 Martin Ezeani
Martin Ezeani
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med. , 20 June 2019
Sec. Cardiac Rhythmology
Volume 6 - 2019 | https://doi.org/10.3389/fcvm.2019.00083
This article is part of the Research Topic Sudden Cardiac Death and Channelopathies View all 15 articles
Ion channel biology offers great opportunity in identifying and learning about cardiac pathophysiology mechanisms. The discovery of transient receptor potential (TRP) channels is an add-on to the opportunity. Interacting with numerous signaling pathways, being activated multimodally, and having prescribed signatures underlining acute hemodynamic control and cardiac remodeling, TRP channels regulate cardiac pathophysiology. Impaired Ca2+-handling cause contractile abnormality. Modulation of intracellular Ca2+ concentration ([Ca2+]i) is a major part of Ca2+-handling processes in cardiac pathophysiology. TRP channels including TRPM4 regulate [Ca2+]i, Ca2+-handling and cardiac contractility. The channels modulate flux of divalent cations, such as Ca2+ during Ca2+-handling and cardiac contractility. Seminal works implicate TRPM4 and TRPC families in intracellular Ca2+ homeostasis. Defective Ca2+-homeostasis through TRP channels interaction with Ca2+-dependent regulatory proteins such as sodium calcium exchanger (NCX) results in abnormal Ca2+ handling, contractile dysfunction and in spontaneous ectopy. This review provides insight into TRP channels mediated pathological Ca2+-handling and spontaneous ectopy.
Ca2+ flux at organellar levels govern excitation and contraction coupling. The ionic movement is important. During action potential propagation, cell membranes depolarize, and Ca2+ enters into the cells. L-type calcium channel passes the Ca2+ influx as inward Ca2+ current (ICa,L), which contributes to the shape of action potential plateau. The Ca2+ entry initiates Ca2+ release from the sarcoplasmic reticulum (SR) by activation of ryanodine receptors (RyRs), thereby elevating [Ca2+]i in the so-called Ca2+-activated Ca2+ release. The raise in the [Ca2+]i, activated by a variety of ways, is a known mechanism of cell signaling. It enables Ca2+ to bind to contractile machineries such as myofilament protein and troponin C, and initiate contraction. The raise in the [Ca2+]i contrarily declines in relaxation. Intracellular Ca2+ is pumped from the SR by sarcoplasmic reticulum calcium pump (SERCA), and sarcolemmal efflux through sodium calcium exchanger (NCX), with little contribution by the SERCA during relaxation (1). This results in a decline of [Ca2+]i, allowing Ca2+ to dissociate from troponin for relaxation to occur at diastole. While decline in [Ca2+]i through Ca2+ efflux leads to relaxation, elevation in [Ca2+]i through Ca2+ influx leads to contraction. The movement of monovalent and divalent cations through the channels is crucial to cardiac excitation and contraction coupling, and in this review, considerations are given to TRPC and TRPM channels in this process.
The process is impaired in contractile abnormality (2) and in general in heart diseases. Abnormal cardiac performance, which characterizes heart failure, cardiomyopathy, and arrhythmia, occurs through impaired Ca2+-homeostasis. Compared with non-failing cardiomyocytes, failing cardiomyocytes SR Ca2+ re-uptake was reduced with no significant changes in the rate of Ca2+ efflux by NCX (3). Frequency-dependent increases in SR Ca2+ load, that was associated with a decline in contractile tone at high heart rates, was absent in failing myocardium (4). In addition, it has been demonstrated at single cell levels that alteration in Ca2+-handling occurred through abnormal Ca2+-homeostasis and increase in [Ca2+]i in familial hypertrophic cardiomyopathy disease (5). The impaired Ca2+-homeostasis may be due to translational and transcriptional changes at the level of expression of the Ca2+-regulatory proteins. Chronic heart failure patients had abnormal changes in the levels of Ca2+-regulatory proteins, such as the SR Ca2+-ATPase (6). Together, impaired Ca2+-homeostasis and altered Ca2+-regulatory proteins orchestrate abnormal Ca2+-handling. The mechanisms responsible for this are not completely elucidated.
Tremendous efforts at understanding the process is ongoing, but it is hampered by the complicated nature of changes in [Ca2+]i. Subcellular Ca2+ concentration is compartmentalized, and [Ca2+]i varies tremendously. Furthermore, intracellular Ca2+ homeostasis can be regulated by local signaling processes within restricted space, rather than by just changes in the global cytosolic [Ca2+]i. With the complexities, it is difficult to completely define distinct Ca2+ homeostasis together with their [Ca2+]i biological networks, and their roles. This may have implied that sources that control subcellular Ca2+ are just too numerous and are yet to be defined completely. This review discusses some transient receptor potential (TRP) channels seminal works in pathologic Ca2+-handling and spontaneous ectopy.
Regulation of [CA2+]i, that is part of CA2+ homeostasis, and governs Ca2+-handling in excitation and contraction coupling is an important process of cardiac contractile performance. Cardiac contractility is the intrinsic ability of the myocardium to contract. Readily occurrence of contraction depends on incremental degrees of binding between myosin (thick) and actin (thin) filaments, when the [Ca2+]i is elevated. Factors that promote Ca2+-handling and contractility do so by inducing increases in the [Ca2+]i. The heart is unable to contract in too little Ca2+ concentration (small Ca2+ transient amplitude) and unable to relax in too much Ca2+ concentration (large Ca2+ transient amplitude), indicating negative and positive inotropy, respectively. TRP channels as cell membrane ion channel fall into a category and were first identified in impaired Drosophila visual adaption, where photoreceptors carrying TRP gene mutations showed a transient voltage response to continuous light (7, 8). Twenty eight mammalian TRP genes have been identified to date and have fallen into six related protein families. Most of them appear to be multimodally gated and interact with multiple signaling pathways and show pertinent trait to cardiac remodeling. While some are voltage dependent others are not, and they regulate a variety of cell functions such as, apoptosis, thermo regulation, cell viability and proliferation, and renal Ca2+ absorption. TRP channels are relatively non-selective permeable cation channels. As such, TRP channels permeate Na+, Ca2+, and Mg2+, and thus regulate intracellular ionic concentrations, including [Ca2+]i. Seminal works (9–13) implicate some TRPM4 and TRPC channels in [Ca2+]i variability. The channels, therefore, modulate Ca2+ transients, inotropy, and action potential wave form (Figure 1).
