 Caroline Mullis
Caroline Mullis Talia H. Swartz
Talia H. Swartz
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Cardiovasc. Med. , 11 June 2020
Sec. Atherosclerosis and Vascular Medicine
Volume 7 - 2020 | https://doi.org/10.3389/fcvm.2020.00095
36.9 million people worldwide are living with HIV-1. The disease remains incurable and HIV-infected patients have increased risk of atherosclerosis. Inflammation is a key driver of atherosclerosis, but no targeted molecular therapies have been developed to reduce cardiovascular risk in people with HIV-1 (PWH). While the mechanism is unknown, there are several important inflammatory signaling events that are implicated in the development of chronic inflammation in PWH and in the inflammatory changes that lead to atherosclerosis. Here we describe the pro-inflammatory state of HIV-1 infection that leads to increased risk of cardiovascular disease, the role of the NLR Family Pyrin Domain Containing 3 (NLRP3) inflammasome in HIV-1 infection, the role of the NLRP3 inflammasome in cardiovascular disease (CVD), and outline a model whereby HIV-1 infection can lead to atherosclerotic disease through NLRP3 inflammasome activation. Our discussion highlights the literature supporting HIV-1 infection as a stimulator of the NLRP3 inflammasome as a driver of atherosclerosis.
Human Immunodeficiency Virus (HIV-1) causes a chronic infection that impacts more than 36 million individuals worldwide. HIV-1 treatment with highly active antiretroviral therapy (ART) can achieve virologic suppression leading to increased life expectancy in PWH. Still, PWH develop age-related co-morbidities including atherosclerosis and cardiovascular disease (CVD) earlier than their HIV-negative counterparts. This phenomenon is multifactorial including contributions from higher behavioral risk factors, ART toxicity, and chronic inflammation (1–6). Inflammatory signaling pathway activation has been described in the generation of chronic inflammation in PWH and in cardiovascular disease. The NLRP3 inflammasome pathway is activated in HIV-1 infection contributing to chronic inflammation and has also been implicated in atherosclerotic plaque formation. We present a potential role for NLRP3 inflammasome activation in chronic inflammation contributing to increased rates of CVD in PWH.
The early development of age-related co-morbidities in PWH can in part be attributable to the observed persistent inflammation and immune dysfunction seen in HIV-1 infection (1, 7, 8). HIV-1 infection creates a pro-inflammatory state marker by an increase in senescent cells that secrete inflammatory mediators resulting in low-level inflammation and increased T lymphocyte cell turnover (1, 9). Senescent cells secrete inflammatory mediators resulting in low-level inflammation and increased cell turnover (1, 10–12). Desai and Landay propose a multifaceted model for accelerated aging in HIV-1 infection (13): residual HIV-1 replication in activated immune cells despite ART; HIV-1 depletion of Th17 lymphocytes at GI epithelial mucosa causes microbial translocation and antigenic burden; thymic dysfunction causes loss of naïve T lymphocytes and regulatory CD4+ T lymphocytes, suppressing T lymphocyte activation; clonal expansion of activated immune cells with T lymphocyte loss of CD28 and telomere shortening, causing non-functional, senescent T lymphocytes (9, 14–17). Klatt et al. describe fibrosis and dysfunction of lymphoid organs and co-infection by pathogens such as cytomegalovirus (CMV) (4).
Both the viral reservoir and immune checkpoint molecule dysregulation mark an important mechanism for HIV-1 persistence and residual inflammation. HIV-1 results in productive infection of a small percentage of permissive cells with abortive infection of non-permissive bystander cells in lymphoid tissues (>95% CD4 T cell population). These bystander cells undergo pyroptosis, a programmed cell death, which could re-activate latently infected cells, causing more uninfected cells to die and sustain chronic inflammation (12, 18–24). Additionally an immune checkpoint molecule, programmed cell death protein 1 (PD-1) is highly expressed on CD4+ and CD8+ T lymphocytes during HIV-1 infection, and does not fully normalize on ART (12). On ART PD-1 levels correlate with CD4+ T lymphocyte count and are upregulated by Interleukin-7 (IL-7). Immune checkpoint molecules, lymphocyte-activation gene 3(LAG-3) and T cell immunoreceptor with Ig and ITIM domains (TIGIT), were also markers of HIV-1 infected CD4+ T cells on ART suggesting a role for immune checkpoint dysregulation in viral persistence.
Cardiovascular disease is a major cause of morbidity and mortality in PWH (25–32). The D:A:D (Data Collection on Adverse Events of Anti-HIV Drugs) study estimates that 11% of deaths among PWH are attributable to CVD (33) and the Clinical and Virological Outcome of European Patients Infected With HIV (EuroSIDA) study estimates that one third of clinical events that are non-AIDS defining are related to cardiovascular disease (34). Risk of CVD in PWH is multifactorial with possible contributions from antiretroviral therapies (ART), increased exposures to traditional risk factors and chronic inflammation (35–39). In a meta-analysis, PWH when compared to HIV-1 uninfected control patients had an increased relative risk of CVD (40, 41). The D:A:D study also showed an increased relative risk of CVD with exposure the protease inhibitor drug class (33, 40) that mostly was driven by the increased CVD risk associated with ritonavir-boosted darunavir (42). The contribution of protease inhibitors to CVD risk has been controversial and limited by observational data. Nucleoside reverse transcriptase inhibitors (NRTIs) such as stavudine and zidovudine are associated with dyslipidemia, impaired insulin resistance, and greater carotid intima-media thickness (cIMT) in the setting of hyperlipidemia contributing to CVD risk (43–45). In the D:A:D trial abacavir was associated with increased risk of CVD; however, this association has not been observed in longitudinal data (40).
Higher rates of smoking, substance use and dyslipidemia contribute to increased risk of CVD in PWH (42, 46–48). PWH had 50% increased risk of acute myocardial infarction after controlling for behavioral and Framingham risk factors such as dyslipidemia, hypertension, and smoking suggesting additional mechanisms for increased CVD in PWH (49). However, some evidence suggests the association between smoking and CVD is even stronger in PWH (48).
Inflammatory biomarkers demonstrate chronic inflammation are associated with cardiovascular disease in PWH in large clinical cohorts (7, 35, 46, 50, 50–54). Compared to uninfected individuals PWH had 50% higher high sensitivity C-reactive protein (hsCRP), 150% higher Interleukin-6 (IL-6), 90% higher D-dimer, and 25% higher cystatin-C level. Inflammatory biomarkers like IL-6, hsCRP, and D-dimer remain elevated in PWH compared to uninfected controls despite ART treatment. D-dimer, hsCRP, and IL-6 have been associated with increased risk of CVD, and soluble CD14 (sCD14) has been associated with microbial translocation (7). In the Multicenter AIDS Cohort Study (MACS) of HIV+ men, IL-6, hsCRP, tumor necrosis factor alpha (TNF-α), soluble CD14 (sCD14), and soluble tumor necrosis factor receptor II (sTNFR II) were markers of frailty in HIV-1 disease, establishing an association with monocyte-macrophage immune activation in PWH (55). Treatment intensification has not resulted in improvement in HIV-1 viremia or inflammatory biomarkers (56, 57). Initiation of ART in patients who are elite controllers, those who spontaneously suppress HIV-1 viral load without ART, remains controversial (58–60).
The HIV-1 pro-inflammatory state outlined in the prior section also contributes to increased rates of CVD observed in PWH. The formation of atherosclerotic plaques contributing to CVD is an inflammatory process involving inflammatory signaling leading to monocyte recruitment, migration, and activation into pro-inflammatory foam cells (61, 62). HIV-1 proteins (nef, tat, and env) can induce inflammatory signals activating macrophages facilitating foam cell transformation and plaque formation (61).
In PWH, CVD has been associated with elevated inflammatory markers. In the Strategies for Management of ART (SMART) trial, a randomized, controlled trial assessing episodic ART therapy guided by CD4+ count compared to continuous ART, the observed hazard ratio of cardiovascular disease between the drug conservation group (episodic ART) against the viral suppression group (continuous ART) was 1.6 (p < 0.05) (50). This increase in CVD risk with episodic ART use was associated with an increase in inflammatory markers such as IL-6 (40, 63). Arterial inflammation is also modestly increased in PWH (59) and correlates with circulating inflammatory biomarkers such hsCRP and IL-6 but not with HIV-1 markers. In PWH initiated on ART, reduction of HIV-1 RNA correlated with decrease in D-dimer and IL-6 but not in hsCRP, while hsCRP was associated with progression of HIV-1 and mortality after adjustment for CD4+ count and HIV-1 viral load (64). These studies suggest a role for HIV-1 related inflammation as a contributor to CVD risk in PWH.
Decreasing inflammation represents an important therapeutic strategy for CVD prevention (26, 65). In the JUPITER trial patients with low LDL and high hsCRP were randomized to statin vs. placebo. Statin treatment had 44% relative risk reduction (66, 67). In the SATURN-HIV trial of rosuvastain in PWH, cIMT reduction on statin compared to placebo was independent of the lipid lowering effects (66, 68). With long-term statin use (48 weeks) almost all inflammatory markers decreased, including sCD14. By contrast aspirin did not decrease markers of inflammation including sCD14, IL-1 and D-dimer in a smaller trial of 12 week follow-up (69). Table 1 summarizes therpies that have been tested to reduce inflammation as a cause of atherosclerosis in HIV-1 disease.
