
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Cardiovasc. Med., 28 September 2018
Sec. Cardiovascular Metabolism
Volume 5 - 2018 | https://doi.org/10.3389/fcvm.2018.00135
This article is part of the Research TopicMechanisms by Which Acute and Chronic Exercise Promote Cardiometabolic HealthView all 9 articles
 Matthew A. Nystoriak*
Matthew A. Nystoriak* Aruni Bhatnagar
Aruni BhatnagarIt is widely accepted that regular physical activity is beneficial for cardiovascular health. Frequent exercise is robustly associated with a decrease in cardiovascular mortality as well as the risk of developing cardiovascular disease. Physically active individuals have lower blood pressure, higher insulin sensitivity, and a more favorable plasma lipoprotein profile. Animal models of exercise show that repeated physical activity suppresses atherogenesis and increases the availability of vasodilatory mediators such as nitric oxide. Exercise has also been found to have beneficial effects on the heart. Acutely, exercise increases cardiac output and blood pressure, but individuals adapted to exercise show lower resting heart rate and cardiac hypertrophy. Both cardiac and vascular changes have been linked to a variety of changes in tissue metabolism and signaling, although our understanding of the contribution of the underlying mechanisms remains incomplete. Even though moderate levels of exercise have been found to be consistently associated with a reduction in cardiovascular disease risk, there is evidence to suggest that continuously high levels of exercise (e.g., marathon running) could have detrimental effects on cardiovascular health. Nevertheless, a specific dose response relationship between the extent and duration of exercise and the reduction in cardiovascular disease risk and mortality remains unclear. Further studies are needed to identify the mechanisms that impart cardiovascular benefits of exercise in order to develop more effective exercise regimens, test the interaction of exercise with diet, and develop pharmacological interventions for those unwilling or unable to exercise.
Cardiovascular disease (CVD) is the leading cause of morbidity and mortality worldwide. In the United States, CVD accounts for ~600,000 deaths (25%) each year (1, 2), and after a continuous decline over the last 5 decades, its incidence is increasing again (3). Among the many risk factors that predispose to CVD development and progression, a sedentary lifestyle, characterized by consistently low levels of physical activity, is now recognized as a leading contributor to poor cardiovascular health. Conversely, regular exercise and physical activity are associated with remarkable widespread health benefits and a significantly lower CVD risk. Several long-term studies have shown that increased physical activity is associated with a reduction in all-cause mortality and may modestly increase life expectancy, an effect which is strongly linked to a decline in the risk of developing cardiovascular and respiratory diseases (4). Consistent with this notion, death rates among men and women have been found to be inversely related to cardiorespiratory fitness levels, even in the presence of other predictors of cardiovascular mortality such as smoking, hypertension, and hyperlipidemia (5). Moreover, better fitness levels in both men and women can partially reverse the elevated rates of all-cause mortality as well as CVD mortality associated with high body mass index (6, 7). Recent work from cardiovascular cohorts shows that sustained physical activity is associated with a more favorable inflammatory marker profile, decreases heart failure risk, and improves survival at 30 years follow-up in individuals with coronary artery disease (8–10).
Despite the robust beneficial effects of physical activity and exercise on cardiovascular health, the processes and mechanisms by which frequent physical activity promotes cardiorespiratory fitness and decreases CVD risk remain unclear. In the past several decades, considerable research effort has aimed to identify the major physiological and biochemical contributors to the cardiovascular benefits of exercise, and as a result, significant advances have been made from observational and interventional studies with human participants. In parallel, valuable mechanistic insights have been garnered from experimental studies in animal models. Thus, in this review, we provide a synopsis of the major known effects of exercise and physical activity on principal factors associated with risk for poor cardiovascular health including blood lipids, hypertension, and arterial stiffness. For the purpose of the review, we follow the definition of exercise as “a subset of physical activity that is planned, structured, and repetitive and has as a final or an intermediate objective the improvement or maintenance of physical fitness (11).” These characteristics distinguish exercise from less structured and planned physical activity, which is often not solely for the purpose of maintaining or improving physical fitness. Most long-term observational studies report levels of physical activity, whereas more controlled and short duration studies examine the effects of exercise. Throughout the text, we distinguish between these two types of activities to the extent possible. We also discuss the means by which a healthy cardiovascular system adapts to exercise conditioning as well as recently proposed mechanisms of adaptation that may work to antagonize cardiovascular disease.
Given the centrality of plasma lipids as key determinants of CVD risk, many studies have tested whether regular engagement in physical activity may lower CVD risk by affecting the levels of circulating lipoproteins. These studies have found that endurance training is associated with elevated levels of circulating high density lipoprotein (HDL) and, to a lesser extent, a reduction in triglyceride levels (12)—both changes that can reduce the risk of coronary heart disease (13). Nonetheless, results concerning the effects of physical activity on plasma lipids have been variable and confounded by an apparent dependence on the type, intensity, and duration of exercise as well as diet (14). In addition, early studies aimed at determining effects of physical activity on low density lipoprotein (LDL) levels did not test the dose-dependence of exercise. However, a study of subjects with mild to moderate dyslipidemia, randomized into high amount/high intensity (23 kcal/kg/wk, jogging), low amount/high intensity (14 kcal/kg/wk, jogging), and low amount/moderate intensity (14 kcal/kg/wk, walking) exercise training groups over a 6 months period, found a dose-dependent effect of exercise on plasma levels of LDL, triglycerides, and large particle, very low density lipoprotein (VLDL) (15). Increasing levels of exercise over time were also found in this study to increase HDL from baseline (pre-exercise regimen) levels. Although higher levels of HDL are associated lower CVD risk (16, 17), recent work suggests that some pharmacological interventions that elevate plasma HDL levels fail to reduce the risk of major cardiovascular events (18, 19). Nevertheless, HDL particle size is a key determinant of ATP binding cassette transporter A1 (ABCA1)-mediated cholesterol efflux (20), indicating that HDL particle size may be an important correlate of CVD risk. Hence, an increase in the size of LDL and HDL particles and a decrease in VLDL particle size, rather than HDL levels per se, upon exercise training (15) may impart CVD risk protection. In agreement with this view a recent study investigating the dose-dependent effects of exercise on cholesterol efflux in 2 randomized trials consisting of six distinct exercise doses reported a significant increase in HDL cholesterol and efflux capacity with exercise, albeit in the high amount/high intensity intervention groups only (21). Thus, even though exercise alters plasma lipid profile and increases HDL concentration and particle size, moderate exercise may produce only limited effects on HDL functionality and the contribution of changes in plasma lipoprotein concentration, structure, and function to overall reduction in CVD risk by exercise remains unclear.
In addition to changes in plasma lipids, exercise could directly impact the homeostasis of the arterial wall to antagonize the progression of atherosclerotic disease and thereby contribute to the well-documented reduction in coronary artery disease in people with active lifestyles, when compared with sedentary individuals (22–25). Even in people with symptomatic coronary artery disease, an increase in regular physical activity can improve VO2 max and, at high doses (~2,200 kcal/week), promote regression of atherosclerotic lesions (26). In patients with stable CAD, 4 weeks of rowing or cycling led to enhanced vasodilatory responses to acetylcholine, which was associated with increased total endothelial nitric oxide synthase (eNOS) expression and eNOS, and protein kinase B (Akt) phosphorylation (27). That exercise stimulates NO production is supported by animal studies. For instance, it has been reported that carotid arteries from exercised ApoE−/− mice exhibit elevated eNOS expression and suppressed neointimal formation after injury when compared with those from sedentary ApoE−/− control mice (28). In contrast, aorta from sedentary mice kept in normal housing conditions exhibit increased vascular lipid peroxidation and superoxide levels, which may contribute to endothelial dysfunction and lesion formation, when compared with mice subjected to 6 weeks of voluntary wheel running (29). Regular, but not intermittent, physical activity in high cholesterol diet-fed LDLR-null mice has also been found to rescue aortic valve endothelial integrity, reduce inflammatory cell recruitment, and prevent aortic valve calcification (30), which raises the possibility that exercise may reduce the development and progression of degenerative aortic valve disease. Despite this evidence, it remains unclear to what extent salutary changes in blood lipids and vascular function contribute to the cardiovascular benefits of exercise and further studies are required to quantify both lipid-dependent and lipid-independent effects of physical activity.
