 Kikkie Poels
Kikkie Poels Winnie G. Vos
Winnie G. Vos Esther Lutgens1,2,3‡
Esther Lutgens1,2,3‡ Tom T. P. Seijkens
Tom T. P. Seijkens- 1Department of Medical Biochemistry, Amsterdam Cardiovascular Sciences (ACS), Amsterdam UMC, Location AMC, University of Amsterdam, Amsterdam, Netherlands
- 2Institute for Cardiovascular Prevention (IPEK), Ludwig Maximilian's University, Munich, Germany
- 3German Centre for Cardiovascular Research (DZHK), Partner Site Munich Heart Alliance, Munich, Germany
- 4Department of Internal Medicine, Amsterdam UMC, Location VUmc, VU University, Amsterdam, Netherlands
- 5Department of Hematology, Amsterdam UMC, Location VUmc, VU University, Amsterdam, Netherlands
Chronic low-grade inflammation drives atherosclerosis and despite optimal pharmacological treatment of classical cardiovascular risk factors, one third of the patients with atherosclerotic cardiovascular disease has elevated inflammatory biomarkers. Additional anti-inflammatory strategies to target this residual inflammatory cardiovascular risk are therefore required. T-cells are a dominant cell type in human atherosclerotic lesions. Modulation of T-cell activation is therefore a potential strategy to target inflammation in atherosclerosis. Ubiquitination is an important regulatory mechanism of T-cell activation and several E3 ubiquitin ligases, including casitas B-lineage lymphoma proto-oncogene B (Cbl-B), itchy homolog (Itch), and gene related to anergy in lymphocytes (GRAIL), function as a natural brake on T-cell activation. In this review we discuss recent insights on the role of Cbl-B, Itch, and GRAIL in atherosclerosis and explore the therapeutic potential of these E3 ubiquitin ligases in cardiovascular medicine.
Introduction
Atherosclerosis, a chronic lipid-driven inflammatory disease of the large arteries, is a major underlying cause of cardiovascular diseases (CVD) (1). Despite optimal primary and secondary pharmacological prevention by lipid lowering therapies and anti-platelet drugs, a significant part of the population develops atherosclerotic CVD, suggesting that additional factors drive atherogenesis in these subjects (2). One third of the patients with stable coronary artery disease and on-target cholesterol levels (<70 mg/dl) have elevated levels of the inflammatory biomarker high-sensitivity C-reactive protein (hsCRP), indicating that inflammation is an independent risk factor for atherosclerotic CVD that is not targeted by current pharmacological interventions (3).
The therapeutic potential of anti-inflammatory strategies in cardiovascular medicine is highlighted by several clinical trials (4–7). For example, the Canakinumab Anti-inflammatory Thrombosis Outcome Study (CANTOS) trial demonstrated that antibody-mediated inhibition of IL1β reduced recurrent CVD in patients with elevated hsCRP levels (>2 mg/l) (4). In the Colchicine Cardiovascular Outcomes Trial (COLCOT), colchicine reduced a composite endpoint of cardiovascular death and various presentations of recurrent atherosclerotic CVD among patients with a recent myocardial infarction (5). In contrast, the Cardiovascular Inflammation Reduction Trial (CIRT) failed to show beneficial cardiovascular effects of methotrexate in patients with type 2 diabetes or the metabolic syndrome and a history of CVD (6). Importantly, patients in CIRT had median hsCRP levels of 1.6 mg/l, reflecting a lower residual inflammatory risk in comparison to patients in CANTOS, who had median hsCRP levels of 4.2 mg/l (4, 6). These landmark trials emphasize that anti-inflammatory interventions have the potential to reduce (recurrent) atherosclerotic CVD in patients with a substantial residual inflammatory risk, which fuels the search for novel immunomodulatory strategies to temper inflammation in atherosclerosis.
Recent studies demonstrated that T-cells are a dominant cell type in human atherosclerotic lesions and comprise 65% of the immune cell content (8). Transcriptional and flow cytometric analyses of human plaques demonstrated that both CD4+ helper T-cells and CD8+ cytotoxic T-cells have an activated profile, especially in patients with symptomatic CVD, which highlights the importance of these cells in atherosclerosis (8, 9). In accordance with many preclinical studies, these findings indicate that modulation of T-cell activation is an attractive strategy to temper inflammation in atherosclerosis (1, 10, 11). The three-step process of T-cell activation is initiated by ligation of the T-cell receptor (TCR) and followed by a second signal that is provided by immune checkpoint proteins (10). Co-stimulatory and co-inhibitory molecules, which are expressed on antigen presenting cells, are the predominant members of the immune checkpoint protein family and may either enhance or hamper T-cell activation (10). The third signal is provided by soluble factors, such as cytokines (10). In the past two decades, the E3 ubiquitin ligases casitas B-lineage lymphoma proto-oncogene B (Cbl-B), itchy homolog (Itch), and gene related to anergy in lymphocytes (GRAIL) have emerged as key regulators of T-cell activation (12–15). Here, we discuss the role of these E3 ubiquitin ligases in atherosclerosis and explore their therapeutic potential in cardiovascular medicine.
E3 Ubiquitin Ligases
Ubiquitination, the post-translational process that results in the conjugation of the peptide ubiquitin to a lysine residue on a substrate protein, is a key regulatory mechanisms of many biological processes, including immune cell activation, as recently reviewed by Tang and colleagues (15). Ubiquitination affects the function, cellular localization, and/or degradation of target proteins and the functional outcome depends on the generated ubiquitin signal, e.g., mono- or poly-ubiquitination (13–15). Ubiquitination starts with the conjugation of ubiquitin to an E1 ubiquitin activating enzyme, then, ubiquitin is transferred to an E2 ubiquitin conjugating enzyme (13–15). Subsequently, the ubiquitin-E2 complex interacts with an E3 ubiquitin ligase, resulting in a covalent bond between ubiquitin and the target protein (Figure 1A) (13–15).