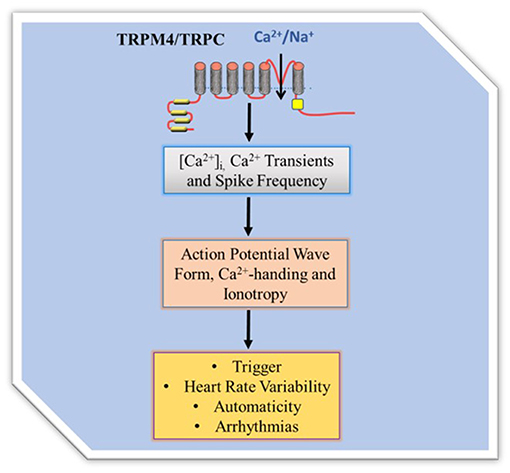
Figure 1. TRP channel regulation of intracellular Ca2+ homeostasis. TRPM4 mediates membrane depolarization dependent Ca2+ entry while TRPC mediates permeability dependent Ca 2+ entry. The Ca2+ influx directly and/or indirectly contributes to Ca2+ transient to regulate Ca2+-handling, action potential wave form and contractility. Since TRPC are Na+/Ca2+ permeable they can directly contribute Ca2+ entry while the Ca2+ non permeable TRPM4 regulates Ca2+entry by modification of the membrane potential and thus driving force.
Transient receptor potential melastatin (TRPM) is a family of TRP ion channels, that consists of eight different subfamilies (TRPM1-TRPM8). TRPM4 subfamily while permeable to Na+ is activated by [Ca2+]i, in Ca2+-induced Ca2+ release process and modulates inotropic β-adrenergic effects on ventricular heart muscle by increasing Ca2+ transients amplitude (11). The activation process is important because it contributes to Ca2+ transients. Ca2+-handling, excitation-contraction coupling and action potential wave forms depend on Ca2+ transients, proofing that the [Ca2+]i activation and inotropic β-adrenergic effects of TRPM4 are both important in contractility. TRPM4 inhibition altered action potential wave form and reduced action potential duration (13). More so, it has been proposed that TRPM4 channel activity might couple to ICa,L functional activity in elevating [Ca2+]i. Trpm4−/− ventricular myocytes had fast repolarization as a result of enhanced driving force of ICa,L for Ca2+ entry (11). Therefore, it appears TRPM4 regulates action potential adaptation and duration, and underpins voltage-gated Ca2+ channel Ca2+ influx that couples its activity and that of ICa,L in the so called Ca2+-induce Ca2+ release that promote contractility.
In addition to TRPM4, TRP canonical (TRPC) is another family of TRP channels. It consists of seven subfamilies (TRPC1-TRPC7). TRPCs are also permeable to but activated by Ca2+, and may be important in Ca2+-handling. TRPC6 promoter gene had two conserved nuclear factor of activated T-cell transcriptional factor (NFAT) consensus sites (14) and NFAT is calcium-dependent regulatory transcriptional factor. Therefore, TRPC6 is an intracellular Ca2+ signaling effector. Increased TRPC1/TRPC4 expression underscores elevated SR Ca2+ content in right ventricular hypertrophied cardiomyocytes during monocrotaline exposure (14). Transgenic mice with inhibition of TRPC3/6/7 and TRPC1/4/5 subfamilies had membrane Ca2+ leak in pathological hypertrophy following either activation of neuroendocrine (phenylephrine and angiotensin II (Ang II) infusion) or pressure overload induction (15). TRPC channels as cation-selective channels can produce pathological cardiac growth of adult myocytes through Ca2+ influx and calcineurin activation, that alter Ca2+-handling and Ca2+-dependent signaling.
Although it is possible that TRPC/Ca2+/calcineurin/NFAT signaling loop might regulate Ca2+-handling in diseases, how the signaling loop couples to [Ca2+]i and L-type Ca2+ channel activity is completely unknown. It appears in smooth muscle that entry of cations through a receptor operated TRPC6 caused membrane depolarization and consequent functional activity of L-type Ca2+ channels, Ca2+ influx, and smooth muscle contraction. However, whether this role is associated with ryanodine and NFAT is not known. Given that TRPC6 can operate as receptor-activated cation channels, which increases [Ca2+]i by Ca2+ entry across plasma membrane and/or by release of Ca2+ from intracellular stores such as the endoplasmic reticulum, TRPC6 contributes to Ca2+-handling and [Ca2+]i. The contribution can also be deduced on store-operation molecular standpoint. This is since store-operated and receptor-operated channels can come from the same proteins of the same family of TRP channels.
