LC3 expression in cardiac muscle without boosting autophagic flux (282). Nevertheless, this increased capacity limited cardiac injury and improved function after myocardial infarction (283, 284). These observations suggest that the regulation of autophagy by exercise is multifaceted and adaptive.
The role of autophagy in cardiac disease has been more challenging to determine, since it can be adaptive or maladaptive depending on the specific pathology and pathogenesis stage. The presence of abnormal protein aggregation in the myocardium of dilated cardiomyopathy patients was associated with impaired cardiac autophagy (12), while the detection of autophagic vacuoles was associated with improved heart failure prognosis (285). In preclinical studies, myocardial ischemia-induced autophagy in mouse (286) and swine models (287), moderating apoptosis, and autophagy induction, in turn, limited myocardial injury (272, 288, 289). Autophagy was reduced after prolonged pressure overload in mouse hearts, and ATG5 deletion aggravated cardiac remodeling and performance (269). These results indicate that autophagy is required to preserve cardiac function in response to pathological stresses. However, excessive BECN1 expression and autophagosome formation were found to be detrimental during reperfusion (290) and pressure overload (291), also in mouse studies. The seemingly confusing outcomes could be explained by the discovery that BECN1 association to the Rubicon protein was able to inhibit autophagic flux by interfering with autophagosome maturation (292), which has been recently termed autosis (293), a form of cell death. In addition, substantial evidence indicate that maladaptive autophagy was observed in atrial fibrillation by the degradation of cardiac troponin I/T (294) and calcium channel CAV1.2 (295), resulting in contractility and electrical alterations. Collectively, adaptive autophagy is essential to cardiac health, whereas either insufficient or excessive autophagy is detrimental.
The elucidation of the role of autophagy in DCM has been intricate due to the complexities of the disease and the duality of the nature of autophagy. In T2DM, autophagic flux is increased in the early stages of disease (296) and later reduced, with cardiac function improvement mostly being associated with therapeutic restoration of the autophagic flux (297–305). Increased expression of autophagy proteins was observed in human atrial samples and obese mouse myocardium, while fractional shortening was maintained (306). However, impaired autophagy by long-term chloroquine administration (300) or cardiac ULK1 deficiency was detrimental for cardiac function in obesity, resulting from fibrosis and apoptosis (297). On the contrary, boosting autophagic flux in a later stage by rapamycin administration improved systolic performance in high-fat diet (HFD)-induced diabetes (302). It has been suggested that autophagy contributes to high fructose-induced cardiomyopathy (307); nonetheless, these samples also displayed signs indicating that autophagic flux might have been blunted. In myocardial samples of obese and T2DM patients, amylin aggregates were detected and found to induce cardiac dysfunction (308). These aggregates, also known as islet amyloid polypeptide (IAPP) oligomers, disrupted autophagy-associated disposal, increasing their toxicity (309, 310). All together, autophagy is stimulated in T2DM stages with preserved cardiac function, while its abnormalities likely cause the onset and development of DCM and heart failure.
In T1DM models, the regulation and function of autophagy in the heart are elusive. Cardiac autophagy could be enhanced at an early time point (311); nonetheless, most evidence indicates it is suppressed. More importantly, preclinical experiments suggest that autophagy inhibition could be therapeutic in this case. Cardiac BECN1 overexpression in streptozotocin (STZ)-induced diabetes aggravated cardiac function (312). Conversely, autophagy reduction by BECN1 insufficiency and hypomorphic ATG16 improved echocardiographic measurements and hemodynamic analysis in the same model and in OVE26 mice, which develop severe early-onset T1DM due to deficient insulin production. Of note, even though autophagic flux was diminished, the functional improvements were accompanied by increased expression of RAB9 (312), which directed a non-canonical form of autophagy (313); therefore, it is speculated that non-canonical autophagy fulfills beneficial effects in T1DM. Akin to observations in T2DM patients (308), STZ-injected mice displayed toxic cardiac protein aggregation that can be improved by boosting autophagy (314). Interestingly, in T1DM and T2DM, CMA was found to be promoted even after autophagy was suppressed; however, evidence showed it could be contributing to metabolic inflexibility (7). As such, the molecular mechanisms underlying cardiac autophagy in DCM require further investigation.
Despite the distinct etiologies and biochemical stresses present in T1DM and T2DM, a few regulatory pathways have been demonstrated to be involved in autophagy suppression and the development of cardiac dysfunction (Figure 7). AMPK phosphorylation is reduced in the hearts of a number of DCM mouse models, including OVE26 (315), STZ-induced (316), diet-induced (302), and genetically obese mice. In OVE26 mice, the stimulation of AMPK phosphorylation by metformin restored autophagy and cardiac dysfunction (315). AMPK-mediated autophagy regulation is attained through numerous molecular mechanisms (316, 317), but mainly through repression of MTORC1 activity (302, 304, 318). MTORC1 suppresses autophagy by phosphorylating ULK1 (319, 320) and transcription factor EB (TFEB) (321), a master regulator of autophagy gene expression. Cardiac TFEB is suppressed in both T1DM and T2DM (322). AKT, mitogen-activated protein kinases 1 and 3 (ERK1/2), and the SIRT family are additional nodal points for autophagy regulation in DCM. AKT (57) and ERK1/2 (323) inhibited autophagic flux in the hearts of obese mice through MTORC1 activation. Strikingly, cardiac Akt2 knockout preserved cardiac function in high-fat diet-induced obesity by rescuing cardiac autophagosome maturation (301). In contrast, SIRT3 and SIRT1 were downregulated in both STZ-induced (324) and HFD-induced diabetic hearts (325). Consistently, SIRT3 (326) and SIRT1 (327) mediated the cardioprotective effects of resveratrol observed in T1DM by enhancing autophagic flux via activation of FOXO3A (324) and FOXO1 (327). As such, MTORC1, AKT, and ERK1/2 act as negative regulators of autophagy during the development of DCM, while AMPK and the SIRT family are considered as the enhancers.
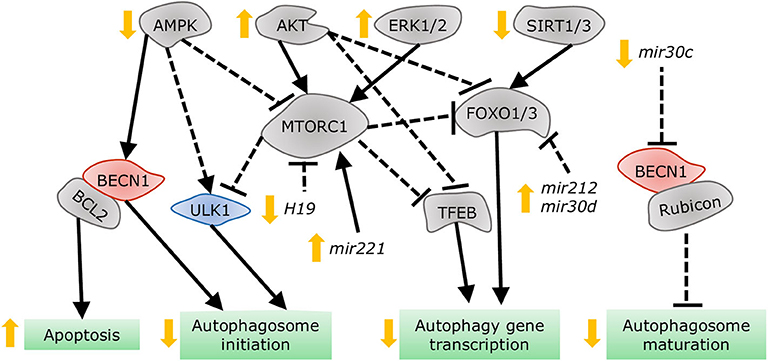
Figure 7. Dysregulation of autophagy in the diabetic heart. AMPK and SIRT1/3 inhibition, in addition to AKT and ERK1/2 stimulation, impair autophagy gene transcription, and autophagosome initiation via MTORC1, TFEB, and FOXO1/3 regulation. Decreased dissociation of the BECN1/BCL2 complex augments apoptosis, while increased association of BECN1 with Rubicon blunts autophagosome maturation. MiRNAs altered in diabetes interfere with autophagy by acting on players such as MTORC1, FOXO1/3, and BECN1.
More recently, non-coding RNAs have been found to be involved in numerous mechanisms underlying the development of DCM, with some of them regulating autophagy. Dysregulation of miRNAs was analyzed in the left ventricle of diabetic mice after STZ-injection, revealing that mir30a, mir133a, mir212, and mir221 are particularly associated with autophagy regulation. Among them, mir212 and mir221 were significantly upregulated in diabetic ventricles and even remained increased after insulin treatment (328), suggesting that they are likely involved in cardiac deterioration even with proper glycemic control. Mir212 targets Foxo3a (329), while mir221 targets p27 mRNA, modulating MTORC1 activity (330), both of which inhibit autophagy. Additionally, mir30d also targets Foxo3a to suppress autophagy in DCM (331). On the other hand, mir30c targets Becn1, and cardiac overexpression of this miRNA improved cardiac function in genetically obese mice (332), possibly by decreasing BECN1-Rubicon association (292) and improving autophagosome clearance. Lastly, the long non-coding RNA H19 is downregulated in the heart of STZ-induced diabetic rats, allowing for increased MTORC1 signaling and suppressed autophagy (333). Therefore, emerging evidence demonstrates that non-coding RNA regulation of autophagy also acts as potential therapeutic targets for treating DCM.
Even though mitochondria possess their own UPR, proteotoxic stress or damage can surpass their capacity, requiring a more robust response. Mitochondria can be selectively targeted for autophagy. Without adequate mitochondrial clearance, remaining damaged mitochondria are unable to meet ATP demand, produce excessive ROS, and promote cell death and inflammation (195). Mitochondrial autophagy, termed mitophagy, is directed by the serine/threonine-protein kinase PINK1 (PINK1) phosphorylating the E3 ubiquitin-protein ligase PRKN (PRKN) and fostering mitochondrial protein ubiquitination. The adaptor proteins SQSTM1, neighbor of BRCA1 (NBR1), nuclear dot protein 52 (NDP52), optineurin (OPTN), TAX1BP1 (TBK1), BNIP3, and NIX link damaged mitochondria to autophagosomes for their degradation (334) and can sometimes induce PINK1-independent mitophagy (Figure 6C). PINK1 deletion results in mitochondrial dysfunction, oxidative stress, and cardiomyopathy (335). Similarly, knocking out cardiac PRKN in mice accelerated the development of systolic dysfunction after HFD-feeding, accompanied by the accumulation of dysfunctional mitochondria and lipids (296). ULK1 (336) and RAB9-dependent (312) mitophagy has also been observed in the hearts of diabetic mice, and its impairment aggravated cardiac function. However, the levels of these proteins continued to increase when diastolic (336) and systolic dysfunction (312) were detected; therefore, there is doubt on whether alternative mitophagy could be sufficient to sustain cardiac function during metabolic stress.
Impaired mitophagy is a major contributor to the pathogenesis of DCM. In a compensated stage of HDF feeding, mitophagy is upregulated by the general autophagy ATG7-dependent mechanism, and disrupting mitophagy by deleting cardiac PRKN expression accelerates the appearance of cardiac dysfunction (296). Consistently, PINK1 and PRKN levels were found to be downregulated in the myocardium of STZ-induced and OVE26 diabetic mice with systolic dysfunction (312). Different mechanisms have been proposed to contribute to the loss of mitophagy after metabolic stress. MST1 was found to inhibit cardiac mitophagy in the hearts of diabetic mice via SIRT3- mediated PRKN suppression (196). SIRT3 and PRKN activities were ameliorated by melatonin (337) and icariin administration (338), resulting in improved mitochondrial function. In pancreatic islets and hepatocytes from obese mice, an increase in P53 protein suppressed mitophagy by direct interaction of P53 and PRKN, blunting mitochondrial uptake by autophagosomes (339, 340). Lipid metabolism was associated to HFD-induced PRKN reduction, given that stimulation of lipid catabolism by overexpressing acetyl-coA 2 (ACC2) restored mitophagy and cardiac function in mice (341). Adequate mitochondrial morphology and dynamics are also vital to facilitate mitophagy during DCM. HFD feeding induced dynamin-related protein 1 (DRP1) activity in the hearts of mice and monkeys. DRP1 is the primary regulator of mitochondrial fission, and its increased activity suppressed mitophagy and resulted in cardiac inflammation and heart failure (342, 343). On the contrary, myocardial samples of ob/ob mice showed reduced levels of mitofusin 2 (MFN2), the lead regulator of mitochondrial fusion. Restoration of MFN2 expression in cardiomyocytes exposed to high-glucose and high-fat treatment recovered mitochondrial membrane potential and function (344). MFN2 also promotes PRKN translocation and mitophagy in cardiomyocytes (345). Albeit the need to continue exploring the mechanisms governing mitophagy during DCM, its role in maintaining mitochondrial quality has been acknowledged.
The different PQC systems act as an integrated stress response. They are interconnected and regulate each other at the transcriptional and protein level, and this interdependence is relevant for health and disease. For instance, the UPS downregulates autophagy by processing transcription factors, such as P53, NFkB, HIF, and FOXO (346), and autophagy components, such as BECN1 (347, 348), LC3, p62, and ULK1(346). In cancer cells, chemotherapy resistance to bortezomib, a proteasomal inhibitor, arises from the induction of autophagy as a compensatory mechanism (349). The combination of bortezomib with hydroxychloroquine improved the treatment outcome (350). In turn, proteasomes can be degraded by autophagy. Amino acid starvation-induced autophagic activity also enhances polyubiquitination of 19S regulatory particle, targeting it for autophagic uptake and decreasing proteasomal activity level (351, 352).
Similarly, the UPRER components, ATF6, CHOP and IRE1 are degraded by the UPS, while two UPRER branches, PERK-ATF4 and IRE1-XBP1 regulate the expression of UPS components (346). IRE1 is handled through direct interaction with the ERAD complex SEL1-HRD1 and during ER stress, this interaction is broken for UPRER initiation (353). At the same time, IRE1-XBP1 pathways stimulate the transcription of SEL1L and HRD1 for further UPS function of misfolded proteins (354). This self-modulating feedback loop prevents overactivation and ER-mediated cell death. In db/db mice, cardiac expression of HRD1 is reduced, suggesting blunted ERAD activity contributes to prolonged ERSR (355). On the contrary, in doxorubicin-induced cardiomyopathy, UPS activity was observed to be increased (168, 169), perhaps furthering the impairment of UPRER function (356). Even though the goal of all PQC system is the restoration of protein homeostasis, the impact of each one in cellular function under stress conditions differs.
Clear links between the UPRER, UPS, and autophagy have been acknowledged; however, few of them have been explored in the diabetic heart. Both IRE1 and PERK branches of the UPRER induce autophagy by promoting BECN1/BCL2 dissociation and upregulating autophagy genes, such as ATG12, BECN1, and LC3. In addition, ER calcium release can stimulate the Ca2+/calmodulin-dependent kinase kinase β (CaCMKKβ) that phosphorylates and activates AMPK, resulting in autophagy stimulation (357). UPS malfunction has also been found to provoke autophagy through NRF2-mediated SQSTM1 upregulation (358) and calcineurin-TFEB activation (165), suggesting that autophagy acts as a compensatory mechanism upon proteasomal insufficiency. Proteasomal insufficiency was detected in diabetic hearts previous to cardiac dysfunction (UPS section); therefore, it is possible that these mechanisms contribute to autophagy induction in early stage of DCM. On the other hand, autophagy suppression inhibits UPS function due to the accumulation of SQSTM1 that drives the excessive sequestration of ubiquitinated proteins in protein aggregates, preventing proteasomal degradation (359). Autophagy inhibition in late DCM could be aggravating UPS malfunction and further damaging cardiac function. The exploration of these crosstalks in the heart of diabetes patients and animal models could clarify the status of PQC components and regulatory mechanisms.
Proper regulation of the PQC system in the myocardium is vital to maintain cardiac physiology and preserve heart performance in response to pathological stresses. Studies on the alterations and regulation of PQC in the heart in the fact of diabetes mellitus have provided new insights into the molecular pathogenesis of DCM, as well as delivered proof-of-concept evidence that the fine-tuned modulation of the UPRER, UPS, and autophagic event is a potential therapeutic strategy to treat DCM and prevent heart failure in the diabetic populations.
As a metabolic disorder, DCM embodies subsequent UPRER events, where protective ER response is dominant in early stages, followed by decreased UPRER signaling, and ultimately, irreversible ERSR associated with structural abnormalities in the myocardium (360). Therefore, potentiating initial UPRER activation to restore protein homeostasis and impeding ER apoptotic response to prevent cell death qualifies as a therapeutic strategy for managing ER stress in DCM (17, 51). Interestingly, exercise, a known physiological UPRER inducer, mitigated apoptotic ERSR by reducing CHOP and caspase 12 expression, and augmented cardiac function in STZ induced type 1 diabetes (361). This suggests that exercise is beneficial by restoring protein homeostasis.
Modulation of the UPRER sensors and GRP78 restores cellular homeostasis and improve heart function in multiple cardiovascular disorders. Although GRP78 overexpression is cardioprotective in hypoxia-induced injury (362), normalizing GRP78 is shown to be beneficial for disorders with overactivated UPRER such as DCM. Moreover, chemically enhancing IRE1-XBP1 (363) and ATF6 activity (364) reduced ER-associated apoptosis following myocardial infarction. Contrastingly, IRE1 and PERK inhibition alleviated atherosclerosis development and decreased cell death in cardiac arrhythmias, respectively. As stated above, PERK and ATF6 hyperactivation are deleterious in the diabetic heart, indicating that our UPRER knowledge is incomplete in the context of different cardiac etiologies. Nonetheless, the pharmacological modulation of UPRER signaling following numerous diabetic stresses has dramatically increased in the past few years (Table 2).
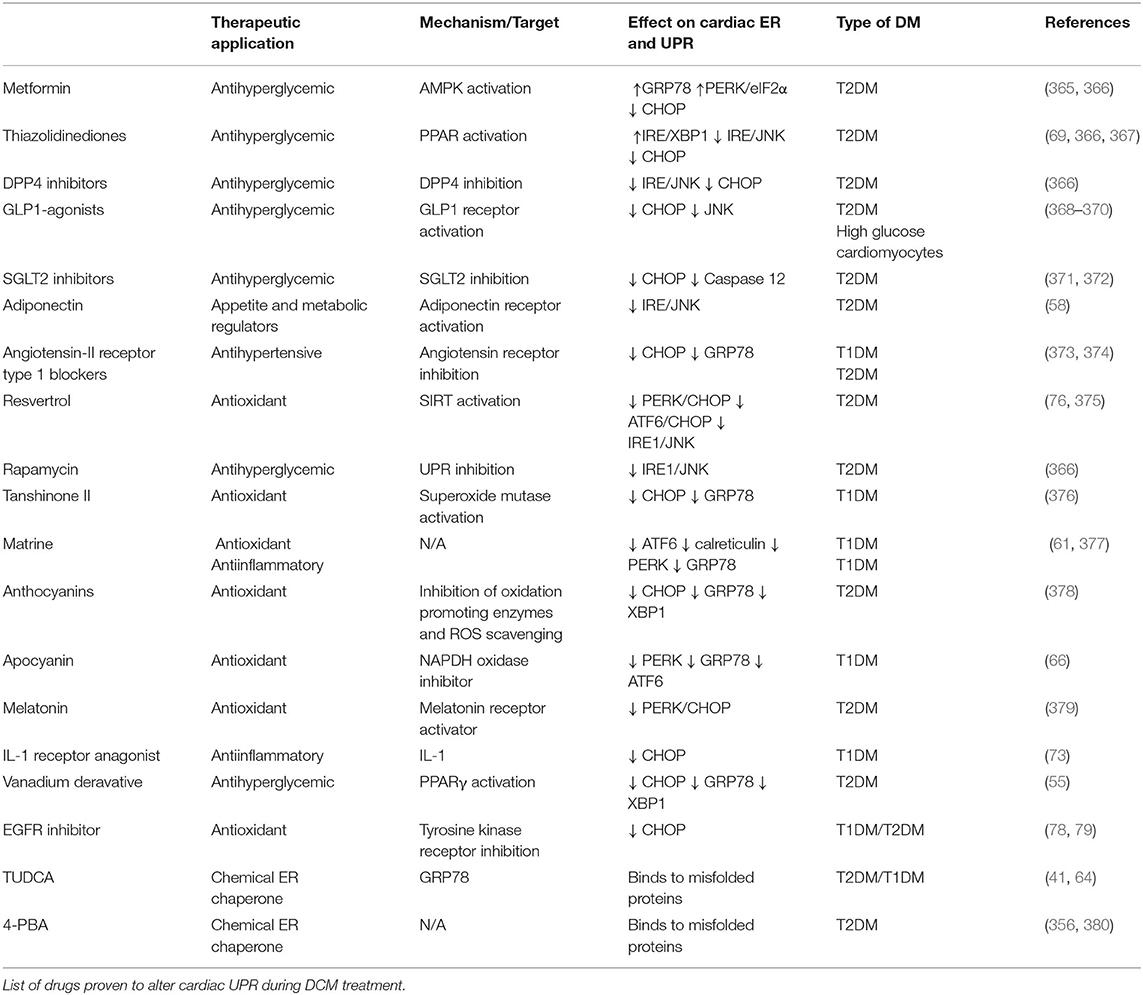
Table 2. UPR-targeting drugs.
