 Przemysław Błyszczuk
Przemysław Błyszczuk
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med. , 16 May 2019
Sec. Cardiovascular Biologics and Regenerative Medicine
Volume 6 - 2019 | https://doi.org/10.3389/fcvm.2019.00064
This article is part of the Research Topic Cutting Edge Methodologies in Experimental Cardiovascular Research View all 10 articles
Myocarditis is defined as an inflammation of the cardiac muscle. In humans, various infectious and non-infectious triggers induce myocarditis with a broad spectrum of histological presentations and clinical symptoms of the disease. Myocarditis often resolves spontaneously, but some patients develop heart failure and require organ transplantation. The need to understand cellular and molecular mechanisms of inflammatory heart diseases led to the development of mouse models for experimental myocarditis. It has been shown that pathogenic agents inducing myocarditis in humans can often trigger the disease in mice. Due to multiple etiologies of inflammatory heart diseases in humans, a number of different experimental approaches have been developed to induce myocarditis in mice. Accordingly, experimental myocarditis in mice can be induced by infection with cardiotropic agents, such as coxsackievirus B3 and protozoan parasite Trypanosoma cruzi or by activating autoimmune responses against heart-specific antigens. In certain models, myocarditis is followed by the phenotype of dilated cardiomyopathy and the end stage of heart failure. This review describes the most commonly used mouse models of experimental myocarditis with a focus on the role of the innate and adaptive immune systems in induction and progression of the disease. The review discusses also advantages and limitations of individual mouse models in the context of the clinical manifestation and the course of the disease in humans. Finally, animal-free alternatives in myocarditis research are outlined.
The World Heart Federation estimated that about 400,000 persons die annually worldwide because of inflammatory heart diseases. Epidemiologic post-mortem studies identified myocarditis as an important cause of unexpected and sudden death. Myocarditis has been implicated in cardiac sudden deaths in young adults at the rate of 8.6–12% (1, 2). The occurrence of myocarditis in sudden death in children was reported at the rate of 17% (3). Considering cardiovascular death in children and young adults, myocarditis was a major cause in 10–42% cases (4).
Myocarditis is classified as an inflammatory disease of the heart muscle. Traditionally, myocarditis is diagnosed based on endomyocardial biopsies. According to the widely used “Dallas” criteria published in 1987, a diagnosis of active myocarditis requires the presence of inflammatory infiltrates of non-ischemic origin in myocardial tissue associated with necrosis and/or degeneration of adjacent cardiomyocytes. Presence of inflammatory infiltrates in the absence of apparent myocyte damage is classified as borderline myocarditis (5). The definition of myocarditis has been more recently enumerated by the ESC Working Group on Myocardial and Pericardial Diseases, which proposed abnormal number of inflammatory infiltrates in myocardial tissue as ≥14 leucocytes/mm2 including up to 4 monocytes/mm2 with the presence of ≥7 cells/mm2 CD3-positive T lymphocytes (6). These criteria have become widely accepted. Beside this common histological feature of inflammatory condition of the heart, there is a high diversity observed in the disease cause, characteristic of inflammatory infiltrates, clinical symptoms, course of inflammation, and the prognosis.
Patients with myocarditis, proven via biopsies, show 55–80% 5-year transplantation-free survival (7–10). Mortality is observed not only during the acute phase, but also during the follow up. Myocarditis is a progressive disease with two post-acute clinical scenarios. In the first scenario, resolution of the inflammation is followed by complete recovery associated with improved heart function. It has been reported that myocarditis resolves spontaneously in ~40–60% of cases (11, 12). In the second scenario, the acute phase is followed by development of stable dilated cardiomyopathy (DCM). Follow-up clinical studies showed development of DCM pathology over a period of several years in 14–52% patients with histologically proven myocarditis (12). DCM is referred to as left ventricular dilation associated with systolic dysfunction in the absence of coronary artery disease. Histologically, DCM is manifested by the extensive replacement of cardiac muscle cells with fibrotic tissue and deposition of collagen (13, 14). DCM patients develop not only heart pump weakening, but also heart valve problems, blood clots, and arrhythmias leading to heart and secondary organ failures. DCM patients show transplantation-free survival at the rate of 50–60% over 5 years in children (15) and in adults (7, 16, 17), however improved medication can increase the survival rate up to 80% (16). Particularly high mortality has been reported for patients with DCM due to Chagas disease (17). DCM can be associated with inflammation of the myocardium. Co-occurrence of myocarditis and DCM is referred to as inflammatory DCM (iDCM) (6). In fact, 16–30% of patients with chronic DCM show immunohistochemical evidences of myocardial inflammation (15, 18). Cardiac deaths in the follow-up myocarditis cohort were predominantly associated with DCM characteristic including systolic dysfunction and left ventricular dilation (9). Accordingly, ventricular dilatation and systolic dysfunction are associated with worse prognosis for myocarditis patients (8).
In Europe and North America myocarditis is often idiopathic. Infections with cardiotropic enteroviruses such as coxsackievirus B3 (CVB3) have been associated with the disease and considered as a causative agent. The prevalence of enteroviruses detected in cardiac biopsies of myocarditis patients was reported at the rate of 14–57% (19). Other viruses such as parvovirus B19, adenoviruses or herpesviruses have also been detected in biopsies of myocarditis patients (19). Over decades, a shift in detection of enterovirus and adenovirus to parvovirus B19 and herpesviruses has been observed. However, the causative role of detected viruses in myocarditis patients is not evident. For example, surprisingly high prevalence of parvovirus B19 has been detected in myocarditis patients, suggesting its pathogenic role in the disease (20). More recent data showed, however, a high prevalence of parvovirus B19 also in myocarditis-negative hearts (21). Thus, the causative or associative role of individual viral infections in pathogenesis of myocarditis is still under investigation. Furthermore, it also remains to be elucidated, whether the persistence of the viral genome in the myocardium influences the clinical outcomes. So far, clinical studies resulted rather in contradicting conclusions (8, 22). In Europe and North America, myocarditis is also diagnosed in patients with Lyme disease (borreliosis). The disease is caused by bacteria Borrelia burgdorferi, which is transmitted by the bite of an infected ticks. It is estimated that up to 10% of Lyme disease patients develop myocarditis (23).
In Latin America, infections with protozoan Trypanosoma cruzi (Chagas disease) are the most common cause of inflammatory heart disease (24). The etiology of Chagas disease is quite well-established. Bites of blood-sucking triatomines (called also kissing bugs) spread the infective forms of the parasite. In humans, trypanosomal infection triggers the disease with two clinically distinct phases. The acute phase lasts several weeks and is usually asymptomatic or is associated with fever and local swelling or skin lesion. 10–30 years later about one third of the infected individuals develop a chronic form of the disease primarily manifested by DCM or iDCM, but also by neurological and/or gastrointestinal track pathologies. The chronic phase of the Chagas disease is usually progressive, leading to permanent heart failure (25). Cardiac dysfunction due to myocarditis and iDCM represents the most frequent and the most severe clinical manifestation of Chagas disease, which is associated with poor prognosis and high mortality rates (24, 26).
Non-infectious causes of myocarditis include mainly systemic autoimmune diseases and certain medications (27). Myocarditis has been observed for example in systemic lupus erythematosus (28) and in myasthenia gravis (29) patients. Recently, numerous cases of fatal myocarditis have been reported in cancer patients shortly after starting treatment with immune checkpoint inhibitors (30, 31). Immune checkpoint inhibitors refer to a category of drugs (antibodies) targeting negative regulators of T cell response, such as cytotoxic T-lymphocyte associated protein-4 (CTLA-4), programmed cell death protein-1 (PD-1), and PD-1 ligand (PD-L1). It is considered that immune checkpoint inhibitors may activate heart-specific autoimmunity in predisposed individuals (32). Today, it is widely accepted that autoimmune mechanisms are involved in the development and/or progression of myocarditis (33). Clinical evidences suggest active autoimmune response in human myocarditis on both, cellular and humoral levels. Initially, the concept of heart-specific autoimmunity came from the observation of high titers of heart-specific autoantibodies in CVB3 infected individuals. Accordingly, 30% of patients with myocarditis and DCM develop high titers of heart-specific autoantibodies (34). Cardiac myosin heavy chain (MyHC) has been identified as the most prominent autoantigen for circulating heart autoantibodies in myocarditis and cardiomyopathy patients (35). In fact, the presence of anti-MyHC autoantibodies has been associated with worse left ventricular systolic function and diastolic stiffness in patients with chronic myocarditis (36). There are strong indications that also antigen-presenting cells play an important role in the pathogenesis of myocarditis in humans by promoting autoimmune mechanisms. For example, histological analysis demonstrated increased levels of major histocompatibility complex (MHC) class I and II, known as human leukocyte antigen (HLA) complexes (37) and co-stimulatory molecules B7-1, B7-2, and CD40 (38) in hearts of myocarditis patients. More recent data pointed also to the importance of the humoral response in myocarditis (39).
Clinical manifestation of myocarditis varies with a broad spectrum of symptoms, ranging from asymptomatic courses through shortness of breath, cardiac arrhythmias to chest pain resembling myocardial infarction (27, 40, 41). Myocarditis is often associated with left ventricular dysfunction (42), in some cases with cardiac arrhythmias (43) and elevated levels of certain biomarkers (6). These clinical symptoms are, however, not specific for myocarditis and the definitive diagnosis requires detection of inflammatory cells in the myocardium, typically on endomyocardial biopsy. In addition to histological analyses of cardiac biopsies, the assessment of myocarditis could be performed using a cardiac magnetic resonance imaging (44, 45). Improved imaging protocols confirmed usefulness of this modern, non-invasive technology in diagnosing myocarditis (46). Magnetic resonance imaging shows excellent diagnostic accuracy in patients with acute symptoms, while its usefulness is limited in patients with suspected chronic myocarditis (45, 47). On the one hand, histological evaluation of myocardial biopsies still represents a “gold standard,” mainly because it allows not only to diagnose myocarditis, but also to identify infective agents and characterize the type of inflammatory cells. These data can be indicative for selection of the personalized treatment strategy and may be predictive for disease outcome (27, 40, 41, 48). In fact, endomyocardial biopsies confirm inflammation in 44–70% of patients with suspected myocarditis (49–51). On the other hand, due to the often patchy pattern of inflammation in the heart, endomyocardial biopsies-based diagnosis of myocarditis yields rather low sensitivity (52–54). Biopsies guided by non-invasive molecular imaging and/or electroanatomic mapping could increase the success rate. It seems, however, that the actual prevalence of myocarditis possibly remains underestimated. It is noteworthy that, incidental inflammation of the myocardium evaluated in a clinicopathological study reported that any inflammatory cells were present in 18% and multifocal inflammation in 9% of total cardiac and non-cardiac deaths (55).
In the clinic, myocarditis can be classified based on the causative, histological, and clinicopathological criteria, which are summarized in Table 1. The causative criteria define infectious agents (virus, protozoa, or bacteria) or non-infectious condition (autoimmune diseases, medications etc.) associated with myocarditis. Identification of the infectious agent or potential non-infectious trigger may be indicative not only for disease etiology, but also helps to choose the most effective therapeutic strategy for the affected patients. In addition to identification of the causative agent, histological and immunohistological analyses are performed to categorize myocarditis based on the presence, morphology and type of inflammatory infiltrates in the myocardium. Lymphocytic myocarditis characterized by extensive infiltration of lymphocytes and monocytes with signs of cardiomyocyte necrosis (active lymphocytic myocarditis) represents the most frequent type of myocarditis (10). Lymphocytic myocarditis is often observed in myocardium tested positive for viral persistence. Less common forms of myocarditis represent giant cell myocarditis and eosinophilic myocarditis. Giant cell myocarditis is characterized by the presence of multinucleated giant cells and lymphocytes on heart biopsies. Presence of giant cells within non-caseating granulomas, usually associated with myocardial fibrosis is referred to as cardiac sarcoidosis (56). The characteristic feature of eosinophilic myocarditis is the presence of eosinophil-rich infiltrates in the myocardium and extensive myocyte necrosis, which is accompanied with elevated level of circulating eosinophils (57). Giant cell myocarditis and eosinophilic myocarditis are associated with particularly poor prognosis (57–60).
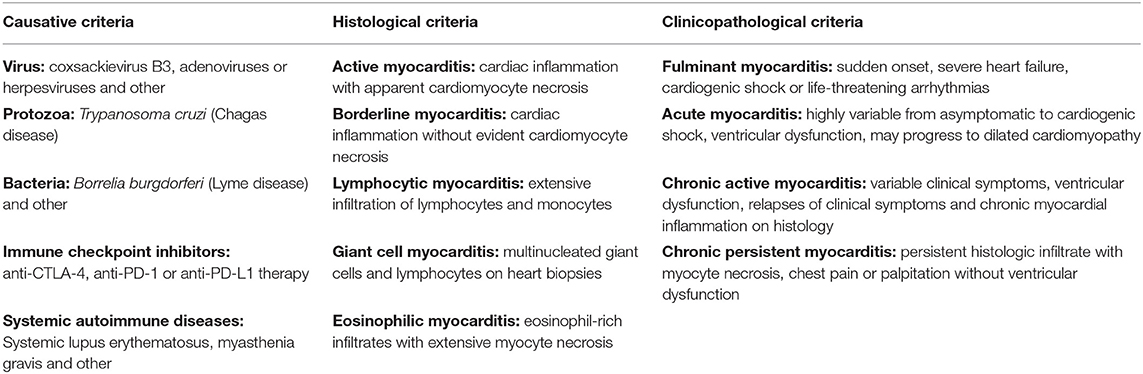
Table 1. Clinical classifications of myocarditis.
Combination of the histologic data and clinical course of the disease resulted in clinicopathologic classification of myocarditis (61). Parameters such as onset of the disease, initial clinical and histological presentation, disease course and cardiac dysfunction define acute, fulminant, chronic active and chronic persistent subtypes of myocarditis. Acute myocarditis represents the most common type of myocarditis, in which symptoms last typically for days or weeks and the acute phase is followed by spontaneous improvement or development of stable DCM (62). In patients with fulminant myocarditis disease progresses rapidly resulting in severe heart failure and cardiogenic shock with mortality rate of 30–40% during the acute phase (63, 64). Patients diagnosed with fulminant myocarditis surviving the acute phase have been instead suggested to have excellent long-term prognosis (65), although a recently published study demonstrated contradictive findings (66). In its chronic form, myocarditis is detected over a period of three or more months. Clinical and histologic relapses and development of ventricular dysfunction is characteristic for chronic active myocarditis, whereas chronic persistent myocarditis is characterized by persistent presence of inflammatory cells in the myocardium, but it is usually not associated with ventricular dysfunction.
