 George E. G. Kluck1
George E. G. Kluck1 Kristina K. Durham
Kristina K. Durham Jeong-Ah Yoo
Jeong-Ah Yoo Bernardo L. Trigatti
Bernardo L. Trigatti
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med. , 28 April 2020
Sec. Cardio-Oncology
Volume 7 - 2020 | https://doi.org/10.3389/fcvm.2020.00065
This article is part of the Research Topic Emerging Challenges of Cardiovascular and Metabolic Dysfunctions in Cardio-oncology: From Bench to Bedside View all 11 articles
Cardiovascular disease and cancer are the leading causes of death in developed societies. Despite their effectiveness, many cancer therapies exhibit deleterious cardiovascular side effects such as cardiotoxicity and heart failure. The cardiotoxic effects of anthracyclines such as doxorubicin are the most well-characterized of cardiotoxic anti-cancer therapies. While other anti-neoplastic drugs also induce cardiotoxicity, often leading to heart failure, they are beyond the scope of this review. This review first summarizes the mechanisms of doxorubicin-induced cardiotoxicity. It then reviews emerging preclinical evidence that high density lipoprotein and its precursor protein apolipoprotein A1, which are known for their protective effects against ischemic cardiovascular disease, may also protect against doxorubicin-induced cardiotoxicity both directly and indirectly, when used therapeutically.
Advances in cancer treatment over the past few decades have led to substantial increases in cancer survivorship (1, 2). As cancer-related survival has improved, an unexpected increase in premature cardiovascular events, including myocardial ischemia, myocardial infarction, congestive heart failure (HF), QT interval prolongation, hypertension, and stroke has occurred (3, 4). A major contributing factor to cardiovascular outcomes is cardiotoxicity related to antitumor drugs, which may become apparent acutely during treatment, or often, well after treatment has ended (4, 5). In general terms, cancer therapy related cardiotoxicity ultimately leads to pathological alterations in the cardiac muscle tissue (5). Many different classes of chemotherapeutic agents have cardiotoxic effects. Anthracyclines such as doxorubicin (DOX) are, perhaps, the most well-studied cardiotoxic chemotherapeutic agents. They have a number of direct cardiotoxic effects, including DNA damage, interfering with mitochondrial function, induction of reactive oxygen species (ROS), alterations in autophagy and induction in apoptosis. Mechanisms of anthracycline induced cardiotoxicity will be discussed in more detail below. Other cardiotoxic chemotherapeutic agents/treatments include fluoropyrimidines, such as 5-fluorouracil, biologicals, such as trastuzumab and radiation therapy, all of which impact cardiomyocyte survival by triggering apoptosis through differing, but overlapping pathways (summarized in Figure 1). For example, fluoropyrimidines, such as 5-fluorouracil (5-FU) and capecitabine, are used to treat different types of tumors, especially those that appear in the head, neck and breast (10, 11). In the case of these agents, cardiotoxicity appears to be due to both direct toxic effects of these drugs on cardiomyocytes, through the generation of reactive oxygen species (ROS) and reactive nitrogen species (RNS), leading to both oxidative and nitrosative stress, and activation of apoptosis and autophagy (12). Furthermore, these agents also appear to have indirect cardiotoxic effects through interaction with the coagulation system and autoimmune responses (8). Trastuzumab, an antibody targeting the Human epidermal growth factor 2 receptor (HER/ErbB2), and used as first choice therapy against breast cancer (13, 14), also inhibits this receptor in cardiomyocytes, affecting myocardial structure and survival pathways, triggering cardiomyocyte apoptosis and leading to asymptomatic decreased left ventricular ejection fraction, and eventually heart failure, particularly when it is used in combination with other agents, such as anthracyclines (15, 16). Radiation therapy also triggers cardiotoxicity through induction of DNA damage as well as ROS in cardiomyocytes, again affecting cell survival pathways and triggering apoptosis (17). Radiation therapy induced cardiovascular effects may manifest as pericarditis, coronary artery disease, myocardial infarction, valve heart disease, changes in rhythm, silent myocardial ischemia and damage to the conduction system (18). The risk of heart disease is mainly related to the total radiation dose and the volume of the heart receiving radiation (19).
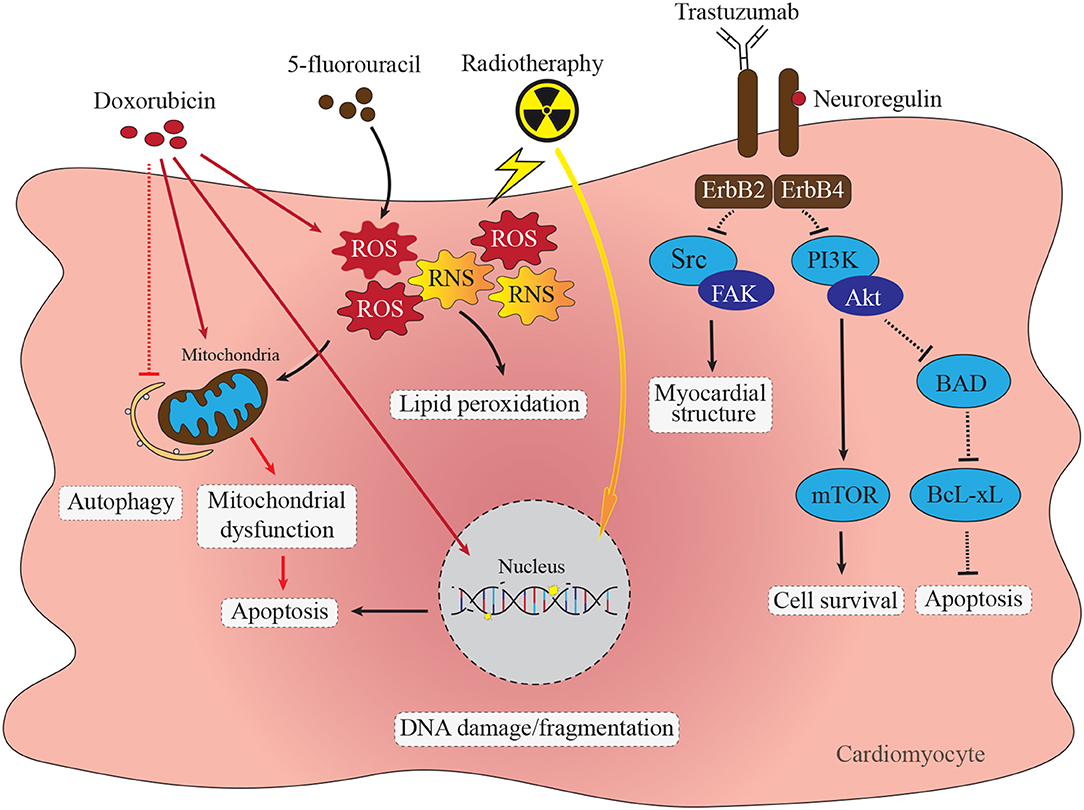
Figure 1. Direct cardiotoxic effects of anti-cancer therapies in cardiomyocytes. Doxorubicin cardiomyocyte cytotoxicity through diverse pathways including DNA damage, accumulation of ROS and RNS, mitochondrial dysfunction, apoptosis and impaired autophagy. Inhibition of ErbB2/4 receptor complex with trastuzumab impacts several signaling pathways resulting in impaired protein synthesis and myocardial structure via the Src/Fak pathway (6). Trastuzumab mediated supression of ErbB2/4 stimulated PI3K-Akt signaling impacts cell survival via mTOR pathway as well as increases apoptosis via BAD/BcL-xL pathway (7). Chemotherapy with fluoropyrimidines, such as 5-fluorouracil (5-FU) cause increase in formation of ROS and RNS leading to mitochondrial dysfunction and activation of caspase-3 and apoptosis (8). Radiotherapy leads to ROS and RNS formation, mitochondrial dysfunction and single-stranded DNA breaks leading to apoptosis activation (9).
As a result of these cardiotoxic effects, the mode of administration, time and dose of these treatments and the presence of pre-existing comorbidities such as cardiovascular and liver disease, diabetes mellitus and hypertension are important factors to be taken into consideration upon treatment (20). Furthermore, long term follow-up for cardiovascular complications is important, particularly in those patients with pre-existing cardiovascular risk factors (21–23). Understanding the mechanisms by which chemotherapeutic agents cause cardiotoxicity, as well as the identification of pathways that can be targeted in the cardiomyocyte to selectively mitigate chemotherapy induced cardiotoxicity is of fundamental importance in decreasing the undesirable impact on normal tissues and improving cancer treatment outcomes.
High density lipoproteins (HDL) have long been associated with cardioprotection, with a major focus being on arteriosclerosis-associated ischemic heart disease and stroke (24–28). This has largely been associated with its role in the transport of cholesterol from the artery wall to the liver for excretion or recycling, a process called reverse cholesterol transport (RCT) (29). However, recent advances in our understanding of HDL properties and biological functions have revealed HDL associated functions extending beyond cholesterol transport, including direct cytoprotective effects on a number of cell types including cardiomyocytes (30, 31). These advances provide key insights into the potential of exploiting these cytoprotective properties for therapeutic approaches to mitigate chemotherapy associated cardiotoxicity.
Here-in, we will first review the mechanisms by which anthracyclines, the most well-characterized of the cardiotoxic chemotherapeutic agents, induce cardiotoxicity, focusing on DOX as a well-studied anthracycline. While other anti-neoplastic drugs also induce cardiotoxicity, often leading to heart failure, they are beyond the scope of this review. We will then review recent advances in our understanding of HDL functions and properties that can be exploited to mitigate DOX-associated cardiotoxicity. These include understanding the mechanisms by which HDL induces cytoprotective responses in cells including cardiomyocytes, the ability of HDL and synthetic particles based on it to encapsulate DOX and serve as delivery vehicles, and findings that therapeutic treatment with HDL's major structural protein, apolipoprotein A1 (ApoA1) appears to have direct antineoplastic effects in preclinical tumor models (32–34).
Anthracyclines are a class of chemotherapeutics commonly prescribed to both adult and pediatric populations (35) and can be used alone or in combination with other cancer treatments. Since its discovery in the late 1960's, DOX (also called adriamycin, the prototypical anthracycline) has become widely prescribed due to its efficacy in treating cancers of both hematologic and solid origin (36, 37). Despite their widespread use, anthracyclines such as DOX are not specific in their cell target and exhibit cytotoxic effects in cardiomyocytes thereby limiting their long-term use due to dose-dependent cardiotoxicity (38). Immediate cardiac side effects of DOX infusion are detectable in the form of arrhythmias (39), and cardiotoxic outcomes can be measured following termination of treatment in both early (weeks to months) and late phases (years). These outcomes range from asymptomatic left ventricular dysfunction, to problematic arrhythmias and severe symptomatic congestive heart failure (37, 38). In a retrospective analysis of three trials of DOX therapy for breast or small cell lung cancers, the estimated cumulative percentage of patients with congestive heart failure was 5% in patients receiving a cumulative DOX dose of 400 mg/m2, which increased to 26% at a dose of 550 mg/m2, and 48% at 700 mg/m2 (40). The incidence of cardiotoxicity is highest within the first year following the termination of chemotherapy in adults, although in cases of childhood cancer, onset of cardiotoxicity has been observed to be delayed in survivors by 4–20 years (40–43).
A number of pathways have been implicated in DOX-mediated cardiomyocyte cytotoxicity. These include the direct and indirect induction of oxidative stress, DNA damage, and mitochondrial dysfunction. These pathways, along with alterations in homeostatic processes such as autophagy directly and indirectly lead to induction of cell death pathways, including apoptosis and necrosis (44–46) (Figure 2). These pathways have been the subject of recent comprehensive reviews (51–55) and will be summarized in the following sections.