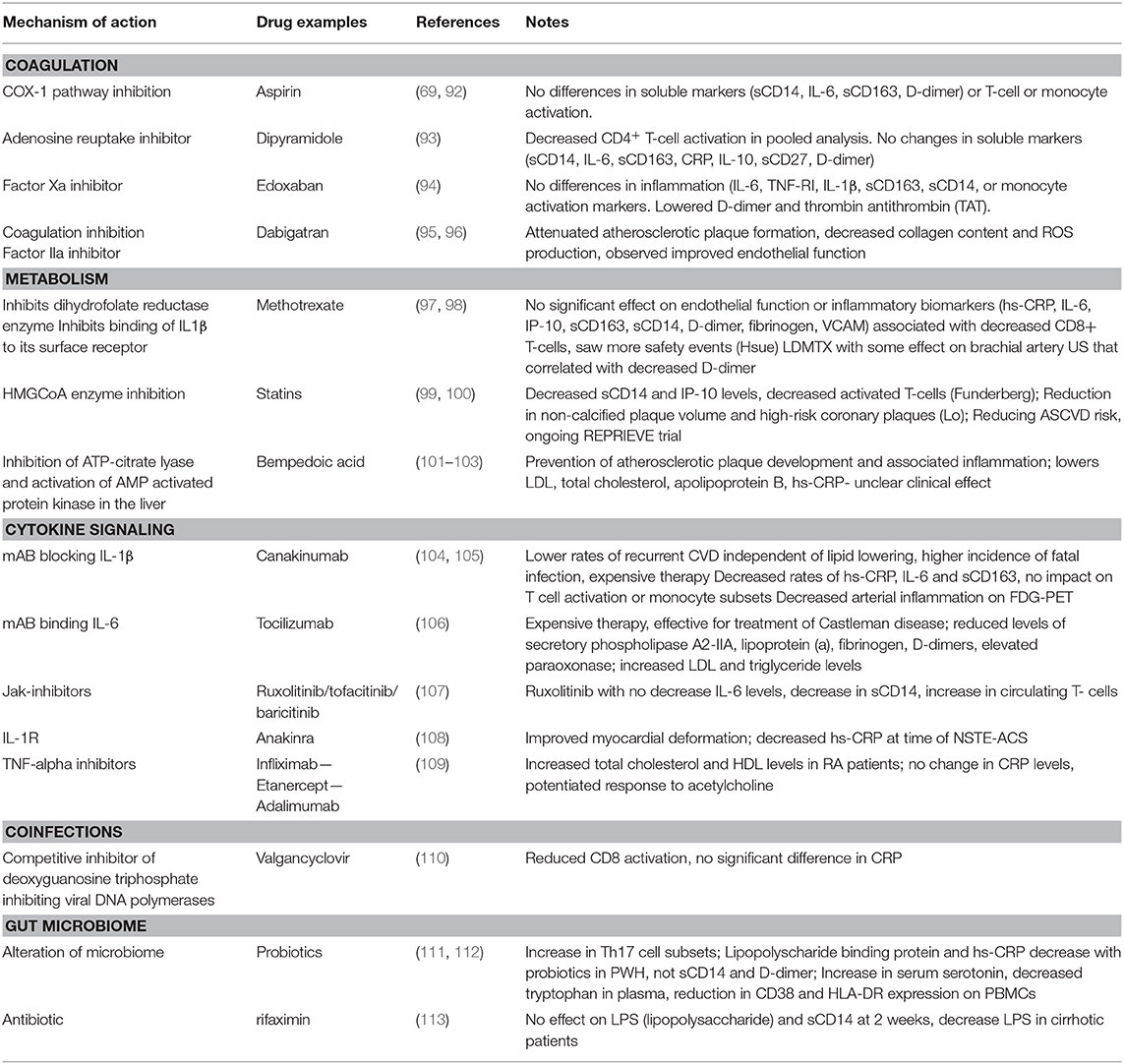
Table 1. Drugs with anti-inflammatory properties to reduce atherosclerosis in HIV disease.
Activity of the NLRP3 inflammasome contributes to the chronic, pro-inflammatory state in PWH (70–75). The inflammasome is part of the disease through the innate immune system activated by pattern recognition receptors (76). The inflammasome activates caspase-1, which cleaves prointerleukin-1β (pro-IL1β) into the mature, secretory interleukin-1β (IL-1β). Inflammasome activation also mediates pyroptosis or programmed cell death of myeloid and lymphoid cells.
HIV-1 infection provides the first of two signals for NLRP3 inflammasome activation (77). Monocyte-derived macrophages primed with HIV-1 have increased IL-1β production after exposure to the second NLRP3 activation signal. HIV-1 virions induce Toll-Like Receptors (TLRs) to stimulate pro-IL-1β expression (71, 78). HIV-1 infection is required for activation as, when exposed to ART, induction of pro-IL-1β and release of IL-1β were decreased (78). HIV-1 and HCV virion induction of TLRs may not be dependent on cell entry as induction of the inflammasome was still seen in the presence of cell entry inhibitors (71).
The NLRP3 inflammasome has been well-studied as a cause of T lymphocyte cell death and activation through pyroptosis (19, 21, 22, 24, 79, 80). This has been demonstrated as infected CD4 T lymphocytes can stimulate bystander cell NLRP3 inflammasome activation and stimulation of pyroptosis (19, 21, 22, 24). The role of the NLRP3 inflammasome has been studied in HIV pathogenesis in lymphoid tissue and in peripheral blood (81). Peripheral blood monocytes in PWH were positive for an inflammasome adaptor protein, ASC speck (apoptosis-associated speck-like protein containing a caspase-recruitment domain), not seen in healthy controls (82). ASC speck protein is a marker for inflammasome activation, suggesting that in PWH activation of pyroptotic cell death is responsible for progressive CD4+ T lymphocyte death and contributes to chronic inflammation.
Inflammasome activation occurs during acute HIV-1 infection (83) and persists in immune non-responders, patients on ART without CD4+ T lymphocyte recovery (CD4 < 350) (74). When stimulated with lipopolysaccharide (LPS), a TLR signal for inflammasome activation, upregulation of inflammasome genes (NLRP3, caspase-1) was seen in both immune non-responders and responders (CD4 > 500). Substance abuse enhances inflammasome activation in PWH. Cocaine exposure to HIV-1 infected macrophages increases activity by potentially priming the NLRP3 inflammasome by potentiating reactive oxygen species (ROS) production (84, 85). By contrast, cannabis has been demonstrated to reduce NLRP3 inflammasome activation (86, 87).
Several studies implicate the NLRP3 inflammasome in the pathogenesis of atherosclerosis (88). The role of the NLRP3 inflammasome in atherosclerosis was demonstrated using low-density lipoprotein (LDL) receptor deficient mice, a model for familial hypercholesterolemia (89). After lethally irradiated bone marrow was reconstituted with wild-type, NLRP3-, ASC-, or IL1α/β-deficient bone marrow, mice had lower levels of IL-18 and IL-1 family cytokines and showed decreased atherosclerosis. IL-1β release was observed after 24-h incubation with LDL even in the absence of other known NLRP3 inflammasome primers suggesting a role for cholesterol in both priming and activation of the inflammasome pathway.
Interestingly, in an in vivo model ApoE−/−, Nlrp3−/−, ApoE−/−, Asc−/−, ApoE−/−, and caspase-1−/− double-deficient mice fed a high-fat diet failed to demonstrate differences in atherosclerosis progression and phenotype of the plaque (90). The differences in these findings could be attributed to the use of different mouse models and a potential role of IL-1α, which can be generated independently of the NLRP3 inflammasome. These contradictory results have also been attributed to different diets as in the Menu et al. study mice were fed 1.25% cholesterol (HFD) diet which could have overwhelmed genetic differences as compared to the 0.15% cholesterol diet (western diet) (91).
Inflammasome activity in plaque generation is further supported by the presence of activated caspase-1 in atheromatous plaques and caspase knockout models in ApoE−/− mice (114–116). Caspase knockout in ApoE−/− mice decreased the size of atherosclerotic lesion size in the aortic arch, intra-lesion IFN-γ and plasma levels of IL-1β and IL-1α. In ASC−/− mice, the NLRP3 inflammasome contributes to atherogenesis by triggering maturity of IL-1β and IL-18 in atherosclerotic plaques after vascular injury (117, 118). In ASC−/− mice neointimal formation was attenuated and decreased IL-1β and IL-18 expression was observed in the plaques compared to wild type. Inhibition of NLRP3 inflammasome with arglabin, a plant-based metabolite inhibitor of the NLRP3 inflammasome, showed decreased atherosclerotic lesions in apolipoE2-Ki mice fed a high fat diet (118, 119).