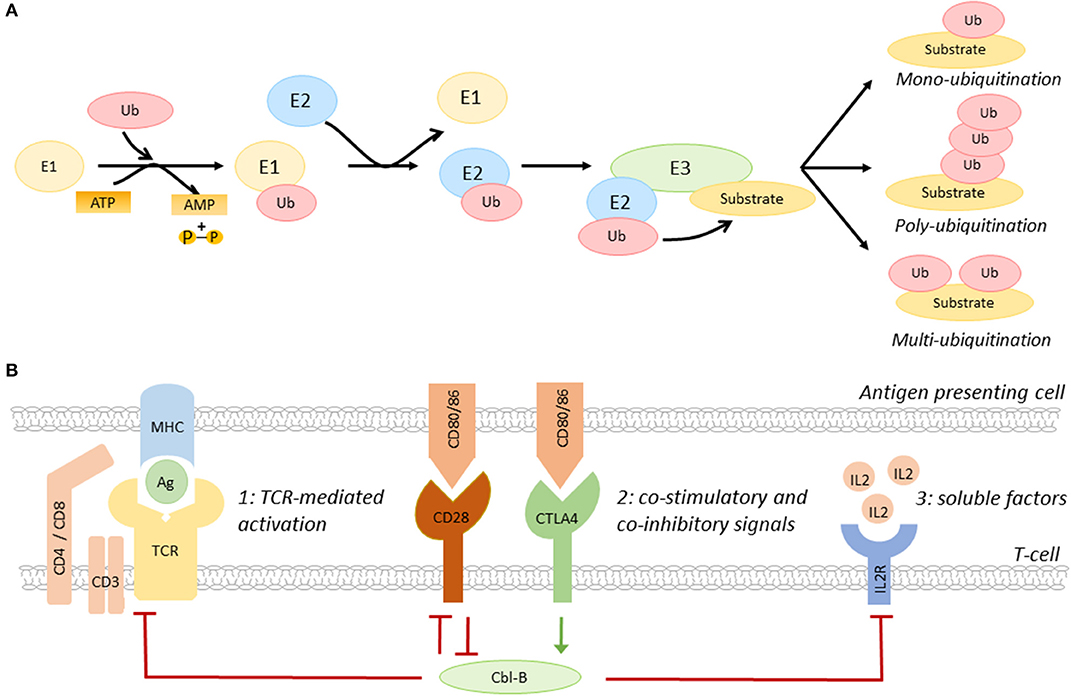
Figure 1. The role of Cbl-B in T-cell activation. (A) Schematic overview of the ubiquitination pathway. (B) Co-inhibitory molecules, such as CTLA4, induce Cbl-B expression, whereas the co-stimulatory molecule CD28 limits the expression of this E3 ubiquitin ligase. Cbl-B negatively regulates the three steps of T-cell activation, including T-cell receptor expression and signaling (1), immune checkpoint protein-induced signaling (2), and the production of IL2 and IL2R-induced signaling pathways (3). IL2R, IL2 receptor; Ag, antigen; MHC, major histocompatibility complex.
E3 ubiquitin ligases are a family of >600 proteins and determine substrate specificity of the ubiquitination process (13–15). Three types of E3 ubiquitin ligases are distinguished; really interesting new gene (RING) E3 ligases, homologous to E6-associated protein carboxyl terminus (HECT) E3 ligases, and RING-between-RING (RBR) ligases (13–15). Whereas RING ligases function as scaffold for E2 ligases and the target, HECT E3 ligases have a catalytic function that facilitates the transfer of ubiquitin to the target protein, and RBR ligases use a hybrid mechanism of RING and HECT ligases to induce ubiquitination (13–15). The removal of ubiquitin by deubiquitinating enzymes makes ubiquitination a highly dynamic process (16).
The E3 ubiquitin ligase family members have a complex and diverse role in the regulation of many inflammatory and metabolic processes that drive atherosclerosis. For example, the RING E3 ligase membrane-associated RING-CH-type finger 1 (MARCH1) limits the number of circulating inflammatory monocytes and hampers atherogenesis, whereas the HECT E3 ligase neural precursor cell-expressed developmentally down-regulated gene 4 (NEDD4-1) limits cholesterol efflux from macrophages, which promotes foam cell formation and atherosclerosis (17, 18). E3 ligases also have an important role in the regulation of the balance between T-cell activation and T-cell tolerance (13). By regulating the expression of the TCR, immune checkpoint proteins, cytokine receptors, and/or their downstream signaling pathways, E3 ligases determine the threshold for T-cell activation (Figure 1B) (10, 13–15). Interestingly, the E3 ligases Cbl-B, Itch, and GRAIL are upregulated during T-cell anergy, a hypo-responsive state in which immune cells do not acquire full effector functions, indicating that these enzymes act as natural inhibitors of T-cell activation (12, 13).
Cbl-B as a Regulator of Immune Cell Activation
The RING-type E3 ubiquitin ligase Cbl-B is highly expressed in CD4+ and CD8+ T-cells in peripheral lymphoid tissues (15). Immune checkpoint proteins have a key role in the regulation of Cbl-B expression in these cells. Whereas, co-stimulatory molecules promote the degradation of Cbl-B, co-inhibitory molecules induce Cbl-B expression, thereby regulating various pathways that amplify T-cell activation, such as TCR- and cytokine-induced signaling (Figure 1B) (15). Cbl-B not only limits the steady-state expression of the TCR, but also promotes its downregulation upon antigen-mediated activation (19). Additionally, Cbl-B promotes ubiquitination-mediated degradation of early downstream signaling proteins of the TCR, such as protein kinase C (PKC) θ and phospholipase C (PLC) γ (15, 20). Loss of Cbl-B also uncouples the requirement for CD28-mediated co-stimulation for T-cell activation, which lowers the activation threshold of these cells (21–23). In addition to this, Cbl-B limits the production of IL2 and inhibits MAPK/ERK-mediated signaling pathways downstream of the IL2 receptor, thereby reducing the proliferation of these cells (23).
The functional relevance of Cbl-B in the regulation of T-cell activation is supported by observations in Cbl-B−/− mice. Genetic deficiency of Cbl-B results in a hyper-responsive T-cell phenotype, characterized by increased proliferation and production of cytokines and granzymes, which triggers spontaneous autoimmune phenomena, e.g., T-cell dependent auto-antibody production and lymphocyte infiltration in various organs, and increases the susceptibility to experimental auto-immune diseases, such as experimental autoimmune encephalomyelitis (24, 25). Decreased expression of Cbl-B also enhances the effector functions of CD4+ T-cells and reduces the suppressive capacity of regulatory T-cells in patients with systemic lupus erythematosus and multiple sclerosis, indicating that Cbl-B is a clinically relevant negative regulator of T-cell driven inflammation (26, 27).