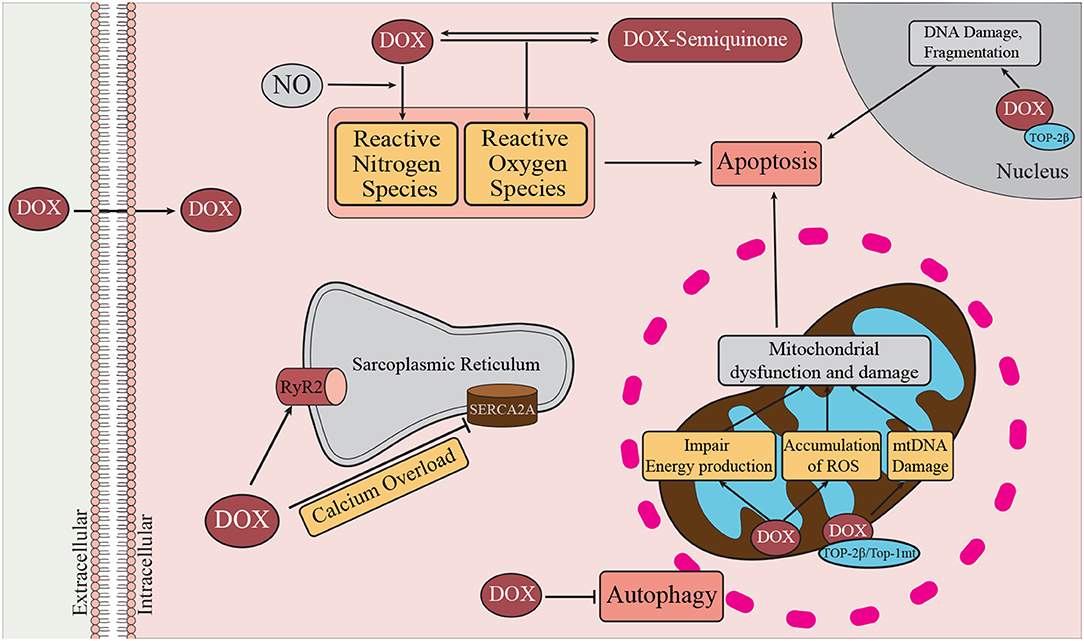
Figure 2. Mechanisms of DOX-induced cardiotoxicity. DOX enters the cell by passive diffusion, and within the cytoplasm undergoes redox cycling resulting in the generation of ROS and RNS (47). Oxidative and nitrosative stress are known to contribute to activation of cell death pathways, such as autophagy, necrosis, and apoptosis. DOX can promote Ca2+ overload by inhibiting SERCA2a and transiently enhancing the activity of RyR2. DOX can bind cardiolipin, a major inner mitochondrial membrane lipid. At the mitochondria DOX promotes damage to mitochondrial DNA (mtDNA) through interactions with Top-2β, and Top-1mt. In the nucleus, DOX interacts with Top-2β and DNA to form the Top-2β–DOX—DNA cleavage complex (48). By binding to Top-2β, DOX disrupts the rejoining of DNA leading to the accumulation of double stranded DNA breaks thereby triggering apoptosis (49). Damage to mitochondrial DNA affects mitochondrial biogenesis and contributes to mitochondrial dysfunction and therefore reduced ATP production (50).
Oxidative stress is the accumulation of oxygen and nitrogen free radicals resulting when their production exceeds the capacity of anti-oxidant enzymes to detoxify them (56). Oxidative stress has been proposed to be a major contributor to cardiomyocyte death and dysfunction following DOX treatment (56) (Figure 2). DOX can directly induce oxidative stress. The quinone moiety of DOX can act as an electron acceptor which can be reduced by a variety of enzymes to a semi-quinone, with the generation of oxygen free radicals. These can react with proteins, lipids and DNA, resulting in protein dysfunction, lipid peroxidation and DNA damage (50). DOX can also trigger reductions in the activities and expression of antioxidant enzymes, including matrix manganese superoxide dismutase (MnSOD) and glutathione (GSH) peroxidase (57), thereby reducing the antioxidant capacity of the cardiomyocyte. DOX can also interact with nitric oxide (NO) to generate reactive nitrogen species, contributing to nitrosative stress (56). DOX can also contribute to free radical formation and oxidative stress in cardiomyocytes through complex formation with iron (54). DOX treatment also increases accumulation of mitochondrial iron, once again manifesting in increased reactive oxygen and nitrogen species production (50). Dexrazoxane (DRZ) is an iron chelator which has seen limited clinical use to mitigate DOX-associated cardiotoxicity. On the other hand, pre-clinical studies of anti-oxidants have shown limited effectiveness at reducing DOX-associated cardiotoxicity, leading to doubt regarding the role of DOX-induced ROS formation in DOX associated cardiotoxicity (53).
DOX also appears to act within the nucleus to trigger cytotoxic effects in cardiomyocytes (Figure 2). DOX can reportedly bind to the cytoplasmic proteasome, which assists in the translocation of DOX to the nucleus by an ATP-dependent nuclear pore-mediated mechanism (58). Topoisomerase (Top)-2β was identified as a molecular mediator of DOX cardiotoxicity (48). Top-2β is located in both the nucleus and mitochondria, and is an important regulator of DNA topology by catalyzing the breaking and rejoining of DNA in order to allow for strands to pass by one another (59). DOX binding to Top-2β and DNA forms the Top-2β–DOX—DNA cleavage complex (48). By binding to Top-2β, DOX reportedly disrupts the rejoining of DNA leading to the accumulation of double stranded DNA breaks thereby triggering apoptosis (49). The importance of this pathway is highlighted by the finding that cardiomyocyte specific deletion of the gene encoding Top-2β protected cultured cardiomyocytes from DOX-induced cytotoxicity and protected mice from DOX-induced cardiotoxicity and heart failure (48). DOX has also been implicated in affecting mitochondrial DNA integrity via similar mechanism of ternary DOX—DNA—topoisomerase complex formation involving Top-2β residing in mitochondria as well as involving a mitochondrial specific topoisomerase, Top-1mt (60, 61).
Cardiomyocytes are highly dependent on mitochondrial function for energy production and proper contractile function (62–64). As outlined above, DOX can lead to the accumulation of iron within mitochondria and to the iron dependent and independent accumulation of reactive oxygen and nitrogen species which, themselves, can impair mitochondrial energy production (50, 65, 66). Furthermore, as mentioned above, through ternary complex formation between DOX, DNA and mitochondrial topoisomerases (Top-2β and Top-1mt), DOX can damage mitochondrial DNA by inducing double strand DNA breaks (48, 59). This can impair mitochondrial biogenesis as well as further impact mitochondrial function leading to insufficient energy production for the needs of the cardiomyocyte (60, 61).
Autophagy is a homeostatic mechanism whereby damaged or dysfunctional organelles are recycled to generate substrates for energy production or anabolic processes. Autophagy contributes to normal physiology of the heart and under conditions of acute cardiac stress can promote cardiac survival by releasing energy substrates and breaking down damaged organelles (67). Stimulation of autophagy results in the recruitment of autophagy-related proteins (ATGs) to a specific subcellular site and nucleation of an isolation membrane forming a structure called a phagophore and then an autophagosome, surrounding the damaged target organelle (68). Under conditions of cellular stress, such as nutrient deprivation, the serine/threonine kinase master cell growth regulator, mTOR, is inhibited, resulting in autophagy (69, 70). ATGs that were phosphorylated by mTOR under normal conditions now become dephosphorylated and recruited for autophagosome formation (71). In addition, under conditions of prolonged stress increased autophagic activity can lead to atrophy (decreased size) of cardiac muscle and activation of cell death pathways (67). Tumor-bearing mice display signs of dysregulated cardiac autophagy (increased expression of autophagic markers) and atrophy (32–34), and several research groups have highlighted the importance of autophagy in cardiotoxicity resulting from treatment of mice with DOX (46, 51, 72).
Autophagy is orchestrated by a complex set of regulatory proteins which identify the target for autophagic disposal, form the limiting membrane and orchestrate fusion with lysosomes [reviewed in Li et al. and Koleini and Kardami (51, 52)]. Recent research has demonstrated DOX dysregulates autophagy in cardiomyocytes and that this appears to play an important role in DOX mediated cardiotoxicity. This has been the subject of a number of recent comprehensive reviews (51, 52). DOX appears to exert a dose-dependent disruption of the normal regulation of autophagy: low, clinically relevant doses, replicating chemotherapy, appear to suppress normal levels of basal autophagy, whereas high doses appear to induce autophagy above normal basal levels (51, 52). The impaired autophagy in cardiomyocytes, resulting from chronic low-dose treatment of mice with DOX, has been reported to involve DOX-mediated interference with lysosome mediated degradation of autophagosome contents (73). On the other hand, DOX, particularly at higher doses, appears to lead to an accumulation of PTEN-induced kinase (PINK) 1 and parkin (an E3 ubiquitin ligase) within mitochondria (74). PINK1 and parkin play a key role in regulating the balance between mitophagy and mitochondrial biogenesis; DOX appears to trigger a depolarization of the mitochondrial membrane potential resulting in an accumulation of PINK1 and parkin on mitochondria. PINK1 is a serine/threonine kinase which phosphorylates ubiquitin and allows parkin to ubiquitinate a number of mitochondrial outer membrane proteins, targeting the mitochondria for mitophagy (52, 74). This, possibly together with DOX-mediated interference with lysosome mediated degradation, results in an accumulation of autophagosomes containing mitochondria (52, 73, 74). An emerging consensus appears to be that DOX interferes with autophagic flux, as a result of simultaneous induction of early stages of mitophagy (triggering mitochondrial dysfunction/membrane depolarization, the marking of mitochondria for mitophagy and formation of autophagosomes surrounding dysfunctional mitochondria) and interference with later stages of autophagy (51, 52). As mentioned above, this may be through DOX-mediated interference with lysosome acidification, thereby preventing the lysosomal degradation of contents of autophagosomes (73). Alternatively, others have reported that DOX mediated mitochondrial damage leads to maladaptive activation of phosphatidylinositol 3-kinase (PI3K) γ signaling, which blocked autophagy, and that this was alleviated by cardiac specific PI3K γ inhibition in mice (75). The consequence of these effects appears to be a build-up of damaged/dysfunctional mitochondria and reactive oxygen species, exacerbating DOX-induced oxidative stress, and leading to activation of apoptotic pathways, triggering cell death (51).
Apoptosis is a form of programmed cell death involving the induction of a caspase proteolytic cascade ultimately leading to nuclear condensation and fragmentation, phosphatidylserine exposure on the cell surface, formation of apoptotic bodies and their clearance by phagocytes by virtue of recognition of exposed phosphatidylserine. Apoptosis is characterized by the activation of caspase proteolytic cascades leading to the activation of effector caspases, such as caspases 3, 6, and 7 (76, 77). These activate nucleases and proteases which degrade DNA, nuclear, cytoplasmic, and cytoskeletal proteins (76, 77). These effector caspases are activated by two main pathways of apoptosis regulation, the extrinsic and intrinsic pathways. In the extrinsic pathway, ligation of “death receptors” such as the tumor necrosis factor (TNF) α receptor by their extrinsic ligands on the cell surface results in receptor dimerization, bringing together “death domains” leading to the formation of a death inducing signaling complex (DISC) that ultimately triggers the activation of caspase 8, which, in turn, goes on to activate the effector caspases 3, 6, and 7 (77).
On the other hand, mitochondria play central roles in the intrinsic apoptosis pathway, which is regulated by a series of soluble proteins that regulate pore formation in the mitochondrial membrane (76, 77). These factors belong to the Bcl-2 family of proteins and comprise 3 groups: the anti-apoptotic Bcl-2 family members, such as Bcl-2 itself, the pro-apoptotic effectors such as Bax and Bak, and the pro-apoptotic regulators such as Bim. Together these comprise the intrinsic apoptosis pathway. In this pathway, the anti-apoptotic family members bind to and sequester the pro-apoptotic effectors, preventing them from assembling as complexes on the mitochondrial membrane. The pro-apoptotic regulators, in turn, bind to the anti-apoptotic regulators, preventing them from sequestering the pro-apoptotic effectors and, additionally, assist the pro-apoptotic effectors in assembly on the mitochondria, where the pro-apoptotic effectors can form pores in the mitochondrial membrane. This leads to the leakage of mitochondrial cytochrome C and formation of a protein complex, called the apoptosome which serves as a platform for the activation of caspase 9, which can then activate effector caspases 3, 6, and 7 (76). In this intrinsic pathway, the relative amounts of pro-apoptotic effectors, regulators and anti-apoptotic factors determine whether apoptosis proceeds (76). Nuclear DNA damage and induction of oxidative stress and damage to mitochondria are all potent activators of the intrinsic apoptosis pathway (76, 77). In addition, DOX treatment of cardiomyocytes has been reported to result in increased transcription of the pro-apoptotic regulator Bim, thus enhancing the intrinsic apoptotic pathway (78). Additionally, recent work has identified increased expression of death receptors (TNF receptor 1, Fas, death receptor 4, death receptor 5) in human induced pluripotent stem cell derived cardiomyocytes following DOX treatment (44).
In addition to causing the death of cardiomyocytes, DOX also triggers cardiomyocyte atrophy, or a reduction in cardiomyocyte size. Ultrastructural changes in the myocardium, including myofibril structural disarray and atrophy due to DOX-cardiotoxicity appear well before clinical manifestations (79). The ubiquitin proteasome system regulates cardiomyocyte size by tagging proteins with polyubiquitin chains for subsequent degradation by the proteasome (80). The tagging of target proteins for proteolysis by the ubiquitin-proteasome system involves the assembly of polyubiquitin chains on target proteins mediated by E1 ubiquitin activating enzymes, E2 ubiquitin conjugating enzymes and E3 ubiquitin ligases. Atrogin-1 is a muscle specific E3 ubiquitin ligase that facilitates atrophic signaling in cardiomyocytes (80). It and other E3 ubiquitin ligases appear to be upregulated in cardiomyocytes by treatment with DOX and/or other anthracyclines (81, 82). This leads to the degradation of contractile proteins, and reduction in cardiomyocyte size (81). As cardiomyocyte size is related to overall contractile force generation, reduced cardiomyocyte size (atrophy) compounds reduced cardiomyocyte numbers in diseased states and eventually manifests as progressive reduction in cardiac function, as in the case of DOX-induced cardiotoxicity (83).
