 Himani Kumari
Himani Kumari Wan-Hong Huang
Wan-Hong Huang Michael W. Y. Chan
Michael W. Y. Chan- 1Department of Biomedical Sciences, National Chung Cheng University, Chiayi, Taiwan
- 2Epigenomics and Human Disease Research Center, National Chung Cheng University, Chiayi, Taiwan
- 3Center for Innovative Research on Aging Society (CIRAS), National Chung Cheng University, Chiayi, Taiwan
Use of anthracyclines such as doxorubicin (DOX), for the treatment of cancer, is known to induce cardiotoxicity, begetting numerous evaluations of this adverse effect. This review emphasizes the mechanism of how consideration of DOX-induced cardiotoxicity is important for the development of cardioprotective agents. As DOX is involved in mitochondrial dysfunction, enzymes involved in epigenetic modifications that use mitochondrial metabolite as substrate are most likely to be affected. Therefore, this review article focuses on the fact that epigenetic modifications, namely, DNA methylation, histone modifications, and noncoding RNA expression, contribute to DOX-associated cardiotoxicity. Early interventions needed for patients undergoing chemotherapy, to treat or prevent heart failure, would, overall, improve the survival, and quality of life of cancer patients. These epigenetic modifications can either be used as molecular markers for cancer prognosis or represent molecular targets to attenuate DOX-induced cardiotoxicity in cancer patients.
Introduction
Cardiotoxicity, in simpler terms, is defined as “toxicity which damages the heart,” often during or after chemotherapeutic treatment (1). Treatment options for cancer have been improving significantly in recent years, and the rates of survival in several human cancers have increased significantly with reduced recurrences (2). However, the applicability of these drugs is limited by the risk of cardiotoxicity (1). Doxorubicin (DOX)–induced cardiomyopathy can occur within a few days of its administration or delayed until decades after chemotherapy, thus affecting morbidity, mortality, and quality of life of cancer patients (3–6). However, the mechanism of DOX-induced cardiotoxicity is not fully understood.
Epigenetic modifications, including DNA methylation, histone modifications, and noncoding RNA (ncRNA) expression, play an important role in regulating gene expression and are considered as a hallmark of several human diseases, such as cardiovascular disease [review in Kimball and Vondriska (7)]. In this review, we discuss the mechanism of how aberrant epigenetic modifications contribute to DOX-induced cardiotoxicity and possible alternative therapeutic options that could forestall or prevent chemotherapy-induced cardiotoxicity (8).
Chemotherapeutic-Associated Cardiotoxicity
In the 1960s, DOX (Adriamycin®), first isolated from Streptomyces actinobacteria, was found as one of the first anthracyclines (9), to be used for several cancer treatments, including breast carcinomas, sarcomas, leukemias, non-Hodgkin and Hodgkin lymphoma, and many other cancers (10, 11). At the molecular level, DOX acts to stabilize topoisomerase DNA isomers and therefore blocks DNA replication and transcription (12, 13). It has been reported in several studies over the last 15 years that despite the successful development of small molecules and targeted therapies, anthracycline-based chemotherapy still plays not only prominent anticancer but also overall detrimental roles in many types of cancer treatment (14). Concerning the latter, DOX causes a cumulative, irreversible, and dose-dependent cardiomyopathy that ultimately leads to congestive heart failure (15). Previous studies have demonstrated that cardiotoxicity is a repercussion of dose-dependent administration of DOX, with those exceeding 500 mg/m2 greatly increasing the risk of congestive heart failure tremendously (16). Understanding the mechanism involved in DOX is important in developing novel preventive measures, and treatment strategies, against DOX-induced cardiotoxicity.
Cardiotoxicity is one of the major adverse effects of chemotherapy, and a leading cause of increased mortality and morbidity, in cancer patients (6, 17). Cardiotoxicity can occur in the early or late stages of the course of the disease and may vary from subclinical myocardial dysfunction to irreversible heart failure or death (18). Documented reports are limited to the mechanism of the appearance of cardiac dysfunction during chemotherapy and the susceptibility of patients to develop cardiotoxicity (1, 19). However, a proposed clinical study demonstrated that among all cancer patients, the overall occurrence of DOX-induced cardiotoxicity was ~9%, and most cases occurred immediately during the first year after the completion of chemotherapy and have even been noticed after a follow-up of 4 years (20). Complications emerging from chemotherapy-induced cardiotoxicity are potentially life-threatening, further limiting the clinical use of various chemotherapeutic agents (particularly anthracyclines) (8), thus strongly supporting the need for improved cardioprotective agents.
Mechanisms Of Dox-Induced Cardiotoxicity
One widely accepted mechanism for DOX-induced cardiotoxicity is the generation of reactive oxygen species (ROS) after DOX treatment in cardiac mitochondria; this occurrence marks as the primary initiating event in the cascade of intracellular modifications (21). In mitochondria, DOX is reduced by NADH dehydrogenase and undergoes redox cycling, generating ROS (22). Elevated levels of ROS result in cellular damage, also known as oxidative stress, which is initiated when the delicate balance between the ROS-generating system and antioxidant measures is disrupted (8). Cardiomyocytes are highly susceptible to oxidative stress, as treatment with DOX reduced the levels of antioxidant enzymes such as glutathione peroxidase, catalase, and superoxide dismutase (23). Cancer patients receiving DOX treatment also undergo immediate systemic oxidative stress, which is due to a decrease in glutathione and total antioxidant capacity of plasma (24).
Production of ROS also affects the DNA, RNA, proteins, and lipids and can also act as secondary signaling molecules in various pathways that are involved in homeostasis, including cell proliferation and cell death (25, 26). Thus, maintenance of a proper level of ROS in the intracellular and extracellular environment is of vital importance. Hence, it could be inferred that oxidative stress could be a leading cause of cellular hypertrophy in the heart (27), due to gene expression alterations (28), cell death activation (29), extracellular matrix transformation (30), ventricular remodeling (29), and calcium transient perturbation (31), all of which could result in the pathophysiological changes that lead to cardiomyopathy and heart failure.
