 Kyle Fulghum
Kyle Fulghum Bradford G. Hill
Bradford G. Hill- 1Department of Medicine, Envirome Institute, Institute of Molecular Cardiology, Diabetes and Obesity Center, Louisville, KY, United States
- 2Department of Physiology, University of Louisville, Louisville, KY, United States
Exercise has a myriad of physiological benefits that derive in part from its ability to improve cardiometabolic health. The periodic metabolic stress imposed by regular exercise appears fundamental in driving cardiovascular tissue adaptation. However, different types, intensities, or durations of exercise elicit different levels of metabolic stress and may promote distinct types of tissue remodeling. In this review, we discuss how exercise affects cardiac structure and function and how exercise-induced changes in metabolism regulate cardiac adaptation. Current evidence suggests that exercise typically elicits an adaptive, beneficial form of cardiac remodeling that involves cardiomyocyte growth and proliferation; however, chronic levels of extreme exercise may increase the risk for pathological cardiac remodeling or sudden cardiac death. An emerging theme underpinning acute as well as chronic cardiac adaptations to exercise is metabolic periodicity, which appears important for regulating mitochondrial quality and function, for stimulating metabolism-mediated exercise gene programs and hypertrophic kinase activity, and for coordinating biosynthetic pathway activity. In addition, circulating metabolites liberated during exercise trigger physiological cardiac growth. Further understanding of how exercise-mediated changes in metabolism orchestrate cell signaling and gene expression could facilitate therapeutic strategies to maximize the benefits of exercise and improve cardiac health.
Introduction
Exercise promotes general metabolic wellness (1–3), improves mental health (4, 5), builds and preserves musculoskeletal function (6), and increases lifespan (7–10). These beneficial effects of exercise are related, in part, to enhanced function and health of cardiovascular tissues as well as to increased resistance of the heart to injury (11, 12). The magnitude of risk reduction for cardiovascular disease and survival afforded by exercise parallels that of not smoking (10, 13). Moreover, exercise is a core component of cardiac rehabilitation regimens, and, in patients with heart disease, it reduces cardiovascular morbidity and mortality (14–18). Nevertheless, the molecular mechanisms by which exercise improves cardiovascular health and prevents tissue injury remain unclear.
The recurrent deviations in whole body homeostasis caused by exercise drive adaptations in several organs, including brain, liver, adipose tissue, skeletal muscle, and, the topic of this review—the heart (6, 19). The idea that metabolic perturbations are important for attaining exercise-induced health benefits is consistent with a paradigm suggested first by Galen (c 129–210 CE), who recognized that not all movement is exercise and that exercise is most beneficial when vigorous, with “the criterion for vigorousness [defined by a] change in respiration…those movements which do not alter respiration are not called exercise” (20). Hence, with Galen, a definition of exercise and the overarching tenet that the salutary effects of exercise require significant deviations in metabolism first became apparent. Although several reviews cover the known mechanisms by which exercise regulates the health and adaptation of the heart and vasculature [e.g., (12, 21–25)], we highlight in this short review knowledge of how cardiac metabolism changes with exercise as well as recent findings of how exercise-induced changes in metabolism may drive cardiac remodeling. Specifically, we address the following questions:
(1) What kinds of exercise elicit changes in cardiac structure and function?
(2) How does cardiac metabolism change during exercise?
(3) How might exercise-induced changes in metabolism promote cardiac adaptation?
What Kinds of Exercise Elicit Changes in Cardiac Structure and Function?
Cardiac adaptations associated with exercise were first documented in 1899. Physical examination using auscultation and percussion revealed that Nordic skiers (26) and university rowers (27) had increased cardiac dimensions. The latter study highlighted that “the period of greatest enlargement corresponded to the period of the most arduous work,” (27) which provided an early indication that relatively high workloads correspond with exercise-induced cardiac growth. Later studies using electrocardiography and chest radiography identified functional and structural cardiac changes caused by exercise (28–31). Subsequent echocardiographic studies further described the degree and proportional features of the exercise-remodeled heart [reviewed in (32)]. Collectively, these studies laid the groundwork for understanding how repetitive bouts of exercise stimulate adaptive changes in the heart.
Acute Cardiac Responses to Exercise
Increases in physical activity require changes in the distribution of oxygen and nutrients throughout the body. The increased work and ATP turnover of skeletal muscle (6) are facilitated by several integrated changes including physiological adjustments in ventilation and cardiac output as well as markedly decreased vascular resistance in skeletal muscle (19). During aerobic exercise, changes in cardiac function occur immediately and are typically associated with several phases. Heart rate and stroke volume increase upon heightened levels of physical activity, and together they augment cardiac output in a relationship defined by the Fick equation (32, 33). After a prolonged period of moderate to high intensity aerobic exercise (e.g., >20 min), cardiac output is maintained; however, heart rate tends to increase further and stroke volume begins to drop due to cardiovascular drift, a phenomenon thought to be associated with vasodilation, hyperthermia, increased blood flow to the skin, decreased filling time, and decreased plasma volume (34–37). Coordinated changes in vascular function combined with sustained augmentation of cardiac function integrate to increase blood flow to skeletal muscle, with cardiac output distribution to working muscle tracking with exercise intensity (38) (Figure 1).