Other activating factors have been identified. Trimethylamine-N-oxide (TMAO), a by-product of choline and L-carnitine metabolism, promotes the formation of foam cells from macrophages in a process mediated by inflammasome activity (120). TMAO stimulated thioredoxin-interactive protein (TXNIP)-NLRP3 inflammasome activity in human umbilical vein endothelial cells causing release of IL-1β and IL-18 in a dose and time dependent manner. LPS-exposed THP-1 macrophages induced Lectin-like oxLDL receptor-1 (LOX-1) expression, generation of ROS, auto-phagosome formation and damage to mitochondrial DNA (121). LOX-1 inhibition resulted in attenuated NLRP3 inflammasome activity consistent with decreased atherosclerotic plaque burden in LOX-1 deletion in mice fed a high fat diet.
Oxidative stress-responsive transcription factor NF-E2 related 2 (Nrf2) also has a role in inflammasome activation and atherosclerosis (122, 123). Nrf2-/ApoE- mice showed attenuation of atherosclerosis without change in lipid metabolism or foam cell transformation when compared to Nrf2+/ApoE− mice. Cholesterol crystals triggered production of IL-1α and IL-1β in Nrf2+/+ dendritic cells, not observed in Nrf2 deficient dendritic cells. In NLRP3-deficient and caspase1−/− macrophages, cholesterol crystal-induced IL-1β production was reduced. Neutralization of IL-1α and IL-1β by induction of neutralizing antibodies resulted in reduced atherosclerosis in Nrf2+/+ ApoE−/− but not in Nrf2−/− ApoE −/− mice. These results suggest that the observed Nrf2 effects on atherogenesis are from its role in inflammasome activation and IL-1 production (122).
Atherosclerosis is also associated with endothelial dysfunction. Endothelial senescence, which is linked to CV diseases, is associated with NLRP3 activation (124). Induction of endothelial cell senescence with bleomycin showed increased IL-1β and caspase-1. IL-1β promoted endothelial cell senescence as indicated by upregulation of p53/p21 expression.
Inflammasome activity in CVD is also modulated by separate cardiovascular risk factors including hyperglycemia, obesity and hyperuricemia (125). IL-1β is elevated in patients with high blood pressure and type 2 diabetes mellitus (118, 126). In type 2 diabetes use of γ-tocotrienol to inhibit the NLRP3 inflammasome can delay disease progression (127). Diabetic rats showed significantly increased NLRP3 inflammasome activation (118, 128). When animals were given rosuvastatin both NLRP3 inflammasome and MAPK expression was decreased, with associated decrease in cardiac fibrosis, suggesting a potential role for the NLRP3 inflammasome in diabetic cardiomyopathy.
There is literature supporting the role of the NLRP3 in hypertension. Attenuation of NLRP3 and caspase by chronic inhibition of NF-kB attenuates high salt induced hypertension (129). In a murine model of hypertension, a highly selective SGK1 inhibitor, EMD638683, was shown to suppress IL-1β release, NLRP3 expression, and caspase-1 activation which was associated by reduced transformation of fibroblasts to myofibroblasts (130). These effects on cardiac fibrosis were not observed with supplementation of exogenous IL-1β suggesting NLRP3 and IL-1β have a role in hypertensive cardiac damage.
The NLRP3 inflammasome and the associated inflammatory response have a role in the pathophysiology of a myocardial infarction (MI) (131, 132). During MI, release of cellular debris and production of reactive oxygen species activate the NLRP3 inflammasome leading to development of cardiomyopathy. Sandanger et al. demonstrate the role of the inflammasome in a murine myocardial ischemia-reperfusion injury, demonstrating NLRP3, IL-1β, and IL-18 mRNA expression was increased in cardiac fibroblasts post-MI (131). Cardiac fibroblasts had dose-dependent mRNA expression of NLRP3 and IL-1β when incubated with TLR2 and TLR4 ligands; this was blocked when incubated with NF-kB inhibitor. Post-MI, NLRP3−/− mice demonstrated improved cardiac function and reduced infarct size compared with wild type after reperfusion. NLRP3 inflammasome activation may play a cardioprotective role in ischemia-reperfusion injury (133). IL-1β neutralizing antibodies and anakinra, an IL-1 receptor antagonist, showed reduced cardiac hypertrophy and myocardial dysfunction post-MI; this suggests potential therapeutic interventions to ameliorate cardiac dysfunction post-MI (118, 134, 135).
Therapeutic interventions targeting reduction of inflammation to reduce CVD risk have been tested. The JUPITER trial was a primary-prevention trial randomizing patients with a low LDL cholesterol (<130 mg/dL) but elevated hsCRP (>2.0 mg/L) to receive rosuvastatin or placebo (67). Rosuvastatin was associated with reduction in cardiovascular risk (hazard ratio 0.56), lower LDL, and hsCRP. Simvastatin reduced NLRP3 activation in a diabetic rat model (136). In addition to lowering LDL, statins may decrease CVD by decreasing inflammasome activity.
The NLRP3 inflammasome is also a potential therapeutic target. NLRP3 inflammasome inhibitors show potential in murine models. Arglabin, showed decreased IL-1β plasma levels and decreased atherosclerotic lesion size in an ApoE knockout (119). MCC950, a selective inhibitor of NLRP3, demonstrated decreased plasma IL-1β levels and decreased atherosclerotic plaque size and volume (137). The CANTOS trial randomized patients with past MI and elevated hsCRP on optimal lipid lowering therapy to receive canakinumab, a monoclonal antibody targeting IL-1β, or placebo (104). A 15% reduction in major CV events was observed without change in LDL cholesterol. The benefit of this therapy associated with higher risk of fatal infection. Subsequently numerous reports have implicated a role of targeting IL-1β in reducing atherosclerotic disease (104, 138–148). Most recently, Li et al. demonstrated that treatment with VX-765, an NLRP3 inflammasome inhibitor, halted progression of atherosclerosis and reduced vascular smooth muscle cells (VSMCs) pyroptosis (149).
An emerging literature supports the role of the NLRP3 inflammasome as a driver of CVD in HIV-1 disease (150, 151). Yearley et al. demonstrated that in SIV-infected rhesus macaques, IL-18 secretion as a marker of NLRP3 inflammasome activity was associated with atherosclerotic progression (152). Further, they observed that the IL-18 was observed to be associated with macrophages, not T lymphocytes, suggesting the activation of alternative cell types in response to HIV-1 infection of T lymphocytes. Kearns et al. demonstrated that expression of HIV-1 transcripts can drive atherosclerosis through activation of caspase-1 in inflammatory monocytes. They further demonstrated that IL-18 levels were higher in HIV-1 infected patients with atherosclerotic disease and that these levels were correlated with monocyte/macrophage activation markers.
In a follow up study to the CANTOS trial, Hsue et al. demonstrated that a single dose of canakinumab reduced numerous inflammatory markers including cytokine production and atrial inflammation in PWH (105). This study and others suggest that inflammasome targeted therapeutic interventions reduction of cardiovascular events demonstrate a crucial role for this pathway in the pathogenesis of CVD in PWH. Hoel et al. evaluated PWH and demonstrated that soluble markers of interleukin 1 (IL-1Ra) levels were associated with a 1.5-fold increased risk of first-time myocardial infarction (153).
Here we describe the evidence supporting HIV-1 infection as a chronic and pro-inflammatory state that is associated with increased risk of atherosclerosis. We review the evidence defining the role of HIV-1 infection in activation of the NLRP3 inflammasome and the role of NLRP3 activation in the development of atherosclerosis. HIV-1 infection can mediate NLRP3 inflammasome activation, thus potentiating a pro-inflammatory state and increasing risk of atherosclerosis. Figure 1 demonstrates a model whereby HIV-1 infection can stimulate the NLRP3 inflammasome to result in IL-1β secretion which impacts on various cell types including endothelial cells, macrophages, monocytes, and smooth muscle, signaling through soluble factors including chemokines, cytokines, adhesion molecules, matrix metalloproteinases to result in progression of atherosclerotic disease. HIV-1 chronic inflammation, in concert with other factors, can drive inflammation including that seen in cardiovascular disease and that targeting this pathway can have important therapeutic benefits.
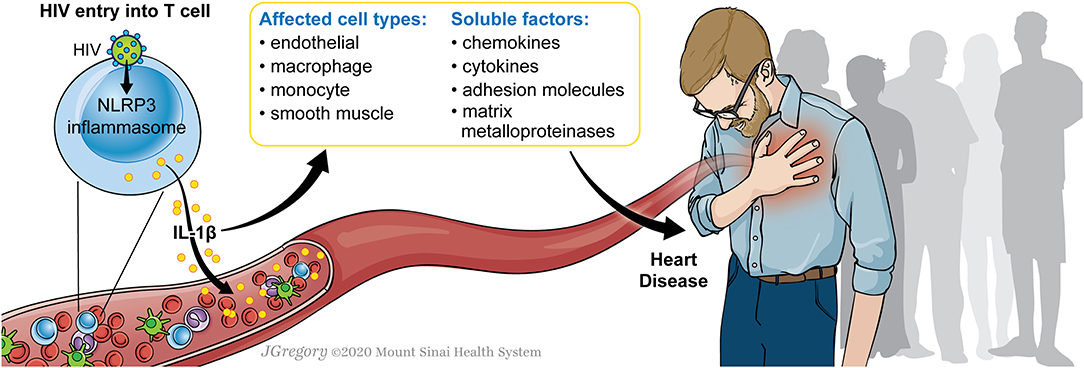
Figure 1. Model for HIV-1 infection and NLRP3 inflammasome activation to drive atherosclerosis. A model is shown in which HIV-1 infection of a CD4+ T cell can stimulate the NLRP3 inflammasome. This results in IL-1β secretion which impacts on various cell types including endothelial cells, macrophages, monocytes, and smooth muscle, signaling through soluble factors including chemokines, cytokines, adhesion molecules, matrix metalloproteinases. The activation of these factors can potentiate progression of atherosclerotic disease.