Poor glycemic control is associated with increased ER stress and decreased function in the diabetic heart. Sodium-glucose cotransporter 2 (SGLT2) inhibitors as an effective-glucose lowering therapy showed robust cardioprotective outcomes in clinical trials (371) and pre-clinical studies (372) by reduced ER-mediated apoptosis following oxidative stress. However, glucagon-like peptide 1 (GLP-1) agonists are shown to exacerbate heart failure or have no significant cardiovascular outcome in T2DM patients (371). GLP-1 agonists are cardioprotective in DCM rodent models by inhibiting UPRER signaling and ER-mediated apoptosis (368–370, 381). Therefore, these drugs fall short of mitigating heart failure in diabetes patients, possibly due to inhibition of adaptive ERSR. Also, metformin (382) and thiazolidinediones (TZD) (367) lower cardiovascular events in T2DM patients by its antihyperglycemic effects. Meanwhile, in pre-clinical studies, metformin also induced the protective UPRER function (365, 366), and TZDs improved insulin sensitivity by upstream mediated attenuation of inflammation and ER-associated apoptosis (69), thereby ameliorating cardiac function in diabetes. Taken together, clinically antihyperglycemic drugs fulfil cardioprotective role in DCM, although their function on UPRER needs to be further confirmed.
Multiple approaches are being employed to improve cardiac function by the administration of anti-ER stress chemicals, which may facilitate UPRER action (60). Chemical chaperones restored the UPRER, which attenuates maladaptive ERSR under pathological stresses (383), including diabetes (71, 384). 4-phenyl butyric acid (4-PBA) and tauroursdoeoxycholic (TUDCA) improved heart function in doxorubicin-induced cardiomyopathy (356), emphasizing their potential as cardioprotective drugs. Moreover, these chaperones can reduce ER protein load in cardiomyocytes by reducing fatty acid uptake (380) and normalizing GRP78 expression (64) in T2DM rat models. Additionally, TUDCA is currently employed in three clinical trials in amyloid cardiomyopathy associated with the onset of type 1 diabetes (385). Of note, given the ubiquitous nature of UPRER signaling, these multi-organ targeting drugs may have off-target effects. Therefore, further clarification of specific drug targets is of considerable significance to improve the efficacy of these drugs as DCM therapy.
Other strategies to target ER stress as DCM therapy include antihypertensives, antioxidants, and antiinflammatory compounds. Besides the metabolic alterations, increased angiotensin II signaling in diabetes also induces ER stress in the heart (60). The antihypertensive drugs, such as valsartan, are shown to downregulate CHOP expression and reduce cardiac remodeling in DCM (373, 374). On the other hand, phytochemicals, such as matrine, have attracted attention in attenuating maladaptive ERSR (376) and preserving UPRER signaling (61, 377), subsequently improving cardiac function in STZ-induced DCM. Moreover, vanadium derivative (55) and endogenous hormones, such as melatonin (379) and FGF21 (386, 387), have been investigated for their cardioprotective role by suppressing oxidative stress-mediated ERSR and cell death in T2DM. Furthermore, targeting upstream regulators of UPRER signaling, such as SIRT1 (76, 375), might be beneficial as DCM therapy. Therefore, these molecules may be further developed as novel therapeutic agents with clinical efficacy to target UPRER signaling in DCM. In conclusion, since targeting UPRER signaling is a two-edged sword, proper UPRER regulation is essential to restore protein homeostasis in the cardiomyocytes, while inappropriate suppression of ERSR may have unpredictable effects on cardiac function in DM populations.
Given its indispensable role in maintaining cellular proteostasis, the proteasome is a potent therapeutic target to treat proteotoxic stress in the heart. Benign enhancement of proteasomal function by overexpression of the 11S proteasomal subunit PA28α markedly reduced aberrant protein aggregation and cardiac hypertrophy in a mouse model of CryABR120G proteinopathy (388). Likewise, cardiac-specific proteasome enhancement partially improved right ventricular dysfunction and survival in mice subjected to pressure overload (389). More recently, Li et al. reported that restoration of proteasome function facilitated by PA28α overexpression preserves cardiac hemodynamics and ameliorates diabetes-induced pathological cardiac remodeling in STZ-induced diabetic mice (222). These salient findings suggest that genetic proteasome enhancement restores PQC and improves cardiac function in response to various pathological conditions, including metabolic stress.
Pharmacological stimulation of cAMP-PKA and cGMP-PKG signaling by phosphodiesterase (PDE) inhibitors can also activate cardiac proteasome activity. The synthesis of cAMP and cGMP is mediated by adenylyl cyclases or guanylyl cyclases, respectively, whereas their degradation is mediated by PDEs (149). Thus, inhibiting PDEs increases cellular levels of cAMP and cGMP. Eleven PDE families have been identified; among them, PDE1, PDE2, PDE3, PDE4, PDE5, and PDE8 are expressed in the heart (390). Ranek et al. (391) demonstrated that sildenafil, an FDA approved PDE5 inhibitor functioning on activation of PKG, stimulates proteasome peptidase activity, enhances the clearance of misfolded proteins, and decreases aberrant protein aggregation, thereby improving cardiac proteostasis. More recently, it has been reported that pharmacological inhibition of PDE1 (IC86430) increases cardiac proteasome function and accelerates proteasomal degradation of aberrant myocardial proteins in a PKA- and PKG-mediated manner (392). Strikingly, the administration of IC86430 at an overt disease stage markedly improved diastolic function and delayed premature death in CryABR120G mice (392). Conclusively, pharmacological enhancement of proteasome activity by stimulating PKA or PKG is likely a novel strategy to treat DCM by eliminating aggregation of damaged proteins and alleviating cellular proteotoxicity.
Due to their ability to regulate UPS, the E3 ubiquitin ligases represent promising drug targets for patients with diabetic heart disease. Hydrogen sulfide (H2S) is a gastotransmitter to maintain cardiovascular homeostasis, which is blunted in various cardiovascular diseases, including DCM (355). H2S primarily signals through a specific protein modification termed sulfhydration, whereby the thiol group of a reactive cysteine is converted to an hydropersulfide (–SSH) group (393, 394). Recent work by Yu et al. (355) has shown that exogenous H2S reduces translocation of the free fatty acid (FFA) transporter CD36 from intracellular stores to the plasma membrane by promoting sulfhydration of the ER-resident ubiquitin ligase, HRD1; thereby attenuating myocardial fatty acid uptake and lipotoxicity in db/db mice. More specifically, HRD1 S-sulfhydration regulates the ubiquitylation of VAMP3 (involved in CD36 trafficking) and promotes its degradation. Interestingly, H2S-generating compounds have been tested in various preclinical models of heart disease (395). For instance, SG-1002, an orally active H2S prodrug, attenuates cardiac dysfunction in HFD-induced type II diabetic mice (394). Of note, SG-1002 has been clinically investigated in patients with cardiovascular disease (396). Thus, H2S may hold therapeutic potential for the treatment of DCM.
Additionally, AMPK has been shown to regulate the transcription of two ubiquitin ligases in the heart; Atrogin-1 and MURF1 (397). AMPK activation leads to increased rates of UPS-mediated protein degradation, thereafter increasing amino acid availability for protein synthesis or ATP production as the heart adapts to a deteriorating metabolic milieu (397). In the diabetic heart, AMPK-mediated protein lysis is cardioprotective due to preserved PQC (22, 397). Numerous studies have reported that metformin improves clinical outcomes in patients with diabetic heart failure by activating AMPK (398–400); however, whether and how metformin regulates cardiac UPS in the progression of DCM requires further investigation.
Autophagy has been targeted for the treatment of cardiovascular disease; however, in DCM, it is not yet determined whether the induction or inhibition of autophagy has potential treatment effects. Preventive and therapeutic advice in diabetes includes lifestyle modifications, such as exercise and caloric restriction, which have been shown to induce cardiac autophagy and diminish the risk of cardiac events (401). Caloric restriction is sometimes supported by the prescription of appetite suppressors, among which adiponectin (302, 402), leptin (403), and GLP1 receptor agonists (404, 405) have also been proved to induce cardiac autophagy in diabetic models.
Pharmacological treatment for DCM relies mostly on the attenuation of the systemic derangements that lead to cardiac stress; nevertheless, several of these treatments are also able to activate autophagy in cardiac cells (Table 3). As mentioned above, one of the most widely prescribed insulin sensitizers is metformin. This AMPK activator improved cardiac function in animal DCM (315) and human heart failure (400). Similarly, improving glycemic control by other insulin sensitizers (302, 408–412), stimulating insulin secretion (413, 414, 416–418), or reducing glucose uptake in the renal tube (419–421) has cardioprotective effects, also restoring myocardial autophagy in DCM models. Lipid-lowering treatments (422, 423) and antihypertensives (424, 428, 452) enhanced myocardial autophagy in preclinical models of T1DM and T2DM through different molecular mechanims, such as calcium-mediated autophagosome-lysosome fusion and receptors modulation. Despite some conflicting results (453), the antioxidant supplements, resveratrol (327, 432, 433, 454, 455), spermidine (437–440), and epigallocatechin gallate (EGCG) (441–444) protected the heart in clinical and preclinical studies in both types of diabetes, also by regulating myocardial autophagy. While the systemic effects of these treatments are essential, the cardiac-specific regulation of autophagy is necessary for the maintenance of cardiac function in diabetes. Further research is required to delineate which drugs in each class are the most beneficial.
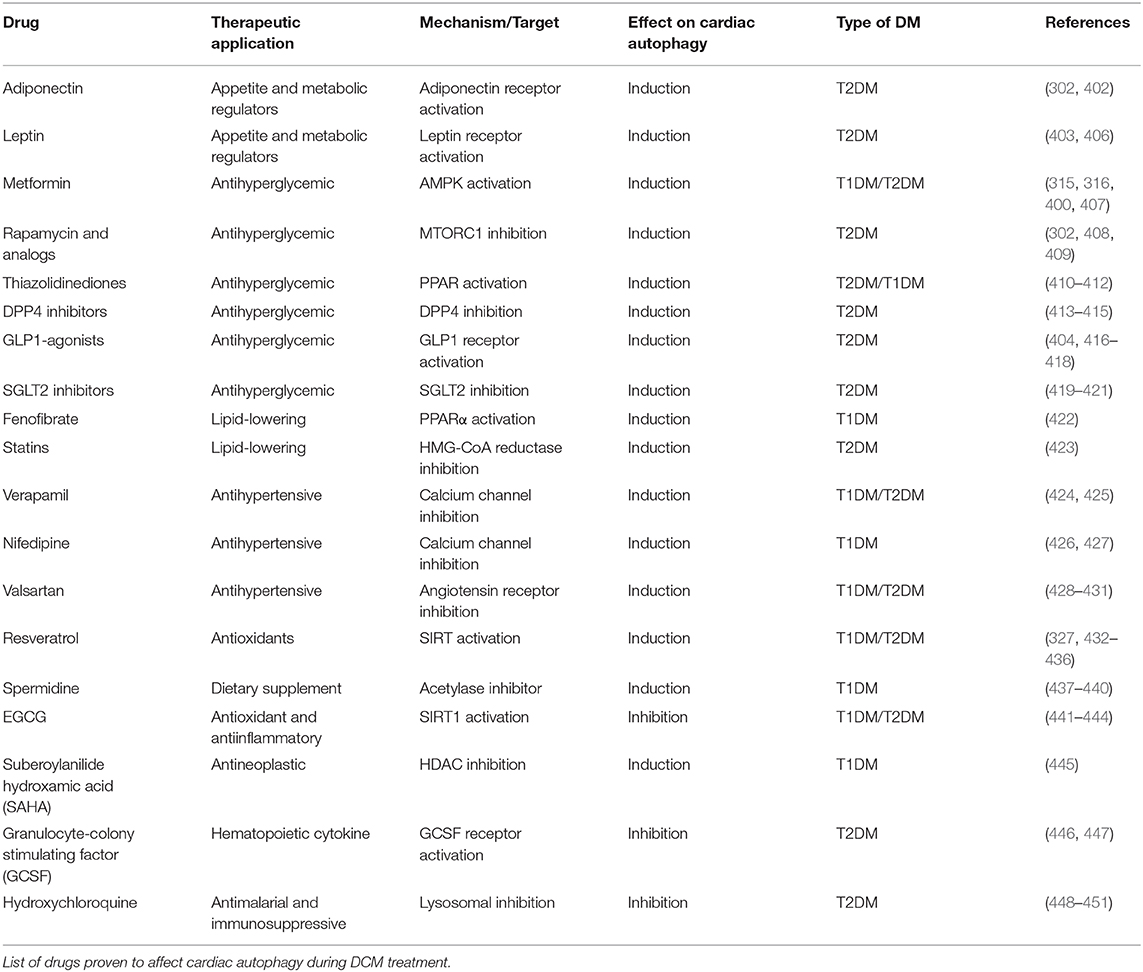
Table 3. Autophagy-targeting drugs.
Some nondiabetic treatments also display benefits in cardiovascular health in diabetes through autophagy regulation. Suberoylanilide hydroxamic acid (SAHA), a histone deacetylase inhibitor, restored cardiomyocyte contractility in STZ-injected rats (445) and can promote cardiac autophagy (446). On the other hand, the granulocyte-colony stimulating factor (GCSF) stimulates bone marrow function and ameliorated diastolic dysfunction in rodent T2DM models (447, 456, 457) by downregulating autophagy (448). The antimalarial drug and lupus treatment hydroxychloroquine is mostly associated with high risks of cardiotoxicity and heart failure (458); nevertheless, it improved β-cell function in obese nondiabetic patients (449) and reduced glycemic levels in T2DM patients (450). Hydroxychloroquine represses autophagy by accumulating in lysosomes and inhibiting autophagosome fusion (459). Considering this, it remains to be explored whether reduced autophagy might be a therapeutic strategy in early stages of disease and whether targeting cardiac autophagy is an adjuvant strategy for the current metabolic treatments to prevent DCM and heart failure in the diabetic populations.
Accumulating evidence on the molecular pathogenesis of DCM has revealed the essential roles of proper cellular protein quality control in diabetes-associated heart disease. Upon diabetic stress, the PQC machinery senses, detects, and disposes of the damaged proteins by multiple processes, including the UPR, the UPS, and autophagy. Concerted action of the three cellular systems can tackle proteotoxicity, subsequently improving the cardiac outcome in diabetes; accordingly, compromised PQC mechanisms appear to contribute to heart disease as a result of impaired cellular homeostasis. Dysregulation of the UPR results in the accumulation of misfolded proteins, while malfunction of the UPS and autophagy lead to aggregation of toxic proteins in the cytosol, all of which triggers cell death and provokes the onset and development of heart failure in diabetes. Therefore, it is suggested that the maintenance of protein homeostasis may be a valuable and promising therapeutic strategy to treat cardiac complications in diabetes patients. Of note, either insufficient or exaggerated action of the principal mechanisms exacerbates cytotoxicity in the face of pathological stresses, including diabetic stress (Figure 8). Given this dual role in the heart, finely tuned manipulation of the UPR, the UPS, and autophagy in the myocardium is mandatory to maintain cellular equilibrium in response to the diabetic condition with increased demand of protein turnover. As such, despite the growing knowledge of the mechanisms underlying cardiac proteostasis networks, further research is indispensable to investigate the therapeutic potential for heart disease by targeting PQC molecules in diabetes mellitus. As the understanding of molecular regulation of PQC function develops, so will our capability to exploit pharmacological interventions to prevent proteotoxicity and cardiac dysfunction in diabetic populations.
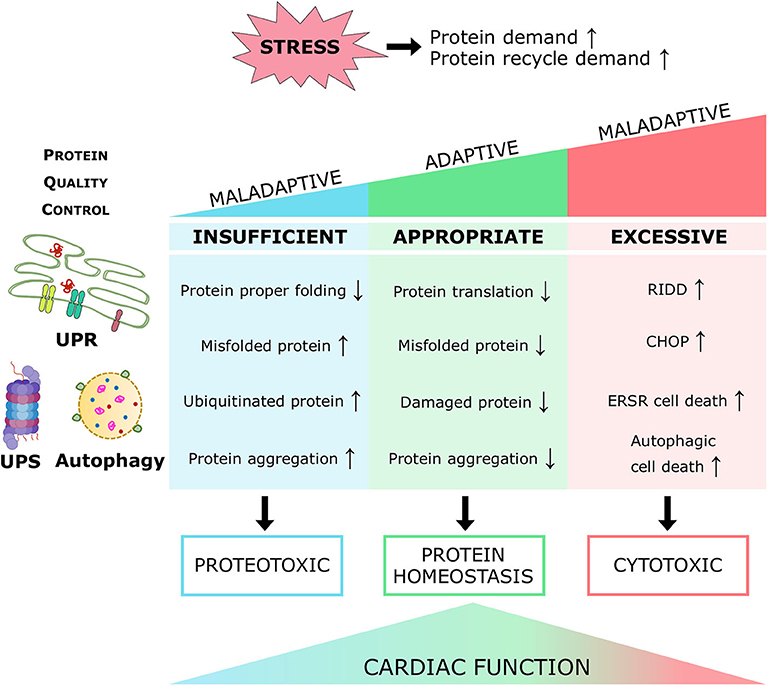
Figure 8. Role of PQC systems in cardiac function under pathological stress. Either insufficient or excessive activity of the main PQC systems is maladaptive. By inducing protein or cellular toxicity, they contribute to cardiac dysfunction. Appropriate level of PQC maintains cardiac protein homeostasis and sustains cardiac function.
NK, RR, and AR-V collected references, generated tables and figures, and drafted the manuscript. AR-V and WL designed the work, wrote, and proofread the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by grants FS/15/16/31477, FS/18/73/33973, PG/19/66/34600, and FS/19/70/34650 to WL from the British Heart Foundation.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. IDF Diabetes Atlas, 9th edn. Federation ID, editor. Brussels: International Diabetes Federation (2019).