On the other hand, DOX can also disrupt cellular and mitochondrial metabolism, a phenomenon not fully explored. For example, DOX can reduce mitochondrial NADH accumulation and impair oxidative phosphorylation in heart tissues, events associated with reduced glucose uptake (32). Doxorubicin can also induce the opening of mitochondrial permeability transition pore, resulting in the loss of mitochondrial membrane potential, thus explaining DOX-mediated apoptosis in cardiomyocytes. Moreover, DOX can reduce both the protein level and AMPK phosphorylation, thus contributing to stress and metabolic dysfunction (33, 34). More recently, one study found that the noncanonical function of the tumor suppressor p53 is involved in DOX-mediated cardiotoxicity (35). Doxorubicin treatment of TP53-depleted mice resulted in left ventricular systolic dysfunction, in association with decreased oxidative metabolism, and reduced mitochondrial volume and DNA transcription. Taken together, induction of oxidative stress and disruption of metabolism in mitochondria are crucial to the development of cardiotoxicity by DOX.
Role of Epigenetic Alterations in Dox-Induced Cardiotoxicity
Mitochondrial metabolites constitute a large number of cofactors for several enzymes involved in human biochemical pathways, including epigenetic modifications (36). For example, S-adenosylmethionine (SAM) is the universal substrate for DNA and histone methylation. It is therefore believed that mitochondrial disruption may likely affect cardiomyocyte genomic chromatin (7). Indeed, DNA methylation and histone modifications, as well as non-coding RNA expression, have recently been found to play a role in DOX-induced cardiotoxicity. Furthermore, in vivo experiments also demonstrated that rat deficient in methyl donors developed cardiomyopathy with disrupted mitochondrial alignment in the myocardium (37). This effect was due to the reduced activity of PGC-1α, the master regulator for mitochondrial biogenesis (38). Interestingly, such reduced PGC-1α activity was found to be due to increased acetylation and a decreased methylation of PGC-1α, through downregulation of the histone modifiers, SIRT1 deacetylase, and PRMT1 methyltransferase, thus further supporting the interplay between metabolism and epigenetic modifications (37). The role of DOX in the alteration of gene expression via epigenetic modifications is illustrated in Figure 1.
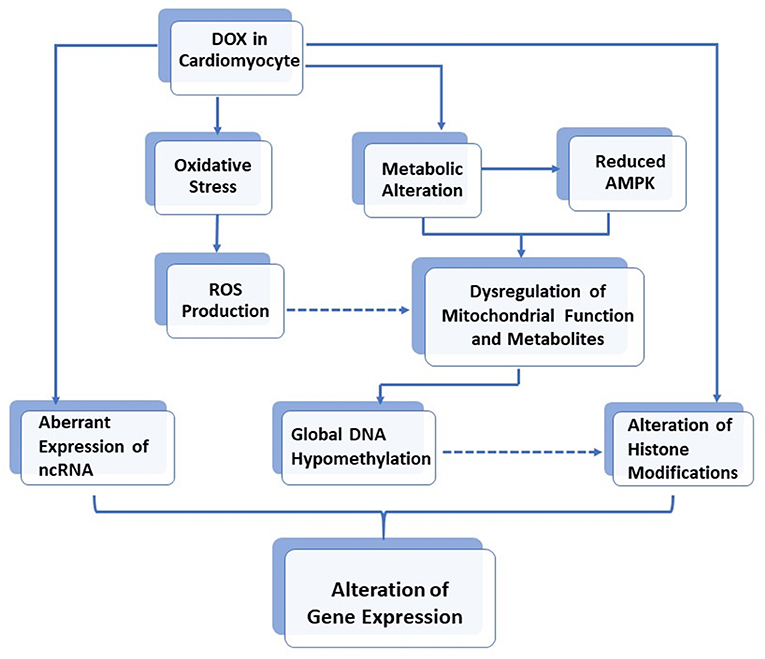
Figure 1. The role of DOX in alteration of gene expressions via epigenetic modifications in cardiomyocytes.
Epigenetic Modification: DNA Methylation
DNA methylation is often referred to as the “fifth” DNA based, because of its ubiquitousness in occurring at the 5′ position of cytosine in CpG dinucleotide (39). 5-Methylcytosine is established, maintained, and removed by several enzymes, including DNA methyltransferases (DNMTs) and Tet (the ten-eleven translocation hydroxylases) family protein. DNA methylation at the promoter region of a gene is associated with transcriptional repression by recruitment of transcriptional repressors and histone modifiers (such as histone deacetylases and histone methyltransferase), resulting in a repressive chromatin. The interplay between DNA methylation and histone modifications has been reviewed elsewhere (40, 41) and will not be discussed here.
DNA methyltransferases and Tet require SAM or α-ketoglutarate (α-KG) for the formation of 5-methylcytosine or 5-hydroxymethylcytosine (5hMC), in the process of DNA methylation and demethylation, respectively (42). In particular, the metabolic pathway from mitochondria generates SAM and α-KG; mitochondrial dysfunction associated with chronic DOX therapy may affect epigenetic machinery.
Indeed, in one of the studies, mouse cardiomyoblast H9c2 cells were used to analyze the effect of DOX (21). Together with a decrease in glycolytic activity and basal respiration in DOX-treated cells, dysregulation of mitochondrial DNA transcripts was observed. Importantly downregulation of DNMT1 (a maintenance methyltransferase), accompanied by a decrease in global DNA methylation, was also observed. This effect is in agreement with a previous animal study that global DNA hypomethylation, accompanied by a dysregulated expression of mitochondrial gene products encoded from both nuclear and mitochondrial genome, was observed in the hearts of rats treated with DOX (15). It is also interesting to point out that Ferreira et al. (21) found that pre-exposure of DOX can confer resistance to subsequent exposure of DOX in H9c2 cells, probably due to mitochondrial adaptation. As DNA methylation of the mitochondrial genome is maintained by DNMT1, the only DNMT member that can be translocated into the mitochondria (43, 44), downregulation of DNMT1 by oxidative stress may eventually affect the methylation of mitochondrial genome. Taken together, these studies thus suggest that DOX may affect global DNA methylation via dysregulation of mitochondrial function and related metabolites.