The need to understand cellular and molecular mechanisms of inflammatory heart diseases led to development of animal models for experimental myocarditis. In general, these models can be categorized based on the causative agents into two major classes: infectious and non-infectious. In infectious models, pathogens associated with myocarditis in humans are used to induce cardiac inflammation in animals. CVB3 and T. cruzi represent two classical infectious pathogens used for induction of experimental myocarditis in mice. In non-infectious models, myocarditis is typically triggered by an autoimmune response against heart-specific antigens. A comparative summary of the selected models is presented in the Table 2.
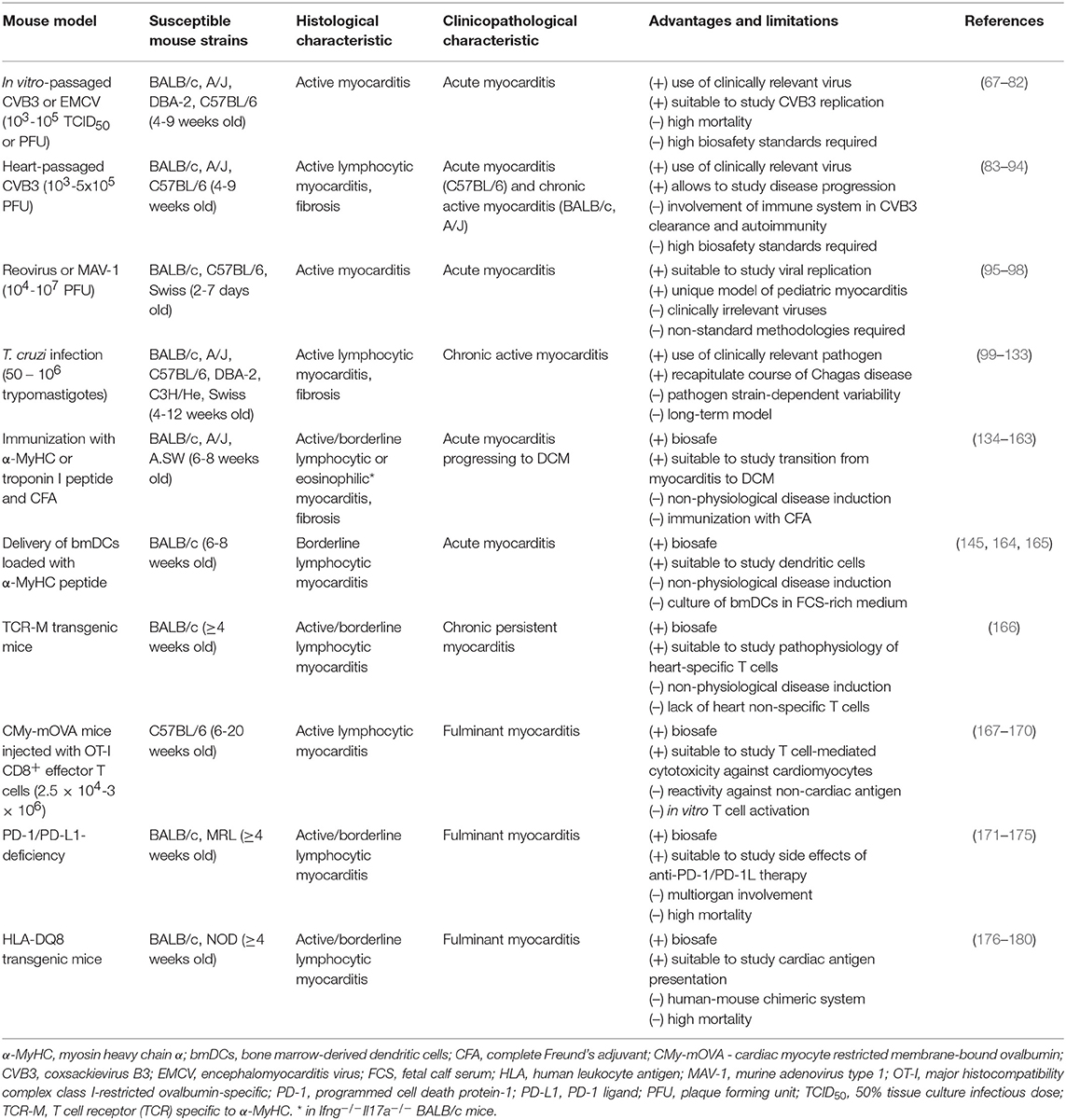
Table 2. Characteristics of commonly used mouse models of experimental myocarditis.
CVB3 has been implicated to the pathogenesis of myocarditis in humans and therefore this virus was used to induce experimental myocarditis in animals. Coxackieviruses belonging to the Picornaviridae family represent positive-sense single-stranded RNA enteroviruses. Coxsackieviruses are typically transmitted by the oral route and for replication require host cells. Unlike other serotypes, CVB3 efficiently infects and replicates in cardiomyocytes leading to their death through apoptosis (181) or necroptosis (182). Effective CVB3 replication has been demonstrated also in cardiac fibroblasts (183). CVB3 infection begins by coupling the virus with host-cell coxsackievirus and adenovirus receptor (CAR), and decay-accelerating factor (DAF). Additionally, recent data pointed to the relevance of NOD2 in CVB3 uptake (184). After entering into the cytoplasm, viral RNA is translated and then transcribed. The viral genome is further translated into viral structural proteins, which assemble with the positive-strand RNA viral genome forming the complete infectious virion (185). Infected cardiomyocytes become ultimately lysed, which results in release of cytosolic proteins and virus progeny. Active viral replication as well as latent viral persistence have been described in hearts of myocarditis patients (186).
The first successful myocarditis induction in mice using purified CVB3 (Nancy strain) was reported in 1974 (67). The Nancy strain of CVB3 is the most commonly used virus to induce myocarditis in mice until today. The virus was passaged in vitro in the host cells. Inoculation of purified, in vitro-passaged CVB3 resulted in high viral replication in hearts of host mice. This model is characterized by substantial cardiomyocyte necrosis, moderate inflammation, pancreatitis, and often high mortality during the acute phase of disease in BALB/c, A/J, and C57BL/6 mice (67–74). Poor survival rate of mice infected with in vitro-passaged CVB3 led to development of the heart-passaged CVB3 model of experimental myocarditis (83). In this model, hearts of mice infected with CVB3 were used for preparation of the infective pathogen. Such heart-passaged CVB3 containing not only the virus, but also cardiac myosin is inoculated into host animals. In this model, viral replication peaks around day 7 and the pathogen is cleared around day 14, post-infection. Infected mice develop acute myocarditis around day 10–14, which is characterized by massive infiltration of cardiac tissue with primarily cells of myeloid lineage accompanied by T (mainly CD4+) and some B lymphocytes in various mouse strains including BALB/c, A/J, ABY/SnJ, and C57BL/6 (83–89). In this model myocarditis is associated with left ventricular dysfunction during the acute phase. In contrast to high mortality rate observed in in vitro-passaged CVB3 model, typically all mice infected with heart-passaged CVB3 survive.
Following the acute myocarditis phase, disease course strongly depends on the genetic background of infected mice. Susceptible BALB/c, ABY/SnJ, and A/J mice progress to a phenotype of iDCM, characterized by chronic myocarditis, myocardial fibrosis, and cardiomyopathy, which is observed at day 28 post-infection and later (83, 89, 90). Whereas, mice with C57BL/6 genetic background do not develop DCM/iDCM phenotype (83, 91, 92), unless they are additionally treated with lipopolysaccharide (LPS) (93, 94). Infection with CVB3 leads to impaired cardiac functionality at later stages, which develops independently of the fibrotic phenotype in the heart (92, 94).
Infection with CVB3 triggers the respective innate and adaptive immune responses. Synthesis of antiviral cytokines such as type I interferons (IFNs) represent the first line of the innate immune defense against CVB3 infection, which aims to inhibit viral replication. Accordingly, treatments with IFN-α or IFN-β were reported to effectively eliminate virus in CVB3 infected mice (75) as well as in myocarditis patients (187). Following CVB3 entry into the target cell, the virus can engage intracellular nucleotide binding and oligomerization domain (NOD)-like receptors (NLRs) and activate certain Toll-like receptors (TLRs) (188). Activation of TRIF-dependent TLR3 has been recognized to be crucial for antiviral type I IFN production (76, 77). Interestingly, activation of other NLR and TLR pathways exacerbate myocarditis in CVB3 infected mice through negative regulation of type I IFN and stimulation of proinflammatory cytokines (73, 74, 184).
The innate immune response is usually followed by the adaptive response against the infective virus. In CVB3-mediated experimental myocarditis, the protective role of the adaptive immune response has been well-established. Studies using immunodeficient mice showed that lack of T and B cells led to viral persistence and enhanced myocarditis upon CVB3 infection (189, 190). Interestingly, in the CVB3 myocarditis model CD4+, but not CD8+ T cells play a pivotal role in viral clearance and thus protect infected mice from persistent cardiac inflammation (191, 192), whilst CD8+ T cells have been implicated mainly in the autoimmune response (191, 193). These paradoxical observations can be explained by the findings that CD4+ T cell response recognizes infected, but not uninfected myocytes, while CD8+ effector T cells react only to uninfected myocytes through recognition of cardiac myosin (193). Furthermore, natural killer (NK) (78) and NK T cells (79, 80) were also reported to play protective roles in CVB3-induced myocarditis.
As stated above, heart-specific autoimmunity has been implicated in the pathogenesis of viral myocarditis. Early data indeed pointed to the development of functional heart-specific autoimmune response in CVB3 infected mice (81). Detection of circulating autoantibodies represents a basic diagnostic assay indicating ongoing autoimmune disorder. Following CVB3 infection, high titers of heart-specific autoantibodies have been detected in host A/J and BALB/c mice (194, 195). Heart-specific autoantibodies are commonly detected also in myocarditis patients (196) pointing to similarity between mouse models and clinical scenario. Infection with CVB3 activates also cardiac myosin reactive CD4+ T cells in mice (69). Data from experimental model provided evidences that cardiac myosin reactive cells functionally contribute to cardiac pathology during chronic stage of the disease in CVB3 infected BALB/c mice (197). Heart specific autoimmunity seems to be a consequence of significant release of cardiac peptides from cardiomyocytes lysed during cardiotropic infection or molecular mimicry (epitope cross-reactivity) between the virus and cardiac proteins. Cardiac autoantigens in the presence of certain co-stimulatory, so called “third signal” cytokines can trigger the effector response of autoreactive T lymphocytes. It has been suggested that proinflammatory cytokines, mainly TNFα and IL-1β, produced during the innate response against viral infection play critical role in induction of the effector autoimmune response (33). Thus, myocarditis is likely a result of not only immune response against the infective virus, but also a consequence of boosted heart-specific autoimmune response. It seems that viral infection primary triggers myocarditis, while autoimmune response contributes to disease progression. In summary, myocarditis and iDCM phenotypes following CVB3 infection is the result of interplay between immune responses against the virus and heart-specific autoimmunity. Published data indicate that CVB3 infectious myocarditis mouse models accurately recapitulate principles of the immune responses in humans.
CVB3 represents the most common, but not the only virus used for induction of experimental myocarditis in mice. Cardiac inflammation associated with cardiomyocyte necrosis can be also induced with encephalomyocarditis virus (EMCV) (82). EMCV, like CVB3, are positive single-stranded RNA viruses belonging to the Picornaviridae family, which induce necrotic myocarditis with the similar mechanism of action (198). Enteroviruses CVB3 and EMVC are used to induce myocarditis in ≥4 weeks old mice. Whereas, the murine adenovirus type 1 (MAV-1) and reovirus 8B have been used to establish the mouse model for pediatric myocarditis. Depending on the delivery route, MAV-1 induces lethal [intraperitoneal injection (95)] or non-lethal [intranasal infection (96)] myocarditis in newborn mice. Infection of newborn mice with reovirus 8B also induces acute viral myocarditis. In this model myocarditis is characterized by marked cardiomyocyte necrosis and mild inflammation leading to death of infected BALB/c (97), but not C57BL/6 (98) mice. In contrast to the CVB3 model, autoimmunity seems not to be involved in myocarditis progression in reovirus 8B infected mice (97). Myocarditis in children is a deadly disease, particularly for newborns and infants and viral infections have been suggested as important causative agents in these young patients (199, 200). In that respect, MAV-1 and reovirus B8 models could be useful to study pathophysiological mechanisms of the disease in children.
Trypanosomal infection can cause myocarditis and iDCM in mouse organisms. Experimental Chagas heart disease has been successfully established in a number of mouse lines using various T. cruzi strains including Colombian, Tulahuen, CL Brener Brazil, the Y, and SylvioX10, but so far, no model has been generally accepted as the classical one. Pathogenic trypanosome strains were isolated from Chagastic patients, insect vectors, and animal reservoir (99). Trypanosoma cruzi is typically passaged in mice and bloodstream trypomastigotes (infective form of the parasite) are transferred into experimental animals by different delivery routes including intraperitoneal, intradermal, and oral transmission. Inbred strains BALB/c, C57BL/6, A/J, DBA-2, or C3H/He are often used as hosts, however many laboratories use outbred Swiss mice to induce experimental Chagas heart disease. Disease course, organ involvement and survival rate in different models are characterized by high variability and strongly depend on the T. cruzi strain, delivery route and genetic background of the recipient mice (99–105). An example of the high variability in mouse Chagastic model was demonstrated in the experiment with Swiss mice infected with different clones of the Colombian strain, which showed mouse mortality ranging from 0 to 100% depending on the clone (102). Apparently, interplay between the host, parasite genetics and environmental factors ultimately determine the outcome of a mouse infection with T. cruzi. Trypanosomal infection may lead to myocarditis development in recipient mice within 1–3 weeks post-infection (101, 102). In the chronic form, experimental Chagas heart disease is associated with progressive inflammation, iDCM phenotype and heart dysfunction. This phenotype is observed several months post-infection (100, 104, 106–110). It seems that chronic models recapitulate not only the end stage heart phenotype, but also the course of the disease observed in Chagastic patients.