2. Kannel WB, Hjortland M, Castelli WP. Role of diabetes in congestive heart failure: the Framingham study. Am J Cardiol. (1974) 34:29–34. doi: 10.1016/0002-9149(74)90089-7
3. Preis SR, Pencina MJ, Hwang S-J, D'Agostino RB, Savage PJ, Levy D, et al. Trends in cardiovascular disease risk factors in individuals with and without diabetes mellitus in the Framingham Heart Study. Circulation. (2009) 120:212–20. doi: 10.1161/CIRCULATIONAHA.108.846519
4. Rubler S, Dlugash J, Yuceoglu YZ, Kumral T, Branwood AW, Grishman A. New type of cardiomyopathy associated with diabetic glomerulosclerosis. Am J Cardiol. (1972) 30:595–602. doi: 10.1016/0002-9149(72)90595-4
5. Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE, Drazner MH, et al. 2013 ACCF/AHA guideline for the management of heart failure. J Am Coll Cardiol. (2013) 62:e147–e239. doi: 10.1016/j.jacc.2013.05.019
6. Jia G, Hill MA, Sowers JR. Diabetic cardiomyopathy. Circ Res. (2018) 122:624–38. doi: 10.1161/CIRCRESAHA.117.311586
7. Tan Y, Zhang Z, Zheng C, Wintergerst KA, Keller BB, Cai L. Mechanisms of diabetic cardiomyopathy and potential therapeutic strategies: preclinical and clinical evidence. Nat Rev Cardiol. (2020) 17:585–607. doi: 10.1038/s41569-020-0339-2
8. Wang ZV, Hill JA. Protein quality control and metabolism: bidirectional control in the heart. Cell Metab. (2015) 21:215–26. doi: 10.1016/j.cmet.2015.01.016
9. Yang L, Zhao D, Ren J, Yang J. Endoplasmic reticulum stress and protein quality control in diabetic cardiomyopathy. Biochim Biophys Acta. (2015) 1852:209–18. doi: 10.1016/j.bbadis.2014.05.006
10. Ciechanover A, Kwon YT. Degradation of misfolded proteins in neurodegenerative diseases: therapeutic targets and strategies. Exp Mol Med. (2015) 47:e147. doi: 10.1038/emm.2014.117
11. Melber A, Haynes CM. UPRmt regulation and output: a stress response mediated by mitochondrial-nuclear communication. Cell Res. (2018) 28:281–95. doi: 10.1038/cr.2018.16
12. Henning RH, Brundel BJJM. Proteostasis in cardiac health and disease. Nat Rev Cardiol. (2017) 14:637–53. doi: 10.1038/nrcardio.2017.89
13. Chen B, Retzlaff M, Roos T, Frydman J. Cellular strategies of protein quality control. Cold Spring Harb Perspect Biol. (2011) 3:a004374. doi: 10.1101/cshperspect.a004374
14. Hwang J, Qi L. Quality control in the endoplasmic reticulum: crosstalk between ERAD and UPR pathways. Trends Biochem Sci. (2018) 43:593–605. doi: 10.1016/j.tibs.2018.06.005
15. Moon HW, Han HG, Jeon YJ. Protein quality control in the endoplasmic reticulum and cancer. Int J Mol Sci. (2018) 19:3020. doi: 10.3390/ijms19103020
16. McLendon PM, Robbins J. Proteotoxicity and cardiac dysfunction. Circ Res. (2015) 116:1863–82. doi: 10.1161/CIRCRESAHA.116.305372
17. Dubnikov T, Ben-Gedalya T, Cohen E. Protein quality control in health and disease. Cold Spring Harb Perspect Biol. (2017) 9. doi: 10.1101/cshperspect.a023523
18. Brodsky JL. Cleaning up: ER-associated degradation to the rescue. Cell. (2012) 151:1163–7. doi: 10.1016/j.cell.2012.11.012
19. Kozutsumi Y, Segal M, Normington K, Gething MJ, Sambrook J. The presence of malfolded proteins in the endoplasmic reticulum signals the induction of glucose-regulated proteins. Nature. (1988) 332:462–4. doi: 10.1038/332462a0
20. Groenendyk J, Agellon LB, Michalak M. Coping with endoplasmic reticulum stress in the cardiovascular system. Annu Rev Physiol. (2013) 75:49–67. doi: 10.1146/annurev-physiol-030212-183707
21. Arrieta A, Blackwood EA, Stauffer WT, Glembotski CC. Integrating ER and mitochondrial proteostasis in the healthy and diseased heart. Front Cardiovasc Med. (2020) 6:193. doi: 10.3389/fcvm.2019.00193
22. Wang X, Robbins J. Heart failure and protein quality control. Circ Res. (2006) 99:1315–28. doi: 10.1161/01.RES.0000252342.61447.a2
23. Tarone G, Brancaccio M. Keep your heart in shape: molecular chaperone networks for treating heart disease. Cardiovasc Res. (2014) 102:346–61. doi: 10.1093/cvr/cvu049
24. Hetz C, Zhang K, Kaufman RJ. Mechanisms, regulation and functions of the unfolded protein response. Nat Rev Mol Cell Biol. (2020) 21:421–38. doi: 10.1038/s41580-020-0250-z
25. Lindholm D, Korhonen L, Eriksson O, Koks S. Recent insights into the role of unfolded protein response in ER stress in health and disease. Front Cell Dev Biol. (2017) 5:48. doi: 10.3389/fcell.2017.00048
26. Ghemrawi R, Battaglia-Hsu SF, Arnold C. Endoplasmic reticulum stress in metabolic disorders. Cells. (2018) 7:63. doi: 10.3390/cells7060063
27. Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. (2011) 334:1081–6. doi: 10.1126/science.1209038
28. Maamoun H, Abdelsalam SS, Zeidan A, Korashy HM, Agouni A. Endoplasmic reticulum stress: a critical molecular driver of endothelial dysfunction and cardiovascular disturbances associated with diabetes. Int J Mol Sci. (2019) 20:1658. doi: 10.3390/ijms20071658
29. Scorrano L, Oakes SA, Opferman JT, Cheng EH, Sorcinelli MD, Pozzan T, et al. BAX and BAK regulation of endoplasmic reticulum Ca2+: a control point for apoptosis. Science. (2003) 300:135–9. doi: 10.1126/science.1081208
30. Nakagawa T, Yuan J. Cross-talk between two cysteine protease families. Activation of caspase-12 by calpain in apoptosis. J Cell Biol. (2000) 150:887–94. doi: 10.1083/jcb.150.4.887
31. Wang X, Bi X, Zhang G, Deng Y, Luo X, Xu L, et al. Glucose-regulated protein 78 is essential for cardiac myocyte survival. Cell Death Differ. (2018) 25:2181–94. doi: 10.1038/s41418-018-0109-4
32. Masaki T, Yoshida M, Noguchi S. Targeted disruption of CRE-binding factor TREB5 gene leads to cellular necrosis in cardiac myocytes at the embryonic stage. Biochem Biophys Res Commun. (1999) 261:350–6. doi: 10.1006/bbrc.1999.0972
33. Shintani-Ishida K, Nakajima M, Uemura K, Yoshida K. Ischemic preconditioning protects cardiomyocytes against ischemic injury by inducing GRP78. Biochem Biophys Res Commun. (2006) 345:1600–5. doi: 10.1016/j.bbrc.2006.05.077
34. Sun Y, Liu G, Song T, Liu F, Kang W, Zhang Y, et al. Upregulation of GRP78 and caspase-12 in diastolic failing heart. Acta Biochim Pol. (2008) 55:511–6. doi: 10.18388/abp.2008_3057
35. Severino A, Campioni M, Straino S, Salloum FN, Schmidt N, Herbrand U, et al. Identification of protein disulfide isomerase as a cardiomyocyte survival factor in ischemic cardiomyopathy. J Am Coll Cardiol. (2007) 50:1029–37. doi: 10.1016/j.jacc.2007.06.006
36. Sawada T, Minamino T, Fu HY, Asai M, Okuda K, Isomura T, et al. X-box binding protein 1 regulates brain natriuretic peptide through a novel AP1/CRE-like element in cardiomyocytes. J Mol Cell Cardiol. (2010) 48:1280–9. doi: 10.1016/j.yjmcc.2010.02.004
37. Castillero E, Akashi H, Pendrak K, Yerebakan H, Najjar M, Wang C, et al. Attenuation of the unfolded protein response and endoplasmic reticulum stress after mechanical unloading in dilated cardiomyopathy. Am J Physiol Heart Circ Physiol. (2015) 309:H459–70. doi: 10.1152/ajpheart.00056.2015
38. Yao Y, Lu Q, Hu Z, Yu Y, Chen Q, Wang QK. A non-canonical pathway regulates ER stress signaling and blocks ER stress-induced apoptosis and heart failure. Nat Commun. (2017) 8:133. doi: 10.1038/s41467-017-00171-w
39. Martindale JJ, Fernandez R, Thuerauf D, Whittaker R, Gude N, Sussman MA, et al. Endoplasmic reticulum stress gene induction and protection from ischemia/reperfusion injury in the hearts of transgenic mice with a tamoxifen-regulated form of ATF6. Circ Res. (2006) 98:1186–93. doi: 10.1161/01.RES.0000220643.65941.8d
40. Zhang G, Wang X, Bi X, Li C, Deng Y, Al-Hashimi AA, et al. GRP78 (Glucose-Regulated Protein of 78 kDa) promotes cardiomyocyte growth through activation of GATA4 (GATA-Binding Protein 4). Hypertension. (2019) 73:390–8. doi: 10.1161/HYPERTENSIONAHA.118.12084
41. Groenendyk J, Lee D, Jung J, Dyck JR, Lopaschuk GD, Agellon LB, et al. Inhibition of the unfolded protein response mechanism prevents cardiac fibrosis. PLoS One. (2016) 11:e0159682. doi: 10.1371/journal.pone.0159682
42. Okada K, Minamino T, Tsukamoto Y, Liao Y, Tsukamoto O, Takashima S, et al. Prolonged endoplasmic reticulum stress in hypertrophic and failing heart after aortic constriction: possible contribution of endoplasmic reticulum stress to cardiac myocyte apoptosis. Circulation. (2004) 110:705–12. doi: 10.1161/01.CIR.0000137836.95625.D4
43. Liu X, Kwak D, Lu Z, Xu X, Fassett J, Wang H, et al. Endoplasmic reticulum stress sensor protein kinase R-like endoplasmic reticulum kinase (PERK) protects against pressure overload-induced heart failure and lung remodeling. Hypertension. (2014) 64:738–44. doi: 10.1161/HYPERTENSIONAHA.114.03811
44. Binder P, Wang S, Radu M, Zin M, Collins L, Khan S, et al. Pak2 as a novel therapeutic target for cardioprotective endoplasmic reticulum stress response. Circ Res. (2019) 124:696–711. doi: 10.1161/CIRCRESAHA.118.312829
45. Steiger D, Yokota T, Li J, Ren S, Minamisawa S, Wang Y. The serine/threonine-protein kinase/endoribonuclease IRE1alpha protects the heart against pressure overload-induced heart failure. J Biol Chem. (2018) 293:9652–61. doi: 10.1074/jbc.RA118.003448
46. Wang ZV, Deng Y, Gao N, Pedrozo Z, Li DL, Morales CR, et al. Spliced X-box binding protein 1 couples the unfolded protein response to hexosamine biosynthetic pathway. Cell. (2014) 156:1179–92. doi: 10.1016/j.cell.2014.01.014
47. Duan Q, Ni L, Wang P, Chen C, Yang L, Ma B, et al. Deregulation of XBP1 expression contributes to myocardial vascular endothelial growth factor-A expression and angiogenesis during cardiac hypertrophy in vivo. Aging Cell. (2016) 15:625–33. doi: 10.1111/acel.12460
48. Jin JK, Blackwood EA, Azizi K, Thuerauf DJ, Fahem AG, Hofmann C, et al. ATF6 decreases myocardial ischemia/reperfusion damage and links ER stress and oxidative stress signaling pathways in the heart. Circ Res. (2017) 120:862–75. doi: 10.1161/CIRCRESAHA.116.310266
49. Minamino T, Kitakaze M. ER stress in cardiovascular disease. J Mol Cell Cardiol. (2010) 48:1105–10. doi: 10.1016/j.yjmcc.2009.10.026
50. Miyazaki Y, Kaikita K, Endo M, Horio E, Miura M, Tsujita K, et al. C/EBP homologous protein deficiency attenuates myocardial reperfusion injury by inhibiting myocardial apoptosis and inflammation. Arterioscler Thromb Vasc Biol. (2011) 31:1124–32. doi: 10.1161/ATVBAHA.111.224519
51. Xu J, Zhou Q, Xu W, Cai L. Endoplasmic reticulum stress and diabetic cardiomyopathy. Exp Diabetes Res. (2012) 2012:827971. doi: 10.1155/2012/827971
52. Ljubkovic M, Gressette M, Bulat C, Cavar M, Bakovic D, Fabijanic D, et al. Disturbed fatty acid oxidation, endoplasmic reticulum stress, and apoptosis in left ventricle of patients with type 2 diabetes. Diabetes. (2019) 68:1924–33. doi: 10.2337/db19-0423
53. Yan R, Hu S, Ming Z, Lin L, Le H, Jin W. GW26-e2443 changes in gene expression of calreticulin in the myocardium of streptozotocin-induced diabetic cardiomyopathy. J Am Coll Cardiol. (2015) 66(16 Suppl):C88–C9. doi: 10.1016/j.jacc.2015.06.350
54. Li Z, Zhang T, Dai H, Liu G, Wang H, Sun Y, et al. Involvement of endoplasmic reticulum stress in myocardial apoptosis of streptozocin-induced diabetic rats. J Clin Biochem Nutr. (2007) 41:58–67. doi: 10.3164/jcbn.2007008
55. Cong XQ, Piao MH, Li Y, Xie L, Liu Y. Bis(maltolato)oxovanadium(IV) (BMOV) attenuates apoptosis in high glucose-treated cardiac cells and diabetic rat hearts by regulating the unfolded protein responses (UPRs). Biol Trace Elem Res. (2016) 173:390–8. doi: 10.1007/s12011-016-0668-5
56. Toldo S, Boccellino M, Rinaldi B, Seropian IM, Mezzaroma E, Severino A, et al. Altered oxido-reductive state in the diabetic heart: loss of cardioprotection due to protein disulfide isomerase. Mol Med. (2011) 17:1012–21. doi: 10.2119/molmed.2011.00100
57. Pei Z, Deng Q, Babcock SA, He EY, Ren J, Zhang Y. Inhibition of advanced glycation endproduct (AGE) rescues against streptozotocin-induced diabetic cardiomyopathy: role of autophagy and ER stress. Toxicol Lett. (2018) 284:10–20. doi: 10.1016/j.toxlet.2017.11.018
58. Dong F, Ren J. Adiponectin improves cardiomyocyte contractile function in db/db diabetic obese mice. Obesity (Silver Spring). (2009) 17:262–8. doi: 10.1038/oby.2008.545
59. Sun X, Zhao D, Lu F, Peng S, Yu M, Liu N, et al. Hydrogen sulfide regulates muscle RING finger-1 protein S-sulfhydration at Cys(44) to prevent cardiac structural damage in diabetic cardiomyopathy. Br J Pharmacol. (2020) 177:836–56. doi: 10.1111/bph.14601
60. Xu J, Wang G, Wang Y, Liu Q, Xu W, Tan Y, et al. Diabetes- and angiotensin II-induced cardiac endoplasmic reticulum stress and cell death: metallothionein protection. J Cell Mol Med. (2009) 13:1499–512. doi: 10.1111/j.1582-4934.2009.00833.x
61. Liu Z, Zhang Y, Tang Z, Xu J, Ma M, Pan S, et al. Matrine attenuates cardiac fibrosis by affecting ATF6 signaling pathway in diabetic cardiomyopathy. Eur J Pharmacol. (2017) 804:21–30. doi: 10.1016/j.ejphar.2017.03.061
62. Liu ZW, Zhu HT, Chen KL, Dong X, Wei J, Qiu C, et al. Protein kinase RNA-like endoplasmic reticulum kinase (PERK) signaling pathway plays a major role in reactive oxygen species (ROS)-mediated endoplasmic reticulum stress-induced apoptosis in diabetic cardiomyopathy. Cardiovasc Diabetol. (2013) 12:158. doi: 10.1186/1475-2840-12-158
63. Lakshmanan AP, Harima M, Suzuki K, Soetikno V, Nagata M, Nakamura T, et al. The hyperglycemia stimulated myocardial endoplasmic reticulum (ER) stress contributes to diabetic cardiomyopathy in the transgenic non-obese type 2 diabetic rats: a differential role of unfolded protein response (UPR) signaling proteins. Int J Biochem Cell Biol. (2013) 45:438–47. doi: 10.1016/j.biocel.2012.09.017
64. Miki T, Miura T, Hotta H, Tanno M, Yano T, Sato T, et al. Endoplasmic reticulum stress in diabetic hearts abolishes erythropoietin-induced myocardial protection by impairment of phospho-glycogen synthase kinase-3beta-mediated suppression of mitochondrial permeability transition. Diabetes. (2009) 58:2863–72. doi: 10.2337/db09-0158
65. Rutkowski DT, Hegde RS. Regulation of basal cellular physiology by the homeostatic unfolded protein response. J Cell Biol. (2010) 189:783–94. doi: 10.1083/jcb.201003138
66. Li J, Zhu H, Shen E, Wan L, Arnold JM, Peng T. Deficiency of rac1 blocks NADPH oxidase activation, inhibits endoplasmic reticulum stress, and reduces myocardial remodeling in a mouse model of type 1 diabetes. Diabetes. (2010) 59:2033–42. doi: 10.2337/db09-1800
67. He Y, Zhou L, Fan Z, Liu S, Fang W. Palmitic acid, but not high-glucose, induced myocardial apoptosis is alleviated by Nacetylcysteine due to attenuated mitochondrial-derived ROS accumulation-induced endoplasmic reticulum stress. Cell Death Dis. (2018) 9:568. doi: 10.1038/s41419-018-0593-y
68. Hotamisligil GS. Role of endoplasmic reticulum stress and c-Jun NH2-terminal kinase pathways in inflammation and origin of obesity and diabetes. Diabetes. (2005) 54 (Suppl 2):S73–8. doi: 10.2337/diabetes.54.suppl_2.S73
69. Palomer X, Capdevila-Busquets E, Botteri G, Salvado L, Barroso E, Davidson MM, et al. PPARbeta/delta attenuates palmitate-induced endoplasmic reticulum stress and induces autophagic markers in human cardiac cells. Int J Cardiol. (2014) 174:110–8. doi: 10.1016/j.ijcard.2014.03.176
70. Boden G, Cheung P, Kresge K, Homko C, Powers B, Ferrer L. Insulin resistance is associated with diminished endoplasmic reticulum stress responses in adipose tissue of healthy and diabetic subjects. Diabetes. (2014) 63:2977–83. doi: 10.2337/db14-0055
71. Gray S, Kim JK. New insights into insulin resistance in the diabetic heart. Trends Endocrinol Metab. (2011) 22:394–403. doi: 10.1016/j.tem.2011.05.001
72. Park SY, Cho YR, Kim HJ, Higashimori T, Danton C, Lee MK, et al. Unraveling the temporal pattern of diet-induced insulin resistance in individual organs and cardiac dysfunction in C57BL/6 mice. Diabetes. (2005) 54:3530–40. doi: 10.2337/diabetes.54.12.3530
73. Liu Z, Zhao N, Zhu H, Zhu S, Pan S, Xu J, et al. Circulating interleukin-1beta promotes endoplasmic reticulum stress-induced myocytes apoptosis in diabetic cardiomyopathy via interleukin-1 receptor-associated kinase-2. Cardiovasc Diabetol. (2015) 14:125. doi: 10.1186/s12933-015-0288-y
74. Cao SS, Luo KL, Shi L. Endoplasmic reticulum stress interacts with inflammation in human diseases. J Cell Physiol. (2016) 231:288–94. doi: 10.1002/jcp.25098
75. Luo W, Jin Y, Wu G, Zhu W, Qian Y, Zhang Y, et al. Blockage of ROS and MAPKs-mediated inflammation via restoring SIRT1 by a new compound LF10 prevents type 1 diabetic cardiomyopathy. Toxicol Appl Pharmacol. (2019) 370:24–35. doi: 10.1016/j.taap.2019.03.005
76. Guo R, Liu W, Liu B, Zhang B, Li W, Xu Y. SIRT1 suppresses cardiomyocyte apoptosis in diabetic cardiomyopathy: an insight into endoplasmic reticulum stress response mechanism. Int J Cardiol. (2015) 191:36–45. doi: 10.1016/j.ijcard.2015.04.245
77. Feng W, Lei T, Wang Y, Feng R, Yuan J, Shen X, et al. GCN2 deficiency ameliorates cardiac dysfunction in diabetic mice by reducing lipotoxicity and oxidative stress. Free Radic Biol Med. (2019) 130:128–39. doi: 10.1016/j.freeradbiomed.2018.10.445
78. Galan M, Kassan M, Choi SK, Partyka M, Trebak M, Henrion D, et al. A novel role for epidermal growth factor receptor tyrosine kinase and its downstream endoplasmic reticulum stress in cardiac damage and microvascular dysfunction in type 1 diabetes mellitus. Hypertension. (2012) 60:71–80. doi: 10.1161/HYPERTENSIONAHA.112.192500
79. Mali V, Haddox S, Hornersmith C, Matrougui K, Belmadani S. Essential role for EGFR tyrosine kinase and ER stress in myocardial infarction in type 2 diabetes. Pflugers Arch. (2018) 470:471–80. doi: 10.1007/s00424-017-2097-5
80. Battiprolu PK, Hojayev B, Jiang N, Wang ZV, Luo X, Iglewski M, et al. Metabolic stress-induced activation of FoxO1 triggers diabetic cardiomyopathy in mice. J Clin Invest. (2012) 122:1109–18. doi: 10.1172/JCI60329
81. Xin Z, Ma Z, Jiang S, Wang D, Fan C, Di S, et al. FOXOs in the impaired heart: new therapeutic targets for cardiac diseases. Biochim Biophys Acta Mol Basis Dis. (2017) 1863:486–98. doi: 10.1016/j.bbadis.2016.11.023
82. Tang Q, Len Q, Liu Z, Wang W. Overexpression of miR-22 attenuates oxidative stress injury in diabetic cardiomyopathy via Sirt 1. Cardiovasc Ther. (2018) 36. doi: 10.1111/1755-5922.12318
83. Xiao H, Wu C, Li P, Tang B. Simultaneous fluorescence visualization of endoplasmic reticulum superoxide anion and polarity in myocardial cells and tissue. Anal Chem. (2018) 90:6081–8. doi: 10.1021/acs.analchem.7b05440
84. Du Toit A. Post-translational modification: sweetening protein quality control. Nat Rev Mol Cell Biol. (2014) 15:295. doi: 10.1038/nrm3787
85. Ducheix S, Magre J, Cariou B, Prieur X. Chronic O-GlcNAcylation and diabetic cardiomyopathy: the bitterness of glucose. Front Endocrinol. (2018) 9:642. doi: 10.3389/fendo.2018.00642
86. Shpilka T, Haynes CM. The mitochondrial UPR: mechanisms, physiological functions and implications in ageing. Nat Rev Mol Cell Biol. (2018) 19:109–20. doi: 10.1038/nrm.2017.110
87. Jovaisaite V, Mouchiroud L, Auwerx J. The mitochondrial unfolded protein response, a conserved stress response pathway with implications in health and disease. J Exp Biol. (2014) 217(Pt 1):137–43. doi: 10.1242/jeb.090738
88. Zhao Q, Wang J, Levichkin IV, Stasinopoulos S, Ryan MT, Hoogenraad NJ. A mitochondrial specific stress response in mammalian cells. EMBO J. (2002) 21:4411–9. doi: 10.1093/emboj/cdf445
89. Martinus RD, Garth GP, Webster TL, Cartwright P, Naylor DJ, Høj PB, et al. Selective induction of mitochondrial chaperones in response to loss of the mitochondrial genome. Eur J Biochem. (1996) 240:98–103. doi: 10.1111/j.1432-1033.1996.0098h.x
90. Haynes CM, Ron D. The mitochondrial UPR - protecting organelle protein homeostasis. J Cell Sci. (2010) 123(Pt 22):3849–55. doi: 10.1242/jcs.075119
91. Li J, Zhang D, Brundel B, Wiersma M. Imbalance of ER and mitochondria interactions: prelude to cardiac ageing and disease? Cells. (2019) 8:1617. doi: 10.3390/cells8121617
92. Yi HS, Chang JY, Shong M. The mitochondrial unfolded protein response and mitohormesis: a perspective on metabolic diseases. J Mol Endocrinol. (2018) 61:R91–R105. doi: 10.1530/JME-18-0005
93. Ravanelli S, den Brave F, Hoppe T. Mitochondrial quality control governed by ubiquitin. Front Cell Dev Biol. (2020) 8:270. doi: 10.3389/fcell.2020.00270
94. Oh CM, Ryu D, Cho S, Jang Y. Mitochondrial quality control in the heart: new drug targets for cardiovascular disease. Korean Circ J. (2020) 50:395–405. doi: 10.4070/kcj.2019.0416
95. Glembotski CC, Arrieta A, Blackwood EA. Unfolding the roles of mitochondria as therapeutic targets for heart disease. J Am Coll Cardiol. (2019) 73:1807–10. doi: 10.1016/j.jacc.2018.12.089
96. Smyrnias I, Gray SP, Okonko DO, Sawyer G, Zoccarato A, Catibog N, et al. Cardioprotective effect of the mitochondrial unfolded protein response during chronic pressure overload. J Am Coll Cardiol. (2019) 73:1795–806. doi: 10.1016/j.jacc.2018.12.087
97. Wang YT, Lim Y, McCall MN, Huang KT, Haynes CM, Nehrke K, et al. Cardioprotection by the mitochondrial unfolded protein response requires ATF5. Am J Physiol Heart Circ Physiol. (2019) 317:H472–H8. doi: 10.1152/ajpheart.00244.2019
98. Williamson CL, Dabkowski ER, Dillmann WH, Hollander JM. Mitochondria protection from hypoxia/reoxygenation injury with mitochondria heat shock protein 70 overexpression. Am J Physiol Heart Circ Physiol. (2008) 294:H249–56. doi: 10.1152/ajpheart.00775.2007
99. Kuo CY, Chiu YC, Lee AY, Hwang TL. Mitochondrial Lon protease controls ROS-dependent apoptosis in cardiomyocyte under hypoxia. Mitochondrion. (2015) 23:7–16. doi: 10.1016/j.mito.2015.04.004
100. Venkatesh S, Li M, Saito T, Tong M, Rashed E, Mareedu S, et al. Mitochondrial LonP1 protects cardiomyocytes from ischemia/reperfusion injury in vivo. J Mol Cell Cardiol. (2019) 128:38–50. doi: 10.1016/j.yjmcc.2018.12.017
101. Lu B, Shangguan F, Huang D, Gong S, Shi Y, Song Z, et al. LonP1 orchestrates UPRmt and UPRER and mitochondrial dynamics to regulate heart function. bioRxiv [Preprint]. (2019). doi: 10.1101/564492
102. Dogan SA, Pujol C, Maiti P, Kukat A, Wang S, Hermans S, et al. Tissue-specific loss of DARS2 activates stress responses independently of respiratory chain deficiency in the heart. Cell Metab. (2014) 19:458–69. doi: 10.1016/j.cmet.2014.02.004
103. Acin-Perez R, Lechuga-Vieco AV, Del Mar Munoz M, Nieto-Arellano R, Torroja C, Sanchez-Cabo F, et al. Ablation of the stress protease OMA1 protects against heart failure in mice. Sci Transl Med. (2018) 10:eaan4935. doi: 10.1126/scitranslmed.aan4935
104. Shepherd DL, Hathaway Q, Nichols CE, Durr AJ, Pinti MV, Hughes KM, et al. Mitochondrial proteome disruption in the diabetic heart through targeted epigenetic regulation at the mitochondrial heat shock protein 70 (mtHsp70) nuclear locus. J Mol Cell Cardiol. (2018) 119:104–15. doi: 10.1016/j.yjmcc.2018.04.016
105. Chen HS, Wu TE, Juan CC, Lin HD. Myocardial heat shock protein 60 expression in insulin-resistant and diabetic rats. J Endocrinol. (2009) 200:151–7. doi: 10.1677/JOE-08-0387
106. Zhang X, Zhong Z, Li W. Downregulation of TRAP1 aggravates injury of H9c2 cardiomyocytes in a hyperglycemic state. Exp Ther Med. (2019) 18:2681–6. doi: 10.3892/etm.2019.7847
107. Itoh T, Kouzu H, Miki T, Tanno M, Kuno A, Sato T, et al. UPR in endoplasmic reticulum vs. UPR in mitochondria in diabetic myocardium: which is responsible for deficient regulation of mitochondrial permeability transition pores? Circulation. (2018) 124:A15050.