Nonetheless, TRP channels store operated and receptor operated calcium entry (SOCE) and (ROCE) are less clear. Discovery has it that stromal interaction molecule 1 (STIM1) and Orai 1 are mediators of SOCE. Fast Ca2+-dependent inactivation kinetics of STIM1 current is known to be critically dependent on cytosolic Ca2+ levels, which also regulates native SOCE and ROCE currents, through changes in the [Ca2+]i, similarly to that of TRP channel currents. Whether TRP channels kinetics are associated to SOCE and ROCE, is not elucidated. Native store-operated and receptor-operated complexes stoichiometry, if distinct from hetero/homo multimer variants Orai or TRP is also not known. Regardless, STIM1 indirectly activates TRPC3/6, but not TRPC7 (16), and TRPC1/4/5 directly bind with STIM1 to activate SOCE (17), and in lipid raft domains, TRPC channels co-localize with STIM1 and Orai (18). There may be a better understanding of TRP channels SOCE attribute arguably in micro-domains (19). In fact, it is known that the Ca2+-activated signaling effectors are either in direct proximity or attached to Ca2+ entry channels in compartmentalized micro-domains (20). Dissociation of TRPC1 from Cav1, following Ca2+-store depletion is an important process in the activation of TRPC1-SOCE (20). Expression of TRPC channels and Ca2+ influx through TRPC within micro-domains affects contractility reserve and contributes to cardiac Ca2+ cycling. Taken together: (1) direct effects of TRP channels and receptor operated calcium entry mechanisms on [Ca2+]i and Ca2+-handling may be questioned. (2) It is more likely that TRP channels regulate the process through effects on micro-domains or through direct interaction with Ca2+-dependent regulatory proteins. (3) It is noted that STIM1 is a Ca2+ sensor that relays Ca2+ load of the endoplasmic reticulum to store operated channels and may navigate some of the TRP channels into micro-domains to govern their roles in cardiovascular diseases through calcium signaling effectors.
In addition to the myocardial TRPC channels, atrial endocardial TRPC-6 channel has been identified to be crucial in TRPC-6-dependent paracrine factors that regulate the amplitude of myocardial Ca2+ transients through mechanotransduction. Mechanical stretch is a determinant of atrial function such as contractility and TRPC-6 is a stretch responsive cation channel that governs mechanotransduction. To understand the roles of stretched-atrial endocardium, associated with physiological conditions and arrhythmogenesis Nikolova-Krstevski et al., tested the consequences of stretch-induced endocardial TRPC-6 activation on myocardial function and hypothesized that endocardial TRPC-6 is required for mechanical stretch responses in the atrium (21). Findings from this investigation, demonstrate that, in acute stretch, Ca2+-mediating function of TRPC-6 was consequentially enhanced via [Ca2+]i, and endocardial TRPC-6 regulatory feedback protects against this stretch-induced myocardial Ca2+ overload process, whereas in chronic stretch, reduced protein expression of endocardial TRPC-6 and irregular Ca2+ transient cyclic events may lead to enhanced susceptibility to arrhythmia. To understand the Ca2+ transient mechano feedback response, atrial endocardial-myocardial (cell-cell) communication/signaling was investigated in cultured human-induced pluripotent stem cell-derived cardiomyocytes before and after the addition of media collected from non-stretched and stretched atrial endocardial cells from porcine. The investigation revealed alteration of Ca2+ transient in human-induced pluripotent stem cell-derived cardiomyocytes with non-stretched but not with stretched atrial endocardial cells (21). The finding suggests that TRPC-6 dependent stretch-induced mechanotransduction can induce changes in global [Ca2+]I that modulate intrinsic atrial endocardial function, such as contractility, as well as affecting myocardial function, through endothelium secreting factors such as endothelin-1, nitric oxide, prostacyclin, and angiotensin II (21).
The question now remains how does the channels regulate [Ca2+]i, and thus control Ca2+-handling and contractile tone? The mechanisms of TRP channel mediated Ca2+ handling remain largely unknown. Ca2+-dependent regulatory proteins such as NCX and SERCA regulate Ca2+-handling and cardiac contractility, and the TRP channels interact directly with NCX and SERCA. Functional and physical interaction of TRPC channels with NCX proteins is a novel principle behind TRPC-mediated increase in intracellular Ca2+ signaling. To assess for the existence of a native TRPC3/NCX1 signaling complex in rat cardiac myocytes, previously identified in HEK 293 cells, the Eder et al conducted reciprocal co-immunoprecipitation to detect TRPC3 and NCX1 interaction by immunoprecipiting solubilized proteins of crude adult rat cardiomyocyte membrane fractions with either anti-TRPC3 or anti-NCX (22). This demonstrated significant TRPC3 and NCX1 co-localization. Glutathione S-transferase pulldown experiment consistently replicated the native TRPC3/NCX1 signaling complex within the cardiomyocyte. To understand the functional consequences of this interaction, phpspholipase C (PLC) was first stimulated in the myocytes, and because TRPC3 is a key component of PLC-dependent Ca2+ signaling, the cardiomyocytes were then transiently transfected with TRPC3 N-terminal fragment to exert dominant negative effects on TRPC3. The activation of PLC promoted reverse mode NCX1-mediated Ca2+ entry, as identified, through [Ca2+]i measurement, that was regulated by TRPC3-mediated Na+ loading in the myocytes through angiotensin-induced activation of the G protein Gq-PLC pathway (22). Measurement of this NCX-mediated cellular Ca2+ signals in the cells expressing dominant negative TRPC3 revealed significant reduction in Ca2+ signals, illustrating the idea of TRPC3-dependent Na+ loading, as part of cardiac NCX operation (22).