During the acute phase of Chagas disease, trypomastigotes spread with the bloodstream throughout the body and enter into target cells, in which they differentiate into amastigotes and multiply causing death of the host cells. Infection of myeloid cells and cardiomyocytes represent two important check points for the progression of the disease. Myeloid cells, like macrophages and dendritic cells actively internalize parasites by phagocytosis. The innate immune response of macrophages and dendritic cells represent the first line of defense against the parasite involving TLR-dependent and TLR-independent mechanisms. Trypomastigote cell surface membrane glycosylphosphatidylinositol-anchored mucin-like glycoproteins and glycoinositolphospholipids as well as secreted Tc52 proteins activate innate immune cells through TLR2-, TLR4-, and TLR9-dependent mechanisms (111–113). The classical TLR-dependent response activates NF-κb and MAPK pathways leading to production of proinflammatory cytokines including TNFα and Th1 polarizing IL-12. Such responses are indeed observed during trypanosomal infections in mice (114, 115). Furthermore, in response to IFN-γ (produced by Th1 cells, but also by activated NK cells) and TNFα macrophages produce nitric oxide. This short-lived free radical effectively suppresses parasite replication and represents the primary defense mechanism during the acute phase of the infection (116). During the acute trypanosomal infection, Th1 polarization is facilitated also through TLR-independent innate mechanisms. For example, a cysteine protease cruzipain released by trypomastigote generates short-lived kinins, which stimulate IL-12 production through the bradykinin B2 receptor on the host cells and subsequently induce the protective Th1 response in infected mice (117).
Furthermore, T. cruzi triggers a robust adaptive immune response in the infected mouse organism. Phenotypically, infected mice show accumulation of lymphocytes in the spleen and subcutaneous lymph nodes associated with thymus atrophy (118). Early reports demonstrated persistent, non-specific polyclonal activation of T and B cells with phenotypic hypergammaglobulinemia (119, 120). Indeed, T. cruzi components such as DNA or glycoproteins have been shown to non-specifically activate T and B cells (121, 122), whereas more recent data pointed to the key role of antigen-specific response during parasite infection in mouse and in human (123, 124, 201). Importantly, the adaptive immune response plays a crucial role in pathogen clearance. Depletion of CD4+ or CD8+ T cells leads to an increase in parasite burden and exacerbation of myocarditis (125). Similarly, B cells and trypanosoma-specific antibodies have been shown to protect infected mice from uncontrolled parasite replication (126–128). Furthermore, interplay between T and B cells is needed for the effective adaptive immune response against trypanosomal infection (127, 128).
During the acute phase of the disease, the immune response eventually eliminates most of the infective pathogens, but not all. It has been suggested that a certain degree of parasite persistence, particularly in cardiac tissue correlates with the development of DCM phenotype and heart failure. Trypanosomal reactivation is commonly observed under immunosuppressive conditions in mouse models (129) and in humans (202) supporting the concept of parasite persistence. In the clinical scenario, Chagastic patients are treated with one of two anti-parasitic medications, benznidazole or nifurtimox, which generate free radicals, killing T. cruzi pathogens. In the chronic model of experimental Chagas heart disease, elimination of the pathogen during post-acute phase with benznidazole was shown to prevent development of severe chronic DCM in infected mice (110, 130). These results indicate that chronic experimental model can recapitulate incomplete eradication of T. cruzi observed in Chagastic patients. It is important to note that effectiveness of the anti-parasitic treatment decreases as the disease progresses. Ultimately, treatment with benznidazole fails to improve cardiac clinical outcomes in Chagastic patients with established DCM (203).
Heart-specific autoimmunity has been suggested as another disease progressing factor in Chagas heart disease. Heart-specific T cells and high titers of heart-specific autoantibodies have been identified in experimental mouse models (131, 204) as well as in Chagastic patients (205). In T. cruzi infected mice, development of heart-specific autoimmunity is associated with the genetic background of the host organism. Prominent humoral and cellular anti-cardiac myosin responses develop in A/J and BALB/c, but not in C57BL/6 mice (132, 206). Such an anti-cardiac myosin autoimmune response was shown to be non-essential for development of the acute phase of myocarditis (133), but has been implicated in the progression of post-acute cardiomyopathy during chronic phase of experimental Chagas heart disease (132). In mouse model of Chagas disease, the adaptive immune response, which plays a crucial role in the host defense against the infecting parasite, seems to contribute also to disease progression.
As presented above, clinical observations and experimental data from infectious models provide strong evidences for involvement of autoimmune mechanisms in the development and progression of myocarditis. In infectious models, T and B cells are primary involved in pathogen clearance. It is therefore practically impossible to uncouple the defense from autoimmune mechanisms using commonly available technologies. The need to understand contribution and molecular mechanisms of autoimmunity led to development of rodent models of experimental autoimmune myocarditis (EAM), in which myocarditis is induced by heart-reactive T cells in the absence of infectious pathogen.
In the context of autoimmune myocarditis, the question arises whether or not heart-specific T cells naturally occur in mouse and in human. In principle, vertebrates are protected from autoreactive T cells by the immune tolerance mechanisms. In the thymus, central tolerance specifically eliminates newly developing T cells, recognizing body's own antigens in a process called “negative selection.” In this process presentation of self-antigens by antigen-presenting medullary cells is essential for maintenance of a central tolerance. Surprisingly, α-isoform of MyHC (α-MyHC), unlike other cardiac proteins, is not expressed in cells implicated in T cell tolerance. This results in undisturbed development of α-MyHC-specific T cells and leads to their physiological presence in the periphery in mice and in human (207). Thus, α-MyHC represents the major cardiac self-antigen. In fact, many currently used EAM models take advantage of this and activate naturally existing α-MyHC-specific T cells in order to trigger autoimmune-mediated myocarditis. Data from experimental animal models clearly demonstrated that autoreactive CD4+ T lymphocytes were able to trigger myocarditis and DCM.
The first attempt to induce heart-specific autoimmunity in animals was reported in 1958 (208), but in 1987 Neu et al. published the basis for the currently most commonly used mouse model of EAM (134). In this publication, authors demonstrated that delivery of cardiac myosin together with the complete Freund's adjuvant (CFA) induced myocarditis with high prevalence and high titers of myosin autoantibodies in genetically predisposed mice (134). Currently, in this “classical” model of EAM susceptible mice are immunized with α-MyHC peptide together with CFA at day 0 and 7. Myocarditis in α-MyHC/CFA immunized mice is characterized by massive infiltration of mainly myeloid cells together with CD4+ T cells and few B and CD8+ T lymphocytes. Inflammation of cardiac tissue occurs typically 14–21 days after the first immunization. Resolution of the inflammation is followed by the progressive accumulation of fibrotic tissue in the myocardium, ventricular dilatation and impaired heart function in some mice (135–146). Thus, this model allows to study not only autoimmune mechanisms, but also transition from myocarditis to DCM phenotype. Of note, α-MyHC/CFA immunization of Ifng−/−Il17a−/− mice results in myocarditis with extensive infiltration of eosinophils in the myocardium representing a unique model of eosinophilic myocarditis (147).
Published data point to a central role of CD4+ T cells in the α-MyHC/CFA model. Depletion of CD4+ T cells prevents induction of myocarditis and the adoptive transfer of purified CD4+ T cells from immunized mice successfully transfers the disease into immunodeficient hosts (148, 149). A simple passive transfer of high-titer myosin autoantibodies failed to transfer myocarditis in the recipient mice (150), however monoclonal anti-myosin antibodies were shown to induce myocarditis in a predisposed mouse strain (151). CD8+ T cells, instead, contribute mainly to myocarditis severity, but are not essential for disease induction (148). However, recent data showed that using the specific α-MyHC peptide for EAM induction, CD8+ T cells were able to limited extends convey myocarditis (152).
Co-delivery of a strong adjuvant, such as CFA represents the second key element of EAM induction in the “classical” model. CFA contains heat-killed Mycobacterium tuberculosis, which can activate TLR2, TLR4, and TLR9 on host cells (209). Activation of TLRs on the innate immune cells triggers secretion of a broad range of cytokines. In the adaptive immune response, the “third signal” cytokines produced by dendritic cells program vitality and expansion potential of antigen-activated T lymphocytes (210). The “third signal” cytokines have been also shown to polarize differentiating T cells toward Th1, Th2, or Th17 lineages. The importance of the “third signal” cytokines in the development of EAM has been demonstrated in a number of studies. Genetic deletion or blockage of the “third signal” cytokine signaling, including TNF-α (153, 154), GM-CSF (155), IL-1 (156), IL-6 (157), or IL-23 (158) resulted in complete resistance or amelioration of EAM. It remains, however, unclear whether the acute response to adjuvant in mice reflects immune processes during myocarditis induction in humans.
Development of EAM is a multifactorial process, which depends not only on the presence of heart-specific T cells and TLR activation, but also strongly on genetic predisposition. α-MyHC/CFA immunization induces myocarditis in susceptible strains only. Mice on BALB/c, A/J or A.SW background are susceptible to EAM, while mice on C57BL/6 background are resistant (134, 159–161). From a practical point of view, the resistance to EAM of widely-used C57BL/6 strain limits use of numerous transgenic models without the need for back-crossing onto the susceptible background. Differences in MHC haplotypes (H-2) of susceptible and resistant strains have been suggested to determine susceptibility of mice to EAM (148, 162). However, differences in susceptibility of A.SW and B10.S mice, which share the same H-2 genes, pointed also to the importance of non-H-2 genes in EAM development (163). Summarizing, the “classical” EAM model offers a well-established, simple and safe method to study heart-specific autoimmunity and progression of cardiac inflammation to DCM phenotype, but is limited to few inbred strains only.
The idea that activation of self-antigen presenting cells is critical for myocarditis induction led to development of “dendritic cell” EAM model. It has been demonstrated that myocarditis could be effectively induced by adoptive transfer of bone marrow-derived dendritic cells (bmDCs) loaded with α-MyHC peptide and activated with LPS - a major component of the outer membrane of Gram-negative bacteria and the anti-CD40 antibody. In the “dendritic cell” EAM model, adoptive transfer of activated α-MyHC-loaded bmDCs at days 0, 3 and 5 results in acute myocarditis at days 8–12 (145, 164, 165). In contrast to the “classical” model, mice receiving α-MyHC-loaded bmDCs develop moderate fibrosis on the follow up. However, additional administration of CFA significantly accelerates fibrosis and induces ventricular dilatation and heart dysfunction in this model (211).
The “classical” and the “dendritic cell” EAM models, both rely on activation of naturally existing α-MyHC-reactive CD4+ T cells. Non-transgenic mice contain physiologically very low prevalence of α-MyHC-reactive T cells and TLRs stimulation with CFA or LPS is needed not only for polarization, but also for expansion of activated α-MyHC-reactive T helper cells. High prevalence of these autoreactive cells can be alternatively obtained by transgenic overexpression of T cell receptor (TCR) specific to α-MyHC (TCR-M). A consequence of the high number of circulating α-MyHC-reactive T cells in the TCR-M transgenic mice is spontaneous development of progressive myocarditis associated with ventricular wall thickening, but without evident systolic dysfunction (166). The TCR-M transgenic model is particularly useful to study pathogenesis of autoreactive T cells in the absence of exogenous TLR agonists.
In contrast to widely studied CD4+ T cells, the role of CD8+ T cells (known also as cytotoxic T lymphocytes) in heart-specific autoimmunity is less understood. Unlike CD4+ T cells, CD8+ T cells recognize antigens presented by MHC class I molecules and directly induce apoptosis of antigen presenting cells by secreting cytotoxins, such as perforins and granzymes. In order to develop CD8+ T cell-dependent myocarditis model, transgenic mice (on C57BL/6 genetic background) expressing cardiomyocyte-restricted membrane-bound ovalbumin (OVA) were injected with in vitro expanded and polarized MHC class I-restricted, OVA-specific OT-I CD8+ T cells. Adoptive transfer of high doses (≥5 × 105) of effector OT-I CD8+ T cells caused massive cardiomyocyte cell death associated with lymphocytic (both CD4+ and CD8+) and myeloid cell infiltration. Severe myocarditis caused death of affected mice 3–7 days post T cell transfer (167, 168). In this model, T cell polarizing factors, such as IL-12 (167) and T-bet (169) played a key role in disease pathogenesis. Disease severity in this model correlates with the number of injected lymphocytes. In contrast to high doses, low doses (2.5 × 104) of effector OT-I CD8+ T cells induce a transient and moderate myocarditis only (170). This model is particularly useful to study CD8+ T cell-mediated cytotoxicity against cardiomyocytes. Reactivity against non-physiological antigen (OVA) seems to be, however, a major drawback of this method.
Under homeostatic conditions, effector functions of CD4+ and CD8+ T cells are controlled by regulatory T cells (Treg). Accordingly, adoptive transfer of Treg-depleted T cells induces multiorgan inflammation including fatal autoimmune myocarditis and high-titer anti-myosin autoantibodies in the recipient mice. Of note, inflamed myocardium displayed multinucleated inflammatory cells resembling giant cell myocarditis in humans (212).
Once T cells become activated, more regulatory mechanisms control their expansion and effector function. Immune checkpoint regulators, such as PD-1 represents an example of regulatory mechanisms. Mechanistically, PD-1-PD-1L signaling inhibits TCR signaling on activated T cells and thus suppresses autoimmune response. Accordingly, mice deficient of PD-1 or PD-1L spontaneously develop systemic or organ-specific inflammations. Progressive myocarditis, iDCM phenotype and heart failures have been described in PD-1- and PD-1L-deficient mice on BALB/c (171, 172) and MRL (173, 174) genetic backgrounds, although for BALB/c not in all housing conditions (175). In mice lacking PD-1, fatal myocarditis caused high mortality in particular on MRL genetic background. Cardiac inflammation in PD-1-deficient mice is entirely dependent on the adaptive immunity (autoimmunity) and could be transferred by splenocytes (171, 174). In mouse models, PD-1-PD-1L signaling protects from myocarditis mediated by CD4+ T (175) and by CD8+ T (170) cells. PD-1 deficiency results in myocarditis also in aged C57BL/6 mice, but these mice are characterized by multiorgan inflammation and represent rather a model of systemic lupus erythematosus (213). It seems that the use of PD-1- and PD-1L-deficient mice developing progressive myocarditis represents a suitable model to study mechanisms of cardiac side effects observed during anti-PD-1-PD-1L therapies in oncological patients.
Although α-MyHC represents a main cardioimmunogenic antigen, other cardiac proteins can also trigger heart-specific autoimmunity. Immunization of A/J mice with recombinant cardiac troponin I peptide together with CFA induces myocarditis, which is followed by myocardial fibrosis, ventricular dilatation, and impaired systolic function (214). Unlike α-MyHC, troponin I is expressed in medullary thymic epithelial cells (207). The occurrence of circulating troponin I-reactive T cells is, instead, a result of inefficient elimination of autoreactive T cells during the “negative selection.” The “leakage” of autoreactive T cells into periphery is a physiological phenomenon and its degree depends on the affinity of the TCR to the antigen-presenting medullary cells. Clinical data show elevated levels of troponin I in around one third of myocarditis patients (215), but high titers of anti-troponin I antibodies were detected only in 7% of DCM patients (216). It seems that troponin I represents rather a secondary autoantigen in heart-specific autoimmunity in myocarditis patients.