In addition to its role in the regulation of T-cells activity, Cbl-B also affects other immune cells. For example, genetic deficiency of Cbl-B increased B-cell proliferation and elicited T-cell-independent (auto)antibody production, possibly due to increased susceptibility to CD40-induced co-stimulation and enhanced Irf4-, NFκB-, and JNK-mediated signaling pathways (28, 29). In monocytes and macrophages, Cbl-B limits LFA1-ICAM1-mediated recruitment of monocytes toward inflamed tissues by inhibiting phosphorylation of the β2-chain of LFA1 (30). Cbl-B also promotes ubiquitination of TLR4, which hampers LPS-induced activation of MyD88 and degradation of IκBα, which enhances NFκB dependent inflammatory pathways in myeloid cells and ameliorates sepsis-induced macrophage-mediated lung inflammation (30–32). Together, these studies indicate that Cbl-B is a critical regulator of both adaptive and innate immune responses.
The Protective Role of Cbl-B in Atherosclerosis
Cbl-B is predominantly expressed in T-cells and macrophages within human and murine atherosclerotic plaques (33). In human atherosclerotic plaques, Cbl-B expression negatively correlates with necrotic core size, which indicates a possible role for Cbl-B in hampering the progression of atherosclerosis. This role was confirmed in murine studies where genetic deficiency of Cbl-B aggravated atherosclerosis in Apoe−/− mice (33). During the initial stages of atherogenesis, absence of Cbl-B increased the expression of chemokine receptors on monocytes, including CCR1, CCR2, and CCR7, which enhanced the recruitment of these cells to sites of vascular inflammation and increased macrophage abundance in the plaque (33). Deficiency of Cbl-B also increased CD36-mediated lipid uptake in bone marrow-derived macrophages, which promoted foam cell formation, and enhanced LPS-induced production of inflammatory mediators, such as TNF and IL6, and reactive oxygen species, indicating that Cbl-B deficiency induced an atherogenic monocyte and macrophage phenotype (33).
When atherosclerosis progresses, adaptive immune cells, especially T-cells, are recruited to the plaque (34). Deficiency of Cbl-B increased the number of CD8+ T-cells in the circulation, spleen, and atherosclerotic plaques of Apoe−/− mice by increasing the production of IL2 by these cells (33). Additionally, Cblb−/− CD8+ T-cells were more resistant to apoptosis and less susceptible to suppression by regulatory T-cells (33). Cbl-B deficiency reduced CD44−CD62L+ naïve CD8+ T-cells and increased CD44+CD62L+ central memory CD8+ T-cells, and these cells produced more effector molecules, reflecting an activated cytotoxic T-cell phenotype (33). During the later stages of atherosclerosis, Cblb−/− CD8+ T-cells provoked macrophage death in the plaque, resulting in increased necrotic core formation, which promoted plaque progression toward clinically unfavorable lesions (33). Although Cbl-B is also expressed in non-hematopoietic cells, such as smooth muscle cells, CD8+ T cell depletion studies in hematopoietic Cblb−/−Apoe−/− mice confirmed that the progression of atherosclerosis in Cblb−/− mice was predominantly driven by CD8+ T-cells (33). Together these data show that Cbl-B puts a brake on cytotoxic T-cell responses during atherogenesis, thereby limiting plaque inflammation and progression (Figure 2). Enhancing the activity Cbl-B may therefore be a therapeutic strategy for atherosclerosis.
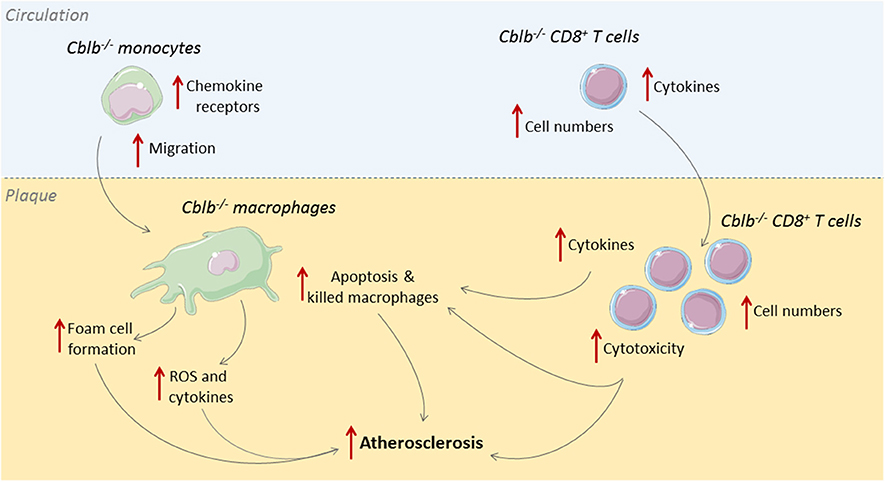
Figure 2. Cbl-B limits CD8+ T-cell and macrophage driven inflammation in atherosclerosis. During the initial stages of atherosclerosis, Cbl-B deficiency increases lesion formation by enhancing monocyte influx into the arterial wall; these monocytes subsequently develop into macrophages with an atherogenic phenotype (33). During the advanced stages of atherosclerosis, CBL-B deficiency increases plaque CD8+ T cell abundance and increases the cytotoxic phenotype of these cells, which aggravates plaque inflammation and provokes macrophage death, thereby enhancing the progression of plaques toward clinically unfavorable high-risk plaques with large necrotic cores (33).
Itch: A Metabolic Regulator of Atherosclerosis
Itch, a HECT E3 ubiquitin ligase, has an important role in the regulation of T-cell activation and loss-of-function mutations in its ubiquitin ligase domain trigger a multisystem autoimmune disease in mice and men (35–39). Various studies in Itch−/− mice identified a regulatory role in T-cell activation (40). For example, upon TCR-mediated activation, Itch induces the ubiquitination-mediated degradation of TCR signal transduction molecules, including PLCγ, PKCθ, and zeta-chain-associated protein kinase (ZAP) 70, thereby increasing the threshold for T-cell activation (36, 41). Itch also promotes the ubiquitin-mediated degradation of the IL2 receptor signaling intermediate Janus kinase 1, which limits T-cell proliferation. Effector functions of CD4+ T-cells are further hampered by the Itch-mediated degradation of the transcription factor JunB, which halts Th2 polarization (42). Additionally, Itch promotes FoxP3 expression in CD4+ T-cells by facilitating the ubiquitination-mediated activation of TGFβ inducible early gene-1 (TIEG1), resulting in increased development of regulatory T-cells (43).