Notably, a recent animal study demonstrated that the involvement of mitochondrial genome was not observed, as genes showing significant differential methylation in DOX-treated rats were all encoded from the nucleus. However, a global DNA hypomethylation in the DOX-treated group was still observed (45). This discrepancy may be due to the methods used in these studies. Study from Nordgren et al. (45) utilized a sequencing-based approach Reduced representation bisulfite sequencing (RRBS) that can only interrogate DNA methylation at the CpG rich region, whereas studies from Ferreira et al. used a candidate gene approach to analyze the change of the mitochondrial genome (21). In this regard, further unbiased experiments are required to analyze the role of DOX in the change of the methylome in mitochondria and nucleus.
Histone Modification
Besides DNA methylation, histone modifications are also involved in DOX-induced cardiotoxicity (Table 1). These modifications can give rise to synergistic or antagonistic interactions with chromatin-associated protein, resulting in dynamic switching between transcriptionally active (accessible euchromatin) and silent (condensed heterochromatin) states (50). For example, histone deacetylase, HDAC6, was found to be upregulated in DOX-treated primary rat cardiomyocytes, in vitro, and mice model, in vivo, resulting in deacetylation of α-tubulin (48). The upregulation of other HDACs (Table 1) has also been observed in the heart tissue of mice treated with DOX (46). In this regard, Song et al. (48) demonstrated that genetic or pharmacological inhibition of HDAC6 in mice showed a cardioprotective effect against DOX by restoring autophagic flux.
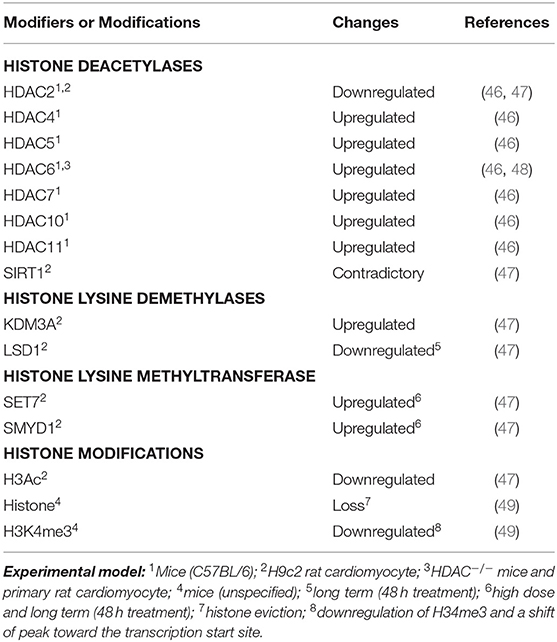
Table 1. Changes of histone modifications and modifiers in DOX treated cardiomyocytes.
Furthermore, a recent study using H9c2 cardiac myoblast cells also demonstrated that expression of several histone modifiers was dysregulated in association with downregulation of global acetylation of histone H3 (Table 1). In this study, Hanf et al. (47) demonstrated that expression levels of histone deacetylases (SIRT1 and HDAC2) were affected upon DOX treatment. In particular, different isoforms of SIRT1 displayed a contradictory expression level. However, pterostilbene, a natural analog of resveratrol and antioxidant, has been found to alleviate DOX-induced cardiotoxicity both in vitro and in vivo (51). This effect is due to enhanced deacetylation activity of SIRT1, suggesting its cardioprotective effect against DOX. In the case of HDAC2, treatment with low-dose DOX resulted in decreased expression of HDAC2, but no significant changes in high-dose treatment, as compared to control. Consistently, HDAC2 downregulation was observed in the heart tissue of mice treated with DOX (46). As most of the HDACs were found to be upregulated in DOX-treated cardiomyocytes, it is reasonable to observe the cardioprotective effect of HDAC inhibitors on DOX (52). Intriguingly, studies found that trichostatin A, a pan-HDAC inhibitor, can enhance DOX-mediated hypertrophy and apoptosis in H9c2 rat cardiomyoblasts (53, 54). In one of the studies, Ma et al. (54) found that DOX-induced cardiotoxicity is mediated through Rac1, a GTP-binding protein, and subunit of NADPH oxidase, resulting in the suppression of HDAC activity and upregulation of p53. Importantly, this process is ROS-independent. In this regard, treatment of HDAC inhibitor further enhances the effect of DOX-mediated cardiotoxicity. The involvement of specific HDAC isoforms in this process, however, remains to be determined.
Moreover, the histone lysine demethylase, KDM3A, was significantly upregulated upon DOX treatment of H9C2 cells; however, long-term DOX treatment also significantly decreased the lysine-specific histone demethylase 1 (i.e., LSD1). In parallel, significant upregulation of the histone lysine methyltransferases, SET7 and SMYD1, was only observed in long-term and high-dose DOX treatment. Notably, a heart-specific transcriptional alteration was only observed in mice treated with DOX, but not etoposide, a nonanthracycline (49). This event was due to the inhibition of topoisomerase 2β (55), as “eviction” of specific histones from chromatin, resulting in a shift of histone modification (H3K4me3), and chromatin structure, around the promoter region of a gene.