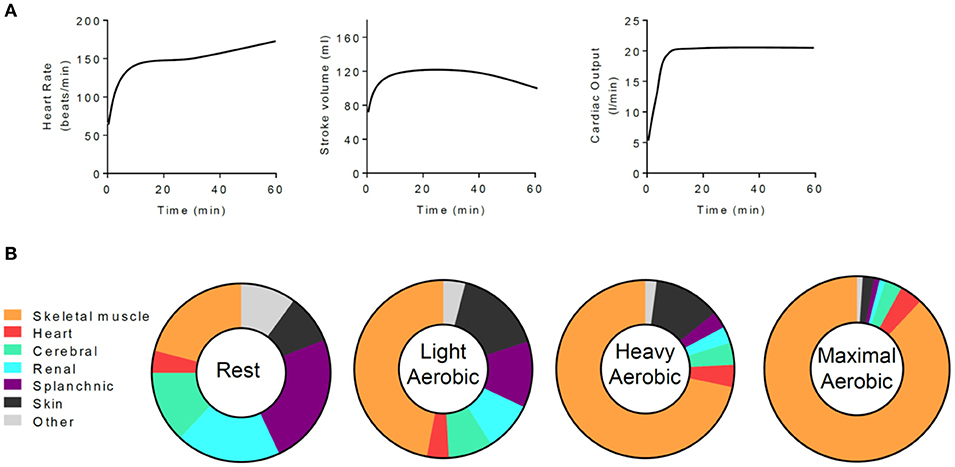
Figure 1. Exercise-mediated changes in cardiac function and in the tissue distribution of cardiac output. (A) Generalized schematic of cardiac responses to a moderate to intense, 1 h session of aerobic exercise. (B) Distribution of cardiac output at rest and with increasingly intense levels of exercise. Data are adapted from Plowman and Smith (38).
Whereas the cardiac responses to endurance exercise are directly associated with the use of oxygen for ATP production in skeletal muscle, resistance exercises are more anaerobic in nature. In addition, resistance exercise generally increases blood pressure, which is due in part to mechanical restriction of blood flow during static contraction. These features of resistance exercise result in markedly different cardiac responses as compared with aerobic exercise. The modest increase in cardiac output initiated by resistance exercise is predominantly due to increases in heart rate, with virtually no change in stroke volume (39, 40). A higher number of repetitions increases heart rate and thus leads to larger increases in cardiac output (41). With heavy weightlifting, the heart must also deal with spikes in blood pressure, which can transiently reach levels of 320/250 mmHg (42) or higher. The degree to which blood pressure changes during resistance exercise appears to be a function of the degree of effort, muscle mass, and the breathing patterns commonly performed during strength training (i.e., the Valsalva maneuver) (41, 43).
Chronic Effects of Exercise on the Heart
Repetitive bouts of strenuous exercise can promote mild cardiac hypertrophy and/or chamber enlargement (32, 44, 45), which is typically reversible upon prolonged cessation of exercise (46–48) (Figure 2). The type and intensity of exercise determines the nature and degree of exercise-induced cardiac remodeling, with hemodynamic changes during exercise providing a stimulus for growth and chamber adaptation. Isometric or static exercises—commonly grouped as strength training (e.g., weightlifting, wrestling)—involve brief, intense periods of increased peripheral vascular resistance with little to no change in cardiac output and are associated with mild concentric hypertrophy and a normal to mildly enlarged left atrium. The increase in cardiac wall thickness appears largely caused by the parallel addition of sarcomeres within cardiomyocytes. In contrast, prolonged isotonic or dynamic aerobic exercise—generally termed endurance exercise (e.g., long distance running, cycling, rowing, or swimming)—requires sustained elevations in cardiac output and is typically associated with normal or diminished peripheral vascular resistance. Endurance exercise promotes eccentric left ventricular hypertrophy, right ventricular dilation, and biatrial enlargement [(49, 50) and reviewed in (32) and (51)]. Addition of cardiomyocyte sarcomeres in series predominates in this form of hypertrophy. Nevertheless, exercise-induced cardiac remodeling caused by endurance training has been suggested to be phasic in nature, with one study showing an initial concentric LV hypertrophy giving way to later eccentric LV hypertrophy (52) and another suggesting early increases in chamber size followed by later increases in wall thickness (53).
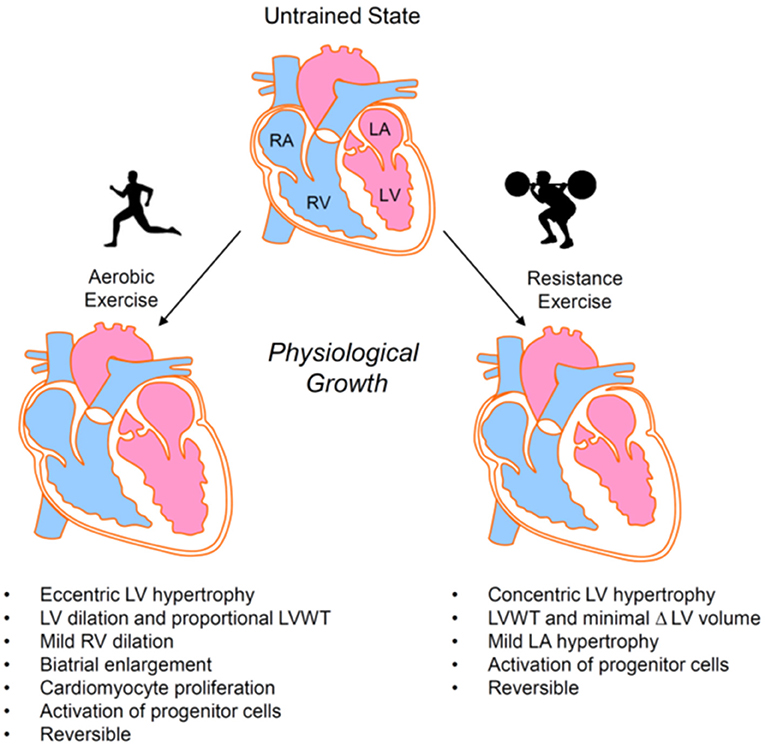
Figure 2. Exercise-induced cardiac growth. Aerobic and resistance exercise elicit different forms of physiological cardiac remodeling. Hypertrophic responses are primarily eccentric in nature for aerobic exercise and concentric in nature for resistance exercise. LA, left atrium; LV, left ventricle; LVWT, left ventricular wall thickness; RA, right atrium; RV, right ventricle.