PLC dependently recruited TRPC3-NCX1 complex into the plasma membrane and regulated Ca2+ homeostasis in rat cardiomyocytes (19, 22). TRPC3 channel may be a major component of PLC-dependent activation of the Ca2+ calcineurin-NFAT signaling pathway, a pathological cardiac hypertrophy pathway, in which NCX1 can mediate Ca2+ influx by reverse mode and contribute to Ca2+ transients and action potential wave forms. Together, TRPC3 may be an important component of NCX regulation of Ca2+-handling. Furthermore, SERCA is an ATP-dependent Ca2+ pump located in the SR membrane. It appears silencing SR Ca2+-ATPase (SERCA)2 with small interfering RNA (siRNA) increases TRPC levels (23), as a compensatory mechanism for SERCA Ca2+ pump. While colocalizing on the skin of Darier's disease patient on immunostaining, TRPC1 had increased immunoreactivity whereas, SERCA2 had reduced immunoreactivity, suggesting reciprocal activity of TRPC1 and SERCA2 (23). Western blots performed on the skin from the DD patient and immunolocalization performed on skin samples from SERCA2+/+ and SERCA2+/− mice showed consistent reciprocal expression of TRPC1 and SERCA2 (23). The DD patients as well as SERCA2+/− mice have only one normal copy of the gene encoding SERCA2. To understand the association of the reciprocal expression, a human keratinocyte (HaCaT) cell line was employed. The keratinocytes from DD patients have also one normal copy of SERCA2. Single copy of the gene is known to be inadequate, to appropriately account for proper amount of SERCA2, leading to changes in Ca2+ signaling in the cells. To examine this SERCA2 reduction in function, HaCaT cells were treated with adenovirus encoding either SERCA2-siRNA or control-siRNA and Ca2+ imaging was performed on the cells upon stimulation with thapsigargin, to deplete endoplasmic reticulum Ca2+ (23). Addition of thapsigargin in HaCaT cells Ca2+-containing media, caused an increase in global [Ca2+]i. Whereas this increase was high in HaCaT cells expressing SERCA2-siRNA, no increase in control-siRNA was documented (23). Consistent with the reciprocal expression of TRPC1 and SERCA2, release of Ca2+ from the internal stores upon the thapsigargin addition was low in cells overexpressing SERCA2-siRNA, whereas no changes were seen in TRPC1-overexpressing cells (23).
Accordingly, SERCA2 silencing was followed by increased transcription of NCX, TRPC4, and TRPC5 in cardiac myocytes (24). Neonatal rat and chicken embryo cardiac myocytes expressing SERCA2-siRNA endogenously had reduced expression of SERCA2 as demonstrated by immunostaining, western blotting, real-time RT-PCR, and microscopy (24). The functional effects of the reduced SERCA2 expression was tested on Ca2+ signaling in rat myocytes by measuring Ca2+ transients, and ICa,L in neonatal rat myocytes under voltage clamp protocol. Whereas, there was reduced Ca2+ transient amplitude (i.e., reduced sarcoplasmic reticulum Ca2+ load) in the myocytes subjected to SERCA2-siRNA, there was no differences in ICa,L under the voltage clamp protocol tested. However, the myocytes were shown to have the ability to initiate transient elevations in cytosolic Ca2+ upon membrane stimulation, in compensatory mechanisms. To understand this process, the presence or absence of Na+ to examine the role of NCX to cytosolic Ca2+ removal under voltage-clamp depolarizations, and Ba2+ influx measurement in fura-2-loaded myocytes treated with thapsigargin to block SERCA pump activity to examine the role of TRPC channels, in another experiment, were together, employed to access the impact of SERCA2-siRNA on NCX, TRPC channels and Ca2+ signaling. This investigation revealed that reduced SERCA2 expression was associated with an elevation in TRPC expression and activity, as well as increased NCX expression and activity, that together may boost and normalize for reduced sarcoplasmic reticulum Ca2+ signaling following SERCA2 depletion (24). This mechanism was attributed to NCX and TRPC transcription, as well as, the upregulation of other transcriptional factors such as stimulating protein 1, myocyte enhancer factor 2, and nuclear factor activated T-cell, cytoplasmic 4 (24). In application of this finding, SR releases 70–80% of the Ca2+ needed for contraction into the cytosol. SERCA, NCX, and ATP-dependent Ca2+pumps remove Ca2+ from the cytosol. If reduction in expression of SERCA2, which can directly affect SERCA/Ca2+ pump, inversely increases NCX and TRPC levels, then TRPC and NCX govern [Ca2+]i, excitation- contraction coupling and inotropy, because TRPC and NCX operated Ca2+ homeostasis will compensate for SERCA Ca2+ pump. Figure 2 illustrates the organellar TRP channels and Ca2+-dependent regulatory proteins regulation of Ca2+-handling.
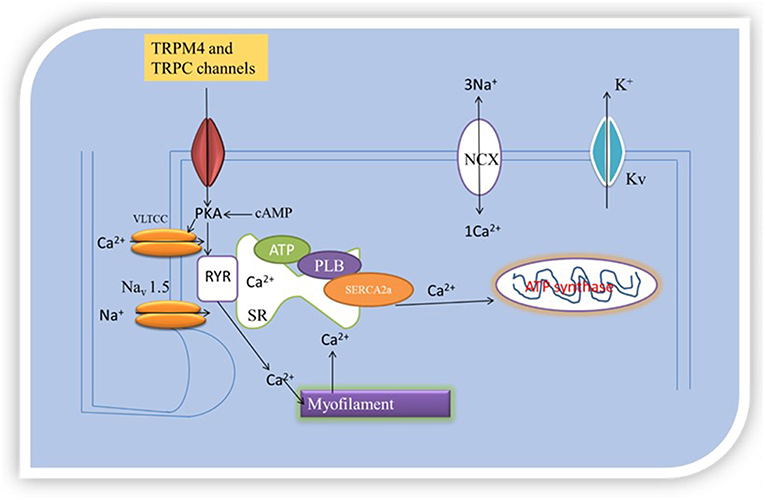
Figure 2. Oganellar Mechanisms of TRP channels mediated pathologic Ca2+ handling. Reduced levels of [Ca2+]i means that the heart cannot contract and pump blood while high levels of [Ca2+]i means that the heart cannot relax. TRPM4 and TRPC interact with Ca2+-dependent regulatory proteins (SERCA2a and NCX). How this interaction occurs either reduces or boosts excitation and contraction coupling (EC) by either reducing or increasing [Ca2+]i. EC is initiated by excitation of the cell membrane which activates Navl.5 leading to the opening of VLTCC. VLTCC opening allows passage of inward Ca2+ current that triggers opening of ryanodine receptor 2 (RyR2) channels by a Ca2+- induced Ca2+ release process. The Ca2+-induced Ca2+ release leads to coordinated release of sarcoplasmic reticulum (SR) Ca2+ caused by PLB phosphorylation. Intracellular free Ca2+ concentration become high, activating the myofilaments after binding with troponin C. TRPM4 and TRPC mediated Ca2+ entry can couple to this process of elevation in [Ca2+]i to cause contraction. This process is part of pathologic Ca2+-handling.