We present the data implicating the NLRP3 inflammasome as a signaling pathway that is both activated by HIV-1 infection and that drives the development of atherosclerosis in PWH. Gaps exist in our understanding to establish clear links between HIV-1 infection, NLRP3 inflammasome activation, and atherosclerotic disease with the approach of developing targeting therapies to reduce the inflammatory signaling that drives this important comorbidity. Animals studies of humanized mice that connect the risk of HIV-1 infection, NLRP3 activation, and the development of atherosclerosis are necessary.
The above studies highlight the importance of cardiovascular risk in PWH and the need for mechanistic understanding behind targeted therapies. Care of PWH should continue to focus on approaches to reduce cardiovascular risk in PWH through lifestyle modifications, tight lipid, hypertension, and glycemic control, while seeking to further identify biomarkers with linked clinical outcomes.
CM and TS conceived the idea and wrote the manuscript.
This work was supported by funding from the National Institutes of Health, 1K08AI120806 (TS) and the Alpha Omega Alpha Research Award, and the Schneider-Lesser Foundation (TS).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
PWH, people with HIV-1; hsCRP, high sensitivity C-reactive protein; CVD, cardiovascular disease; ART, Antiretroviral therapy; NLRP3, NLR Family Pyrin Domain Containing 3.
1. Deeks SG. HIV infection, inflammation, immunosenescence, and aging. Annu Rev Med. (2011) 62:141–55. doi: 10.1146/annurev-med-042909-093756
2. Hazenberg MD, Otto SA, van Benthem BH, Roos MT, Coutinho RA, Lange JM, et al. Persistent immune activation in HIV-1 infection is associated with progression to AIDS. AIDS. (2003) 17:1881–8. doi: 10.1097/00002030-200309050-00006
3. Deeks SG, Kitchen CM, Liu L, Guo H, Gascon R, Narvaez AB, et al. Immune activation set point during early HIV infection predicts subsequent CD4+ T-cell changes independent of viral load. Blood. (2004) 104:942–7. doi: 10.1182/blood-2003-09-3333
4. Klatt NR, Chomont N, Douek DC, Deeks SG. Immune activation and HIV persistence: implications for curative approaches to HIV infection. Immunol Rev. (2013) 254:326–42. doi: 10.1111/imr.12065
5. Vassallo M, Mercie P, Cottalorda J, Ticchioni M, Dellamonica P. The role of lipopolysaccharide as a marker of immune activation in HIV-1 infected patients: a systematic literature review. Virol J. (2012) 9:174. doi: 10.1186/1743-422X-9-174
6. Burdo TH, Lentz MR, Autissier P, Krishnan A, Halpern E, Letendre S, et al. Soluble CD163 made by monocyte/macrophages is a novel marker of HIV activity in early and chronic infection prior to and after anti-retroviral therapy. J Infect Dis. (2011) 204:154–63. doi: 10.1093/infdis/jir214
8. Guaraldi G, Orlando G, Zona S, Menozzi M, Carli F, Garlassi E, et al. Premature age-related comorbidities among HIV-infected persons compared with the general population. Clin Infect Dis. (2011) 53:1120–6. doi: 10.1093/cid/cir627
9. Hunt PW, Martin JN, Sinclair E, Bredt B, Hagos E, Lampiris H, et al. T cell activation is associated with lower CD4+ T cell gains in human immunodeficiency virus-infected patients with sustained viral suppression during antiretroviral therapy. J Infect Dis. (2003) 187:1534–43. doi: 10.1086/374786
10. Lederman MM, Calabrese L, Funderburg NT, Clagett B, Medvik K, Bonilla H, et al. Immunologic failure despite suppressive antiretroviral therapy is related to activation and turnover of memory CD4 cells. J Infect Dis. (2011) 204:1217–26. doi: 10.1093/infdis/jir507
11. Massanella M, Negredo E, Perez-Alvarez N, Puig J, Ruiz-Hernandez R, Bofill M, et al. CD4 T-cell hyperactivation and susceptibility to cell death determine poor CD4 T-cell recovery during suppressive HAART. AIDS. (2010) 24:959–68. doi: 10.1097/QAD.0b013e328337b957
12. Massanella M, Fromentin R, Chomont N. Residual inflammation and viral reservoirs: alliance against an HIV cure. Curr Opin HIV AIDS. (2016) 11:234–41. doi: 10.1097/COH.0000000000000230
13. Desai S, Landay A. Early immune senescence in HIV disease. Curr HIV/AIDS Rep. (2010) 7:4–10. doi: 10.1007/s11904-009-0038-4
14. Hunt PW, Sinclair E, Rodriguez B, Shive C, Clagett B, Funderburg N, et al. Gut epithelial barrier dysfunction and innate immune activation predict mortality in treated HIV infection. J Infect Dis. (2014) 210:1228–38. doi: 10.1093/infdis/jiu238
15. Brenchley JM, Schacker TW, Ruff LE, Price DA, Taylor JH, Beilman GJ, et al. CD4+ T cell depletion during all stages of HIV disease occurs predominantly in the gastrointestinal tract. J Exp Med. (2004) 200:749–59. doi: 10.1084/jem.20040874
16. Brenchley JM, Price DA, Douek DC. HIV disease: fallout from a mucosal catastrophe? Nat Immunol. (2006) 7:235–9. doi: 10.1038/ni1316
17. Mehandru S, Poles MA, Tenner-Racz K, Horowitz A, Hurley A, Hogan C, et al. Primary HIV-1 infection is associated with preferential depletion of CD4+ T lymphocytes from effector sites in the gastrointestinal tract. J Exp Med. (2004) 200:761–70. doi: 10.1084/jem.20041196
18. Doitsh G, Cavrois M, Lassen KG, Zepeda O, Yang Z, Santiago ML, et al. Abortive HIV infection mediates CD4 T cell depletion and inflammation in human lymphoid tissue. Cell. (2010) 143:789–801. doi: 10.1016/j.cell.2010.11.001
19. Doitsh G, Galloway NL, Geng X, Yang Z, Monroe KM, Zepeda O, et al. Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection. Nature. (2014) 505:509–14. doi: 10.1038/nature12940
20. Doitsh G, Greene WC. Dissecting how CD4 T cells are lost during HIV infection. Cell Host Microbe. (2016) 19:280–91. doi: 10.1016/j.chom.2016.02.012
21. Doitsh G, Galloway NL, Geng X, Yang Z, Monroe KM, Zepeda O, et al. Corrigendum: Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection. Nature. (2017) 544:124. doi: 10.1038/nature22066
22. Galloway NL, Doitsh G, Monroe KM, Yang Z, Muñoz-Arias I, Levy DN, et al. Cell-to-cell transmission of HIV-1 is required to trigger pyroptotic death of lymphoid-tissue-derived CD4 T cells. Cell Rep. (2015) 12:1555–63. doi: 10.1016/j.celrep.2015.08.011
23. Monroe KM, Yang Z, Johnson JR, Geng X, Doitsh G, Krogan NJ, et al. IFI16 DNA sensor is required for death of lymphoid CD4 T cells abortively infected with HIV. Science. (2014) 343:428–32. doi: 10.1126/science.1243640
24. Muñoz-Arias I, Doitsh G, Yang Z, Sowinski S, Ruelas D, Greene WC. Blood-derived CD4 T cells naturally resist pyroptosis during abortive HIV-1 infection. Cell Host Microbe. (2015) 18:463–70. doi: 10.1016/j.chom.2015.09.010
25. Feinstein MJ, Hsue PY, Benjamin LA, Bloomfield GS, Currier JS, Freiberg MS, et al. Characteristics, prevention, and management of cardiovascular disease in people living with HIV: a scientific statement from the american heart association. Circulation. (2019) 140:e98–124. doi: 10.1161/CIR.0000000000000695
26. Hsue PY, Waters DD. HIV infection and coronary heart disease: mechanisms and management. Nat Rev Cardiol. (2019) 16:745–59. doi: 10.1038/s41569-019-0219-9
27. Currier JS, Hsue P. The role of inflammation in HIV-associated atherosclerosis - one size may not fit all. J Infect Dis. (2019) 221:495–97. doi: 10.1093/infdis/jiz256
28. Vachiat A, McCutcheon K, Tsabedze N, Zachariah D, Manga P. HIV and ischemic heart disease. J Am Coll Cardiol. (2017) 69:73–82. doi: 10.1016/j.jacc.2016.09.979
29. Hasse B, Tarr PE, Marques-Vidal P, Waeber G, Preisig M, Mooser V, et al. Strong impact of smoking on multimorbidity and cardiovascular risk among human immunodeficiency virus-infected individuals in comparison with the general population. Open Forum Infect Dis. (2015) 2:ofv108. doi: 10.1093/ofid/ofv108
30. Sinha A, Feinstein MJ. Coronary artery disease manifestations in HIV: what, how, and why. Can J Cardiol. (2019) 35:270–9. doi: 10.1016/j.cjca.2018.11.029
31. Hsue PY, Waters DD. Time to recognize HIV infection as a major cardiovascular risk factor. Circulation. (2018) 138:1113–5. doi: 10.1161/CIRCULATIONAHA.118.036211
32. Triant VA. Cardiovascular disease and HIV infection. Curr HIV/AIDS Rep. (2013) 10:199–206. doi: 10.1007/s11904-013-0168-6
33. Smith C, Sabin CA, Lundgren JD, Thiebaut R, Weber R, Law M, et al. Factors associated with specific causes of death amongst HIV-positive individuals in the D:A:D Study. AIDS. (2010) 24:1537–48.