Noncoding RNA Expression
Another recognized epigenetic modification is the regulation of ncRNAs, including long noncoding RNAs (lncRNAs) and microRNAs (miRNAs). Noncoding RNAs are involved in numerous human biological processes, as well as human diseases (56). Up to 30% of gene expression in humans is regulated by ~1,000 known miRNAs, ranging from 18 to 25 nucleotides. MicroRNAs may originate from either independent genes or introns of protein coding genes and are transcribed by RNA polymerase II. Subsequently, these “primary miRNAs” are processed into mature miRNAs and then assembled into argonaute family proteins containing ribonucleoprotein complexes called miRNA-induced silencing complexes. These complexes then bind to their mRNA target sequences in 3′ UTR (untranslated region) of mRNA transcripts, resulting in either translational blockage or mRNA degradation.
Aberrant expression of several miRNAs has been shown involved in DOX-mediated cardiotoxicity (Table 2). For example, upregulation of miR-15 was observed in DOX-induced apoptotic H9c2 cardiomyocytes (57). This effect was probably due to suppression of Bmpr1a, a target of miR-15 and BMP receptor, previously found to be involved in cardiac contractility (68). Activation of BMP signaling by Bmpr1a agonist is therefore able to rescue DOX-mediated cardiotoxicity in H9c2 cells (57). Similarly, upregulation of miR-23a (58), miR-34a (61, 62), miR-140 (63), miR-146a (64), and miR-532 (66) were observed either in vitro or in vivo models of DOX-induced cardiotoxicity. Interestingly, upregulation of miR-34a, a well-known tumor suppressive miRNA, could epigenetically suppress SIRT1 (61, 62), thus partially explaining the downregulation of this HDAC, by DOX, in the aforementioned study (47).
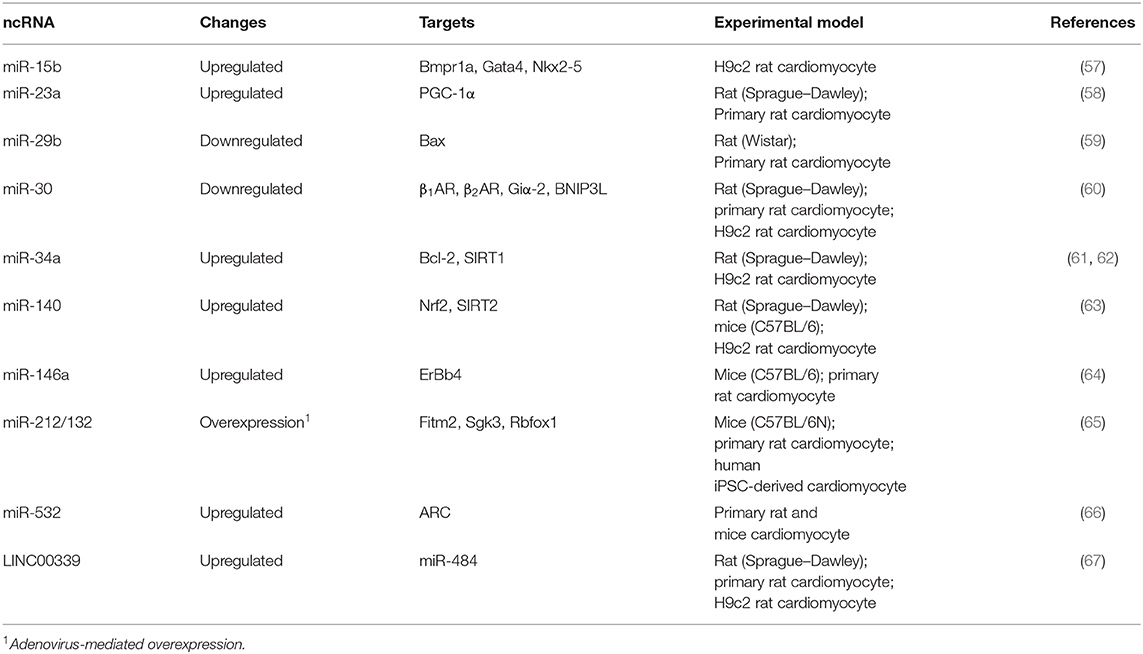
Table 2. Expression changes of ncRNA in cardiomyocytes treated with DOX.
Therapeutically, adenovirus-mediated overexpression of miR-212/132 cluster has been shown to prevent DOX-induced cardiotoxicity in a mouse model (65). This effect may be partially due to direct targeting of Fitm2, a transmembrane protein involved in fat storage, by miR-232/132. Moreover, downregulation of miR-29b (59) and miR-30 (60) was also observed in DOX-treated cardiomyocytes in an animal model, leading to de-repression of BAX, a proapoptotic protein, and β-adrenoceptor (β1- and β2AR), involved in myocyte contraction, respectively.
On the other hand, lncRNAs, which are more than 200 nucleotides long, regulate gene expression by diverse mechanisms (69). For example, lncRNAs can serve as a scaffold to recruit activators or repressors to regulate gene expression. The molecular function and clinical application of lncRNAs in cardiovascular disease have been recently reviewed (70, 71). Particularly, several studies have provided evidence to demonstrate that lncRNA can directly “sponge” or bind to miRNAs, thus regulating the activity of those miRNAs through a competing endogenous RNA (ceRNA) mechanism (71–74). For example, DOX can upregulate the lncRNA, LINC00339 (Table 2), resulting in the suppressing of miR-484 by ceRNA mechanism, in cardiomyocytes in vitro and in an animal model (67).