Further support is delineated from the study of Oslon's group in 2006. It appears from the study that TRPC6-calcineurin-NFAT pathway, inversely, regulates the expression of SERCA2 (14), suggesting reciprocal relationship between the channel and the protein/gene. What is not known is whether downregulation of TRPC4/5 would upregulate SERCA2 expression in reverse. Understanding the precision of this relationship is pertinent and vital. It is pertinent, since the Seth et al., Eder et al., and the Pani et al. studies highlight novel association between TRP channels and Ca2+-dependent regulatory proteins that governs [Ca2+]i. It is vital since Ca2+-dependent regulatory proteins that associate with the TRP channels are potential therapeutic targets for Ca2+ -mishandling (25), and Ca2+-dependent regulatory gene therapy approaches are being understood to increase myocardial SERCA2a in heart failure contractile abnormality (26). This in part emphasis the importance of this review in development of therapeutic agents that can benefit the heart in contractile dysfunction beyond short-term.
For instance, it is known that Trpm4−/− mice and their control littermate had no differences in cardiac contractile parameters under basal conditions, but infusion of ≤300 ng/kg·min isoprenaline resulted in robust increases (11). This is an observation of incremental physiological contractile activity, which may be rather beneficial in heart failure, cardiomyopathy and arrhythmias, characterized by diminished contractile tone. Thus, genetic downregulation of TRPM4 proteins might be a novel therapeutic approach. Trpm4−/− mice with ischaemic heart failure had improved survival rate and enhanced β-adrenergic cardiac response, indicating better contractile performance (27). Together, TRP channels have pleiotropic roles in the heart (28), and are part of excellent potential targets in gene therapy approach being appreciated to increase myocardial SERCA2a expression in heart failure contractile abnormality.
Given that SERCA2a regulates Ca2+ homeostasis, it has impact on systolic and diastolic functions. An elegant approach to improve left ventricular systolic function can be through activation of cardiac myosin, as well. Besides conventional inotropic agents that modulate Ca2+ homeostasis in the myocardium, molecular motor (myosin) and sarcomeric scaffolding, and energetic agents referred to as cardiac myotropes and mitotropes, respectively, have been proposed as pharmacological agents that improve myocardial performance (29). The framework of this proposal is to better understand the clinical approaches of agents that ameliorate worse myocardial performance and accelerate appropriate testing of therapeutic agents that can improve cardiac contractility and contraction.
TRP channels regulate Ca2+-handling through interaction with Ca2+-dependent regulatory proteins. This interaction can lead to spontaneous ectopy, but how this may occur is not known. Spontaneous ectopy may be regarded as uncontrolled variability in heart rhythm during repolarization, which may or may not be pathological. Cycles of Ca2+ fluxes during normal heart beat characterize excitation- contraction coupling and permit homogenous action of cardiac sarcomeres. Increased Ca2+ influx, Ca2+ leaks, spontaneous oscillatory of Ca2+ and Ca2+ overload disturb systole and diastole and cause arrhythmias (30). As stated in previous sections, this implicates the regulation and mechanisms of Ca2+-handling. These processes are schematically summarized in Figure 2, organellar mechanisms of TRP channel mediated pathological Ca2+-handling. TRPM4 and TRPC channels trigger Ca2+ entry, through membrane depolarization and permeability, respectively, consequently leading to Ca2+ overload. Ca2+ overload promote alteration in action potential wave form, Ca2+-mishandling, inotropy, and sustain electrical remodeling, through Ca2+ transients. Ca2+ overload and large Ca2+ transients increase repolarization (31). Prolonged action potential duration leads to early afterdepolarizations (EAD) and delayed afterdepolarizations (DAD) ectopic firing. EADs and DADs are classes of ectopy (32).
EADs are abnormally secondary cell membrane depolarizations during repolarization phases of action potential. Prolongation of APD is the principle factor that causes EAD, because reduced repolarization enables ICa,L to recover from inactivation, which leads to depolarizing inward movement of Ca2+ ions along the plateau phase of action potential to produce Ca2+ inward current. EAD is also caused by increases in NCX current. DADs are also abnormally secondary cell membrane depolarizations during the repolarization phases but are known to be caused by abnormally diastolic Ca2+ release from sarcoplasmic reticulum Ca2+ stores, which NCX also contributes to. In response to transmembrane Ca2+ entry, a specialized Ca2+-dependent regulatory apparatus, SR Ca2+ channels known as RyRs release Ca2+. RyRs are closed in diastolic condition but are open if functionally defective or if the SR Ca2+ is above physiological arrange (33). Under physiological conditions and in healthy hearts, exercise-induced activation of sympathetic nervous system increases catecholamines produced from the chromaffin cells, which bind to and stimulate the G protein-coupled β-adrenergic receptors. This activation, in turn, stimulates adenylate cyclase to activate the cAMP-dependent protein kinase A (PKA). PKA phosphorylates major Ca2+-handling proteins, such PLB, RYR2, and L-type Ca2+ channel. Increased RyR2 Ca2+ release in the SR and enhanced Ca2+ by SERCA2a, enhances [Ca2+]i and contractility. PKA hyperphosphorylation of RyR2 reduces the binding affinity of the RYR2-stabilizing subunit, calstabin2, producing robust activity of the RYR2 channel to Ca2+-dependent activation (34). PKA hyperphosphorylation of RyR2 produces a diastolic SR Ca2+ leak in cardiomyocytes leading to persistently diminished SR Ca2+ content and contractility (34). RYR2 channel diastolic calcium leak contributes to abnormal contractility in arrhythmogenesis and sudden cardiac death (35). Furthermore, SERCA2a diminished Ca2+ loading and enhanced Ca2+ exflux through NCX also results in SR reduced Ca2+ loading and reduced cardiac contractility. 1 Ca2+ release during the diastole is exchanged for 3 extracellular Na+ by NCX. The result of this is a net depolarizing inward positive-ion movement called transient inward current (Iti) that underlies the DADs, and contractile dysfunction (36).
