
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med. , 05 November 2019
Sec. General Cardiovascular Medicine
Volume 6 - 2019 | https://doi.org/10.3389/fcvm.2019.00158
This article is part of the Research Topic Cardiovascular Disease Modeling in Large Animals View all 7 articles
 Stefan Michael Sattler1,2*†
Stefan Michael Sattler1,2*† Lasse Skibsbye3†
Lasse Skibsbye3† Dominik Linz4,5
Dominik Linz4,5 Anniek Frederike Lubberding6Jacob Tfelt-Hansen1,7
Anniek Frederike Lubberding6Jacob Tfelt-Hansen1,7 Thomas Jespersen6
Thomas Jespersen6Ventricular arrhythmia and subsequent sudden cardiac death (SCD) due to acute myocardial infarction (AMI) is one of the most frequent causes of death in humans. Lethal ventricular arrhythmias like ventricular fibrillation (VF) prior to hospitalization have been reported to occur in more than 10% of all AMI cases and survival in these patients is poor. Identification of risk factors and mechanisms for VF following AMI as well as implementing new risk stratification models and therapeutic approaches is therefore an important step to reduce mortality in people with high cardiovascular risk. Studying spontaneous VF following AMI in humans is challenging as it often occurs unexpectedly in a low risk subgroup. Large animal models of AMI can help to bridge this knowledge gap and are utilized to investigate occurrence of arrhythmias, involved mechanisms and therapeutic options. Comparable anatomy and physiology allow for this translational approach. Through experimental focus, using state-of-the-art technologies, including refined electrical mapping equipment and novel pharmacological investigations, valuable insights into arrhythmia mechanisms and possible interventions for arrhythmia-induced SCD during the early phase of AMI are now beginning to emerge. This review describes large experimental animal models of AMI with focus on first AMI-associated ventricular arrhythmias. In this context, epidemiology of first AMI, arrhythmogenic mechanisms and various potential therapeutic pharmacological targets will be discussed.
Despite a decrease in overall cardiovascular mortality over the past decades, ~17 million deaths a year occur worldwide as a result of cardiovascular disease (1) and ~50% of these are reported to be sudden cardiac deaths (SCD) (2). In the elderly, constituting the major age group at risk, SCD is often associated with chronic degenerative diseases, such as coronary artery disease (with acute myocardial infarction (AMI) as its ultimate consequence), valvular diseases, and heart failure.
In the early stages of AMI ventricular fibrillation (VF) is a frequent cause of SCD, which often occurs within minutes after onset of clinical symptoms, often even before the patient has established contact to medical health care systems. As out of hospital VF following AMI is sudden and unexpected, only sparse information concerning risk factors and protective mechanisms are available from human studies.
Hence, to understand the mechanisms of ventricular arrhythmias developing during AMI and to discover new treatment modalities for these patients, investigations through translational experimental models, mimicking the clinical situation, are needed. While numerous experimental models of myocardial infarction exist, most of them focus on mechanisms and on options to prevent manifestation and expansion of infarction and scar formation in order to improve tissue survival in post-AMI patients. Only a relatively limited number of studies focus on the acute arrhythmogenic risk of AMI. This in spite of the fact, that there is a critical need for fast acting acute antiarrhythmic treatments in AMI patients. Treatment that is safe to use on site or in an ambulance without severe negative hemodynamic or proarrhythmic adverse effects. Hence, there is an unmet medical need for such therapeutic options, which calls for investigations of novel pharmacological targets in clinical and preclinical antiarrhythmic research.
This review will present the published literature on experimental large animal models of AMI with focus on translatability to the epidemiology of first AMI-associated ventricular arrhythmias. Various therapeutic pharmacological targets will be discussed in this context. Along with this, a brief overview of the involved arrhythmogenic mechanisms leading to VF during AMI is given.
Ischemia-related VF is by far the leading cause of sudden cardiac arrest and the most frequent occurring arrhythmia in SCD (2–5). In AMI, occlusion of a coronary artery induces an imbalance between oxygen supply and demand leading to ischemia-induced cell death of cardiac myocytes. Transmural AMI, seen as ST-elevation myocardial infarction (STEMI) in the electrocardiogram (ECG) is present in 25–40% of all AMI cases (6). AMI rapidly changes electrophysiological properties of the ventricular myocardium and, in a significant proportion of patients, promote electrical disturbances in conduction and repolarization, leading to ventricular tachycardia (VT) or VF and subsequently cardiac arrest (Figure 1) (6). Due to development of myocardial scar tissue and heart failure, ventricular arrhythmias and subsequent SCD can still occur after transition from acute to chronic myocardial infarction.
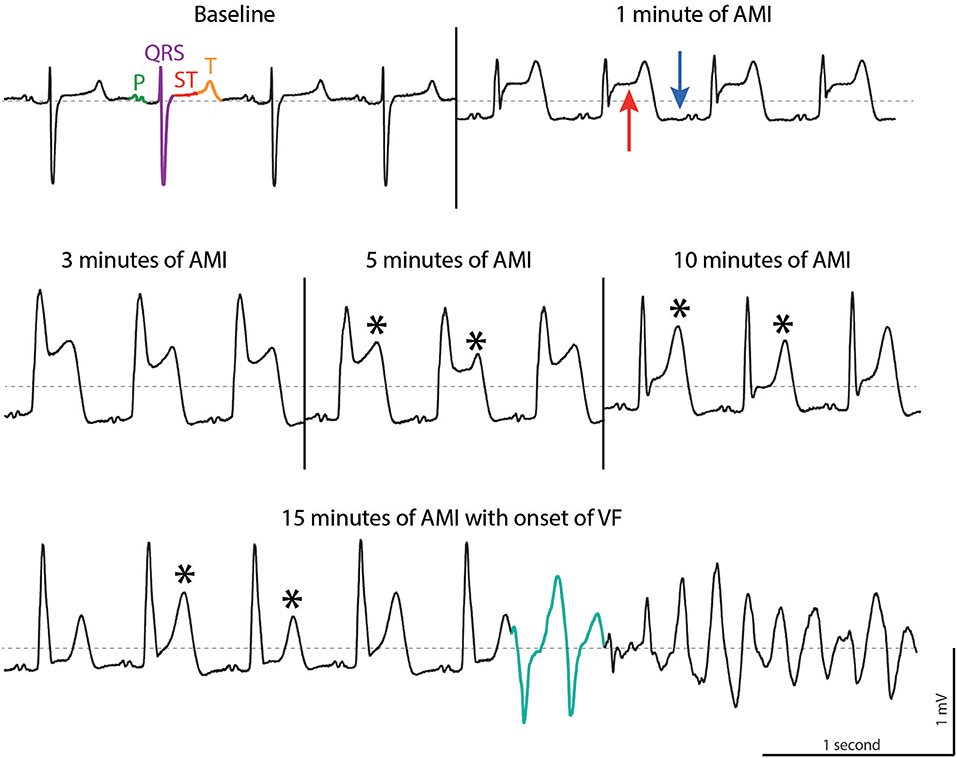
Figure 1. ECG tracings during acute myocardial infarction (AMI) in a porcine model. At baseline, sinus rhythm is present with P wave (green), QRS complex (purple), ST segment (red) and T wave (orange). AMI leads to elevation of the ST segment (red arrow), depression of the TQ segment (blue arrow) followed by T wave alternans (asterisk, note the alternating morphologies). In this example, ventricular fibrillation is triggered by two short coupled ectopic beats (turquoise). Dotted line, 0 mV. Reproduced with permission from Sattler (7).
By nature, it is difficult to establish the cause of death to be VF caused by AMI when it happens out of hospital. The numbers in the following studies likely underestimate the true burden of VF during first AMI because individuals experiencing SCD are not reported. Many patients with ischemia-induced VF will be found dead, or, after being resuscitated, die or survive with or without neurological sequelae (8, 9). Even if medical contact is established, only 4.3–19.4% of the patients experiencing out-of-hospital cardiac arrest (OHCA) survive the first 30 days (8). In patients with STEMI, 30-days survival decreases from 94% in patients without VF to 79% in those with VF (10). In our own Danish nationwide prospective case-control study, the GEVAMI (GEnetic causes of Ventricular Arrhythmias in patients with first ST-elevation Myocardial Infarction) study, we enrolled 660 consecutive STEMI patients and found 11.6% to develop VF before primary PCI (11). The French e-MUST (Evaluation en Médecine d'Urgence des Stratégies Thérapeutiques des infarctus du myocarde) study reported that 5.6% of patients with STEMI experienced VF witnessed by emergency medical service (EMS) on site. Of these patients, 9.1% died out-of-hospital and a further 29% in-hospital (12).
The assessment of risk factors of VF due to AMI is non-trivial: Studies must be designed to distinguish between risk factors for AMI without VF and those for AMI with VF. Further, patients suffering from SCD before AMI is diagnosed are often not included in such studies. Finally, these events occur in low risk subgroups without any known risk factors, like structural heart disease (4). Possible risk factors for VF can either be present before AMI takes place or can be directly associated with specific factors of the AMI itself. The former are determined by heritability, comorbidity, medication, lifestyle, and gender, while the latter, mostly observed as changes in the ECG, includes infarct size and location of coronary obstruction, as well as individual ventricular and coronary anatomy (11, 13).
The Dutch AGNES (Arrhythmia Genetics in the Netherlands) study was the first designed to differentiate between risk factors for AMI in general and AMI complicated by VF (13). This study compared first AMI patients who developed VF with AMI patients without VF. Those with VF had a family history of SCD, a lower body mass index, and a lower prevalence of hypercholesterolemia and diabetes compared to controls.
The GEVAMI study compared first STEMI with and without VF and was able to identify several independent risk factors for VF including age below 60 years, family history of SCD, absence of pre-infarction angina, use of statins, history of atrial fibrillation (AF) and alcohol intake (> 7 units per week) (11). Whereas, alcohol has favorable effects on the development of thrombosis and atherosclerosis and lowers the risk of AMI (14), a u-shaped relationship exists in SCD. A moderate consumption of alcohol (2–6 units a week) reduces the risk for SCD (15, 16), while higher alcohol intake increases the risk (17).
Several studies support the theory that a genetic predisposition to SCD and VF exists in the setting of AMI (13, 18). A family history of SCD among siblings or parents increased the risk of VF (odds ratio of 1.6–3.2) (11, 13, 19). However, little is known about the specific genetic factors affecting VF during AMI. A genome wide association study (GWAS) in the AGNES study cohort revealed a strong association between VF during AMI and a gene locus near the CXADR gene, that encodes for the coxsackie- and adenovirus receptor (20). Smaller studies point at genetic variations in the SCN5A gene, coding for the cardiac sodium channel Nav1.5, as a predisposing factor for the development of VF upon AMI (21). Epidemiology and genetics of VF in AMI have been reviewed recently by Glinge et al. (22).