At present, monitoring and screening for cardiotoxicity following anthracycline therapy is imperative for timely treatment. In a recent prospective study of anthracycline-treated patients, of the 9% of patients who developed heart failure, 98% of cases occurred within a year following anthracycline treatment (40). Countless prophylactic therapeutics are currently under study in animal models but few have been assessed clinically. Furthermore, only a small number cardioprotective therapeutics that have been tested in humans reduce the cardiotoxic effects of anthracyclines and currently no clear guidelines or worldwide accepted therapies exist. β-blockers, angiotensin converting enzyme (ACE) inhibitors, statins, and dexrazoxane (DRZ) are drugs that have been assessed in small trials for protection against anthracycline cardiotoxicity. Results are promising, but data is limited by small study sizes and variability in study methods such as follow up time. Early identification of reduced cardiac function and immediate treatment with heart failure medication such as enalapril (an ACE inhibitor) alone or in combination with β-blockers (carvedilol, or bisoprolol) provided either full or partial improvement of cardiac function in 82% of patients (40). A small group of non-Hodgkins lymphoma patients receiving ramipril and/or bisoprolol as a prophylactic during anthracycline treatment also exhibited reductions in new symptoms of cardiotoxicity and projected prolonged survival (84).
Statins are commonly prescribed to reduce morbidity and mortality associated with atherosclerosis. Given the cardioprotective nature of statins, and the fact that cancer patients receiving chemotherapy may also be concurrently treated with statins for atherosclerosis, the prophylactic effect of statins on anthracycline cardiotoxicity has been examined in a number of small trials. Breast cancer patients on continuous statin therapy had reduced risk of heart failure following anthracycline treatment compared to those with non-continuous statin therapy (85), and similarly, anthracycline induced decline in left ventricular ejection fraction (LVEF) was reduced by statin treatment as compared to no statin treatment (86). In a small study of 40 patients, those receiving atorvastatin prior to chemotherapy infusion showed no significant change in LVEF at 6 months post therapy compared to a reduction in LVEF in those receiving anthracycline alone (87).
While statins and ACE inhibitors have shown promising results in mitigating resultant anthracycline cardiotoxicity or preventing cardiotoxicity when individuals happen to be taking them concurrently with chemotherapy for treatment of comorbidities, DRZ is currently the only United States Food and Drug Administration, and Health Canada approved prophylactic drug for use in combination with DOX to specifically limit cardiotoxicity in adults (88, 89). DRZ acts as an iron chelator, interferes with ROS production, can bind to Top-2β to inhibit complex formation with DOX, and also reduce Top-2β expression (90–92). Early multi-center randomized double blind trials of breast cancer patients receiving combination chemotherapy which included a cumulative DOX dose of 300 mg/m2 reported a hazard ratio of cardiac events of 2.63 (placebo vs. DRZ); however, despite this promising effect of DRZ on heart function, patient survival was not improved (93). A systematic review also identified an association of DRZ with reduced risk of cardiovascular complications, but increased risk of secondary malignant neoplasms in children receiving chemotherapy (94). Given these results, Health Canada cautions against use of DRZ in children, as well as in elderly populations with reduced cardiac, hepatic, or renal function.
Identification of a treatment that protects against the cardiotoxic side effects without impacting the chemotherapeutic effects of DOX remains of utmost importance. Potential for development or further study of effective primary prevention therapies exists given the expansion of research uncovering the broad nature of mechanisms in the pathogenesis of anthracycline mediated heart failure.
HDL has long been associated epidemiologically with reduced risk for cardiovascular disease. The main mechanistic explanation has traditionally been its apparent protection against atherosclerotic narrowing of arteries, thereby combating ischemic cardiovascular disease. However, recent research in pre-clinical models have increasingly suggested that HDL may exert direct cardioprotective effects on the heart itself (24). Furthermore, HDL based nanospheres have been developed as delivery vehicles for a variety of drugs including chemotherapeutic agents. Finally, pre-clinical studies have shown that delivery of supra-physiological amounts of HDL's major apolipoprotein, ApoA1, may itself attenuate tumor growth. In the following sections, we provide an overview of HDL structure, composition, formation and function and then discuss recent findings demonstrating direct cardioprotective effects of HDL and/or its precursor, ApoA1 against DOX-induced cardiotoxicity, advances in the use of HDL based nanospheres for encapsulation of DOX for therapeutic delivery and direct anti-tumor effects of ApoA1.
Lipoproteins are diverse biological particles that provide a means of transport for lipids between cells, tissues, and other lipoproteins, and can activate intracellular signaling pathways (95). Lipoproteins are separated into five classes (chylomicron, VLDL, IDL, LDL, and HDL) based on criteria including density, size, and relative content of lipids (cholesterol and triglyceride), and apolipoproteins (95). HDL represents one of the five major classes of lipoproteins, distinguishable from others based on their small particle size (5–11 nm), high density (1.063–1.21 g/ml), and unique apolipoprotein content (96). Unlike other lipoproteins, HDLs are unique in their cytoprotective actions and initiate anti-oxidative, anti-apoptotic, and anti-inflammatory effects. HDL can inhibit the oxidation of LDL and enhance endothelial function by inhibiting the expression of endothelial adhesion molecules (97). In addition, HDL can suppress atherosclerosis progression and inflammation by modulating production of monocytes and neutrophils (98).
HDL represents a class of particles of distinct protein and lipid composition. The proteome of HDL is diverse and can contain close to 100 proteins, more than can fit on a single particle, underscoring the notion that HDL represents a class of particles of distinct compositions. The HDL proteome includes apolipoproteins, diverse enzymes, lipid transfer proteins, acute phase proteins, and proteinase inhibitors, and other proteins of distinct functions (99). ApoA1 (243 amino acids, 28 kD) is the most abundant protein in HDL, comprising 70% of the protein carried by HDL (100). ApoA1 has over 90% amphipathic α-helical content, allowing for formation and stabilization of the HDL (101). ApoA1 is linked to several beneficial effects of HDL. Therefore, several research groups have reported that HDL quality is highly dependent on the abundance and function of ApoA1 (102).
ApoA1 is secreted by the liver (70%) and small intestine (30%) in a lipid-poor state, and is assembled into HDL by the addition of phospholipids and unesterified cholesterol aided by the ATP-binding cassette transporter A1 (ABCA1) at the cell surface, forming nascent HDL (an immature form of HDL). Next, the nascent HDL particle becomes mature HDL by activating lecithin: cholesterol acyltransferase (LCAT), which converts unesterified cholesterol to cholesteryl esters. The esterification of cholesterol increases its hydrophobicity, resulting in its movement into the core of the HDL particle and the particle itself adopting a spherical shape (103, 104).
HDL particles are continuously remodeled and catabolized by plasma and membrane proteins, thereby giving rise to dynamic subfractions. Membrane receptors including ATP-binding cassette transporter G1 (ABCG1) and the scavenger receptor class B type 1 (SR-B1) promote movement of lipids between cells and HDL, and plasma proteins such as cholesteryl ester transfer protein (CETP) assist in movement of lipids between lipoproteins. SR-B1 is a high affinity receptor for HDL, is highly expressed in liver and steroidogenic tissues, and plays a critical role in tissue uptake of HDL cholesterol, a key step in reverse cholesterol transport, which is the transport of cholesterol from peripheral tissues to the liver for repackaging into nascent lipoproteins or excretion (105). ApoA1 appears to play an important role in this process as adenoviral mediated or transgenic overexpression of human ApoA1 in mice leads to enhanced reverse cholesterol transport (106).
The Framingham Heart Study shows strong relationships between levels of HDL and the incidence of developing heart disease (25). Statins, inhibitors of hydroxymethylglutaryl coenzyme A (HMG-CoA), a key enzyme in the cholesterol biosynthetic pathway, have long been major therapeutic tools in the reduction of cardiovascular events. This is due to their ability to reduce production and increase clearance of VLDL and LDL, thereby reducing blood cholesterol levels. Many statins have also been shown to modestly raise HDL cholesterol levels by between 3 and 15% (107–109). The mechanisms and clinical benefit of statin triggered increases in HDL cholesterol are unclear and still the subject of debate (107–111). In part this may be due to the difficulty in evaluating the contribution of the relatively modest statin-induced increases in HDL-cholesterol levels in the context of dramatic reductions in LDL cholesterol.
The epidemiological association of increased HDL cholesterol with reduced risk for cardiovascular disease has, over the years, led to efforts to increase HDL levels pharmacologically. The focus, however, has been in increasing HDL-cholesterol as opposed to increasing HDL particles or, more subtly, to increasing functional HDL particles. This focus on raising HDL cholesterol levels has led to the development of CETP inhibitors which block the CETP mediated transfer of cholesterol from HDL to triglyceride rich lipoproteins such as VLDL. These drugs increase HDL cholesterol levels by more than 25% (up to 60%), but largely have not reduced cardiovascular events (107, 109, 112–118). More recent attempts at increasing HDL particle number and/or function have focused on infusion of reconstituted HDL, administration of ApoA1 mimetics, or upregulation of ApoA1 production by liver (119–124). CSL112 is a new reconstituted HDL (rHDL) made with human ApoA1. Clinical trials of CSL112 showed that it enhances cholesterol efflux capacity an important measure of HDL mediated reverse cholesterol transport mediated cardiovascular protection (124, 125). Also, the AEGIS-I trial suggests that CSL112 has advantages over other rHDL formulations (e.g., CSL-111, CER-001) or ApoA-I Milano because it is well-tolerated with no side effects in major organs (such as liver or kidney toxicity) or immunogenicity (126–128). However, the potential benefit of CSL112 in reducing major adverse cardiovascular events in this group of high-risk patients still remains to be shown in the large phase III AEGIS-II study that is expected to be concluded in 2022 (124, 125).
The protective effects of HDL against cardiovascular disease have long been the subject of intensive research. While most of the focus has been on the ability of HDL to protect against atherosclerosis and vascular dysfunction, and the impacts of that on ischemic cardiovascular disease, more recent focus has increasingly been placed on the direct cardioprotective effects of HDL, through its interactions with cardiomyocytes themselves (24). Epidemiological studies, as well as pre-clinical studies in animal models, have demonstrated that HDL can protect against cardiac disease independent of effects on coronary artery atherosclerosis, suggesting that it may also exert direct effects on the heart itself (129–132). For example, HDL treatment has been shown to protect hearts (in vivo and ex vivo) and isolated cardiomyocytes, from ischemia/reperfusion injury and infarction (130–132). In the context of DOX-induced cardiotoxicity, recent studies using isolated cardiomyocytes in culture (133–136) and in preclinical animal models (135, 137) demonstrate that HDL is able to protect against cardiomyocyte apoptosis and myocardial atrophy. For example pre-treatment of cultured cardiomyocytes with HDL prior to subsequent treatment with DOX, protects them against DOX induced apoptosis (137, 138). Similarly, HDL pretreatment of cultured cardiomyocytes protects them against other stresses leading to cytotoxicity, including necrosis resulting from oxygen and glucose deprivation (134), suggesting that HDL may protect cardiomyocytes against diverse forms of cell death. HDL mediated protection against DOX-induced cardiomyocyte apoptosis has been reported in different studies to involve the activation of AKT (134, 137) or the activation of the signal transducer and activator of transcription (STAT) 3 (138). Pharmacological or genetic inhibition of these signaling mediators has been shown to impair HDL mediated protection of cardiomyocytes against DOX-induced apoptosis (137, 138). The importance of the AKT pathway in cardioprotection against DOX has also been demonstrated by the finding that expression of constitutively active AKT1 in the myocardium inhibits DOX induced cardiotoxicity by preventing left ventricular dysfunction and cardiac atrophy (139). Furthermore, cardiac restricted overexpression of STAT3 in mice led to protection against DOX-induced atrophy and congestive heart failure, whereas cardiac specific knockout of STAT3 in mice was accompanied by increased cardiac fibrosis and age-dependent heart failure (140, 141). AKT and STAT3 form the respective cornerstones of the RISK (reperfusion injury salvage kinase) and SAFE (survivor activating factor enhancement) signaling pathways known to play important roles in cardioprotection, for example in the setting of ischemia/reperfusion injury (142, 143). These data also suggest that AKT and STAT3 are critical mediators of cardioprotection against DOX-induced cardiotoxicity (Figure 3). HDL dependent activation of AKT and STAT3 and other signaling pathways has been reported in different cell types to involve HDL mediated delivery of the bioactive lipid, sphingosine-1-phosphate (S1P), acting via the S1P receptors (136, 144–148) (Figure 3). In the case of cardiomyocytes, HDL and S1P mediated activation of STAT3 appears to be mediated by the S1P receptor 2 (S1PR2) (136). On the other hand, the involvement of S1P/S1P receptors in HDL mediated activation of AKT signaling in cardiomyocytes has not been demonstrated (134, 135, 137). However, HDL mediated activation of AKT signaling in cardiomyocytes and protection of cardiomyocytes against DOX-induced apoptosis appears to require the HDL receptor, SR-B1. SR-B1 is expressed by both mouse and human cardiomyocytes in culture and mouse cardiac tissue (137, 149). The ability of HDL to induce AKT phosphorylation and protection against DOX induced cytotoxicity in cultured mouse or human cardiomyocytes was lost when the gene for SR-B1 was either knocked out or knocked down (135, 137). SR-B1 mediates lipid transport between bound HDL particles and cells via a hydrophobic channel, suggesting that SR-B1 mediated transport of HDL bound, water insoluble S1P molecules from HDL, into the cell membrane, where they can access S1P receptors may be a potential mechanism for the involvement of SR-B1, and HDL associated S1P and S1P receptors in cardioprotection. This, however, remains to be demonstrated experimentally. The potential role for HDL associated S1P in cardioprotection against DOX-induced cardiotoxicity highlights the importance of understanding the role of HDL composition in evaluating HDL function and designing HDL based therapeutics such as reconstituted HDL-like particles (150).