108. Dikic I. Proteasomal and autophagic degradation systems. Annu Rev Biochem. (2017) 86:193–224. doi: 10.1146/annurev-biochem-061516-044908
109. Rock KL, Gramm C, Rothstein L, Clark K, Stein R, Dick L, et al. Inhibitors of the proteasome block the degradation of most cell proteins and the generation of peptides presented on MHC class I molecules. Cell. (1994) 78:761–71. doi: 10.1016/S0092-8674(94)90462-6
110. Schwartz AL, Ciechanover A. The ubiquitin-proteasome pathway and pathogenesis of human diseases. Annu Rev Med. (1999) 50:57–74. doi: 10.1146/annurev.med.50.1.57
111. Deshaies RJ. Make it or break it: the role of ubiquitin-dependent proteolysis in cellular regulation. Trends Cell Biol. (1995) 5:428–34. doi: 10.1016/S0962-8924(00)89102-3
112. Hochstrasser M. Ubiquitin-dependent protein degradation. Annu Rev Genet. (1996) 30:405–39. doi: 10.1146/annurev.genet.30.1.405
113. Jentsch S, Schlenker S. Selective protein degradation: a journey's end within the proteasome. Cell. (1995) 82:881–4. doi: 10.1016/0092-8674(95)90021-7
114. Vijay-Kumar S, Bugg CE, Cook WJ. Structure of ubiquitin refined at 1.8 A resolution. J Mol Biol. (1987) 194:531–44. doi: 10.1016/0022-2836(87)90679-6
115. Zheng Q, Huang T, Zhang L, Zhou Y, Luo H, Xu H, et al. Dysregulation of ubiquitin-proteasome system in neurodegenerative diseases. Front Aging Neurosci. (2016) 8:303. doi: 10.3389/fnagi.2016.00303
116. Finley D. Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu Rev Biochem. (2009) 78:477–513. doi: 10.1146/annurev.biochem.78.081507.101607
117. Kulathu Y, Komander D. Atypical ubiquitylation - the unexplored world of polyubiquitin beyond Lys48 and Lys63 linkages. Nat Rev Mol Cell Biol. (2012) 13:508–23. doi: 10.1038/nrm3394
118. Lopez-Mosqueda J, Dikic I. Deciphering functions of branched ubiquitin chains. Cell. (2014) 157:767–9. doi: 10.1016/j.cell.2014.04.026
119. Yau R, Rape M. The increasing complexity of the ubiquitin code. Nat Cell Biol. (2016) 18:579–86. doi: 10.1038/ncb3358
120. Thrower JS, Hoffman L, Rechsteiner M, Pickart CM. Recognition of the polyubiquitin proteolytic signal. EMBO J. (2000) 19:94–102. doi: 10.1093/emboj/19.1.94
121. Kwon YT, Ciechanover A. The ubiquitin code in the ubiquitin-proteasome system and autophagy. Trends Biochem Sci. (2017) 42:873–86. doi: 10.1016/j.tibs.2017.09.002
122. Komander D. The emerging complexity of protein ubiquitination. Biochem Soc Trans. (2009) 37(Pt 5):937–53. doi: 10.1042/BST0370937
123. Reyes-Turcu FE, Ventii KH, Wilkinson KD. Regulation and cellular roles of ubiquitin-specific deubiquitinating enzymes. Annu Rev Biochem. (2009) 78:363–97. doi: 10.1146/annurev.biochem.78.082307.091526
124. Voges D, Zwickl P, Baumeister W. The 26S proteasome: a molecular machine designed for controlled proteolysis. Annu Rev Biochem. (1999) 68:1015–68. doi: 10.1146/annurev.biochem.68.1.1015
125. Groll M, Bajorek M, Kohler A, Moroder L, Rubin DM, Huber R, et al. A gated channel into the proteasome core particle. Nat Struct Biol. (2000) 7:1062–7. doi: 10.1038/80992
126. Groll M, Ditzel L, Lowe J, Stock D, Bochtler M, Bartunik HD, et al. Structure of 20S proteasome from yeast at 2.4 A resolution. Nature. (1997) 386:463–71. doi: 10.1038/386463a0
127. Glickman MH, Ciechanover A. The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev. (2002) 82:373–428. doi: 10.1152/physrev.00027.2001
128. Elsasser S, Chandler-Militello D, Muller B, Hanna J, Finley D. Rad23 and Rpn10 serve as alternative ubiquitin receptors for the proteasome. J Biol Chem. (2004) 279:26817–22. doi: 10.1074/jbc.M404020200
129. Husnjak K, Elsasser S, Zhang N, Chen X, Randles L, Shi Y, et al. Proteasome subunit Rpn13 is a novel ubiquitin receptor. Nature. (2008) 453:481–8. doi: 10.1038/nature06926
130. Navon A, Goldberg AL. Proteins are unfolded on the surface of the ATPase ring before transport into the proteasome. Mol Cell. (2001) 8:1339–49. doi: 10.1016/S1097-2765(01)00407-5
131. Rabl J, Smith DM, Yu Y, Chang SC, Goldberg AL, Cheng Y. Mechanism of gate opening in the 20S proteasome by the proteasomal ATPases. Mol Cell. (2008) 30:360–8. doi: 10.1016/j.molcel.2008.03.004
132. Baumeister W, Walz J, Zuhl F, Seemuller E. The proteasome: paradigm of a self-compartmentalizing protease. Cell. (1998) 92:367–80. doi: 10.1016/S0092-8674(00)80929-0
133. Hartl FU, Bracher A, Hayer-Hartl M. Molecular chaperones in protein folding and proteostasis. Nature. (2011) 475:324–32. doi: 10.1038/nature10317
134. Hipp MS, Kasturi P, Hartl FU. The proteostasis network and its decline in ageing. Nat Rev Mol Cell Biol. (2019) 20:421–35. doi: 10.1038/s41580-019-0101-y
135. Kettern N, Dreiseidler M, Tawo R, Hohfeld J. Chaperone-assisted degradation: multiple paths to destruction. Biol Chem. (2010) 391:481–9. doi: 10.1515/bc.2010.058
136. Arndt V, Rogon C, Hohfeld J. To be, or not to be–molecular chaperones in protein degradation. Cell Mol Life Sci. (2007) 64:2525–41. doi: 10.1007/s00018-007-7188-6
137. Wang X, Su H, Ranek MJ. Protein quality control and degradation in cardiomyocytes. J Mol Cell Cardiol. (2008) 45:11–27. doi: 10.1016/j.yjmcc.2008.03.025
138. Ranek MJ, Zheng H, Huang W, Kumarapeli AR, Li J, Liu J, et al. Genetically induced moderate inhibition of 20S proteasomes in cardiomyocytes facilitates heart failure in mice during systolic overload. J Mol Cell Cardiol. (2015) 85:273–81. doi: 10.1016/j.yjmcc.2015.06.014
139. Takayama S, Bimston DN, Matsuzawa S, Freeman BC, Aime-Sempe C, Xie Z, et al. BAG-1 modulates the chaperone activity of Hsp70/Hsc70. EMBO J. (1997) 16:4887–96. doi: 10.1093/emboj/16.16.4887
140. Alberti S, Demand J, Esser C, Emmerich N, Schild H, Hohfeld J. Ubiquitylation of BAG-1 suggests a novel regulatory mechanism during the sorting of chaperone substrates to the proteasome. J Biol Chem. (2002) 277:45920–7. doi: 10.1074/jbc.M204196200
141. Alberti S, Bohse K, Arndt V, Schmitz A, Hohfeld J. The cochaperone HspBP1 inhibits the CHIP ubiquitin ligase and stimulates the maturation of the cystic fibrosis transmembrane conductance regulator. Mol Biol Cell. (2004) 15:4003–10. doi: 10.1091/mbc.e04-04-0293
142. Townsend PA, Cutress RI, Carroll CJ, Lawrence KM, Scarabelli TM, Packham G, et al. BAG-1 proteins protect cardiac myocytes from simulated ischemia/reperfusion-induced apoptosis via an alternate mechanism of cell survival independent of the proteasome. J Biol Chem. (2004) 279:20723–8. doi: 10.1074/jbc.M400399200
143. Zhang C, Xu Z, He XR, Michael LH, Patterson C. CHIP, a cochaperone/ubiquitin ligase that regulates protein quality control, is required for maximal cardioprotection after myocardial infarction in mice. Am J Physiol Heart Circ Physiol. (2005) 288:H2836–42. doi: 10.1152/ajpheart.01122.2004
144. Bartha E, Solti I, Szabo A, Olah G, Magyar K, Szabados E, et al. Regulation of kinase cascade activation and heat shock protein expression by poly(ADP-ribose) polymerase inhibition in doxorubicin-induced heart failure. J Cardiovasc Pharmacol. (2011) 58:380–91. doi: 10.1097/FJC.0b013e318225c21e
145. Ke X, Chen J, Peng L, Zhang W, Yang Y, Liao X, et al. Heat shock protein 90/Akt pathway participates in the cardioprotective effect of exogenous hydrogen sulfide against high glucose-induced injury to H9c2 cells. Int J Mol Med. (2017) 39:1001–10. doi: 10.3892/ijmm.2017.2891
146. Fan GC, Ren X, Qian J, Yuan Q, Nicolaou P, Wang Y, et al. Novel cardioprotective role of a small heat-shock protein, Hsp20, against ischemia/reperfusion injury. Circulation. (2005) 111:1792–9. doi: 10.1161/01.CIR.0000160851.41872.C6
147. Macario AJ, Grippo TM, Conway de Macario E. Genetic disorders involving molecular-chaperone genes: a perspective. Genet Med. (2005) 7:3–12. doi: 10.1097/01.GIM.0000151351.11876.C3
148. Wang X, Osinska H, Klevitsky R, Gerdes AM, Nieman M, Lorenz J, et al. Expression of R120G-alphaB-crystallin causes aberrant desmin and alphaB-crystallin aggregation and cardiomyopathy in mice. Circ Res. (2001) 89:84–91. doi: 10.1161/hh1301.092688
149. Wang X, Wang H. Priming the proteasome to protect against proteotoxicity. Trends Mol Med. (2020) 26:639–48. doi: 10.1016/j.molmed.2020.02.007
150. Christianson JC, Ye Y. Cleaning up in the endoplasmic reticulum: ubiquitin in charge. Nat Struct Mol Biol. (2014) 21:325–35. doi: 10.1038/nsmb.2793
151. Qi L, Tsai B, Arvan P. New insights into the physiological role of endoplasmic reticulum-associated degradation. Trends Cell Biol. (2017) 27:430–40. doi: 10.1016/j.tcb.2016.12.002
152. Olzmann JA, Kopito RR, Christianson JC. The mammalian endoplasmic reticulum-associated degradation system. Cold Spring Harb Perspect Biol. (2013) 5:a013185. doi: 10.1101/cshperspect.a013185
153. Locke M, Toth JI, Petroski MD. Lys11- and Lys48-linked ubiquitin chains interact with p97 during endoplasmic-reticulum-associated degradation. Biochem J. (2014) 459:205–16. doi: 10.1042/BJ20120662
154. Ye Y, Meyer HH, Rapoport TA. Function of the p97-Ufd1-Npl4 complex in retrotranslocation from the ER to the cytosol: dual recognition of nonubiquitinated polypeptide segments and polyubiquitin chains. J Cell Biol. (2003) 162:71–84. doi: 10.1083/jcb.200302169
155. Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol. (2012) 13:89–102. doi: 10.1038/nrm3270
156. Hetz C, Chevet E, Harding HP. Targeting the unfolded protein response in disease. Nat Rev Drug Discov. (2013) 12:703–19. doi: 10.1038/nrd3976
157. Hetz C, Papa FR. The unfolded protein response and cell fate control. Mol Cell. (2018) 69:169–81. doi: 10.1016/j.molcel.2017.06.017
158. Kleiger G, Mayor T. Perilous journey: a tour of the ubiquitin-proteasome system. Trends Cell Biol. (2014) 24:352–9. doi: 10.1016/j.tcb.2013.12.003
159. Gilda JE, Gomes AV. Proteasome dysfunction in cardiomyopathies. J Physiol. (2017) 595:4051–71. doi: 10.1113/JP273607
160. Day SM. The ubiquitin proteasome system in human cardiomyopathies and heart failure. Am J Physiol Heart Circ Physiol. (2013) 304:H1283–H93. doi: 10.1152/ajpheart.00249.2012
161. Weekes J, Morrison K, Mullen A, Wait R, Barton P, Dunn MJ. Hyperubiquitination of proteins in dilated cardiomyopathy. Proteomics. (2003) 3:208–16. doi: 10.1002/pmic.200390029
162. Willis MS, Bevilacqua A, Pulinilkunnil T, Kienesberger P, Tannu M, Patterson C. The role of ubiquitin ligases in cardiac disease. J Mol Cell Cardiol. (2014) 71:43–53. doi: 10.1016/j.yjmcc.2013.11.008
163. Predmore MJ, Wang VP, Davis BF, Bartolone RS, Westfall MM, Dyke MD, et al. Ubiquitin proteasome dysfunction in human hypertrophic and dilated cardiomyopathies. Circulation. (2010) 121:997–1004. doi: 10.1161/CIRCULATIONAHA.109.904557
164. Liu J, Chen Q, Huang W, Horak KM, Zheng H, Mestril R, et al. Impairment of the ubiquitin-proteasome system in desminopathy mouse hearts. FASEB J. (2006) 20:362–4. doi: 10.1096/fj.05-4869fje
165. Pan B, Li J, Parajuli N, Tian Z, Wu P, Lewno MT, et al. The calcineurin-TFEB-p62 pathway mediates the activation of cardiac macroautophagy by proteasomal malfunction. Circ Res. (2020) 127:502–18. doi: 10.1101/2019.12.27.889519
166. Herrmann J, Wohlert C, Saguner AM, Flores A, Nesbitt LL, Chade A, et al. Primary proteasome inhibition results in cardiac dysfunction. Eur J Heart Failure. (2013) 15:614–23. doi: 10.1093/eurjhf/hft034
167. Cole DC, Frishman WH. Cardiovascular complications of proteasome inhibitors used in multiple myeloma. Cardiol Rev. (2018) 26:122–9. doi: 10.1097/CRD.0000000000000183
168. Kumarapeli AR, Horak KM, Glasford JW, Li J, Chen Q, Liu J, et al. A novel transgenic mouse model reveals deregulation of the ubiquitin-proteasome system in the heart by doxorubicin. FASEB J. (2005) 19:2051–3. doi: 10.1096/fj.05-3973fje
169. Liu J, Zheng H, Tang M, Ryu YC, Wang X. A therapeutic dose of doxorubicin activates ubiquitin-proteasome system-mediated proteolysis by acting on both the ubiquitination apparatus and proteasome. Am J Physiol Heart Circ Physiol. (2008) 295:H2541–50. doi: 10.1152/ajpheart.01052.2008
170. Lindsten K, Menendez-Benito V, Masucci MG, Dantuma NP, Kumarapeli AR, Horak KM, et al. GFP reporter mouse models of UPS proteolytic function. FASEB J. (2006) 20:1027; author reply 8. doi: 10.1096/fj.06-0504ufm
171. Xu N, Gulick J, Osinska H, Yu Y, McLendon PM, Shay-Winkler K, et al. Ube2v1 positively regulates protein aggregation by modulating ubiquitin proteasome system performance partially through K63 ubiquitination. Circ Res. (2020) 126:907–22. doi: 10.1161/CIRCRESAHA.119.316444
172. Maejima Y, Usui S, Zhai P, Takamura M, Kaneko S, Zablocki D, et al. Muscle-specific RING finger 1 negatively regulates pathological cardiac hypertrophy through downregulation of calcineurin A. Circ Heart Failure. (2014) 7:479–90. doi: 10.1161/CIRCHEARTFAILURE.113.000713
173. Li H-H, Du J, Fan Y-N, Zhang M-L, Liu D-P, Li L, et al. The ubiquitin ligase MuRF1 protects against cardiac ischemia/reperfusion injury by its proteasome-dependent degradation of phospho-c-Jun. Am J Pathol. (2011) 178:1043–58. doi: 10.1016/j.ajpath.2010.11.049
174. Mattox TA, Young ME, Rubel CE, Spaniel C, Rodriguez JE, Grevengoed TJ, et al. MuRF1 activity is present in cardiac mitochondria and regulates reactive oxygen species production in vivo. J Bioenerg Biomembr. (2014) 46:173–87. doi: 10.1007/s10863-014-9549-9
175. Willis MS, Schisler JC, Portbury AL, Patterson C. Build it up-tear it down: protein quality control in the cardiac sarcomere. Cardiovasc Res. (2009) 81:439–48. doi: 10.1093/cvr/cvn289
176. Willis MS, Ike C, Li L, Wang DZ, Glass DJ, Patterson C. Muscle ring finger 1, but not muscle ring finger 2, regulates cardiac hypertrophy in vivo. Circ Res. (2007) 100:456–9. doi: 10.1161/01.RES.0000259559.48597.32
177. He J, Quintana MT, Sullivan J, T LP, T JG, Schisler JC, et al. MuRF2 regulates PPARgamma1 activity to protect against diabetic cardiomyopathy and enhance weight gain induced by a high fat diet. Cardiovasc Diabetol. (2015) 14:97. doi: 10.1186/s12933-015-0252-x
178. Fielitz J, van Rooij E, Spencer JA, Shelton JM, Latif S, van der Nagel R, et al. Loss of muscle-specific RING-finger 3 predisposes the heart to cardiac rupture after myocardial infarction. Proc Natl Acad Sci U S A. (2007) 104:4377–82. doi: 10.1073/pnas.0611726104
179. Quintana MT, He J, Sullivan J, Grevengoed T, Schisler J, Han Y, et al. Muscle ring finger-3 protects against diabetic cardiomyopathy induced by a high fat diet. BMC Endocr Disord. (2015) 15:36. doi: 10.1186/s12902-015-0028-z
180. Li HH, Kedar V, Zhang C, McDonough H, Arya R, Wang DZ, et al. Atrogin-1/muscle atrophy F-box inhibits calcineurin-dependent cardiac hypertrophy by participating in an SCF ubiquitin ligase complex. J Clin Invest. (2004) 114:1058–71. doi: 10.1172/JCI200422220
181. Xie P, Guo SB, Fan Y, Zhang H, Gu D, Li H. Atrogin-1/MAFbx enhances simulated ischemia/reperfusion-induced apoptosis in cardiomyocytes through degradation of MAPK phosphatase-1 and sustained JNK activation. J Biol Chem. (2009) 284:5488–96. doi: 10.1074/jbc.M806487200
182. Galasso G, De Rosa R, Piscione F, Iaccarino G, Vosa C, Sorriento D, et al. Myocardial expression of FOXO3a-Atrogin-1 pathway in human heart failure. Eur J Heart Fail. (2010) 12:1290–6. doi: 10.1093/eurjhf/hfq102