Another important confounder to VF in AMI is AF (11, 13). Data from the FAST-MI (French ST-elevation and non-ST-elevation Myocardial Infarction) 2005 registry point at AF observed on the initial ECG obtained during STEMI to be an independent risk factor for early VF (odds ratio 2.5) (23). Increased heart rate in AF can shorten ventricular refractoriness creating a substrate for VF (24). Additionally, this proarrhythmic substrate can be aggravated by hemodynamic changes that can decrease parasympathetic and increase sympathetic tone in AF (25).
Properties of AMI itself including infarct size and location can alter the incidence of VF. Electrocardiograms obtained in the early phase of AMI are therefore not only important for diagnosing AMI, but also in early risk stratification. Patients presenting with high ST elevation score (11, 26), short RR intervals (27) or prolonged PR and QRS intervals (26) in their ECGs carry a higher risk of VF. Additionally, patients with ST-segment elevation accompanied with distortion of the terminal QRS complex had increased ischemia burden, faster progression of necrosis (28), and a higher risk of VF (26).
Incidence of VF is also influenced by the affected artery or arteries. A Belgian case-control study, investigating cardiac arrest, found an increased risk of out-of-hospital VF in patients with acute occlusion on the left coronary artery (left anterior descending (LAD) or left circumflex (LCx) artery) compared to the right coronary artery. However, the exact occlusion site (proximal or distal part) within the left coronary artery did not affect VF burden (29). Similar results were found in the Dutch AGNES study (13). This is somehow conflicting as proximal occlusion sites result in larger areas of myocardium at risk and have been shown to be a risk factor in animal models (30, 31). Reasons for these discrepancies could be manifold including time to reopening of the occluded vessel, residual flow in the coronary artery, or the limited number of patients.
AMI causes a variety of electrophysiological changes generating triggering factors and a proarrhythmic substrate that can induce and sustain VF. In this section we give a brief introduction to important concepts on cardiac electrophysiology and arrhythmogenesis during ischemia. For a more in depth review we refer to the reviews by Janse and Wit (32) and Carmeliet (33).
The cardiac action potential (AP) is formed following fine-tuned temporal and spatial openings of depolarizing sodium (Na+) and calcium (Ca2+) and repolarizing potassium (K+) channels (Figure 2, left) (34). AMI alters the metabolic state (mainly via acidosis and ATP depletion) and causes electrical changes in both the ischemic area and the border zone between the ischemic and non-ischemic area. Numerous factors have been proposed to support the generation of arrhythmias: The resting membrane potential is depolarized, conduction velocity in the ventricles slows down, and AP duration shortens by alterations in the K+ and Cl− currents during AMI (Figure 2, right) (32, 35).
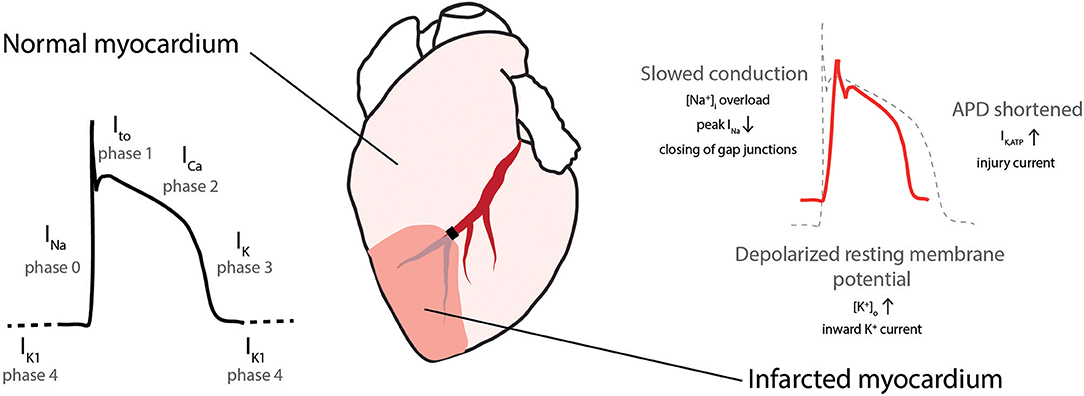
Figure 2. Cardiac action potential in normal and ischemic myocardium. (Left) Action potential with depolarizing sodium (INa), transient outward potassium (Ito), calcium (ICa), and repolarizing potassium (IK) currents. (Right) During ischemia conduction is slowed, the resting membrane potential depolarized and action potential duration (APD) is shortened. [Na+]i, intracellular sodium concentration, [K+]o extracellular potassium concentration, IK,ATP, ATP-sensitive potassium current. Modified with permission after Sattler (7).
During ischemia three underlying mechanisms, automaticity, triggered activity, and reentry can be involved in inducing or sustaining arrhythmias (premature beats, VT and VF; Figure 3) (33). Automaticity is caused by myocardial stretch, or short-circuit and injury currents in the border zone leading to spontaneous depolarizations outside the sinus node. Triggered activity is caused by Ca2+ overload in Purkinje fibers or cardiomyocytes. The Purkinje system is thought to play a role in the transition of a stable reentrant VT into VF by rapid and irregular conduction of reentrant waves into non-ischemic tissue (36). Finally, reentry is caused by wave fronts following a pathway around an area of unidirectional block and reexcitement of previously activated cardiomyocytes instead of dying out. Reentry occurs in the presence of a functional disturbance in impulse conduction and heterogeneity in conduction as present during AMI (32).
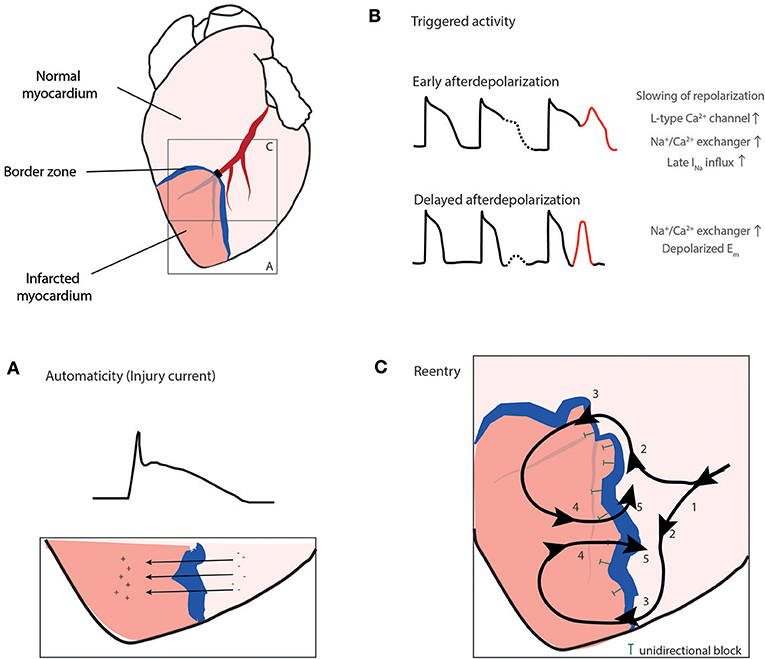
Figure 3. The three mechanisms, automaticity (A), triggered activity (B), and reentry (C) can play a role in arrhythmogenesis during ischemia. (A) Injury current across the border zone leading to ST elevation in the electrocardiogram, (B) Triggered activity mainly caused by Ca2+ overload in cardiomyocytes or Purkinje fibers. (C) Reentry. Electrical activation wave front (1) is deflected at the border zone due to unidirectional block (T) into two wave fronts (2), eventually passing the border zone (3) and exciting the infarct zone (4) and finally passing the unidirectional block re-exciting the area in front of the block (5). Ito, transient outward potassium current; [K+]o, extracellular potassium concentration; [Na+]i, intracellular sodium concentration. Reproduced with permission from Sattler (7).
Early studies on VF used rather simplified concepts to explain VF initiation, including a single stable reentry circuit traveling along an anatomical obstacle or the leading circle theory in which a wave front travels around a functional obstacle that is constantly kept depolarized (37). Rotors, a form of functional reentry where the curved wave front and wave tail meet each other in an excitable singularity, have emerged as an explanatory theory for VF in the last decades: A propagating wave front with a curvature R (source) travels along an obstacle (for example an unidirectional block) and is about to enter excitable but non-excited tissue (sink). The curvature R is dependent on electrophysiological properties like conduction velocity. At the edge of the obstacle, the curvature R determines whether the wave will detach from the obstacle or follow it. If R is smaller than the critical radius of the obstacle, the wave will eventually detach, curl and rotate around its broken tip (37). In VF initiation and maintenance, a small number of driving rotors (mother rotor) and multiple wavelets have been detected in humans with VF and are thought to be important (38).
Autonomic imbalance, characterized by increased sympathetic activation and parasympathetic withdrawal, may also contribute to the early manifestation of ventricular arrhythmia in the acute phase of AMI as well as following AMI (39, 40).
Changes in ionic currents and the development of an arrhythmic substrate during early AMI have a defined time course and ventricular arrhythmias (premature beats, VT and VF) occur in two distinct phases, 1a and 1b (32, 41).
Phase 1a takes place between 2 and 10 min after AMI and is induced by focal activation due to injury currents in a substrate of profound slowing in conduction velocity and delayed activation due to decreased resting membrane potential (41). Reentry is the predominant mechanism during this phase, but triggered activity can also be involved in the generation of premature beats (32, 42).
Phase 1b occurs 18–30 min after coronary occlusion. Arrhythmias are likely related to mechanical stretch in the border zone, increased catecholamine levels and cellular uncoupling (43–45).
Overall incidence of arrhythmias is higher during phase 1a while VF occurs more often during phase 1b (46). Data in humans on the importance of phase 1a and 1b on SCD are scarce, since ECG recordings in patients are rare, while animal models suggest larger mortality in phase 1b than in phase 1a (47–49).