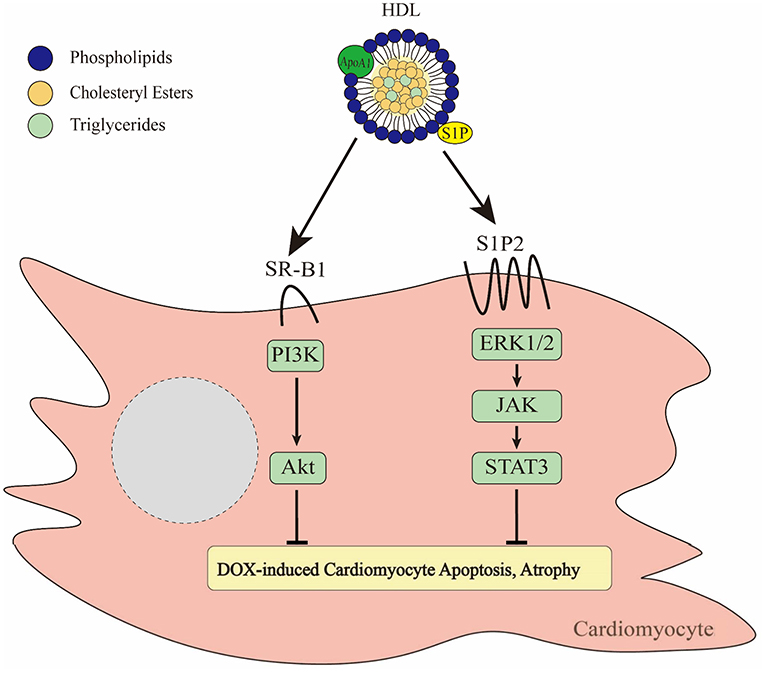
Figure 3. Effects of HDL on DOX-induced cytotoxicity of cardiomyocytes. HDL binds to SR-B1 leading to activation of the PI3K/Akt pathway (137). In addition, HDL is able to activate ERK1/2-JAK-STAT3 signaling via a pathway involving HDL mediated delivery of the bioactive lipid, S1P acting via the S1P2 receptor (136). Together these pathways prevent DOX-induced apoptosis and atrophy.
These in vitro studies of HDL mediated protection against DOX-induced cardiotoxicity have recently been extended to in vivo models (135, 137) by examining the effects of increased circulating HDL levels on DOX-induced cardiotoxicity in mice. We first tested the effects of genetic overexpression of human ApoA1, on cardiotoxicity induced by repeated weekly DOX dosing in mice. Overexpression of transgenic human ApoA1 in mice has been shown to trigger dramatically increased circulating HDL levels by seeding the formation of new mature HDL particles (151). In one study, transgenic overexpression of human ApoA1 in mice virtually completely prevented chronic low dose DOX treatment from triggering myocardial apoptosis and atrophy, and protected mice from DOX-treatment induced reduction in left ventricular function (137). A drawback of this study was that although it represented a proof of concept, transgenic overexpression of ApoA1 led to levels of ApoA1 and HDL that were extremely high and therefore not likely to be therapeutically relevant (137). A more recent study, however, demonstrated that intraperitoneal injection of purified ApoA1 similarly prevented cardiotoxicity associated with chronic low dose DOX treatment in mice (135). Mice that were treated with five weekly injections of DOX alone exhibited substantial apoptosis in cardiomyocytes in hearts, and substantially reduced left ventricular function, whereas control mice that did not receive DOX displayed little myocardial apoptosis and normal left ventricular function (135). On the other hand mice that were treated with injection of ApoA1 alongside DOX were virtually completely protected against DOX-induced myocardial apoptosis and left ventricular dysfunction (135). Regardless of means of HDL increase (ApoA1 transgenic expression or ApoA1 injection) cardioprotection was lost if mice lacked SR-B1 (135, 137). In fact, SR-B1 knockout mice were more susceptible to DOX induced cardiotoxicity than corresponding wild type mice. This effect of SR-B1 appeared to be associated with SR-B1 expression in cardiac tissue, consistent with observations that SR-B1 expression in cultured cardiomyocytes was required for HDL mediated protection against DOX-induced apoptosis (135, 137). These findings clearly demonstrate that in pre-clinical models, HDL-therapies such as injection of the HDL precursor ApoA1 have the potential to protect against DOX induced cardiotoxicity but are dependent on the expression of cardiomyocyte SR-B1 (Figure 3).
In addition to HDL's ability to protect cardiomyocytes against cytotoxicity induced by anti-cancer agents, reconstituted HDL (rHDL)-based nanoparticles have also been explored as drug delivery vehicles for chemotherapeutic agents such as DOX. The use of rHDL as a drug delivery system for DOX has been studied using both in vitro and in vivo methods. Yuan et al. showed that DOX encapsulated in HDL particles (rHDL-DOX) is more efficiently taken up by and more effective at inducing apoptosis in hepatocellular carcinoma cells, when compared to DOX alone or encapsulated in liposomes (45). Furthermore, in preclinical mouse tumor models, treatment with rHDL-DOX resulted in greater tumor regression than DOX alone (45). Wang et al. confirmed that incorporation of DOX into rHDL-based particles enhanced the cytotoxic effects of DOX on tumors in vivo and cancer cells in vitro (152). Furthermore, they demonstrated that the HDL receptor SR-B1 was required in tumor cells for rHDL mediated delivery of the encapsulated DOX (152). Interestingly, the authors measured DOX tissue distribution after treating mice with rHDL-DOX and showed that DOX uptake by the heart was low (152). Others have tested the effects of using rHDL to deliver paclitaxel (PTX) either alone or in combination with DOX. Co-delivery of PTX and DOX encapsulated in rHDL was shown to improve their anti-cancer effects over co-administration of non-encapsulated PTX and DOX (153). When used to treat preclinical models of liver cancer, the majority of PTX and DOX delivered via rHDL was found in the liver tumors (attributed to uptake via SR-B1) with little accumulation in the heart and very little cardiac damage (153). These findings suggest that, at least for liver cancer rHDL encapsulation can provide a means for targeted delivery of anti-cancer agents to tumor cells, sparing cardiac tissues. Whether the reduced cardiac damage was solely due to targeted delivery of the anti-cancer agents to the hepatic tumor over the heart or whether it also involved induction of survival signaling at the heart (PI3K/AKT and STAT3 signaling as described above) remains to be determined. It also remains to be determined whether rHDL-mediated chemotherapeutic delivery is effective against other types of cancer or against tumor cells which do not express high levels of SR-B1. Nevertheless, these studies suggest the potential for rHDL based drug delivery systems to confer tissue selective delivery to at least some types of tumors, sparing the heart from cardiotoxic damage. More research is required to determine the full potential of this.
In addition to research showing that HDL can protect cardiomyocytes from chemotherapy-induced cytotoxicity both directly by inducing survival signaling in the cardiomyocytes, and indirectly by acting as a targeted delivery system for anti-cancer agents, sparing the heart, other research has suggested that HDL and its precursor ApoA1 may also have direct anti-tumor effects themselves.
Results of epidemiological studies of endogenous HDL cholesterol levels and the incidence of cancer are mixed with some studies reporting an inverse correlation between HDL cholesterol and cancer risk and/or mortality, while other studies report minimal association, particularly when corrected for confounding factors (152, 154–159). Contributing to this is uncertainty over the cause-vs.-effect relationship between low HDL-cholesterol and cancer, with some studies suggesting that tumor cells may drive the lowering of HDL-cholesterol levels by utilizing HDL-cholesterol to support tumor growth (159). Complicating matters further are reports that HDL prepared from cancer patients or from patients with other co-morbidities, such as type 2 diabetes or obesity exhibit altered functions as compared to HDL from unaffected individuals, for example, promoting rather than inhibiting migration and invasion of tumor cells in in vitro assays (160–166). Therefore, it is presently unclear what, if any, effects levels of endogenous HDL or variations in those levels have on cancer development.
On the other hand, preclinical studies in mouse models have suggested that supra-physiological levels of ApoA1 may have therapeutic potential against tumor growth and metastasis. For example, Zamanian-Daryoush et al. reported that transgenic overexpression of human ApoA1 reduced, while complete knockout of endogenous ApoA1 increased tumor growth and metastasis in mice compared to control mice with normal levels of endogenous ApoA1 (167). They also demonstrated that pharmacological treatment with purified ApoA1 similarly attenuated both primary tumor development and metastasis in mouse models (167). They provided evidence that ApoA1 reduced tumor angiogenesis and recruited tumor cell targeting macrophages and CD8+ cytotoxic T cells, thereby altering the tumor microenvironment to one less permissive for tumor development (167, 168). By using different tumor cell lines, including a human melanoma cell line (A375), they demonstrated that supra-physiological levels of ApoA1 may have general anti-neoplastic effects including toward human tumors (167, 168). Others have reported that synthetic ApoA1 mimetic peptides, which replicate the amphipathic properties of ApoA1, also exhibit anti-tumor properties, when used at pharmacological concentrations. For example, the ApoA1 mimetic peptide, L-5F was reported to prevent angiogenesis, suggesting that it may have therapeutic potential against angiogenesis associated diseases such as cancer (169). In preclinical studies, the ApoA1 mimetic peptide 4F has been reported to suppress ovarian tumorigenesis (170). Similarly, preclinical studies demonstrated that the recombinant ApoA1 mimetic peptide 6F reduced tumor burden in mouse models of metastatic lung cancer (171). However, ApoA1 mimetic peptides may exert anti-tumor effects via mechanisms distinct from ApoA1. For example ApoA1 mimetic peptides are thought to strongly bind and neutralize lysophosphatidic acid (LPA), which is known to stimulate cell proliferation, oncogenesis, and metastasis (172). On the other hand, neither transgenic overexpression of human ApoA1 nor ApoA1 knockout affected LPA levels in tumor-bearing mice (167). Whether ApoA1 (injected or overexpressed) or ApoA1 mimetic peptides exert anti-tumor effects by driving the increased formation of HDL-like particles or whether their anti-neoplastic effects are independent of HDL particle formation has not been examined. Other pre-clinical studies have reported that in certain cases, HDL may drive the development of breast cancer, particularly in circumstances when breast tumor cells overexpress the HDL receptor, SR-B1, since this receptor can mediate both survival signaling and uptake of cholesterol fueling tumor growth (173–177). Therefore, direct anti-tumor effects of ApoA1 or ApoA1-mimetic peptides may be restricted to tumors that do not overexpress SR-B1; although SR-B1 overexpression in tumors could be exploited by strategies that encapsulate chemotherapeutic agents like DOX in HDL based nanoparticles (see previous section), which may be readily and preferentially taken up by tumor cells overexpressing SR-B1.