Despite its important role in the regulation of T-cell activation and regulatory T-cell development, genetic deficiency of Itch reduced atherosclerotic lesion size in Apoe−/− mice by ~75% (44). Although this study did not include a detailed analysis of T-cell populations in the plaque, circulation or lymphoid tissues, a striking metabolic phenotype, characterized by a ~50% decrease in plasma cholesterol levels, was observed in Itch−/−Apoe−/− mice (44). The lower cholesterol levels resulted from an increased expression of key lipid metabolism regulators, including SIRT6 and SREBP2, which enhanced low density lipoprotein receptor-mediated cholesterol uptake in the liver (44). In contrast to full body knock-out mice, hematopoietic deficiency of Itch did not affect cholesterol levels or atherosclerosis in Apoe−/− mice, indicating that the atheroprotective effect of Itch depended on the atheroprotective lipid profile (44). Although it would be interesting to investigate T-cell populations and activity in atherosclerotic Itch−/− mice, the current data suggest that therapeutic modulation of Itch will not be a suitable strategy to target the residual inflammatory risk in patients with CVD.
Grail: A Regulator of Immune Cell Activation in Atherosclerosis?
The RING-type E3 ubiquitin ligase GRAIL is well-known for its role in the regulation of T-cell activity (12, 13, 45). Genetic deficiency of GRAIL increases the expression of the TCR and CD3 on T-cells and Grail−/− T-cells do not depend on co-stimulatory signals for their activation (45). Following TCR-mediated activation, GRAIL promotes the ubiquitination-mediated degradation of the endocytosolic TCR-CD3 complex and restricts the expression of the transcription factor nuclear factor of activated T-cells (NFATc1), thereby limiting T-cell activation, IL2-induced proliferation, and the expression of Th1 (IFNγ) and Th17 (IL17, IL21, IL22) cytokines (45). In accordance with the negative regulatory role of GRAIL in T-cell activation, aged Grail−/− mice are more susceptible to experimental auto-immune diseases and spontaneously develop autoimmune phenomena, e.g., immune cell infiltration in organs and auto-antibody formation (45). Adoptive transfer of Grail−/− CD4+ T-cells into lymphocyte deficient Rag1−/− mice aggravated experimental autoimmunity, indicating that the autoimmune phenotype of Grail−/− mice was predominantly driven by aberrant CD4+ T-cell responses (45). In accordance, polymicrobial sepsis in mice induces the upregulation of GRAIL in CD4+ T-cells, which limits their proliferation and effector functions and contributes to the T-cell immunoparalysis that is common in sepsis (46, 47).
Whether GRAIL has a similar immunomodulatory role in atherosclerosis is currently unknown, but several lines of evidence implicate a role for this E3 ligase in atherogenesis. First, GRAIL regulates the expression of co-stimulatory and co-inhibitory molecules on T-cells, which determines the threshold for T-cell activation and steers T-cell effector functions (10, 48). For example, GRAIL promotes ubiquitination-mediated degradation of the co-stimulatory molecules CD40L, OX-40, and CD137 in CD4+ and CD8+ T-cells and enhances the expression of the co-inhibitory molecules cytotoxic T-lymphocyte–associated antigen 4 (CTLA4) and glucocorticoid-induced TNFR-related protein (GITR) on T-cells, which resulted in an anti-inflammatory immune checkpoint protein landscape that limits the activation of these cells (48–51). Additionally, Grail−/− CD8+ cytotoxic T-cell exhibit an increased migratory potential and enhanced production of IFNγ and granzyme B, which promote the progression of atherosclerosis (33, 51). Finally, GRAIL enhances regulatory T-cell responses by increasing FoxP3 and TGFβ expression in CD4+ T-cells, which may boost the protective role of regulatory T-cells in atherogenesis (45, 49). Together, these data implicate a protective role for GRAIL in atherosclerosis by limiting effector T-cell responses and enhancing the anti-inflammatory effects of regulatory T-cells, a concept that should be addressed in future studies.
The Therapeutic Potential of E3 Ubiquitin Ligases
Insights in the anti-inflammatory potential of E3 ligases in humans come from observations in patients with leprosy, an infectious disease caused by Mycobacterium leprae, which is associated with a hyporesponsive T-cell phenotype (52). An 80% increase in Cbl-B expression is observed in circulating CD4+ and CD8+ T-cells of leprosy patients, which enhances the expression of the co-inhibitory molecule CTLA4 and downregulates CD28, thereby increasing the threshold for T-cell activation (52, 53). Moreover, these cells produce less IL2, which further limits T-cell-mediated immunity against M. leprae (53, 54). Similarly, Trypanosoma cruzi, Mycobacterium tuberculosis, and Helminth infections are associated with severe CD4+ T-cell hyporesponsiveness due to increased expression of GRAIL (55–57). Phenotypically, T-cells that upregulate GRAIL during an infection have an exhausted, anti-inflammatory phenotype, characterized by high expression of CTLA4 and programmed cell death protein 1 (PD1), and low IFNγ and IL2 production (55). Although immunological responses in patients with infectious diseases undoubtedly differ from the inflammatory response in patients with atherosclerotic CVD, these data at least indicate that increased expression of Cbl-B and GRAIL limits the inflammatory propensity of T-cells in humans.
Enhancing T-cell effector functions is a potent anti-cancer therapy and CRISPR screens of human T-cells identified Cbl-B as a primary candidate to boost their activity (58–60). Accordingly, adoptive transfer of Cbl-B−/− CD8+ T-cells improved anti-tumor immunity in various models (61–63). Interestingly, siRNA-mediated silencing of Cbl-B in CD8+ T-cells also improved the efficacy of dendritic cell-based tumor vaccines, indicating that inhibition of Cbl-B may become an adjuvant strategy to improve the efficacy of cancer immunotherapy (64–66). Whether enhanced activity of Cbl-B is associated with increased tumorigenesis is currently unknown and should be investigated in future studies.