Animal models are essential in understanding the mechanisms of identified risk factors on arrhythmogenesis and subsequent SCD. Mimicking the exact pathophysiological conditions in experimental models translatable to AMI in humans can be challenging. Ideally the experimentally-induced AMI results in the same pathophysiological changes that would be expected in a diseased human, however, experiments in smaller animals have limitations in translatability to humans. This is mainly due to differences in the dimensions of the ventricles, the heart and the cardiovascular system as a whole, the morphology and duration of the AP, as well as differences in hemodynamic and cardiac electrophysiological responses to ventricular ischemia (50). In contrast, large animal models provide a better opportunity to study the pathophysiology and pathogenesis of AMI and associated arrhythmias (32) and allow the use of established invasive techniques also applied in the clinic, such as coronary angiograms, multisite electrogram recordings, stimulation protocols, electrode arrays, or whole electrode mapping-socks covering the entire heart (51, 52).
Besides the applied species, the occurrence of studied ventricular arrhythmias depends on a number of factors, including anesthetic regimen, mode of coronary occlusion, occlusion site and size of the ischemic area, coronary collaterals, and the regulation of the autonomic nervous system.
The vast majority of animal models developed for understanding AMI-induced ventricular arrhythmias have concerned fully anesthetized animals, although, notable differences can be observed between conscious and anesthetized animals (53). Numerous different anesthetic drug-regimens have been used, as reviewed by Hamlin (54). Anesthetics have different abilities to sustain an adequate anesthesia but often come with unwanted effects on susceptibility of ventricular arrhythmias, sympathetic and parasympathetic regulation, and on the cardiovascular system itself.
Briefly, many studies investigating arrhythmias in animals have used barbiturates. These drugs have strong parasympatholytic effects, reducing transmural dispersion of ventricular repolarization (55), leading to a less frequent occurrence of ventricular arrhythmias (56, 57). A comparison of the volatile anesthetics isoflurane and sevoflurane in pigs revealed a higher VF and mortality rate in the isoflurane group (58). Propofol is an anesthetic drug acting mainly on the gamma-aminobutyric acid (GABA) receptor and is widely used nowadays as it is easy to dose. It comes with notable effects on hemodynamics, including reduced cardiac output, venous return, systemic pressure, and vascular resistance (59). In studies that investigate autonomic regulation of the cardiovascular system and effects of cardiovascular drugs the use of morphine and alpha chloralose (60, 61) or urethane and alpha chloralose (62) might be considered. The use of morphine and alpha chloralose has lower effects on the cardiovascular- and autonomic nervous system compared to other regimens and create a state comparable to sleep (63). Thus, when the focus is placed on assessing risk predictors and effects of antiarrhythmic treatments, the effects of the used anesthetic regimen must be taken into account.
A response similar to AMI caused by atherosclerosis or thromboembolic events in otherwise healthy hearts can be provoked by occluding a coronary artery in an experimental animal. This can be performed surgically by coronary vessel ligation in an open chest configuration (Figure 4A) or, less invasively, by catheter based techniques in closed-chest models including percutaneous balloon occlusion (Figure 4B) or microsphere injection (65, 66). Surgical ligation and balloon occlusion allow for ischemia/reperfusion models and correspond to transmural myocardial infarction seen as STEMI in patients (67). Microspheres induce local clotting in the heart microcirculation and lead to subsequent chronic myocardial ischemia (66). These techniques model AMI, the ultimate consequence of coronary artery disease. Less distinctive effects of coronary artery disease preceding AMI, including marginal effects of heart failure with chronic remodeling of autonomic innervation, inflammatory activation with its direct effect on the arrhythmic substrate (68), as well as development of collaterals are often missed.
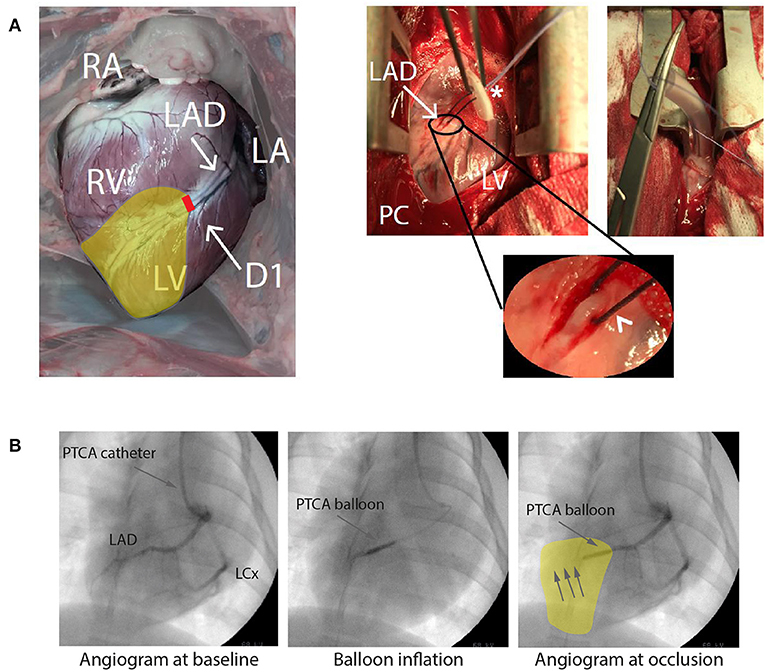
Figure 4. (A) Open chest approach. Visualization of a heart from a Danish Landrace pig after mid-thoracotomy in anterior-posterior view. Right (RA) and left atrium (LA), right (RV), and left ventricle (LV) as well as the left anterior descending artery (LAD) with its diagonal branch (D1) are visible. Pericardium (PC) is opened, allowing the dissection of the LAD, a silk snare with a counter bearing (asterisk) is placed around the artery (arrowhead, round magnification) and tightened for coronary occlusion. For more stable hemodynamic conditions the thorax is closed to a minimum. (B) Closed chest approach presenting an angiogram of a pig heart of the left coronary artery with LAD and left circumflex artery (LCx) in anterior-posterior view. Anatomical details of the coronary arteries can be identified. Placing a percutaneous transluminal coronary angioplasty (PTCA) balloon and inflating this leads to an occlusion downstream the LAD, here shown for a mid-LAD occlusion. RA, Right atrium; LA, Left atrium. Yellow area indicates area at risk during ligation/occlusion. Modified with permission after Sattler (7) and Sattler et al. (64).
The development of canine and porcine infarct models started with two-staged open chest models. A coronary artery was occluded to 50% of its diameter and occlusion was kept for 30 min before the artery was occluded completely and transmural AMI was induced (65, 69). These two-staged experiments often showed decreased arrhythmias and infarct size, most likely due to myocardial preconditioning (70). Two-staged models can be used to mimic the correlate of pre-infarction angina. Schwartz et al. moved from modeling the “healthy” human heart with first AMI toward acute-on-chronic myocardial infarction. In a first procedure, myocardial infarction by occluding the LAD was induced. Over a 2- to 4-week period, the animals developed anatomical and structural substrates, similar to those observed in clinical chronic artery disease, due to the chronic infarction in the LAD area. In a second procedure AMI was induced by LCx occlusion and the arrhythmic effects could be investigated (71).
While open chest models have been crucial in understanding AMI-induced arrhythmias, they come with several limitations: thoracotomy and subsequent opening of the pericardium influences thoracic pressure, cardiac filling and blood pressure and the surgery itself can lead to global ischemic preconditioning. In contrast, when percutaneous angioplasty is performed, the thorax and pericardium can be left intact, allowing for more stable hemodynamic conditions (72, 73). Further, anatomical limitations in choice of coronary occlusion site, such as vessels on the posterior side of the heart or intramural vessels, can be overcome.
Infarct location and occlusion sites within the coronary arteries can alter the incidence of ectopic beats and ventricular arrhythmias. Variation of occlusion site in the LAD in a porcine model (30) and anatomical variations of the LCx in an ovine model resulted in varying infarcted areas and different incidences of VF (31).
Since infarct size is dependent on the individual anatomical variation of coronary vessels and on the presence of collaterals, an important aspect when aiming for reproducible infarct sizes is the presence and degree of collateral flow. Humans have a well-developed network of coronary collaterals (74) that can be recruited rapidly during acute ischemia when direct flow is interrupted (75). Although only 20 to 25% of humans with healthy coronary arteries have sufficient collateral flow to prevent signs of myocardial ischemia during short vascular occlusions (76), those with coronary artery disease or prior myocardial infarction have a collateral flow that can be sufficient to prevent myocardial ischemia during coronary occlusion (77). In both humans and pigs it has been found that the degree of coronary flow reduction has direct influence on ventricular electrical and mechanical parameters (74, 78). Among animal models there is a large diversity in collateral perfusion: Non-diseased pigs, rabbits, and ferrets have low collateral flow. Dogs, cats, and guinea pigs, on the contrary, have a high collateral flow (75, 79).
Differences between human and animal exist in evolution of myocardial infarction within the myocardium at risk. Humans experiencing AMI show significantly slower infarct progression than animals (pigs, rats and dogs) with induced ischemia (80).
Several different large animal models have been used over the last few decades in the setting of AMI and VF, during which some have become more popular than others. A PubMed search in May 2019 on studies conducted in dogs, pigs, sheep or primates found 448, 77, 9, and 7 publications, respectively [MeSH search terms used: “myocardial infarction” AND “ventricular fibrillation” AND (“dogs” OR “swine” OR “sheep” OR “primates NOT hominidae”)].
Dog models have provided deep insights into the complex electrophysiological processes and arrhythmias during AMI (32). The cardiac electrophysiology of the dog quite closely resembles that of man. Its anatomy and distribution of Purkinje fibers is similar to humans, providing a similar sequence of cardiac activation and ventricular depolarization, as can be observed in the ECG (54). However, ventricular repolarization shows marked differences in dogs, with much greater variability and inhomogeneity of repolarization across the ventricular wall (55, 81). The dog heart has been found to possess a rich collateral circulation (82), in contrast to pigs and humans, with the exception of patients with coronary artery disease. Recently, the use of dogs has been restricted by animal welfare laws, accompanied by rising expenses and the publications including dogs in the field have consequently declined markedly.
Pigs are widely used since their cardiovascular system in general, and the anatomy of the heart in particular, resembles that of the human. Despite the similarities, Landrace pigs with similar size to humans are only adolescents; full-grown adults weigh several 100 kg. In contrast to Landrace pigs, minipigs show slower growth rates accounting for a more stable heart/body weight ratio (83) and providing a more cost-effective option, especially in long term experiments (84). Cardiac similarity of pig to man is evident in many aspects, such as structure, electrical properties, and metabolic response to AMI (85). Since collateral flow is largely absent in pigs, a close modeling of AMI with a more controlled and solid infarct zone is possible (75). There are differences in electrical properties, Purkinje fibers and propagation speed of ventricular excitation (86). Pigs have a higher number of Purkinje fibers, spreading almost completely from endo- to epicardium, which is different to the predominantly endocardial presence in humans. Purkinje fibers have a 5–10 times higher conduction velocity compared to humans, leading to narrower QRS complexes when compared to humans (87). This results in a different orientation in excitation of the ventricles (87), which may impact the onset and maintenance of ventricular arrhythmias, especially during later phases of VF (88). In addition, when compared to humans, pigs show higher VF thresholds upon electrical stimulation (89).