34. Mocroft A, Reiss P, Gasiorowski J, Ledergerber B, Kowalska J, Chiesi A, et al. Serious fatal and nonfatal non-AIDS-defining illnesses in Europe. J Acquir Immune Defic Syndr. (2010) 55:262–70. doi: 10.1097/QAI.0b013e3181e9be6b
35. Neuhaus J, Jacobs DR Jr, Baker JV, Calmy A, Duprez D, La Rosa A, et al. Markers of inflammation, coagulation, and renal function are elevated in adults with HIV infection. J Infect Dis. (2010) 201:1788–95. doi: 10.1086/652749
36. Hart BB, Nordell AD, Okulicz JF, Palfreeman A, Horban A, Kedem E, et al. Inflammation-related morbidity and mortality among HIV-positive adults: how extensive is it? J Acquir Immune Defic Syndr. (2018) 77:1–7. doi: 10.1097/QAI.0000000000001554
37. Duprez DA, Neuhaus J, Kuller LH, Tracy R, Belloso W, De Wit S, et al. Inflammation, coagulation and cardiovascular disease in HIV-infected individuals. PLoS ONE. (2012) 7:e44454. doi: 10.1371/journal.pone.0044454
38. Nordell AD, McKenna M, Borges Á, Duprez D, Neuhaus J, Neaton JD, et al. Severity of cardiovascular disease outcomes among patients with HIV is related to markers of inflammation and coagulation. J Am Heart Assoc. (2014) 3:e000844. doi: 10.1161/JAHA.114.000844
39. Hsue PY, Hunt PW, Sinclair E, Bredt B, Franklin A, Killian M, et al. Increased carotid intima-media thickness in HIV patients is associated with increased cytomegalovirus-specific T-cell responses. AIDS. (2006) 20:2275–83. doi: 10.1097/QAD.0b013e3280108704
40. Cerrato E, Calcagno A, D'Ascenzo F, Biondi-Zoccai G, Mancone M, Grosso Marra W, et al. Cardiovascular disease in HIV patients: from bench to bedside and backwards. Open Heart. (2015) 2:e000174. doi: 10.1136/openhrt-2014-000174
41. Islam FM, Wu J, Jansson J, Wilson DP. Relative risk of cardiovascular disease among people living with HIV: a systematic review and meta-analysis. HIV Med. (2012) 13:453–68. doi: 10.1111/j.1468-1293.2012.00996.x
42. Ryom L, Lundgren JD, El-Sadr W, Reiss P, Kirk O, Law M, et al. Cardiovascular disease and use of contemporary protease inhibitors: the D:A:D international prospective multicohort study. Lancet HIV. (2018) 5:e291–300. doi: 10.1016/S2352-3018(18)30043-2
43. Di Biagio A, Del Bono V, Rosso R, Viscoli C. HIV and accelerated atheroprogression: role of antiretroviral therapy. Curr Pharm Biotechnol. (2012) 13:88–96. doi: 10.2174/138920112798868520
44. Seminari E, Pan A, Voltini G, Carnevale G, Maserati R, Minoli L, et al. Assessment of atherosclerosis using carotid ultrasonography in a cohort of HIV-positive patients treated with protease inhibitors. Atherosclerosis. (2002) 162:433–8. doi: 10.1016/S0021-9150(01)00736-5
45. Chironi G, Escaut L, Gariepy J, Cogny A, Teicher E, Monsuez JJ, et al. Brief report: carotid intima-media thickness in heavily pretreated HIV-infected patients. J Acquir Immune Defic Syndr. (2003) 32:490–3. doi: 10.1097/00126334-200304150-00004
46. Kaplan-Lewis E, Aberg JA, Lee M. Aging with HIV in the ART era. Semin Diagn Pathol. (2017) 34:384–97. doi: 10.1053/j.semdp.2017.04.002
47. Crane HM, Paramsothy P, Drozd DR, Nance RM, Delaney JA, Heckbert SR, et al. Types of myocardial infarction among human immunodeficiency virus-infected individuals in the United States. JAMA Cardiol. (2017) 2:260–7. doi: 10.1001/jamacardio.2016.5139
49. Freiberg MS, Chang CC, Kuller LH, Skanderson M, Lowy E, Kraemer KL, et al. HIV infection and the risk of acute myocardial infarction. JAMA Intern Med. (2013) 173:614–22. doi: 10.1001/jamainternmed.2013.3728
50. El-Sadr WM, Lundgren J, Neaton JD, Gordin F, Abrams D, Arduino RC, et al. CD4+ count-guided interruption of antiretroviral treatment. N Engl J Med. (2006) 355:2283–96. doi: 10.1056/NEJMoa062360
51. Bild DE, Bluemke DA, Burke GL, Detrano R, Diez Roux AV, Folsom AR, et al. Multi-Ethnic Study of Atherosclerosis: objectives and design. Am J Epidemiol. (2002) 156:871–81. doi: 10.1093/aje/kwf113
52. Tan HY, Yong YK, Shankar EM, Paukovics G, Ellegard R, Larsson M, et al. Aberrant inflammasome activation characterizes tuberculosis-associated immune reconstitution inflammatory syndrome. J Immunol. (2016) 196:4052–63. doi: 10.4049/jimmunol.1502203
53. Tenorio AR, Zheng Y, Bosch RJ, Krishnan S, Rodriguez B, Hunt PW, et al. Soluble markers of inflammation and coagulation but not T-cell activation predict non-AIDS-defining morbid events during suppressive antiretroviral treatment. J Infect Dis. (2014) 210:1248–59. doi: 10.1093/infdis/jiu254
54. Cutter GR, Burke GL, Dyer AR, Friedman GD, Hilner JE, Hughes GH, et al. Cardiovascular risk factors in young adults. the CARDIA baseline monograph. Control Clin Trials. (1991) 12:1S−77S. doi: 10.1016/0197-2456(91)90002-4
55. Leng SX, Margolick JB. Understanding frailty, aging, and inflammation in HIV infection. Curr HIV AIDS Rep. (2015) 12:25–32. doi: 10.1007/s11904-014-0247-3
56. Dinoso JB, Kim SY, Wiegand AM, Palmer SE, Gange SJ, Cranmer L, et al. Treatment intensification does not reduce residual HIV-1 viremia in patients on highly active antiretroviral therapy. Proc Natl Acad Sci USA. (2009) 106:9403–8. doi: 10.1073/pnas.0903107106
57. Hatano H, Hayes TL, Dahl V, Sinclair E, Lee TH, Hoh R, et al. A randomized, controlled trial of raltegravir intensification in antiretroviral-treated, HIV-infected patients with a suboptimal CD4+ T cell response. J Infect Dis. (2011) 203:960–8. doi: 10.1093/infdis/jiq138
58. Koay WLA, Siems LV, Persaud D. The microbiome and HIV persistence: implications for viral remission and cure. Curr Opin HIV AIDS. (2018) 13:61–8. doi: 10.1097/COH.0000000000000434
59. Tawakol A, Ishai A, Li D, Takx RA, Hur S, Kaiser Y, et al. Association of arterial and lymph node inflammation with distinct inflammatory pathways in human immunodeficiency virus infection. JAMA Cardiol. (2017) 2:163–71. doi: 10.1001/jamacardio.2016.4728
60. Promer K, Karris MY. Current treatment options for HIV elite controllers: a review. Curr Treat Options Infect Dis. (2018) 10:302–9. doi: 10.1007/s40506-018-0158-8
61. Maniar A, Ellis C, Asmuth D, Pollard R, Rutledge J. HIV infection and atherosclerosis: evaluating the drivers of inflammation. Eur J Prev Cardiol. (2013) 20:720–8. doi: 10.1177/2047487312447843
62. Shrestha S, Irvin MR, Grunfeld C, Arnett DK. HIV, inflammation, and calcium in atherosclerosis. Arterioscler Thromb Vasc Biol. (2014) 34:244–50. doi: 10.1161/ATVBAHA.113.302191
63. Hsu DC, Ma YF, Hur S, Li D, Rupert A, Scherzer R, et al. Plasma IL-6 levels are independently associated with atherosclerosis and mortality in HIV-infected individuals on suppressive antiretroviral therapy. AIDS. (2016) 30:2065–74. doi: 10.1097/QAD.0000000000001149
64. Fichtenbaum CJ. Inflammatory markers associated with coronary heart disease in persons with HIV infection. Curr Infect Dis Rep. (2011) 13:94–101. doi: 10.1007/s11908-010-0153-9
65. Titanji B, Gavegnano C, Hsue P, Schinazi R, Marconi VC. Targeting inflammation to reduce atherosclerotic cardiovascular risk in people with HIV infection. J Am Heart Assoc. (2020) 9:e014873. doi: 10.1161/JAHA.119.014873
66. Eckard AR, Meissner EG, Singh I, McComsey GA. Cardiovascular disease, statins, and HIV. J Infect Dis. (2016) 214(Suppl. 2):S83–92. doi: 10.1093/infdis/jiw288