Although regular, intensive endurance exercise can decrease resting and submaximal heart rates [e.g., (44, 54)], the effects of exercise on other indices of cardiac function are less conspicuous. A meta-analysis of athletes participating in endurance, strength, or combined dynamic and static sports showed no major changes in systolic or diastolic function between sport type or when compared with control subjects (55). However, several studies have identified changes in diastolic function in exercise-adapted subjects. For example, endurance exercise appears to enhance diastolic function modestly (54, 56–61). In contrast, strength training could actually diminish diastolic function, as evinced by studies showing impairment of LV relaxation in American football players (61). In general, in the rested state, individuals that engage in regular exercise do not show remarkably different ejection fractions or fractional shortening values when measured by conventional echocardiography under resting conditions (54, 62–65); however, more subtle changes captured by tissue Doppler and speckle-tracking echocardiography suggest modestly enhanced systolic function in exercise-adapted subjects (66–68).
Cardiac remodeling in response to exercise appears also to involve processes beyond cardiomyocyte hypertrophy. For example, exercise increases levels of circulating progenitor cells (69–75) and cardiac-resident stem/progenitor cells (76–79), which have been implicated in augmentation of vascular density and cardiac repair (80–82). It appears that both resistance and endurance exercises activate progenitor cells [e.g., (83, 84)] and that exercise duration and/or intensity are important in the amplitude and kinetics of their activation (85–88). While the extent to which progenitor/stem cell subtypes regulate physiological cardiac growth remains unclear, their exercise-mediated activation is consistent with the angiogenesis and coronary vascular remodeling (25, 89, 90) and the improved responses to injury (91, 92) associated with exercise-induced cardiac remodeling. In addition, exercise promotes modest cardiomyocyte proliferation (78, 93), which may be important for physiological cardiac adaptation as well as for understanding the mechanisms that trigger cardiomyogenesis in the adult, mammalian heart.
Potential Deleterious Effects of Exercise on the Heart
Although too little exercise is currently a much more serious health problem than too much exercise (94), the popularity of intense exercise (e.g., ultramarathon, CrossFit) has increased remarkably over the past 30 years (95–98). High levels of exercise can transiently increase the risk of acute cardiovascular events such as sudden cardiac death, and it can acutely diminish cardiac function, cause atrial fibrillation, trigger arrhythmias, and lead to pathological remodeling of the heart and vasculature [reviewed in (95)]. Exercise may also markedly change right ventricular morphology and function, contributing to arrhythmogenesis (99). Although young individuals that die during exercise commonly bear inherited or conditional abnormalities such as hypertrophic cardiomyopathy (100), older individuals more commonly die during exercise as a consequence of acute coronary thrombosis and myocardial infarction (101). Nevertheless, sudden cardiac death during exercise is relatively rare and has been estimated to occur in 1 per 15,000–18,000 formerly asymptomatic adults per year (102, 103).
Prolonged endurance exercise can promote “cardiac fatigue,” characterized by decreased cardiac output and ejection fraction (104, 105), although changes in cardiac function typically recover within 2 days after exercise (106). Acute decreases in cardiac function could be due to multiple factors including decreased sensitivity to catecholamines, blood volume redistribution leading to decreased venous return, and cardiomyocyte damage (95). With respect to the last possibility, mild cardiac injury during intense exercise (e.g., marathons, triathlons) is suggested by elevated levels of circulating cardiac troponins [reviewed in (95)], which are typically used to diagnose acute myocardial infarction (107), and exercise intensity is a strong predictor of elevated circulating cardiac troponin levels (108). Other biochemical indicators of cardiac dysfunction, such as B-type natriuretic peptide (BNP) and its cleaved N-terminal fragment (NT-proBNP) may be elevated up to 10-fold after endurance exercise events, but typically return to baseline levels within a few days [reviewed in (95)]. It is hypothesized that exercise-induced BNP/NT-proBNP is indicative of mild myocardial injury (109) or may be a physiological phenomenon important for cardiac adaptation (110). Endurance exercise may also promote myocardial fibrosis and increase coronary artery calcification (95), although the clinical significance of these effects in athletes remains unclear.
How Does Cardiac Metabolism Change During Exercise?
The heart has a high energy demand, which requires continuous ATP generation to sustain contractile function, ion homeostasis, anabolic processes, and signaling (111–114). In normoxia, the heart fuels ATP turnover by generating >95% of its ATP from mitochondrial oxidative phosphorylation, with the remaining 5% derived from substrate level phosphorylation in glycolysis (113, 115). Although the majority of generated ATP supports contractile function, relatively large quantities of ATP are also necessary to maintain ionic homeostasis through ion pumps (116, 117). Below we review some fundamental aspects of cardiac metabolism, followed by the acute metabolic changes in the heart caused by exercise (Figure 3).