Preclinical studies suggest that HDL targeted therapies involving pharmacological treatment with supra-physiological levels of ApoA1, peptides based on ApoA1 (ApoA1 mimetic peptides) or rHDL like particles may show promise in the protection against chemotherapy related cardiotoxicity via a number of mechanisms (Figure 4). These include (1) direct HDL mediated survival signaling in cardiomyocytes leading to protection against cytotoxicity, as exemplified by studies using DOX as a cardiotoxic agent; (2) indirect protection afforded to the heart by utilizing rHDL-based nanoparticles as targeted delivery vehicles for chemotherapeutic agents which spare the heart and have the potential to target tumor cells which may overexpress SR-B1; and (3) indirect protection resulting from direct ApoA1 mediated tumor suppression (Figure 4). It remains to be demonstrated experimentally whether these mechanisms broadly impact diverse malignancies and chemotherapeutic agents or are specific for those that have been tested to date. Importantly, these mechanisms may not necessarily be mutually exclusive. For example, in the case of treatment with ApoA1 along with chemotherapeutic agents such as DOX, the ApoA1 may be acting by seeding the formation of new HDL particles which may incorporate the chemotherapeutic agent, act as targeted delivery systems for certain types of tumors, directly attenuate tumor growth, and directly induce survival signaling in cardiomyocytes, thus inducing both direct and indirect mechanisms of cardioprotection simultaneously. However, studies need to be designed to test whether these mechanisms do occur simultaneously, in the same preclinical models. Whether or not they do occur simultaneously, much more work remains to be done do determine the full potential for HDL targeted therapies as therapeutic approaches to prevent chemotherapy induced cardiotoxicity in human disease.
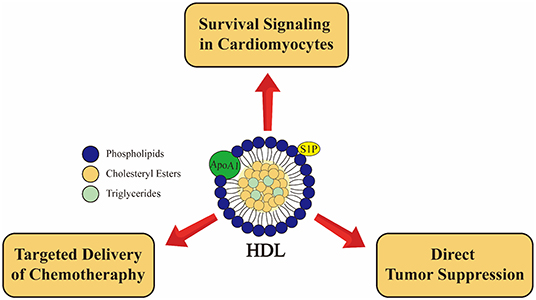
Figure 4. Emerging roles of high density lipoprotein (HDL) in cancer and cancer therapy associated cardiotoxicity from preclinical studies. HDL induces cardioprotective effects in cardiomyocytes via SR-B1 receptor and activation of PI3K-Akt pathway as well as via S1P2 receptor leading activation of STAT3-JAK-ERK1/2 pathway (136, 137). rHDL used as a drug delivery system may allow for targeted delivery to at least some types of tumors, sparing cardiomyocytes (45, 152). Furthermore, the major apoliprotein of HDL (ApoA1) directly attenuates tumor growth and metastasis in preclinical models (167). These pathways are not necessarily muatually exclusive.
All authors listed above made substantial, direct and intellectual contributions to the work, and have approved the final version.
Research on HDL in the Trigatti Lab was funded by grants from the Canadian Institutes of Health Research (MOP−74753 and PJT−162272).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. de Moor JS, Mariotto AB, Parry C, Alfano CM, Padgett L, Kent EE, et al. Cancer survivors in the United States: prevalence across the survivorship trajectory and implications for care. Cancer Epidemiol Biomarkers Prev. (2013) 22:561–70. doi: 10.1158/1055-9965.EPI-12-1356
2. DeSantis CE, Lin CC, Mariotto AB, Siegel RL, Stein KD, Kramer JL, et al. Cancer treatment and survivorship statistics, 2014. CA Cancer J Clin. (2014) 64:252–71. doi: 10.3322/caac.21235
3. Moslehi JJ. Cardiovascular toxic effects of targeted cancer therapies. N Engl J Med. (2016) 375:1457–67. doi: 10.1056/NEJMra1100265
4. Jain D, Russell RR, Schwartz RG, Panjrath GS, Aronow W. Cardiac complications of cancer therapy: pathophysiology, identification, prevention, treatment, and future directions. Curr Cardiol Rep. (2017) 19:36. doi: 10.1007/s11886-017-0846-x
5. Tocchetti CG, Cadeddu C, Di Lisi D, Femminò S, Madonna R, Mele D, et al. From molecular mechanisms to clinical management of antineoplastic drug-induced cardiovascular toxicity: a translational overview. Antioxidants Redox Signal. (2019) 30:2110–53. doi: 10.1089/ars.2016.6930
6. Kuramochi Y, Guo X, Sawyer DB. Neuregulin activates erbB2-dependent src/FAK signaling and cytoskeletal remodeling in isolated adult rat cardiac myocytes. J Mol Cell Cardiol. (2006) 41:228–35. doi: 10.1016/j.yjmcc.2006.04.007
7. Gassmann M, Casagranda F, Orloli D, Simon H, Lai C, Kleint R, et al. Aberrant neural and cardiac development in mice lacking the ErbB4 neuregulin receptor. Nature. (1995) 378:390–4. doi: 10.1038/378390a0
8. Sara JD, Kaur J, Khodadadi R, Rehman M, Lobo R, Chakrabarti S, et al. 5-fluorouracil and cardiotoxicity: a review. Ther Adv Med Oncol. (2018) 10:1758835918780140. doi: 10.1177/1758835918780140
9. Sylvester CB, Abe J, Patel ZS, Grande-Allen KJ. Radiation-induced cardiovascular disease: mechanisms and importance of linear energy transfer. Front Cardiovasc Med. (2018) 5:5. doi: 10.3389/fcvm.2018.00005
11. Labianca R, Beretta G, Clerici M, Fraschini P, Luporini G. Cardiac toxicity of 5-fluorouracil: a study on 1083 patients. Tumori. (1982) 68:505–10. doi: 10.1177/030089168206800609
12. Lamberti M, Porto S, Marra M, Zappavigna S, Grimaldi A, Feola D, et al. 5-Fluorouracil induces apoptosis in rat cardiocytes through intracellular oxidative stress. J Exp Clin Cancer Res. (2012) 31:60. doi: 10.1186/1756-9966-31-60
13. Wahler J, Suh N. Targeting HER2 positive breast cancer with chemopreventive agents. Curr Pharmacol Reports. (2015) 1:324–35. doi: 10.1007/s40495-015-0040-z
14. Wang J, Xu B. Targeted therapeutic options and future perspectives for HER2-positive breast cancer. Signal Transduct Target Ther. (2019) 4:34. doi: 10.1038/s41392-019-0069-2
15. Ahmed S, Talat A, Iqbal N, Emara M, Le D, Sami A, Xiang J. Cardiac toxicity of HER2-directed therapy in women with breast cancer: epidemiology, etiology, risk factors, and management. In: Pham PV, editor. Breast Cancer - From Biology to Medicine. London: IntechOpen (2017). doi: 10.5772/66437
16. Guo S, Wong S. Cardiovascular toxicities from systemic breast cancer therapy. Front Oncol. (2014) 4:346. doi: 10.3389/fonc.2014.00346
17. Soucy KG, Lim HK, Kim JH, Oh Y, Attarzadeh DO, Sevinc B, et al. HZE 56 Fe-Ion irradiation induces endothelial dysfunction in rat aorta: role of xanthine oxidase. Radiat Res. (2011) 176:474–85. doi: 10.1667/RR2598.1
18. Rygiel K. Cardiotoxic effects of radiotherapy and strategies to reduce them in patients with breast cancer: An overview. J Cancer Res Ther. (2017) 13:186–92. doi: 10.4103/0973-1482.187303
19. Lewis GD, Farach A. Cardiovascular toxicities of radiation therapy. Methodits Debakey Cardiovasc J. (2019) 15:274–81. doi: 10.14797/mdcj-15-4-274
20. Hershman DL, McBride RB, Eisenberger A, Tsai WY, Grann VR, Jacobson JS. Doxorubicin, cardiac risk factors, and cardiac toxicity in elderly patients with diffuse B-cell Non-Hodgkin's Lymphoma. J Clin Oncol. (2008) 26:3159–65. doi: 10.1200/JCO.2007.14.1242
21. Einarson TR, Acs A, Ludwig C, Panton UH. Prevalence of cardiovascular disease in type 2 diabetes: a systematic literature review of scientific evidence from across the world in 2007-2017. Cardiovasc Diabetol. (2018) 17:1–19. doi: 10.1186/s12933-018-0728-6
22. Lee SI, Patel M, Jones CM, Narendran P. Cardiovascular disease and type 1 diabetes: Prevalence, prediction and management in an ageing population. Ther Adv Chronic Dis. (2015) 6:347–74. doi: 10.1177/2040622315598502
23. Kalofoutis C, Piperi C, Kalofoutis A, Harris F, Phoenix D, Singh J. Type II diabetes mellitus and cardiovascular risk factors: current therapeutic approaches. Exp Clin Cardiol. (2007) 12:17−28.
24. Van Linthout S, Frias M, Singh N, De Geest B. Therapeutic potential of HDL in cardioprotection and tissue repair. In: Handbook of Experimental Pharmacology. Newyork, NY: Springer New York LLC. pp. 527–65.
25. Castelli WP. Cholesterol and lipids in the risk of coronary artery disease. The Framingham Heart Study. Can J Cardiol. (1988) 4:5A−10A.
26. He Y, Kothari V, Bornfeldt KE. High-density lipoprotein function in cardiovascular disease and diabetes mellitus. Arterioscler Thromb Vasc Biol. (2018) 38:e10–6. doi: 10.1161/ATVBAHA.117.310222
27. Rye KA, Barter PJ. Thematic review series: high density lipoprotein structure, function, and metabolism cardioprotective functions of HDLs 1. J Lipid Res. (2014) 55:168–79. doi: 10.1194/jlr.R039297
28. Annema W, von Eckardstein A, Kovanen PT. HDL and atherothrombotic vascular disease. In: von Eckardstein A, Kardassis D, editors. High Density Lipoproteins. Handbook of Experimental Pharmacology. Vol 224. Heidelberg: Springer (2015). p. 371–403. doi: 10.1007/978-3-319-09665-_11
29. Lewis GF, Rader DJ. New insights into the regulation of HDL metabolism and reverse cholesterol transport. Circ Res. (2005) 96:1221–32. doi: 10.1161/01.RES.0000170946.56981.5c
30. Mineo C, Shaul PW. Novel biological functions of high-density lipoprotein cholesterol. Circ Res. (2012) 111:1079–90. doi: 10.1161/CIRCRESAHA.111.258673
31. Kosmas CE, Martinez I, Sourlas A, Bouza KV, Campos FN, Torres V, et al. High-density lipoprotein (HDL) functionality and its relevance to atherosclerotic cardiovascular disease. Drugs Context. (2018) 7:212525. doi: 10.7573/dic.212525
32. Cosper PF, Leinwand LA. Cancer causes cardiac atrophy and autophagy in a sexually dimorphic manner. Cancer Res. (2011) 71:1710–20. doi: 10.1158/0008-5472.CAN-10-3145
33. Musolino V, Palus S, Tschirner A, Drescher C, Gliozzi M, Carresi C, et al. Megestrol acetate improves cardiac function in a model of cancer cachexia-induced cardiomyopathy by autophagic modulation. J Cachexia Sarcopenia Muscle. (2016) 7:555–66. doi: 10.1002/jcsm.12116
34. Manne NDPK, Lima M, Enos RT, Wehner P, Carson JA, Blough E. Altered cardiac muscle mTOR regulation during the progression of cancer cachexia in the ApcMin/+ mouse. Int J Oncol. (2013) 42:2134–40. doi: 10.3892/ijo.2013.1893
35. Volkova M, Russell R. Anthracycline cardiotoxicity: prevalence, pathogenesis and treatment. Curr Cardiol Rev. (2012) 7:214–20. doi: 10.2174/157340311799960645
36. Arcamone F, Cassinelli G, Fantini G, Grein A, Orezzi P, Pol C, et al. Adriamycin, 14-hydroxydaimomycin, a new antitumor antibiotic from S. Peucetius var. caesius. Biotechnol Bioeng. (1969) 11:1101–10. doi: 10.1002/bit.260110607
37. Shakir D. Chemotherapy induced cardiomyopathy: pathogenesis, monitoring and management. J Clin Med Res. (2009) 1:8–12. doi: 10.4021/jocmr2009.02.1225
38. Swain SM, Whaley FS, Ewer MS. Congestive heart failure in patients treated with doxorubicin: a retrospective analysis of three trials. Cancer. (2003) 97:2869–79. doi: 10.1002/cncr.11407
39. Nousiainen T, Vanninen E, Rantala A, Jantunen E, Hartikainen J. QT dispersion and late potentials during doxorubicin therapy for non-Hodgkin's lymphoma. J Intern Med. (1999) 245:359–64. doi: 10.1046/j.1365-2796.1999.00480.x
40. Cardinale D, Colombo A, Bacchiani G, Tedeschi I, Meroni CA, Veglia F, et al. Early detection of anthracycline cardiotoxicity and improvement with heart failure therapy. Circulation. (2015) 131:1981–8. doi: 10.1161/CIRCULATIONAHA.114.013777
41. Lipshultz SE, Lipsitz SR, Sallan SE, Dalton VM, Mone SM, Gelber RD, et al. Chronic progressive cardiac dysfunction years after doxorubicin therapy for childhood acute lymphoblastic leukemia. J Clin Oncol. (2005) 23:2629–36. doi: 10.1200/JCO.2005.12.121
42. Feijen EAM, Leisenring WM, Stratton KL, Ness KK, Van Der Pal HJH, Caron HN, et al. Equivalence ratio for daunorubicin to doxorubicin in relation to late heart failure in survivors of childhood cancer. J Clin Oncol. (2015) 33:3774–80. doi: 10.1200/JCO.2015.61.5187
43. Hilfiker-Kleiner D, Ardehali H, Fischmeister R, Burridge P, Hirsch E, Lyon AR. Late onset heart failure after childhood chemotherapy. Eur Heart J. (2019) 40:798–800. doi: 10.1093/eurheartj/ehz046
44. Zhao L, Zhang B. Doxorubicin induces cardiotoxicity through upregulation of death receptors mediated apoptosis in cardiomyocytes. Sci Rep. (2017) 7:44735. doi: 10.1038/srep44735
45. Yuan Y, Wang W, Wang B, Zhu H, Zhang B, Feng M. Delivery of hydrophilic drug doxorubicin hydrochloride-targeted liver using apoAI as carrier. J Drug Target. (2013) 21:367–74. doi: 10.3109/1061186X.2012.757769
46. Zhang YW, Shi J, Li YJ, Wei L. Cardiomyocyte death in doxorubicin-induced cardiotoxicity. Arch Immunol Ther Exp. (2009) 57:435–45. doi: 10.1007/s00005-009-0051-8
47. Doroshow JH. Effect of anthracycline antibiotics on oxygen radical formation in rat heart. Cancer Res. (1983) 43:460–72.