The association between blood lipids and cardiovascular health is highly influenced by systemic insulin sensitivity, and resistance to insulin signaling is known to promote the development of heart disease, in part by altering the blood lipid profile (31). Resistance of adipocytes to the effects of insulin and resulting reduction in glucose uptake leads to the increased release of free fatty acids and greater production and release of triglycerides, and VLDL by the liver (32). In addition, reduced HDL in the insulin resistant state, resulting in part from increased activity of cholesteryl ester transfer protein (CETP), and transfer of cholesteryl esters from HDL to triglyceride-rich lipoproteins (33), suppresses reverse cholesterol transport from the arterial wall and promotes atherosclerotic plaque formation.
Insulin signaling within the vascular endothelium promotes Akt-dependent phosphorylation and activation of eNOS, which produces the vasodilator - NO. This, however, is antagonized by the activation of the Ras-RAF-MAPK pathway that stimulates cell growth and differentiation and increases the production of the potent vasoconstrictor - endothelin-1 (ET-1) (34, 35). During diabetes, selective inhibition of the PI3K-Akt-eNOS pathway, together with compensatory hyperinsulinemia leads to unmasking and stimulation of the MAPK-mediated production of endothelin-1 (ET-1) (36, 37), and vascular smooth muscle proliferation, which could contribute to atherosclerotic plaque development and peripheral artery disease (38, 39). Enhanced endothelial production and secretion of ET-1, along with heightened sympathetic activity may represent key contributing factors in enhanced vasoconstriction of small diameter arteries and arterioles in the insulin-resistant state, thereby increasing systemic vascular resistance to blood flow and elevating arterial blood pressure. In addition, as a hallmark of diabetes and insulin-resistance, elevated blood glucose levels also accelerate the formation of advanced glycation end products (AGEs), proteins and lipids that have undergone non-enzymatic glycation and oxidation, leading to cross-linking of collagen and elastin fibers and loss of vascular compliance (i.e., arterial stiffening) (40, 41).
A number of studies have shown that individuals with insulin-dependent and non-insulin-dependent diabetes mellitus have improved sensitivity to insulin and improved glycemic control after exercise training (42–44). Indeed, it has been found that even a single low-intensity (50% VO2 max, 350 kcal expended) exercise session results in significantly improved insulin sensitivity and fatty acid uptake upon examination on the following day (45). Studies in animal models of exercise suggest that increased physical activity can improve insulin sensitivity in adipose tissue, skeletal muscle, and endothelium (46–49), which are major contributors to systemic insulin resistance in individuals with type 2 diabetes. While our understanding of the precise cellular and molecular mechanisms involved in the enhancement of insulin signaling following exercise has been hampered by inconsistent results across species and exercise protocols, it appears that exercise conditioning is associated with adaptive remodeling in the expression or regulation of one or more components of the insulin receptor/insulin receptor substrate (IRS)/PI3K/Akt signaling cascade (50–52). During exercise, insulin levels are slightly reduced and frequently contracting muscle exhibits greater glucose uptake via enhanced insulin-independent sarcolemmal translocation of GLUT4 glucose transporters (53–55). Moreover, muscle damage associated with eccentric exercise can paradoxically cause insulin resistance via TNF-α-mediated reductions in PI3K activity (56–59). Thus, further research is required to elucidate how certain exercise regimens can promote tissue-specific adaptations in insulin-signaling and how these pathways may be targeted to reverse insulin-resistance and associated cardiovascular complications of diabetes.
During exercise, increases in cardiac stroke volume and heart rate raise cardiac output, which coupled with a transient increase in systemic vascular resistance, elevate mean arterial blood pressure (60). However, long-term exercise can promote a net reduction in blood pressure at rest. A meta-analysis of randomized controlled interventional studies found that regular moderate to intense exercise performed 3–5 times per week lowers blood pressure by an average of 3.4/2.4 mmHg (61). While this change may appear small, recent work shows that even a 1 mmHg reduction in systolic BP is associated with 20.3 fewer (blacks) or 13.3 fewer (whites) heart failure events per 100,000 person-years (62). Thus, reductions in blood pressure observed when exercise is included as a behavioral intervention along with dietary modification and weight loss (63, 64) could have a significant impact on CVD incidence.
Lower ambulatory blood pressure, associated with chronic aerobic and resistance exercise, is thought to be driven largely by a chronic reduction in systemic vascular resistance (65). Contributing to this effect, shear forces, as well as released metabolites from active skeletal muscle during exercise, signal the production and release of nitric oxide (NO) and prostacyclin from the vascular endothelium, which promotes enhanced vasodilation via relaxation of vascular smooth muscle cells (66). This effect is especially significant because a reduction in eNOS activity that occurs with aging or due to NOS3 polymorphism, has been reported to contribute to hypertension (67–69). Long-term exercise training increases eNOS expression as well as NO production in hypertensive individuals, consistent with the blood pressure lowering effect of physical activity (70). An important role of NO in mediating the vascular effects of exercise is further supported by results showing that rats with hypertension induced by chronic NOS inhibition undergoing a swimming exercise regimen for 6 weeks have significantly elevated eNOS protein expression and improved acetylcholine-induced vasodilation (71). Thus, improvements in NO production and bioavailability appear to represent significant factors that contribute to improved endothelium-dependent vasodilation following exercise training, which can reduce resting vascular resistance and lower blood pressure. However, in addition to NO-mediated reductions in resistance vascular tone, adaptive reductions in sympathetic nerve activity, prevention or reversal of arterial stiffening, and suppression of inflammation are also likely contributors to the blood pressure lowering effects of exercise, although the impact of exercise on these outcomes may be population specific (e.g., at-risk versus healthy adults) (72–74). As with changes in blood lipid profile, it remains unclear to what extent the blood pressure lowering effects of exercise can account for the beneficial effects of exercise on CVD risk and mortality.
During exercise, the heart is subjected to intermittent hemodynamic stresses of pressure overload, volume overload, or both. To normalize such stress and to meet the systemic demand for an increased blood supply, the heart undergoes morphological adaptation to recurrent exercise by increasing its mass, primarily through an increase in ventricular chamber wall thickness. This augmentation of heart size is primarily the result of an increase in the size of individual terminally differentiated cardiac myocytes (75). Adaptive remodeling of the heart in response to exercise typically occurs with preservation or enhancement of contractile function. This contrasts with pathologic remodeling due to chronic sustained pressure overload (e.g., during hypertension or aortic stenosis), which can proceed to a loss of contractile function and heart failure (76).
Recent work in experimental animal exercise models has identified several cellular and molecular alterations involved in the physiologic growth program of the heart that accompanies exercise conditioning. Whereas pathologic remodeling of the heart is associated with a reduction in oxidative energy production via fatty acid oxidation and more reliance on glucose utilization, mitochondrial biogenesis and capacity for fatty acid oxidation are enhanced following exercise (77, 78). A recent study suggests that changes in myocardial glycolytic activity during acute exercise and the subsequent recovery period can also play an important role in regulating the expression of metabolic genes and cardiac remodeling (79). Possibly upstream of these metabolic changes, studies have also revealed a dominant role for IGF-1 and insulin receptor signaling, via the PI3K/Akt1 pathway leading to the activation of transcriptional pathways associated with protein synthesis and hypertrophy (80, 81). Untargeted approaches have identified other major determinants of transcriptional programs that drive the exercise-induced hypertrophic response. For instance, it has been reported that exercise-induced reduction in the expression of CCAAT-enhancer binding protein β (C/EBPβ) relieves its negative regulation by CBP/p300-interactive transactivator with ED-rich carboxy-terminal domain-4 (Cited4) (82). Activation of Cited4 has been found to be necessary for exercise-induced cardiac hypertrophy, and cardiac-specific overexpression of the gene is sufficient to increase heart mass and protect against ischemia/reperfusion injury (83). Other transcriptional pathways known to be activated by pathologic stimuli and cardiac hypertrophy, such as NFATc2, are decreased in exercise models (79, 84), suggesting that some signaling pathways activated during exercise-induced growth program may directly antagonize specific factors that promote pathological remodeling.
In addition to metabolic and molecular remodeling, exercise can also promote functional adaptation of the heart, which may ultimately increase cardiac output and reduce the risk of arrhythmia. Clinical studies have shown that exercise-trained individuals have improved systolic and diastolic function (85, 86), while results of studies using animal models of exercise show that endurance exercise promotes enhanced cardiomyocyte contraction-relaxation velocities and force generation (87–90). This effect of exercise on cardiomyocyte contractile function may be related to alterations in the rise and decay rates of intracellular Ca2+ transients, possibly due to enhanced coupling efficiency between L-type Ca2+ channel-mediated Ca2+ entry and activation of subsarcolemmal ryanodine receptors (RyR; i.e., calcium-induced calcium release), and increased expression and activity of the sarcoendoplasmic reticulum Ca2+ ATPase (SERCA2a) and sodium-calcium exchanger (NCX) (88, 91, 92). In addition, the sensitivity of the cardiomyocyte contractile apparatus may also become more sensitive to Ca2+, thus producing a greater force of contraction at a given [Ca2+]i, following exercise, (93). These changes may at least partially depend on upregulation of the Na+/H+ antiporter and altered regulation of intracellular pH.