183. Liu W, Wang G, Zhang C, Ding W, Cheng W, Luo Y, et al. MG53, a novel regulator of KChIP2 and Ito, plays a critical role in electrophysiological remodeling in cardiac hypertrophy. Circulation. (2019) 139:2142–56. doi: 10.1161/CIRCULATIONAHA.118.029413
184. Cao CM, Zhang Y, Weisleder N, Ferrante C, Wang X, Lv F, et al. MG53 constitutes a primary determinant of cardiac ischemic preconditioning. Circulation. (2010) 121:2565–74. doi: 10.1161/CIRCULATIONAHA.110.954628
185. Zhang Y, Lv F, Jin L, Peng W, Song R, Ma J, et al. MG53 participates in ischaemic postconditioning through the RISK signalling pathway. Cardiovasc Res. (2011) 91:108–15. doi: 10.1093/cvr/cvr029
186. Liu F, Song R, Feng Y, Guo J, Chen Y, Zhang Y, et al. Upregulation of MG53 induces diabetic cardiomyopathy through transcriptional activation of peroxisome proliferation-activated receptor alpha. Circulation. (2015) 131:795–804. doi: 10.1161/CIRCULATIONAHA.114.012285
187. He B, Tang RH, Weisleder N, Xiao B, Yuan Z, Cai C, et al. Enhancing muscle membrane repair by gene delivery of MG53 ameliorates muscular dystrophy and heart failure in delta-Sarcoglycan-deficient hamsters. Mol Ther. (2012) 20:727–35. doi: 10.1038/mt.2012.5
188. Pan JA, Sun Y, Jiang YP, Bott AJ, Jaber N, Dou Z, et al. TRIM21 ubiquitylates SQSTM1/p62 and suppresses protein sequestration to regulate redox homeostasis. Mol Cell. (2016) 62:149–51. doi: 10.1016/j.molcel.2016.03.015
189. Hauck L, Stanley-Hasnain S, Fung A, Grothe D, Rao V, Mak TW, et al. Cardiac-specific ablation of the E3 ubiquitin ligase Mdm2 leads to oxidative stress, broad mitochondrial deficiency and early death. PLoS One. (2017) 12:e0189861. doi: 10.1371/journal.pone.0189861
190. Fu W, Ma Q, Chen L, Li P, Zhang M, Ramamoorthy S, et al. MDM2 acts downstream of p53 as an E3 ligase to promote FOXO ubiquitination and degradation. J Biol Chem. (2009) 284:13987–4000. doi: 10.1074/jbc.M901758200
191. Rafiq K, Kolpakov MA, Seqqat R, Guo J, Guo X, Qi Z, et al. c-Cbl inhibition improves cardiac function and survival in response to myocardial ischemia. Circulation. (2014) 129:2031–43. doi: 10.1161/CIRCULATIONAHA.113.007004
192. Schisler JC, Rubel CE, Zhang C, Lockyer P, Cyr DM, Patterson C. CHIP protects against cardiac pressure overload through regulation of AMPK. J Clin Invest. (2013) 123:3588–99. doi: 10.1172/JCI69080
193. Kubli DA, Zhang X, Lee Y, Hanna RA, Quinsay MN, Nguyen CK, et al. Parkin protein deficiency exacerbates cardiac injury and reduces survival following myocardial infarction. J Biol Chem. (2013) 288:915–26. doi: 10.1074/jbc.M112.411363
194. Sun T, Ding W, Xu T, Ao X, Yu T, Li M, et al. Parkin regulates programmed necrosis and myocardial ischemia/reperfusion injury by targeting cyclophilin-D. Antioxid Redox Signal. (2019) 31:1177–93. doi: 10.1089/ars.2019.7734
195. Tang Y, Liu J, Long J. Phosphatase and tensin homolog-induced putative kinase 1 and Parkin in diabetic heart: role of mitophagy. J Diabetes Investig. (2015) 6:250–5. doi: 10.1111/jdi.12302
196. Wang S, Zhao Z, Fan Y, Zhang M, Feng X, Lin J, et al. Mst1 inhibits Sirt3 expression and contributes to diabetic cardiomyopathy through inhibiting Parkin-dependent mitophagy. Biochim Biophys Acta. (2019) 1865:1905–14. doi: 10.1016/j.bbadis.2018.04.009
197. Doroudgar JS, Völkers LM, Thuerauf JD, Khan HTM, Mohsin AS, Respress CJ, et al. Hrd1 and ER-associated protein degradation, ERAD, are critical elements of the adaptive ER stress response in cardiac myocytes. Circ Res. (2015) 117:536–46. doi: 10.1161/CIRCRESAHA.115.306993
198. Wang Y, Gao P, Wei C, Li H, Zhang L, Zhao Y, et al. Calcium sensing receptor protects high glucose-induced energy metabolism disorder via blocking gp78-ubiquitin proteasome pathway. Cell Death Dis. (2017) 8:e2799. doi: 10.1038/cddis.2017.193
199. Bi HL, Zhang XL, Zhang YL, Xie X, Xia YL, Du J, et al. The deubiquitinase UCHL1 regulates cardiac hypertrophy by stabilizing epidermal growth factor receptor. Sci Adv. (2020) 6:eaax4826. doi: 10.1126/sciadv.aax4826
200. Wang H, Lai Y, Mathis BJ, Wang W, Li S, Qu C, et al. Deubiquitinating enzyme CYLD mediates pressure overload-induced cardiac maladaptive remodeling and dysfunction via downregulating Nrf2. J Mol Cell Cardiol. (2015) 84:143–53. doi: 10.1016/j.yjmcc.2015.04.012
201. Huang H, Tang QZ, Wang AB, Chen M, Yan L, Liu C, et al. Tumor suppressor A20 protects against cardiac hypertrophy and fibrosis by blocking transforming growth factor-beta-activated kinase 1-dependent signaling. Hypertension. (2010) 56:232–9. doi: 10.1161/HYPERTENSIONAHA.110.149963
202. Li HL, Zhuo ML, Wang D, Wang AB, Cai H, Sun LH, et al. Targeted cardiac overexpression of A20 improves left ventricular performance and reduces compensatory hypertrophy after myocardial infarction. Circulation. (2007) 115:1885–94. doi: 10.1161/CIRCULATIONAHA.106.656835
203. He B, Zhao YC, Gao LC, Ying XY, Xu LW, Su YY, et al. Ubiquitin-specific protease 4 is an endogenous negative regulator of pathological cardiac hypertrophy. Hypertension. (2016) 67:1237–48. doi: 10.1161/HYPERTENSIONAHA.116.07392
204. Chen S, Czernuszewicz G, Lombardi R, Jin JP, Willerson J, Marian A. Human molecular genetic and functional studies identify TRIM63, encoding muscle RING ringer protein 1, as a novel gene for human hypertrophic cardiomyopathy. Circulation. (2012) 126:907–16. doi: 10.1161/CIRCRESAHA.112.270207
205. Belmont JP, Chen JW, San Pedro NM, Thuerauf JD, Lowe GN, Gude AN, et al. Roles for endoplasmic reticulum-associated degradation and the novel endoplasmic reticulum stress response gene Derlin-3 in the ischemic heart. Circ Res. (2010) 106:307–16. doi: 10.1161/CIRCRESAHA.109.203901
206. Bai T, Wang F, Mellen N, Zheng Y, Cai L. Diabetic cardiomyopathy: role of the E3 ubiquitin ligase. Am J Physiol Endocrinol Metab. (2016) 310:E473–83. doi: 10.1152/ajpendo.00467.2015
207. Kato S, Ding J, Pisck E, Jhala US, Du K. COP1 functions as a FoxO1 ubiquitin E3 ligase to regulate FoxO1-mediated gene expression. J Biol Chem. (2008) 283:35464–73. doi: 10.1074/jbc.M801011200
208. Li F, Xie P, Fan Y, Zhang H, Zheng L, Gu D, et al. C terminus of Hsc70-interacting protein promotes smooth muscle cell proliferation and survival through ubiquitin-mediated degradation of FoxO1. J Biol Chem. (2009) 284:20090–8. doi: 10.1074/jbc.M109.017046
209. Oka T, Maillet M, Watt AJ, Schwartz RJ, Aronow BJ, Duncan SA, et al. Cardiac-specific deletion of Gata4 reveals its requirement for hypertrophy, compensation, and myocyte viability. Circ Res. (2006) 98:837–45. doi: 10.1161/01.RES.0000215985.18538.c4
210. Kobayashi S, Mao K, Zheng H, Wang X, Patterson C, O'Connell TD, et al. Diminished GATA4 protein levels contribute to hyperglycemia-induced cardiomyocyte injury. J Biol Chem. (2007) 282:21945–52. doi: 10.1074/jbc.M703048200
211. Madrazo JA, Kelly DP. The PPAR trio: regulators of myocardial energy metabolism in health and disease. J Mol Cell Cardiol. (2008) 44:968–75. doi: 10.1016/j.yjmcc.2008.03.021
212. Park SY, Cho YR, Finck BN, Kim HJ, Higashimori T, Hong EG, et al. Cardiac-specific overexpression of peroxisome proliferator-activated receptor-alpha causes insulin resistance in heart and liver. Diabetes. (2005) 54:2514–24. doi: 10.2337/diabetes.54.9.2514
213. Rodriguez JE, Liao JY, He J, Schisler JC, Newgard CB, Drujan D, et al. The ubiquitin ligase MuRF1 regulates PPARα activity in the heart by enhancing nuclear export via monoubiquitination. Mol Cell Endocrinol. (2015) 413:36–48. doi: 10.1016/j.mce.2015.06.008
214. Kim HJ, Zheng M, Kim SK, Cho JJ, Shin CH, Joe Y, et al. CO/HO-1 induces NQO-1 expression via Nrf2 activation. Immune Netw. (2011) 11:376–82. doi: 10.4110/in.2011.11.6.376
215. Gong P, Stewart D, Hu B, Li N, Cook J, Nel A, et al. Activation of the mouse heme oxygenase-1 gene by 15-deoxy-Delta(12,14)-prostaglandin J(2) is mediated by the stress response elements and transcription factor Nrf2. Antioxid Redox Signal. (2002) 4:249–57. doi: 10.1089/152308602753666307
216. Li J, Ichikawa T, Villacorta L, Janicki JS, Brower GL, Yamamoto M, et al. Nrf2 protects against maladaptive cardiac responses to hemodynamic stress. Arterioscler Thromb Vasc Biol. (2009) 29:1843–50. doi: 10.1161/ATVBAHA.109.189480
217. Wang W, Li S, Wang H, Li B, Shao L, Lai Y, et al. Nrf2 enhances myocardial clearance of toxic ubiquitinated proteins. J Mol Cell Cardiol. (2014) 72:305–15. doi: 10.1016/j.yjmcc.2014.04.006
218. Yamamoto M, Kensler TW, Motohashi H. The KEAP1-NRF2 system: a thiol-based sensor-effector apparatus for maintaining redox homeostasis. Physiol Rev. (2018) 98:1169–203. doi: 10.1152/physrev.00023.2017
219. Kobayashi A, Kang MI, Okawa H, Ohtsuji M, Zenke Y, Chiba T, et al. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol Cell Biol. (2004) 24:7130–9. doi: 10.1128/MCB.24.16.7130-7139.2004
220. Tan Y, Ichikawa T, Li J, Si Q, Yang H, Chen X, et al. Diabetic downregulation of Nrf2 activity via ERK contributes to oxidative stress-induced insulin resistance in cardiac cells in vitro and in vivo. Diabetes. (2011) 60:625–33. doi: 10.2337/db10-1164
221. Xing Y, Niu T, Wang W, Li J, Li S, Janicki JS, et al. Triterpenoid dihydro-CDDO-trifluoroethyl amide protects against maladaptive cardiac remodeling and dysfunction in mice: a critical role of Nrf2. PLoS One. (2012) 7:e44899. doi: 10.1371/journal.pone.0044899
222. Li J, Ma W, Yue G, Tang Y, Kim I-M, Weintraub NL, et al. Cardiac proteasome functional insufficiency plays a pathogenic role in diabetic cardiomyopathy. J Mol Cell Cardiol. (2017) 102:53–60. doi: 10.1016/j.yjmcc.2016.11.013
223. Powell SR, Samuel SM, Wang P, Divald A, Thirunavukkarasu M, Koneru S, et al. Upregulation of myocardial 11S-activated proteasome in experimental hyperglycemia. J Mol Cell Cardiol. (2008) 44:618–21. doi: 10.1016/j.yjmcc.2007.12.009
224. Smith DM, Fraga H, Reis C, Kafri G, Goldberg AL. ATP binds to proteasomal ATPases in pairs with distinct functional effects, implying an ordered reaction cycle. Cell. (2011) 144:526–38. doi: 10.1016/j.cell.2011.02.005
225. Kabakov AE, Budagova KR, Latchman DS, Kampinga HH. Stressful preconditioning and HSP70 overexpression attenuate proteotoxicity of cellular ATP depletion. Am J Physiol Cell Physiol. (2002) 283:C521–34. doi: 10.1152/ajpcell.00503.2001
226. Boudina S, Abel E. Mitochondrial uncoupling: a key contributor to reduced cardiac efficiency in diabetes. Physiology. (2006) 21:250–8. doi: 10.1152/physiol.00008.2006
227. Boudina S, Sena S, Theobald H, Sheng X, Wright JJ, Hu XX, et al. Mitochondrial energetics in the heart in obesity-related diabetes: direct evidence for increased uncoupled respiration and activation of uncoupling proteins. Diabetes. (2007) 56:2457–66. doi: 10.2337/db07-0481
228. Boudina TS, Sena ES, O'neill DB, Tathireddy DP, Young DM, Abel DE. Reduced mitochondrial oxidative capacity and increased mitochondrial uncoupling impair myocardial energetics in obesity. Circulation. (2005) 112:2686–95. doi: 10.1161/CIRCULATIONAHA.105.554360
229. Aiken CT, Kaake RM, Wang X, Huang L. Oxidative stress-mediated regulation of proteasome complexes. Mol Cell Proteomics. (2011) 10:R110.006924. doi: 10.1074/mcp.M110.006924
230. Divald A, Kivity S, Wang P, Hochhauser E, Roberts B, Teichberg S, et al. Myocardial ischemic preconditioning preserves postischemic function of the 26S proteasome through diminished oxidative damage to 19S regulatory particle subunits. Circ Res. (2010) 106:1829–38. doi: 10.1161/CIRCRESAHA.110.219485
231. Bulteau AL, Szweda LI, Friguet B. Age-dependent declines in proteasome activity in the heart. Arch Biochem Biophys. (2002) 397:298–304. doi: 10.1006/abbi.2001.2663
232. Ghosh R, Hwang SM, Cui Z, Gilda JE, Gomes AV. Different effects of the nonsteroidal anti-inflammatory drugs meclofenamate sodium and naproxen sodium on proteasome activity in cardiac cells. J Mol Cell Cardiol. (2016) 94:131–44. doi: 10.1016/j.yjmcc.2016.03.016
233. Campos JC, Queliconi BB, Dourado PM, Cunha TF, Zambelli VO, Bechara LR, et al. Exercise training restores cardiac protein quality control in heart failure. PLoS One. (2012) 7:e52764. doi: 10.1371/journal.pone.0052764
234. Bouchard RA, Bose D. Influence of experimental diabetes on sarcoplasmic reticulum function in rat ventricular muscle. Am J Physiol. (1991) 260(2 Pt 2):H341–54. doi: 10.1152/ajpheart.1991.260.2.H341
235. Lagadic-Gossmann D, Buckler KJ, Le Prigent K, Feuvray D. Altered Ca2+ handling in ventricular myocytes isolated from diabetic rats. Am J Physiol. (1996) 270(5 Pt 2):H1529–H37. doi: 10.1152/ajpheart.1996.270.5.H1529
236. Shen X, Valencia CA, Szostak JW, Dong B, Liu R. Scanning the human proteome for calmodulin-binding proteins. Proc Natl Acad Sci U S A. (2005) 102:5969–74. doi: 10.1073/pnas.0407928102
237. Cui Z, Scruggs SB, Gilda JE, Ping P, Gomes AV. Regulation of cardiac proteasomes by ubiquitination, SUMOylation, and beyond. J Mol Cell Cardiol. (2014) 71:32–42. doi: 10.1016/j.yjmcc.2013.10.008
238. Ferreira JC, Boer BN, Grinberg M, Brum PC, Mochly-Rosen D. Protein quality control disruption by PKCbetaII in heart failure; rescue by the selective PKCbetaII inhibitor, betaIIV5-3. PLoS One. (2012) 7:e33175. doi: 10.1371/journal.pone.0033175
239. Wang X, Li J, Zheng H, Su H, Powell SR. Proteasome functional insufficiency in cardiac pathogenesis. Am J Physiol Heart Circ Physiol. (2011) 301:H2207–19. doi: 10.1152/ajpheart.00714.2011
240. Hnia K, Clausen T, Moog-Lutz C. Shaping striated muscles with ubiquitin proteasome system in health and disease. Trends Mol Med. (2019) 25:760–74. doi: 10.1016/j.molmed.2019.05.008
241. Tang M, Li J, Huang W, Su H, Liang Q, Tian Z, et al. Proteasome functional insufficiency activates the calcineurin-NFAT pathway in cardiomyocytes and promotes maladaptive remodelling of stressed mouse hearts. Cardiovasc Res. (2010) 88:424–33. doi: 10.1093/cvr/cvq217
242. Wilkins BJ, Molkentin JD. Calcium-calcineurin signaling in the regulation of cardiac hypertrophy. Biochem Biophys Res Commun. (2004) 322:1178–91. doi: 10.1016/j.bbrc.2004.07.121
243. Dong X, Liu J, Zheng H, Glasford JW, Huang W, Chen QH, et al. In situ dynamically monitoring the proteolytic function of the ubiquitin-proteasome system in cultured cardiac myocytes. Am J Physiol Heart Circ Physiol. (2004) 287:H1417–25. doi: 10.1152/ajpheart.01233.2003
244. Tsukamoto O, Minamino T, Okada K, Shintani Y, Takashima S, Kato H, et al. Depression of proteasome activities during the progression of cardiac dysfunction in pressure-overloaded heart of mice. Biochem Biophys Res Commun. (2006) 340:1125–33. doi: 10.1016/j.bbrc.2005.12.120
245. Ding W-X, Yin X-M. Sorting, recognition and activation of the misfolded protein degradation pathways through macroautophagy and the proteasome. Autophagy. (2008) 4:141–50. doi: 10.4161/auto.5190
246. Tannous P, Zhu H, Nemchenko A, Berry JM, Johnstone JL, Shelton JM, et al. Intracellular protein aggregation is a proximal trigger of cardiomyocyte autophagy. Circulation. (2008) 117:3070–8. doi: 10.1161/CIRCULATIONAHA.107.763870
247. Zhang T, Shen S, Qu J, Ghaemmaghami S. Global analysis of cellular protein flux quantifies the selectivity of basal autophagy. Cell Rep. (2016) 14:2426–39. doi: 10.1016/j.celrep.2016.02.040
248. Zheng Q, Su H, Tian Z, Wang X. Proteasome malfunction activates macroautophagy in the heart. Am J Cardiovasc Dis. (2011) 1:214–26.