Hearts of sheep have fairly similar anatomical characteristics and physiological function as those of man. Their coronary circulation, as well as the anatomy of their conduction system is comparable to that of humans making sheep an interesting model for human AMI (90–92). However, only a few studies addressing AMI have been performed on sheep.
Whereas some earlier studies tried to exploit the close genetic similarity of non-human primates to man by using rhesus monkeys and baboons in AMI research (93, 94), today the use of monkeys is very limited. This is mostly due to logistical, ethical and monetary difficulties. LAD occlusion in unanaesthetized rhesus monkeys provoked cardiovascular response similar to man. VF was seen in 3/9 individuals (94). Additionally, despite close anatomical resemblance to humans, the hearts of these monkey species are small and have much faster sinus rhythm than man, making them less favorable in respect to electrophysiological resemblance (54).
Despite survival rate being markedly decreased in AMI patients experiencing VF before or during primary PCI (8, 10) the prophylactic use of antiarrhythmic drugs is not recommended in the latest ACC/AHA guideline on STEMI therapy from 2013 in lack of effective therapy options (95). Only initiation of beta blocker therapy within 24 h after AMI onset is recommended as it has beneficial effects on VT/VF occurrence (96). However, AMI leads to a variety of pro-arrhythmic changes that can potentially pose pharmacological targets.
In the following section, amiodarone, save and effective in arrhythmia protection in the setting of AMI, and new drugs or experimental compounds in the setting of AMI induced VF that have been tested in large animal models or humans are reviewed. Drugs that have been proven harmful to human patients, like the class Ic antiarrhythmic drugs encainide and flecainide (97) or the class III drug d-sotalol (98) are not discussed here. An overview is given in Table 1. It is noteworthy that nearly all tested drugs were applied either before AMI was induced or during VF, while almost no experiment or clinical studies tested drug effects immediately after AMI but before coronary blood flow could be re-established. The temporal relationship between drug and ischemia can be crucial when aiming for translatability as the drug in human patients will not at all, or in lower concentrations, be present in the infarcted myocardium. The importance of this temporal relationship was shown for the class 1b agent aprindine were pre-treatment with the drug resulted in higher rates of VF during AMI than treatment applied after onset of AMI (117).
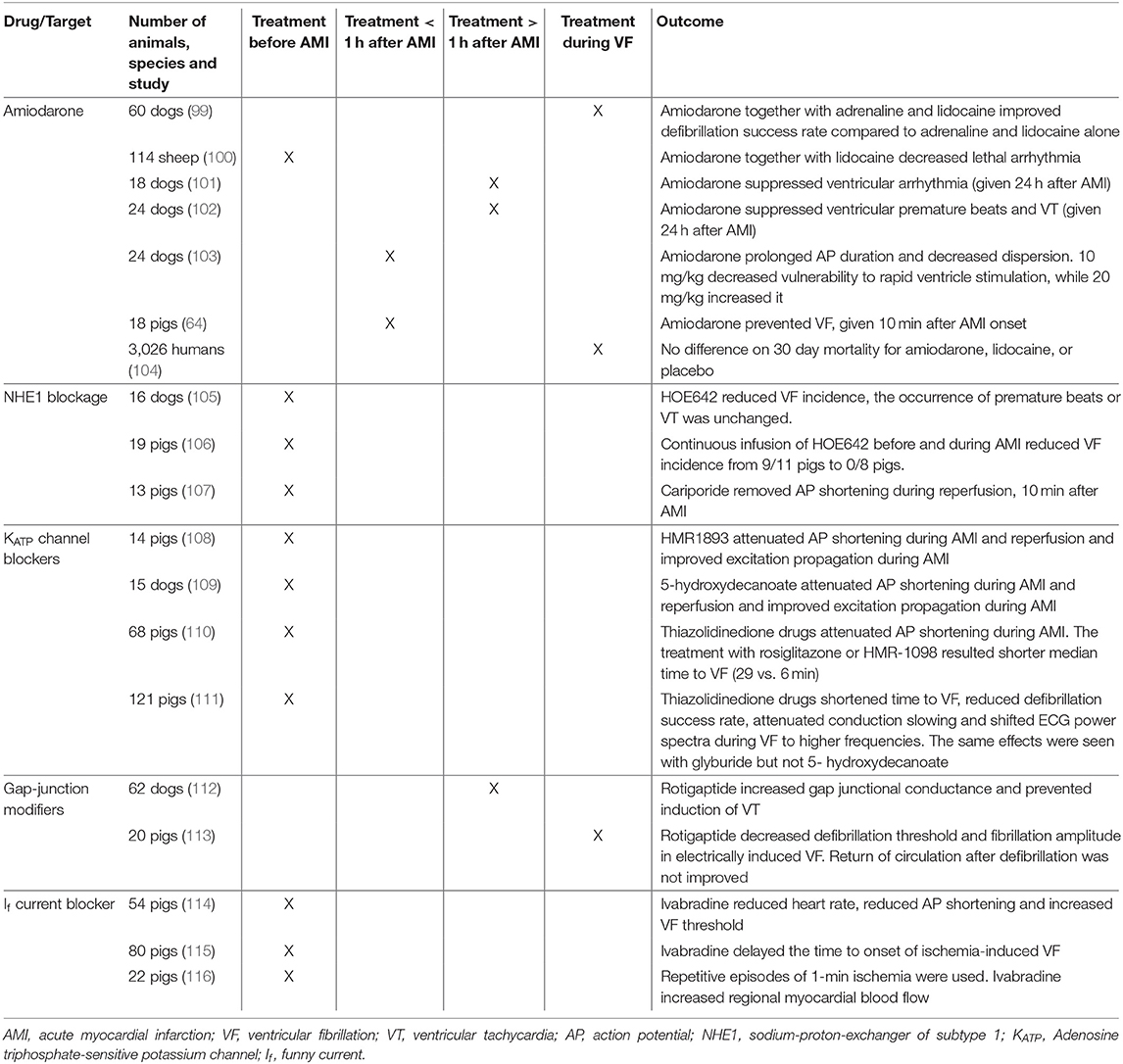
Table 1. Studies conducted on pharmacological targets in first acute myocardial infarction to prevent ventricular fibrillation including species and temporal relationship between drug treatment and ischemia.
Amiodarone is believed to be one of the most effective antiarrhythmic drugs for the treatment of a variety of different arrhythmias. Effects of amiodarone differ between acute and chronic treatment. In acute treatment the drug inhibits inward Na+ and Ca2+ currents and outward K+ currents (IKr, IKs, IK,Ach, and IK,Na) (118) and has non-competitive beta blocking effects (119). Acute intravenous applications mainly lead to slowing in atrioventricular conduction and have only mild effects on the effective refractory period and little effect on QTc intervals, whereas chronic treatment slows conduction in almost all cardiac tissues and prolongs QTc interval (118).
Treatment with amiodarone has been tested in a variety of animal models before and after AMI, as well as during VF. Overall, animal models showed reduced ventricular arrhythmia (100–102), decreased dispersion (103) and improved defibrillation success rate (99). We showed reduced VF incidence rates with amiodarone when given 10 min after AMI onset (64). In humans, little evidence is available for the prophylactic use of amiodarone in the acute setting of AMI since randomized trials mainly looked at unselected OHCA cohorts (120, 121). As reported in a multicenter study on 3,026 patients, survival in OHCA patients was not improved by the use of amiodarone or lidocaine (a Na+ channel blocker) when administered during VF (104). Yet data from a retrospective analysis focusing on VT and VF during AMI suggest a positive effect of treatment with amiodarone or lidocaine on survival (122). No randomized trials in humans, investigating prophylactic treatment with amiodarone after onset of AMI to prevent VT/VF in the early phase, exist.
The Na+/H+ exchanger of subtype 1 (NHE1) is an important proton-extruding mechanism in cells including cardiomyocytes (123). During AMI, intracellular pH decreases due to anaerobic metabolism, leading to an activation of NHE1 (124). Exchange of the excessive proton load for Na+ causes intracellular Na+ accumulation that could indirectly affect the activity of other ion transporters, particularly the NCX, which may lead to ventricular arrhythmogenicity.
Pharmacological NHE1 inhibition in AMI animal models has only assessed pre-treatment (105–107). These reports showed reduced VF incidence (105, 106) without changes in other ventricular arrhythmias (105), and eliminated reperfusion induced AP shortening (107). In humans, NHE1 inhibition during AMI has not been assessed.
The ATP-sensitive K+ (KATP) channels generate an inwardly rectifying outward current that is activated when the intracellular ATP concentration is low, such as during AMI. KATP activation during myocardial ischemia is partly responsible for the abbreviation of the ventricular AP and protects the heart from damage during ischemia by preventing Ca2+ build-up (125). KATP channels may represent an intriguing inhibitory target for antiarrhythmic therapy by reducing AMI-induced AP shortening. In several large animal studies, inhibition of KATP suppressed AMI-induced AP shortening (108–110) and improved excitation propagation (108, 109), however, time to VF was shortened (110, 111). This suggests that the protective effect of preventing intracellular Ca2+ build-up outweighs any possible anti-arrhythmic effect of reducing AP shortening.
Cardiac gap-junctions, with connexin 43 (Cx43) as their main isoform, provide communication and connections between cells, allow the exchange of small molecules, and play an important role in the propagation of electrical excitation in the heart (126). Gap junction inhibition during AMI reduces conduction velocity and increases defibrillation thresholds (127). In contrast, gap junction modifiers, such as rotigaptide, increase conduction velocity by increasing functional coupling of gap junctions at the intercalated disc (128). Rotigaptide prevented programmed stimulation-induced reentrant VT during MI (112) and decreased defibrillation threshold during prolonged VF (113). This, however, did not translate to improved rates of return of spontaneous circulation (113). Mitochondrial Cx43 is involved in the energy metabolism in the heart (129) and Ca2+ handling (130). Affecting these processes could produce unwanted side effects.
Sinus node tissue shows spontaneous depolarization by a complex interaction of multiple inwardly and outwardly directed ion currents, including the funny current (If). This current is carried primarily by the hyperpolarization-activated cyclic nucleotide-gated (HCN4) channels primarily conducting Na+. In the setting of AMI, extra-nodal If in the left ventricle can be increased leading to proarrhythmic tendencies (131).