During pathologic remodeling of the heart, electrical instability can result from a lack of upregulation of key cardiac ion channel subunits associated with action potential repolarization relative to an increase in myocyte size (94). In contrast, increased myocyte size in physiological hypertrophy is associated with the upregulation of depolarizing and repolarizing currents, which may be protective against abnormal electrical signaling in the adapted heart (95, 96). For example, cardiac myocytes isolated from mice after 4 weeks of swim training were found to have elevated outward K+ current densities (i.e., Ito,f, IK,slow, Iss, and IK1) and increased expression of underlying molecular component Kv and Kir subunits in parallel with increases in total protein levels (96). Interestingly, a follow up study found that while increases in K+ channel subunit expression following exercise training requires PI3K, these changes occur independently of Akt1 and hypertrophy (97).
The oxygen carrying capacity of blood, determined by the number of circulating erythrocytes and their associated intracellular hemoglobin concentration, is an important determinant of exercise performance and resistance to fatigue (98). High endurance athletes commonly have “athlete's anemia,” possibly due to loss of erythrocytes, or low hematocrit secondary to an expansion of plasma volume (99). Yet, overall total erythrocyte mass is increased in athletes, especially those who train at high altitude (100). This is in part due to a dose-dependent effect of O2 on hypoxia-inducible factor (HIF)-mediated erythropoietin production as well as upregulation of erythropoietin receptors, iron transporters, and transferrins (101). Multiple studies have shown that hematopoiesis is enhanced immediately following exercise (102, 103). Intense exercise is associated with the release of a variety of stress and inflammatory factors that are active on the bone marrow such as cortisol, IL-6, TNF-α, PMN elastase, and granulocyte colony stimulating factor (104–106). Although HPCs appear to modestly decline in the period immediately following an exercise session in conditioned runners, one study found that circulating CD34+ hematopoietic progenitor cell counts were 3- to 4-fold higher in runners vs. non-runners at baseline (102), which may represent an adaptive response that facilitates tissue repair. A subsequent study found that a bout of intense exercise was associated with a release of CD34+/KDR+ endothelial progenitor cells from the bone marrow and that this effect was enhanced in individuals with elevated LDL/HDL and LDL/TC profiles (107). Likewise, a significant increase in the number of circulating EPCs, associated with increased levels of VEGF, HIF-1α, and EPO was found within hours after varying intensities of resistance training in women (108). Nonetheless, the physiological significance of these responses remains unclear, as the effects of exercise on angiogenesis and the wound healing response have not been systematically studied.
The resistance arterial vascular network also undergoes functional and structural adaptation to exercise (109). During acute exercise, small arteries and pre-capillary arterioles that supply blood to the skeletal muscles must dilate to increase blood flow through the release of vasodilatory signals (e.g., adenosine, lactate, K+, H+, CO2) from active surrounding muscle (110–112). Repeated exercise leads to an adaptive response in skeletal muscle arterioles that includes increased vascular density coupled with greater vasodilatory capacity, such that enhanced perfusion can occur after conditioning (113–116). This may be partly due to adaptation of the endothelium to the complex interplay of recurrent variations in hemodynamic stresses and vasodilatory stimuli of exercise. Endothelial synthesis of NO is greatly increased at rest and during exercise in conditioned individuals/animals (117). A similar adaptive response to exercise has also been noted in the coronary vasculature, which must dilate to meet the increased metabolic demands of the myocardium (118). Exercise-trained humans and animals demonstrate reduced myocardial blood flow at rest, which may reflect a reduction in cardiac oxygen consumption primarily as a result of lower resting heart rate (119, 120). However, a large body of evidence suggests that multiple mechanisms converge to enhance the ability of the coronary circulation to deliver a greater supply of oxygen to the conditioned myocardium during exercise. This includes structural adaptations consisting of an expansion in the density of intramyocardial arterioles and capillaries as well as enhanced microvascular collateral formation (121–124). Additionally, like skeletal muscle arterioles, coronary arterial network enhances its responsiveness to vasoactive stimuli via a number of distinct mechanisms including, but not limited to, augmentation of endothelial NO production, altered responsiveness to adrenergic stimuli, or changes in the metabolic regulation of vascular tone (125–127). In addition, some studies implicate hydrogen peroxide (H2O2)-mediated vasodilation in opposing exertion-induced arterial dysfunction in overweight obese adults after a period of exercise training (128, 129), suggesting enhanced contribution of NO-independent mechanisms to improved microvascular endothelial function with exercise. Collectively, these adaptations may act to support enhanced myocardial function and increased cardiac output during repeated exercise, and increased total body oxygen demand following exercise conditioning. Further advancement of our understanding of how blood flow is improved in response to exercise could lead to novel therapeutic strategies to prevent or reverse organ failure in patients resulting from inadequate blood flow.
Despite the extensive body of knowledge documenting the unequivocal health benefits of exercise, a vast majority of Americans do not engage in sufficient physical activity (130). Nonetheless, mortality risk reduction appears with even small bouts of daily exercise and peak at 50–60 min of vigorous exercise each day (131). However, the question remains as to how much exercise is optimal for cardiovascular health benefit. Studies in endurance runners show that the frequency of adverse cardiovascular events in marathoners is equivalent to that in a population with established CHD, suggesting that too much exercise may be detrimental (132). An upper limit for the cardiovascular benefits of exercise is further supported by a recent study showing that individuals who completed at least 25 marathons over a period of 25 years have higher than expected levels of coronary artery calcification (CAC) and calcified coronary plaque volume when compared with sedentary individuals (133). A recent investigation also showed that individuals who maintain very high levels of physical activity (~3 times recommended levels) have higher odds of developing CAC, particularly in white males (134). In contrast, other studies report greater plaque stability due to calcification in exercisers, thus indicating that with higher levels of physical activity, plaque quality may be favorably impacted to lower the risk of cardiovascular events, despite a higher incidence of plaques and normal CAC scores (135, 136). Nevertheless, as with other effects of exercise, the shape of the dose-response curve remains obscure and it is not clear at what levels of intensity and duration the effects of exercise begin to taper and where they start to become detrimental. It is also unknown how this threshold of transition from benefit to harm is affected by personal demographic features such as age, sex, ethnicity, and baseline CVD risk.
Other remaining questions are: can initiation of regular exercise, later in life, reverse the consequences of lifestyle choices made during earlier years of life (e.g., sedentarism, smoking), and whether the beneficial effects of exercise show circadian or seasonal dependence such that exercising during a particular time of day or a particular season imparts more benefit than under other conditions. A recent study showing that adherence to a two-year, high-intensity exercise program decreases left ventricular stiffness in previously sedentary middle-aged participants (137) suggests that to some extent, beginning exercise, even late in life can be effective in reversing structural and functional changes in the cardiovascular system associated with aging and/or disease states such as heart failure with preserved ejection fraction. Yet, perhaps the most important questions relate to the mechanisms by which exercise imparts it remarkable benefits to cardiovascular health. As discussed above and summarized in Figure 1, regular physical activity can ameliorate a variety of CVD risk factors such as dyslipidemia or hypertension, but a well-powered analysis of the cardiovascular effects of exercise revealed that reduction in the burden of classical risk factors can account for only about 59% of the total reduction in cardiovascular mortality (138). What accounts for the remaining 41% reduction in risk remains unclear, but it may be related to changes in systemic inflammation as well as favorable responses to acute inflammatory challenge. Indeed exercise has pervasive effects on immune cells—natural killer cells, neutrophils, monocytes, regulatory T cells, as well as the balance of T-cell types are all affected by exercise (139) and it promotes a healthy anti-inflammatory milieu (140). Nevertheless, how exercise affects inflammation and immunity and how these changes could account for the salubrious effects of exercise on cardiovascular disease risk and mortality are important questions that require additional careful investigations. Additional work is also required to assess how nutrition affects exercise capacity as well as the cardiovascular benefits of exercise and how exercise affects the gut and the microbiome (139, 140). In this regard, it is important to clearly delineate the extent to which nutritional supplements such as β-alanine and carnosine, which enhance the buffering capacity of muscle (141) affect exercise capacity as well as muscle growth and hypertrophy. Such work is essential and important not only for a basic understanding of the mechanisms of exercise-induced protection, but also for developing more effective exercise regimens, testing the efficacy of combined treatments involving exercise and dietary supplements, and for devising appropriate pharmacological interventions for those who would not or cannot exercise.