249. Orogo AM, Gustafsson ÅB. Therapeutic targeting of autophagy. Circ Res. (2015) 116:489–503. doi: 10.1161/CIRCRESAHA.116.303791
250. Kirchner P, Bourdenx M, Madrigal-Matute J, Tiano S, Diaz A, Bartholdy BA, et al. Proteome-wide analysis of chaperone-mediated autophagy targeting motifs. PLOS Biol. (2019) 17:e3000301. doi: 10.1371/journal.pbio.3000301
251. Anding AL, Baehrecke EH. Cleaning house: selective autophagy of organelles. Dev Cell. (2017) 41:10–22. doi: 10.1016/j.devcel.2017.02.016
252. Kraft C, Peter M, Hofmann K. Selective autophagy: ubiquitin-mediated recognition and beyond. Nat Cell Biol. (2010) 12:836–41. doi: 10.1038/ncb0910-836
253. Shaid S, Brandts CH, Serve H, Dikic I. Ubiquitination and selective autophagy. Cell Death Differ. (2013) 20:21–30. doi: 10.1038/cdd.2012.72
254. Zientara-Rytter K, Subramani S. The roles of ubiquitin-binding protein shuttles in the degradative fate of ubiquitinated proteins in the ubiquitin-proteasome system and autophagy. Cells. (2019) 8:40. doi: 10.3390/cells8010040
255. Dikic I, Elazar Z. Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol. (2018) 19:349–64. doi: 10.1038/s41580-018-0003-4
256. Bandyopadhyay U, Kaushik S, Varticovski L, Cuervo AM. The chaperone-mediated autophagy receptor organizes in dynamic protein complexes at the lysosomal membrane. Mol Cell Biol. (2008) 28:5747–63. doi: 10.1128/MCB.02070-07
257. Sahu R, Kaushik S, Clement CC, Cannizzo ES, Scharf B, Follenzi A, et al. Microautophagy of cytosolic proteins by late endosomes. Dev Cell. (2011) 20:131–9. doi: 10.1016/j.devcel.2010.12.003
258. Arndt V, Dick N, Tawo R, Dreiseidler M, Wenzel D, Hesse M, et al. Chaperone-assisted selective autophagy is essential for muscle maintenance. Curr Biol. (2010) 20:143–8. doi: 10.1016/j.cub.2009.11.022
259. Stürner E, Behl C. The role of the multifunctional BAG3 protein in cellular protein quality control and in disease. Front Mol Neurosci. (2017) 10:177. doi: 10.3389/fnmol.2017.00177
260. Kaushik S, Cuervo AM. Chaperones in autophagy. Pharmacol Res. (2012) 66:484–93. doi: 10.1016/j.phrs.2012.10.002
261. D'Souza RS, Levandowski C, Slavov D, Graw SL, Allen LA, Adler E, et al. Danon disease. Circulation. (2014) 7:843–9. doi: 10.1161/CIRCHEARTFAILURE.114.001105
262. Cullup T, Kho AL, Dionisi-Vici C, Brandmeier B, Smith F, Urry Z, et al. Recessive mutations in EPG5 cause Vici syndrome, a multisystem disorder with defective autophagy. Nat Genet. (2013) 45:83–7. doi: 10.1038/ng.2497
263. Muhammad E, Levitas A, Singh SR, Braiman A, Ofir R, Etzion S, et al. PLEKHM2 mutation leads to abnormal localization of lysosomes, impaired autophagy flux and associates with recessive dilated cardiomyopathy and left ventricular noncompaction. Hum Mol Genet. (2015) 24:7227–40. doi: 10.1093/hmg/ddv423
264. Norton N, Li D, Mark Jill, Rampersaud E, Züchner S, et al. Genome-wide studies of copy number variation and exome sequencing identify rare variants in BAG3 as a cause of dilated cardiomyopathy. Am J Hum Genet. (2011) 88:273–82. doi: 10.1016/j.ajhg.2011.01.016
265. Villard E, Perret C, Gary F, Proust C, Dilanian G, Hengstenberg C, et al. A genome-wide association study identifies two loci associated with heart failure due to dilated cardiomyopathy. Eur Heart J. (2011) 32:1065–76. doi: 10.1093/eurheartj/ehr105
266. Inomata Y, Nagasaka S, Miyate K, Goto Y, Hino C, Toukairin C, et al. Bcl-2-associated athanogene 3 (BAG3) is an enhancer of small heat shock protein turnover via activation of autophagy in the heart. Biochem Biophys Res Commun. (2018) 496:1141–7. doi: 10.1016/j.bbrc.2018.01.158
267. Bhuiyan MS, Pattison JS, Osinska H, James J, Gulick J, McLendon PM, et al. Enhanced autophagy ameliorates cardiac proteinopathy. J Clin Invest. (2013) 123:5284–97. doi: 10.1172/JCI70877
268. Taneike M, Yamaguchi O, Nakai A, Hikoso S, Takeda T, Mizote I, et al. Inhibition of autophagy in the heart induces age-related cardiomyopathy. Autophagy. (2010) 6:600–6. doi: 10.4161/auto.6.5.11947
269. Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M, et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med. (2007) 13:619–24. doi: 10.1038/nm1574
270. Pyo J-O, Yoo S-M, Ahn H-H, Nah J, Hong S-H, Kam T-I, et al. Overexpression of Atg5 in mice activates autophagy and extends lifespan. Nat Commun. (2013) 4:2300. doi: 10.1038/ncomms3300
271. Fernández ÁF, Sebti S, Wei Y, Zou Z, Shi M, McMillan KL, et al. Disruption of the beclin 1-BCL2 autophagy regulatory complex promotes longevity in mice. Nature. (2018) 558:136–40. doi: 10.1038/s41586-018-0162-7
272. Godar RJ, Ma X, Liu H, Murphy JT, Weinheimer CJ, Kovacs A, et al. Repetitive stimulation of autophagy-lysosome machinery by intermittent fasting preconditions the myocardium to ischemia-reperfusion injury. Autophagy. (2015) 11:1537–60. doi: 10.1080/15548627.2015.1063768
273. Wohlgemuth SE, Julian D, Akin DE, Fried J, Toscano K, Leeuwenburgh C, et al. Autophagy in the heart and liver during normal aging and calorie restriction. Rejuvenation Res. (2004) 10:281–92. doi: 10.1089/rej.2006.0535
274. Li H, Miao W, Ma J, Xv Z, Bo H, Li J, et al. Acute exercise-induced mitochondrial stress triggers an inflammatory response in the myocardium via NLRP3 inflammasome activation with mitophagy. Oxid Med Cell Longev. (2016) 2016:1–11. doi: 10.1155/2016/1987149
275. Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, et al. The role of autophagy during the early neonatal starvation period. Nature. (2004) 432:1032–6. doi: 10.1038/nature03029
276. Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol. (2005) 169:425–34. doi: 10.1083/jcb.200412022
277. Yan Z, Kronemberger A, Blomme J, Call JA, Caster HM, Pereira RO, et al. Exercise leads to unfavourable cardiac remodelling and enhanced metabolic homeostasis in obese mice with cardiac and skeletal muscle autophagy deficiency. Sci Rep. (2017) 7:7894. doi: 10.1038/s41598-017-08480-2
278. Hariharan N, Maejima Y, Nakae J, Paik J, Depinho RA, Sadoshima J. Deacetylation of FOXO by SIRT1 plays an essential role in mediating starvation-induced autophagy in cardiac myocytes. Circ Res. (2010) 107:1470–82. doi: 10.1161/CIRCRESAHA.110.227371
279. Riehle C, Wende AR, Sena S, Pires KM, Pereira RO, Zhu Y, et al. Insulin receptor substrate signaling suppresses neonatal autophagy in the heart. J Clin Invest. (2013) 123:5319–33. doi: 10.1172/JCI71171
280. Russell RC, Yuan H-X, Guan K-L. Autophagy regulation by nutrient signaling. Cell Research. (2014) 24:42–57. doi: 10.1038/cr.2013.166
281. He C, Bassik MC, Moresi V, Sun K, Wei Y, Zou Z, et al. Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature. (2012) 481:511–5. doi: 10.1038/nature10758
282. Tam BT, Pei XM, Yung BY, Yip SP, Chan LW, Wong CS, et al. Autophagic adaptations to long-term habitual exercise in cardiac muscle. Int J Sports Med. (2015) 36:526–34. doi: 10.1055/s-0034-1398494
283. Campos JC, Queliconi BB, Bozi LHM, Bechara LRG, Dourado PMM, Andres AM, et al. Exercise reestablishes autophagic flux and mitochondrial quality control in heart failure. Autophagy. (2017) 13:1304–17. doi: 10.1080/15548627.2017.1325062
284. Chen CY, Hsu HC, Lee BC, Lin HJ, Chen YH, Huang HC, et al. Exercise training improves cardiac function in infarcted rabbits: involvement of autophagic function and fatty acid utilization. Eur J Heart Fail. (2010) 12:323–30. doi: 10.1093/eurjhf/hfq028
285. Saito T, Asai K, Sato S, Hayashi M, Adachi A, Sasaki Y, et al. Autophagic vacuoles in cardiomyocytes of dilated cardiomyopathy with initially decompensated heart failure predict improved prognosis. Autophagy. (2016) 12:579–87. doi: 10.1080/15548627.2016.1145326
286. Matsui Y, Takagi H, Qu X, Abdellatif M, Sakoda H, Asano T, et al. Distinct roles of autophagy in the heart during ischemia and reperfusion. Circ Res. (2007) 100:914–22. doi: 10.1161/01.RES.0000261924.76669.36
287. Yan L, Vatner DE, Kim SJ, Ge H, Masurekar M, Massover WH, et al. Autophagy in chronically ischemic myocardium. Proc Natl Acad Sci U S A. (2005) 102:13807–12. doi: 10.1073/pnas.0506843102
288. Kanamori H, Takemura G, Goto K, Maruyama R, Ono K, Nagao K, et al. Autophagy limits acute myocardial infarction induced by permanent coronary artery occlusion. Am J Physiol Heart Circ Physiol. (2011) 300:H2261–H71. doi: 10.1152/ajpheart.01056.2010
289. Sciarretta S, Yee D, Nagarajan N, Bianchi F, Saito T, Valenti V, et al. Trehalose-induced activation of autophagy improves cardiac remodeling after myocardial infarction. J Am Coll Cardiol. (2018) 71:1999–2010. doi: 10.1016/j.jacc.2018.02.066
290. Ma X, Liu H, Foyil SR, Godar RJ, Weinheimer CJ, Hill JA, et al. Impaired autophagosome clearance contributes to cardiomyocyte death in ischemia/reperfusion injury. Circulation. (2012) 125:3170–81. doi: 10.1161/CIRCULATIONAHA.111.041814
291. Zhu H, Tannous P, Johnstone JL, Kong Y, Shelton JM, Richardson JA, et al. Cardiac autophagy is a maladaptive response to hemodynamic stress. J Clin Invest. (2007) 117:1782–93. doi: 10.1172/JCI27523
292. Zhong Y, Wang QJ, Li X, Yan Y, Backer JM, Chait BT, et al. Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1-phosphatidylinositol-3-kinase complex. Nat Cell Biol. (2009) 11:468–76. doi: 10.1038/ncb1854
293. Nah J, Zhai P, Huang C-Y, Fernández ÁF, Mareedu S, Levine B, et al. Upregulation of Rubicon promotes autosis during myocardial ischemia/reperfusion injury. J Clin Invest. (2020) 130:2978–991. doi: 10.1172/JCI132366
294. Wiersma M, Meijering RAM, Qi XY, Zhang D, Liu T, Hoogstra-Berends F, et al. Endoplasmic reticulum stress is associated with autophagy and cardiomyocyte remodeling in experimental and human atrial fibrillation. J Am Heart Assoc. (2017) 6:e006458. doi: 10.1161/JAHA.117.006458
295. Yuan Y, Zhao J, Gong Y, Wang D, Wang X, Yun F, et al. Autophagy exacerbates electrical remodeling in atrial fibrillation by ubiquitin-dependent degradation of L-type calcium channel. Cell Death Dis. (2018) 9:873. doi: 10.1038/s41419-018-0860-y
296. Tong M, Saito T, Zhai P, Oka S-I, Mizushima W, Nakamura M, et al. Mitophagy is essential for maintaining cardiac function during high fat diet-induced diabetic cardiomyopathy. Circ Res. (2019) 124:1360–71. doi: 10.1161/CIRCRESAHA.118.314607
297. An M, Ryu D-R, Won Park J, Ha Choi J, Park E-M, Eun Lee K, et al. ULK1 prevents cardiac dysfunction in obesity through autophagy-meditated regulation of lipid metabolism. Cardiovasc Res. (2017) 113:1137–47. doi: 10.1093/cvr/cvx064
298. Cao L, Qin X, Peterson MR, Haller SE, Wilson KA, Hu N, et al. CARD9 knockout ameliorates myocardial dysfunction associated with high fat diet-induced obesity. J Mol Cell Cardiol. (2016) 92:185–95. doi: 10.1016/j.yjmcc.2016.02.014
299. Sciarretta S, Zhai P, Shao D, Maejima Y, Robbins J, Volpe M, et al. Rheb is a critical regulator of autophagy during myocardial ischemia. Circulation. (2012) 125:1134–46. doi: 10.1161/CIRCULATIONAHA.111.078212
300. Kanamori H, Takemura G, Goto K, Tsujimoto A, Mikami A, Ogino A, et al. Autophagic adaptations in diabetic cardiomyopathy differ between type 1 and type 2 diabetes. Autophagy. (2015) 11:1146–60. doi: 10.1080/15548627.2015.1051295
301. Xu X, Hua Y, Sreejayan N, Zhang Y, Ren J. Akt2 knockout preserves cardiac function in high-fat diet-induced obesity by rescuing cardiac autophagosome maturation. J Mol Cell Biol. (2013) 5:61–3. doi: 10.1093/jmcb/mjs055
302. Guo R, Zhang Y, Turdi S, Ren J. Adiponectin knockout accentuates high fat diet-induced obesity and cardiac dysfunction: Role of autophagy. Biochim Biophys Acta. (2013) 1832:1136–48. doi: 10.1016/j.bbadis.2013.03.013
303. Hu N, Zhang Y. TLR4 knockout attenuated high fat diet-induced cardiac dysfunction via NF-κB/JNK-dependent activation of autophagy. Biochim Biophys Acta. (2017) 1863:2001–11. doi: 10.1016/j.bbadis.2017.01.010
304. Kandadi MR, Panzhinskiy E, Roe ND, Nair S, Hu D, Sun A. Deletion of protein tyrosine phosphatase 1B rescues against myocardial anomalies in high fat diet-induced obesity: role of AMPK-dependent autophagy. Biochim Biophys Acta. (2015) 1852:299–309. doi: 10.1016/j.bbadis.2014.07.004
305. Xu X, Ren J. Macrophage migration inhibitory factor (MIF) knockout preserves cardiac homeostasis through alleviating Akt-mediated myocardial autophagy suppression in high-fat diet-induced obesity. Int J Obesity. (2015) 39:387–96. doi: 10.1038/ijo.2014.174
306. Munasinghe PE, Riu F, Dixit P, Edamatsu M, Saxena P, Hamer NSJ, et al. Type-2 diabetes increases autophagy in the human heart through promotion of Beclin-1 mediated pathway. Int J Cardiol. (2016) 202:13–20. doi: 10.1016/j.ijcard.2015.08.111
307. Mellor KM, Bell JR, Young MJ, Ritchie RH, Delbridge LMD. Myocardial autophagy activation and suppressed survival signaling is associated with insulin resistance in fructose-fed mice. J Mol Cell Cardiol. (2011) 50:1035–43. doi: 10.1016/j.yjmcc.2011.03.002
308. Despa S, Margulies KB, Chen L, Knowlton AA, Havel PJ, Taegtmeyer H, et al. Hyperamylinemia contributes to cardiac dysfunction in obesity and diabetes: a study in humans and rats. Circ Res. (2012) 110:598–608. doi: 10.1161/CIRCRESAHA.111.258285
309. Rivera JF, Gurlo T, Daval M, Huang CJ, Matveyenko AV, Butler PC, et al. Human-IAPP disrupts the autophagy/lysosomal pathway in pancreatic β-cells: protective role of p62-positive cytoplasmic inclusions. Cell Death Differ. (2011) 18:415–26. doi: 10.1038/cdd.2010.111
310. Shigihara N, Fukunaka A, Hara A, Komiya K, Honda A, Uchida T, et al. Human IAPP-induced pancreatic β cell toxicity and its regulation by autophagy. J Clin Invest. (2014) 124:3634–44. doi: 10.1172/JCI69866
311. Eguchi M, Kim YH, Kang KW, Shim CY, Jang Y, Dorval T, et al. Ischemia-reperfusion injury leads to distinct temporal cardiac remodeling in normal versus diabetic mice. PLoS One. (2012) 7:e30450. doi: 10.1371/journal.pone.0030450
312. Xu X, Kobayashi S, Chen K, Timm D, Volden P, Huang Y, et al. Diminished autophagy limits cardiac injury in mouse models of type 1 diabetes. J Biol Chem. (2013) 288:18077–92. doi: 10.1074/jbc.M113.474650
313. Nishida Y, Arakawa S, Fujitani K, Yamaguchi H, Mizuta T, Kanaseki T, et al. Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature. (2009) 461:654–8. doi: 10.1038/nature08455
314. Zhang M, Zhang L, Hu J, Lin J, Wang T, Duan Y, et al. MST1 coordinately regulates autophagy and apoptosis in diabetic cardiomyopathy in mice. Diabetologia. (2016) 59:2435–47. doi: 10.1007/s00125-016-4070-9
315. Xie Z, Lau K, Eby B, Lozano P, He C, Pennington B, et al. Improvement of cardiac functions by chronic metformin treatment is associated with enhanced cardiac autophagy in diabetic OVE26 mice. Diabetes. (2011) 60:1770–8. doi: 10.2337/db10-0351
316. He C, Zhu H, Li H, Zou MH, Xie Z. Dissociation of Bcl-2-Beclin1 complex by activated AMPK enhances cardiac autophagy and protects against cardiomyocyte apoptosis in diabetes. Diabetes. (2013) 62:1270–81. doi: 10.2337/db12-0533
317. Guo Y, Yu W, Sun D, Wang J, Li C, Zhang R, et al. A novel protective mechanism for mitochondrial aldehyde dehydrogenase (ALDH2) in type i diabetes-induced cardiac dysfunction: role of AMPK-regulated autophagy. (2015) 1852:319–31. doi: 10.1016/j.bbadis.2014.05.017
318. Liang L, Shou X-L, Zhao H-K, Ren G-Q, Wang J-B, Wang X-H, et al. Antioxidant catalase rescues against high fat diet-induced cardiac dysfunction via an IKKβ-AMPK-dependent regulation of autophagy. Biochim Biophys Acta. (2015) 1852:343–52. doi: 10.1016/j.bbadis.2014.06.027
319. Kim J, Kundu M, Viollet B, Guan K-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. (2011) 13:132–41. doi: 10.1038/ncb2152
320. Kobayashi S, Xu X, Chen K, Liang Q. Suppression of autophagy is protective in high glucose-induced cardiomyocyte injury. Autophagy. (2012) 8:577–92. doi: 10.4161/auto.18980
321. Yang M, Liu E, Tang L, Lei Y, Sun X, Hu J, et al. Emerging roles and regulation of MiT/TFE transcriptional factors. Cell Commun Signal. (2018) 16:31. doi: 10.1186/s12964-018-0242-1
322. Trivedi PC, Bartlett JJ, Perez LJ, Brunt KR, Legare JF, Hassan A, et al. Glucolipotoxicity diminishes cardiomyocyte TFEB and inhibits lysosomal autophagy during obesity and diabetes. Biochim Biophys Acta. (2016) 1861:1893–910. doi: 10.1016/j.bbalip.2016.09.004