EADs and DADs appear to be mediated by TRPM4 protein. TRPM mediated Ca2+ signaling can modulate action potential wave form and cause EADs. TRPM can also modulate diastolic Ca2+ release from sarcoplasmic reticulum Ca2+ stores and cause DADs. 9-Phenanthrol, an inhibitor of TRPM4 channel abolished hypoxia and re-oxygenation-induced EADs in a mouse model (37). TRPM4-induced ectopy can be drawn on ion channel determinant of cell membrane depolarization and action potential morphology. TRPM4b a calcium activated non-selective (CAN) channel regulates cell membrane depolarization (38). TRPM4 activation is a process that contributes to the controls of the magnitude of Ca2+ influx by regulating membrane potential, and intracellular Ca2+ increased through ICa,L upon TRPM4 protein depletion (11).
CAN channel activity had been suggested to contribute to Iti initiated by Ca2+ waves that underline DAD. Iti has been described in Purkinje fibers, atrial and ventricular cardiomyocytes, and in sinoatrial node cells (39). In sinoatrial node (SAN), cardiac automaticity resulted from a CAN current that is attributed in part to TRPM4 (40), which slowed diastolic depolarizations lope due to the nature of “funny” current, and NCX activity (39). The molecular identity of Iti is controversial, but it appears to reflect 3 Ca2+- dependent components: NCX, Ca2+-activated chloride current and current mediated by CAN channels such as TRPM4 (40). Put together, these studies support the hypothesis that Iti is mediated by TRPM4. However, that Iti is mediated by TRPM4 may be questioned by the fact that the single channel conductance of the Iti-mediating channel was 120 Ps (41), which is much larger than the ~25 pS of TRPM4 (38).
The past few decades have witnessed tremendous evolution in characterization of the molecular and genetic mechanisms of acquired and inherited arrhythmias. The mechanisms are as numerous as the phenotypes with which the arrhythmias present. Identifying the sources that control Ca2+ is novel in understanding arrhythmogenesis. The TRP channels mediate Ca2+ flux and voltage changes across membranes. Regulation of Ca2+-handling by the TRP channels indicate they can potentially boost Ca2+ cycling disorders. Plasma membrane sensory and metabotropic TRPM4 subgroup is a drug candidate for Brugada syndrome and familial heart blocker (42). Better understanding of the TRP channels, Ca2+-handling and contractility is crucial.
Essentially, studies illustrating the roles of TRP channels in pathological Ca2+-handling and spontaneous ectopy are lacking. This study reflects on TRPM and TRPC seminal works, based on their biophysical properties, to state mechanistically their potential implications in Ca2+ signaling dynamics. Intracellular Ca2+ homeostasis show tremendous changes during cardiac cycle. Intracellular Ca2+ homeostasis is regulated by spatial signaling processes within restricted regions, rather than by changes in global cytosolic [Ca2+]i. [Ca2+]i is mediated by Ca2+ release from intracellular organelles, Ca2+ entry across plasma membrane through receptor agonists, receptor-activated Ca2+ channels, voltage-gated Ca2+ channels, and by ligand-gated cation channels. It appears that while overexpression of the TRPCs increase [Ca2+]i, TRPM4 depletion increases [Ca2+]i, through Ca2+ influx. This can be explained by the biophysical properties of the channels. Mechanization of [Ca2+]i, is defective in heart failure, failing hearts, cardiomyopathy, arrhythmia and sudden cardiac deaths. Defective intracellular Ca2+ homeostasis is responsible for pathological Ca2+-handling and contractile dysfunction. Hearts in these conditions show abnormal contractility, characterized by decreased SR Ca2+ sequestration, diminished intracellular Ca2+ transients, and enhanced diastolic SR Ca2+ leak activity. Sources that regulate these processes are unknown completely, and may include TRP channels and TRP channels SOCE associated mechanisms. Highlighting a potential therapeutic opportunity of the channels, this work discussed TRP channels mediated pathologic Ca2+-handling in spontaneous ectopy, and stated further scopes for TRP channels functions in cardiac pathophysiology.
The author confirms being the sole contributor of this work and has approved it for publication.