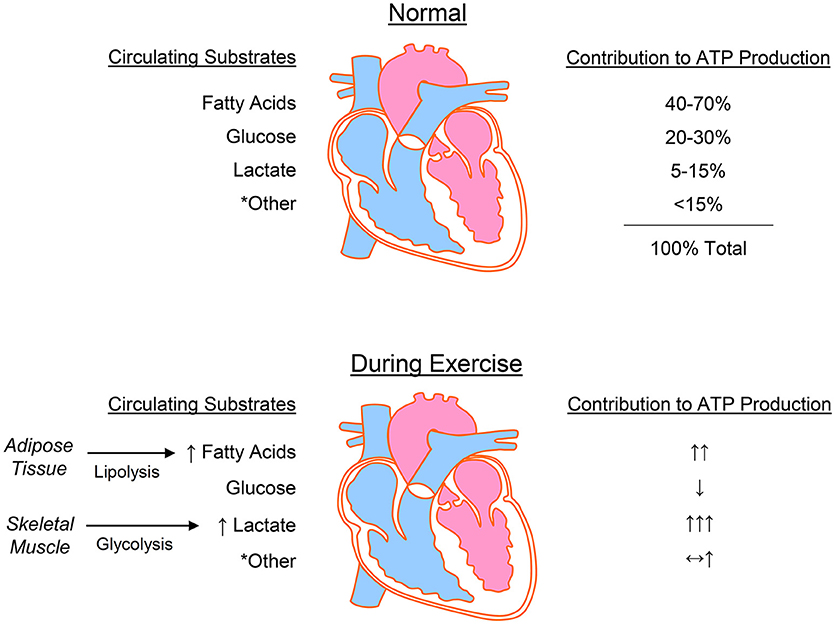
Figure 3. Cardiac metabolism at rest and during exercise. The heart uses numerous substrates for energy provision, with the predominant sources for ATP production being fatty acids, glucose, and lactate. During exercise, lipolysis in adipose tissue and glycolysis in working skeletal muscle increase the circulating levels of fatty acids and lactate, respectively, which are used by the heart to fuel increased energy demands. *Other = ketone bodies, pyruvate, acetate, and branched chain amino acids.
Some Fundamental Aspects of Cardiac Metabolism
Oxidation of fatty acids is the primary contributor to ATP production in the heart, with catabolism of lactate, glucose, ketones, and amino acids fulfilling the remaining energy demand (118–120). This ability of the heart to use a myriad of substrates has led to classification of the heart as a metabolic “omnivore” capable of modulating substrate utilization in a manner dependent on numerous factors, including substrate availability, hormonal stimuli, and myocardial demand. Isotopic labeling studies in humans indicate that 84% of the FFAs entering myocytes are oxidized, with ~16% entering the triacylglycerol (TAG) pool (121). This TAG pool may contribute to ~10% of cardiac ATP production (122, 123) and also plays a central role in signaling and gene expression (124). Although fat oxidation supplies 40–70% cardiac ATP (125–131), it is also less efficient, which is due in part to fatty acid-induced uncoupling of oxidative phosphorylation (123, 132). The relative ATP yield of fats appears dependent on chain length, with long chain fatty acids yielding ~4 mol ATP/mol acetyl CoA and the shortest chain fatty acid, i.e., acetate, costing 2 mol of ATP/mol of acetyl CoA (133). Acetate is usually low in circulation (i.e., below 0.2 mM) and is unlikely to contribute meaningfully to metabolism in the normal heart; however, high alcohol consumption can increase circulating acetate levels to low millimolar concentrations (134, 135) and may under extreme circumstances contribute to cardiac energy deficits (136–139).
Substrates such as glucose, lactate, and pyruvate are generally more efficient energy sources for the heart. In the normal mammalian heart, glucose metabolism via glycolysis supplies approximately 2–8% and glucose oxidation contributes up to 30% to the ATP yield (125). Interestingly, carbon deriving from nearly half of the glucose extracted by the heart is allocated to ancillary pathways of glucose metabolism, which are important for energy storage (glycogen) or biosynthesis of cellular building blocks (e.g., nucleotides, phospholipids, amino acids) (140–145). Lactate is also a major fuel source for the heart, contributing up to 15% of ATP production (125). Lactate tracer studies indicate that the heart is a net lactate consumer (140, 146–148) and that only ~13% of glucose extracted by the heart is converted to lactate (140). Moreover, arterial lactate concentration correlates positively with myocardial lactate uptake and oxidation (141, 149, 150). In humans, lactate is a significant contributor to cardiac ATP production (141, 146), and, in dogs, it can account for up to 87% of cardiac substrate oxidation (151). In rat heart, high lactate levels contribute to nearly 40% of ATP production (150). The myocardium can also use pyruvate readily when extracellular levels are in the millimolar range; however, circulating concentrations of pyruvate are typically less than 150 μM (118), which make it an unlikely source of myocardial energy in vivo.
Ketone bodies such as acetoacetate and β-hydroxybutyrate have received recent attention due to their potential importance in heart failure (152–154); however, should circulating ketone bodies become highly abundant, the normal heart would be expected to increase ketone body oxidation as well. Early studies showed that high concentrations of ketone bodies (e.g., 1–10 mM) can account for nearly 80% of cardiac oxygen consumption (155), and that ketone body provision has a pronounced inhibitory effect on glucose (143, 156, 157) and fat catabolism (158, 159). Interestingly, when provided alone, ketone bodies appear to cause contractile failure (160–162); however, their availability in the presence of other substrates such as glucose may increase efficiency of the working heart (163). Such findings have advanced the idea that ketone bodies are a “superfuel” that enable efficient ATP production (164, 165). Although it has been suggested that 5–15% of ATP production in normal heart is via ketone body oxidation (125), this would depend on the levels of circulating ketones, which in the healthy, fed state are typically less than 500 μM. Although it remains to be clarified whether constitutively high levels of ketone bodies or their oxidation are healthy for the heart (166), ketone diets and ketone body supplements have been suggested to improve exercise performance and augment cardiac energy provision (167, 168).
Amino acids have a relatively minor role in ATP production in the heart; however, they are essential for processes such as protein synthesis and cell signaling. In particular, branched chain amino acids (BCAAs; comprising leucine, isoleucine, and valine) are major amino acids taken up by the heart, with uptake dependent primarily on circulating BCAA concentration (169). Because they are essential amino acids, their intracellular levels are largely dependent on import, with the L-type amino acid transporters and bidirectional amino acid transporters likely contributing to their abundance in the heart (170–172). BCAA catabolism contributes to less than 5% of myocardial oxygen consumption (173), in part because the heart expresses relatively low levels the branched chain aminotransferase enzyme and the branched chain α-keto acid dehydrogenase complex (174, 175). Nevertheless, BCAAs are important regulators of mTOR, which coordinates anabolism and processes such as proliferation, survival, and autophagy (176). Indeed, high intramyocardial levels of BCAAs are associated with cardiac hypertrophy and heart failure (177–179), and recent findings indicate that intracellular accumulation of BCAAs, via a glucose-KLF15-BCAA degradation axis, is required for mTOR activation and cardiomyocyte hypertrophy (180). High intracellular levels of BCAAs may negatively influence cardiac health by inhibiting mitochondrial metabolism (179, 181–183).