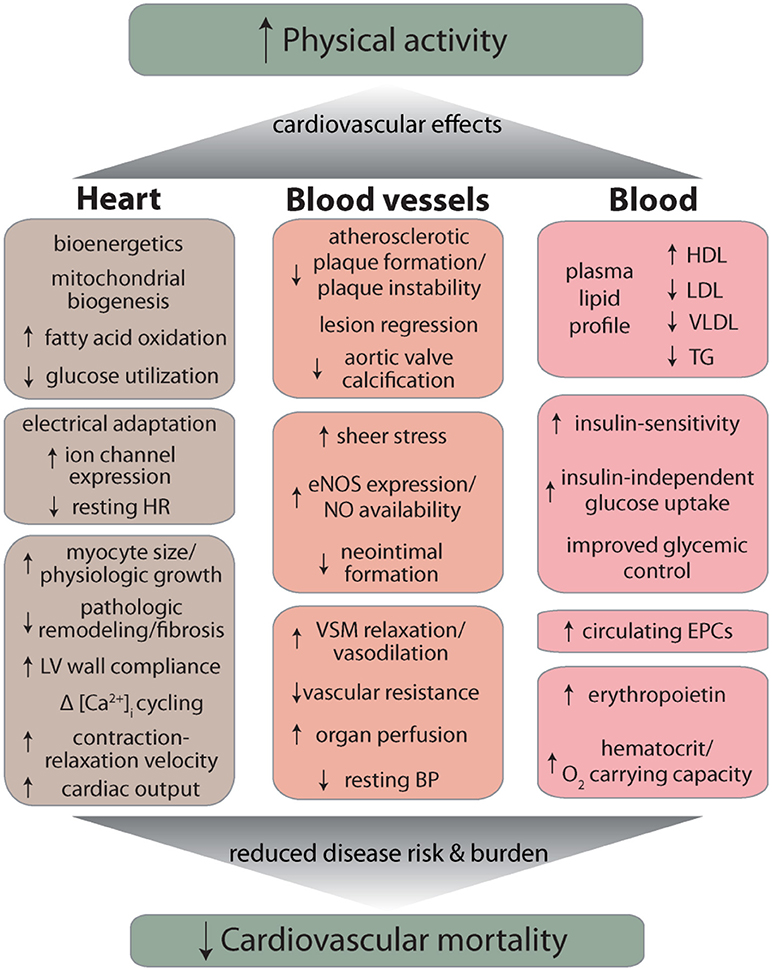
Figure 1. Overview of major cardiovascular effects of exercise. Abbreviations: HR, heart rate; LV, left ventricle; eNOS, endothelial nitric oxide synthase; NO, nitric oxide; VSM, vascular smooth muscle; BP, blood pressure; HDL, high density lipoprotein; LDL, low density lipoprotein; VLDL, very low density lipoprotein; TG, triglycerides; EPC, endothelial progenitor cell.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
This work was supported in part by grants from the National Institutes of Health (GM103492, HL142710) and the American Heart Association (16SDG27260070).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. CDC, N. (2015). Underlying Cause of Death 1999-2013 on CDC WONDER Online Database, Released 2015. Data are From the Multiple Cause of Death Files, 1999-2013, as Compiled From Data Provided by the 57 Vital Statistics Jurisdictions Through the Vital Statistics Cooperative Program (Accessed Feb. 3, 2015).
2. Benjamin EJ, Blaha MJ, Chiuve SE, Cushman M, Das SR, Deo R, et al. Heart disease and stroke statistics-2017 update: a report from the American Heart Association. Circulation (2017) 135:e146–603. doi: 10.1161/CIR.0000000000000485
3. Roth GA, Forouzanfar MH, Moran AE, Barber R, Nguyen G, Feigin VL, et al. Demographic and epidemiologic drivers of global cardiovascular mortality. N Engl J Med. (2015) 372:1333–41. doi: 10.1056/NEJMoa1406656
4. Paffenbarger RS Jr, Hyde RT, Wing AL, Hsieh CC. Physical activity, all-cause mortality, and longevity of college alumni. N Engl J Med. (1986) 314:605–13. doi: 10.1056/NEJM198603063141003
5. Blair SN, Kampert JB, Kohl HW III, Barlow CE, Macera CA, Paffenbarger RS Jr, et al. Influences of cardiorespiratory fitness and other precursors on cardiovascular disease and all-cause mortality in men and women. JAMA (1996) 276:205–10. doi: 10.1001/jama.1996.03540030039029
6. Stevens J, Cai J, Evenson KR, Thomas R. Fitness and fatness as predictors of mortality from all causes and from cardiovascular disease in men and women in the lipid research clinics study. Am J Epidemiol. (2002) 156:832–41. doi: 10.1093/aje/kwf114
7. Hu FB, Willett WC, Li T, Stampfer MJ, Colditz GA, Manson JE. Adiposity as compared with physical activity in predicting mortality among women. N Engl J Med. (2004) 351:2694–703. doi: 10.1056/NEJMoa042135
8. Vella CA, Allison MA, Cushman M, Jenny NS, Miles MP, Larsen B, et al. Physical activity and adiposity-related inflammation: the MESA. Med Sci Sports Exerc. (2017) 49:915–21. doi: 10.1249/MSS.0000000000001179
9. Florido R, Kwak L, Lazo M, Nambi V, Ahmed HM, Hegde SM, et al. Six-year changes in physical activity and the risk of incident heart failure: ARIC study. Circulation (2018) 137:2142–51. doi: 10.1161/CIRCULATIONAHA.117.030226
10. Moholdt T, Lavie CJ, Nauman J. Sustained physical activity, not weight loss, associated with improved survival in coronary heart disease. J Am Coll Cardiol. (2018) 71:1094–101. doi: 10.1016/j.jacc.2018.01.011
11. Caspersen CJ, Powell KE, Christenson GM. Physical activity, exercise, and physical fitness: definitions and distinctions for health-related research. Public Health Rep. (1985) 100:126–31.