Ivabradine selectively blocks the If current in a use-dependent manner, resulting in reduced heart rate without negatively affecting hemodynamics (132). Ivabradine pre-treatment in AMI reduced heart rate, increased VF threshold and prevented AP shortening in the ischemic area without changing myocardial contractility (114). Ivabradine also delayed the time to ischemia-induced VF by preserving myocardial metabolic energy status (115) and lowering heart rate (116).
Besides pharmacological interventions on ion channels, techniques to modulate autonomic dysfunction in AMI by increasing vagal activity and reducing sympathetic hyperactivation are arising (133). The different techniques have been reviewed recently by Lai et al. (134). Briefly, vagal activity can for example be increased by the tragus nerve, carotid baroreceptor and vagus nerve stimulation, sympathetic activity can for example be decreased by spinal cord stimulation, stellate ganglion denervation or neuromodulation and renal sympathetic denervation.
Modulation of the sympathetic nervous system by renal denervation reduces ventricular ectopy and VF in a porcine model of AMI. During reperfusion AP shortening and spontaneous arrhythmias were not affected by this modulation of the autonomic nervous system (135). Direct stimulation of cervical pre-ganglionic parasympathetic fibers activates overall cardiac vagal tone (136). Vagal stimulation, performed shortly after the onset of an acute ischemic episode in conscious animals with a healed MI, can effectively prevent VF independent of heart rate reduction (137). Spinal cord stimulation also has an antiarrhythmic effect on spontaneous non-sustained VT and sustained VT during ischemia-reperfusion in association with a reduction of repolarization alterations independent of any effect on infarct size (138).
AMI complicated by VF is one of the most common causes of SCD while treatment options still remain limited. Even if SCD is averted, VF in AMI patients is associated with decreased survival rates. Several risk factors like sex, family history, life style factors and alcohol consumption are known from epidemiological studies. As studying AMI-induced VF is difficult in humans, large animal models provide an important translational approach to study AMI complicated by VF.
A variety of large animal models resembles human cardiac physiology and can be used to investigate mechanisms of risk factors or new treatment modalities. There are several important factors to take into account when studying spontaneous VF during AMI: the animal with its specific cardiac anatomy and electrophysiology; the anesthesia and its effect on heart rate and autonomic regulation; and the method, size and location of coronary occlusion and the resulting ischemic area.
Although a number of promising pharmacological treatment regimens such as KATP blockers, gap-junction modifiers, If channel blockers, and NHE1 blockers have been investigated in large animal models, the applied experimental design is often insufficient to be successfully translated into clinical trials. Many of the experiments evaluating new antiarrhythmic drugs are performed pre-treatment before AMI onset, thereby results in drug accumulation also in the infarcted area. This can both lead to electrical and metabolic stabilization or destabilization of the infarcted area. For a successful translation into clinical studies it is of importance to adjust the experimental design according to human situation.
SS and LS: concept, design, drafting article, critical revision of article, and approval of article. DL, AL, JT-H, and TJ: drafting article, critical revision of article, and approval of article. AL: critical revision of article and approval of article.
This work was supported by Novo Nordisk Foundation Synergy program (to JT-H and TJ).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Mendis S, Puska P, Norrving B. Global Atlas on Cardiovascular Disease Prevention and Control. Geneva: World Health Organization (2011). p. 2–13.
2. Deo R, Albert CM. Epidemiology and genetics of sudden cardiac death. Circulation. (2012) 125:620–37. doi: 10.1161/CIRCULATIONAHA.111.023838
3. Zipes DP, Wellens HJJ. Sudden cardiac death. Circulation. (1998) 98:2334–51. doi: 10.1161/01.CIR.98.21.2334
4. Huikuri HV, Castellanos A, Myerburg RJ. Sudden death due to cardiac arrhythmias. N Engl J Med. (2001) 345:1473–82. doi: 10.1056/NEJMra000650
5. de Vreede-Swagemakers JJ, Gorgels AP, Dubois-Arbouw WI, van Ree JW, Daemen MJA, Houben LG, et al. Out-of-hospital cardiac arrest in the 1990s: a population-based study in the maastricht area on incidence, characteristics and survival. J Am Coll Cardiol. (1997) 30:1500–5. doi: 10.1016/S0735-1097(97)00355-0
6. Thygesen K, Alpert JS, White HD. Joint ESC/ACCF/AHA/WHF Task Force for the Redefinition of Myocardial Infarction. Universal definition of myocardial infarction. Eur Heart J. (2007) 28:2525–38. doi: 10.1093/eurheartj/ehm355
7. Sattler SM. Acute myocardial infarction and ventricular fibrillation in a porcine model (Ph.D. thesis). University of Copenhagen, Copenhagen, Denmark (2019).
8. Wissenberg M, Lippert FK, Folke F, Weeke P, Hansen CM, Christensen EF, et al. Association of National initiatives to improve cardiac arrest management with rates of bystander intervention and patient survival after out-of-hospital cardiac arrest. JAMA. (2013) 310:1377–84. doi: 10.1001/jama.2013.278483
9. Risgaard B, Winkel BG, Jabbari R, Behr ER, Ingemann-Hansen O, Thomsen JL, et al. Burden of sudden cardiac death in persons aged 1 to 49 years: nationwide study in Denmark. Circ Arrhythmia Electrophysiol. (2014) 7:205–11. doi: 10.1161/CIRCEP.113.001421
10. Jabbari R, Risgaard B, Fosbøl EL, Scheike T, Philbert BT, Winkel BG, et al. Factors associated with and outcomes after ventricular fibrillation before and during primary angioplasty in patients with ST-segment elevation myocardial infarction. Am J Cardiol. (2015) 116:678–85. doi: 10.1016/j.amjcard.2015.05.037
11. Jabbari R, Engstrom T, Glinge C, Risgaard B, Jabbari J, Winkel BG, et al. Incidence and risk factors of ventricular fibrillation before primary angioplasty in patients with first ST-elevation myocardial infarction: a nationwide study in Denmark. J Am Heart Assoc. (2015) 4:e001399. doi: 10.1161/JAHA.114.001399
12. Karam N, Bataille S, Marijon E, Giovannetti O, Tafflet M, Savary D, et al. Identifying patients at risk for prehospital sudden cardiac arrest at the early phase of myocardial infarction: the e-MUST Study (Evaluation en Médecine d'Urgence des Stratégies Thérapeutiques des infarctus du myocarde). Circulation. (2016) 134:2074–83. doi: 10.1161/CIRCULATIONAHA.117.027775
13. Dekker LRC, Bezzina CR, Henriques JPS, Tanck MW, Koch KT, Alings MW, et al. Familial sudden death is an important risk factor for primary ventricular fibrillation: a case-control study in acute myocardial infarction patients. Circulation. (2006) 114:1140–5. doi: 10.1161/CIRCULATIONAHA.105.606145
14. Walsh CR, Larson MG, Evans JC, Djousse L, Ellison RC, Vasan RS, et al. Alcohol consumption and risk for congestive heart failure in the Framingham Heart Study. Ann Intern Med. (2002) 136:181–91. doi: 10.7326/0003-4819-136-3-200202050-00005
15. Albert CM, Manson JE, Cook NR, Ajani UA, Gaziano JM, Hennekens CH. Moderate alcohol consumption and the risk of sudden cardiac death among US male physicians. Circulation. (1999) 100:944–50. doi: 10.1161/01.CIR.100.9.944
16. Chiuve SE, Rimm EB, Mukamal KJ, Rexrode KM, Stampfer MJ, Manson JE, et al. Light-to-moderate alcohol consumption and risk of sudden cardiac death in women. Hear Rhythm. (2010) 7:1374–80. doi: 10.1016/j.hrthm.2010.05.035
17. Wannamethee G, Shaper AG. Alcohol and sudden cardiac death. Heart. (1992) 68:443–8. doi: 10.1136/hrt.68.11.443
18. Kaikkonen KS, Kortelainen M-L, Linna E, Huikuri HV. Family history and the risk of sudden cardiac death as a manifestation of an acute coronary event. Circulation. (2006) 114:1462–7. doi: 10.1161/CIRCULATIONAHA.106.624593
19. Friedlander Y, Siscovick DS, Arbogast P, Psaty BM, Weinmann S, Lemaitre RN, et al. Sudden death and myocardial infarction in first degree relatives as predictors of primary cardiac arrest. Atherosclerosis. (2002)162:211–6. doi: 10.1016/S0021-9150(01)00701-8
20. Bezzina CR, Pazoki R, Bardai A, Marsman RF, de Jong JSSG, Blom MT, et al. Genome-wide association study identifies a susceptibility locus at 21q21 for ventricular fibrillation in acute myocardial infarction. Nat Genet. (2010) 42:688–91. doi: 10.1038/ng.623
21. Boehringer T, Bugert P, Borggrefe M, Elmas E. SCN5A mutations and polymorphisms in patients with ventricular fibrillation during acute myocardial infarction. Mol Med Rep. (2014) 10:2039–44. doi: 10.3892/mmr.2014.2401