Glutamine, a “conditionally essential” amino acid, also appears to regulate the metabolism and health of the heart. In particular, it can activate mTOR in cardiomyocytes (184), and it can protect the heart from injury (185–188). Although many proliferating cells use glutamine as an oxidative fuel (189–191), the normal heart appears to produce glutamine by amidation of glutamate rather than oxidize it for energy provision (169, 192). Nevertheless, glutamine can augment myocardial oleate oxidation and triglyceride formation (193) as well as activate the hexosamine biosynthetic pathway (HBP) (194, 195).
Cardiac Intermediary Metabolism in Exercise
An acute increase in workload during exercise has robust effects on the metabolism of striated muscle (196). In the heart, exercise increases contractile power and oxygen consumption up to 10-fold above resting rates (24, 123). Changes in substrate utilization and ATP production during exercise are a product of the integrated effects of physiologic cues that occur with changes in circulating hormones, metabolic substrates, and hemodynamics.
An increase in myocardial workload is accompanied by increases in the catabolism of multiple substrates, in particular, fatty acids and lactate (141, 149, 197–200). During exercise, hormone-activated lipolysis in adipose tissue increases circulating FFA to levels up to 2.4 mM (201), which enhances FFA uptake and utilization (121, 147, 202). However, heightened levels of circulating FFAs are only partially responsible for increasing fatty acid oxidation because higher cardiac workloads appear sufficient to increase fat oxidation in the heart (203). Cardiac TAG utilization rates also increase considerably with exercise (198) and appear to be further stimulated by lactate availability, suggesting that lactate may stimulate TAG turnover (204). Furthermore, after exercise adaptation, genes responsible for fatty acid transport and catabolism are elevated, which may help optimize fat utilization in the heart (205–207).
Similar to free fatty acids, plasma lactate levels increase during exercise. The increase in lactate is dependent on the type of exercise, with intense exercise (e.g., 60–80% of VO2max) resulting in large increases in arterial lactate levels (208). During intense exercise, circulating lactate levels can increase 5–10-fold (to nearly 10 mM), which is primarily due to lactate extrusion by skeletal muscle. Under these conditions, the contribution of lactate to total oxidative metabolism may account for 60–90% of substrate utilization (149, 151, 209, 210). Although low to moderate intensity exercise (e.g., 40% of VO2 max) does not increase circulating lactate levels remarkably (141), the contribution of lactate oxidation to overall myocardial oxidative metabolism is higher than that compared with the sedentary state (141). Lactate may also enhance fat oxidation in the heart (199), which would increase the capacity of the heart to generate ATP under high workloads.
Although circulating levels of glucose are fairly stable compared with levels of lactate and FFAs, weightlifting and prolonged endurance exercise can decrease arterial glucose concentrations (201, 211), whereas high intensity aerobic exercise may increase blood glucose levels (197). Hemodynamic changes and increases in local and circulating catecholamines can increase the oxidation of stored glucose (glycogen) (212). Although moderate intensity exercise and increases in cardiac workload have been associated with elevations in myocardial glucose uptake and oxidation (141, 197, 199, 200), elevations in circulating concentrations of competing substrates such as lactate and FFAs may decrease glucose catabolism (197–199, 213). Moreover, studies in both humans and animal models suggest that exercise can lower oxygen extraction ratios for glucose and decrease glucose uptake and utilization (198, 213). Recent findings suggest that relatively prolonged, intense endurance exercise can decrease glucose catabolism in the heart by diminishing the activity of phosphofructokinase (214, 215). Collectively, these findings suggest that exercise can acutely increase or decrease both circulating glucose levels and myocardial use in a manner dependent on the type, intensity, or duration of exercise.
Regular exercise also promotes adaptive metabolic remodeling in the heart. Perfused mouse heart studies suggest that adaptation to exercise increases the rates of basal glycolysis (214), glucose oxidation, and fat oxidation (216); however, compared with hearts from sedentary controls, basal cardiac glycolysis has been suggested to be diminished in exercise-adapted rats, despite increases in myocardial glucose and palmitate oxidation (91). That exercise-induced changes in cardiac metabolic remodeling are dependent on exercise intensity is suggested by studies in mice in which moderate-intensity treadmill regimen showed no effect on basal glucose oxidation, palmitate oxidation, or myocardial oxygen consumption, whereas a high-intensity, interval style regimen increased glucose oxidation, diminished palmitate oxidation, and led to a net decrease in resting myocardial oxygen consumption (217). The reasons for discrepancies between studies could be due to model-specific factors (e.g., rodent strain, type of exercise) or differences in cardiac perfusion protocols (e.g., substrate levels, addition of hormones). Circadian rhythm may also account for disparate findings because it influences cardiac metabolism (218), stress responses and protein turnover (219), and inflammatory processes (220). Chronobiology remains an important consideration for understanding how exercise influences cardiac biochemistry and physiology (221, 222).
How do Exercise-Induced Changes in Metabolism Promote Cardiac Adaptation?
Understanding how changes in metabolism regulate cardiac adaptation to exercise presents a challenge. Metabolic pathways coordinate not only ATP production and biosynthesis, but modulate cell signaling and redox state as well (118). Nevertheless, it is clear that repetitive bouts of exercise elicit changes in metabolism that are important for coordinating gene expression in other tissues such as skeletal muscle (6). The mechanisms by which exercise-induced metabolic changes may promote cardiac adaptation are reviewed below and are summarized in Figure 4.