Conclusion
Although DOX is still the mainstay anthracyclines (9) for the treatment of several human cancers, a major concern is the side effect of cardiotoxicity. In this review, we have summarized recent findings that epigenetic modifications were observed in cardiomyocytes treated with DOX, both in vitro and in vivo. Although the causal relationship between cardiotoxicity and epigenetic modifications has not been fully explored, epigenetic modifications may contribute to either a cardiotoxic or cardioprotective process. Whether this process is contributed by DOX-mediated ROS or specific signaling pathways may require further investigation (54). Therapeutically, combinations of chemotherapeutic agents with epigenetic therapies, such as small molecule inhibitor of epigenetic writer/reader/eraser or miRNAs manipulations, may confer protection of patients from cardiotoxicity. However, whether such a potential cardioprotective agent will affect the efficacy of DOX or create other side effects requires further clinical investigation (i.e., the colloquial “double-edged sword”). For example, dexrazoxane, the only Food and Drug Administration–approved cardioprotective agent, has been shown to prevent DOX-mediated cardiotoxicity (75). However, the beneficial effect of dexrazoxane is still debated because of the risk for the development of acute myeloid leukemia and myelodysplastic syndrome in children (76, 77). In conclusion, epigenetic modifications may play a role in DOX-mediated apoptosis and atrophy in cardiomyocytes. Delineation of specific epigenetic therapies as detrimental vs. beneficial cardioprotective merits further investigation.
Author Contributions
W-HH and MC performed literature search. HK and MC wrote the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This study was supported by grants from the Ministry of Science and Technology, Taiwan (MOST 106-2923-B-194-001-MY3), and the Center for Innovative Research on Aging Society (CIRAS) from The Featured Areas Research Center Program within the framework of the Higher Education Sprout Project by Ministry of Education (MOE) in Taiwan.
References
2. Falzone L, Salomone S, Libra M. Evolution of cancer pharmacological treatments at the turn of the third millennium. Front Pharmacol. (2018) 9:1300. doi: 10.3389/fphar.2018.01300
3. Octavia Y, Tocchetti CG, Gabrielson KL, Janssens S, Crijns HJ, Moens AL. Doxorubicin-induced cardiomyopathy: from molecular mechanisms to therapeutic strategies. J Mol Cell Cardiol. (2012) 52:1213–25. doi: 10.1016/j.yjmcc.2012.03.006
4. Chatterjee K, Zhang J, Honbo N, Karliner JS. Doxorubicin cardiomyopathy. Cardiology. (2010) 115:155–62. doi: 10.1159/000265166
5. Pugazhendhi A, Edison T, Velmurugan BK, Jacob JA, Karuppusamy I. Toxicity of Doxorubicin (Dox) to different experimental organ systems. Life Sci. (2018) 200:26–30. doi: 10.1016/j.lfs.2018.03.023
6. Steinherz LJ, Steinherz PG, Tan CT, Heller G, Murphy ML. Cardiac toxicity 4 to 20 years after completing anthracycline therapy. JAMA. (1991) 266:1672–7. doi: 10.1001/jama.266.12.1672
7. Kimball TH, Vondriska T. Metabolism epigenetics, and causal inference in heart failure. Trends Endocrinol Metab. (2019) 3:181–91.
8. Angsutararux P, Luanpitpong S, Issaragrisil S. Chemotherapy-induced cardiotoxicity: overview of the roles of oxidative stress. Oxid Med Cell Longev. (2015) 2015:795602. doi: 10.1155/2015/795602
9. Arcamone F, Cassinelli G, Fantini G, Grein A, Orezzi P, Pol C, et al. Peucetius var. caesius. Reprinted from Biotechnology and Bioengineering, Vol. XI, 1969, Issue 6, Pages 1101–1110. Biotechnol Bioeng. (2000) 67:704–13. doi: 10.1002/(SICI)1097-0290(20000320)67:6<704::AID-BIT8>3.0.CO;2-L
10. Simunek T, Sterba M, Popelova O, Adamcova M, Hrdina R, Gersl V. Anthracycline-induced cardiotoxicity: overview of studies examining the roles of oxidative stress and free cellular iron. Pharmacol Rep. (2009) 61:154–71. doi: 10.1016/S1734-1140(09)70018-0
11. Sterba M, Popelova O, Vavrova A, Jirkovsky E, Kovarikova P, Gersl V, et al. Oxidative stress, redox signaling, and metal chelation in anthracycline cardiotoxicity and pharmacological cardioprotection. Antioxid Redox Signal. (2013) 18:899–929. doi: 10.1089/ars.2012.4795
12. Tewey KM, Rowe TC, Yang L, Halligan BD, Liu LF. Adriamycin-induced DNA damage mediated by mammalian DNA topoisomerase II. Science. (1984) 226:466–8. doi: 10.1126/science.6093249
13. Marinello J, Delcuratolo M, Capranico G. Anthracyclines as topoisomerase II poisons: from early studies to new perspectives. Int J Mol Sci. (2018) 19: doi: 10.3390/ijms19113480
14. McGowan JV, Chung R, Maulik A, Piotrowska I, Walker JM, Yellon DM. Anthracycline chemotherapy and cardiotoxicity. Cardiovasc Drugs Ther. (2017) 31:63–75. doi: 10.1007/s10557-016-6711-0
15. Ferreira A, Cunha-Oliveira T, Simoes RF, Carvalho FS, Burgeiro A, Nordgren K, et al. Altered mitochondrial epigenetics associated with subchronic doxorubicin cardiotoxicity. Toxicology. (2017) 390:63–73. doi: 10.1016/j.tox.2017.08.011
16. Lefrak EA, Pitha J, Rosenheim S, Gottlieb JA. A clinicopathologic analysis of adriamycin cardiotoxicity. Cancer. (1973) 32:302–14. doi: 10.1002/1097-0142(197308)32:2<302::AID-CNCR2820320205>3.0.CO;2-2
17. Cardinale D, Colombo A, Lamantia G, Colombo N, Civelli M, De Giacomi G, et al. Anthracycline-induced cardiomyopathy: clinical relevance and response to pharmacologic therapy. J Am Coll Cardiol. (2010) 55:213–20. doi: 10.1016/j.jacc.2009.03.095
18. Mercuro G, Cadeddu C, Piras A, Dessi M, Madeddu C, Deidda M, et al. Early epirubicin-induced myocardial dysfunction revealed by serial tissue Doppler echocardiography: correlation with inflammatory and oxidative stress markers. Oncologist. (2007) 12:1124–33. doi: 10.1634/theoncologist.12-9-1124
19. Khakoo AY, Liu PP, Force T, Lopez-Berestein G, Jones LW, Schneider J, et al. Cardiotoxicity due to cancer therapy. Tex Heart Inst J. (2011) 38:253–6.