Presentation of cardiac antigen represent another important element of autoimmunity. Clinical studies suggested that specific HLA haplotypes are associated with heart-specific autoimmunity. This idea was functionally confirmed by introducing specific HLA complexes into a mouse. Replacement of mouse MHC class II with the specific HLA-DQ8 in NOD or BALB/c mice resulted in spontaneous development of myocarditis and iDCM phenotype (without evident fibrosis), and were associated with cardiac arrhythmias and high mortality of the transgenic mice (176–180). This model resembles a course of fatal fulminant myocarditis in humans. Adoptive transfer experiments pointed to the key role of CD4+ T cells in pathogenesis of the disease in this mouse model (178, 179). Interestingly, introduction of HLA-DR3 or HLA-DQ6 failed to induce cardiac pathology pointing the specific role of HLA-DQ8 in heart-specific autoimmunity (177, 178). The HLA-DQ8 transgenic mice seem to be particular useful to study mechanisms of cardiac antigen presentation and induction of heart-specific autoimmunity.
Taking together clinical observations as well as data from animal models, a triphasic model of myocarditis development and progression could be proposed (Figure 1). The initial phase is associated with heart injury, caused usually by cardiotropic infections or non-infectious triggers. Damaged myocardium induces primary inflammatory response and development of heart-specific autoimmunity, which results in the development of myocarditis. In certain cases, myocarditis can be directly induced by heart-specific autoimmunity. Cardiac inflammation can be transient or chronic. In the transient form, acute phase of myocarditis is followed by complete resolution of inflammation or development of stable DCM. In case of chronic myocarditis, many patients develop also DCM phenotype (iDCM). Cardiac dysfunction in DCM and in iDCM is in most cases progressive leading to end stage organ failure.
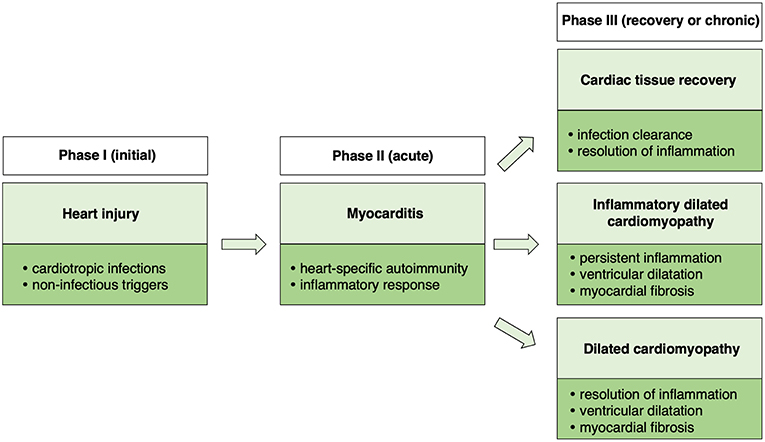
Figure 1. Schematic presentation of a triphasic model of myocarditis.
As presented above, a number of different animal models of myocarditis have been developed. Experimental myocarditis in mice can be induced with the whole spectrum of disease triggers ranging from clinically relevant agents, such as infective pathogens up to artificial models like transgenic animals. Depending on the model, myocarditis is characterized by different composition of inflammatory infiltrates and different extent of necrotic myocardium. In many models, DCM or iDCM phenotype associated with heart dysfunction represent the end stage of the disease. The question arises, which model reflects human myocarditis in the most relevant way. As myocarditis in humans shows high diversity in terms of causative agents, autoimmune response, course of inflammation, and progression to DCM/iDCM, it becomes evident that one animal model cannot mirror all aspects of the disease in humans.
For modeling of myocarditis with known etiology, as for example CVB3-mediated myocarditis or Chagas disease, use of the same infectious agent seems to be the most obvious choice. Infectious pathogens spread and induce myocarditis in mice and in humans using similar mechanisms, therefore these models are particularly useful to study early phase of disease. However, it is difficult to uncouple immune mechanisms involved in pathogen clearance and in autoimmune responses. Furthermore, handling the human infective pathogens is potentially dangerous for experimentators and requires exceptionally high biosafety standards.
In contrast, non-infectious models are safe for experimentators, but induce myocarditis in rather non-physiologic way. Nevertheless, lack of infectious agents allows to better study autoimmune mechanisms and involvement of the immune system to progression of myocarditis to DCM/iDCM phenotype. Summarizing, the usefulness of the respective mouse model is mostly limited to certain aspects of the disease in humans. It is therefore very important to address scientific question by choosing the relevant model.
In developed countries, public approval to conduct animal experimentations is today low as never before. Activists raise ethical concerns and appeal to minimize or even to stop performing experiments involving animals. Some of these postulates pointing to protocol optimization and to reduction of severity in animal experimentation are rational and have been already implemented in the form of 3R (replacement, reduction, and refinement) guidelines, but is there a realistic animal-free alternative in myocarditis research?
It seems that certain aspects of the disease, for example replication of cardiotropic pathogens, fibroblast-to-myofibroblast transition, endothelial cell activation, can be effectively addressed using conventional ex vivo or in vitro systems. These systems are suitable for molecular studies, but rather poorly reflect biomechanical and biochemical microenvironment of cardiac tissue. Dynamic development of three-dimensional cell culture methodologies, like organoids or microtissues represents a recent advancement, which can address some of these concerns. Furthermore, development of the human induced pluripotent stem cell (iPSC) technology represents another important milestone toward animal-free research. The use of human iPSC-derived cardiomyocytes in combination with other cell types allows today for a simplistic modeling of a human heart (217, 218). Furthermore, de novo tissue fabrication opens new possibilities to integrate bioscaffolds for improved architecture and microelectronics for live monitoring of cardiac tissue (219). These advances offer potentially very attractive replacements for animal models (220). However, generation of physiologically-relevant human cardiac tissue faces a number of serious problems, which include immature state of iPSC-derived cardiomyocytes, limited availability of non-cardiac primary cells (fibroblasts, endothelial cells) and high costs of de novo tissue fabrication. Moreover, in myocarditis active migration of inflammatory cells into the tissue plays a key role in the disease development. Currently, modeling of inflammatory response in terms of influx of inflammatory cells into cardiac tissue in vitro is not available.
Summarizing, it seems that the whole process of myocarditis is too complex to reproduce it outside of a body by using today's technologies. However, certain aspects of the disease can be reliably studied in vitro and upcoming advances may allow to reduce animal research in the future.
Animal models represent an important platform in preclinical research. As presented above, developed models of experimental myocarditis appear to reliably mirror many specific aspects of the disease in humans. Currently, these animal models are commonly used to get an insight into pathophysiology of myocarditis on molecular and cellular level and to test pharmaceutic compounds for treatment efficacy and safety. However, animal studies are commonly unicenter, involving mostly low group sizes and experiments are rarely reproduced by others. Furthermore, in some cases, published papers lack detailed description of used methodologies. Thus, the power of such exploratory studies is usually low.
In laboratory practice, typical large scale, multicenter animal studies are not possible due to ethical and economic reasons. It seems that synthetic research integrating data from independent studies is needed to increase the power of experimental findings. In this case, the use of the same or similar procedures is a basic prerequisite to analyse data from different laboratories. In the area of experimental myocarditis, some models show high reproducibility in terms of used protocols and obtained results. In particularly, published data by independent laboratories on disease course and severity of myocarditis induced in specific inbred strains by infection with CVB3 or by immunization with α-MyHC/CFA show high consistency. In contrast, other models like experimental Chagas disease show high variability due to inconsistencies in methodologies. The use of standardized procedures would allow to more effectively plan experimentations and more accurately interpret obtained data. Furthermore, the use of unified methodologies would be important to effectively share omics resources and to implement meta-analyses in animal research. Combined efforts are therefore needed to more efficiently use the potential of animal models in order to translate this knowledge into innovative, more effective treatment therapies.
The author confirms being the sole contributor of this work and has approved it for publication.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work has been supported by the National Science Centre (Poland) grants 2014/14/E/NZ5/00175 and 2016/21/B/NZ5/01397.
1. Fabre A, Sheppard MN. Sudden adult death syndrome and other non-ischaemic causes of sudden cardiac death. Heart. (2006) 92:316–20. doi: 10.1136/hrt.2004.045518
2. Doolan A, Langlois N, Semsarian C. Causes of sudden cardiac death in young Australians. Med J Aust. (2004) 180:110–2. doi: 10.5694/j.1326-5377.2004.tb05830.x
3. Noren GR, Staley NA, Bandt CM, Kaplan EL. Occurrence of myocarditis in sudden death in children. J Forensic Sci. (1977) 22:188–96. doi: 10.1520/JFS10385J
4. Basso C, Calabrese F, Corrado D, Thiene G. Postmortem diagnosis in sudden cardiac death victims: macroscopic, microscopic and molecular findings. Cardiovasc Res. (2001) 50:290–300. doi: 10.1016/S0008-6363(01)00261-9
5. Aretz HT, Billingham ME, Edwards WD, Factor SM, Fallon JT, Fenoglio JJ Jr, et al. Myocarditis. A histopathologic definition and classification. Am J Cardiovasc Pathol. (1987) 1:3–14.
6. Caforio AL, Pankuweit S, Arbustini E, Basso C, Gimeno-Blanes J, Felix SB, et al. Current state of knowledge on aetiology, diagnosis, management, and therapy of myocarditis: a position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J. (2013) 34:2636–48:2648a–d. doi: 10.1093/eurheartj/eht210
7. Grogan M, Redfield MM, Bailey KR, Reeder GS, Gersh BJ, Edwards WD, et al. Long-term outcome of patients with biopsy-proved myocarditis: comparison with idiopathic dilated cardiomyopathy. J Am Coll Cardiol. (1995) 26:80–4. doi: 10.1016/0735-1097(95)00148-S
8. Kindermann I, Kindermann M, Kandolf R, Klingel K, Bultmann B, Muller T, et al. Predictors of outcome in patients with suspected myocarditis. Circulation. (2008) 118:639–48. doi: 10.1161/CIRCULATIONAHA.108.769489
9. Grun S, Schumm J, Greulich S, Wagner A, Schneider S, Bruder O, et al. Long-term follow-up of biopsy-proven viral myocarditis: predictors of mortality and incomplete recovery. J Am Coll Cardiol. (2012) 59:1604–15. doi: 10.1016/j.jacc.2012.01.007
10. Magnani JW, Danik HJ, Dec GW Jr, DiSalvo TG. Survival in biopsy-proven myocarditis: a long-term retrospective analysis of the histopathologic, clinical, and hemodynamic predictors. Am Heart J. (2006) 151:463–70. doi: 10.1016/j.ahj.2005.03.037
11. Dec GW Jr, Palacios IF, Fallon JT, Aretz HT, Mills J, Lee DC, et al. Active myocarditis in the spectrum of acute dilated cardiomyopathies. Clinical features, histologic correlates, and clinical outcome. N Engl J Med. (1985) 312:885–90. doi: 10.1056/NEJM198504043121404
12. D'Ambrosio A, Patti G, Manzoli A, Sinagra G, Di Lenarda A, Silvestri F, et al. The fate of acute myocarditis between spontaneous improvement and evolution to dilated cardiomyopathy: a review. Heart. (2001) 85:499–504. doi: 10.1136/heart.85.5.499
13. Jefferies JL, Towbin JA. Dilated cardiomyopathy. Lancet. (2010) 375:752–62. doi: 10.1016/S0140-6736(09)62023-7