22. Glinge C, Sattler S, Jabbari R, Tfelt-hansen J. Epidemiology and genetics of ventricular fibrillation during acute myocardial infarction. J Geriatr Cardiol. (2016) 13:789–97. doi: 10.11909/j.issn.1671-5411.2016.09.006
23. Bougouin W, Marijon E, Puymirat E, Defaye P, Celermajer DS, Le Heuzey J-Y, et al. Incidence of sudden cardiac death after ventricular fibrillation complicating acute myocardial infarction: a 5-year cause-of-death analysis of the FAST-MI 2005 registry. Eur Heart J. (2014) 35:116–22. doi: 10.1093/eurheartj/eht453
24. Denes P, Wu D, Dhingra R, Pietras RJ, Rosen KM. The effects of cycle length on cardiac refractory periods in man. Circulation. (1974) 49:32–41. doi: 10.1161/01.CIR.49.1.32
25. Shen MJ, Zipes DP. Role of the autonomic nervous system in modulating cardiac arrhythmias. Circ Res. (2014) 114:1004–21. doi: 10.1161/CIRCRESAHA.113.302549
26. Lemmert ME, de Jong JSSG, van Stipdonk AMW, Crijns HJGM, Wellens HJJ, Krucoff MW, et al. Electrocardiographic factors playing a role in ischemic ventricular fibrillation in ST elevation myocardial infarction are related to the culprit artery. Hear Rhythm. (2008) 5:71–8. doi: 10.1016/j.hrthm.2007.09.011
27. Lemmert ME, Majidi M, Krucoff MW, Bekkers SCAM, Crijns HJGM, Wellens HJJ, et al. RR-interval irregularity precedes ventricular fibrillation in ST elevation acute myocardial infarction. Hear Rhythm. (2010) 7:65–71. doi: 10.1016/j.hrthm.2009.09.024
28. Birnbaum Y, Sclarovsky S. The grades of ischemia on the presenting electrocardiogram of patients with ST elevation acute myocardial infarction. J Electrocardiol. (2001) 34(Suppl.):17–26. doi: 10.1054/jelc.2001.28819
29. Gheeraert PJ, Henriques JP, De Buyzere ML, Voet J, Calle P, Taeymans Y, et al. Out-of-hospital ventricular fibrillation in patients with acute myocardial infarction. J Am Coll Cardiol. (2000) 35:144–50. doi: 10.1016/S0735-1097(99)00490-8
30. Li X, Shao D, Wang G, Jiang T, Wu H, Gu B, et al. Effects of different LAD-blocked sites on the development of acute myocardial infarction and malignant arrhythmia in a swine model. J Thorac Dis. (2014) 6:1271–7. doi: 10.3978/j.issn.2072-1439.2014.07.22
31. Euler DE, Spear JF, Moore EN. Effect of coronary occlusion on arrhythmias and conduction in the ovine heart. Am J Physiol Circ Physiol. (1983) 245:H82–9. doi: 10.1152/ajpheart.1983.245.1.H82
32. Janse MJ, Wit AL. Electrophysiological mechanisms of ventricular arrhythmias resulting from myocardial ischemia and infarction. Physiol Rev. (1989) 69:1049–169. doi: 10.1152/physrev.1989.69.4.1049
33. Carmeliet E. Cardiac ionic currents and acute ischemia: from channels to arrhythmias. Physiol Rev. (1999) 79:917–1017. doi: 10.1152/physrev.1999.79.3.917
34. Nerbonne JM, Kass RS. Molecular physiology of cardiac repolarization. Physiol Rev. (2005) 85:1205–53. doi: 10.1152/physrev.00002.2005
35. Yang K-C, Kyle JW, Makielski JC, Dudley SC. Mechanisms of sudden cardiac death: oxidants and metabolism. Circ Res. (2015) 116:1937–55. doi: 10.1161/CIRCRESAHA.116.304691
36. Janse MJ. Vulnerability to ventricular fibrillation. Chaos. (1998) 8:149–56. doi: 10.1063/1.166295
37. Pandit SV, Jalife J. Rotors and the dynamics of cardiac fibrillation. Circ Res. (2013) 112:849–62. doi: 10.1161/CIRCRESAHA.111.300158
38. Nash MP, Mourad A, Clayton RH, Sutton PM, Bradley CP, Hayward M, et al. Evidence for multiple mechanisms in human ventricular fibrillation. Circulation. (2006) 114:536–42. doi: 10.1161/CIRCULATIONAHA.105.602870
39. Ripplinger CM, Noujaim SF, Linz D. The nervous heart. Prog Biophys Mol Biol. (2016) 120:199–209. doi: 10.1016/j.pbiomolbio.2015.12.015
40. Fukuda K, Kanazawa H, Aizawa Y, Ardell JL, Shivkumar K. Cardiac innervation and sudden cardiac death. Circ Res. (2015) 116:2005–19. doi: 10.1161/CIRCRESAHA.116.304679
41. Janse MJ, Coronel R, Wilms-Schopman FJ, de Groot JR. Mechanical effects on arrhythmogenesis: from pipette to patient. Prog Biophys Mol Biol. (2003) 82:187–95. doi: 10.1016/S0079-6107(03)00015-4
42. Pogwizd SM, Corr PB. Mechanisms underlying the development of ventricular fibrillation during early myocardial ischemia. Circ Res. (1990) 66:672–95. doi: 10.1161/01.RES.66.3.672
43. Coronel R, Wilms-Schopman FJ, DeGroot JR. Origin of ischemia-induced phase 1b ventricular arrhythmias in pig hearts. J Am Coll Cardiol. (2002) 39:166–76. doi: 10.1016/S0735-1097(01)01686-2
44. de Groot J. Late ventricular arrhythmias during acute regional ischemia in the isolated blood perfused pig heart Role of electrical cellular coupling. Cardiovasc Res. (2001) 50:362–72. doi: 10.1016/S0008-6363(01)00222-X
45. Penny WJ. The deleterious effects of myocardial catecholamines on cellular electrophsiology and arrhythmias during ischaemia and reperfusion. Eur Heart J. (1984) 5:960–73. doi: 10.1093/oxfordjournals.eurheartj.a061616
46. Smith WT, Fleet WF, Johnson TA, Engle CL, Cascio WE. The Ib phase of ventricular arrhythmias in ischemic in situ porcine heart is related to changes in cell-to-cell electrical coupling. Circulation. (1995) 92:3051–60. doi: 10.1161/01.CIR.92.10.3051
47. Kaplinsky E, Ogawa S, Balke CW, Dreifus LS. Two periods of early ventricular arrhythmia in the canine acute myocardial infarction model. Circulation. (1979) 60:397–403. doi: 10.1161/01.CIR.60.2.397
48. Kabell G, Scherlag BJ, Hope RR, Lazzara R. Regional myocardial blood flow and ventricular arrhythmias following one-stage and two-stage coronary artery occlusion in anesthetized dogs. Am Heart J. (1982) 104:537–44. doi: 10.1016/0002-8703(82)90224-1
49. Cinca J, Warren M, Carreno A, Tresanchez M, Armadans L, Gomez P, et al. Changes in myocardial electrical impedance induced by coronary artery occlusion in pigs with and without preconditioning: correlation with local ST-segment potential and ventricular arrhythmias. Circulation. (1997) 96:3079–86. doi: 10.1161/01.CIR.96.9.3079
50. Gussak I, Chaitman BR, Kopecky SL, Nerbonne JM. Rapid ventricular repolarization in rodents: electrocardiographic manifestations, molecular mechanisms, and clinical insights. J Electrocardiol. (2000) 33:159–70. doi: 10.1016/S0022-0736(00)80072-2
51. Taggart P, Orini M, Hanson B, Hayward M, Clayton R, Dobrzynski H, et al. Developing a novel comprehensive framework for the investigation of cellular and whole heart electrophysiology in the in situ human heart: historical perspectives, current progress and future prospects. Prog Biophys Mol Biol. (2014) 115:252–60. doi: 10.1016/j.pbiomolbio.2014.06.004
52. Watanabe I, Gettes LS. Initial and secondary ST-T alternans during acute myocardial ischemia in the in-situ pig heart. Int Heart J. (2016) 57:327–35. doi: 10.1536/ihj.15-337
53. Bolli R, Myers ML, Zhu WX, Roberts R. Disparity of reperfusion arrhythmias after reversible myocardial ischemia in open chest and conscious dogs. J Am Coll Cardiol. (1986) 7:1047–56. doi: 10.1016/S0735-1097(86)80222-4
54. Hamlin RL. Animal models of ventricular arrhythmias. Pharmacol Ther. (2007) 113:276–95. doi: 10.1016/j.pharmthera.2006.08.006
55. Weissenburger J, Nesterenko VV, Antzelevitch C. Transmural heterogeneity of ventricular repolarization under baseline and long QT conditions in the canine heart in vivo: torsades de pointes develops with halothane but not pentobarbital anesthesia. J Cardiovasc Electrophysiol. (2000) 11:290–304. doi: 10.1111/j.1540-8167.2000.tb01798.x
56. Collins MN, Billman GE. Autonomic response to coronary occlusion in animals susceptible to ventricular fibrillation. Am J Physiol Circ Physiol. (1989) 257:H1886–94. doi: 10.1152/ajpheart.1989.257.6.H1886
57. Halliwill JR, Billman GE. Effect of general anesthesia on cardiac vagal tone. Am J Physiol Circ Physiol. (1992) 262:H1719–24. doi: 10.1152/ajpheart.1992.262.6.H1719
58. Regueiro-Purriños M, Fernández-Vázquez F, de Prado AP, Altónaga JR, Cuellas-Ramón C, Ajenjo-Silverio JM, et al. Ventricular arrhythmias and mortality associated with isoflurane and sevoflurane in a porcine model of myocardial infarction. J Am Assoc Lab Anim Sci. (2011) 50:73–8.
59. de Wit F, van Vliet AL, de Wilde RB, Jansen JR, Vuyk J, Aarts LP, et al. The effect of propofol on haemodynamics: cardiac output, venous return, mean systemic filling pressure, and vascular resistances. Br J Anaesth. (2016) 116:784–9. doi: 10.1093/bja/aew126
60. Van Citters RL, Franklin DL, Rushmer RF. Left ventricular dynamics in dogs during anesthesia with alpha-chloralose and sodium pentobarbital. Am J Cardiol. (1964) 13:349–54. doi: 10.1016/0002-9149(64)90451-5
61. Silverman J, Muir WW. A review of laboratory animal anesthesia with chloral hydrate and chloralose. Lab Anim Sci. (1993) 43:210–6.