12. Haskell WL. The influence of exercise on the concentrations of triglyceride and cholesterol in human plasma. Exerc Sport Sci Rev. (1984) 12:205–44. doi: 10.1249/00003677-198401000-00009
13. Fuster V, Gotto AM, Libby P, Loscalzo J, McGill HC. 27th Bethesda Conference: matching the intensity of risk factor management with the hazard for coronary disease events. Task Force 1. Pathogenesis of coronary disease: the biologic role of risk factors. J Am Coll Cardiol. (1996) 27:964–76. doi: 10.1016/0735-1097(96)00014-9
14. Leon AS, Sanchez OA. Response of blood lipids to exercise training alone or combined with dietary intervention. Med Sci Sports Exerc. (2001) 33(Suppl. 6):S502–15; discussion S528–509. doi: 10.1097/00005768-200106001-00021
15. Kraus WE, Houmard JA, Duscha BD, Knetzger KJ, Wharton MB, McCartney JS, et al. Effects of the amount and intensity of exercise on plasma lipoproteins. N Engl J Med. (2002) 347:1483–92. doi: 10.1056/NEJMoa020194
16. Gordon T, Castelli WP, Hjortland MC, Kannel WB, Dawber TR. High density lipoprotein as a protective factor against coronary heart disease. The Framingham Study. Am J Med. (1977) 62:707–14. doi: 10.1016/0002-9343(77)90874-9
17. Assmann G, Schulte H. Relation of high-density lipoprotein cholesterol and triglycerides to incidence of atherosclerotic coronary artery disease (the PROCAM experience). Prospective Cardiovascular Munster study. Am J Cardiol. (1992) 70:733–7. doi: 10.1016/0002-9149(92)90550-I
18. Schwartz GG, Olsson AG, Abt M, Ballantyne CM, Barter PJ, Brumm J, et al. Effects of dalcetrapib in patients with a recent acute coronary syndrome. N Engl J Med. (2012) 367:2089–99. doi: 10.1056/NEJMoa1206797
19. Group HTC, Landray MJ, Haynes R, Hopewell JC, Parish S, Aung T, et al. Effects of extended-release niacin with laropiprant in high-risk patients. N Engl J Med. (2014) 371:203–12. doi: 10.1056/NEJMoa1300955
20. Du XM, Kim MJ, Hou L, Le Goff W, Chapman MJ, Van Eck M, et al. HDL particle size is a critical determinant of ABCA1-mediated macrophage cellular cholesterol export. Circ Res. (2015) 116:1133–42. doi: 10.1161/CIRCRESAHA.116.305485
21. Sarzynski MA, Ruiz-Ramie JJ, Barber JL, Slentz CA, Apolzan JW, McGarrah RW, et al. Effects of increasing exercise intensity and dose on multiple measures of HDL (High-Density Lipoprotein) function. Arterioscler Thromb Vasc Biol. (2018) 38:943–52. doi: 10.1161/ATVBAHA.117.310307
22. Lee IM, Paffenbarger RS Jr, Hennekens CH. Physical activity, physical fitness and longevity. Aging (1997) 9:2–11. doi: 10.1007/BF03340123
23. Sesso HD, Paffenbarger RS Jr, Lee IM. Physical activity and coronary heart disease in men: the Harvard Alumni Health Study. Circulation (2000) 102:975–80. doi: 10.1161/01.CIR.102.9.975
24. Blair SN, Jackson AS. Physical fitness and activity as separate heart disease risk factors: a meta-analysis. Med Sci Sports Exerc. (2001) 33:762–4. doi: 10.1097/00005768-200105000-00013
25. Thompson PD, Buchner D, Pina IL, Balady GJ, Williams MA, Marcus BH, et al. Exercise and physical activity in the prevention and treatment of atherosclerotic cardiovascular disease: a statement from the Council on Clinical Cardiology (Subcommittee on Exercise, Rehabilitation, and Prevention) and the Council on Nutrition, Physical Activity, and Metabolism (Subcommittee on Physical Activity). Circulation (2003) 107:3109–16. doi: 10.1161/01.CIR.0000075572.40158.77
26. Hambrecht R, Niebauer J, Marburger C, Grunze M, Kalberer B, Hauer K, et al. Various intensities of leisure time physical activity in patients with coronary artery disease: effects on cardiorespiratory fitness and progression of coronary atherosclerotic lesions. J Am Coll Cardiol. (1993) 22:468–77. doi: 10.1016/0735-1097(93)90051-2
27. Hambrecht R, Adams V, Erbs S, Linke A, Krankel N, Shu Y, et al. Regular physical activity improves endothelial function in patients with coronary artery disease by increasing phosphorylation of endothelial nitric oxide synthase. Circulation (2003) 107:3152–8. doi: 10.1161/01.CIR.0000074229.93804.5C
28. Pynn M, Schafer K, Konstantinides S, Halle M. Exercise training reduces neointimal growth and stabilizes vascular lesions developing after injury in apolipoprotein e-deficient mice. Circulation (2004) 109:386–92. doi: 10.1161/01.CIR.0000109500.03050.7C
29. Laufs U, Wassmann S, Czech T, Munzel T, Eisenhauer M, Bohm M, et al. Physical inactivity increases oxidative stress, endothelial dysfunction, and atherosclerosis. Arterioscler Thromb Vasc Biol. (2005) 25:809–14. doi: 10.1161/01.ATV.0000158311.24443.af
30. Matsumoto Y, Adams V, Jacob S, Mangner N, Schuler G, Linke A. Regular exercise training prevents aortic valve disease in low-density lipoprotein-receptor-deficient mice. Circulation (2010) 121:759–67. doi: 10.1161/CIRCULATIONAHA.109.892224
31. Ginsberg HN. Insulin resistance and cardiovascular disease. J Clin Invest. (2000) 106:453–8. doi: 10.1172/JCI10762
32. Lewis GF. Fatty acid regulation of very low density lipoprotein production. Curr Opin Lipidol. (1997) 8:146–53. doi: 10.1097/00041433-199706000-00004
33. Borggreve SE, De Vries R, Dullaart RP. Alterations in high-density lipoprotein metabolism and reverse cholesterol transport in insulin resistance and type 2 diabetes mellitus: role of lipolytic enzymes, lecithin:cholesterol acyltransferase and lipid transfer proteins. Eur J Clin Invest. (2003) 33:1051–69. doi: 10.1111/j.1365-2362.2003.01263.x
34. Steinberg HO, Brechtel G, Johnson A, Fineberg N, Baron AD. Insulin-mediated skeletal muscle vasodilation is nitric oxide dependent. A novel action of insulin to increase nitric oxide release. J Clin Invest. (1994) 94:1172–9. doi: 10.1172/JCI117433
35. Zeng G, Quon MJ. Insulin-stimulated production of nitric oxide is inhibited by wortmannin. Direct measurement in vascular endothelial cells. J Clin Invest. (1996) 98:894–8. doi: 10.1172/JCI118871
36. Potenza MA, Marasciulo FL, Chieppa DM, Brigiani GS, Formoso G, Quon MJ, et al. Insulin resistance in spontaneously hypertensive rats is associated with endothelial dysfunction characterized by imbalance between NO and ET-1 production. Am J Physiol Heart Circ Physiol. (2005) 289:H813–22. doi: 10.1152/ajpheart.00092.2005
37. Marasciulo FL, Montagnani M, Potenza MA. Endothelin-1: the yin and yang on vascular function. Curr Med Chem. (2006) 13:1655–65. doi: 10.2174/092986706777441968
38. Beckman JA, Creager MA, Libby P. Diabetes and atherosclerosis: epidemiology, pathophysiology, and management. JAMA (2002) 287:2570–81. doi: 10.1001/jama.287.19.2570
39. Wang CC, Gurevich I, Draznin B. Insulin affects vascular smooth muscle cell phenotype and migration via distinct signaling pathways. Diabetes (2003) 52:2562–9. doi: 10.2337/diabetes.52.10.2562
40. Schleicher ED, Wagner E, Nerlich AG. Increased accumulation of the glycoxidation product N(epsilon)-(carboxymethyl)lysine in human tissues in diabetes and aging. J Clin Invest. (1997) 99:457–68. doi: 10.1172/JCI119180
41. Sell DR, Monnier VM. Molecular basis of arterial stiffening: role of glycation - a mini-review. Gerontology (2012) 58:227–37. doi: 10.1159/000334668
42. Wallberg-Henriksson H, Gunnarsson R, Henriksson J, DeFronzo R, Felig P, Ostman J, et al. Increased peripheral insulin sensitivity and muscle mitochondrial enzymes but unchanged blood glucose control in type I diabetics after physical training. Diabetes (1982) 31:1044–50. doi: 10.2337/diacare.31.12.1044
43. Trovati M, Carta Q, Cavalot F, Vitali S, Banaudi C, Lucchina PG, et al. Influence of physical training on blood glucose control, glucose tolerance, insulin secretion, and insulin action in non-insulin-dependent diabetic patients. Diabetes Care (1984) 7:416–20. doi: 10.2337/diacare.7.5.416
44. Koivisto VA, Yki-Jarvinen H, DeFronzo RA. Physical training and insulin sensitivity. Diabetes Metab Rev (1986) 1:445–81. doi: 10.1002/dmr.5610010407
45. Newsom SA, Everett AC, Hinko A, Horowitz JF. A single session of low-intensity exercise is sufficient to enhance insulin sensitivity into the next day in obese adults. Diabetes Care (2013) 36:2516–22. doi: 10.2337/dc12-2606
46. Richter EA, Garetto LP, Goodman MN, Ruderman NB. Muscle glucose metabolism following exercise in the rat: increased sensitivity to insulin. J Clin Invest. (1982) 69:785–93. doi: 10.1172/JCI110517
47. Craig BW, Garthwaite SM, Holloszy JO. Adipocyte insulin resistance: effects of aging, obesity, exercise, and food restriction. J Appl Physiol. (1987) 62:95–100. doi: 10.1152/jappl.1987.62.1.95
48. Zheng C, Liu Z. Vascular function, insulin action, and exercise: an intricate interplay. Trends Endocrinol Metab. (2015) 26:297–304. doi: 10.1016/j.tem.2015.02.002
49. Olver TD, McDonald MW, Klakotskaia D, Richardson RA, Jasperse JL, Melling CWJ, et al. A chronic physical activity treatment in obese rats normalizes the contributions of ET-1 and NO to insulin-mediated posterior cerebral artery vasodilation. J Appl Physiol. (2017) 122:1040–50. doi: 10.1152/japplphysiol.00811.2016
50. Kim Y, Inoue T, Nakajima R, Nakae K, Tamura T, Tokuyama K, et al. Effects of endurance training on gene expression of insulin signal transduction pathway. Biochem Biophys Res Commun. (1995) 210:766–73. doi: 10.1006/bbrc.1995.1725
51. Houmard JA, Shaw CD, Hickey MS, Tanner CJ. Effect of short-term exercise training on insulin-stimulated PI 3-kinase activity in human skeletal muscle. Am J Physiol. (1999) 277(6 Pt 1):E1055–60. doi: 10.1152/ajpendo.1999.277.6.E1055
52. Kirwan JP, del Aguila LF, Hernandez JM, Williamson DL, O'Gorman DJ, Lewis R, et al. Regular exercise enhances insulin activation of IRS-1-associated PI3-kinase in human skeletal muscle. J Appl Physiol (2000) 88:797–803. doi: 10.1152/jappl.2000.88.2.797
53. Richter EA, Mikines KJ, Galbo H, Kiens B. Effect of exercise on insulin action in human skeletal muscle. J Appl Physiol. (1989) 66:876–85. doi: 10.1152/jappl.1989.66.2.876
54. Goodyear LJ, King PA, Hirshman MF, Thompson CM, Horton ED, Horton ES. Contractile activity increases plasma membrane glucose transporters in absence of insulin. Am J Physiol. (1990) 258(4 Pt 1):E667–72. doi: 10.1152/ajpendo.1990.258.4.E667
55. Gao J, Ren J, Gulve EA, Holloszy JO. Additive effect of contractions and insulin on GLUT-4 translocation into the sarcolemma. J Appl Physiol. (1994) 77:1597–601. doi: 10.1152/jappl.1994.77.4.1597
56. Hotamisligil GS, Murray DL, Choy LN, Spiegelman BM. Tumor necrosis factor alpha inhibits signaling from the insulin receptor. Proc Natl Acad Sci USA. (1994) 91:4854–8. doi: 10.1073/pnas.91.11.4854
57. del Aguila LF, Claffey KP, Kirwan JP. TNF-alpha impairs insulin signaling and insulin stimulation of glucose uptake in C2C12 muscle cells. Am J Physiol. (1999) 276(Pt 1):E849–55.