20. Cardinale D, Colombo A, Bacchiani G, Tedeschi I, Meroni CA, Veglia F, et al. Early detection of anthracycline cardiotoxicity and improvement with heart failure therapy. Circulation. (2015) 131:1981–8. doi: 10.1161/CIRCULATIONAHA.114.013777
21. Ferreira LL, Cunha-Oliveira T, Veloso CD, Costa CF, Wallace KB, Oliveira PJ. Single nanomolar doxorubicin exposure triggers compensatory mitochondrial responses in H9c2 cardiomyoblasts. Food Chem Toxicol. (2019) 124:450–61. doi: 10.1016/j.fct.2018.12.017
22. Doroshow JH, Davies KJ. Redox cycling of anthracyclines by cardiac mitochondria. II. Formation of superoxide anion, hydrogen peroxide, hydroxyl radical. J Biol Chem. (1986) 261:3068–74.
23. Babaei-Kouchaki S, Babapour V, Panahi N, Badalzadeh R. Effect of troxerutin on oxidative stress and expression of genes regulating mitochondrial biogenesis in doxorubicin-induced myocardial injury in rats. Naunyn-Schmiedeb Arch Pharmacol. (2020) doi: 10.1007/s00210-020-01818-0. [Epub ahead of print].
24. Panis C, Herrera AC, Victorino VJ, Campos FC, Freitas LF, De Rossi T, et al. Oxidative stress and hematological profiles of advanced breast cancer patients subjected to paclitaxel or doxorubicin chemotherapy. Breast Cancer Res Treat. (2012) 133:89–97. doi: 10.1007/s10549-011-1693-x
25. Deavall DG, Martin EA, Horner JM, Roberts R. Drug-induced oxidative stress and toxicity. J Toxicol. (2012) 2012:645460. doi: 10.1155/2012/645460
26. Devasagayam TP, Tilak JC, Boloor KK, Sane KS, Ghaskadbi SS, Lele RD. Free radicals and antioxidants in human health: current status and future prospects. J Assoc Phys. (2004) 52:794–804.
27. Sabri A, Hughie HH, Lucchesi PA. Regulation of hypertrophic and apoptotic signaling pathways by reactive oxygen species in cardiac myocytes. Antioxid Redox Signal. (2003) 5:731–40. doi: 10.1089/152308603770380034
28. Siwik DA, Tzortzis JD, Pimental DR, Chang DL, Pagano PJ, Singh K, et al. Inhibition of copper-zinc superoxide dismutase induces cell growth, hypertrophic phenotype, and apoptosis in neonatal rat cardiac myocytes in vitro. Circ Res. (1999) 85:147–53. doi: 10.1161/01.RES.85.2.147
29. Kwon SH, Pimentel DR, Remondino A, Sawyer DB, Colucci WS. H(2)O(2) regulates cardiac myocyte phenotype via concentration-dependent activation of distinct kinase pathways. J Mol Cell Cardiol. (2003) 35:615–21. doi: 10.1016/S0022-2828(03)00084-1
30. Siwik DA, Pagano PJ, Colucci WS. Oxidative stress regulates collagen synthesis and matrix metalloproteinase activity in cardiac fibroblasts. Am J Physiol Cell Physiol. (2001) 280:C53–60. doi: 10.1152/ajpcell.2001.280.1.C53
31. Dhalla NS, Temsah RM, Netticadan T. Role of oxidative stress in cardiovascular diseases. J Hypertens. (2000) 18:655–73. doi: 10.1097/00004872-200018060-00002
32. Montaigne D, Marechal X, Baccouch R, Modine T, Preau S, Zannis K, et al. Stabilization of mitochondrial membrane potential prevents doxorubicin-induced cardiotoxicity in isolated rat heart. Toxicol Appl Pharmacol. (2010) 244:300–7. doi: 10.1016/j.taap.2010.01.006
33. Gratia S, Kay L, Potenza L, Seffouh A, Novel-Chate V, Schnebelen C, et al. Inhibition of AMPK signalling by doxorubicin: at the crossroads of the cardiac responses to energetic, oxidative, genotoxic stress. Cardiovasc Res. (2012) 95:290–9. doi: 10.1093/cvr/cvs134
34. Tokarska-Schlattner M, Zaugg M, da Silva R, Lucchinetti E, Schaub MC, Wallimann T, et al. Acute toxicity of doxorubicin on isolated perfused heart: response of kinases regulating energy supply. Am J Physiol Heart Circ Physiol. (2005) 289:H37–47. doi: 10.1152/ajpheart.01057.2004
35. Li J, Wang PY, Long NA, Zhuang J, Springer DA, Zou J, et al. p53 prevents doxorubicin cardiotoxicity independently of its prototypical tumor suppressor activities. Proc Natl Acad Sci USA. (2019) 116:19626–34. doi: 10.1073/pnas.1904979116
36. Bahat A, Gross A. Mitochondrial plasticity in cell fate regulation. J Biol Chem. (2019) 294:13852–63. doi: 10.1074/jbc.REV118.000828
37. Garcia MM, Gueant-Rodriguez RM, Pooya S, Brachet P, Alberto JM, Jeannesson E, et al. Methyl donor deficiency induces cardiomyopathy through altered methylation/acetylation of PGC-1alpha by PRMT1 and SIRT1. J Pathol. (2011) 225:324–35. doi: 10.1002/path.2881
38. Handschin C, Spiegelman BM. Peroxisome proliferator-activated receptor gamma coactivator 1 coactivators, energy homeostasis, and metabolism. Endocr Rev. (2006) 27:728–35. doi: 10.1210/er.2006-0037