As with any anti-inflammatory intervention, immune suppression is a potential concern. A specific concern of strategies that enhance Cbl-B activity is candidiasis, as experimental studies demonstrated that a deficiency of Cbl-B improved survival rates in mice subjected to Candida albicans infection (67–69). As the beneficial effects of Cbl-B predominantly depended on improved myeloid-driven antifungal immune responses, cell type-specific modulation of E3 ligases, for example by antibody-drug conjugates that target T-cells, may circumvent these potential immunosuppressive side effects (67–69). Another potential concern is the complex role of Cbl-B in anti-viral immunity. For example, Cbl-B limits excessive immune activation in mice that are infected with an intermediate dose of lymphocytic choriomeningitis virus (LCMV), which improves their survival (15, 70, 71). On the other hand, increased Cbl-B activity promotes the entry of the hepatitis C virus (HCV) into hepatocytes and contributes to the development of HCV-related autoimmunity by enhancing the formation of anergic B cells (72, 73). Future studies should therefore scrutinize the role of E3 ligases in antimicrobial immunity before strategies that enhance the function of these enzymes are developed toward the clinic.
Future Directions
Pharmacological strategies to enhance the activity of Cbl-B or GRAIL do currently not exist, but several strategies can be applied to boost the activity of these E3 ligases. For example, small molecule-mediated blockage of intramolecular inhibitory regions, such as the unphosphorylated N-terminal region of Cbl-B that covers the E2 ligase binding site of the RING domain, can be used to enhance the activity of Cbl-B (74). Additionally, small molecules or peptides that support the open and active conformation of Cbl-B may increase its activity. Alternatively, natural inhibitors of E3 ligases can be targeted to enhance their function. For example, upon TCR-mediated activation, Src homology region 2 domain-containing phosphatase-1 (SHP-1) binds to Cbl-B, which abolishes its ubiquitin ligase activity, suggesting that inhibition of this interaction may enhance Cbl-B activity, a concept that should be investigated in future studies (75).
Conclusions
Given the abundant presence of activated T-cells in human atherosclerotic lesions, modulation of T-cell activity is a promising strategy to target the residual inflammatory risk in patients with atherosclerotic CVD. Although the number of studies on the role of Cbl-B and GRAIL in experimental atherosclerosis and human atherosclerotic CVD is limited, these findings, in conjunction with studies from the immunological field, identify Cbl-B and GRAIL as natural brakes on T-cell activation, increasing the expression and/or activity of these E3 ubiquitin ligases may therefore be an attractive strategy to temper T-cell driven inflammation in atherosclerotic CVD.
Author Contributions
TS wrote the first draft of the manuscript. TS, KP, and WV wrote the sections of the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
Funding
This work was supported by the Dutch Heart Foundation (Dr. Dekker Junior Clinical Scientist grant to TS), The Netherlands CardioVascular Research Initiative: the Dutch Heart Foundation, Dutch Federation of University Medical Centers, the Netherlands, Organization for Health Research and Development, and the Royal Netherlands Academy of Sciences for the GENIUS-II project Generating the best evidence-based pharmaceutical targets for atherosclerosis-II (CVON2018-19 to EL). This study was also supported by the Netherlands Organization for Scientific Research (NWO) (VICI grant 016.130.676 to EL), the European Research Council (ERC consolidator grant CD40-INN 681492 to EL), and the German Science Foundation (DFG, CRC1123, project A5 to EL). The funders had no role in the design and writing of this manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Servier Medical Art for the artwork used in the figures.
References
1. Lutgens E, Atzler D, Doring Y, Duchene J, Steffens S, Weber C. Immunotherapy for cardiovascular disease. Eur Heart J. (2019) 40:3937–46. doi: 10.1093/eurheartj/ehz283
2. Aday AW, Ridker PM. Targeting residual inflammatory risk: a shifting paradigm for atherosclerotic disease. Front Cardiovasc Med. (2019) 6:16. doi: 10.3389/fcvm.2019.00016
3. Peikert A, Kaier K, Merz J, Manhart L, Schafer I, Hilgendorf I, et al. Residual inflammatory risk in coronary heart disease: incidence of elevated high-sensitive CRP in a real-world cohort. Clin Res Cardiol. (2020) 109:315–23. doi: 10.1007/s00392-019-01511-0
4. Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. (2017) 377:1119–31. doi: 10.1056/NEJMoa1707914
5. Tardif JC, Kouz S, Waters DD, Bertrand OF, Diaz R, Maggioni AP, et al. Efficacy and safety of low-dose colchicine after myocardial infarction. N Engl J Med. (2019) 381:2497–505. doi: 10.1056/NEJMoa1912388
6. Ridker PM, Everett BM, Pradhan A, MacFadyen JG, Solomon DH, Zaharris E, et al. Low-dose methotrexate for the prevention of atherosclerotic events. N Engl J Med. (2019) 380:752–62. doi: 10.1056/NEJMoa1809798
7. Ridker PM. From CANTOS to CIRT to COLCOT to clinic: will all atherosclerosis patients soon be treated with combination lipid-lowering and inflammation-inhibiting agents? Circulation. (2020) 141:787–9. doi: 10.1161/CIRCULATIONAHA.119.045256
8. Fernandez DM, Rahman AH, Fernandez NF, Chudnovskiy A, Amir ED, Amadori L, et al. Single-cell immune landscape of human atherosclerotic plaques. Nat Med. (2019) 25:1576–88. doi: 10.1038/s41591-019-0590-4