14. Richardson P, McKenna W, Bristow M, Maisch B, Mautner B, O'Connell J, et al. Report of the 1995 World Health Organization/International Society and Federation of Cardiology Task Force on the Definition and Classification of cardiomyopathies. Circulation. (1996) 93:841–2. doi: 10.1161/01.CIR.93.5.841
15. Towbin JA, Lowe AM, Colan SD, Sleeper LA, Orav EJ, Clunie S, et al. Incidence, causes, and outcomes of dilated cardiomyopathy in children. JAMA. (2006) 296:1867–76. doi: 10.1001/jama.296.15.1867
16. Matsumura Y, Takata J, Kitaoka H, Kubo T, Baba Y, Hoshikawa E, et al. Long-term prognosis of dilated cardiomyopathy revisited: an improvement in survival over the past 20 years. Circ J. (2006) 70:376–83. doi: 10.1253/circj.70.376
17. Pereira Nunes Mdo C, Barbosa MM, Ribeiro AL, Amorim Fenelon LM, Rocha MO. Predictors of mortality in patients with dilated cardiomyopathy: relevance of chagas disease as an etiological factor. Rev Esp Cardiol. (2010) 63:788–97. doi: 10.1016/S1885-5857(10)70163-8
18. Wojnicz R, Nowalany-Kozielska E, Wojciechowska C, Glanowska G, Wilczewski P, Niklewski T, et al. Randomized, placebo-controlled study for immunosuppressive treatment of inflammatory dilated cardiomyopathy: two-year follow-up results. Circulation. (2001) 104:39–45. doi: 10.1161/01.CIR.104.1.39
19. Pankuweit S, Klingel K. Viral myocarditis: from experimental models to molecular diagnosis in patients. Heart Fail Rev. (2013) 18:683–702. doi: 10.1007/s10741-012-9357-4
20. Mahrholdt H, Wagner A, Deluigi CC, Kispert E, Hager S, Meinhardt G, et al. Presentation, patterns of myocardial damage, and clinical course of viral myocarditis. Circulation. (2006) 114:1581–90. doi: 10.1161/CIRCULATIONAHA.105.606509
21. Schenk T, Enders M, Pollak S, Hahn R, Huzly D. High prevalence of human parvovirus B19 DNA in myocardial autopsy samples from subjects without myocarditis or dilative cardiomyopathy. J Clin Microbiol. (2009) 47:106–10. doi: 10.1128/JCM.01672-08
22. Kuhl U, Pauschinger M, Seeberg B, Lassner D, Noutsias M, Poller W, et al. Viral persistence in the myocardium is associated with progressive cardiac dysfunction. Circulation. (2005) 112:1965–70. doi: 10.1161/CIRCULATIONAHA.105.548156
23. Kostic T, Momcilovic S, Perisic ZD, Apostolovic SR, Cvetkovic J, Jovanovic A, et al. Manifestations of Lyme carditis. Int J Cardiol. (2017) 232:24–32. doi: 10.1016/j.ijcard.2016.12.169
24. Hidron A, Vogenthaler N, Santos-Preciado JI, Rodriguez-Morales AJ, Franco-Paredes C, Rassi A Jr. Cardiac involvement with parasitic infections. Clin Microbiol Rev. (2010) 23:324–49. doi: 10.1128/CMR.00054-09
25. Parada H, Carrasco HA, Anez N, Fuenmayor C, Inglessis I. Cardiac involvement is a constant finding in acute Chagas' disease: a clinical, parasitological and histopathological study. Int J Cardiol. (1997) 60:49–54. doi: 10.1016/S0167-5273(97)02952-5
26. Bestetti RB, Muccillo G. Clinical course of Chagas' heart disease: a comparison with dilated cardiomyopathy. Int J Cardiol. (1997) 60:187–93. doi: 10.1016/S0167-5273(97)00083-1
27. Sagar S, Liu PP, Cooper LT Jr. Myocarditis. Lancet. (2012) 379:738–47. doi: 10.1016/S0140-6736(11)60648-X
28. Moder KG, Miller TD, Tazelaar HD. Cardiac involvement in systemic lupus erythematosus. Mayo Clin Proc. (1999) 74:275–84. doi: 10.4065/74.3.275
29. Suzuki S, Utsugisawa K, Yoshikawa H, Motomura M, Matsubara S, Yokoyama K, et al. Autoimmune targets of heart and skeletal muscles in myasthenia gravis. Arch Neurol. (2009) 66:1334–8. doi: 10.1001/archneurol.2009.229
30. Johnson DB, Balko JM, Compton ML, Chalkias S, Gorham J, Xu Y, et al. Fulminant Myocarditis with Combination Immune Checkpoint Blockade. N Engl J Med. (2016) 375:1749–55. doi: 10.1056/NEJMoa1609214
31. Mahmood SS, Fradley MG, Cohen JV, Nohria A, Reynolds KL, Heinzerling LM, et al. Myocarditis in patients treated with immune checkpoint inhibitors. J Am Coll Cardiol. (2018) 71:1755–64. doi: 10.1016/j.jacc.2018.02.037
32. Grabie N, Lichtman AH, Padera R. T Cell checkpoint regulators in the heart. Cardiovasc Res. (2019) 115:869–77. doi: 10.1093/cvr/cvz025
33. Rose NR. Myocarditis: infection versus autoimmunity. J Clin Immunol. (2009) 29:730–7. doi: 10.1007/s10875-009-9339-z
34. Caforio AL, Mahon NJ, Tona F, McKenna WJ. Circulating cardiac autoantibodies in dilated cardiomyopathy and myocarditis: pathogenetic and clinical significance. Eur J Heart Fail. (2002) 4:411–7. doi: 10.1016/S1388-9842(02)00010-7
35. Neumann DA, Burek CL, Baughman KL, Rose NR, Herskowitz A. Circulating heart-reactive antibodies in patients with myocarditis or cardiomyopathy. J Am Coll Cardiol. (1990) 16:839–46. doi: 10.1016/S0735-1097(10)80331-6
36. Lauer B, Schannwell M, Kuhl U, Strauer BE, Schultheiss HP. Antimyosin autoantibodies are associated with deterioration of systolic and diastolic left ventricular function in patients with chronic myocarditis. J Am Coll Cardiol. (2000) 35:11–8. doi: 10.1016/S0735-1097(99)00485-4
37. Herskowitz A, Ahmed-Ansari A, Neumann DA, Beschorner WE, Rose NR, Soule LM, et al. Induction of major histocompatibility complex antigens within the myocardium of patients with active myocarditis: a nonhistologic marker of myocarditis. J Am Coll Cardiol. (1990) 15:624–32. doi: 10.1016/0735-1097(90)90637-5
38. Seko Y, Takahashi N, Ishiyama S, Nishikawa T, Kasajima T, Hiroe M, et al. Expression of costimulatory molecules B7-1, B7-2, and CD40 in the heart of patients with acute myocarditis and dilated cardiomyopathy. Circulation. (1998) 97:637–9. doi: 10.1161/01.CIR.97.7.637
39. Caforio AL, Angelini A, Blank M, Shani A, Kivity S, Goddard G, et al. Passive transfer of affinity-purified anti-heart autoantibodies (AHA) from sera of patients with myocarditis induces experimental myocarditis in mice. Int J Cardiol. (2015) 179:166–77. doi: 10.1016/j.ijcard.2014.10.165
41. Kindermann I, Barth C, Mahfoud F, Ukena C, Lenski M, Yilmaz A, et al. Update on myocarditis. J Am Coll Cardiol. (2012) 59:779–92. doi: 10.1016/j.jacc.2011.09.074
42. Felker GM, Boehmer JP, Hruban RH, Hutchins GM, Kasper EK, Baughman KL, et al. Echocardiographic findings in fulminant and acute myocarditis. J Am Coll Cardiol. (2000) 36:227–32. doi: 10.1016/S0735-1097(00)00690-2
43. Ukena C, Mahfoud F, Kindermann I, Kandolf R, Kindermann M, Bohm M. Prognostic electrocardiographic parameters in patients with suspected myocarditis. Eur J Heart Fail. (2011) 13:398–405. doi: 10.1093/eurjhf/hfq229
44. Friedrich MG, Sechtem U, Schulz-Menger J, Holmvang G, Alakija P, Cooper LT, et al. Cardiovascular magnetic resonance in myocarditis: a JACC White Paper. J Am Coll Cardiol. (2009) 53:1475–87. doi: 10.1016/j.jacc.2009.02.007
45. Lurz P, Luecke C, Eitel I, Fohrenbach F, Frank C, Grothoff M, et al. Comprehensive cardiac magnetic resonance imaging in patients with suspected myocarditis: the myoracer-trial. J Am Coll Cardiol. (2016) 67:1800–11. doi: 10.1016/j.jacc.2016.02.013
46. Ferreira VM, Schulz-Menger J, Holmvang G, Kramer CM, Carbone I, Sechtem U, et al. Cardiovascular magnetic resonance in nonischemic myocardial inflammation: expert recommendations. J Am Coll Cardiol. (2018) 72:3158–76. doi: 10.1016/j.jacc.2018.09.072
47. Lurz P, Eitel I, Adam J, Steiner J, Grothoff M, Desch S, et al. Diagnostic performance of CMR imaging compared with EMB in patients with suspected myocarditis. JACC Cardiovasc Imaging. (2012) 5:513–24. doi: 10.1016/j.jcmg.2011.11.022
48. Leone O, Veinot JP, Angelini A, Baandrup UT, Basso C, Berry G, et al. 2011 consensus statement on endomyocardial biopsy from the Association for European Cardiovascular Pathology and the Society for Cardiovascular Pathology. Cardiovasc Pathol. (2012) 21:245–74. doi: 10.1016/j.carpath.2011.10.001
49. Yilmaz A, Kindermann I, Kindermann M, Mahfoud F, Ukena C, Athanasiadis A, et al. Comparative evaluation of left and right ventricular endomyocardial biopsy: differences in complication rate and diagnostic performance. Circulation. (2010) 122:900–9. doi: 10.1161/CIRCULATIONAHA.109.924167
50. Chimenti C, Frustaci A. Contribution and risks of left ventricular endomyocardial biopsy in patients with cardiomyopathies: a retrospective study over a 28-year period. Circulation. (2013) 128:1531–41. doi: 10.1161/CIRCULATIONAHA.13.001414
51. Stiermaier T, Fohrenbach F, Klingel K, Kandolf R, Boudriot E, Sandri M, et al. Biventricular endomyocardial biopsy in patients with suspected myocarditis: Feasibility, complication rate and additional diagnostic value. Int J Cardiol. (2017) 230:364–70. doi: 10.1016/j.ijcard.2016.12.103
52. Hauck AJ, Kearney DL, Edwards WD. Evaluation of postmortem endomyocardial biopsy specimens from 38 patients with lymphocytic myocarditis: implications for role of sampling error. Mayo Clin Proc. (1989) 64:1235–45. doi: 10.1016/S0025-6196(12)61286-5
53. Chow LH, Radio SJ, Sears TD, McManus BM. Insensitivity of right ventricular endomyocardial biopsy in the diagnosis of myocarditis. J Am Coll Cardiol. (1989) 14:915–20. doi: 10.1016/0735-1097(89)90465-8
54. Khan T, Selvakumar D, Trivedi S, Rao K, Harapoz M, Thiagalingam A, et al. The value of endomyocardial biopsy in diagnosis and guiding therapy. Pathology. (2017) 49:750–6. doi: 10.1016/j.pathol.2017.08.004
55. Zhang M, Tavora F, Zhang Y, Ripple M, Fowler D, Li L, et al. The role of focal myocardial inflammation in sudden unexpected cardiac and noncardiac deaths–a clinicopathological study. Int J Legal Med. (2013) 127:131–8. doi: 10.1007/s00414-011-0634-x
56. Blauwet LA, Cooper LT. Idiopathic giant cell myocarditis and cardiac sarcoidosis. Heart Fail Rev. (2013) 18:733–46. doi: 10.1007/s10741-012-9358-3
57. Ginsberg F, Parrillo JE. Eosinophilic myocarditis. Heart Fail Clin. (2005) 1:419–29. doi: 10.1016/j.hfc.2005.06.013
58. Cooper LT Jr, Berry GJ, Shabetai R. Idiopathic giant-cell myocarditis–natural history and treatment. Multicenter Giant Cell Myocarditis Study Group Investigators. N Engl J Med. (1997) 336:1860–6. doi: 10.1056/NEJM199706263362603
59. Okura Y, Dec GW, Hare JM, Kodama M, Berry GJ, Tazelaar HD, et al. A clinical and histopathologic comparison of cardiac sarcoidosis and idiopathic giant cell myocarditis. J Am Coll Cardiol. (2003) 41:322–9. doi: 10.1016/S0735-1097(02)02715-8
60. Magnani JW, Dec GW. Myocarditis: current trends in diagnosis and treatment. Circulation. (2006) 113:876–90. doi: 10.1161/CIRCULATIONAHA.105.584532
61. Lieberman EB, Hutchins GM, Herskowitz A, Rose NR, Baughman KL. Clinicopathologic description of myocarditis. J Am Coll Cardiol. (1991) 18:1617–26. doi: 10.1016/0735-1097(91)90493-S
62. Dennert R, Crijns HJ, Heymans S. Acute viral myocarditis. Eur Heart J. (2008) 29:2073–82. doi: 10.1093/eurheartj/ehn296
63. Asaumi Y, Yasuda S, Morii I, Kakuchi H, Otsuka Y, Kawamura A, et al. Favourable clinical outcome in patients with cardiogenic shock due to fulminant myocarditis supported by percutaneous extracorporeal membrane oxygenation. Eur Heart J. (2005) 26:2185–92. doi: 10.1093/eurheartj/ehi411
64. Nakamura T, Ishida K, Taniguchi Y, Nakagawa T, Seguchi M, Wada H, et al. Prognosis of patients with fulminant myocarditis managed by peripheral venoarterial extracorporeal membranous oxygenation support: a retrospective single-center study. J Intensive Care. (2015) 3:5. doi: 10.1186/s40560-014-0069-9
65. McCarthy RE III, Boehmer JP, Hruban RH, Hutchins GM, Kasper EK, Hare JM, et al. Long-term outcome of fulminant myocarditis as compared with acute (nonfulminant) myocarditis. N Engl J Med. (2000) 342:690–5. doi: 10.1056/NEJM200003093421003
66. Ammirati E, Cipriani M, Lilliu M, Sormani P, Varrenti M, Raineri C, et al. Survival and left ventricular function changes in fulminant versus nonfulminant acute myocarditis. Circulation. (2017) 136:529–45. doi: 10.1161/CIRCULATIONAHA.117.026386
67. Woodruff JF, Woodruff JJ. Involvement of T lymphocytes in the pathogenesis of coxsackie virus B3 heart disease. J Immunol. (1974) 113:1726–34.