39. Ehrlich M, Lacey M. DNA methylation and differentiation: silencing, upregulation and modulation of gene expression. Epigenomics. (2013) 5:553–68. doi: 10.2217/epi.13.43
40. Flavahan WA, Gaskell E, Bernstein BE. Epigenetic plasticity and the hallmarks of cancer. Science. (2017) 357: doi: 10.1126/science.aal2380
41. Baylin SB, Jones PA. Epigenetic determinants of cancer. Cold Spring Harb Perspect Biol. (2016) 8: doi: 10.1101/cshperspect.a019505
42. Clare CE, Brassington AH, Kwong WY, Sinclair KD. One-Carbon metabolism: linking nutritional biochemistry to epigenetic programming of long-term development. Ann Rev Anim Biosci. (2019) 7:263–87. doi: 10.1146/annurev-animal-020518-115206
43. Shock LS, Thakkar PV, Peterson EJ, Moran RG, Taylor SM. DNA methyltransferase 1, cytosine methylation, and cytosine hydroxymethylation in mammalian mitochondria. Proc Natl Acad Sci USA. (2011) 108:3630–5. doi: 10.1073/pnas.1012311108
44. Sharma N, Pasala MS, Prakash A. Mitochondrial DNA: epigenetics and environment. Environ Mol Mutag. (2019) 60:668–82. doi: 10.1002/em.22319
45. Nordgren KKS, Hampton M, Wallace KB. Editor's highlight: the altered dna methylome of chronic doxorubicin exposure in sprague dawley rats. Toxicol Sci. (2017) 159:470–9. doi: 10.1093/toxsci/kfx150
46. Piotrowska I, Isalan M, Mielcarek M. Early transcriptional alteration of histone deacetylases in a murine model of doxorubicin-induced cardiomyopathy. PLoS ONE. (2017) 12:e0180571. doi: 10.1371/journal.pone.0180571
47. Hanf A, Oelze M, Manea A, Li H, Münzel T, Daiber A. The anti-cancer drug doxorubicin induces substantial epigenetic changes in cultured cardiomyocytes. Chemico-Biol Inter. (2019) 313:108834. doi: 10.1016/j.cbi.2019.108834
48. Song R, Yang Y, Lei H, Wang G, Huang Y, Xue W, et al. HDAC6 inhibition protects cardiomyocytes against doxorubicin-induced acute damage by improving alpha-tubulin acetylation. J Mol Cell Cardiol. (2018) 124:58–69. doi: 10.1016/j.yjmcc.2018.10.007
49. Pang B, Qiao X, Janssen L, Velds A, Groothuis T, Kerkhoven R, et al. Drug-induced histone eviction from open chromatin contributes to the chemotherapeutic effects of doxorubicin. Nat Commun. (2013) 4:1908. doi: 10.1038/ncomms2921
50. Al-Hasani K, Mathiyalagan P, El-Osta A. Epigenetics, cardiovascular disease, cellular reprogramming. J Mol Cell Cardiol. (2019) 128:129–33. doi: 10.1016/j.yjmcc.2019.01.019
51. Liu D, Ma Z, Xu L, Zhang X, Qiao S, Yuan J. PGC1alpha activation by pterostilbene ameliorates acute doxorubicin cardiotoxicity by reducing oxidative stress via enhancing AMPK and SIRT1 cascades. Aging. (2019) 11:10061–73. doi: 10.18632/aging.102418
52. Tarasenko N, Kessler-Icekson G, Boer P, Inbal A, Schlesinger H, Phillips DR, et al. The histone deacetylase inhibitor butyroyloxymethyl diethylphosphate (AN-7) protects normal cells against toxicity of anticancer agents while augmenting their anticancer activity. Invest New Drugs. (2012) 30:130–43. doi: 10.1007/s10637-010-9542-z
53. Karagiannis TC, Lin AJ, Ververis K, Chang L, Tang MM, Okabe J, et al. Trichostatin A accentuates doxorubicin-induced hypertrophy in cardiac myocytes. Aging. (2010) 2:659–68. doi: 10.18632/aging.100203
54. Ma J, Wang Y, Zheng D, Wei M, Xu H, Peng T. Rac1 signalling mediates doxorubicin-induced cardiotoxicity through both reactive oxygen species-dependent and -independent pathways. Cardiovasc Res. (2013) 97:77–87. doi: 10.1093/cvr/cvs309
55. Zhang S, Liu X, Bawa-Khalfe T, Lu LS, Lyu YL, Liu LF, et al. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat Med. (2012) 18:1639–42. doi: 10.1038/nm.2919
56. Dykxhoorn DM, Novina CD, Sharp PA. Killing the messenger: short RNAs that silence gene expression. Nat Rev Mol Cell Biol. (2003) 4:457–67. doi: 10.1038/nrm1129
57. Wan GX, Cheng L, Qin HL, Zhang YZ, Wang LY, Zhang YG. MiR-15b-5p is involved in doxorubicin-induced cardiotoxicity via inhibiting Bmpr1a signal in H9c2 cardiomyocyte. Cardiovasc Toxicol. (2019) 19:264–75. doi: 10.1007/s12012-018-9495-6
58. Du J, Hang P, Pan Y, Feng B, Zheng Y, Chen T, et al. Inhibition of miR-23a attenuates doxorubicin-induced mitochondria-dependent cardiomyocyte apoptosis by targeting the PGC-1alpha/Drp1 pathway. Toxicol Appl Pharmacol. (2019) 369:73–81. doi: 10.1016/j.taap.2019.02.016
59. Jing X, Yang J, Jiang L, Chen J, Wang H. MicroRNA-29b regulates the mitochondria-dependent apoptotic pathway by targeting bax in doxorubicin cardiotoxicity. Cell Physiol Biochem. (2018) 48:692–704. doi: 10.1159/000491896