9. Grivel JC, Ivanova O, Pinegina N, Blank PS, Shpektor A, Margolis LB, et al. Activation of T lymphocytes in atherosclerotic plaques. Arterioscler Thromb Vasc Biol. (2011) 31:2929–37. doi: 10.1161/ATVBAHA.111.237081
10. Kusters PJH, Lutgens E, Seijkens TTP. Exploring immune checkpoints as potential therapeutic targets in atherosclerosis. Cardiovasc Res. (2018) 114:368–77. doi: 10.1093/cvr/cvx248
11. Bullenkamp J, Dinkla S, Kaski JC, Dumitriu IE. Targeting T cells to treat atherosclerosis: odyssey from bench to bedside. Eur Heart J Cardiovasc Pharmacother. (2016) 2:194–9. doi: 10.1093/ehjcvp/pvw001
12. Mueller DL. E3 ubiquitin ligases as T cell anergy factors. Nat Immunol. (2004) 5:883–90. doi: 10.1038/ni1106
13. Ebner P, Versteeg GA, Ikeda F. Ubiquitin enzymes in the regulation of immune responses. Crit Rev Biochem Mol Biol. (2017) 52:425–60. doi: 10.1080/10409238.2017.1325829
14. Morreale FE, Walden H. Types of ubiquitin ligases. Cell. (2016) 165:248–248.e1. doi: 10.1016/j.cell.2016.03.003
15. Tang R, Langdon WY, Zhang J. Regulation of immune responses by E3 ubiquitin ligase Cbl-b. Cell Immunol. (2019) 340:103878. doi: 10.1016/j.cellimm.2018.11.002
16. Yang XD, Sun SC. Deubiquitinases as pivotal regulators of T cell functions. Front Med. (2018) 12:451–62. doi: 10.1007/s11684-018-0651-y
17. Galbas T, Raymond M, Sabourin A, Bourgeois-Daigneault MC, Guimont-Desrochers F, Yun TJ, et al. MARCH1 E3 ubiquitin ligase dampens the innate inflammatory response by modulating monocyte functions in mice. J Immunol. (2017) 198:852–61. doi: 10.4049/jimmunol.1601168
18. Aleidi SM, Howe V, Sharpe LJ, Yang A, Rao G, Brown AJ, et al. The E3 ubiquitin ligases, HUWE1 and NEDD4-1, are involved in the post-translational regulation of the ABCG1 and ABCG4 lipid transporters. J Biol Chem. (2015) 290:24604–13. doi: 10.1074/jbc.M115.675579
19. Shamim M, Nanjappa SG, Singh A, Plisch EH, LeBlanc SE, Walent J, et al. Cbl-b regulates antigen-induced TCR down-regulation and IFN-gamma production by effector CD8 T cells without affecting functional avidity. J Immunol. (2007) 179:7233–43. doi: 10.4049/jimmunol.179.11.7233
20. Liu Q, Zhou H, Langdon WY, Zhang J. E3 ubiquitin ligase Cbl-b in innate and adaptive immunity. Cell Cycle. (2014) 13:1875–84. doi: 10.4161/cc.29213
21. Zhang J, Bardos T, Li D, Gal I, Vermes C, Xu J, et al. Cutting edge: regulation of T cell activation threshold by CD28 costimulation through targeting Cbl-b for ubiquitination. J Immunol. (2002) 169:2236–40. doi: 10.4049/jimmunol.169.5.2236
22. Stromnes IM, Blattman JN, Tan X, Jeevanjee S, Gu H, Greenberg PD. Abrogating Cbl-b in effector CD8(+) T cells improves the efficacy of adoptive therapy of leukemia in mice. J Clin Invest. (2010) 120:3722–34. doi: 10.1172/JCI41991
23. Zhao MF, Qu XJ, Qu JL, Jiang YH, Zhang Y, Hou KZ, et al. The role of E3 ubiquitin ligase Cbl proteins in interleukin-2-induced Jurkat T-cell activation. Biomed Res Int. (2013) 2013:430861. doi: 10.1155/2013/430861
24. Chiang YJ, Kole HK, Brown K, Naramura M, Fukuhara S, Hu RJ, et al. Cbl-b regulates the CD28 dependence of T-cell activation. Nature. (2000) 403:216–20. doi: 10.1038/35003235
25. Jeon MS, Atfield A, Venuprasad K, Krawczyk C, Sarao R, Elly C, et al. Essential role of the E3 ubiquitin ligase Cbl-b in T cell anergy induction. Immunity. (2004) 21:167–77. doi: 10.1016/j.immuni.2004.07.013
26. Gomez-Martin D, Ibarra-Sanchez M, Romo-Tena J, Cruz-Ruiz J, Esparza-Lopez J, Galindo-Campos M, et al. Casitas B lineage lymphoma b is a key regulator of peripheral tolerance in systemic lupus erythematosus. Arthritis Rheum. (2013) 65:1032–42. doi: 10.1002/art.37833
27. Romo-Tena J, Rajme-Lopez S, Aparicio-Vera L, Alcocer-Varela J, Gomez-Martin D. Lys63-polyubiquitination by the E3 ligase casitas B-lineage lymphoma-b (Cbl-b) modulates peripheral regulatory T cell tolerance in patients with systemic lupus erythematosus. Clin Exp Immunol. (2018) 191:42–9. doi: 10.1111/cei.13054
28. Qiao G, Lei M, Li Z, Sun Y, Minto A, Fu YX, et al. Negative regulation of CD40-mediated B cell responses by E3 ubiquitin ligase Casitas-B-lineage lymphoma protein-B. J Immunol. (2007) 179:4473–9. doi: 10.4049/jimmunol.179.7.4473
29. Li X, Gadzinsky A, Gong L, Tong H, Calderon V, Li Y, et al. Cbl ubiquitin ligases control B cell exit from the germinal-center reaction. Immunity. (2018) 48:530–41.e6. doi: 10.1016/j.immuni.2018.03.006
30. Choi EY, Orlova VV, Fagerholm SC, Nurmi SM, Zhang L, Ballantyne CM, et al. Regulation of LFA-1-dependent inflammatory cell recruitment by Cbl-b and 14-3-3 proteins. Blood. (2008) 111:3607–14. doi: 10.1182/blood-2007-07-103077
31. Bachmaier K, Toya S, Gao X, Triantafillou T, Garrean S, Park GY, et al. E3 ubiquitin ligase Cblb regulates the acute inflammatory response underlying lung injury. Nat Med. (2007) 13:920–6. doi: 10.1038/nm1607
32. Collins PE, Mitxitorena I, Carmody RJ. The ubiquitination of NF-kappaB subunits in the control of transcription. Cells. (2016) 5:E23. doi: 10.3390/cells5020023
33. Seijkens TTP, Poels K, Meiler S, van Tiel CM, Kusters PJH, Reiche M, et al. Deficiency of the T cell regulator Casitas B-cell lymphoma-B aggravates atherosclerosis by inducing CD8+ T cell-mediated macrophage death. Eur Heart J. (2019) 40:372–82. doi: 10.1093/eurheartj/ehy714
34. Gistera A, Hansson GK. The immunology of atherosclerosis. Nat Rev Nephrol. (2017) 13:368–80. doi: 10.1038/nrneph.2017.51
35. Liu YC, The E3 ubiquitin ligase Itch in T cell activation, differentiation, tolerance. Semin Immunol. (2007) 19:197–205. doi: 10.1016/j.smim.2007.02.003
36. Moser EK, Oliver PM. Regulation of autoimmune disease by the E3 ubiquitin ligase Itch. Cell Immunol. (2019) 340:103916. doi: 10.1016/j.cellimm.2019.04.004