68. Nakamura H, Kunitsugu I, Fukuda K, Matsuzaki M, Sano M. Diverse stage-dependent effects of glucocorticoids in a murine model of viral myocarditis. J Cardiol. (2013) 61:237–42. doi: 10.1016/j.jjcc.2012.11.006
69. Gangaplara A, Massilamany C, Brown DM, Delhon G, Pattnaik AK, Chapman N, et al. Coxsackievirus B3 infection leads to the generation of cardiac myosin heavy chain-alpha-reactive CD4 T cells in A/J mice. Clin Immunol. (2012) 144:237–49. doi: 10.1016/j.clim.2012.07.003
70. Zheng C, Wu SM, Lian H, Lin YZ, Zhuang R, Thapa S, et al. Low-intensity pulsed ultrasound attenuates cardiac inflammation of CVB3-induced viral myocarditis via regulation of caveolin-1 and MAPK pathways. J Cell Mol Med. (2018) 23:1963–75. doi: 10.1111/jcmm.14098
71. Gou W, Zhang Z, Yang C, Li Y. MiR-223/Pknox1 axis protects mice from CVB3-induced viral myocarditis by modulating macrophage polarization. Exp Cell Res. (2018) 366:41–8. doi: 10.1016/j.yexcr.2018.03.004
72. Massilamany C, Gangaplara A, Basavalingappa RH, Rajasekaran RA, Vu H, Riethoven JJ, et al. Mutations in the 5' NTR and the non-structural protein 3A of the coxsackievirus B3 selectively attenuate myocarditogenicity. PLoS ONE. (2015) 10:e0131052. doi: 10.1371/journal.pone.0131052
73. Fuse K, Chan G, Liu Y, Gudgeon P, Husain M, Chen M, et al. Myeloid differentiation factor-88 plays a crucial role in the pathogenesis of Coxsackievirus B3-induced myocarditis and influences type I interferon production. Circulation. (2005) 112:2276–85. doi: 10.1161/CIRCULATIONAHA.105.536433
74. Valaperti A, Nishii M, Liu Y, Naito K, Chan M, Zhang L, et al. Innate immune interleukin-1 receptor-associated kinase 4 exacerbates viral myocarditis by reducing CCR5(+) CD11b(+) monocyte migration and impairing interferon production. Circulation. (2013) 128:1542–54. doi: 10.1161/CIRCULATIONAHA.113.002275
75. Matsumori A, Tomioka N, Kawai C. Protective effect of recombinant alpha interferon on coxsackievirus B3 myocarditis in mice. Am Heart J. (1988) 115:1229–32. doi: 10.1016/0002-8703(88)90013-0
76. Negishi H, Osawa T, Ogami K, Ouyang X, Sakaguchi S, Koshiba R, et al. A critical link between Toll-like receptor 3 and type II interferon signaling pathways in antiviral innate immunity. Proc Natl Acad Sci USA. (2008) 105:20446–51. doi: 10.1073/pnas.0810372105
77. Riad A, Westermann D, Zietsch C, Savvatis K, Becher PM, Bereswill S, et al. TRIF is a critical survival factor in viral cardiomyopathy. J Immunol. (2011) 186:2561–70. doi: 10.4049/jimmunol.1002029
78. Yuan J, Liu Z, Lim T, Zhang H, He J, Walker E, et al. CXCL10 inhibits viral replication through recruitment of natural killer cells in coxsackievirus B3-induced myocarditis. Circ Res. (2009) 104:628–38. doi: 10.1161/CIRCRESAHA.108.192179
79. Huber SA, Roberts B, Moussawi M, Boyson JE. Slam haplotype 2 promotes NKT but suppresses Vgamma4+ T-cell activation in coxsackievirus B3 infection leading to increased liver damage but reduced myocarditis. Am J Pathol. (2013) 182:401–9. doi: 10.1016/j.ajpath.2012.10.019
80. Liu W, Moussawi M, Roberts B, Boyson JE, Huber SA. Cross-regulation of T regulatory-cell response after coxsackievirus B3 infection by NKT and gammadelta T cells in the mouse. Am J Pathol. (2013) 183:441–9. doi: 10.1016/j.ajpath.2013.04.015
81. Nakamura H, Yamamura T, Umemoto S, Fukuta S, Shioi T, Matsumori A, et al. Autoimmune response in chronic ongoing myocarditis demonstrated by heterotopic cardiac transplantation in mice. Circulation. (1996) 94:3348–54. doi: 10.1161/01.CIR.94.12.3348
82. Yamada T, Matsumori A, Sasayama S. Therapeutic effect of anti-tumor necrosis factor-alpha antibody on the murine model of viral myocarditis induced by encephalomyocarditis virus. Circulation. (1994) 89:846–51. doi: 10.1161/01.CIR.89.2.846
83. Fairweather D, Rose NR. Coxsackievirus-induced myocarditis in mice: a model of autoimmune disease for studying immunotoxicity. Methods. (2007) 41:118–22. doi: 10.1016/j.ymeth.2006.07.009
84. Fairweather D, Frisancho-Kiss S, Yusung SA, Barrett MA, Davis SE, Steele RA, et al. IL-12 protects against coxsackievirus B3-induced myocarditis by increasing IFN-gamma and macrophage and neutrophil populations in the heart. J Immunol. (2005) 174:261–9. doi: 10.4049/jimmunol.174.1.261
85. Van Linthout S, Savvatis K, Miteva K, Peng J, Ringe J, Warstat K, et al. Mesenchymal stem cells improve murine acute coxsackievirus B3-induced myocarditis. Eur Heart J. (2011) 32:2168–78. doi: 10.1093/eurheartj/ehq467
86. Pappritz K, Savvatis K, Miteva K, Kerim B, Dong F, Fechner H, et al. Immunomodulation by adoptive regulatory T-cell transfer improves Coxsackievirus B3-induced myocarditis. FASEB J. (2018) 32:6066–78. doi: 10.1096/fj.201701408R
87. Savvatis K, Muller I, Frohlich M, Pappritz K, Zietsch C, Hamdani N, et al. Interleukin-6 receptor inhibition modulates the immune reaction and restores titin phosphorylation in experimental myocarditis. Basic Res Cardiol. (2014) 109:449. doi: 10.1007/s00395-014-0449-2
88. Miteva K, Pappritz K, El-Shafeey M, Dong F, Ringe J, Tschope C, et al. Mesenchymal stromal cells modulate monocytes trafficking in coxsackievirus B3-induced myocarditis. Stem Cells Transl Med. (2017) 6:1249–61. doi: 10.1002/sctm.16-0353
89. Kraft L, Erdenesukh T, Sauter M, Tschope C, Klingel K. Blocking the IL-1beta signalling pathway prevents chronic viral myocarditis and cardiac remodeling. Basic Res Cardiol. (2019) 114:11. doi: 10.1007/s00395-019-0719-0
90. Afanasyeva M, Georgakopoulos D, Belardi DF, Bedja D, Fairweather D, Wang Y, et al. Impaired up-regulation of CD25 on CD4+ T cells in IFN-gamma knockout mice is associated with progression of myocarditis to heart failure. Proc Natl Acad Sci USA. (2005) 102:180–5. doi: 10.1073/pnas.0408241102
91. Klingel K, Hohenadl C, Canu A, Albrecht M, Seemann M, Mall G, et al. Ongoing enterovirus-induced myocarditis is associated with persistent heart muscle infection: quantitative analysis of virus replication, tissue damage, and inflammation. Proc Natl Acad Sci USA. (1992) 89:314–8. doi: 10.1073/pnas.89.1.314
92. Becher PM, Gotzhein F, Klingel K, Escher F, Blankenberg S, Westermann D, et al. Cardiac function remains impaired despite reversible cardiac remodeling after acute experimental viral myocarditis. J Immunol Res. (2017) 2017:6590609. doi: 10.1155/2017/6590609
93. Lane JR, Neumann DA, Lafond-Walker A, Herskowitz A, Rose NR. LPS promotes CB3-induced myocarditis in resistant B10.A mice. Cell Immunol. (1991) 136:219–33. doi: 10.1016/0008-8749(91)90396-S
94. Heymans S, Pauschinger M, De Palma A, Kallwellis-Opara A, Rutschow S, Swinnen M, et al. Inhibition of urokinase-type plasminogen activator or matrix metalloproteinases prevents cardiac injury and dysfunction during viral myocarditis. Circulation. (2006) 114:565–73. doi: 10.1161/CIRCULATIONAHA.105.591032
95. Blailock ZR, Rabin ER, Melnick JL. Adenovirus endocarditis in mice. Science. (1967) 157:69–70. doi: 10.1126/science.157.3784.69
96. McCarthy MK, Procario MC, Twisselmann N, Wilkinson JE, Archambeau AJ, Michele DE, et al. Proinflammatory effects of interferon gamma in mouse adenovirus 1 myocarditis. J Virol. (2015) 89:468–79. doi: 10.1128/JVI.02077-14
97. Sherry B, Schoen FJ, Wenske E, Fields BN. Derivation and characterization of an efficiently myocarditic reovirus variant. J Virol. (1989) 63:4840–9.
98. Holm GH, Pruijssers AJ, Li L, Danthi P, Sherry B, Dermody TS. Interferon regulatory factor 3 attenuates reovirus myocarditis and contributes to viral clearance. J Virol. (2010) 84:6900–8. doi: 10.1128/JVI.01742-09
99. Sanchez-Guillen Mdel C, Bernabe C, Tibayrenc M, Zavala-Castro J, Totolhua JL, Mendez-Lopez J, et al. Trypanosoma cruzi strains isolated from human, vector, and animal reservoir in the same endemic region in Mexico and typed as T. cruzi I, discrete typing unit 1 exhibit considerable biological diversity. Mem Inst Oswaldo Cruz. (2006) 101:585–90. doi: 10.1590/S0074-02762006000600002
100. Michailowsky V, Silva NM, Rocha CD, Vieira LQ, Lannes-Vieira J, Gazzinelli RT. Pivotal role of interleukin-12 and interferon-gamma axis in controlling tissue parasitism and inflammation in the heart and central nervous system during Trypanosoma cruzi infection. Am J Pathol. (2001) 159:1723–33. doi: 10.1016/S0002-9440(10)63019-2
101. Camandaroba EL, Pinheiro Lima CM, Andrade SG. Oral transmission of Chagas disease: importance of Trypanosoma cruzi biodeme in the intragastric experimental infection. Rev Inst Med Trop Sao Paulo. (2002) 44:97–103. doi: 10.1590/S0036-46652002000200008
102. Camandaroba E, The TS, Pessina DH, Andrade SG. Trypanosoma cruzi: clones isolated from the Colombian strain, reproduce the parental strain characteristics, with ubiquitous histotropism. Int J Exp Pathol. (2006) 87:209–17. doi: 10.1111/j.1365-2613.2006.00476.x
103. Marino AP, Azevedo MI, Lannes-Vieira J. Differential expression of adhesion moleculesshaping the T-cell subset prevalence during the early phase of autoimmune and Trypanosoma cruzi-elicited myocarditis. Mem Inst Oswaldo Cruz. (2003) 98:945–52. doi: 10.1590/S0074-02762003000700015
104. Andrade LO, Machado CR, Chiari E, Pena SD, Macedo AM. Trypanosoma cruzi: role of host genetic background in the differential tissue distribution of parasite clonal populations. Exp Parasitol. (2002) 100:269–75. doi: 10.1016/S0014-4894(02)00024-3
105. Sanches TL, Cunha LD, Silva GK, Guedes PM, Silva JS, Zamboni DS. The use of a heterogeneously controlled mouse population reveals a significant correlation of acute phase parasitemia with mortality in Chagas disease. PLoS ONE. (2014) 9:e91640. doi: 10.1371/journal.pone.0091640
106. Bustamante JM, Rivarola HW, Fretes R, Paglini-Oliva PA. Weekly electrocardiographic pattern in mice infected with two different Trypanosoma cruzi strains. Int J Cardiol. (2005) 102:211–7. doi: 10.1016/j.ijcard.2004.05.014
107. Marinho CR, Nunez-Apaza LN, Bortoluci KR, Bombeiro AL, Bucci DZ, Grisotto MG, et al. Infection by the Sylvio X10/4 clone of Trypanosoma cruzi: relevance of a low-virulence model of Chagas' disease. Microbes Infect. (2009) 11:1037–45. doi: 10.1016/j.micinf.2009.07.011
108. Ponce NE, Sanmarco LM, Eberhardt N, Garcia MC, Rivarola HW, Cano RC, et al. CD73 inhibition shifts cardiac macrophage polarization toward a microbicidal phenotype and ameliorates the outcome of experimental chagas cardiomyopathy. J Immunol. (2016) 197:814–23. doi: 10.4049/jimmunol.1600371
109. Vilar-Pereira G, Carneiro VC, Mata-Santos H, Vicentino AR, Ramos IP, Giarola NL, et al. Resveratrol reverses functional chagas heart disease in mice. PLoS Pathog. (2016) 12:e1005947. doi: 10.1371/journal.ppat.1005947
110. Garcia S, Ramos CO, Senra JF, Vilas-Boas F, Rodrigues MM, Campos-de-Carvalho AC, et al. Treatment with benznidazole during the chronic phase of experimental Chagas' disease decreases cardiac alterations. Antimicrob Agents Chemother. (2005) 49:1521–8. doi: 10.1128/AAC.49.4.1521-1528.2005
111. Campos MA, Almeida IC, Takeuchi O, Akira S, Valente EP, Procopio DO, et al. Activation of Toll-like receptor-2 by glycosylphosphatidylinositol anchors from a protozoan parasite. J Immunol. (2001) 167:416–23. doi: 10.4049/jimmunol.167.1.416
112. Oliveira AC, Peixoto JR, de Arruda LB, Campos MA, Gazzinelli RT, Golenbock DT, et al. Expression of functional TLR4 confers proinflammatory responsiveness to Trypanosoma cruzi glycoinositolphospholipids and higher resistance to infection with T. cruzi. J Immunol. (2004) 173:5688–96. doi: 10.4049/jimmunol.173.9.5688
113. Bafica A, Santiago HC, Goldszmid R, Ropert C, Gazzinelli RT, Sher A. Cutting edge: TLR9 and TLR2 signaling together account for MyD88-dependent control of parasitemia in Trypanosoma cruzi infection. J Immunol. (2006) 177:3515–9. doi: 10.4049/jimmunol.177.6.3515
114. Huang H, Petkova SB, Cohen AW, Bouzahzah B, Chan J, Zhou JN, et al. Activation of transcription factors AP-1 and NF-kappa B in murine Chagasic myocarditis. Infect Immun. (2003) 71:2859–67. doi: 10.1128/IAI.71.5.2859-2867.2003
115. Huang H, Petkova SB, Pestell RG, Bouzahzah B, Chan J, Magazine H, et al. Trypanosoma cruzi infection (Chagas' disease) of mice causes activation of the mitogen-activated protein kinase cascade and expression of endothelin-1 in the myocardium. J Cardiovasc Pharmacol. (2000) 36:S148–50. doi: 10.1097/00005344-200036001-00046
116. Vespa GN, Cunha FQ, Silva JS. Nitric oxide is involved in control of Trypanosoma cruzi-induced parasitemia and directly kills the parasite in vitro. Infect Immun. (1994) 62:5177–82.
117. Monteiro AC, Schmitz V, Svensjo E, Gazzinelli RT, Almeida IC, Todorov A, et al. Cooperative activation of TLR2 and bradykinin B2 receptor is required for induction of type 1 immunity in a mouse model of subcutaneous infection by Trypanosoma cruzi. J Immunol. (2006) 177:6325–35. doi: 10.4049/jimmunol.177.9.6325
118. de Meis J, Morrot A, Farias-de-Oliveira DA, Villa-Verde DM, Savino W. Differential regional immune response in Chagas disease. PLoS Negl Trop Dis. (2009) 3:e417. doi: 10.1371/journal.pntd.0000417
119. Minoprio PM, Eisen H, Forni L, D'Imperio Lima MR, Joskowicz M, et al. Polyclonal lymphocyte responses to murine Trypanosoma cruzi infection. I. Quantitation of both T- and B-cell responses. Scand J Immunol. (1986) 24:661–8. doi: 10.1111/j.1365-3083.1986.tb02185.x
120. Minoprio P, Bandeira A, Pereira P, Mota Santos T, Coutinho A. Preferential expansion of Ly-1 B and CD4- CD8- T cells in the polyclonal lymphocyte responses to murine T. cruzi infection. Int Immunol. (1989) 1:176–84. doi: 10.1093/intimm/1.2.176
121. Todeschini AR, Nunes MP, Pires RS, Lopes MF, Previato JO, Mendonca-Previato L, et al. Costimulation of host T lymphocytes by a trypanosomal trans-sialidase: involvement of CD43 signaling. J Immunol. (2002) 168:5192–8. doi: 10.4049/jimmunol.168.10.5192
122. Shoda LK, Kegerreis KA, Suarez CE, Roditi I, Corral RS, Bertot GM, et al. DNA from protozoan parasites Babesia bovis, Trypanosoma cruzi, and T. brucei is mitogenic for B lymphocytes and stimulates macrophage expression of interleukin-12, tumor necrosis factor alpha, and nitric oxide. Infect Immun. (2001) 69:2162–71. doi: 10.1128/IAI.69.4.2162-2171.2001
123. Kumar S, Tarleton RL. Antigen-specific Th1 but not Th2 cells provide protection from lethal Trypanosoma cruzi infection in mice. J Immunol. (2001) 166:4596–603. doi: 10.4049/jimmunol.166.7.4596
124. Martin DL, Tarleton RL. Antigen-specific T cells maintain an effector memory phenotype during persistent Trypanosoma cruzi infection. J Immunol. (2005) 174:1594–601. doi: 10.4049/jimmunol.174.3.1594
125. Tarleton RL, Sun J, Zhang L, Postan M. Depletion of T-cell subpopulations results in exacerbation of myocarditis and parasitism in experimental Chagas' disease. Infect Immun. (1994) 62:1820–9.