58. Del Aguila LF, Krishnan RK, Ulbrecht JS, Farrell PA, Correll PH, Lang CH, et al. Muscle damage impairs insulin stimulation of IRS-1, PI 3-kinase, and Akt-kinase in human skeletal muscle. Am J Physiol Endocrinol Metab. (2000) 279:E206–12. doi: 10.1152/ajpendo.2000.279.1.E206
59. Kirwan JP, del Aguila LF. Insulin signalling, exercise and cellular integrity. Biochem Soc Trans. (2003). 31(Pt 6):1281–5. doi: 10.1042/bst0311281
61. Fagard RH. Exercise characteristics and the blood pressure response to dynamic physical training. Med Sci Sports Exerc. (2001) 33(Suppl. 6):S484–92; discussion S493–484. doi: 10.1097/00005768-200106001-00018
62. Hardy ST, Loehr LR, Butler KR, Chakladar S, Chang PP, Folsom AR, et al. Reducing the blood pressure-related burden of cardiovascular disease: impact of achievable improvements in blood pressure prevention and control. J Am Heart Assoc. (2015) 4:e002276. doi: 10.1161/JAHA.115.002276
63. Cox KL, Puddey IB, Morton AR, Burke V, Beilin LJ, McAleer M. Exercise and weight control in sedentary overweight men: effects on clinic and ambulatory blood pressure. J Hypertens. (1996) 14:779–90. doi: 10.1097/00004872-199606000-00015
64. Bacon SL, Sherwood A, Hinderliter A, Blumenthal JA. Effects of exercise, diet and weight loss on high blood pressure. Sports Med. (2004) 34:307–16. doi: 10.2165/00007256-200434050-00003
65. Fagard RH. Exercise is good for your blood pressure: effects of endurance training and resistance training. Clin Exp Pharmacol Physiol. (2006) 33:853–6. doi: 10.1111/j.1440-1681.2006.04453.x
66. Niebauer J, Cooke JP. Cardiovascular effects of exercise: role of endothelial shear stress. J Am Coll Cardiol. (1996) 28:1652–60. doi: 10.1016/S0735-1097(96)00393-2
67. Dominiczak AF, Bohr DF. Nitric oxide and its putative role in hypertension. Hypertension (1995) 25:1202–11. doi: 10.1161/01.HYP.25.6.1202
68. Kim IJ, Bae J, Lim SW, Cha DH, Cho HJ, Kim S, et al. Influence of endothelial nitric oxide synthase gene polymorphisms (-786T>C, 4a4b, 894G>T) in Korean patients with coronary artery disease. Thromb Res. (2007) 119:579–85. doi: 10.1016/j.thromres.2006.06.005
69. Cruz-Gonzalez I, Corral E, Sanchez-Ledesma M, Sanchez-Rodriguez A, Martin-Luengo C, Gonzalez-Sarmiento R. Association between -T786C NOS3 polymorphism and resistant hypertension: a prospective cohort study. BMC Cardiovasc Disord. (2009) 9:35. doi: 10.1186/1471-2261-9-35
70. Zago AS, Park JY, Fenty-Stewart N, Kokubun E, Brown MD. Effects of aerobic exercise on the blood pressure, oxidative stress and eNOS gene polymorphism in pre-hypertensive older people. Eur J Appl Physiol. (2010) 110:825–32. doi: 10.1007/s00421-010-1568-6
71. Kuru O, Senturk UK, Kocer G, Ozdem S, Baskurt OK, Cetin A, et al. Effect of exercise training on resistance arteries in rats with chronic NOS inhibition. J Appl Physiol. (2009) 107:896–902. doi: 10.1152/japplphysiol.91180.2008
72. Wilund KR. Is the anti-inflammatory effect of regular exercise responsible for reduced cardiovascular disease? Clin Sci. (2007) 112:543–55. doi: 10.1042/CS20060368
73. Fleenor BS, Marshall KD, Durrant JR, Lesniewski LA, Seals DR. Arterial stiffening with ageing is associated with transforming growth factor-beta1-related changes in adventitial collagen: reversal by aerobic exercise. J Physiol. (2010) 588(Pt 20):3971–82. doi: 10.1113/jphysiol.2010.194753
74. Carter JR, Ray CA. Sympathetic neural adaptations to exercise training in humans. Auton Neurosci. (2015) 188:36–43. doi: 10.1016/j.autneu.2014.10.020
75. Breisch EA, White FC, Nimmo LE, McKirnan MD, Bloor CM. Exercise-induced cardiac hypertrophy: a correlation of blood flow and microvasculature. J Appl Physiol. (1986) 60:1259–67. doi: 10.1152/jappl.1986.60.4.1259
76. Borlaug BA, Lam CS, Roger VL, Rodeheffer RJ, Redfield MM. Contractility and ventricular systolic stiffening in hypertensive heart disease insights into the pathogenesis of heart failure with preserved ejection fraction. J Am Coll Cardiol. (2009) 54:410–8. doi: 10.1016/j.jacc.2009.05.013
77. Burelle Y, Wambolt RB, Grist M, Parsons HL, Chow JC, Antler C, et al. Regular exercise is associated with a protective metabolic phenotype in the rat heart. Am J Physiol Heart Circ Physiol. (2004) 287:H1055–63. doi: 10.1152/ajpheart.00925.2003
78. Riehle C, Wende AR, Zhu Y, Oliveira KJ, Pereira RO, Jaishy BP, et al. Insulin receptor substrates are essential for the bioenergetic and hypertrophic response of the heart to exercise training. Mol Cell Biol. (2014) 34:3450–60. doi: 10.1128/MCB.00426-14
79. Gibb AA, Epstein PN, Uchida S, Zheng Y, McNally LA, Obal D, et al. Exercise-induced changes in glucose metabolism promote physiological cardiac growth. Circulation (2017) 136:2144–57. doi: 10.1161/CIRCULATIONAHA.117.028274
80. McMullen JR, Shioi T, Huang WY, Zhang L, Tarnavski O, Bisping E, et al. The insulin-like growth factor 1 receptor induces physiological heart growth via the phosphoinositide 3-kinase(p110alpha) pathway. J Biol Chem. (2004) 279:4782–93. doi: 10.1074/jbc.M310405200
81. Kim J, Wende AR, Sena S, Theobald HA, Soto J, Sloan C, et al. Insulin-like growth factor I receptor signaling is required for exercise-induced cardiac hypertrophy. Mol Endocrinol. (2008) 22:2531–43. doi: 10.1210/me.2008-0265
82. Bostrom P, Mann N, Wu J, Quintero PA, Plovie ER, Panakova D, et al. C/EBPbeta controls exercise-induced cardiac growth and protects against pathological cardiac remodeling. Cell (2010) 143:1072–83. doi: 10.1016/j.cell.2010.11.036
83. Bezzerides VJ, Platt C, Lerchenmuller C, Paruchuri K, Oh NL, Xiao C, et al. CITED4 induces physiologic hypertrophy and promotes functional recovery after ischemic injury. JCI Insight (2016) 1:e85904. doi: 10.1172/jci.insight.85904
84. Wilkins BJ, Dai YS, Bueno OF, Parsons SA, Xu J, Plank DM, et al. Calcineurin/NFAT coupling participates in pathological, but not physiological, cardiac hypertrophy. Circ Res. (2004) 94:110–8. doi: 10.1161/01.RES.0000109415.17511.18
85. Ferguson S, Gledhill N, Jamnik VK, Wiebe C, Payne N. Cardiac performance in endurance-trained and moderately active young women. Med Sci Sports Exerc. (2001) 33:1114–9. doi: 10.1097/00005768-200107000-00008
86. Esch BT, Scott JM, Haykowsky MJ, McKenzie DC, Warburton DE. Diastolic ventricular interactions in endurance-trained athletes during orthostatic stress. Am J Physiol Heart Circ Physiol. (2007) 293:H409–15. doi: 10.1152/ajpheart.00928.2006