48. Zhang S, Liu X, Bawa-Khalfe T, Lu LS, Lyu YL, Liu LF, et al. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat Med. (2012) 18:1639–42. doi: 10.1038/nm.2919
49. Minotti G, Menna P, Salvatorelli E, Cairo G, Gianni L. Anthracyclines: molecular advances and pharmacologie developments in antitumor activity and cardiotoxicity. Pharmacol Rev. (2004) 56:185–29. doi: 10.1124/pr.56.2.6
50. Ichikawa Y, Ghanefar M, Bayeva M, Wu R, Khechaduri A, Naga Prasad SV, et al. Cardiotoxicity of doxorubicin is mediated through mitochondrial iron accumulation. J Clin Invest. (2014) 124:617–30. doi: 10.1172/JCI72931
51. Li M, Russo M, Pirozzi F, Tocchetti CG, Ghigo A. Autophagy and cancer therapy cardiotoxicity: from molecular mechanisms to therapeutic opportunities. Biochim Biophys Acta Mol Cell Res. (2020) 1867:118493. doi: 10.1016/j.bbamcr.2019.06.007
52. Koleini N, Kardami E. Autophagy and mitophagy in the context of doxorubicin-induced cardiotoxicity. Oncotarget. (2017) 8:46663–80. doi: 10.18632/oncotarget.16944
53. Kalyanaraman B. Teaching the basics of the mechanism of doxorubicin-induced cardiotoxicity: have we been barking up the wrong tree? Redox Biol. (2020) 29:101394. doi: 10.1016/j.redox.2019.101394
54. Damiani RM, Moura DJ, Viau CM, Caceres RA, Henriques JAP, Saffi J. Pathways of cardiac toxicity: comparison between chemotherapeutic drugs doxorubicin and mitoxantrone. Arch Toxicol. (2016) 90:2063–76. doi: 10.1007/s00204-016-1759-y
55. Hahn VS, Lenihan DJ, Ky B. Cancer therapy-induced cardiotoxicity: basic mechanisms and potential cardioprotective therapies. J Am Heart Assoc. (2014) 3:e000665. doi: 10.1161/JAHA.113.000665
56. Chen Y, Jungsuwadee P, Vore M, Butterfield DA St. Clair DK. Collateral damage in cancer chemotherapy: oxidative stress in nontargeted tissues. Mol Interv. (2007) 7:147–56. doi: 10.1124/mi.7.3.6
57. Li T, Singal PK. Adriamycin-induced early changes in myocardial antioxidant enzymes and their modulation by probucol. Circulation. (2000) 102:2105–10. doi: 10.1161/01.CIR.102.17.2105
58. Kiyomiya KI, Matsuo S, Kurebe M. Mechanism of specific nuclear transport of adriamycin: the mode of nuclear translocation of adriamycin-proteasome complex. Cancer Res. (2001) 61:2467–71.
59. Sobek S, Boege F. DNA topoisomerases in mtDNA maintenance and age. Exp Gerontol. (2014) 56:135–41. doi: 10.1016/j.exger.2014.01.009
60. Nitiss KC, Nitiss JL. Twisting and ironing: doxorubicin cardiotoxicity by mitochondrial DNA damage. Clin Cancer Res. (2014) 20:4737–9. doi: 10.1158/1078-0432.CCR-14-0821
61. Khiati S, Rosa ID, Sourbier C, Ma X, Rao VA, Neckers LM, et al. Mitochondrial topoisomerase i (Top1mt) is a novel limiting factor of doxorubicin cardiotoxicity. Clin Cancer Res. (2014) 20:4873–81. doi: 10.1158/1078-0432.CCR-13-3373
62. Piquereau J, Caffin F, Novotova M, Lemaire C, Veksler V, Garnier A, et al. Mitochondrial dynamics in the adult cardiomyocytes: which roles for a highly specialized cell? Front Physiol. (2013) 4:102. doi: 10.3389/fphys.2013.00102
63. Martín-Fernández B, Gredilla R. Mitochondria and oxidative stress in heart aging. Age. (2016) 38:225–38. doi: 10.1007/s11357-016-9933-y
64. Zhao Q, Sun Q, Zhou L, Liu K, Jiao K. Complex regulation of mitochondrial function during cardiac development. J Am Heart Assoc. (2019) 8:e012731. doi: 10.1161/JAHA.119.012731
65. Gorini S, Angelis A De, Berrino L, Malara N, Rosano G, Ferraro E. Chemotherapeutic drugs and mitochondrial dysfunction: focus on doxorubicin, trastuzumab, and sunitinib. Oxid Med Cell Longev. (2018) 2018:7582730. doi: 10.1155/2018/7582730
66. Guven C, Sevgiler Y, Taskin E. Mitochondrial dysfunction associated with doxorubicin. In: Taskin E, Guven C, Sevgiler Y, editors. Mitochondrial Diseases. London: IntechOpen (2018). doi: 10.5772/intechopen.80284
67. Lavandero S, Troncoso R, Rothermel BA, Martinet W, Sadoshima J, Hill JA. Cardiovascular autophagy. Autophagy. (2013) 9:1455–66. doi: 10.4161/auto.25969
68. Glick D, Barth S, Macleod KF. Autophagy: cellular and molecular mechanisms. J Pathol. (2010) 221:3–12. doi: 10.1002/path.2697
69. González A, Hall MN. Nutrient sensing and TOR signaling in yeast and mammals. EMBO J. (2017) 36:397–408. doi: 10.15252/embj.201696010
70. Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell. (2017) 168:960–76. doi: 10.1016/j.cell.2017.02.004
71. Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y, et al. Nutrient-dependent mTORCl association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell. (2009) 20:1981–91. doi: 10.1091/mbc.e08-12-1248
72. Lu L, Wu W, Yan J, Li X, Yu H, Yu X. Adriamycin-induced autophagic cardiomyocyte death plays a pathogenic role in a rat model of heart failure. Int J Cardiol. (2009) 134:82–90. doi: 10.1016/j.ijcard.2008.01.043
73. Li DL, Wang ZV, Ding G, Tan W, Luo X, Criollo A, et al. Doxorubicin blocks cardiomyocyte autophagic flux by inhibiting lysosome acidification. Circulation. (2016) 133:1668–87. doi: 10.1161/CIRCULATIONAHA.115.017443
74. Yin J, Guo J, Zhang Q, Cui L, Zhang L, Zhang T, et al. Doxorubicin-induced mitophagy and mitochondrial damage is associated with dysregulation of the PINK1/parkin pathway. Toxicol Vitr. (2018) 51:1–10. doi: 10.1016/j.tiv.2018.05.001
75. Li M, Sala V, De Santis MC, Cimino J, Cappello P, Pianca N, et al. Phosphoinositide 3-kinase gamma inhibition protects from anthracycline cardiotoxicity and reduces tumor growth. Circulation. (2018) 138:696–711. doi: 10.1161/CIRCULATIONAHA.117.030352
76. Singh R, Letai A, Sarosiek K. Regulation of apoptosis in health and disease: the balancing act of BCL-2 family proteins. Nat Rev Mol Cell Biol. (2019) 20:175–93. doi: 10.1038/s41580-018-0089-8
77. Cavalcante GC, Schaan AP, Cabral GF, Santana-da-Silva MN, Pinto P, Vidal AF, et al. A cell's fate: an overview of the molecular biology and genetics of apoptosis. Int J Mol Sci. (2019) 20:4133. doi: 10.3390/ijms20174133
78. Xia P, Chen J, Liu Y, Fletcher M, Jensen BC, Cheng Z. Doxorubicin induces cardiomyocyte apoptosis and atrophy through cyclin-dependent kinase 2-mediated activation of forkhead box O1. J Biol Chem. (2020) 295:4265–76. doi: 10.1074/jbc.RA119.011571
79. Zhuo C, Wang Y, Wang X, Wang Y, Chen Y. Cardioprotection by ischemic postconditioning is abolished in depressed rats: role of Akt and signal transducer and activator of transcription-3. Mol Cell Biochem. (2011) 346:39–47. doi: 10.1007/s11010-010-0589-0
80. Willis MS, Bevilacqua A, Pulinilkunnil T, Kienesberger P, Tannu M, Patterson C. The role of ubiquitin ligases in cardiac disease. J Mol Cell Cardiol. (2014) 71:43–53. doi: 10.1016/j.yjmcc.2013.11.008
81. Yamamoto Y, Hoshino Y, Ito T, Nariai T, Mohri T, Obana M, et al. Atrogin-1 ubiquitin ligase is upregulated by doxorubicin via p38-MAP kinase in cardiac myocytes. Cardiovasc Res. (2008) 79:89–96. doi: 10.1093/cvr/cvn076
82. Sishi BJN, Bester DJ, Wergeland A, Loos B, Jonassen AK, van Rooyen J, et al. Daunorubicin therapy is associated with upregulation of E3 ubiquitin ligases in the heart. Exp Biol Med. (2012) 237:219–26. doi: 10.1258/ebm.2011.011106
83. Burchfield JS, Xie M, Hill JA. Pathological ventricular remodeling: mechanisms: part 1 of 2. Circulation. (2013) 128:388–400. doi: 10.1161/CIRCULATIONAHA.113.001878
84. Długosz-Danecka M, Gruszka AM, Szmit S, Olszanecka A, Ogórka T, Sobocinski M, et al. Primary cardioprotection reduces mortality in lymphoma patients with increased risk of anthracycline cardiotoxicity, treated by R-CHOP regimen. Chemotherapy. (2018) 63:238–45. doi: 10.1159/000492942
85. Seicean S, Seicean A, Plana JC, Budd GT, Marwick TH. Effect of statin therapy on the risk for incident heart failure in patients with breast cancer receiving anthracycline chemotherapy: an observational Clinical Cohort Study. J Am Coll Cardiol. (2012) 60:2384–90. doi: 10.1016/j.jacc.2012.07.067
86. Chotenimitkhun R, D'Agostino R, Lawrence JA, Hamilton CA, Jordan JH, Vasu S, et al. Chronic statin administration may attenuate early anthracycline-associated declines in left ventricular ejection function. Can J Cardiol. (2015) 31:302–7. doi: 10.1016/j.cjca.2014.11.020
87. Acar Z, Kale A, Turgut M, Demircan S, Durna K, Demir S, et al. Efficiency of atorvastatin in the protection of anthracycline-induced cardiomyopathy. J Am Coll Cardiol. (2011) 58:988–9. doi: 10.1016/j.jacc.2011.05.025
88. Kutateladze TG. Translation of the phosphoinositide code by PI effectors. Nat Chem Biol. (2010) 6:507–13. doi: 10.1038/nchembio.390
89. Cancer Drug Manual. Available online at: http://www.bccancer.bc.ca/health-professionals/clinical-resources/cancer-drug-manual (accessed December 3, 2019).