60. Roca-Alonso L, Castellano L, Mills A, Dabrowska AF, Sikkel MB, Pellegrino L, et al. Myocardial MiR-30 downregulation triggered by doxorubicin drives alterations in beta-adrenergic signaling and enhances apoptosis. Cell Death Dis. (2015) 6:e1754. doi: 10.1038/cddis.2015.89
61. Piegari E, Russo R, Cappetta D, Esposito G, Urbanek K, Dell'Aversana C, et al. De angelis, microRNA-34a regulates doxorubicin-induced cardiotoxicity in rat. Oncotarget. (2016) 7:62312–26. doi: 10.18632/oncotarget.11468
62. Zhu JN, Fu YH, Hu ZQ, Li WY, Tang CM, Fei HW, et al. Activation of miR-34a-5p/Sirt1/p66shc pathway contributes to doxorubicin-induced cardiotoxicity. Sci Rep. (2017) 7:11879. doi: 10.1038/s41598-017-12192-y
63. Zhao L, Qi Y, Xu L, Tao X, Han X, Yin L, et al. MicroRNA-140-5p aggravates doxorubicin-induced cardiotoxicity by promoting myocardial oxidative stress via targeting Nrf2 and Sirt2. Redox Biol. (2018) 15:284–96. doi: 10.1016/j.redox.2017.12.013
64. Horie T, Ono K, Nishi H, Nagao K, Kinoshita M, Watanabe S, et al. Acute doxorubicin cardiotoxicity is associated with miR-146a-induced inhibition of the neuregulin-ErbB pathway. Cardiovasc Res. (2010) 87:656–64. doi: 10.1093/cvr/cvq148
65. Gupta SK, Garg A, Avramopoulos P, Engelhardt S, Streckfuss-Bomeke K, Batkai S, et al. miR-212/132 cluster modulation prevents doxorubicin-mediated atrophy and cardiotoxicity. Mol Ther. (2019) 27:17–28. doi: 10.1016/j.ymthe.2018.11.004
66. Wang JX, Zhang XJ, Feng C, Sun T, Wang K, Wang Y, et al. MicroRNA-532-3p regulates mitochondrial fission through targeting apoptosis repressor with caspase recruitment domain in doxorubicin cardiotoxicity. Cell Death Dis. (2015) 6:e1677. doi: 10.1038/cddis.2015.41
67. Li J, Li L, Li X, Wu S. Long noncoding RNA LINC00339 aggravates doxorubicin-induced cardiomyocyte apoptosis by targeting MiR-484. Biochem Biophys Res Commun. (2018) 503:3038–3043. doi: 10.1016/j.bbrc.2018.08.090
68. El-Bizri N, Wang L, Merklinger SL, Guignabert C, Desai T, Urashima T, et al. Smooth muscle protein 22alpha-mediated patchy deletion of Bmpr1a impairs cardiac contractility but protects against pulmonary vascular remodeling. Circ Res. (2008) 102:380–8. doi: 10.1161/CIRCRESAHA.107.161059
69. Wang KC, Chang HY. Molecular mechanisms of long noncoding RNAs. Mol Cell. (2011) 43:904–14. doi: 10.1016/j.molcel.2011.08.018
70. Hobuss L, Bar C, Thum T. Long Non-coding RNAs: At the Heart of Cardiac Dysfunction? Front Physiol. (2019) 10:30. doi: 10.3389/fphys.2019.00030
71. Zhou H, Wang B, Yang YX, Jia QJ, Zhang A, Qi ZW, et al. Long noncoding RNAs in pathological cardiac remodeling: a review of the update literature. Biomed Res Int. (2019) 2019:7159592. doi: 10.1155/2019/7159592
72. Salmena L, Poliseno L, Tay Y, Kats L, Pandolfi PP. A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language? Cell. (2011) 146:353–8. doi: 10.1016/j.cell.2011.07.014
73. Yuan JH, Yang F, Wang F, Ma JZ, Guo YJ, Tao QF, et al. A long noncoding RNA activated by TGF-beta promotes the invasion-metastasis cascade in hepatocellular carcinoma. Cancer Cell. (2014) 25:666–81. doi: 10.1016/j.ccr.2014.03.010
74. Tay Y, Rinn J, Pandolfi PP. The multilayered complexity of ceRNA crosstalk and competition. Nature. (2014) 505:344–52. doi: 10.1038/nature12986
75. Kalam K, Marwick TH. Role of cardioprotective therapy for prevention of cardiotoxicity with chemotherapy: a systematic review and meta-analysis. Eur J Cancer. (2013) 49:2900–9. doi: 10.1016/j.ejca.2013.04.030
76. Shaikh F, Dupuis LL, Alexander S, Gupta A, Mertens L, Nathan PC. Cardioprotection and second malignant neoplasms associated with dexrazoxane in children receiving anthracycline chemotherapy: a systematic review and meta-analysis. J Natl Cancer Inst. (2016) 108:djv357. doi: 10.1093/jnci/djv357
Keywords: cardiotoxicity, chemotherapy, epigenetics, cancer, doxorubicin
Citation: Kumari H, Huang W-H and Chan MWY (2020) Review on the Role of Epigenetic Modifications in Doxorubicin-Induced Cardiotoxicity. Front. Cardiovasc. Med. 7:56. doi: 10.3389/fcvm.2020.00056
Received: 11 December 2019; Accepted: 20 March 2020;
Published: 07 May 2020.
Edited by:
Carlo Gabriele Tocchetti, University of Naples Federico II, ItalyReviewed by:
Valentina Sala, University of Turin, ItalyEdoardo Lazzarini, University of Zurich, Switzerland
Copyright © 2020 Kumari, Huang and Chan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michael W. Y. Chan, Ymlvd3ljQGNjdS5lZHUudHc=