ME is supported by Government of Australia Training Research Program Scholarship.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Bers DM. Calcium cycling and signaling in cardiac myocytes. Annu Rev Physiol. (2008) 70:23–49. doi: 10.1146/annurev.physiol.70.113006.100455
2. Pogwizd SM, Schlotthauer K, Li L, Yuan W, Bers DM. Arrhythmogenesis and contractile dysfunction in heart failure: roles of sodium–calcium exchange, inward rectifier potassium current and residual b-adrenergic responsiveness. Circ Res. (2001) 88:1159–67. doi: 10.1161/hh1101.091193
3. Piacentino V III, Weber RC, Chen X, Weisser-Thomas J, Margulies BK, Bers MD, et al. Cellular basis of abnormal calcium transients of failing human ventricular myocytes. Circ Res. (2003) 92:651–8. doi: 10.1161/01.RES.0000062469.83985.9B
4. Hasenfuss G, Pieske B. Calcium cycling in congestive heart failure. J Mol Cell Cardiol. (2002) 34:951–69. doi: 10.1006/jmcc.2002.2037
5. Lan F, Lee SA, Liang P, Sanchez-Freire V, Nguyen KP, Wang L, et al. Abnormal calcium handling properties underlie familial hypertrophic cardiomyopathy pathology in patient-specific induced pluripotent stem cells. Cell. (2012) 12:101–13. doi: 10.1016/j.stem.2012.10.010
6. Kubo H, Margulies BK, Piacentino V III, Gaughan PJ, Houser RS. Patients with end-stage congestive heart failure treated with b-adrenergic receptor antagonists have improved ventricular myocyte calcium regulatory protein abundance. Circulation. (2001) 104:1012–8. doi: 10.1161/hc3401.095073
8. Montell C, Jones K, Hafen E, Rubin G. Rescue of the Drosophila phototransduction mutation trp by germline transformation. Science. (1985) 230:1040–3. doi: 10.1126/science.3933112
9. Du J, Xie J, Zhang Z, Tsujikawa H, Fusco D, Silverman D, et al. TRPM7-mediated Ca2+ signals confer fibrogenesis in human atrial fibrillation. Circ Res. (2010) 106:992–1003. doi: 10.1161/CIRCRESAHA.109.206771
10. Yu Y, Chen S, Xiao C, Jia Y, Guo J, Jiang J, et al. TRPM7 is involved in angiotensin II induced cardiac fibrosis development by mediating calcium and magnesium influx. Cell Calcium. (2014) 55:252–60. doi: 10.1016/j.ceca.2014.02.019
11. Mathar I, Kecskes M, Mieren G, Jacobs G, Londoño CJ, Uhl S, et al. Increased β-adrenergic inotropy in ventricular myocardium from Trpm4–/– mice. Circ Res. (2014) 114:283–94. doi: 10.1161/CIRCRESAHA.114.302835
12. Zhou Y, Yi X, Wang T, Li M. Effects of angiotensin II on transient receptor potential melastatin 7 channel function in cardiac fibroblasts. Exp Ther Med. (2015) 2008:2008–12. doi: 10.3892/etm.2015.2362
13. Simard C, Hof T, Keddache Z, Launay P, Guinamard R. The TRPM4 non-selective cation channel contributes to the mammalian atrial action potential. J Mol Cell Cardiol. (2013) 59:11–9. doi: 10.1016/j.yjmcc.2013.01.019
14. Kuwahara K, Wang Y, McAnally J, Richardson AJ, Bassel-Duby R, Hill AJ, et al. TRPC6 fulfills a calcineurin signaling circuit during pathologic cardiac remodeling. J Clin Invest. (2006) 116:3114–26. doi: 10.1172/JCI27702
15. Wu X, Edera P, Changa B, Molkentina DJ. TRPC channels are necessary mediators of pathologic cardiac hypertrophy. Proc Natl Acad Sci USA. (2010) 107:7000–5. doi: 10.1073/pnas.1001825107
16. Liao Y, Plummer NW, George MD, Abramowitz J, Zhu MX, Birnbaumer L. A role for Oraiin TRPC-mediated Ca2+entry suggests that a TRPC: orai com- plex may mediate store and receptor operated Ca2+entry. Proc Natl Acad Sci USA. (2009) 106:3202–6. doi: 10.1073/pnas.0813346106
17. Yuan JP, Zeng W, Huang GN, Worley PF, Muallem S. STIM1 heteromul- timerizes TRPC channels to determine their function as store- operated channels. Nat Cell Biol. (2007) 9:636–45. doi: 10.1038/ncb1590
18. Pani B, Ong HL, Liu X, Rauser K, Ambudkar IS, Singh BB. Lipid rafts determine clustering of STIM1inendoplas- micreticulum-plasma membrane junctions and regulation of store- operated Ca2+ entry (SOCE). J Biol Chem. (2008) 283:17333–40. doi: 10.1074/jbc.M800107200
19. Eder P, Molkentin JD. TRPC channels as effectors of cardiac hypertrophy. Circ Res. (2011) 108:265–72. doi: 10.1161/CIRCRESAHA.110.225888
20. Pani B, Ong HL, Brazer SC, Liu X, Rauser K, Singh BB, et al. Activation of TRPC1 by STIM1 in ER– PM microdomains involves release of the channel from its scaffold caveolin-1. Proc Natl Acad Sci USA. (2009) 106:20087–92. doi: 10.1073/pnas.0905002106
21. Nikolova-Krstevski V, Wagner S, Yu ZY, Cox CD, Cvetkovska J, Hill AP, Huttner IG, et al. Endocardial TRPC-6 channels act as atrial mechanosensors and load-dependent modulators of endocardial/myocardial cross-talk. JACC Basic Transl Sci. (2017) 2:575–90. doi: 10.1016/j.jacbts.2017.05.006
22. Eder P, Probst D, Rosker C, Poteser M, Wolinski H, Kohlwein SD, et al. Phospholipase C-dependent control of cardiac calcium homeostasis involves a TRPC3-NCX1 signaling complex. Cardiovasc Res. (2006) 73:111–9. doi: 10.1016/j.cardiores.2006.10.016