323. Pires KM, Buffolo M, Schaaf C, David Symons J, Cox J, Abel ED, et al. Activation of IGF-1 receptors and Akt signaling by systemic hyperinsulinemia contributes to cardiac hypertrophy but does not regulate cardiac autophagy in obese diabetic mice. J Mol Cell Cardiol. (2017) 113:39–50. doi: 10.1016/j.yjmcc.2017.10.001
324. Yu W, Gao B, Li N, Wang J, Qiu C, Zhang G, et al. Sirt3 deficiency exacerbates diabetic cardiac dysfunction: role of Foxo3A-Parkin-mediated mitophagy. Biochim Biophys Acta. (2017) 1863:1973–83. doi: 10.1016/j.bbadis.2016.10.021
325. Wang S, Wang C, Turdi S, Richmond KL, Zhang Y, Ren J. ALDH2 protects against high fat diet-induced obesity cardiomyopathy and defective autophagy: role of CaM kinase II, histone H3K9 methyltransferase SUV39H, Sirt1, and PGC-1α deacetylation. Int J Obes. (2018) 42:1073–87. doi: 10.1038/s41366-018-0030-4
326. Zhang M, Wang S, Cheng Z, Xiong Z, Lv J, Yang Z, et al. Polydatin ameliorates diabetic cardiomyopathy via Sirt3 activation. Biochem Biophys Res Commun. (2017) 493:1280–7. doi: 10.1016/j.bbrc.2017.09.151
327. Wang B, Yang Q, Sun Y-Y, Xing Y-F, Wang Y-B, Lu X-T, et al. Resveratrol-enhanced autophagic flux ameliorates myocardial oxidative stress injury in diabetic mice. J Cell Mol Med. (2014) 18:1599–611. doi: 10.1111/jcmm.12312
328. Costantino S, Paneni F, Lüscher TF, Cosentino F. MicroRNA profiling unveils hyperglycaemic memory in the diabetic heart. Eur Heart J. (2016) 37:572–6. doi: 10.1093/eurheartj/ehv599
329. Ucar A, Gupta SK, Fiedler J, Erikci E, Kardasinski M, Batkai S, et al. The miRNA-212/132 family regulates both cardiac hypertrophy and cardiomyocyte autophagy. Nat Commun. (2012) 3:1078. doi: 10.1038/ncomms2090
330. Su M, Wang J, Wang C, Wang X, Dong W, Qiu W, et al. MicroRNA-221 inhibits autophagy and promotes heart failure by modulating the p27/CDK2/mTOR axis. Cell Death Differ. (2015) 22:986–99. doi: 10.1038/cdd.2014.187
331. Li X, Du N, Zhang Q, Li J, Chen X, Liu X, et al. MicroRNA-30d regulates cardiomyocyte pyroptosis by directly targeting foxo3a in diabetic cardiomyopathy. Cell Death Dis. (2014) 5:e1479. doi: 10.1038/cddis.2014.430
332. Chen C, Yang S, Li H, Yin Z, Fan J, Zhao Y, et al. Mir30c is involved in diabetic cardiomyopathy through regulation of cardiac autophagy via BECN1. Mol Ther Nucleic Acids. (2017) 7:127–39. doi: 10.1016/j.omtn.2017.03.005
333. Zhuo C, Jiang R, Lin X, Shao M. LncRNA H19 inhibits autophagy by epigenetically silencing of DIRAS3 in diabetic cardiomyopathy. Oncotarget. (2017) 8:1429–37. doi: 10.18632/oncotarget.13637
334. Morciano G, Patergnani S, Bonora M, Pedriali G, Tarocco A, Bouhamida E, et al. Mitophagy in cardiovascular diseases. J Clin Med. (2020) 9:892. doi: 10.3390/jcm9030892
335. Billia F, Hauck L, Konecny F, Rao V, Shen J, Mak TW. PTEN-inducible kinase 1 (PINK1)/Park6 is indispensable for normal heart function. Proc Natl Acad Sci U S A. (2011) 108:9572–7. doi: 10.1073/pnas.1106291108
336. Tong M, Saito T, Zhai P, Oka S, Sadoshima J. ULK1-dependent mitophagy is essential for maintaining cardiac function during high fat diet-induced diabetic cardiomyopathy. FASEB J. (2019) 33:660.13. doi: 10.1096/fasebj.2019.33.1_supplement.660.13
337. Wang S, Zhao Z, Feng X, Cheng Z, Xiong Z, Wang T, et al. Melatonin activates Parkin translocation and rescues the impaired mitophagy activity of diabetic cardiomyopathy through Mst1 inhibition. J Cell Mol Med. (2018) 22:5132–44. doi: 10.1111/jcmm.13802
338. Ni T, Lin N, Huang X, Lu W, Sun Z, Zhang J, et al. Icariin ameliorates diabetic cardiomyopathy through Apelin/Sirt3 signalling to improve mitochondrial dysfunction. Front Pharmacol. (2020) 11:256. doi: 10.3389/fphar.2020.00256
339. Song Y, Lee W, Lee Y-H, Kang E, Cha B-S, Lee B-W. Metformin restores Parkin-mediated mitophagy, suppressed by cytosolic p53. Int J Mol Sci. (2016) 17:122. doi: 10.3390/ijms17010122
340. Hoshino A, Ariyoshi M, Okawa Y, Kaimoto S, Uchihashi M, Fukai K, et al. Inhibition of p53 preserves Parkin-mediated mitophagy and pancreatic β-cell function in diabetes. Proc Natl Acad Sci U S A. (2014) 111:3116–21. doi: 10.1073/pnas.1318951111
341. Shao D, Kolwicz Stephen C, Wang P, Roe Nathan D, Villet O, Nishi K, et al. Increasing fatty acid oxidation prevents high fat diet induced cardiomyopathy through regulating parkin mediated mitophagy. Circulation. (2020) 42:983–97. doi: 10.1016/j.yjmcc.2019.11.128
342. Cahill TJ, Leo V, Kelly M, Stockenhuber A, Kennedy NW, Bao L, et al. Resistance of dynamin-related protein 1 oligomers to disassembly impairs mitophagy, resulting in myocardial inflammation and heart failure. J Biol Chem. (2015) 290:25907–19. doi: 10.1074/jbc.M115.665695
343. Hu Q, Zhang H, Gutiérrez Cortés N, Wu D, Wang P, Zhang J, et al. Increased Drp1 acetylation by lipid overload induces cardiomyocyte death and heart dysfunction. Circ Res. (2020) 126:456–70. doi: 10.1161/CIRCRESAHA.119.315252
344. Hu L, Ding M, Tang D, Gao E, Li C, Wang K, et al. Targeting mitochondrial dynamics by regulating Mfn2 for therapeutic intervention in diabetic cardiomyopathy. Theranostics. (2019) 9:3687–706. doi: 10.7150/thno.33684
345. Xiong W, Ma Z, An D, Liu Z, Cai W, Bai Y, et al. Mitofusin 2 participates in mitophagy and mitochondrial fusion against angiotensin II-induced cardiomyocyte injury. Front Physiol. (2019) 10:411. doi: 10.3389/fphys.2019.00411
346. Kocaturk NM, Gozuacik D. Crosstalk between mammalian autophagy and the ubiquitin-proteasome system. Front Cell Dev Biol. (2018) 6:128. doi: 10.3389/fcell.2018.00128
347. Xu C, Feng K, Zhao X, Huang S, Cheng Y, Qian L, et al. Regulation of autophagy by E3 ubiquitin ligase RNF216 through BECN1 ubiquitination. Autophagy. (2014) 10:2239–50. doi: 10.4161/15548627.2014.981792
348. Jin S, Tian S, Chen Y, Zhang C, Xie W, Xia X, et al. USP19 modulates autophagy and antiviral immune responses by deubiquitinating Beclin-1. EMBO J. (2016) 35:866–80. doi: 10.15252/embj.201593596
349. Obeng EA, Carlson LM, Gutman DM, Harrington WJ Jr., Lee KP, et al. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood. (2006) 107:4907–16. doi: 10.1182/blood-2005-08-3531
350. Vogl DT, Stadtmauer EA, Tan K-S, Heitjan DF, Davis LE, Pontiggia L, et al. Combined autophagy and proteasome inhibition: a phase 1 trial of hydroxychloroquine and bortezomib in patients with relapsed/refractory myeloma. Autophagy. (2014) 10:1380–90. doi: 10.4161/auto.29264
351. Cuervo AM, Palmer A, Rivett AJ, Knecht E. Degradation of proteasomes by lysosomes in rat liver. Eur J Biochem. (1995) 227:792–800. doi: 10.1111/j.1432-1033.1995.tb20203.x
352. Cohen-Kaplan V, Livneh I, Avni N, Fabre B, Ziv T, Kwon YT, et al. p62- and ubiquitin-dependent stress-induced autophagy of the mammalian 26S proteasome. Proc Natl Acad Sci U S A. (2016) 113:E7490–E9. doi: 10.1073/pnas.1615455113
353. Sun S, Shi G, Sha H, Ji Y, Han X, Shu X, et al. IRE1α is an endogenous substrate of endoplasmic-reticulum-associated degradation. Nat Cell Biol. (2015) 17:1546–55. doi: 10.1038/ncb3266
354. Travers KJ, Patil CK, Wodicka L, Lockhart DJ, Weissman JS, Walter P. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell. (2000) 101:249–58. doi: 10.1016/S0092-8674(00)80835-1
355. Yu M, Du H, Wang B, Chen J, Lu F, Peng S, et al. Exogenous H(2)S induces Hrd1 S-sulfhydration and prevents CD36 translocation via VAMP3 ubiquitylation in diabetic hearts. Aging Dis. (2020) 11:286–300. doi: 10.14336/AD.2019.0530
356. Fu HY, Sanada S, Matsuzaki T, Liao Y, Okuda K, Yamato M, et al. Chemical endoplasmic reticulum chaperone alleviates doxorubicin-induced cardiac dysfunction. Circ Res. (2016) 118:798–809. doi: 10.1161/CIRCRESAHA.115.307604
357. Zhang C, Syed TW, Liu R, Yu J. Role of endoplasmic reticulum stress, autophagy, and inflammation in cardiovascular disease. Front Cardiovasc Med. (2017) 4:29. doi: 10.3389/fcvm.2017.00029
358. Su H, Wang X. p62 stages an interplay between the ubiquitin-proteasome system and autophagy in the heart of defense against proteotoxic stress. Trends Cardiovasc Med. (2011) 21:224–8. doi: 10.1016/j.tcm.2012.05.015
359. Tian Z, Wang C, Hu C, Tian Y, Liu J, Wang X. Autophagic-lysosomal inhibition compromises ubiquitin-proteasome system performance in a p62 dependent manner in cardiomyocytes. PLoS ONE. (2014) 9:e100715. doi: 10.1371/journal.pone.0100715
360. Yang Q, Gao H, Dong R, Wu YQ. Sequential changes of endoplasmic reticulum stress and apoptosis in myocardial fibrosis of diabetes mellitus-induced rats. Mol Med Rep. (2016) 13:5037–44. doi: 10.3892/mmr.2016.5180
361. Chengji W, Xianjin F. Exercise protects against diabetic cardiomyopathy by the inhibition of the endoplasmic reticulum stress pathway in rats. J Cell Physiol. (2019) 234:1682–8. doi: 10.1002/jcp.27038
362. Bi X, Zhang G, Wang X, Nguyen C, May HI, Li X, et al. Endoplasmic reticulum chaperone GRP78 protects heart from ischemia/reperfusion injury through Akt activation. Circ Res. (2018) 122:1545–54. doi: 10.1161/CIRCRESAHA.117.312641
363. Wang S, Wang Z, Fan Q, Guo J, Galli G, Du G, et al. Ginkgolide K protects the heart against endoplasmic reticulum stress injury by activating the inositol-requiring enzyme 1alpha/X box-binding protein-1 pathway. Br J Pharmacol. (2016) 173:2402–18. doi: 10.1111/bph.13516
364. Zhang G, Wang X, Gillette TG, Deng Y, Wang ZV. Unfolded protein response as a therapeutic target in cardiovascular disease. Curr Top Med Chem. (2019) 19:1902–17. doi: 10.2174/1568026619666190521093049
365. Quentin T, Steinmetz M, Poppe A, Thoms S. Metformin differentially activates ER stress signaling pathways without inducing apoptosis. Dis Model Mech. (2012) 5:259–69. doi: 10.1242/dmm.008110
366. Jung TW, Choi KM. Pharmacological modulators of endoplasmic reticulum stress in metabolic diseases. Int J Mol Sci. (2016) 17:192. doi: 10.3390/ijms17020192
367. DeFronzo RA, Inzucchi S, Abdul-Ghani M, Nissen SE. Pioglitazone: the forgotten, cost-effective cardioprotective drug for type 2 diabetes. Diab Vasc Dis Res. (2019) 16:133–43. doi: 10.1177/1479164118825376
368. Liu J, Liu Y, Chen L, Wang Y, Li J. Glucagon-Like Peptide-1 analog Liraglutide protects against diabetic cardiomyopathy by the inhibition of the endoplasmic reticulum stress pathway. J Diabetes Res. (2013) 2013:630537. doi: 10.1155/2013/630537
369. Dong Y, Yan S, Li GY, Wang MN, Leng L, Li Q. Identification of key candidate genes and pathways revealing the protective effect of liraglutide on diabetic cardiac muscle by integrated bioinformatics analysis. Ann Transl Med. (2020) 8:181. doi: 10.21037/atm.2020.01.94
370. Younce CW, Burmeister MA, Ayala JE. Exendin-4 attenuates high glucose-induced cardiomyocyte apoptosis via inhibition of endoplasmic reticulum stress and activation of SERCA2a. Am J Physiol Cell Physiol. (2013) 304:C508–18. doi: 10.1152/ajpcell.00248.2012
371. Ritchie RH, Abel ED. Basic mechanisms of diabetic heart disease. Circ Res. (2020) 126:1501–25. doi: 10.1161/CIRCRESAHA.120.315913
372. Zhou Y, Wu W. The sodium-glucose co-transporter 2 inhibitor, Empagliflozin, protects against diabetic cardiomyopathy by inhibition of the endoplasmic reticulum stress pathway. Cell Physiol Biochem. (2017) 41:2503–12. doi: 10.1159/000475942
373. Wu T, Dong Z, Geng J, Sun Y, Liu G, Kang W, et al. Valsartan protects against ER stress-induced myocardial apoptosis via CHOP/Puma signaling pathway in streptozotocin-induced diabetic rats. Eur J Pharm Sci. (2011) 42:496–502. doi: 10.1016/j.ejps.2011.02.005
374. Liu X, Xu Q, Wang X, Zhao Z, Zhang L, Zhong L, et al. Irbesartan ameliorates diabetic cardiomyopathy by regulating protein kinase D and ER stress activation in a type 2 diabetes rat model. Pharmacol Res. (2015) 93:43–51. doi: 10.1016/j.phrs.2015.01.001
375. Wu H, Li GN, Xie J, Li R, Chen QH, Chen JZ, et al. Resveratrol ameliorates myocardial fibrosis by inhibiting ROS/ERK/TGF-beta/periostin pathway in STZ-induced diabetic mice. BMC Cardiovasc Disord. (2016) 16:5. doi: 10.1186/s12872-015-0169-z
376. Tao S, Chen L, Song J, Zhu N, Song X, Shi R, et al. Tanshinone IIA ameliorates diabetic cardiomyopathy by inhibiting Grp78 and CHOP expression in STZ-induced diabetes rats. Exp Ther Med. (2019) 18:729–34. doi: 10.3892/etm.2019.7580
377. Hou H, Zhang Q, Dong H, Ge Z. Matrine improves diabetic cardiomyopathy through TGF-beta-induced protein kinase RNA-like endoplasmic reticulum kinase signaling pathway. J Cell Biochem. (2019) 120:13573–82. doi: 10.1002/jcb.28632
378. Zhao R, Xie X, Le K, Li W, Moghadasian MH, Beta T, et al. Endoplasmic reticulum stress in diabetic mouse or glycated LDL-treated endothelial cells: protective effect of Saskatoon berry powder and cyanidin glycans. J Nutr Biochem. (2015) 26:1248–53. doi: 10.1016/j.jnutbio.2015.05.015
379. Xiong FY, Tang ST, Su H, Tang HQ, Jiang P, Zhou Q, et al. Melatonin ameliorates myocardial apoptosis by suppressing endoplasmic reticulum stress in rats with longterm diabetic cardiomyopathy. Mol Med Rep. (2018) 17:374–81. doi: 10.3892/mmr.2017.7841
380. Ge CX, Xu MX, Qin YT, Gu TT, Lou DS, Li Q, et al. Endoplasmic reticulum stress-induced iRhom2 up-regulation promotes macrophage-regulated cardiac inflammation and lipid deposition in high fat diet (HFD)-challenged mice: intervention of fisetin and metformin. Free Radic Biol Med. (2019) 141:67–83. doi: 10.1016/j.freeradbiomed.2019.05.031
381. Ravassa S, Zudaire A, Carr RD, Diez J. Antiapoptotic effects of GLP-1 in murine HL-1 cardiomyocytes. Am J Physiol Heart Circ Physiol. (2011) 300:H1361–72. doi: 10.1152/ajpheart.00885.2010
382. Griffin SJ, Leaver JK, Irving GJ. Impact of metformin on cardiovascular disease: a meta-analysis of randomised trials among people with type 2 diabetes. Diabetologia. (2017) 60:1620–9. doi: 10.1007/s00125-017-4337-9
383. Park CS, Cha H, Kwon EJ, Sreenivasaiah PK, Kim DH. The chemical chaperone 4-phenylbutyric acid attenuates pressure-overload cardiac hypertrophy by alleviating endoplasmic reticulum stress. Biochem Biophys Res Commun. (2012) 421:578–84. doi: 10.1016/j.bbrc.2012.04.048
384. Ozcan U, Yilmaz E, Ozcan L, Furuhashi M, Vaillancourt E, Smith RO, et al. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science. (2006) 313:1137–40. doi: 10.1126/science.1128294
385. Malick R. Endoplasmic reticulum stress and heart complication in diabetes. J Diabetes Metab. (2015) 6:12. doi: 10.4172/2155-6156.1000630
386. Zhang X, Yang L, Xu X, Tang F, Yi P, Qiu B, et al. A review of fibroblast growth factor 21 in diabetic cardiomyopathy. Heart Fail Rev. (2019) 24:1005–17. doi: 10.1007/s10741-019-09809-x
387. Yang H, Feng A, Lin S, Yu L, Lin X, Yan X, et al. Fibroblast growth factor-21 prevents diabetic cardiomyopathy via AMPK-mediated antioxidation and lipid-lowering effects in the heart. Cell Death Dis. (2018) 9:227. doi: 10.1038/s41419-018-0307-5
388. Li J, Horak KM, Su H, Sanbe A, Robbins J, Wang X. Enhancement of proteasomal function protects against cardiac proteinopathy and ischemia/reperfusion injury in mice. J Clin Invest. (2011) 121:3689–700. doi: 10.1172/JCI45709
389. Rajagopalan V, Zhao M, Reddy S, Fajardo G, Wang X, Dewey S, et al. Altered ubiquitin-proteasome signaling in right ventricular hypertrophy and failure. Am J Physiol Heart Circ Physiol. (2013) 305:H551–H62. doi: 10.1152/ajpheart.00771.2012
390. Miller CL, Yan C. Targeting cyclic nucleotide phosphodiesterase in the heart: therapeutic implications. J Cardiovasc Transl Res. (2010) 3:507–15. doi: 10.1007/s12265-010-9203-9
391. Ranek MJ, Terpstra EJ, Li J, Kass DA, Wang X. Protein kinase g positively regulates proteasome-mediated degradation of misfolded proteins. Circulation. (2013) 128:365–76. doi: 10.1161/CIRCULATIONAHA.113.001971
392. Zhang H, Pan B, Wu P, Parajuli N, Rekhter MD, Goldberg AL, et al. PDE1 inhibition facilitates proteasomal degradation of misfolded proteins and protects against cardiac proteinopathy. Sci Adv. (2019) 5:eaaw5870. doi: 10.1126/sciadv.aaw5870
393. Paul BD, Snyder SH. H(2)S signalling through protein sulfhydration and beyond. Nat Rev Mol Cell Biol. (2012) 13:499–507. doi: 10.1038/nrm3391
394. Barr LA, Shimizu Y, Lambert JP, Nicholson CK, Calvert JW. Hydrogen sulfide attenuates high fat diet-induced cardiac dysfunction via the suppression of endoplasmic reticulum stress. Nitric Oxide. (2015) 46:145–56. doi: 10.1016/j.niox.2014.12.013