126. Kumar S, Tarleton RL. The relative contribution of antibody production and CD8+ T cell function to immune control of Trypanosoma cruzi. Parasite Immunol. (1998) 20:207–16. doi: 10.1046/j.1365-3024.1998.00154.x
127. Sullivan NL, Eickhoff CS, Sagartz J, Hoft DF. Deficiency of antigen-specific B cells results in decreased Trypanosoma cruzi systemic but not mucosal immunity due to CD8 T cell exhaustion. J Immunol. (2015) 194:1806–18. doi: 10.4049/jimmunol.1303163
128. Cardillo F, Postol E, Nihei J, Aroeira LS, Nomizo A, Mengel J. B cells modulate T cells so as to favour T helper type 1 and CD8+ T-cell responses in the acute phase of Trypanosoma cruzi infection. Immunology. (2007) 122:584–95. doi: 10.1111/j.1365-2567.2007.02677.x
129. Pereira ME, Santos LM, Araujo MS, Brener Z. Recrudescence induced by cyclophosphamide of chronic Trypanosoma cruzi infection in mice is influenced by the parasite strain. Mem Inst Oswaldo Cruz. (1996) 91:71–4. doi: 10.1590/S0074-02761996000100011
130. Santos DM, Martins TA, Caldas IS, Diniz LF, Machado-Coelho GL, Carneiro CM, et al. Benznidazole alters the pattern of Cyclophosphamide-induced reactivation in experimental Trypanosoma cruzi-dependent lineage infection. Acta Trop. (2010) 113:134–8. doi: 10.1016/j.actatropica.2009.10.007
131. Rizzo LV, Cunha-Neto E, Teixeira AR. Autoimmunity in Chagas' disease: specific inhibition of reactivity of CD4+ T cells against myosin in mice chronically infected with Trypanosoma cruzi. Infect Immun. (1989) 57:2640–4.
132. Pontes-de-Carvalho L, Santana CC, Soares MB, Oliveira GG, Cunha-Neto E, Ribeiro-dos-Santos R. Experimental chronic Chagas' disease myocarditis is an autoimmune disease preventable by induction of immunological tolerance to myocardial antigens. J Autoimmun. (2002) 18:131–8. doi: 10.1006/jaut.2001.0574
133. Leon JS, Wang K, Engman DM. Myosin autoimmunity is not essential for cardiac inflammation in acute Chagas' disease. J Immunol. (2003) 171:4271–7. doi: 10.4049/jimmunol.171.8.4271
134. Neu N, Rose NR, Beisel KW, Herskowitz A, Gurri-Glass G, Craig SW. Cardiac myosin induces myocarditis in genetically predisposed mice. J Immunol. (1987) 139:3630–6.
135. Blyszczuk P, Behnke S, Luscher TF, Eriksson U, Kania G. GM-CSF promotes inflammatory dendritic cell formation but does not contribute to disease progression in experimental autoimmune myocarditis. Biochim Biophys Acta. (2013) 1833:934–44. doi: 10.1016/j.bbamcr.2012.10.008
136. Blyszczuk P, Berthonneche C, Behnke S, Glonkler M, Moch H, Pedrazzini T, et al. Nitric oxide synthase 2 is required for conversion of pro-fibrogenic inflammatory CD133+ progenitors into F4/80+ macrophages in experimental autoimmune myocarditis. Cardiovasc Res. (2013) 97:219–29. doi: 10.1093/cvr/cvs317
137. Kania G, Blyszczuk P, Stein S, Valaperti A, Germano D, Dirnhofer S, et al. Heart-infiltrating prominin-1+/CD133+ progenitor cells represent the cellular source of transforming growth factor beta-mediated cardiac fibrosis in experimental autoimmune myocarditis. Circ Res. (2009) 105:462–70. doi: 10.1161/CIRCRESAHA.109.196287
138. Kania G, Blyszczuk P, Valaperti A, Dieterle T, Leimenstoll B, Dirnhofer S, et al. Prominin-1+/CD133+ bone marrow-derived heart-resident cells suppress experimental autoimmune myocarditis. Cardiovasc Res. (2008) 80:236–45. doi: 10.1093/cvr/cvn190
139. Kania G, Siegert S, Behnke S, Prados-Rosales R, Casadevall A, Luscher TF, et al. Innate signaling promotes formation of regulatory nitric oxide-producing dendritic cells limiting T-cell expansion in experimental autoimmune myocarditis. Circulation. (2013) 127:2285–94. doi: 10.1161/CIRCULATIONAHA.112.000434
140. van Heeswijk RB, De Blois J, Kania G, Gonzales C, Blyszczuk P, Stuber M, et al. Selective in-vivo visualization of immune-cell infiltration in a mouse model of autoimmune myocarditis by fluorine-19 cardiac magnetic resonance. Circ Cardiovasc Imaging. (2013) 6:277–84. doi: 10.1161/CIRCIMAGING.112.000125
141. Blyszczuk P, Muller-Edenborn B, Valenta T, Osto E, Stellato M, Behnke S, et al. Transforming growth factor-beta-dependent Wnt secretion controls myofibroblast formation and myocardial fibrosis progression in experimental autoimmune myocarditis. Eur Heart J. (2017) 38:1413–25. doi: 10.1093/eurheartj/ehw116
142. Baldeviano GC, Barin JG, Talor MV, Srinivasan S, Bedja D, Zheng D, et al. Interleukin-17A is dispensable for myocarditis but essential for the progression to dilated cardiomyopathy. Circ Res. (2010) 106:1646–55. doi: 10.1161/CIRCRESAHA.109.213157
143. Ma W, Wang Y, Lu S, Yan L, Hu F, Wang Z. Targeting androgen receptor with ASC-J9 attenuates cardiac injury and dysfunction in experimental autoimmune myocarditis by reducing M1-like macrophage. Biochem Biophys Res Commun. (2017) 485:746–52. doi: 10.1016/j.bbrc.2017.02.123
144. Thelemann C, Haller S, Blyszczuk P, Kania G, Rosa M, Eriksson U, et al. Absence of nonhematopoietic MHC class II expression protects mice from experimental autoimmune myocarditis. Eur J Immunol. (2016) 46:656–64. doi: 10.1002/eji.201545945
145. Machino-Ohtsuka T, Tajiri K, Kimura T, Sakai S, Sato A, Yoshida T, et al. Tenascin-C aggravates autoimmune myocarditis via dendritic cell activation and Th17 cell differentiation. J Am Heart Assoc. (2014) 3:e001052. doi: 10.1161/JAHA.114.001052
146. Martin R, Cordova C, San Roman JA, Gutierrez B, Cachofeiro V, Nieto ML. Oleanolic acid modulates the immune-inflammatory response in mice with experimental autoimmune myocarditis and protects from cardiac injury. Therapeutic implications for the human disease. J Mol Cell Cardiol. (2014) 72:250–62. doi: 10.1016/j.yjmcc.2014.04.002
147. Barin JG, Baldeviano GC, Talor MV, Wu L, Ong S, Fairweather D, et al. Fatal eosinophilic myocarditis develops in the absence of IFN-gamma and IL-17A. J Immunol. (2013) 191:4038–47. doi: 10.4049/jimmunol.1301282
148. Smith SC, Allen PM. Myosin-induced acute myocarditis is a T cell-mediated disease. J Immunol. (1991) 147:2141–7.
149. Valaperti A, Marty RR, Kania G, Germano D, Mauermann N, Dirnhofer S, et al. CD11b+ monocytes abrogate Th17 CD4+ T cell-mediated experimental autoimmune myocarditis. J Immunol. (2008) 180:2686–95. doi: 10.4049/jimmunol.180.4.2686
150. Neu N, Ploier B, Ofner C. Cardiac myosin-induced myocarditis. Heart autoantibodies are not involved in the induction of the disease. J Immunol. (1990) 145:4094–100.
151. Liao L, Sindhwani R, Rojkind M, Factor S, Leinwand L, Diamond B. Antibody-mediated autoimmune myocarditis depends on genetically determined target organ sensitivity. J Exp Med. (1995) 181:1123–31. doi: 10.1084/jem.181.3.1123
152. Massilamany C, Gangaplara A, Basavalingappa RH, Rajasekaran RA, Khalilzad-Sharghi V, Han Z, et al. Localization of CD8 T cell epitope within cardiac myosin heavy chain-alpha334-352 that induces autoimmune myocarditis in A/J mice. Int J Cardiol. (2016) 202:311–21. doi: 10.1016/j.ijcard.2015.09.016
153. Smith SC, Allen PM. Neutralization of endogenous tumor necrosis factor ameliorates the severity of myosin-induced myocarditis. Circ Res. (1992) 70:856–63. doi: 10.1161/01.RES.70.4.856
154. Bachmaier K, Pummerer C, Kozieradzki I, Pfeffer K, Mak TW, Neu N, et al. Low-molecular-weight tumor necrosis factor receptor p55 controls induction of autoimmune heart disease. Circulation. (1997) 95:655–61. doi: 10.1161/01.CIR.95.3.655
155. Sonderegger I, Iezzi G, Maier R, Schmitz N, Kurrer M, Kopf M. GM-CSF mediates autoimmunity by enhancing IL-6-dependent Th17 cell development and survival. J Exp Med. (2008) 205:2281–94. doi: 10.1084/jem.20071119
156. Eriksson U, Kurrer MO, Sonderegger I, Iezzi G, Tafuri A, Hunziker L, et al. Activation of dendritic cells through the interleukin 1 receptor 1 is critical for the induction of autoimmune myocarditis. J Exp Med. (2003) 197:323–31. doi: 10.1084/jem.20021788
157. Eriksson U, Kurrer MO, Schmitz N, Marsch SC, Fontana A, Eugster HP, et al. Interleukin-6-deficient mice resist development of autoimmune myocarditis associated with impaired upregulation of complement C3. Circulation. (2003) 107:320–5. doi: 10.1161/01.CIR.0000043802.38699.66
158. Wu L, Diny NL, Ong S, Barin JG, Hou X, Rose NR, et al. Pathogenic IL-23 signaling is required to initiate GM-CSF-driven autoimmune myocarditis in mice. Eur J Immunol. (2016) 46:582–92. doi: 10.1002/eji.201545924
159. Pummerer CL, Luze K, Grassl G, Bachmaier K, Offner F, Burrell SK, et al. Identification of cardiac myosin peptides capable of inducing autoimmune myocarditis in BALB/c mice. J Clin Invest. (1996) 97:2057–62. doi: 10.1172/JCI118642
160. Massilamany C, Gangaplara A, Steffen D, Reddy J. Identification of novel mimicry epitopes for cardiac myosin heavy chain-alpha that induce autoimmune myocarditis in A/J mice. Cell Immunol. (2011) 271:438–49. doi: 10.1016/j.cellimm.2011.08.013
161. Li HS, Ligons DL, Rose NR, Guler ML. Genetic differences in bone marrow-derived lymphoid lineages control susceptibility to experimental autoimmune myocarditis. J Immunol. (2008) 180:7480–4. doi: 10.4049/jimmunol.180.11.7480
162. Smith SC, Allen PM. Expression of myosin-class II major histocompatibility complexes in the normal myocardium occurs before induction of autoimmune myocarditis. Proc Natl Acad Sci USA. (1992) 89:9131–5. doi: 10.1073/pnas.89.19.9131
163. Guler ML, Ligons DL, Wang Y, Bianco M, Broman KW, Rose NR. Two autoimmune diabetes loci influencing T cell apoptosis control susceptibility to experimental autoimmune myocarditis. J Immunol. (2005) 174:2167–73. doi: 10.4049/jimmunol.174.4.2167
164. Eriksson U, Ricci R, Hunziker L, Kurrer MO, Oudit GY, Watts TH, et al. Dendritic cell-induced autoimmune heart failure requires cooperation between adaptive and innate immunity. Nat Med. (2003) 9:1484–90. doi: 10.1038/nm960
165. Van der Borght K, Scott CL, Martens L, Sichien D, Van Isterdael G, Nindl V, et al. Myocarditis elicits dendritic cell and monocyte infiltration in the heart and self-antigen presentation by conventional type 2 dendritic cells. Front Immunol. (2018) 9:2714. doi: 10.3389/fimmu.2018.02714
166. Nindl V, Maier R, Ratering D, De Giuli R, Zust R, Thiel V, et al. Cooperation of Th1 and Th17 cells determines transition from autoimmune myocarditis to dilated cardiomyopathy. Eur J Immunol. (2012) 42:2311–21. doi: 10.1002/eji.201142209
167. Grabie N, Delfs MW, Westrich JR, Love VA, Stavrakis G, Ahmad F, et al. IL-12 is required for differentiation of pathogenic CD8+ T cell effectors that cause myocarditis. J Clin Invest. (2003) 111:671–80. doi: 10.1172/JCI16867
168. Grabie N, Hsieh DT, Buono C, Westrich JR, Allen JA, Pang H, et al. Neutrophils sustain pathogenic CD8+ T cell responses in the heart. Am J Pathol. (2003) 163:2413–20. doi: 10.1016/S0002-9440(10)63596-1
169. Taqueti VR, Grabie N, Colvin R, Pang H, Jarolim P, Luster AD, et al. T-bet controls pathogenicity of CTLs in the heart by separable effects on migration and effector activity. J Immunol. (2006) 177:5890–901. doi: 10.4049/jimmunol.177.9.5890
170. Grabie N, Gotsman I, DaCosta R, Pang H, Stavrakis G, Butte MJ, et al. Endothelial programmed death-1 ligand 1 (PD-L1) regulates CD8+ T-cell mediated injury in the heart. Circulation. (2007) 116:2062–71. doi: 10.1161/CIRCULATIONAHA.107.709360
171. Nishimura H, Okazaki T, Tanaka Y, Nakatani K, Hara M, Matsumori A, et al. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science. (2001) 291:319–22. doi: 10.1126/science.291.5502.319
172. Okazaki T, Tanaka Y, Nishio R, Mitsuiye T, Mizoguchi A, Wang J, et al. Autoantibodies against cardiac troponin I are responsible for dilated cardiomyopathy in PD-1-deficient mice. Nat Med. (2003) 9:1477–83. doi: 10.1038/nm955