90. Seifert CF, Nesser ME, Thompson DF. Dexrazoxane in the prevention of doxorubicin-induced cardiotoxicity. Ann Pharmacother. (1994) 28:1063–72. doi: 10.1177/106002809402800912
91. Hasinoff BB, Kuschak TI, Yalowich JC, Creighton AM. A QSAR study comparing the cytotoxicity and DNA topoisomerase II inhibitory effects of bisdioxopiperazine analogs of ICRF-187 (dexrazoxane). Biochem Pharmacol. (1995) 50:953–8. doi: 10.1016/0006-2952(95)00218-O
92. Deng S, Yan T, Jendrny C, Nemecek A, Vincetic M, Gödtel-Armbrust U, et al. Dexrazoxane may prevent doxorubicin-induced DNA damage via depleting both Topoisomerase II isoforms. BMC Cancer. (2014) 14:842. doi: 10.1186/1471-2407-14-842
93. Swain SM, Whaley FS, Gerber MC, Weisberg S, York M, Spicer D, et al. Cardioprotection with dexrazoxane for doxorubicin-containing therapy in advanced breast cancer. J Clin Oncol. (1997) 15:1318–32. doi: 10.1200/JCO.1997.15.4.1318
94. Shaikh F, Dupuis LL, Alexander S, Gupta A, Mertens L, Nathan PC. Cardioprotection and second malignant neoplasms associated with dexrazoxane in children receiving anthracycline chemotherapy: a systematic review and meta-analysis. J Natl Cancer Inst. (2016) 108:djv357. doi: 10.1093/jnci/djv357
95. Mahley RW, Innerarity TL, Rall SC, Weisgraber KH. Plasma lipoproteins: apolipoprotein structure and function. J Lipid Res. (1984) 25:1277–94.
96. Silverman DI, Ginsburg GS, Pasternak RC. High-density lipoprotein subfractions. Am J Med. (1993) 94:636–45. doi: 10.1016/0002-9343(93)90217-D
97. Tso C, Martinic G, Fan WH, Rogers C, Rye KA, Barter PJ. High-density lipoproteins enhance progenitor-mediated endothelium repair in mice. Arterioscler Thromb Vasc Biol. (2006) 26:1144–9. doi: 10.1161/01.ATV.0000216600.37436.cf
98. Seetharam D, Mineo C, Gormley AK, Gibson LL, Vongpatanasin W, Chambliss KL, et al. High-density lipoprotein promotes endothelial cell migration and reendothelialization via scavenger receptor-B Type I. Circ Res. (2006) 98:63–72. doi: 10.1161/01.RES.0000199272.59432.5b
99. Shah AS, Tan L, Long JL, Davidson WS. Proteomic diversity of high density lipoproteins: our emerging understanding of its importance in lipid transport and beyond. J Lipid Res. (2013) 54:2575–85. doi: 10.1194/jlr.R035725
100. Rader DJ. Molecular regulation of HDL metabolism and function: implications for novel therapies. J Clin Invest. (2006) 116:3090–100. doi: 10.1172/JCI30163
101. Mei X, Atkinson D. Lipid-free apolipoprotein A-I structure: insights into HDL formation and atherosclerosis development. Arch Med Res. (2015) 46:351–60. doi: 10.1016/j.arcmed.2015.05.012
102. Smith JD. Dysfunctional HDL as a diagnostic and therapeutic target. Arterioscler Thromb Vasc Biol. (2010) 30:151–5. doi: 10.1161/ATVBAHA.108.179226
103. Ahsan L, Ossoli AF, Freeman L, Vaisman B, Amar MJ, Shamburek RD, et al. Role of lecithin: cholesterol acyltransferase in HDL metabolism and atherosclerosis. In: Komoda T, editor. The HDL Handbook: Biological Functions and Clinical Implications. 2nd ed. Elsevier Inc. (2013). p. 159–94.
104. Thacker SG, Rousset X, Esmail S, Zarzour A, Jin X, Collins HL, et al. Increased plasma cholesterol esterification by LCAT reduces diet-induced atherosclerosis in SR-BI knockout mice. J Lipid Res. (2015) 56:1282–95. doi: 10.1194/jlr.M048629
105. Rigotti A, Trigatti B, Babitt J, Penman M, Xu S, Krieger M. Scavenger receptor BI–a cell surface receptor for high density lipoprotein. Curr Opin Lipidol. (1997) 8:181–8. doi: 10.1097/00041433-199706000-00009
106. Zhang YZ, Zanotti I, Reilly MP, Glick JM, Rothblat GH, Rader DJ. Overexpression of apolipoprotein A-I promotes reverse transport of cholesterol from macrophages to feces in vivo. Circulation. (2003) 108:661–3. doi: 10.1161/01.CIR.0000086981.09834.E0
107. McTaggart F, Jones P. Effects of statins on high-density lipoproteins: a potential contribution to cardiovascular benefit. Cardiovasc Drugs Ther. (2008) 22:321–38. doi: 10.1007/s10557-008-6113-z
108. Chapman MJ. Are the effects of statins on HDL-cholesterol clinically relevant? Eur Hear J. (2004) 6 (Suppl.):58–63. doi: 10.1016/j.ehjsup.2004.04.002
109. Barter P. Options for therapeutic intervention: how effective are the different agents? Eur Hear J. (2006) 8 (Suppl.):47–53. doi: 10.1093/eurheartj/sul041
110. Sacks FM, Tonkin AM, Shepherd J, Braunwald E, Cobbe S, Hawkins CM, et al. Effect of pravastatin on coronary disease events in subgroups defined by coronary risk factors: the prospective pravastatin pooling project. Circulation. (2000) 102:1893–900. doi: 10.1161/01.CIR.102.16.1893
111. Scandinavian Simvastatin Survival Study Group. Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S). Lancet. (1994) 344:1383–9. doi: 10.1016/S0140-6736(94)90566-5
112. Doggrell SA. No cardiovascular benefit with evacetrapib–is this the end of the road for the ‘cetrapibs’? Expert Opin Pharmacother. (2017) 18:1439–42. doi: 10.1080/14656566.2017.1365838
113. Grabie M, Tai CH, Frishman WH. Is anacetrapib better than its CETP inhibitor counterparts? Cardiol Rev. (2019) 27:242–8. doi: 10.1097/CRD.0000000000000245
114. Garg A, Sharma A, Krishnamoorthy P, Garg J, Virmani D, Sharma T, et al. Role of niacin in current clinical practice: a systematic review. Am J Med. (2017) 130:173–87. doi: 10.1016/j.amjmed.2016.07.038
115. Batuca JR, Amaral MC, Favas C, Paula FS, Ames PRJ, Papoila AL, et al. Extended-release niacin increases anti-apolipoprotein A-I antibodies that block the antioxidant effect of high-density lipoprotein–cholesterol: the EXPLORE clinical trial. Br J Clin Pharmacol. (2017) 83:1002–10. doi: 10.1111/bcp.13198
116. Parhofer KG. Increasing HDL-cholesterol and prevention of atherosclerosis: a critical perspective. Atheroscler Suppl. (2015) 18:109–11. doi: 10.1016/j.atherosclerosissup.2015.02.020
117. McCarthy M. Niacin fails to reduce vascular events in large randomised trial. BMJ. (2014) 349:2–3. doi: 10.1136/bmj.g4774
118. AIM-HIGH Investigators, Boden WE, Probstfield JL, Anderson T, Chaitman BR, Desvignes-Nickens P, et al. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med. (2011) 365:2255–67. doi: 10.1056/NEJMoa1107579
119. Chyu KY, Shah PK. HDL/ApoA-1 infusion and ApoA-1 gene therapy in atherosclerosis. Front Pharmacol. (2015) 6:1–9. doi: 10.3389/fphar.2015.00187
120. Shah PK. Apolipoprotein A-I / HDL infusion therapy for plaque stabilization- regression : a novel therapeutic approach stimulation of reverse cholesterol transport. Current. (2007) 13:1031–8. doi: 10.2174/138161207780487520
121. Dunbar RL, Movva R, Bloedon LAT, Duffy D, Norris RB, Navab M, et al. Oral apolipoprotein A-I mimetic D-4F lowers HDL-inflammatory index in high-risk patients: a first-in-human multiple-dose, randomized controlled trial. Clin Transl Sci. (2017) 10:455–69. doi: 10.1111/cts.12487
122. Gilham D, Wasiak S, Tsujikawa LM, Halliday C, Norek K, Patel RG, et al. RVX-208, a BET-inhibitor for treating atherosclerotic cardiovascular disease, raises ApoA-I/HDL and represses pathways that contribute to cardiovascular disease. Atherosclerosis. (2016) 247:48–57. doi: 10.1016/j.atherosclerosis.2016.01.036
123. Bloedon LAT, Dunbar R, Duffy D, Pinell-Salles P, Norris R, DeGroot BJ, et al. Safety, pharmacokinetics, and pharmacodynamics of oral apoA-I mimetic peptide D-4F in high-risk cardiovascular patients. J Lipid Res. (2008) 49:1344–52. doi: 10.1194/jlr.P800003-JLR200
124. Valanti EK, Dalakoura-Karagkouni K, Sanoudou D. Current and emerging reconstituted hdl-apoa-i and hdl-apoe approaches to treat atherosclerosis. J Pers Med. (2018) 8:1–12. doi: 10.3390/jpm8040034
125. Gille A, D'Andrea D, Tortorici MA, Hartel G, Wright SD. CSL112 (Apolipoprotein A-I [Human]) enhances cholesterol efflux similarly in healthy individuals and stable atherosclerotic disease patients. Arterioscler Thromb Vasc Biol. (2018) 38:953–63. doi: 10.1161/ATVBAHA.118.310538
126. Chenevard R, Hürlimann D, Spieker L, Béchir M, Enseleit F, Hermann M, et al. Reconstituted HDL in acute coronary syndromes. Cardiovasc Ther. (2012) 30:e51–7. doi: 10.1111/j.1755-5922.2010.00221.x
127. Tardif J-C, Ballantyne CM, Barter P, Dasseux J-L, Fayad ZA, Guertin M-C, et al. Effects of the high-density lipoprotein mimetic agent CER-001 on coronary atherosclerosis in patients with acute coronary syndromes: a randomized trial. Eur Heart J. (2014) 35:3277–86. doi: 10.1093/eurheartj/ehu171
128. Kempen HJ, Gomaraschi M, Simonelli S, Calabresi L, Moerland M, Otvos J, et al. Persistent changes in lipoprotein lipids after a single infusion of ascending doses of MDCO-216 (apoA-IMilano/POPC) in healthy volunteers and stable coronary artery disease patients. Atherosclerosis. (2016) 255:17–24. doi: 10.1016/j.atherosclerosis.2016.10.042
129. Berge KG, Canner PL, Hainline A. High-density lipoprotein cholesterol and prognosis after myocardial infarction. Circulation. (1982) 66:1176–8. doi: 10.1161/01.CIR.66.6.1176
130. Calabresi L, Rossoni G, Gomaraschi M, Sisto F, Berti F, Franceschini G. High-density lipoproteins protect isolated rat hearts from ischemia-reperfusion injury by reducing cardiac tumor necrosis factor-α content and enhancing prostaglandin release. Circ Res. (2003) 92:330–37. doi: 10.1161/01.RES.0000054201.60308.1A
131. Frias MA, Pedretti S, Hacking D, Somers S, Lacerda L, Opie LH, et al. HDL protects against ischemia reperfusion injury by preserving mitochondrial integrity. Atherosclerosis. (2013) 228:110–6. doi: 10.1016/j.atherosclerosis.2013.02.003
132. Gordts SC, Muthuramu I, Nefyodova E, Jacobs F, Van Craeyveld E, De Geest B. Beneficial effects of selective HDL-raising gene transfer on survival, cardiac remodelling and cardiac function after myocardial infarction in mice. Gene Ther. (2013) 20:1053–61. doi: 10.1038/gt.2013.30
133. Brinck JW, Thomas A, Brulhart-Meynet MC, Lauer E, Frej C, Dahlbäck B, et al. High-density lipoprotein from end-stage renal disease patients exhibits superior cardioprotection and increase in sphingosine-1-phosphate. Eur J Clin Invest. (2018) 48:e12866. doi: 10.1111/eci.12866
134. Durham KK, Chathely KM, Trigatti BL. High-density lipoprotein protects cardiomyocytes against necrosis induced by oxygen and glucose deprivation through SR-B1, PI3K, and AKT1 and 2. Biochem J. (2018) 475:1253–65. doi: 10.1042/BCJ20170703
135. Durham KK, Kluck G, Mak KC, Deng YD, Trigatti BL. Treatment with apolipoprotein A1 protects mice against doxorubicin-induced cardiotoxicity in a scavenger receptor class B, type I-dependent manner. Am J Physiol Circ Physiol. (2019) 316:H1447–57. doi: 10.1152/ajpheart.00432.2018
136. Frias MA, James RW, Gerber-Wicht C, Lang U. Native and reconstituted HDL activate Stat3 in ventricular cardiomyocytes via ERK1/2: Role of sphingosine-1-phosphate. Cardiovasc Res. (2009) 82:313–23. doi: 10.1093/cvr/cvp024