395. Li Z, Polhemus DJ, Lefer DJ. Evolution of hydrogen sulfide therapeutics to treat cardiovascular disease. Circ Res. (2018) 123:590–600. doi: 10.1161/CIRCRESAHA.118.311134
396. Polhemus DJ, Li Z, Pattillo CB, Gojon G Sr, Gojon G, Giordano T, et al. A novel hydrogen sulfide prodrug, SG1002, promotes hydrogen sulfide and nitric oxide bioavailability in heart failure patients. Cardiovasc Ther. (2015) 33:216–26. doi: 10.1111/1755-5922.12128
397. Baskin KK, Taegtmeyer H. AMP-activated protein kinase regulates E3 ligases in rodent heart. Circ Res. (2011) 109:1153–61. doi: 10.1161/CIRCRESAHA.111.252742
398. Masoudi FA, Inzucchi SE, Wang Y, Havranek EP, Foody JM, Krumholz HM. Thiazolidinediones, metformin, and outcomes in older patients with diabetes and heart failure: an observational study. Circulation. (2005) 111:583–90. doi: 10.1161/01.CIR.0000154542.13412.B1
399. Shah DD, Fonarow GC, Horwich TB. Metformin therapy and outcomes in patients with advanced systolic heart failure and diabetes. J Card Fail. (2010) 16:200–6. doi: 10.1016/j.cardfail.2009.10.022
400. Wong AK, Symon R, AlZadjali MA, Ang DS, Ogston S, Choy A, et al. The effect of metformin on insulin resistance and exercise parameters in patients with heart failure. Eur J Heart Fail. (2012) 14:1303–10. doi: 10.1093/eurjhf/hfs106
401. Gulsin GS, Athithan L, McCann GP. Diabetic cardiomyopathy: prevalence, determinants and potential treatments. Ther Adv Endocrinol Metab. (2019) 10:2042018819834869. doi: 10.1177/2042018819834869
402. Liu Y, Palanivel R, Rai E, Park M, Gabor TV, Scheid MP, et al. Adiponectin stimulates autophagy and reduces oxidative stress to enhance insulin sensitivity during high-fat diet feeding in mice. Diabetes. (2015) 64:36–48. doi: 10.2337/db14-0267
403. Malik SA, Mariño G, Benyounès A, Shen S, Harper F, Maiuri MC, et al. Neuroendocrine regulation of autophagy by leptin. Cell Cycle. (2011) 10:2917–23. doi: 10.4161/cc.10.17.17067
404. Yu W, Zha W, Ren J. Exendin-4 and liraglutide attenuate glucose toxicity-induced cardiac injury through mTOR/ULK1-dependent autophagy. Oxid Med Cell Longev. (2018) 2018:5396806. doi: 10.1155/2018/5396806
405. Margulies KB, Anstrom KJ, Hernandez AF, Redfield MM, Shah MR, Braunwald E, et al. GLP-1 agonist therapy for advanced heart failure with reduced ejection fraction. Circ Heart Failure. (2014) 7:673–9. doi: 10.1161/CIRCHEARTFAILURE.114.000346
406. McGaffin KR, Sun C-K, Rager JJ, Romano LC, Zou B, Mathier MA, et al. Leptin signalling reduces the severity of cardiac dysfunction and remodelling after chronic ischaemic injury. Cardiovasc Res. (2008) 77:54–63. doi: 10.1093/cvr/cvm023
407. Johnson JA, Majumdar SR, Simpson SH, Toth EL. Decreased mortality associated with the use of metformin compared with dulfonylurea monotherapy in type 2 diabetes. Diabetes Care. (2002) 25:2244–8. doi: 10.2337/diacare.25.12.2244
408. Rovira J, Marcelo Arellano E, Burke JT, Brault Y, Moya-Rull D, Bañón-Maneus E, et al. Effect of mTOR inhibitor on body weight: from an experimental rat model to human transplant patients. Transpl Int. (2008) 21:992–8. doi: 10.1111/j.1432-2277.2008.00710.x
409. Das A, Durrant D, Koka S, Salloum FN, Xi L, Kukreja RC. Mammalian target of rapamycin (mTOR) inhibition with rapamycin improves cardiac function in type 2 diabetic mice. J Biol Chem. (2014) 289:4145–60. doi: 10.1074/jbc.M113.521062
410. Hsiao P-J, Chiou H-YC, Jiang H-J, Lee M-Y, Hsieh T-J, Kuo K-K. Pioglitazone enhances cytosolic lipolysis, β-oxidation and autophagy to ameliorate hepatic steatosis. Sci Rep. (2017) 7:9030. doi: 10.1038/s41598-017-09702-3
411. Elrashidy RA, Asker ME, Mohamed HE. Beneficial effects of pioglitazone against cardiovascular injury are enhanced by combination with aliskiren in a rat model of diabetic nephropathy. J Pharm Pharmacol. (2012) 64:862–71. doi: 10.1111/j.2042-7158.2012.01508.x
412. Bhandari U, Kumar V, Kumar P, Tripathi CD, Khanna G. Protective effect of pioglitazone on cardiomyocyte apoptosis in low-dose streptozotocin & high-fat diet-induced type-2 diabetes in rats. Indian J Med Res. (2015) 142:598–605. doi: 10.4103/0971-5916.171290
413. Murase H, Kuno A, Miki T, Tanno M, Yano T, Kouzu H, et al. Inhibition of DPP-4 reduces acute mortality after myocardial infarction with restoration of autophagic response in type 2 diabetic rats. Cardiovasc Diabetol. (2015) 14:103. doi: 10.1186/s12933-015-0264-6
414. Bostick B, Habibi J, Ma L, Aroor A, Rehmer N, Hayden MR, et al. Dipeptidyl peptidase inhibition prevents diastolic dysfunction and reduces myocardial fibrosis in a Mouse model of Western diet induced obesity. Metabolism. (2014) 63:1000–11. doi: 10.1016/j.metabol.2014.04.002
415. Packer M. Worsening heart failure during the use of DPP-4 inhibitors. JACC Heart Failure. (2018) 6:445–51. doi: 10.1016/j.jchf.2017.12.016
416. Marso SP, Bain SC, Consoli A, Eliaschewitz FG, Jódar E, Leiter LA, et al. Semaglutide and cardiovascular outcomes in patients with type 2 diabetes. N Engl J Med. (2016) 375:1834–44. doi: 10.1056/NEJMoa1607141
417. Marso SP, Daniels GH, Brown-Frandsen K, Kristensen P, Mann JFE, Nauck MA, et al. Liraglutide and cardiovascular outcomes in type 2 diabetes. N Engl J Med. (2016) 375:311–22. doi: 10.1056/NEJMoa1603827
418. Noyan-Ashraf MH, Shikatani EA, Schuiki I, Mukovozov I, Wu J, Li RK, et al. A glucagon-like peptide-1 analog reverses the molecular pathology and cardiac dysfunction of a mouse model of obesity. Circulation. (2013) 127:74–85. doi: 10.1161/CIRCULATIONAHA.112.091215
419. Zinman B, Wanner C, Lachin JM, Fitchett D, Bluhmki E, Hantel S, et al. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med. (2015) 373:2117–28. doi: 10.1056/NEJMoa1504720
420. Hammoudi N, Jeong D, Singh R, Farhat A, Komajda M, Mayoux E, et al. Empagliflozin improves left ventricular diastolic dysfunction in a genetic model of type 2 diabetes. Cardiovasc Drugs Therapy. (2017) 31:233–46. doi: 10.1007/s10557-017-6734-1
421. Aragón-Herrera A, Feijóo-Bandín S, Otero Santiago M, Barral L, Campos-Toimil M, Gil-Longo J, et al. Empagliflozin reduces the levels of CD36 and cardiotoxic lipids while improving autophagy in the hearts of Zucker diabetic fatty rats. Biochem Pharmacol. (2019) 170:113677. doi: 10.1016/j.bcp.2019.113677
422. Zhang J, Cheng Y, Gu J, Wang S, Zhou S, Wang Y, et al. Fenofibrate increases cardiac autophagy via FGF21/SIRT1 and prevents fibrosis and inflammation in the hearts of Type 1 diabetic mice. Clin Sci. (2016) 130:625–41. doi: 10.1042/CS20150623
423. Sabe AA, Elmadhun NY, Sadek AA, Chu LM, Bianchi C, Sellke FW. Differential effects of atorvastatin on autophagy in ischemic and nonischemic myocardium in Ossabaw swine with metabolic syndrome. J Thorac Cardiovasc Surg. (2014) 148:3172–8. doi: 10.1016/j.jtcvs.2014.07.104
424. Park H-W, Park H, Semple IA, Jang I, Ro S-H, Kim M, et al. Pharmacological correction of obesity-induced autophagy arrest using calcium channel blockers. Nat Commun. (2014) 5:4834. doi: 10.1038/ncomms5834
425. Afzal N, Ganguly PK, Dhalla KS, Pierce GN, Singal PK, Dhalla NS. Beneficial effects of verapamil in diabetic cardiomyopathy. Diabetes. (1988) 37:936–42. doi: 10.2337/diabetes.37.7.936
426. Brown RA, Lee MM, Sundareson AM, Woodbury DJ, Savage AO. Influence of calcium channel blocker treatment on the mechanical properties of diabetic rat myocardium. Acta Diabetol. (1996) 33:7–14. doi: 10.1007/BF00571933
427. Pushparaj C, Das A, Purroy R, Nàger M, Herreros J, Pamplona R, et al. Voltage-gated calcium channel blockers deregulate macroautophagy in cardiomyocytes. Int J Biochem Cell Biol. (2015) 68:166–75. doi: 10.1016/j.biocel.2015.09.010
428. Zhang X, Li Z-L, Crane JA, Jordan KL, Pawar AS, Textor SC, et al. Valsartan regulates myocardial autophagy and mitochondrial turnover in experimental hypertension. Hypertension. (2014) 64:87–93. doi: 10.1161/HYPERTENSIONAHA.113.02151
429. Guan J, Liu W-Q, Xing M-Q, Shi Y, Tan X-Y, Jiang C-Q, et al. Elevated expression of periostin in diabetic cardiomyopathy and the effect of valsartan. BMC Cardiovasc Disord. (2015) 15:90. doi: 10.1186/s12872-015-0084-3
430. Yang Z-h, Peng X-d. Effects of valsartan on diabetic cardiomyopathy in rats with type 2 diabetes mellitus. Chin Med J. (2010) 123:3640–3.
431. Brandenburg VM, Rocca H-PB-L, Marx N. Herzinsuffizienztherapie bei Patienten mit Diabetes. Dtsch Med Wochenschr. (2016) 141:1647–9. doi: 10.1055/s-0042-000001
432. Zhu X, Wu C, Qiu S, Yuan X, Li L. Effects of resveratrol on glucose control and insulin sensitivity in subjects with type 2 diabetes: systematic review and meta-analysis. Nutr Metab. (2017) 14:60. doi: 10.1186/s12986-017-0217-z
433. Sai M, Dong H, Jiangwei C, Xiujuan L, Tao G, Feng C. GW25-e3514 resveratrol alleviates diabetic cardiomyopathy through improvement of mitochondrial biogenesis and function in a SIRT1-dependent manner. J Am Coll Cardiol. (2014) 64:C24–C5. doi: 10.1016/j.jacc.2014.06.120
434. Timmers S, Konings E, Bilet L, Riekelt Tineke Gijs, et al. Calorie restriction-like effects of 30 days of resveratrol supplementation on energy metabolism and metabolic profile in obese humans. Cell Metab. (2011) 14:612–22. doi: 10.1016/j.cmet.2011.10.002
435. Zordoky BNM, Robertson IM, Dyck JRB. Preclinical and clinical evidence for the role of resveratrol in the treatment of cardiovascular diseases. Biochim Biophys Acta Mol Basis Dis. (2015) 1852:1155–77. doi: 10.1016/j.bbadis.2014.10.016
436. Lv W, Zhang J, Jiao A, Wang B, Chen B, Lin J. Resveratrol attenuates hIAPP amyloid formation and restores the insulin secretion ability in hIAPP-INS1 cell line via enhancing autophagy. Can J Physiol Pharmacol. (2018) 97:82–9. doi: 10.1139/cjpp-2016-0686
437. Yan J, Yan JY, Wang YX, Ling YN, Song XD, Wang SY, et al. Spermidine-enhanced autophagic flux improves cardiac dysfunction following myocardial infarction by targeting the AMPK/mTOR signalling pathway. Br J Pharmacol. (2019) 176:3126–42. doi: 10.1111/bph.14706
438. Hu J, Lu X, Zhang X, Shao X, Wang Y, Chen J, et al. Exogenous spermine attenuates myocardial fibrosis in diabetic cardiomyopathy by inhibiting endoplasmic reticulum stress and the canonical Wnt signaling pathway. Cell Biol Int. (2020) 44:1660–70. doi: 10.1002/cbin.11360
439. Wang Y, Chen J, Li S, Zhang X, Guo Z, Hu J, et al. Exogenous spermine attenuates rat diabetic cardiomyopathy via suppressing ROS-p53 mediated downregulation of calcium-sensitive receptor. Redox Biol. (2020) 32:101514. doi: 10.1016/j.redox.2020.101514
440. Eisenberg T, Abdellatif M, Schroeder S, Primessnig U, Stekovic S, Pendl T, et al. Cardioprotection and lifespan extension by the natural polyamine spermidine. Nat Med. (2016) 22:1428–38. doi: 10.1038/nm.4222
441. Zhou H, Chen Y, Huang S-W, Hu P-F, Tang L-J. Regulation of autophagy by tea polyphenols in diabetic cardiomyopathy. J Zhejiang Univ Sci B. (2018) 19:333–41. doi: 10.1631/jzus.B1700415
442. Othman AI, El-Sawi MR, El-Missiry MA, Abukhalil MH. Epigallocatechin-3-gallate protects against diabetic cardiomyopathy through modulating the cardiometabolic risk factors, oxidative stress, inflammation, cell death and fibrosis in streptozotocin-nicotinamide-induced diabetic rats. Biomed Pharmacother. (2017) 94:362–73. doi: 10.1016/j.biopha.2017.07.129
443. Meng J-M, Cao S-Y, Wei X-L, Gan R-Y, Wang Y-F, Cai S-X, et al. Effects and mechanisms of tea for the prevention and management of diabetes mellitus and diabetic complications: an updated review. Antioxidants. (2019) 8:170. doi: 10.3390/antiox8060170
444. Wu Y, Xia Z-Y, Zhao B, Leng Y, Dou J, Meng Q-T, et al. (–)-Epigallocatechin-3-gallate attenuates myocardial injury induced by ischemia/reperfusion in diabetic rats and in H9c2 cells under hyperglycemic conditions. Int J Mol Med. (2017) 40:389–99. doi: 10.3892/ijmm.2017.3014
445. Bocchi L, Motta BM, Savi M, Vilella R, Meraviglia V, Rizzi F, et al. The histone deacetylase inhibitor suberoylanilide hydroxamic acid (SAHA) restores cardiomyocyte contractility in a rat model of early diabetes. Int J Mol Sci. (2019) 20:1873. doi: 10.3390/ijms20081873
446. Xie M, Kong Y, Tan W, May H, Battiprolu PK, Pedrozo Z, et al. Histone deacetylase inhibition blunts ischemia/reperfusion injury by inducing cardiomyocyte autophagy. Circulation. (2014) 129:1139–51. doi: 10.1161/CIRCULATIONAHA.113.002416
447. Shin JH, Lim Y-H, Song Y-S, So B-I, Park J-Y, Fang C-H, et al. Granulocyte-colony stimulating factor reduces cardiomyocyte apoptosis and ameliorates diastolic dysfunction in Otsuka long-evans tokushima fatty rats. Cardiovasc Drugs Therapy. (2014) 28:211–20. doi: 10.1007/s10557-014-6519-8
448. Miyata S, Takemura G, Kawase Y, Li Y, Okada H, Maruyama R, et al. Autophagic cardiomyocyte death in cardiomyopathic hamsters and its prevention by granulocyte colony-stimulating factor. Am J Pathol. (2006) 168:386–97. doi: 10.2353/ajpath.2006.050137
449. Wasko MCM, McClure CK, Kelsey SF, Huber K, Orchard T, Toledo FGS. Antidiabetogenic effects of hydroxychloroquine on insulin sensitivity and beta cell function: a randomised trial. Diabetologia. (2015) 58:2336–43. doi: 10.1007/s00125-015-3689-2
450. Gerstein HC, Thorpe KE, Wayne Taylor D, Brian Haynes R. The effectiveness of hydroxychloroquine in patients with type 2 diabetes mellitus who are refractory to sulfonylureas-a randomized trial. Diabetes Res Clin Pract. (2002) 55:209–19. doi: 10.1016/S0168-8227(01)00325-4
451. Bourke L, McCormick J, Taylor V, Pericleous C, Blanchet B, Costedoat-Chalumeau N, et al. Hydroxychloroquine protects against cardiac ischaemia/reperfusion injury in vivo via enhancement of ERK1/2 phosphorylation. PLoS One. (2015) 10:e0143771. doi: 10.1371/journal.pone.0143771
452. Qian L-B, Jiang S-Z, Tang X-Q, Zhang J, Liang Y-Q, Yu H-T, et al. Exacerbation of diabetic cardiac hypertrophy in OVE26 mice by angiotensin II is associated with JNK/c-Jun/miR-221-mediated autophagy inhibition. Oncotarget. (2017) 8:106661–71. doi: 10.18632/oncotarget.21302
453. Yoshino J, Conte C, Fontana L, Mittendorfer B, Imai S-I, Kenneth, et al. Resveratrol supplementation does not improve metabolic function in nonobese women with normal glucose tolerance. Cell Metab. (2012) 16:658–64. doi: 10.1016/j.cmet.2012.09.015
454. Szkudelski T, Szkudelska K. Resveratrol and diabetes: from animal to human studies. Biochim Biophys Acta. (2015) 1852:1145–54. doi: 10.1016/j.bbadis.2014.10.013
455. Song Y-J, Zhong C-B, Wu W. Resveratrol and diabetic cardiomyopathy: focusing on the protective signaling mechanisms. Oxid Med Cell Longev. (2020) 2020:1–19. doi: 10.1155/2020/7051845
456. Lim Y-H, Joe J-H, Jang K-S, Song Y-S, So B-I, Fang C-H, et al. Effects of granulocyte-colony stimulating factor (G-CSF) on diabetic cardiomyopathy in Otsuka Long-Evans Tokushima Fatty rats. Cardiovasc Diabetol. (2011) 10:92. doi: 10.1186/1475-2840-10-92
457. Daltro PS, Alves PS, Castro MF, Azevedo CM, Vasconcelos JF, Allahdadi KJ, et al. Administration of granulocyte-colony stimulating factor accompanied with a balanced diet improves cardiac function alterations induced by high fat diet in mice. BMC Cardiovasc Disord. (2015) 15:162. doi: 10.1186/s12872-015-0154-6
458. Tönnesmann E, Stroehmann I, Kandolf R, Wolburg H, Strach K, Musshoff F, et al. Cardiomyopathy caused by longterm treatment with chloroquine: a rare disease, or a rare diagnosis? J Rheumatol. (2012) 39:1099–103. doi: 10.3899/jrheum.110959
Keywords: diabetic cardiomyopathy (DCM), cardiovascular disease, protein quality control (PQC), autophagy, proteostasis, unfolded protein response, proteasome
Citation: Kaur N, Raja R, Ruiz-Velasco A and Liu W (2020) Cellular Protein Quality Control in Diabetic Cardiomyopathy: From Bench to Bedside. Front. Cardiovasc. Med. 7:585309. doi: 10.3389/fcvm.2020.585309
Received: 20 July 2020; Accepted: 09 September 2020;
Published: 15 October 2020.
Edited by:
Venkatesh Sundararajan, The State University of New Jersey, United StatesReviewed by:
Jie Li, Augusta University, United StatesCopyright © 2020 Kaur, Raja, Ruiz-Velasco and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wei Liu, d2VpLmxpdUBtYW5jaGVzdGVyLmFjLnVr; Andrea Ruiz-Velasco, YW5kcmVhLnJ1aXpAbWFuY2hlc3Rlci5hYy51aw==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.