67. Ridker PM, Danielson E, Fonseca FA, Genest J, Gotto AM Jr., et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med. (2008) 359:2195–207. doi: 10.1056/NEJMoa0807646
68. Funderburg NT, Jiang Y, Debanne SM, Storer N, Labbato D, Clagett B, et al. Rosuvastatin treatment reduces markers of monocyte activation in HIV-infected subjects on antiretroviral therapy. Clin Infect Dis. (2014) 58:588–95. doi: 10.1093/cid/cit748
69. O'Brien MP, Hunt PW, Kitch DW, Klingman K, Stein JH, Funderburg NT, et al. A randomized placebo controlled trial of aspirin effects on immune activation in chronically human immunodeficiency virus-infected adults on virologically suppressive antiretroviral therapy. Open Forum Infect Dis. (2017) 4:ofw278. doi: 10.1093/ofid/ofw278
70. Nasi M, De Biasi S, Bianchini E, Digaetano M, Pinti M, Gibellini L, et al. Analysis of inflammasomes and antiviral sensing components reveals decreased expression of NLRX1 in HIV-positive patients assuming efficient antiretroviral therapy. AIDS. (2015) 29:1937–41. doi: 10.1097/QAD.0000000000000830
71. Chattergoon MA, Latanich R, Quinn J, Winter ME, Buckheit RW 3rd, Blankson JN, et al. HIV and HCV activate the inflammasome in monocytes and macrophages via endosomal Toll-like receptors without induction of type 1 interferon. PLoS Pathog. (2014) 10:e1004082. doi: 10.1371/journal.ppat.1004082
72. Chivero ET, Guo ML, Periyasamy P, Liao K, Callen SE, Buch S. HIV-1 tat primes and activates microglial NLRP3 inflammasome-mediated neuroinflammation. J Neurosci. (2017) 37:3599–609. doi: 10.1523/JNEUROSCI.3045-16.2017
73. Swartz TH, Dubyak GR, Chen BK. Purinergic receptors: key mediators of HIV-1 infection and inflammation. Front Immunol. (2015) 6:585. doi: 10.3389/fimmu.2015.00585
74. Bandera A, Masetti M, Fabbiani M, Biasin M, Muscatello A, Squillace N, et al. The NLRP3 inflammasome is upregulated in HIV-infected antiretroviral therapy-treated individuals with Defective Immune Recovery. Front Immunol. (2018) 9:214. doi: 10.3389/fimmu.2018.00214
75. Leal VNC, Reis EC, Pontillo A. Inflammasome in HIV infection: lights and shadows. Mol Immunol. (2020) 118:9–18. doi: 10.1016/j.molimm.2019.12.001
76. Guo H, Callaway JB, Ting JP. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med. (2015) 21:677–87. doi: 10.1038/nm.3893
77. Hernandez JC, Latz E, Urcuqui-Inchima S. HIV-1 induces the first signal to activate the NLRP3 inflammasome in monocyte-derived macrophages. Intervirology. (2014) 57:36–42. doi: 10.1159/000353902
78. Guo H, Gao J, Taxman DJ, Ting JP, Su L. HIV-1 infection induces interleukin-1beta production via TLR8 protein-dependent and NLRP3 inflammasome mechanisms in human monocytes. J Biol Chem. (2014) 289:21716–26. doi: 10.1074/jbc.M114.566620
79. Lu W, Demers AJ, Ma F, Kang G, Yuan Z, Wan Y, et al. Next-generation mRNA sequencing reveals pyroptosis-induced CD4+ T cell death in early simian immunodeficiency virus-infected lymphoid tissues. J Virol. (2016) 90:1080–7. doi: 10.1128/JVI.02297-15
80. Cai R, Liu L, Luo B, Wang J, Shen J, Shen Y, et al. Caspase-1 activity in CD4 T cells is downregulated following antiretroviral therapy for HIV-1 infection. AIDS Res Hum Retroviruses. (2017) 33:164–71. doi: 10.1089/aid.2016.0234
81. Feria MG, Taborda NA, Hernandez JC, Rugeles MT. HIV replication is associated to inflammasomes activation, IL-1β, IL-18 and caspase-1 expression in GALT and peripheral blood. PLoS ONE. (2018) 13:e0192845. doi: 10.1371/journal.pone.0192845
82. Ahmad F, Mishra N, Ahrenstorf G, Franklin BS, Latz E, Schmidt RE, et al. Evidence of inflammasome activation and formation of monocyte-derived ASC specks in HIV-1 positive patients. AIDS. (2018) 32:299–307. doi: 10.1097/QAD.0000000000001693
83. Song J, Jiao Y, Zhang T, Zhang Y, Huang X, Li H, et al. Longitudinal changes in plasma caspase-1 and caspase-3 during the first 2 years of HIV-1 infection in CD4Low and CD4High patient groups. PLoS ONE. (2015) 10:e0121011. doi: 10.1371/journal.pone.0121011
84. Atluri VS, Pilakka-Kanthikeel S, Garcia G, Jayant RD, Sagar V, Samikkannu T, et al. Effect of cocaine on HIV infection and inflammasome gene expression profile in HIV infected macrophages. Sci Rep. (2016) 6:27864. doi: 10.1038/srep27864
85. Xu E, Liu J, Wang X, Xiong H. Inflammasome in drug abuse. Int J Physiol Pathophysiol Pharmacol. (2017) 9:165–77.
86. Ke P, Shao BZ, Xu ZQ, Wei W, Han BZ, Chen XW, et al. Activation of cannabinoid receptor 2 ameliorates DSS-induced colitis through inhibiting NLRP3 inflammasome in macrophages. PLoS ONE. (2016) 11:e0155076. doi: 10.1371/journal.pone.0155076
87. Yu W, Jin G, Zhang J, Wei W. Selective activation of cannabinoid receptor 2 attenuates myocardial infarction via suppressing NLRP3 inflammasome. Inflammation. (2019) 42:904–14. doi: 10.1007/s10753-018-0945-x
88. Qamar A, Rader DJ. Effect of interleukin 1β inhibition in cardiovascular disease. Curr Opin Lipidol. (2012) 23:548–53. doi: 10.1097/MOL.0b013e328359b0a6
89. Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. (2010) 464:1357–61. doi: 10.1038/nature08938
90. Menu P, Pellegrin M, Aubert JF, Bouzourene K, Tardivel A, Mazzolai L, et al. Atherosclerosis in ApoE-deficient mice progresses independently of the NLRP3 inflammasome. Cell Death Dis. (2011) 2:e137. doi: 10.1038/cddis.2011.18
91. Skeldon AM, Faraj M, Saleh M. Caspases and inflammasomes in metabolic inflammation. Immunol Cell Biol. (2014) 92:304–13. doi: 10.1038/icb.2014.5
92. Eidelman RS, Hebert PR, Weisman SM, Hennekens CH. An update on aspirin in the primary prevention of cardiovascular disease. Arch Intern Med. (2003) 163:2006–10. doi: 10.1001/archinte.163.17.2006
93. Macatangay BJC, Jackson EK, Abebe KZ, Comer D, Cyktor J, Klamar-Blain C, et al. A randomized, placebo-controlled, pilot clinical trial of dipyridamole to decrease Hiv-associated chronic inflammation. J Infect Dis. (2019) 221:1598–606. doi: 10.1093/infdis/jiz344
94. Baker JV, Wolfson J, Peterson T, Mooberry M, Gissel M, Mystakelis H, et al. Factor Xa inhibition reduces coagulation activity but not inflammation among people with HIV: a randomized clinical trial. Open Forum Infect Dis. (2020) 7:ofaa026. doi: 10.1093/ofid/ofaa026
95. Kadoglou NP, Moustardas P, Katsimpoulas M, Kapelouzou A, Kostomitsopoulos N, Schafer K, et al. The beneficial effects of a direct thrombin inhibitor, dabigatran etexilate, on the development and stability of atherosclerotic lesions in apolipoprotein E-deficient mice : dabigatran etexilate and atherosclerosis. Cardiovasc Drugs Ther. (2012) 26:367–74. doi: 10.1007/s10557-012-6411-3
96. Lee IO, Kratz MT, Schirmer SH, Baumhäkel M, Böhm M. The effects of direct thrombin inhibition with dabigatran on plaque formation and endothelial function in apolipoprotein E-deficient mice. J Pharmacol Exp Ther. (2012) 343:253–7. doi: 10.1124/jpet.112.194837
97. Hsue PY, Ribaudo HJ, Deeks SG, Bell T, Ridker PM, Fichtenbaum C, et al. Safety and impact of low-dose methotrexate on endothelial function and inflammation in individuals with treated human immunodeficiency virus: AIDS clinical trials group study A5314. Clin Infect Dis. (2019) 68:1877–86. doi: 10.1093/cid/ciy781