62. Linz D, Mahfoud F, Linz B, Hohl M, Schirmer SH, Wirth KJ, et al. Effect of obstructive respiratory events on blood pressure and renal perfusion in a pig model for sleep apnea. Am J Hypertens. (2014) 27:1293–300. doi: 10.1093/ajh/hpu036
63. Rath DP, Little CM, Zhang H, Jiang Z, Abduljalil AM, Zhu H, et al. Sodium pentobarbital versus -chloralose anesthesia : experimental production of substantially different slopes in the transmural CP/ATP ratios within the left ventricle of the canine Myocardium. Circulation. (1995) 91:471–5. doi: 10.1161/01.CIR.91.2.471
64. Sattler SM, Lubberding AF, Skibsbye L, Jabbari R, Wakili R, Jespersen T, et al. Amiodarone treatment in the early phase of acute myocardial infarction protects against ventricular fibrillation in a porcine model. J Cardiovasc Transl Res. (2019) 12:321–30. doi: 10.1007/s12265-018-9861-6
65. Harris AS. Delayed development of ventricular ectopic rhythms following experimental coronary occlusion. Circulation. (1950) 1:1318–28. doi: 10.1161/01.CIR.1.6.1318
66. Heusch G, Kleinbongard P, Bose D, Levkau B, Haude M, Schulz R, et al. Coronary microembolization: from bedside to bench and back to bedside. Circulation. (2009) 120:1822–36. doi: 10.1161/CIRCULATIONAHA.109.888784
67. Janse MJ, Opthof T, Kléber AG. Animal models of cardiac arrhythmias. Cardiovasc Res. (1998) 39:165–77. doi: 10.1016/S0008-6363(97)00313-1
68. Lazzerini PE, Bertolozzi I, Acampa M, Fulceri R, Laghi-Pasini F, Capecchi PL. Torsades de pointes in patients with polymyalgia rheumatica. Curr Pharm Des. (2018) 24:323–40. doi: 10.2174/1381612824666180111111124
69. Arora RB, Sivappa DS. Ectopic ventricular rhythms and myocardial infarction in the domestic pig and their response to nialamide, a monoamine oxidase inhibitor. Br J Pharmacol Chemother. (1962) 19:394–404. doi: 10.1111/j.1476-5381.1962.tb01444.x
70. Taggart P, Yellon DM. Preconditioning and arrhythmias. Circulation. (2002) 106:2999–3001. doi: 10.1161/01.CIR.0000041803.03687.7A
71. Schwartz PJ, Billman GE, Stone HL. Autonomic mechanisms in ventricular fibrillation induced by myocardial ischemia during exercise in dogs with healed myocardial infarction. An experimental preparation for sudden cardiac death. Circulation. (1984) 69:790–800. doi: 10.1161/01.CIR.69.4.790
72. Näslund U, Häggmark S, Johansson G, Marklund SL, Reiz S. A closed-chest myocardial occlusion-reperfusion model in the pig: techniques, morbidity and mortality. Eur Heart J. (1992) 13:1282–9. doi: 10.1093/oxfordjournals.eurheartj.a060350
73. van Hout GPJ, Teuben MPJ, Heeres M, de Maat S, de Jong R, Maas C, et al. Invasive surgery reduces infarct size and preserves cardiac function in a porcine model of myocardial infarction. J Cell Mol Med. (2015) 19:2655–63. doi: 10.1111/jcmm.12656
74. Helfant RH, Vokonas, Pantel S, Gorlin R. Functional importance of the human coronary collateral circulation. N Engl J Med. (1971) 284:1277–81. doi: 10.1056/NEJM197106102842301
75. Teunissen PFA, Horrevoets AJG, van Royen N. The coronary collateral circulation: genetic and environmental determinants in experimental models and humans. J Mol Cell Cardiol. (2012) 52:897–904. doi: 10.1016/j.yjmcc.2011.09.010
76. Wustmann K, Zbinden S, Windecker S, Meier B, Seiler C. Is there functional collateral flow during vascular occlusion in angiographically normal coronary arteries? Circulation. (2003) 107:2213–20. doi: 10.1161/01.CIR.0000066321.03474.DA
77. Seiler C, Stoller M, Pitt B, Meier P. The human coronary collateral circulation: development and clinical importance. Eur Heart J. (2013) 34:2674–82. doi: 10.1093/eurheartj/eht195
78. Watanabe I, Johnson TA, Buchanan J, Engle CL, Gettes LS. Effect of graded coronary flow reduction on ionic, electrical, and mechanical indexes of ischemia in the pig. Circulation. (1987) 76:1127–34. doi: 10.1161/01.CIR.76.5.1127
79. Hearse DJ. The elusive coypu: the importance of collateral flow and the search for an alternative to the dog. Cardiovasc Res. (2000) 45:215–9. doi: 10.1016/S0008-6363(99)00331-4
80. Hedström E, Engblom H, Frogner F, Ålström-Olsson K, Öhlin H, Jovinge S, et al. Infarct evolution in man studied in patients with first-time coronary occlusion in comparison to different species - implications for assessment of myocardial salvage. J Cardiovasc Magn Reson. (2009) 11:1–10. doi: 10.1186/1532-429X-11-38
81. Liu DW, Gintant GA, Antzelevitch C. Ionic bases for electrophysiological distinctions among epicardial, midmyocardial, and endocardial myocytes from the free wall of the canine left ventricle. Circ Res. (1993) 72:671–87. doi: 10.1161/01.RES.72.3.671
82. Blumgart HL, Zoll PM, Freedberg AS, Gilligan DR. The experimental production of intercoronary arterial anastomoses and their functional significance. Circulation. (1950) 1:10–27. doi: 10.1161/01.CIR.1.1.10
83. Köhn F, Sharifi AR, Simianer H. Modeling the growth of the Goettingen minipig1. J Anim Sci. (2007) 85:84–92. doi: 10.2527/jas.2006-271
84. Schuleri KH, Boyle AJ, Centola M, Amado LC, Evers R, Zimmet JM, et al. The adult Göttingen minipig as a model for chronic heart failure after myocardial infarction: focus on cardiovascular imaging and regenerative therapies. Comp Med. (2008) 58:568–79.
85. Crick SJ, Sheppard MN, Ho SY, Gebstein L, Anderson RH. Anatomy of the pig heart: comparisons with normal human cardiac structure. J Anat. (1998) 193(Pt 1):105–19. doi: 10.1046/j.1469-7580.1998.19310105.x
86. Laursen M, Olesen S-P, Grunnet M, Mow T, Jespersen T. Characterization of cardiac repolarization in the Göttingen minipig. J Pharmacol Toxicol Methods. (2011) 63:186–95. doi: 10.1016/j.vascn.2010.10.001
87. Hamlin RL. The QRS electrocardiogram, epicardiogram, vectorcardiogram and ventricular excitation of swine. Am J Physiol Content. (1960) 198:537–42. doi: 10.1152/ajplegacy.1960.198.3.537
88. Huang J, Dosdall DJ, Cheng K-A, Li L, Rogers JM, Ideker RE. the importance of purkinje activation in long duration ventricular fibrillation. J Am Heart Assoc. (2014) 3:e000495–e000495. doi: 10.1161/JAHA.113.000495
89. Walcott GP, Kroll MW, Ideker RE. Ventricular fibrillation: are swine a sensitive species? J Interv Card Electrophysiol. (2015) 42:83–9. doi: 10.1007/s10840-014-9964-1
90. Rabbani S, Ahmadi H, Fayazzadeh E, Sahebjam M, Boroumand MA, Sotudeh M, et al. Development of an ovine model of myocardial infarction. ANZ J Surg. (2008) 78:78–81. doi: 10.1111/j.1445-2197.2007.04359.x
91. Jardine DL, Charles CJ, Frampton CM, Richards AM. Cardiac sympathetic nerve activity and ventricular fibrillation during acute myocardial infarction in a conscious sheep model. Am J Physiol Circ Physiol. (2007) 293:H433–9. doi: 10.1152/ajpheart.01262.2006
92. Shofti R, Zaretzki A, Cohen E, Engel A, Bar-El Y. The sheep as a model for coronary artery bypass surgery. Lab Anim. (2004) 38:149–57. doi: 10.1258/002367704322968821
93. Bruyneel KJJ. Use of moving epicardial electrodes in defining ST-segment changes after acute coronary occlusion in the baboon. Relation to primary ventricular fibrillation. Am Heart J. (1975) 89:731–41. doi: 10.1016/0002-8703(75)90188-X
94. Randall DC, Hasson DM, Brady JV. Acute cardiovascular consequences of anterior descending coronary artery occlusion in unanesthetized monkey. Exp Biol Med. (1978) 158:135–40. doi: 10.3181/00379727-158-40157
95. O'Gara PT, Kushner FG, Ascheim DD, Casey DE, Chung MK, de Lemos JA, et al. 2013 ACCF/AHA guideline for the management of ST-elevation myocardial infarction: a report of the american college of cardiology foundation/american heart association task force on practice guidelines. Circulation. (2013) 127:e362–425. doi: 10.1161/CIR.0b013e3182742cf6
96. Chen ZM, Pan HC, Chen YP, Peto R, Collins R, Jiang LX, et al. Early intravenous then oral metoprolol in 45,852 patients with acute myocardial infarction: randomised placebo-controlled trial. Lancet. (2005) 366:1622–32. doi: 10.1016/S0140-6736(05)67661-1
97. Echt DS, Liebson PR, Mitchell LB, Peters RW, Obias-Manno D, Barker AH, et al. Mortality and morbidity in patients receiving Encainide, Flecainide, or Placebo. N Engl J Med. (1991) 324:781–8. doi: 10.1056/NEJM199103213241201
98. Waldo AL, Camm AJ, DeRuyter H, Friedman PL, MacNeil DJ, Pauls JF, et al. Effect of d-sotalol on mortality in patients with left ventricular dysfunction after recent and remote myocardial infarction. Lancet. (1996) 348:7–12. doi: 10.1016/S0140-6736(96)02149-6
99. Anastasiou-Nana MI, Nanas JN, Nanas SN, Rapti A, Poyadjis A, Stathaki S, et al. Effects of amiodarone on refractory ventricular fibrillation in acute myocardial infarction: experimental study. J Am Coll Cardiol. (1994) 23:253–8. doi: 10.1016/0735-1097(94)90528-2
100. Li T, Wei X, Watkins AC, Sanchez PG, Wu ZJ, Griffith BP. Prophylactic amiodarone and lidocaine improve survival in an ovine model of large size myocardial infarction. J Surg Res. (2013) 185:152–8. doi: 10.1016/j.jss.2013.05.050
101. Nattel S, Davies M, Quantz M. The antiarrhythmic efficacy of amiodarone and desethylamiodarone, alone and in combination, in dogs with acute myocardial infarction. Circulation. (1988) 77:200–8. doi: 10.1161/01.CIR.77.1.200
102. Somani P, Simon VA, McManus K. Pharmacokinetics, antiarrhythmic effects, and tissue concentrations of amiodarone and desethylamiodarone in dogs with acute coronary artery occlusion. Can J Cardiol. (1985) 1:56–65.