87. Moore RL, Musch TI, Yelamarty RV, Scaduto RC Jr, Semanchick AM, Elensky M, et al. Chronic exercise alters contractility and morphology of isolated rat cardiac myocytes. Am J Physiol. (1993) 264(5 Pt 1):C1180–9. doi: 10.1152/ajpcell.1993.264.5.C1180
88. Wisloff U, Loennechen JP, Currie S, Smith GL, Ellingsen O. Aerobic exercise reduces cardiomyocyte hypertrophy and increases contractility, Ca2+ sensitivity and SERCA-2 in rat after myocardial infarction. Cardiovasc Res. (2002) 54:162–74. doi: 10.1016/S0008-6363(01)00565-X
89. Diffee GM, Chung E. Altered single cell force-velocity and power properties in exercise-trained rat myocardium. J Appl Physiol. (2003) 94:1941–8. doi: 10.1152/japplphysiol.00889.2002
90. Kemi OJ, Ellingsen O, Smith GL, Wisloff U. Exercise-induced changes in calcium handling in left ventricular cardiomyocytes. Front Biosci. (2008) 13:356–68. doi: 10.2741/2685
91. Natali AJ, Wilson LA, Peckham M, Turner DL, Harrison SM, White E. Different regional effects of voluntary exercise on the mechanical and electrical properties of rat ventricular myocytes. J Physiol. (2002) 541(Pt 3):863–75. doi: 10.1113/jphysiol.2001.013415
92. Kemi OJ, Wisloff U. Mechanisms of exercise-induced improvements in the contractile apparatus of the mammalian myocardium. Acta Physiol. (2010) 199:425–39. doi: 10.1111/j.1748-1716.2010.02132.x
93. Wisloff U, Loennechen JP, Falck G, Beisvag V, Currie S, Smith G, et al. Increased contractility and calcium sensitivity in cardiac myocytes isolated from endurance trained rats. Cardiovasc Res. (2001) 50:495–508. doi: 10.1016/S0008-6363(01)00210-3
94. Marionneau C, Brunet S, Flagg TP, Pilgram TK, Demolombe S, Nerbonne JM. Distinct cellular and molecular mechanisms underlie functional remodeling of repolarizing K+ currents with left ventricular hypertrophy. Circ Res. (2008) 102:1406–15. doi: 10.1161/CIRCRESAHA.107.170050
95. Biffi A, Maron BJ, Di Giacinto B, Porcacchia P, Verdile L, Fernando F, et al. Relation between training-induced left ventricular hypertrophy and risk for ventricular tachyarrhythmias in elite athletes. Am J Cardiol. (2008) 101:1792–5. doi: 10.1016/j.amjcard.2008.02.081
96. Yang KC, Foeger NC, Marionneau C, Jay PY, McMullen JR, Nerbonne JM. Homeostatic regulation of electrical excitability in physiological cardiac hypertrophy. J Physiol. (2010) 588(Pt 24):5015–32. doi: 10.1113/jphysiol.2010.197418
97. Yang KC, Tseng YT, Nerbonne JM. Exercise training and PI3Kalpha-induced electrical remodeling is independent of cellular hypertrophy and Akt signaling. J Mol Cell Cardiol. (2012) 53:532–41. doi: 10.1016/j.yjmcc.2012.07.004
98. Buick FJ, Gledhill N, Froese AB, Spriet L, Meyers EC. Effect of induced erythrocythemia on aerobic work capacity. J Appl Physiol Respir Environ Exerc Physiol. (1980) 48:636–42. doi: 10.1152/jappl.1980.48.4.636
99. Weight LM, Klein M, Noakes TD, Jacobs P. 'Sports anemia'–a real or apparent phenomenon in endurance-trained athletes? Int J Sports Med. (1992) 13:344–7. doi: 10.1055/s-2007-1021278
100. Mairbaurl H. Red blood cells in sports: effects of exercise and training on oxygen supply by red blood cells. Front Physiol. (2013) 4:332. doi: 10.3389/fphys.2013.00332
101. Semenza GL. Regulation of oxygen homeostasis by hypoxia-inducible factor 1. Physiology (2009) 24:97–106. doi: 10.1152/physiol.00045.2008
102. Bonsignore MR, Morici G, Santoro A, Pagano M, Cascio L, Bonanno A, et al. Circulating hematopoietic progenitor cells in runners. J Appl Physiol. (2002) 93:1691–7. doi: 10.1152/japplphysiol.00376.2002
103. Morici G, Zangla D, Santoro A, Pelosi E, Petrucci E, Gioia M, et al. Supramaximal exercise mobilizes hematopoietic progenitors and reticulocytes in athletes. Am J Physiol Regul Integr Comp Physiol. (2005) 289:R1496–503. doi: 10.1152/ajpregu.00338.2005
104. Dufaux B, Order U. Plasma elastase-alpha 1-antitrypsin, neopterin, tumor necrosis factor, and soluble interleukin-2 receptor after prolonged exercise. Int J Sports Med. (1989) 10:434–8. doi: 10.1055/s-2007-1024939
105. Ostrowski K, Rohde T, Asp S, Schjerling P, Pedersen BK. Pro- and anti-inflammatory cytokine balance in strenuous exercise in humans. J Physiol. (1999) 515 (Pt 1):287–91. doi: 10.1111/j.1469-7793.1999.287ad.x
106. Suzuki K, Yamada M, Kurakake S, Okamura N, Yamaya K, Liu Q, et al. Circulating cytokines and hormones with immunosuppressive but neutrophil-priming potentials rise after endurance exercise in humans. Eur J Appl Physiol. (2000) 81:281–7. doi: 10.1007/s004210050044
107. Van Craenenbroeck EM, Vrints CJ, Haine SE, Vermeulen K, Goovaerts I, Van Tendeloo VF, et al. A maximal exercise bout increases the number of circulating CD34+/KDR+ endothelial progenitor cells in healthy subjects. Relation with lipid profile. J Appl Physiol. (2008) 104:1006–13. doi: 10.1152/japplphysiol.01210.2007
108. Ribeiro F, Ribeiro IP, Goncalves AC, Alves AJ, Melo E, Fernandes R, et al. Effects of resistance exercise on endothelial progenitor cell mobilization in women. Sci Rep. (2017) 7:17880. doi: 10.1038/s41598-017-18156-6
109. Green DJ, Hopman MT, Padilla J, Laughlin MH, Thijssen DH. Vascular Adaptation to Exercise in Humans: Role of Hemodynamic Stimuli. Physiol Rev. (2017) 97:495–528. doi: 10.1152/physrev.00014.2016
110. Hallen J. K+ balance in humans during exercise. Acta Physiol Scand. (1996) 156:279–86. doi: 10.1046/j.1365-201X.1996.187000.x
111. Radegran G, Calbet JA. Role of adenosine in exercise-induced human skeletal muscle vasodilatation. Acta Physiol Scand. (2001) 171:177–85. doi: 10.1046/j.1365-201x.2001.00796.x
112. Sarelius I, Pohl U. Control of muscle blood flow during exercise: local factors and integrative mechanisms. Acta Physiol. (2010) 199:349–65. doi: 10.1111/j.1748-1716.2010.02129.x
113. Sun D, Huang A, Koller A, Kaley G. Short-term daily exercise activity enhances endothelial NO synthesis in skeletal muscle arterioles of rats. J Appl Physiol. (1994) 76:2241–7. doi: 10.1152/jappl.1994.76.5.2241
114. Huonker M, Schmid A, Schmidt-Trucksass A, Grathwohl D, Keul J. Size and blood flow of central and peripheral arteries in highly trained able-bodied and disabled athletes. J Appl Physiol. (2003) 95:685–91. doi: 10.1152/japplphysiol.00710.2001
115. Joyner MJ, Casey DP. Regulation of increased blood flow (hyperemia) to muscles during exercise: a hierarchy of competing physiological needs. Physiol Rev. (2015) 95:549–601. doi: 10.1152/physrev.00035.2013
116. Laughlin MH, Yang HT, Tharp DL, Rector RS, Padilla J, Bowles DK. Vascular cell transcriptomic changes to exercise training differ directionally along and between skeletal muscle arteriolar trees. Microcirculation (2017) 24:e12336. doi: 10.1111/micc.12336