98. Stein JH, Yeh E, Weber JM, Korcarz C, Ridker PM, Tawakol A, et al. Brachial artery echogenicity and grayscale texture changes in HIV-infected individuals receiving low-dose methotrexate. Arterioscler Thromb Vasc Biol. (2018) 38:2870–8. doi: 10.1161/ATVBAHA.118.311807
99. Funderburg NT, Jiang Y, Debanne SM, Labbato D, Juchnowski S, Ferrari B, et al. Rosuvastatin reduces vascular inflammation and T-cell and monocyte activation in HIV-infected subjects on antiretroviral therapy. J Acquir Immune Defic Syndr. (2015) 68:396–404. doi: 10.1097/QAI.0000000000000478
100. Lo J, Lu MT, Ihenachor EJ, Wei J, Looby SE, Fitch KV, et al. Effects of statin therapy on coronary artery plaque volume and high-risk plaque morphology in HIV-infected patients with subclinical atherosclerosis: a randomised, double-blind, placebo-controlled trial. Lancet HIV. (2015) 2:e52–63. doi: 10.1016/S2352-3018(14)00032-0
101. Samsoondar JP, Burke AC, Sutherland BG, Telford DE, Sawyez CG, Edwards JY, et al. Prevention of diet-induced metabolic dysregulation, inflammation, and atherosclerosis in. Arterioscler Thromb Vasc Biol. (2017) 37:647–56. doi: 10.1161/ATVBAHA.116.308963
102. Honigberg MC, Natarajan P. Bempedoic acid for lowering LDL cholesterol. JAMA. (2019) 322:1769–71. doi: 10.1001/jama.2019.16598
103. Goldberg AC, Leiter LA, Stroes ESG, Baum SJ, Hanselman JC, Bloedon LT, et al. Effect of bempedoic acid vs placebo added to maximally tolerated statins on low-density lipoprotein cholesterol in patients at high risk for cardiovascular disease: the CLEAR wisdom randomized clinical trial. JAMA. (2019) 322:1780–8. doi: 10.1001/jama.2019.16585
104. Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. (2017) 377:1119–31. doi: 10.1056/NEJMoa1707914
105. Hsue PY, Li D, Ma Y, Ishai A, Manion M, Nahrendorf M, et al. IL-1β inhibition reduces atherosclerotic inflammation in HIV infection. J Am Coll Cardiol. (2018) 72:2809–11. doi: 10.1016/j.jacc.2018.09.038
106. Nagao A, Nakazawa S, Hanabusa H. Short-term efficacy of the IL6 receptor antibody tocilizumab in patients with HIV-associated multicentric Castleman disease: report of two cases. J Hematol Oncol. (2014) 7:10. doi: 10.1186/1756-8722-7-10
107. Gavegnano C, Haile WB, Hurwitz S, Tao S, Jiang Y, Schinazi RF, et al. Baricitinib reverses HIV-associated neurocognitive disorders in a SCID mouse model and reservoir seeding in vitro. J Neuroinflammation. (2019) 16:182. doi: 10.1186/s12974-019-1565-6
108. Ikonomidis I, Tzortzis S, Lekakis J, Paraskevaidis I, Andreadou I, Nikolaou M, et al. Lowering interleukin-1 activity with anakinra improves myocardial deformation in rheumatoid arthritis. Heart. (2009) 95:1502–7. doi: 10.1136/hrt.2009.168971
109. Pasquereau S, Kumar A, Herbein G. Targeting TNF and TNF receptor pathway in HIV-1 infection: from immune activation to viral reservoirs. Viruses. (2017) 9:64. doi: 10.3390/v9040064
110. Hunt PW, Martin JN, Sinclair E, Epling L, Teague J, Jacobson MA, et al. Valganciclovir reduces T cell activation in HIV-infected individuals with incomplete CD4+ T cell recovery on antiretroviral therapy. J Infect Dis. (2011) 203:1474–83. doi: 10.1093/infdis/jir060
111. d'Ettorre G, Rossi G, Scagnolari C, Andreotti M, Giustini N, Serafino S, et al. Probiotic supplementation promotes a reduction in T-cell activation, an increase in Th17 frequencies, and a recovery of intestinal epithelium integrity and mitochondrial morphology in ART-treated HIV-1-positive patients. Immun Inflamm Dis. (2017) 5:244–60. doi: 10.1002/iid3.160
112. d'Ettorre G, Ceccarelli G, Giustini N, Serafino S, Calantone N, De Girolamo G, et al. Probiotics reduce inflammation in antiretroviral treated, HIV-infected individuals: results of the “Probio-HIV” clinical trial. PLoS ONE. (2015) 10:e0137200. doi: 10.1371/journal.pone.0137200
113. Tenorio AR, Chan ES, Bosch RJ, Macatangay BJ, Read SW, Yesmin S, et al. Rifaximin has a marginal impact on microbial translocation, T-cell activation and inflammation in HIV-positive immune non-responders to antiretroviral therapy - ACTG A5286. J Infect Dis. (2015) 211:780–90. doi: 10.1093/infdis/jiu515
114. Gage J, Hasu M, Thabet M, Whitman SC. Caspase-1 deficiency decreases atherosclerosis in apolipoprotein E-null mice. Can J Cardiol. (2012) 28:222–9. doi: 10.1016/j.cjca.2011.10.013
115. Usui F, Shirasuna K, Kimura H, Tatsumi K, Kawashima A, Karasawa T, et al. Critical role of caspase-1 in vascular inflammation and development of atherosclerosis in Western diet-fed apolipoprotein E-deficient mice. Biochem Biophys Res Commun. (2012) 425:162–8. doi: 10.1016/j.bbrc.2012.07.058
116. Hendrikx T, Jeurissen ML, van Gorp PJ, Gijbels MJ, Walenbergh SM, Houben T, et al. Bone marrow-specific caspase-1/11 deficiency inhibits atherosclerosis development in Ldlr(-/-) mice. FEBS J. (2015) 282:2327–38. doi: 10.1111/febs.13279
117. Yajima N, Takahashi M, Morimoto H, Shiba Y, Takahashi Y, Masumoto J, et al. Critical role of bone marrow apoptosis-associated speck-like protein, an inflammasome adaptor molecule, in neointimal formation after vascular injury in mice. Circulation. (2008) 117:3079–87. doi: 10.1161/CIRCULATIONAHA.107.746453
118. Liu D, Zeng X, Li X, Mehta JL, Wang X. Role of NLRP3 inflammasome in the pathogenesis of cardiovascular diseases. Basic Res Cardiol. (2017) 113:5. doi: 10.1007/s00395-017-0663-9
119. Abderrazak A, Couchie D, Mahmood DF, Elhage R, Vindis C, Laffargue M, et al. Anti-inflammatory and antiatherogenic effects of the NLRP3 inflammasome inhibitor arglabin in ApoE2.Ki mice fed a high-fat diet. Circulation. (2015) 131:1061–70. doi: 10.1161/CIRCULATIONAHA.114.013730
120. Sun X, Jiao X, Ma Y, Liu Y, Zhang L, He Y, et al. Trimethylamine N-oxide induces inflammation and endothelial dysfunction in human umbilical vein endothelial cells via activating ROS-TXNIP-NLRP3 inflammasome. Biochem Biophys Res Commun. (2016) 481:63–70. doi: 10.1016/j.bbrc.2016.11.017
121. Ding Z, Liu S, Wang X, Dai Y, Khaidakov M, Deng X, et al. LOX-1, mtDNA damage, and NLRP3 inflammasome activation in macrophages: implications in atherogenesis. Cardiovasc Res. (2014) 103:619–28. doi: 10.1093/cvr/cvu114
122. Freigang S, Ampenberger F, Spohn G, Heer S, Shamshiev AT, Kisielow J, et al. Nrf2 is essential for cholesterol crystal-induced inflammasome activation and exacerbation of atherosclerosis. Eur J Immunol. (2011) 41:2040–51. doi: 10.1002/eji.201041316
123. Pavillard LE, Marin-Aguilar F, Bullon P, Cordero MD. Cardiovascular diseases, NLRP3 inflammasome, and western dietary patterns. Pharmacol Res. (2018) 131:44–50. doi: 10.1016/j.phrs.2018.03.018
124. Yin Y, Zhou Z, Liu W, Chang Q, Sun G, Dai Y. Vascular endothelial cells senescence is associated with NOD-like receptor family pyrin domain-containing 3 (NLRP3) inflammasome activation via reactive oxygen species (ROS)/thioredoxin-interacting protein (TXNIP) pathway. Int J Biochem Cell Biol. (2017) 84:22–34. doi: 10.1016/j.biocel.2017.01.001
125. Li X, Deroide N, Mallat Z. The role of the inflammasome in cardiovascular diseases. J Mol Med. (2014) 92:307–19. doi: 10.1007/s00109-014-1144-3