173. Lucas JA, Menke J, Rabacal WA, Schoen FJ, Sharpe AH, Kelley VR. Programmed death ligand 1 regulates a critical checkpoint for autoimmune myocarditis and pneumonitis in MRL mice. J Immunol. (2008) 181:2513–21. doi: 10.4049/jimmunol.181.4.2513
174. Wang J, Okazaki IM, Yoshida T, Chikuma S, Kato Y, Nakaki F, et al. PD-1 deficiency results in the development of fatal myocarditis in MRL mice. Int Immunol. (2010) 22:443–52. doi: 10.1093/intimm/dxq026
175. Tarrio ML, Grabie N, Bu DX, Sharpe AH, Lichtman AH. PD-1 protects against inflammation and myocyte damage in T cell-mediated myocarditis. J Immunol. (2012) 188:4876–84. doi: 10.4049/jimmunol.1200389
176. Elliott JF, Liu J, Yuan ZN, Bautista-Lopez N, Wallbank SL, Suzuki K, et al. Autoimmune cardiomyopathy and heart block develop spontaneously in HLA-DQ8 transgenic IAbeta knockout NOD mice. Proc Natl Acad Sci USA. (2003) 100:13447–52. doi: 10.1073/pnas.2235552100
177. Taneja V, Behrens M, Cooper LT, Yamada S, Kita H, Redfield MM, et al. Spontaneous myocarditis mimicking human disease occurs in the presence of an appropriate MHC and non-MHC background in transgenic mice. J Mol Cell Cardiol. (2007) 42:1054–64. doi: 10.1016/j.yjmcc.2007.03.898
178. Hayward SL, Bautista-Lopez N, Suzuki K, Atrazhev A, Dickie P, Elliott JF. CD4 T cells play major effector role and CD8 T cells initiating role in spontaneous autoimmune myocarditis of HLA-DQ8 transgenic IAb knockout nonobese diabetic mice. J Immunol. (2006) 176:7715–25. doi: 10.4049/jimmunol.176.12.7715
179. Taylor JA, Havari E, McInerney MF, Bronson R, Wucherpfennig KW, Lipes MA. A spontaneous model for autoimmune myocarditis using the human MHC molecule HLA-DQ8. J Immunol. (2004) 172:2651–8. doi: 10.4049/jimmunol.172.4.2651
180. Boldizsar F, Tarjanyi O, Olasz K, Hegyi A, Mikecz K, Glant TT, et al. FTY720 (Gilenya) treatment prevents spontaneous autoimmune myocarditis and dilated cardiomyopathy in transgenic HLA-DQ8-BALB/c mice. Cardiovasc Pathol. (2016) 25:353–61. doi: 10.1016/j.carpath.2016.05.003
181. Saraste A, Arola A, Vuorinen T, Kyto V, Kallajoki M, Pulkki K, et al. Cardiomyocyte apoptosis in experimental coxsackievirus B3 myocarditis. Cardiovasc Pathol. (2003) 12:255–62. doi: 10.1016/S1054-8807(03)00077-2
182. Zhou F, Jiang X, Teng L, Yang J, Ding J, He C. Necroptosis may be a novel mechanism for cardiomyocyte death in acute myocarditis. Mol Cell Biochem. (2017) 442:11–8. doi: 10.1007/s11010-017-3188-5
183. Lindner D, Li J, Savvatis K, Klingel K, Blankenberg S, Tschope C, et al. Cardiac fibroblasts aggravate viral myocarditis: cell specific coxsackievirus B3 replication. Mediators Inflamm. (2014) 2014:519528. doi: 10.1155/2014/519528
184. Tschope C, Muller I, Xia Y, Savvatis K, Pappritz K, Pinkert S, et al. NOD2 (Nucleotide-Binding Oligomerization Domain 2) is a major pathogenic mediator of coxsackievirus B3-induced myocarditis. Circ Heart Fail. (2017) 10:e003870. doi: 10.1161/CIRCHEARTFAILURE.117.003870
185. Garmaroudi FS, Marchant D, Hendry R, Luo H, Yang D, Ye X, et al. Coxsackievirus B3 replication and pathogenesis. Future Microbiol. (2015) 10:629–53. doi: 10.2217/fmb.15.5
186. Pauschinger M, Doerner A, Kuehl U, Schwimmbeck PL, Poller W, Kandolf R, et al. Enteroviral RNA replication in the myocardium of patients with left ventricular dysfunction and clinically suspected myocarditis. Circulation. (1999) 99:889–95. doi: 10.1161/01.CIR.99.7.889
187. Kuhl U, Pauschinger M, Schwimmbeck PL, Seeberg B, Lober C, Noutsias M, et al. Interferon-beta treatment eliminates cardiotropic viruses and improves left ventricular function in patients with myocardial persistence of viral genomes and left ventricular dysfunction. Circulation. (2003) 107:2793–8. doi: 10.1161/01.CIR.0000072766.67150.51
188. Epelman S, Liu PP, Mann DL. Role of innate and adaptive immune mechanisms in cardiac injury and repair. Nat Rev Immunol. (2015) 15:117–29. doi: 10.1038/nri3800
189. Chow LH, Beisel KW, McManus BM. Enteroviral infection of mice with severe combined immunodeficiency. Evidence for direct viral pathogenesis of myocardial injury. Lab Invest. (1992) 66:24–31.
190. Merkle I, Tonew M, Gluck B, Schmidtke M, Egerer R, Stelzner A. Coxsackievirus B3-induced chronic myocarditis in outbred NMRI mice. J Hum Virol. (1999) 2:369–79.
191. Henke A, Huber S, Stelzner A, Whitton JL. The role of CD8+ T lymphocytes in coxsackievirus B3-induced myocarditis. J Virol. (1995) 69:6720–8.
192. Leipner C, Borchers M, Merkle I, Stelzner A. Coxsackievirus B3-induced myocarditis in MHC class II-deficient mice. J Hum Virol. (1999) 2:102–14.
193. Huber SA, Sartini D, Exley M. Vgamma4(+) T cells promote autoimmune CD8(+) cytolytic T-lymphocyte activation in coxsackievirus B3-induced myocarditis in mice: role for CD4(+) Th1 cells. J Virol. (2002) 76:10785–90. doi: 10.1128/JVI.76.21.10785-10790.2002
194. Takata S, Nakamura H, Umemoto S, Yamaguchil K, Sekine T, Kato T, et al. Identification of autoantibodies with the corresponding antigen for repetitive coxsackievirus infection-induced cardiomyopathy. Circ J. (2004) 68:677–82. doi: 10.1253/circj.68.677
195. Latva-Hirvela J, Kyto V, Saraste A, Eriksson S, Vuorinen T, Pettersson K, et al. Development of troponin autoantibodies in experimental coxsackievirus B3 myocarditis. Eur J Clin Invest. (2009) 39:457–62. doi: 10.1111/j.1365-2362.2009.02113.x
196. Caforio AL, Tona F, Bottaro S, Vinci A, Dequal G, Daliento L, et al. Clinical implications of anti-heart autoantibodies in myocarditis and dilated cardiomyopathy. Autoimmunity. (2008) 41:35–45. doi: 10.1080/08916930701619235
197. Fousteri G, Dave A, Morin B, Omid S, Croft M, von Herrath MG. Nasal cardiac myosin peptide treatment and OX40 blockade protect mice from acute and chronic virally-induced myocarditis. J Autoimmun. (2011) 36:210–20. doi: 10.1016/j.jaut.2011.01.006
198. Hardarson HS, Baker JS, Yang Z, Purevjav E, Huang CH, Alexopoulou L, et al. Toll-like receptor 3 is an essential component of the innate stress response in virus-induced cardiac injury. Am J Physiol Heart Circ Physiol. (2007) 292:H251–8. doi: 10.1152/ajpheart.00398.2006
199. Bowles NE, Ni J, Kearney DL, Pauschinger M, Schultheiss HP, McCarthy R, et al. Detection of viruses in myocardial tissues by polymerase chain reaction. evidence of adenovirus as a common cause of myocarditis in children and adults. J Am Coll Cardiol. (2003) 42:466–72. doi: 10.1016/S0735-1097(03)00648-X
200. Kuhl U, Schultheiss HP. Myocarditis in children. Heart Fail Clin. (2010) 6:483–96, viii–ix. doi: 10.1016/j.hfc.2010.05.009
201. Wizel B, Palmieri M, Mendoza C, Arana B, Sidney J, Sette A, et al. Human infection with Trypanosoma cruzi induces parasite antigen-specific cytotoxic T lymphocyte responses. J Clin Invest. (1998) 102:1062–71. doi: 10.1172/JCI3835
202. Lattes R, Lasala MB. Chagas disease in the immunosuppressed patient. Clin Microbiol Infect. (2014) 20:300–9. doi: 10.1111/1469-0691.12585
203. Morillo CA, Marin-Neto JA, Avezum A, Sosa-Estani S, Rassi A Jr, Rosas F, et al. Randomized trial of benznidazole for chronic chagas' cardiomyopathy. N Engl J Med. (2015) 373:1295–306. doi: 10.1056/NEJMoa1507574
204. Tibbetts RS, McCormick TS, Rowland EC, Miller SD, Engman DM. Cardiac antigen-specific autoantibody production is associated with cardiomyopathy in Trypanosoma cruzi-infected mice. J Immunol. (1994) 152:1493–9.
205. Cunha-Neto E, Coelho V, Guilherme L, Fiorelli A, Stolf N, Kalil J. Autoimmunity in Chagas' disease. Identification of cardiac myosin-B13 Trypanosoma cruzi protein crossreactive T cell clones in heart lesions of a chronic Chagas' cardiomyopathy patient. J Clin Invest. (1996) 98:1709–12. doi: 10.1172/JCI118969
206. Leon JS, Godsel LM, Wang K, Engman DM. Cardiac myosin autoimmunity in acute Chagas' heart disease. Infect Immun. (2001) 69:5643–9. doi: 10.1128/IAI.69.9.5643-5649.2001
207. Lv H, Havari E, Pinto S, Gottumukkala RV, Cornivelli L, Raddassi K, et al. Impaired thymic tolerance to alpha-myosin directs autoimmunity to the heart in mice and humans. J Clin Invest. (2011) 121:1561–73. doi: 10.1172/JCI44583
208. Kaplan MH. Immunologic studies of heart tissue. I. Production in rabbits of antibodies reactive with an autologous myocardial antigen following immunization with heterologous heart tissue. J Immunol. (1958) 80: 254-67.
209. Rose NR. The adjuvant effect in infection and autoimmunity. Clin Rev Allergy Immunol. (2008) 34:279–82. doi: 10.1007/s12016-007-8049-7
210. Mitchell TC, Hildeman D, Kedl RM, Teague TK, Schaefer BC, White J, et al. Immunological adjuvants promote activated T cell survival via induction of Bcl-3. Nat Immunol. (2001) 2:397–402. doi: 10.1038/87692
211. Blyszczuk P, Kania G, Dieterle T, Marty RR, Valaperti A, Berthonneche C, et al. Myeloid differentiation factor-88/interleukin-1 signaling controls cardiac fibrosis and heart failure progression in inflammatory dilated cardiomyopathy. Circ Res. (2009) 105:912–20. doi: 10.1161/CIRCRESAHA.109.199802
212. Ono M, Shimizu J, Miyachi Y, Sakaguchi S. Control of autoimmune myocarditis and multiorgan inflammation by glucocorticoid-induced TNF receptor family-related protein(high), Foxp3-expressing CD25+ and CD25- regulatory T cells. J Immunol. (2006) 176:4748–56. doi: 10.4049/jimmunol.176.8.4748
213. Nishimura H, Nose M, Hiai H, Minato N, Honjo T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity. (1999) 11:141–51. doi: 10.1016/S1074-7613(00)80089-8
214. Goser S, Andrassy M, Buss SJ, Leuschner F, Volz CH, Ottl R, et al. Cardiac troponin I but not cardiac troponin T induces severe autoimmune inflammation in the myocardium. Circulation. (2006) 114:1693–702. doi: 10.1161/CIRCULATIONAHA.106.635664
215. Smith SC, Ladenson JH, Mason JW, Jaffe AS. Elevations of cardiac troponin I associated with myocarditis. Experimental and clinical correlates. Circulation. (1997) 95:163–8. doi: 10.1161/01.CIR.95.1.163
216. Leuschner F, Li J, Goser S, Reinhardt L, Ottl R, Bride P, et al. Absence of auto-antibodies against cardiac troponin I predicts improvement of left ventricular function after acute myocardial infarction. Eur Heart J. (2008) 29:1949–55. doi: 10.1093/eurheartj/ehn268
217. Mummery CL. Perspectives on the use of human induced pluripotent stem cell-derived cardiomyocytes in biomedical research. Stem Cell Rep. (2018) 11:1306–11. doi: 10.1016/j.stemcr.2018.11.011
218. Karakikes I, Ameen M, Termglinchan V, Wu JC. Human induced pluripotent stem cell-derived cardiomyocytes: insights into molecular, cellular, and functional phenotypes. Circ Res. (2015) 117:80–8. doi: 10.1161/CIRCRESAHA.117.305365
219. Zuppinger C. 3D culture for cardiac cells. Biochim Biophys Acta. (2016) 1863:1873–81. doi: 10.1016/j.bbamcr.2015.11.036
Keywords: myocarditis, animal models, coxsackievirus B3, Chagas disease, heart-specific autoimmunity, experimental autoimmune myocarditis
Citation: Błyszczuk P (2019) Myocarditis in Humans and in Experimental Animal Models. Front. Cardiovasc. Med. 6:64. doi: 10.3389/fcvm.2019.00064
Received: 20 December 2018; Accepted: 30 April 2019;
Published: 16 May 2019.
Edited by:
JeanSébastien Silvestre, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceReviewed by:
Sophie Van Linthout, Charité Medical University of Berlin, GermanyCopyright © 2019 Błyszczuk. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Przemysław Błyszczuk, cHJ6ZW15c2xhdy5ibHlzemN6dWtAdWouZWR1LnBs
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.