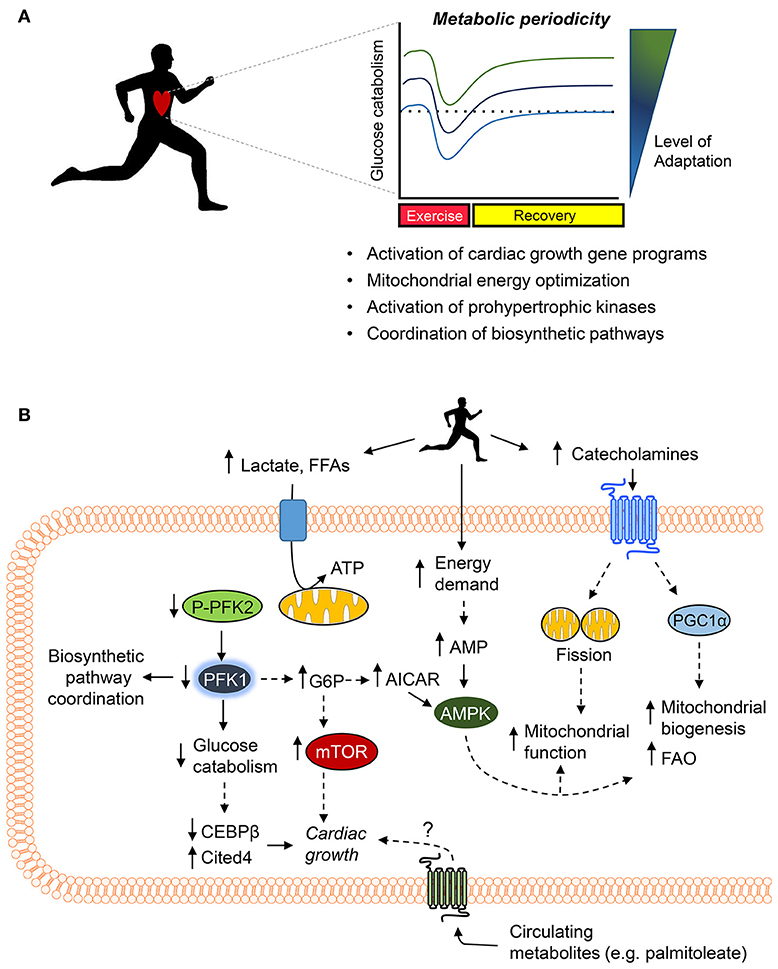
Figure 4. Working model of the metabolic mechanisms of exercise-induced cardiac growth. (A) Periodic changes in glucose metabolism and mitochondrial activity (i.e., metabolic periodicity) occurring with regular exercise promote activation of gene programs responsible for cardiac growth, regulate mitochondrial quality control and function, activate prohypertrophic kinases, and coordinate biosynthetic pathways, all of which integrate to promote cardiac growth. (B) Exercise increases levels of circulating cardiac substrates and catecholamines, which orchestrate changes in cardiomyocyte metabolism. Decreases in the phosphorylation of phosphofructokinase 2 (PFK2) lower phosphofructokinase 1 (PFK1) activity, which decreases glucose catabolism, coordinates ancillary biosynthetic pathways, and increases the levels of upstream glycolytic intermediates (e.g., glucose 6-phosphate, G6P) as well as increases products in the pentose phosphate pathway (e.g., AICAR). Decreases in PFK activity and glucose catabolism appear sufficient to decrease expression of Cebpb and upregulate Cited4, which promote cardiac growth. In addition, elevated levels of G6P, AMP, and AICAR could activate the prohypertrophic signaling kinase mTOR and AMPK. Catecholamine-triggered signaling cascades promote mitochondrial fission and upregulate PGC1α, which acutely increase mitochondrial function and chronically elevate mitochondrial abundance and fatty acid oxidation (FAO) capacity. Last, circulating metabolites (e.g., palmitoleate) may also contribute to exercise-induced physiological cardiac growth.
Importance of Metabolic Periodicity in Cardiac Adaptation
Although episodic changes in metabolism that occur with exercise are known to play an important role in skeletal muscle adaptation (6), relatively less is known about how exercise-induced metabolic periodicity affects adaptive responses in the heart. Nevertheless, it is clear that periodic bouts of exercise stimulate metabolic processes in both cardiac mitochondria and the cytosol. For example, in mice, exercise acutely promotes fission of cardiac mitochondria, which enhances mitochondrial function; these mitochondrial changes were shown to occur in a manner dependent on adrenergic signaling (223). A relatively intense bout of exercise also decreases the activity of phosphofructokinase in mouse heart (214); however, upon adaptation to the exercise regimen and in the rested state (i.e., 24 h after the last exercise bout), apparent myocardial phosphofructokinase activity and glycolytic rate were found to be higher compared with sedentary controls (214). The acute, exercise-induced decreases in myocardial glycolytic rate appear important for cardiac growth because low phosphofructokinase activity brought forth by expression of a cardiac-specific, kinase-deficient 6-phosphofructokinase/fructose-2,6-bisphosphatase transgene in mice (GlycoLo mice) appears sufficient to partially phenocopy the exercise-adapted heart and regulate genes [e.g., Cebpb, Cited4 (224, 225)] required for exercise-induced cardiac growth (214). Moreover, activation of the exercise gene program in GlycoLo mice occurred in the absence of Akt activation, which is thought to be required for regulating physiologic cardiac growth (21, 22, 45). These findings suggest that exercise-induced decreases in glycolysis are a proximal regulator of the cardiac growth program. Collectively, these findings indicate that exercise induces metabolic periodicity in the mitochondrial and cytosolic compartments, which regulate exercise capacity and myocardial growth.