117. Taddei S, Galetta F, Virdis A, Ghiadoni L, Salvetti G, Franzoni F, et al. Physical activity prevents age-related impairment in nitric oxide availability in elderly athletes. Circulation (2000) 101:2896–901. doi: 10.1161/01.CIR.101.25.2896
119. Heiss HW, Barmeyer J, Wink K, Hell G, Cerny FJ, Keul J, et al. Studies on the regulation of myocardial blood flow in man. I: Training effects on blood flow and metabolism of the healthy heart at rest and during standardized heavy exercise Basic Res Cardiol. (1976) 71:658–75. doi: 10.1007/BF01906411
120. Barnard RJ, Duncan HW, Baldwin KM, Grimditch G, Buckberg GD. Effects of intensive exercise training on myocardial performance and coronary blood flow. J Appl Physiol Respir Environ Exerc Physiol. (1980) 49:444–9. doi: 10.1152/jappl.1980.49.3.444
121. Tomanek RJ. Effects of age and exercise on the extent of the myocardial capillary bed. Anat Rec. (1970) 167:55–62. doi: 10.1002/ar.1091670106
122. Thomas DP. Effects of acute and chronic exercise on myocardial ultrastructure. Med Sci Sports Exerc. (1985) 17:546–53. doi: 10.1249/00005768-198510000-00007
123. White FC, Bloor CM, McKirnan MD, Carroll SM. Exercise training in swine promotes growth of arteriolar bed and capillary angiogenesis in heart. J Appl Physiol. (1998) 85:1160–8. doi: 10.1152/jappl.1998.85.3.1160
124. Mobius-Winkler S, Uhlemann M, Adams V, Sandri M, Erbs S, Lenk K, et al. Coronary Collateral growth induced by physical exercise: results of the impact of intensive exercise training on coronary collateral circulation in patients with stable coronary artery disease (EXCITE) trial. Circulation (2016) 133:1438–48; discussion 1448. doi: 10.1161/CIRCULATIONAHA.115.016442
125. DiCarlo SE, Blair RW, Bishop VS, Stone HL. Daily exercise enhances coronary resistance vessel sensitivity to pharmacological activation. J Appl Physiol. (1989) 66:421–8. doi: 10.1152/jappl.1989.66.1.421
126. Bowles DK, Laughlin MH, Sturek M. Exercise training increases K+-channel contribution to regulation of coronary arterial tone. J Appl Physiol. (1998) 84:1225–33. doi: 10.1152/jappl.1998.84.4.1225
127. Laughlin MH, Pollock JS, Amann JF, Hollis ML, Woodman CR, Price EM. Training induces nonuniform increases in eNOS content along the coronary arterial tree. J Appl Physiol. (2001) 90:501–10. doi: 10.1152/jappl.2001.90.2.501
128. Durand MJ, Dharmashankar K, Bian JT, Das E, Vidovich M, Gutterman DD, et al. Acute exertion elicits a H2O2-dependent vasodilator mechanism in the microvasculature of exercise-trained but not sedentary adults. Hypertension (2015) 65:140–5. doi: 10.1161/HYPERTENSIONAHA.114.04540
129. Robinson AT, Franklin NC, Norkeviciute E, Bian JT, Babana JC, Szczurek MR, et al. Improved arterial flow-mediated dilation after exertion involves hydrogen peroxide in overweight and obese adults following aerobic exercise training. J Hypertens. (2016) 34:1309–16. doi: 10.1097/HJH.0000000000000946
130. Simpson ME, Serdula M, Galuska DA, Gillespie C, Donehoo R, Macera C, et al. Walking trends among U.S. adults: the Behavioral Risk Factor Surveillance System, 1987-2000. Am J Prev Med. (2003) 25:95–100. doi: 10.1016/S0749-3797(03)00112-0
131. Wen CP, Wai JP, Tsai MK, Yang YC, Cheng TY, Lee MC, et al. Minimum amount of physical activity for reduced mortality and extended life expectancy: a prospective cohort study. Lancet (2011) 378:1244–53. doi: 10.1016/S0140-6736(11)60749-6
132. O'Keefe JH, Patil HR, Lavie CJ, Magalski A, Vogel RA, McCullough PA. Potential adverse cardiovascular effects from excessive endurance exercise. Mayo Clin Proc. (2012) 87:587–95. doi: 10.1016/j.mayocp.2012.04.005
133. Roberts WO, Schwartz RS, Garberich RF, Carlson S, Knickelbine T, Schwartz JG, et al. Fifty men, 3510 marathons, cardiac risk factors, and coronary artery calcium scores. Med Sci Sports Exerc. (2017) 49:2369–73. doi: 10.1249/MSS.0000000000001373
134. Laddu DR, Rana JS, Murillo R, Sorel ME, Quesenberry CPJr, Allen NB, et al. 25-Year physical activity trajectories and development of subclinical coronary artery disease as measured by coronary artery calcium: the Coronary Artery Risk Development in Young Adults (CARDIA) study. Mayo Clin Proc. (2017) 92:1660–70. doi: 10.1016/j.mayocp.2017.07.016
135. Aengevaeren VL, Mosterd A, Braber TL, Prakken NHJ, Doevendans PA, Grobbee DE, et al. Relationship between lifelong exercise volume and coronary atherosclerosis in athletes. Circulation (2017) 136:138–48. doi: 10.1161/CIRCULATIONAHA.117.027834
136. Merghani A, Maestrini V, Rosmini S, Cox AT, Dhutia H, Bastiaenan R, et al. Prevalence of subclinical coronary artery disease in masters endurance athletes with a low atherosclerotic risk profile. Circulation (2017) 136:126–37. doi: 10.1161/CIRCULATIONAHA.116.026964
137. Howden EJ, Sarma S, Lawley JS, Opondo M, Cornwell W, Stoller D, et al. Reversing the cardiac effects of sedentary aging in middle age-a randomized controlled trial: implications for heart failure prevention. Circulation (2018) 137:1549–60. doi: 10.1161/CIRCULATIONAHA.117.030617
138. Mora S, Cook N, Buring JE, Ridker PM, Lee IM. Physical activity and reduced risk of cardiovascular events: potential mediating mechanisms. Circulation (2007) 116:2110–8. doi: 10.1161/CIRCULATIONAHA.107.729939
139. Fernandez DM, Clemente JC, Giannarelli C. Physical activity, immune system, and the microbiome in cardiovascular disease. Front Physiol. (2018) 9:763. doi: 10.3389/fphys.2018.00763
140. Fiuza-Luces C, Santos-Lozano A, Joyner M, Carrera-Bastos P, Picazo O, Zugaza JL, et al. Exercise benefits in cardiovascular disease: beyond attenuation of traditional risk factors. Nat Rev Cardiol. (2018). doi: 10.1038/s41569-018-0065-1
Keywords: physical activity, endothelium, blood flow, atherosclerosis, coronary artery disease
Citation: Nystoriak MA and Bhatnagar A (2018) Cardiovascular Effects and Benefits of Exercise. Front. Cardiovasc. Med. 5:135. doi: 10.3389/fcvm.2018.00135
Received: 14 June 2018; Accepted: 07 September 2018;
Published: 28 September 2018.
Edited by:
Jacob Haus, University of Michigan, United StatesReviewed by:
Abbi D. Lane-Cordova, University of South Carolina, United StatesCopyright © 2018 Nystoriak and Bhatnagar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Matthew A. Nystoriak, bWF0dGhldy5ueXN0b3JpYWtAbG91aXN2aWxsZS5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.