137. Durham KK, Chathely KM, Mak KC, Momen A, Thomas CT, Zhao Y-Y, et al. HDL protects against doxorubicin-induced cardiotoxicity in a scavenger receptor class B type 1-, PI3K-, and Akt-dependent manner. Am J Physiol Circ Physiol. (2018) 314:H31–44. doi: 10.1152/ajpheart.00521.2016
138. Frias MA, Lang U, Gerber-Wicht C, James RW. Native and reconstituted HDL protect cardiomyocytes from doxorubicin-induced apoptosis. Cardiovasc Res. (2010) 85:118–26. doi: 10.1093/cvr/cvp289
139. Taniyama Y, Walsh K. Elevated myocardial Akt signaling ameliorates doxorubicin-induced congestive heart failure and promotes heart growth. J Mol Cell Cardiol. (2002) 34:1241–7. doi: 10.1006/jmcc.2002.2068
140. Jacoby JJ, Kalinowski A, Liu MG, Zhang SSM, Gao Q, Chai GX, et al. Cardiomyocyte-restricted knockout of STAT3 results in higher sensitivity to inflammation, cardiac fibrosis, and heart failure with advanced age. Proc Natl Acad Sci USA. (2003) 100:12929–34. doi: 10.1073/pnas.2134694100
141. Kunisada K, Negoro S, Tone E, Funamoto M, Osugi T, Yamada S, et al. Signal transducer and activator of transcription 3 in the heart transduces not only a hypertrophic signal but a protective signal against doxorubicin-induced cardiomyopathy. Proc Natl Acad Sci USA. (2000) 97:315–9. doi: 10.1073/pnas.97.1.315
142. Lecour S. Activation of the protective Survivor Activating Factor Enhancement (SAFE) pathway against reperfusion injury: does it go beyond the RISK pathway? J Mol Cell Cardiol. (2009) 47:32–40. doi: 10.1016/j.yjmcc.2009.03.019
143. Sussman MA, Völkers M, Fischer K, Bailey B, Cottage CT, Din S, et al. Myocardial AKT: the omnipresent nexus. Physiol Rev. (2011) 91:1023–70. doi: 10.1152/physrev.00024.2010
144. Kimura T, Tomura H, Mogi C, Kuwabara A, Damirin A, Ishizuka T, et al. Role of scavenger receptor class B type I and sphingosine 1-phosphate receptors in high density lipoprotein-induced inhibition of adhesion molecule expression in endothelial cells. J Biol Chem. (2006) 281:37457–67. doi: 10.1074/jbc.M605823200
145. Ruiz M, Frej C, Holmér A, Guo LJ, Tran S, Dahlbäck B. High-density lipoprotein-associated apolipoprotein M limits endothelial inflammation by delivering sphingosine-1-phosphate to the sphingosine-1-phosphate receptor 1. Arterioscler Thromb Vasc Biol. (2017) 37:118–29. doi: 10.1161/ATVBAHA.116.308435
146. Gonzalez L, Qian AS, Tahir U, Yu P, Trigatti BL. Sphingosine-1-phosphate receptor 1, expressed in myeloid cells, slows diet-induced atherosclerosis and protects against macrophage apoptosis in ldlr KO mice. Int J Mol Sci. (2017) 18:2721. doi: 10.3390/ijms18122721
147. Al-Jarallah A, Chen X, González L, Trigatti BL. High density lipoprotein stimulated migration of macrophages depends on the scavenger receptor class B, type I, PDZK1 and Akt1 and is blocked by sphingosine 1 phosphate receptor antagonists. PLoS ONE. (2014) 9:e106487. doi: 10.1371/journal.pone.0106487
148. Galvani S, Sanson M, Blaho VA, Swendeman SL, Conger H, Dahlbäck B, et al. HDL-bound sphingosine 1-phosphate acts as a biased agonist for the endothelial cell receptor S1P1 to limit vascular inflammation. Sci Signal. (2015) 8:ra79. doi: 10.1126/scisignal.aaa2581
149. Acton S, Rigotti A, Landschulz KT, Xu S, Hobbs HH, Krieger M. Identification of scavenger receptor SR-BI as a high density lipoprotein receptor. Science. (1996) 271:518–20. doi: 10.1126/science.271.5248.518
150. Galvani S, Hla T. Quality versus quantity: making HDL great again. Arterioscler Thromb Vasc Biol. (2017) 37:1018–19. doi: 10.1161/ATVBAHA.117.309441
151. Rubin EM, Ishida BY, Clift SM, Krauss RM. Expression of human apolipoprotein A-I in transgenic mice results in reduced plasma levels of murine apolipoprotein A-I and the appearance of two new high density lipoprotein size subclasses. Proc Natl Acad Sci USA. (1991) 88:434–8. doi: 10.1073/pnas.88.2.434
152. Wang B, Yuan Y, Han L, Ye L, Shi X, Feng M. Recombinant lipoproteins reinforce cytotoxicity of doxorubicin to hepatocellular carcinoma. J Drug Target. (2014) 22:76–85. doi: 10.3109/1061186X.2013.839687
153. Rui M, Xin Y, Li R, Ge Y, Feng C, Xu X. Targeted biomimetic nanoparticles for synergistic combination chemotherapy of paclitaxel and doxorubicin. Mol Pharm. (2017) 14:107–23. doi: 10.1021/acs.molpharmaceut.6b00732
154. Kuai R, Li D, Chen YE, Moon JJ, Schwendeman A. High-density lipoproteins: nature's multifunctional nanoparticles. ACS Nano. (2016) 10:3015–41. doi: 10.1021/acsnano.5b07522
155. Ahn J, Lim U, Weinstein SJ, Schatzkin A, Hayes RB, Virtamo J AD. Prediagnostic total and high-density lipoprotein cholesterol and risk of cancer. Cancer Epidemiol Biomarkers Prev. (2009) 2814–21. doi: 10.1158/1055-9965.EPI-08-1248
156. Jafri H, Alsheikh-Ali AA, Karas RH. Baseline and on-treatment high-density lipoprotein cholesterol and the risk of cancer in randomized controlled trials of lipid-altering therapy. J Am Coll Cardiol. (2010) 55:2846–54. doi: 10.1016/j.jacc.2009.12.069
157. Penson P, Long DL, Howard G, Howard VJ, Jones SR, Martin SS, et al. Associations between cardiovascular disease, cancer, and very low high-density lipoprotein cholesterol in the REasons for Geographical and Racial Differences in Stroke (REGARDS) study. Cardiovasc Res. (2019) 115:204–12. doi: 10.1093/cvr/cvy198
158. Sirniö P, Väyrynen JP, Klintrup K, Mäkelä J, Mäkinen MJ, Karttunen TJ, et al. Decreased serum apolipoprotein A1 levels are associated with poor survival and systemic inflammatory response in colorectal cancer. Sci Rep. (2017) 7:1–8. doi: 10.1038/s41598-017-05415-9
159. Pirro M, Ricciuti B, Rader DJ, Catapano AL, Sahebkar A, Banach M. High density lipoprotein cholesterol and cancer: marker or causative? Prog Lipid Res. (2018) 71:54–69. doi: 10.1016/j.plipres.2018.06.001
160. Pan B, Ren H, Ma Y, Liu D, Yu B, Ji L, et al. High-density lipoprotein of patients with type 2 diabetes mellitus elevates the capability of promoting migration and invasion of breast cancer cells. Int J Cancer. (2012) 131:70–82. doi: 10.1002/ijc.26341
161. Furberg AS, Jasienska G, Bjurstam N, Torjesen PA, Emaus A, Lipson SF, et al. Metabolic and hormonal profiles: HDL cholesterol as a plausible biomarker of breast cancer risk. The Norwegian EBBA study. Cancer Epidemiol Biomarkers Prev. (2005) 14:33−40.
162. Pan B, Ren H, Lv X, Zhao Y, Yu B, He Y, et al. Hypochlorite-induced oxidative stress elevates the capability of HDL in promoting breast cancer metastasis. J Transl Med. (2012) 10:1–3. doi: 10.1186/1479-5876-10-65
163. Besler C, Heinrich K, Rohrer L, Doerries C, Riwanto M, Shih DM, et al. Mechanisms underlying adverse effects of HDL on eNOS-activating pathways in patients with coronary artery disease. J Clin Invest. (2011) 121:2693–708. doi: 10.1172/JCI42946
164. Hansel B, Giral P, Nobecourt E, Chantepie S, Bruckert E, Chapman MJ, et al. Metabolic syndrome is associated with elevated oxidative stress and dysfunctional dense high-density lipoprotein particles displaying impaired antioxidative activity. J Clin Endocrinol Metab. (2004) 89:4963–71. doi: 10.1210/jc.2004-0305
165. Riwanto M, Rohrer L, Roschitzki B, Besler C, Mocharla P, Mueller M, et al. Altered activation of endothelial anti-and proapoptotic pathways by high-density lipoprotein from patients with coronary artery disease: role of high-density lipoprotein-proteome remodeling. Circulation. (2013) 127:891–904. doi: 10.1161/CIRCULATIONAHA.112.108753
166. Navab R, Strumpf D, Bandarchi B, Zhu CQ, Pintilie M, Ramnarine VR, et al. Prognostic gene-expression signature of carcinoma-associated fibroblasts in non-small cell lung cancer. Proc Natl Acad Sci USA. (2011) 108:7160–5. doi: 10.1073/pnas.1014506108
167. Zamanian-Daryoush M, Lindner D, Tallant TC, Wang Z, Buffa J, Klipfell E, et al. The cardioprotective protein apolipoprotein a1 promotes potent anti-tumorigenic effects. J Biol Chem. (2013) 288:21237–52. doi: 10.1074/jbc.M113.468967
168. Zamanian-Daryoush M, DiDonato JA. Apolipoprotein A-I and cancer. Front Pharmacol. (2015) 6:265. doi: 10.3389/fphar.2015.00265
169. Gao F, Vasquez SX, Su F, Roberts S, Shah N, Grijalva V, et al. L-5F, an apolipoprotein A-I mimetic, inhibits tumor angiogenesis by suppressing VEGF/basic FGF signaling pathways. Integr Biol. (2011) 3:479–89. doi: 10.1039/c0ib00147c
170. Marinho AT, Lu H, Pereira SA, Monteiro E, Gabra H, Recchi C. Anti-tumorigenic and platinum-sensitizing effects of apolipoprotein A1 and apolipoprotein A1 mimetic peptides in ovarian cancer. Front Pharmacol. (2019) 9:1–13. doi: 10.3389/fphar.2018.01524
171. Chattopadhyay A, Yang X, Mukherjee P, Sulaiman D, Fogelman HR, Grijalva V, et al. Treating the intestine with oral ApoA-I mimetic Tg6F reduces tumor burden in mouse models of metastatic lung cancer. Sci Rep. (2018) 8:1–12. doi: 10.1038/s41598-018-26755-0
172. Mills GB, Moolenaar WH. The emerging role of lysophosphatidic acid in cancer. Nat Rev Cancer. (2003) 3:582–91. doi: 10.1038/nrc1143
173. Cao WM, Murao K, Imachi H, Yu X, Abe H, Yamauchi A, et al. A mutant high-density lipoprotein receptor inhibits proliferation of human breast cancer cells. Cancer Res. (2004) 64:1515–21. doi: 10.1158/0008-5472.CAN-03-0675
174. Pussinen PJ, Karten B, Wintersperger A, Reicher H, McLean M, Malle E, et al. The human breast carcinoma cell line HBL-100 acquires exogenous cholesterol from high-density lipoprotein via CLA-1 (CD-36 and LIMPII analogous 1)-mediated selective cholesteryl ester uptake. Biochem J. (2000) 349:559–66. doi: 10.1042/bj3490559
175. Danilo C, Gutierrez-Pajares JL, Mainieri MA, Mercier I, Lisanti MP, Frank PG. Scavenger receptor class B type I regulates cellular cholesterol metabolism and cell signaling associated with breast cancer development. Breast Cancer Res. (2013) 15:R87. doi: 10.1186/bcr3483
176. Yuan B, Wu C, Wang X, Wang D, Liu H, Guo L, et al. High scavenger receptor class B type I expression is related to tumor aggressiveness and poor prognosis in breast cancer. Tumor Biol. (2016) 37:3581–8. doi: 10.1007/s13277-015-4141-4
Keywords: HDL, ApoA1, anthracyclin, chemotherapy, cardiotoxicity, cardioprotective, doxorubicin
Citation: Kluck GEG, Durham KK, Yoo J-A and Trigatti BL (2020) High Density Lipoprotein and Its Precursor Protein Apolipoprotein A1 as Potential Therapeutics to Prevent Anthracycline Associated Cardiotoxicity. Front. Cardiovasc. Med. 7:65. doi: 10.3389/fcvm.2020.00065
Received: 15 December 2019; Accepted: 06 April 2020;
Published: 28 April 2020.
Edited by:
Canan G. Nebigil, École Supérieure de Biotechnologie Strasbourg (ESBS), FranceReviewed by:
Alessandra Ghigo, University of Turin, ItalyCopyright © 2020 Kluck, Durham, Yoo and Trigatti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bernardo L. Trigatti, dHJpZ2F0dEBtY21hc3Rlci5jYQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.