37. Lohr NJ, Molleston JP, Strauss KA, Torres-Martinez W, Sherman EA, Squires RH, et al. Human ITCH E3 ubiquitin ligase deficiency causes syndromic multisystem autoimmune disease. Am J Hum Genet. (2010) 86:447–53. doi: 10.1016/j.ajhg.2010.01.028
38. Kleine-Eggebrecht N, Staufner C, Kathemann S, Elgizouli M, Kopajtich R, Prokisch H, et al. Mutation in ITCH gene can cause syndromic multisystem autoimmune disease with acute liver failure. Pediatrics. (2019) 143:e20181554. doi: 10.1542/peds.2018-1554
39. Qiu L, Joazeiro C, Fang N, Wang HY, Elly C, Altman Y, et al. Recognition and ubiquitination of Notch by Itch, a hect-type E3 ubiquitin ligase. J Biol Chem. (2000) 275:35734–7. doi: 10.1074/jbc.M007300200
40. Moser EK, Roof J, Dybas JM, Spruce LA, Seeholzer SH, Cancro MP, et al. The E3 ubiquitin ligase Itch restricts antigen-driven B cell responses. J Exp Med. (2019) 216:2170–83. doi: 10.1084/jem.20181953
41. Heissmeyer V, Macian F, Im SH, Varma R, Feske S, Venuprasad K, et al. Calcineurin imposes T cell unresponsiveness through targeted proteolysis of signaling proteins. Nat Immunol. (2004) 5:255–65. doi: 10.1038/ni1047
42. Hartenstein B, Teurich S, Hess J, Schenkel J, Schorpp-Kistner M, Angel P, et al. Th2 cell-specific cytokine expression and allergen-induced airway inflammation depend on JunB. EMBO J. (2002) 21:6321–9. doi: 10.1093/emboj/cdf648
43. Jin HS, Park Y, Elly C, Liu YC. Itch expression by Treg cells controls Th2 inflammatory responses. J Clin Invest. (2013) 123:4923–34. doi: 10.1172/JCI69355
44. Stohr R, Mavilio M, Marino A, Casagrande V, Kappel B, Mollmann J, et al. ITCH modulates SIRT6 and SREBP2 to influence lipid metabolism and atherosclerosis in ApoE null mice. Sci Rep. (2015) 5:9023. doi: 10.1038/srep09023
45. Nurieva RI, Zheng S, Jin W, Chung Y, Zhang Y, Martinez GJ, et al. The E3 ubiquitin ligase GRAIL regulates T cell tolerance and regulatory T cell function by mediating T cell receptor-CD3 degradation. Immunity. (2010) 32:670–80. doi: 10.1016/j.immuni.2010.05.002
46. Aziz M, Yang WL, Matsuo S, Sharma A, Zhou M, Wang P. Upregulation of GRAIL is associated with impaired CD4 T cell proliferation in sepsis. J Immunol. (2014) 192:2305–14. doi: 10.4049/jimmunol.1302160
47. Jensen IJ, Sjaastad FV, Griffith TS, Badovinac VP. Sepsis-induced T cell immunoparalysis: the Ins and Outs of impaired T cell immunity. J Immunol. (2018) 200:1543–53. doi: 10.4049/jimmunol.1701618
48. Lineberry NB, Su LL, Lin JT, Coffey GP, Seroogy CM, Fathman CG. Cutting edge: The transmembrane E3 ligase GRAIL ubiquitinates the costimulatory molecule CD40 ligand during the induction of T cell anergy. J Immunol. (2008) 181:1622–6. doi: 10.4049/jimmunol.181.3.1622
49. MacKenzie DA, Schartner J, Lin J, Timmel A, Jennens-Clough M, Fathman CG, et al. GRAIL is up-regulated in CD4+ CD25+ T regulatory cells and is sufficient for conversion of T cells to a regulatory phenotype. J Biol Chem. (2007) 282:9696–702. doi: 10.1074/jbc.M604192200
50. Meiler S, Smeets E, Winkels H, Shami A, Pascutti MF, Nolte MA, et al. Constitutive GITR activation reduces atherosclerosis by promoting regulatory CD4+ T-cell responses-brief report. Arterioscler Thromb Vasc Biol. (2016) 36:1748–52. doi: 10.1161/ATVBAHA.116.307354
51. Haymaker C, Yang Y, Wang J, Zou Q, Sahoo A, Alekseev A, et al. Absence of GRAIL promotes CD8(+) T cell anti-tumour activity. Nat Commun. (2017) 8:239. doi: 10.1038/s41467-017-00252-w
52. Kumar S, Naqvi RA, Khanna N, Pathak P, Rao DN. Th3 immune responses in the progression of leprosy via molecular cross-talks of TGF-beta, CTLA-4 and Cbl-b. Clin Immunol. (2011) 141:133–42. doi: 10.1016/j.clim.2011.06.007
53. Kumar S, Naqvi RA, Khanna N, Rao DN. Disruption of HLA-DR raft, deregulations of Lck-ZAP-70-Cbl-b cross-talk and miR181a towards T cell hyporesponsiveness in leprosy. Mol Immunol. (2011) 48:1178–90. doi: 10.1016/j.molimm.2011.02.012
54. Chattree V, Khanna N, Rao DN, Alterations in T cell signal transduction by M. leprae antigens is associated with downregulation of second messengers PKC, calcium, calcineurin, MAPK and various transcription factors in leprosy patients. Mol Immunol. (2007) 44:2066–77. doi: 10.1016/j.molimm.2006.09.008
55. Stempin CC, Rojas Marquez JD, Ana Y, Cerban FM. GRAIL and Otubain-1 are related to T cell hyporesponsiveness during Trypanosoma cruzi infection. PLoS Negl Trop Dis. (2017) 11:e0005307. doi: 10.1371/journal.pntd.0005307
56. Sande OJ, Karim AF, Li Q, Ding X, Harding CV, Rojas RE, et al. Mannose-capped lipoarabinomannan from Mycobacterium tuberculosis induces CD4+ T cell anergy via GRAIL. J Immunol. (2016) 196:691–702. doi: 10.4049/jimmunol.1500710
57. Taylor JJ, Krawczyk CM, Mohrs M, Pearce EJ. Th2 cell hyporesponsiveness during chronic murine schistosomiasis is cell intrinsic and linked to GRAIL expression. J Clin Invest. (2009) 119:1019–28. doi: 10.1172/JCI36534
58. Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Rutkowski P, Lao CD, et al. Five-year survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med. (2019) 381:1535–46. doi: 10.1056/NEJMoa1910836
59. Hellmann MD, Paz-Ares L, Bernabe Caro R, Zurawski B, Kim SW, Carcereny Costa E, et al. Nivolumab plus ipilimumab in advanced non-small-cell lung cancer. N Engl J Med. (2019) 381:2020–31. doi: 10.1056/NEJMoa1910231
60. Shifrut E, Carnevale J, Tobin V, Roth TL, Woo JM, Bui CT, et al. Genome-wide CRISPR screens in primary human T cells reveal key regulators of immune function. Cell. (2018) 175:1958–71.e15. doi: 10.1016/j.cell.2018.10.024
61. Shi ZD, Li XF, Hao L, Zhao Y, Wang YX, Dong BZ, et al. Cbl-b gene silencing in splenic T lymphocytes as a therapeutic strategy to target the prostate cancer RM-1 cell tumors in immune competent mice. Eur Rev Med Pharmacol Sci. (2014) 18:3819–30.