It is likely that periodicity in mitochondrial fission and in intermediary metabolism are interconnected phenomena. In other cell systems, mitochondrial fission is important for regulating glucose and lipid metabolism (226, 227). Moreover, mitochondrial fission is important for regulating mitochondrial quality control by facilitating distribution of mitochondrial components to daughter organelles and by culling defective mitochondria via autophagy (228–230), which is increased the heart during and early after a bout of exercise (231, 232). Exercise-induced periodicity in glucose metabolism appears important for maintaining mitochondrial health because loss of periodicity, either by constitutively increasing or decreasing glucose catabolism, leads to mitochondrial dysfunction (214). Nevertheless, some mechanisms underlying mitochondrial adaptations to exercise appear to diverge from those required for cardiac growth (216, 233), which suggest the presence of distinct circuits by which metabolic changes activate the exercise gene program versus how they modulate mitochondrial health.
Metabolic Changes as a Material Cause of Adaptation
Insight gleaned from bacteria suggest that cells coordinate growth and function via interconversion of glycolytic metabolites to biomass (234), which highlights the obvious role of metabolism as a material cause for structural maintenance and modification. It is likely that changes in ancillary biosynthetic pathway activity are also important for coupling the synthesis of structural materials to activation of the cardiac gene programs responsible for exercise-induced cardiac adaptation.
Rate-limiting steps of glycolysis, e.g., the hexokinase, phosphofructokinase and pyruvate kinase steps, are likely important for modulating biosynthetic pathways in the heart (118). These enzymes are regulated at multiple levels, with allosterism being important for acute changes in activity (235). In several cell types, the phosphofructokinase step of glycolysis regulates the pentose phosphate pathway (PPP), which is important for nucleotide synthesis and redox regulation (236–239). Modeling studies in the adult heart demonstrate that phosphofructokinase activity is particularly important for modulating the activities of the PPP and the polyol pathway (240). In cardiac myocytes, phosphofructokinase activity modulates several ancillary biosynthetic pathways, such as the PPP, the HBP, and the glycerophospholipid synthesis pathway (GLP) by directly modulating glucose carbon entry into the pathways and by indirectly regulating mitochondria-derived molecules important for building block synthesis (e.g., aspartate) (145). Furthermore, metabolomic studies indicate that phosphofructokinase activity also regulates the abundance of several amino acid and lipid metabolites in the heart (214). Much less is known about how exercise affects the hexokinase and pyruvate kinase steps of glycolysis; however, pyruvate kinase activity has been shown to be elevated in the exercise-adapted rat (241) and dog (242) heart.
There is relatively little direct knowledge of how other biosynthetic pathways change with exercise. Transient changes in readouts of HBP activity, i.e., UDP-N-acetylhexosamines or O-GlcNAcylated proteins, occur with exercise (243–246). Changes in the HBP appear important because they may regulate the function and survival of cardiomyocytes (247, 248) as well as reparative cardiac cells (249). To our knowledge, nothing is known regarding how the PPP, GLP, and SBP are influenced in the heart by exercise. While the PPP and the GLP would be thought important for regulating redox state, nucleotide biosynthesis, and phospholipid biosynthesis, the SBP modulates the levels of methyl donors required for DNA methylation reactions and could represent a critical link between metabolism, epigenetic programming, and changes in cardiac structure and function (250).
Signaling Pathways Influencing Cardiac Adaptation
Several signaling pathways integrate to modulate cardiac metabolism and adaptive responses to exercise. Exercise-mediated increases in catecholamines promote upregulation of peroxisome proliferator-activated receptor γ coactivator 1 α (PGC1α) via β-adrenergic signaling and activation of endothelial nitric oxide synthase [reviewed in (21)]. The actions of PGC1α may be mediated via activation of nuclear receptors such as peroxisome proliferator activated receptor α (PPARα) and estrogen-related receptor (ERR) as well as nuclear receptor factor 1 (NRF1), which are known to integrate to increase fatty acid oxidation and to promote mitochondrial biogenesis. Moreover, the metabolic, structural, and functional changes occurring in the exercise-adapted heart are influenced by receptor signaling triggered by insulin-like growth factor-1 (IGF-1) (251, 252) and neuregulin-1 (78, 253), which activate the phosphoinositide 3-kinase (PI3K)/Akt pathway to promote physiologic cardiac growth (254–257) or activate a cardiomyocyte proliferative response (78, 258–260). Interestingly, cardiac glucose metabolism is influenced by catecholamines (130, 261, 262), IGF-1 (263–265), and Nrg-1 (265), which suggests that these hormones may provide additional regulation to acute or chronic metabolic changes induced by exercise.
Metabolite Signaling
Metabolite signaling is another mechanism that connects exercise-induced changes in metabolism to cardiac adaptation. In particular, glucose-derived metabolites regulate the activities of the prohypertrophic kinases mammalian target of rapamycin (mTOR) and AMP-activated kinase (AMPK) (45). The intracellular levels of glucose 6-phosphate (G6P) regulate mTOR activity in the heart (266–268), and 5-amino-4-imidazolecarboxamide ribonucleotide (AICAR), which is an intermediate of the PPP (269), stimulates AMPK (270). It is anticipated that G6P, AMP, and AICAR increase in the heart with exercise. Predictions from crossover theorem (271–274) and modeling studies (240) suggest that acute decreases in phosphofructokinase activity, such as occurs during exercise (118), would increase G6P as well as augment PPP activity, which could increase AICAR levels. In addition, the large increase in myocardial ATP demand would be thought to increase intracellular AMP levels.