126. Krishnan SM, Sobey CG, Latz E, Mansell A, Drummond GR. IL-1beta and IL-18: inflammatory markers or mediators of hypertension? Br J Pharmacol. (2014) 171:5589–602. doi: 10.1111/bph.12876
127. Kim Y, Wang W, Okla M, Kang I, Moreau R, Chung S. Suppression of NLRP3 inflammasome by gamma-tocotrienol ameliorates type 2 diabetes. J Lipid Res. (2016) 57:66–76. doi: 10.1194/jlr.M062828
128. Luo B, Li B, Wang W, Liu X, Liu X, Xia Y, et al. Rosuvastatin alleviates diabetic cardiomyopathy by inhibiting NLRP3 inflammasome and MAPK pathways in a type 2 diabetes rat model. Cardiovasc Drugs Ther. (2014) 28:33–43. doi: 10.1007/s10557-013-6498-1
129. Qi J, Yu XJ, Shi XL, Gao HL, Yi QY, Tan H, et al. NF-kappaB blockade in hypothalamic paraventricular nucleus inhibits high-salt-induced hypertension through NLRP3 and caspase-1. Cardiovasc Toxicol. (2016) 16:345–54. doi: 10.1007/s12012-015-9344-9
130. Gan W, Ren J, Li T, Lv S, Li C, Liu Z, et al. The SGK1 inhibitor EMD638683, prevents angiotensin II-induced cardiac inflammation and fibrosis by blocking NLRP3 inflammasome activation. Biochim Biophys Acta Mol Basis Dis. (2018) 1864:1–10. doi: 10.1016/j.bbadis.2017.10.001
131. Sandanger O, Ranheim T, Vinge LE, Bliksoen M, Alfsnes K, Finsen AV, et al. The NLRP3 inflammasome is up-regulated in cardiac fibroblasts and mediates myocardial ischaemia-reperfusion injury. Cardiovasc Res. (2013) 99:164–74. doi: 10.1093/cvr/cvt091
132. Alcocer-Gomez E, Castejon-Vega B, Cordero MD. Stress-induced NLRP3 inflammasome in human diseases. Adv Protein Chem Struct Biol. (2017) 108:127–62. doi: 10.1016/bs.apcsb.2017.02.002
133. Sandanger O, Gao E, Ranheim T, Bliksoen M, Kaasboll OJ, Alfsnes K, et al. NLRP3 inflammasome activation during myocardial ischemia reperfusion is cardioprotective. Biochem Biophys Res Commun. (2016) 469:1012–20. doi: 10.1016/j.bbrc.2015.12.051
134. Abbate A, Van Tassell BW, Biondi-Zoccai G, Kontos MC, Grizzard JD, Spillman DW, et al. Effects of interleukin-1 blockade with anakinra on adverse cardiac remodeling and heart failure after acute myocardial infarction [from the Virginia Commonwealth University-Anakinra Remodeling Trial (2) (VCU-ART2) pilot study]. Am J Cardiol. (2013) 111:1394–400. doi: 10.1016/j.amjcard.2013.01.287
135. Abbate A, Van Tassell BW, Seropian IM, Toldo S, Robati R, Varma A, et al. Interleukin-1beta modulation using a genetically engineered antibody prevents adverse cardiac remodelling following acute myocardial infarction in the mouse. Eur J Heart Fail. (2010) 12:319–22. doi: 10.1093/eurjhf/hfq017
136. Lv ZH, Phuong TA, Jin SJ, Li XX, Xu M. Protection by simvastatin on hyperglycemia-induced endothelial dysfunction through inhibiting NLRP3 inflammasomes. Oncotarget. (2017) 8:91291–305. doi: 10.18632/oncotarget.20443
137. van der Heijden T, Kritikou E, Venema W, van Duijn J, van Santbrink PJ, Slutter B, et al. NLRP3 inflammasome inhibition by MCC950 reduces atherosclerotic lesion development in apolipoprotein E-deficient mice-brief report. Arterioscler Thromb Vasc Biol. (2017) 37:1457–61. doi: 10.1161/ATVBAHA.117.309575
138. Everett BM, Cornel JH, Lainscak M, Anker SD, Abbate A, Thuren T, et al. Anti-inflammatory therapy with canakinumab for the prevention of hospitalization for heart failure. Circulation. (2019) 139:1289–99. doi: 10.1161/CIRCULATIONAHA.118.038010
139. Ridker PM. Anticytokine agents: targeting interleukin signaling pathways for the treatment of atherothrombosis. Circ Res. (2019) 124:437–50. doi: 10.1161/CIRCRESAHA.118.313129
140. Aday AW, Ridker PM. Antiinflammatory therapy in clinical care: the CANTOS trial and beyond. Front Cardiovasc Med. (2018) 5:62. doi: 10.3389/fcvm.2018.00062
141. Rothman AM, MacFadyen J, Thuren T, Webb A, Harrison DG, Guzik TJ, et al. Effects of interleukin-1β inhibition on blood pressure, incident hypertension, and residual inflammatory risk: a secondary analysis of CANTOS. Hypertension. (2020) 75:477–82. doi: 10.1161/HYPERTENSIONAHA.119.13642
142. Khambhati J, Engels M, Allard-Ratick M, Sandesara PB, Quyyumi AA, Sperling L. Immunotherapy for the prevention of atherosclerotic cardiovascular disease: promise and possibilities. Atherosclerosis. (2018) 276:1–9. doi: 10.1016/j.atherosclerosis.2018.07.007
143. Gomez D, Baylis RA, Durgin BG, Newman AAC, Alencar GF, Mahan S, et al. Interleukin-1β has atheroprotective effects in advanced atherosclerotic lesions of mice. Nat Med. (2018) 24:1418–29. doi: 10.1038/s41591-018-0124-5
144. Ridker PM. Mortality differences associated with treatment responses in CANTOS and FOURIER: insights and implications. Circulation. (2018) 137:1763–6. doi: 10.1161/CIRCULATIONAHA.117.033254
145. Grebe A, Hoss F, Latz E. NLRP3 inflammasome and the IL-1 pathway in atherosclerosis. Circ Res. (2018) 122:1722–40. doi: 10.1161/CIRCRESAHA.118.311362
146. Ridker PM, MacFadyen JG, Thuren T, Libby P. Residual inflammatory risk associated with interleukin-18 and interleukin-6 after successful interleukin-1β inhibition with canakinumab: further rationale for the development of targeted anti-cytokine therapies for the treatment of atherothrombosis. Eur Heart J. (2019). doi: 10.1093/eurheartj/ehz542. [Epub ahead of print].
147. Aday AW, Ridker PM. Targeting residual inflammatory risk: a shifting paradigm for atherosclerotic disease. Front Cardiovasc Med. (2019) 6:16. doi: 10.3389/fcvm.2019.00016
148. Kottoor SJ, Arora RR. The utility of anti-inflammatory agents in cardiovascular disease: a novel perspective on the treatment of atherosclerosis. J Cardiovasc Pharmacol Ther. (2018) 23:483–93. doi: 10.1177/1074248418778548
149. Li Y, Niu X, Xu H, Li Q, Meng L, He M, et al. VX-765 attenuates atherosclerosis in ApoE deficient mice by modulating VSMCs pyroptosis. Exp Cell Res. (2020) 389:111847. doi: 10.1016/j.yexcr.2020.111847
150. Kearns A, Gordon J, Burdo TH, Qin X. HIV-1-associated atherosclerosis: unraveling the missing link. J Am Coll Cardiol. (2017) 69:3084–98. doi: 10.1016/j.jacc.2017.05.012
151. Kearns AC, Liu F, Dai S, Robinson JA, Kiernan E, Tesfaye Cheru L, et al. Caspase-1 activation is related with HIV-associated atherosclerosis in an HIV transgenic mouse model and HIV patient cohort. Arterioscler Thromb Vasc Biol. (2019) 39:1762–75. doi: 10.1161/ATVBAHA.119.312603
152. Yearley JH, Xia D, Pearson CB, Carville A, Shannon RP, Mansfield KG. Interleukin-18 predicts atherosclerosis progression in SIV-infected and uninfected rhesus monkeys (Macaca mulatta) on a high-fat/high-cholesterol diet. Lab Invest. (2009) 89:657–67. doi: 10.1038/labinvest.2009.29
Keywords: inflammasome, HIV, NLRP3, atheroclerosis, inflammation
Citation: Mullis C and Swartz TH (2020) NLRP3 Inflammasome Signaling as a Link Between HIV-1 Infection and Atherosclerotic Cardiovascular Disease. Front. Cardiovasc. Med. 7:95. doi: 10.3389/fcvm.2020.00095
Received: 24 January 2020; Accepted: 06 May 2020;
Published: 11 June 2020.
Edited by:
Carlos Fernández-Hernando, Yale University, United StatesReviewed by:
Teresa Padro, Sant Pau Institute for Biomedical Research, SpainCopyright © 2020 Mullis and Swartz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Talia H. Swartz, dGFsaWEuc3dhcnR6QG1zc20uZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.