62. Chiang JY, Jang IK, Hodes R, Gu H. Ablation of Cbl-b provides protection against transplanted and spontaneous tumors. J Clin Invest. (2007) 117:1029–36. doi: 10.1172/JCI29472
63. Han S, Chung DC, St Paul M, Liu ZQ, Garcia-Batres C, Elford AR, et al. Overproduction of IL-2 by Cbl-b deficient CD4(+) T cells provides resistance against regulatory T cells. Oncoimmunology. (2020) 9:1737368. doi: 10.1080/2162402X.2020.1737368
64. Hinterleitner R, Gruber T, Pfeifhofer-Obermair C, Lutz-Nicoladoni C, Tzankov A, Schuster M, et al. Adoptive transfer of siRNA Cblb-silencedT CD8+ lymphocytes augments tumor vaccine efficacy in a B16 melanoma model. PLoS ONE. (2012) 7:e44295. doi: 10.1371/journal.pone.0044295
65. Peer S, Baier G, Gruber T. Cblb-deficient T cells are less susceptible to PD-L1-mediated inhibition. Oncotarget. (2017) 8:41841–53. doi: 10.18632/oncotarget.18360
66. Sitaram P, Uyemura B, Malarkannan S, Riese MJ. Beyond the cell surface: targeting intracellular negative regulators to enhance T cell anti-tumor activity. Int J Mol Sci. (2019) 20:E5821. doi: 10.3390/ijms20235821
67. van de Veerdonk FL, Netea MG. CBLB ubiquitin ligase: a major regulator of antifungal immunity. Nat Med. (2016) 22:834–5. doi: 10.1038/nm.4160
68. Xiao Y, Tang J, Guo H, Zhao Y, Tang R, Ouyang S, et al. Targeting CBLB as a potential therapeutic approach for disseminated candidiasis. Nat Med. (2016) 22:906–14. doi: 10.1038/nm.4141
69. Wirnsberger G, Zwolanek F, Asaoka T, Kozieradzki I, Tortola L, Wimmer RA, et al. Inhibition of CBLB protects from lethal Candida albicans sepsis. Nat Med. (2016) 22:915–23. doi: 10.1038/nm.4134
70. Ou R, Zhang M, Huang L, Moskophidis D. Control of virus-specific CD8+ T-cell exhaustion and immune-mediated pathology by E3 ubiquitin ligase Cbl-b during chronic viral infection. J Virol. (2008) 82:3353–68. doi: 10.1128/JVI.01350-07
71. Lin HT, Chen CC, Liu PY, Wu HL, Wu TH, Huang CH, et al. GRAIL attenuates influenza A virus infection and pathogenesis by inhibiting viral nucleoprotein. Sci Rep. (2018) 8:17242. doi: 10.1038/s41598-018-35722-8
72. Terrier B, Joly F, Vazquez T, Benech P, Rosenzwajg M, Carpentier W, et al. Expansion of functionally anergic CD21-/low marginal zone-like B cell clones in hepatitis C virus infection-related autoimmunity. J Immunol. (2011) 187:6550–63. doi: 10.4049/jimmunol.1102022
73. Bruening J, Lasswitz L, Banse P, Kahl S, Marinach C, Vondran FW, et al. Hepatitis C virus enters liver cells using the CD81 receptor complex proteins calpain-5 CBLB. PLoS Pathog. (2018) 14:e1007111. doi: 10.1371/journal.ppat.1007111
74. Kobashigawa Y, Tomitaka A, Kumeta H, Noda NN, Yamaguchi M, Inagaki F. Autoinhibition and phosphorylation-induced activation mechanisms of human cancer and autoimmune disease-related E3 protein Cbl-b. Proc Natl Acad Sci USA. (2011) 108:20579–84. doi: 10.1073/pnas.1110712108
Keywords: atherosclerosis, inflammation, E3 ubiquitin ligases, Cbl-B, Itch, GRAIL
Citation: Poels K, Vos WG, Lutgens E and Seijkens TTP (2020) E3 Ubiquitin Ligases as Immunotherapeutic Target in Atherosclerotic Cardiovascular Disease. Front. Cardiovasc. Med. 7:106. doi: 10.3389/fcvm.2020.00106
Received: 20 March 2020; Accepted: 20 May 2020;
Published: 05 June 2020.
Edited by:
Rory R. Koenen, Maastricht University, NetherlandsReviewed by:
Ingrid E. Dumitriu, University of London, United KingdomJürgen Bernhagen, Ludwig Maximilian University of Munich, Germany
Copyright © 2020 Poels, Vos, Lutgens and Seijkens. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tom T. P. Seijkens, dC50LnNlaWprZW5zQGFtc3RlcmRhbXVtYy5ubA==
†These authors share first authorship
‡These authors share last authorship