23. Pani B, Cornatzer E, Cornatzer W, Shin D, Pittelkow RM, Hovnanian A, et al. Up-regulation of transient receptor potential canonical 1(TRPC1) following Sarco(endo)plasmic reticulum Ca2+-ATPase 2 gene silencing promotes cell survival: a potential role for TRPC1 in Darier's disease. Mol Biol Cell. (2006) 17:4446–58. doi: 10.1091/mbc.e06-03-0251
24. Seth M, Sumbilla C, Mullen SP, Lewis D, Klein GM, Hussain A, et al. Sarco(endo)plasmic reticulum Ca2+ ATPase (SERCA) gene silencing and remodeling of the Ca2+ signaling mechanism in cardiac myocytes. Proc Natl Acad Sci USA. (2004) 101:16683–8. doi: 10.1073/pnas.0407537101
25. Jessup M, Greenberg B, Mancini D, Cappola T, Pauly FD, Jaski B, et al. Calcium upregulation by percutaneous administration of gene therapy in cardiac disease (CUPID) a phase 2 trial of intracoronary gene therapy of sarcoplasmic reticulum Ca2_-ATPase in patients with advanced heart failure. Circulation. (2011) 124:304–13. doi: 10.1161/CIRCULATIONAHA.111.022889
26. Kho C, Lee A, Jeong D, Oh GJ, Chaanine HA, Kizana E, et al. SUMO1-dependent modulation of SERCA2a in heart failure. Nature. (2011) 477:601–5. doi: 10.1038/nature10407
27. Jacobs G, Oosterlinck W, Dresselaers T, Geenens R, Kerselaers S, Himmelreich U, et al. Enhanced β-adrenergic cardiac reserve in Trpm42/2 mice with ischaemic heart failure. Cardiovasc Res. (2015) 105:330–9. doi: 10.1093/cvr/cvv009
28. Demion M, Thireau J, Gueffier M, Finan A, Khoueiry Z, Cassan C, et al. Trpm4 gene invalidation leads to cardiac hypertrophy and electrophysiological alterations PLoS ONE. (2014) 9:e115256. doi: 10.1371/journal.pone.0115256
29. Psotka MA, Gottlieb SS, Francis GS, Allen LA, Teerlink JR, Adams KF Jr., et al. Cardiac calcitropes, myotropes, and mitotropes: JACC review topic of the week. J Am Coll Cardiol. (2019) 73:2345–53. doi: 10.1016/j.jacc.2019.02.051
30. Bers DM. Cardiac sarcoplasmic reticulum calcium leak: basis and roles in cardiac dysfunction. Annu Rev Physiol. (2014) 76:107–27. doi: 10.1146/annurev-physiol-020911-153308
31. Venetucci LA, Trafford AW, O'Neill SC, Eisner DA. The sarcoplasmic reticulum and arrhythmogenic calcium release. Cardiovasc Res. (2008) 77:285–92. doi: 10.1093/cvr/cvm009
32. Weiss NJ, Garfinkel A, Karagueuzian SH, Chen P, Qu Z. Early afterdepolarizations and cardiac arrhythmias Heart Rhythm. (2010) 7:1891–9. doi: 10.1016/j.hrthm.2010.09.017
33. Fischer TH, Maier LS, Sossalla S. The ryanodine receptor leak: how a tattered receptor plunges the failing heart into crisis. Heart Fail Rev. (2013) 18:475–83. doi: 10.1007/s10741-012-9339-6
34. Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N, Marks AR. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell. (2000) 101:365–76. doi: 10.1016/S0092-8674(00)80847-8
35. Dobrev D, Wehrens XH. Role of RyR2 phosphorylation in heart failure and arrhythmias: controversies around ryanodine receptor phosphorylation in cardiac disease. Circ Res. (2014) 114:1311–9. doi: 10.1161/CIRCRESAHA.114.300568
36. Bers DM, Pogwizd SM, Schlotthauer K. Upregulated Na/Ca exchange is involved in both contractile dysfunction and arrhythmogenesis in heart failure. Basic Res Cardiol. (2002) 97(Suppl. 1):I36–42. doi: 10.1007/s003950200027
37. Simard C, Sallé L, Rouet R, Guinamard R. Transient receptor potential melastatin 4 inhibitor 9-phenanthrol abolishes arrhythmias induced by hypoxia and re-oxygenation in mouse ventricle. Br J Pharmacol. (2012) 165:2354–64. doi: 10.1111/j.1476-5381.2011.01715.x
38. Launay P, Fleig A, Perraud AL, Scharenberg AM, Penner R, Kinet JP. TRPM4 is a Ca2+-activated nonselective cation channel mediating cell membrane depolarization. Cell. (2002) 109:397–407. doi: 10.1016/S0092-8674(02)00719-5
39. Guinamard R, Demion M, Chatelier A, Bois P. Calcium-activated nonselective cation channels in mammalian cardiomyocytes. Trends Cardiovasc Med. (2006) 16:245–50. doi: 10.1016/j.tcm.2006.04.007
40. Hof T, Simard C, Rouet R, Sallé L, Guinamard R. Implication of the TRPM4 nonselective cation channel in mammalian sinus rhythm. Heart Rhythm. (2013) 10:1683–9. doi: 10.1016/j.hrthm.2013.08.014
41. Hill JA Jr., Coronado R, Strauss HC. Reconstitution and characterization of a calcium-activated channel from heart. Circ Res. (1988) 62:411–5.
Keywords: TRP channels, SR Ca2+-ATPase, NCX, ectopy, Ca2+-handling
Citation: Ezeani M (2019) TRP Channels Mediated Pathological Ca2+-Handling and Spontaneous Ectopy. Front. Cardiovasc. Med. 6:83. doi: 10.3389/fcvm.2019.00083
Received: 04 November 2018; Accepted: 04 June 2019;
Published: 20 June 2019.
Edited by:
Giannis G. Baltogiannis, Vrije University Brussel, BelgiumReviewed by:
Osmar Antonio Centurion, Universidad Nacional de Asunción, ParaguayCopyright © 2019 Ezeani. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Martin Ezeani, bWFydGluLmV6ZWFuaUBtb25hc2guZWR1; bWV6ZWFuaTU1NEBnbWFpbC5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.