103. Mayuga RD, Singer DH. Effects of intravenous amiodarone on electrical dispersion in normal and ischaemic tissues and on arrhythmia inducibility: monophasic action potential studies. Cardiovasc Res. (1992) 26:571–9. doi: 10.1093/cvr/26.6.571
104. Kudenchuk PJ, Brown SP, Daya M, Rea T, Nichol G, Morrison LJ, et al. Amiodarone, Lidocaine, or Placebo in out-of-hospital cardiac arrest. N Engl J Med. (2016) 374:1711–22. doi: 10.1056/NEJMoa1514204
105. Xue YX, Aye NN, Hashimoto K. Antiarrhythmic effects of HOE642, a novel Na+-H+ exchange inhibitor, on ventricular arrhythmias in animal hearts. Eur J Pharmacol. (1996) 317:309–16. doi: 10.1016/S0014-2999(96)00755-8
106. Sack S, Mohri M, Schwarz ER, Arras M, Schaper J, Ballagi-Pordány G, et al. Effects of a new Na+/H+ antiporter inhibitor on postischemic reperfusion in pig heart. J Cardiovasc Pharmacol. (1994) 23:72–8. doi: 10.1097/00005344-199401000-00009
107. Wirth KJ, Maier T, Busch AE. NHE1-inhibitor cariporide prevents the transient reperfusion-induced shortening of the monophasic action potential after coronary ischemia in pigs. Basic Res Cardiol. (2001) 96:192–7. doi: 10.1007/s003950170070
108. Wirth KJ, Uhde J, Rosenstein B, Englert HC, Gögelein H, Schölkens BA, et al. K(ATP) channel blocker HMR 1883 reduces monophasic action potential shortening during coronary ischemia in anesthetised pigs. Naunyn Schmiedebergs Arch Pharmacol. (2000) 361:155–60. doi: 10.1007/s002109900166
109. Moritani K, Miyazaki T, Miyoshi S, Asanagi M, Zhao L-S, Mitamura H, et al. Blockade of ATP-sensitive potassium channels by 5-hydroxydecanoate suppresses monophasic action potential shortening during regional myocardial ischemia. Cardiovasc Drugs Ther. (1994) 8:749–56. doi: 10.1007/BF00877122
110. Lu L, Reiter MJ, Xu Y, Chicco A, Greyson CR, Schwartz GG. Thiazolidinedione drugs block cardiac KATP channels and may increase propensity for ischaemic ventricular fibrillation in pigs. Diabetologia. (2008) 51:675–85. doi: 10.1007/s00125-008-0924-0
111. Sarraf M, Lu L, Ye S, Reiter MJ, Greyson CR, Schwartz GG. Thiazolidinedione drugs promote onset, alter characteristics, and increase mortality of ischemic ventricular fibrillation in pigs. Cardiovasc Drugs Ther. (2012) 26:195–204. doi: 10.1007/s10557-012-6384-2
112. Xing D, Kjølbye AL, Nielsen MS, Petersen JS, Harlow KW, Holstein-Rathlou N-H, et al. ZP123 increases gap junctional conductance and prevents reentrant ventricular tachycardia during myocardial ischemia in open chest dogs. J Cardiovasc Electrophysiol. (2003) 14:510–20. doi: 10.1046/j.1540-8167.2003.02329.x
113. Li J, Zhong J, Zeng Q, Liu H, Su G, Zhang Y. Effect of ZP123, a gap junction modifier, on prolonged ventricular fibrillation in swine. Cardiology. (2011) 118:147–52. doi: 10.1159/000328016
114. Vaillant F, Timour Q, Descotes J, Manati W, Belhani D, Bui-Xuan B, et al. Ivabradine induces an increase in ventricular fibrillation threshold during acute myocardial ischemia: an experimental study. J Cardiovasc Pharmacol. (2008) 52:548–54. doi: 10.1097/FJC.0b013e3181913df4
115. Vaillant F, Dehina L, Dizerens N, Bui-Xuan B, Tabib A, Lauzier B, et al. Ivabradine but not propranolol delays the time to onset of ischaemia-induced ventricular fibrillation by preserving myocardial metabolic energy status. Resuscitation. (2013) 84:384–90. doi: 10.1016/j.resuscitation.2012.07.041
116. Vaillant F, Dehina L, Mazzadi A, Descotes J, Chevalier P, Tabib A, et al. Heart rate reduction with ivabradine increases ischaemia-induced ventricular fibrillation threshold: role of myocyte structure and myocardial perfusion. Resuscitation. (2011) 82:1092–9. doi: 10.1016/j.resuscitation.2011.03.032
117. Nattel S, Pedersen DH, Zipes DP. Alterations in regional myocardial distribution and arrhythmogenic effects of aprindine produced by coronary artery occlusion in the dog. Cardiovasc Res. (1981) 15:80–5. doi: 10.1093/cvr/15.2.80
118. Kodama I, Kamiya K, Toyama J. Cellular electropharmacology of amiodarone. Cardiovasc Res. (1997) 35:13–29. doi: 10.1016/S0008-6363(97)00114-4
119. Polster P, Broekhuysen J. The adrenergic antagonism of amiodarone. Biochem Pharmacol. (1976) 25:131–4. doi: 10.1016/0006-2952(76)90279-3
120. Connolly SJ. Evidence-based analysis of amiodarone efficacy and safety. Circulation. (1999) 100:2025–34. doi: 10.1161/01.CIR.100.19.2025
121. Zipes DP, Camm AJ, Borggrefe M, Buxton AE, Chaitman B, Fromer M, et al. ACC/AHA/ESC 2006 guidelines for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death–executive summary: a report of the American College of Cardiology/American Heart Association Task Force and the European Societ. Eur Heart J. (2006) 27:2099–140. doi: 10.1093/eurheartj/ehl199
122. Piccini JP, Schulte PJ, Pieper KS, Mehta RH, White HD, Van de Werf F, et al. Antiarrhythmic drug therapy for sustained ventricular arrhythmias complicating acute myocardial infarction. Crit Care Med. (2011) 39:78–83. doi: 10.1097/CCM.0b013e3181fd6ad7
123. Karmazyn M, Gan XT, Humphreys RA, Yoshida H, Kusumoto K. The myocardial Na+-H+ exchange : structure, regulation, and its role in heart disease. Circ Res. (1999) 85:777–86. doi: 10.1161/01.RES.85.9.777
124. Scholz W, Albus U. Na+/H+ exchange and its inhibition in cardiac ischemia and reperfusion. Basic Res Cardiol. (1993) 88:443–55. doi: 10.1007/BF00795411
125. Zhang H, Flagg TP, Nichols CG. Cardiac sarcolemmal KATP channels: latest twists in a questing tale! J Mol Cell Cardiol. (2010) 48:71–5. doi: 10.1016/j.yjmcc.2009.07.002
126. Del Ry S, Moscato S, Bianchi F, Morales MA, Dolfi A, Burchielli S, et al. Altered expression of connexin 43 and related molecular partners in a pig model of left ventricular dysfunction with and without dipyrydamole therapy. Pharmacol Res. (2015) 95–96:92–101. doi: 10.1016/j.phrs.2015.03.015
127. Sims JJ, Schoff KL, Loeb JM, Wiegert NA. Regional gap junction inhibition increases defibrillation thresholds. Am J Physiol Circ Physiol. (2003) 285:H10–6. doi: 10.1152/ajpheart.01074.2002
128. Stroemlund LW, Jensen CF, Qvortrup K, Delmar M, Nielsen MS. Gap junctions–guards of excitability. Biochem Soc Trans. (2015) 43:508–12. doi: 10.1042/BST20150059
129. Schultz JG, Andersen S, Andersen A, Erik Nielsen-Kudsk J, Nielsen JM. Evaluation of cardiac electrophysiological properties in an experimental model of right ventricular hypertrophy and failure. Cardiol Young. (2016) 26:451–8. doi: 10.1017/S1047951115000402
130. Srisakuldee W, Makazan Z, Nickel BE, Zhang F, Thliveris JA, Pasumarthi KBS, et al. The FGF-2-triggered protection of cardiac subsarcolemmal mitochondria from calcium overload is mitochondrial connexin 43-dependent. Cardiovasc Res. (2014) 103:72–80. doi: 10.1093/cvr/cvu066
131. Sartiani L, De Paoli P, Stillitano F, Aimond F, Vassort G, Mugelli A, et al. Functional remodeling in post-myocardial infarcted rats: focus on beta-adrenoceptor subtypes. J Mol Cell Cardiol. (2006) 40:258–66. doi: 10.1016/j.yjmcc.2005.11.011
132. DiFrancesco D, Borer JS. The funny current: cellular basis for the control of heart rate. Drugs. (2007) 67(Suppl. 2):15–24. doi: 10.2165/00003495-200767002-00003
133. Linz D, Elliott AD, Hohl M, Malik V, Schotten U, Dobrev D, et al. Role of autonomic nervous system in atrial fibrillation. Int J Cardiol. (2019) 287:181–8. doi: 10.1016/j.ijcard.2018.11.091
134. Lai Y, Yu L, Jiang H. Autonomic neuromodulation for preventing and treating ventricular arrhythmias. Front Physiol. (2019) 10:200. doi: 10.3389/fphys.2019.00200
135. Linz D, Wirth K, Ukena C, Mahfoud F, Pöss J, Linz B, et al. Renal denervation suppresses ventricular arrhythmias during acute ventricular ischemia in pigs. Hear Rhythm. (2013) 10:1525–30. doi: 10.1016/j.hrthm.2013.07.015
136. Linz D, Ukena C, Mahfoud F, Neuberger H-R, Böhm M. Atrial autonomic innervation. J Am Coll Cardiol. (2014) 63:215–24. doi: 10.1016/j.jacc.2013.09.020
137. Vanoli E, De Ferrari GM, Stramba-Badiale M, Hull SS, Foreman RD, Schwartz PJ. Vagal stimulation and prevention of sudden death in conscious dogs with a healed myocardial infarction. Circ Res. (1991) 68:1471–81. doi: 10.1161/01.RES.68.5.1471
138. Odenstedt J, Linderoth B, Bergfeldt L, Ekre O, Grip L, Mannheimer C, et al. Spinal cord stimulation effects on myocardial ischemia, infarct size, ventricular arrhythmia, and noninvasive electrophysiology in a porcine ischemia-reperfusion model. Hear Rhythm. (2011) 8:892–8. doi: 10.1016/j.hrthm.2011.01.029
Keywords: acute myocardial infarction, ventricular arrhythmia, animal models, anti-arrhythmia agents, sudden cardiac death, STEMI, ischemia, ventricular fibrillation
Citation: Sattler SM, Skibsbye L, Linz D, Lubberding AF, Tfelt-Hansen J and Jespersen T (2019) Ventricular Arrhythmias in First Acute Myocardial Infarction: Epidemiology, Mechanisms, and Interventions in Large Animal Models. Front. Cardiovasc. Med. 6:158. doi: 10.3389/fcvm.2019.00158
Received: 25 May 2019; Accepted: 18 October 2019;
Published: 05 November 2019.
Edited by:
Daphne Merkus, Erasmus Medical Center, NetherlandsReviewed by:
Maurizio Acampa, Siena University Hospital, ItalyCopyright © 2019 Sattler, Skibsbye, Linz, Lubberding, Tfelt-Hansen and Jespersen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stefan Michael Sattler, c3RlZmFuLnNhdHRsZXJAbWUuY29t
†These authors share the first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.