Circulating metabolites are also important regulators of exercise-induced cardiac growth. Hormone-mediated adipose tissue lipolysis during exercise liberates palmitoleate (C16:1n7), which promotes cardiac growth potentially by activating G-protein-coupled receptors (GPCRs), Akt, or nuclear receptors (275). The cardiac growth-stimulating effect of palmitoleate is similar to the fatty acid-induced cardiac hypertrophy that occurs in the python heart after a large meal (276). Interestingly, FFAs not only increase acutely with exercise (201), but they appear to remain elevated in the exercise-adapted state as well (277, 278); hence, they could stimulate the signaling required to sustain cardiac adaptations. Given that numerous metabolites have cognate GPCRs (279), it is likely that other metabolites elevated during or after exercise have important roles to play in tissue adaptation. Understanding how circulating metabolites trigger structural and functional changes in the heart could lead to the development of novel therapies to improve cardiac health.
Summary
Metabolic changes caused by exercise are important for cardiac remodeling and adaptation. The integrative metabolic changes brought forth by exercise combine with changes in cardiac workload to regulate cardiac metabolism. In particular, exercise alters levels of competing substrates, and it changes the abundance of circulating hormones, which cue metabolic pathways that are critical for transcriptional changes and cardiac growth. In addition, changes in circulating and endogenous metabolites can trigger physiologic growth by activating prohypertrophic signaling pathways. Nevertheless, numerous questions remain, including questions of how to optimize the amount of exercise to produce beneficial, as opposed to deleterious, effects on cardiovascular health (280) as well as mechanistic questions of how exercise-induced changes in metabolism couple the synthesis of structural materials to activation of the physiological cardiac growth program (118). While this knowledge is acquired, it appears that we would be best served by sticking to the advice of the ancient Greeks—“Exercise till the mind feels delight in reposing from the fatigue.”—Socrates.
Author Contributions
All authors listed have made substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
The authors would like to acknowledge grants from the National Institutes of Health (HL130174, HL78825, ES028268) and the American Diabetes Association Pathway to Stop Diabetes Grant (1-16-JDF-041).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Boule NG, Haddad E, Kenny GP, Wells GA, Sigal RJ. Effects of exercise on glycemic control and body mass in type 2 diabetes mellitus: a meta-analysis of controlled clinical trials. JAMA (2001) 286:1218–27. doi: 10.1001/jama.286.10.1218
2. Helmrich SP, Ragland DR, Leung RW, Paffenbarger RS Jr. Physical activity and reduced occurrence of non-insulin-dependent diabetes mellitus. New Engl J Med. (1991) 325:147–52. doi: 10.1056/NEJM199107183250302
3. Knowler WC, Barrett-Connor E, Fowler SE, Hamman RF, Lachin JM, Walker EA, et al. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. New Engl J Med. (2002) 346:393–403. doi: 10.1056/NEJMoa012512
4. Lawlor DA, Hopker SW. The effectiveness of exercise as an intervention in the management of depression: systematic review and meta-regression analysis of randomised controlled trials. BMJ (2001) 322:763–7. doi: 10.1136/bmj.322.7289.763
5. Chalder M, Wiles NJ, Campbell J, Hollinghurst SP, Haase AM, Taylor AH, et al. Facilitated physical activity as a treatment for depressed adults: randomised controlled trial. BMJ (2012) 344:e2758. doi: 10.1136/bmj.e2758
6. Egan B, Zierath JR. Exercise metabolism and the molecular regulation of skeletal muscle adaptation. Cell Metab. (2013) 17:162–84. doi: 10.1016/j.cmet.2012.12.012
7. Paffenbarger RS Jr, Hyde RT, Wing AL, Hsieh CC. Physical activity, all-cause mortality, and longevity of college alumni. New Engl J Med. (1986) 314:605–13. doi: 10.1056/NEJM198603063141003
8. Blair SN, Kohl HW III, Paffenbarger RS Jr, Clark DG, Cooper KH, Gibbons LW. Physical fitness and all-cause mortality. A prospective study of healthy men and women. JAMA J Am Med Assoc. (1989) 262:2395–401. doi: 10.1001/jama.1989.03430170057028
9. Myers J, Prakash M, Froelicher V, Do D, Partington S, Atwood JE. Exercise capacity and mortality among men referred for exercise testing. New Engl J Med. (2002) 346:793–801. doi: 10.1056/NEJMoa011858
10. Holme I, Anderssen SA. Increases in physical activity is as important as smoking cessation for reduction in total mortality in elderly men: 12 years of follow-up of the Oslo II study. Br J Sports Med. (2015) 49:743–8. doi: 10.1136/bjsports-2014-094522
11. Wei X, Liu X, Rosenzweig A. What do we know about the cardiac benefits of exercise? Trends Cardiovasc Med. (2015) 25:529–36. doi: 10.1016/j.tcm.2014.12.014
12. Platt C, Houstis N, Rosenzweig A. Using exercise to measure and modify cardiac function. Cell Metab. (2015) 21:227–36. doi: 10.1016/j.cmet.2015.01.014
13. Blair SN, Kampert JB, Kohl HW III, Barlow CE, Macera CA, Paffenbarger R, et al. (1996) Influences of cardiorespiratory fitness and other precursors on cardiovascular disease and all-cause mortality in men and women. JAMA 276:205–10. doi: 10.1001/jama.1996.03540030039029
14. Gayda M, Ribeiro PA, Juneau M, Nigam A. Comparison of different forms of exercise training in patients with cardiac disease: where does high-intensity interval training fit? Can J Cardiol. (2016) 32:485–94. doi: 10.1016/j.cjca.2016.01.017
16. Ribeiro PA, Boidin M, Juneau M, Nigam A, Gayda M. High-intensity interval training in patients with coronary heart disease: prescription models and perspectives. Ann Phys Rehabil Med. (2017) 60:50–7. doi: 10.1016/j.rehab.2016.04.004
17. Sharma S, Firoozi S, McKenna WJ. Value of exercise testing in assessing clinical state and prognosis in hypertrophic cardiomyopathy. Cardiol Rev. (2001) 9:70–6. doi: 10.1097/00045415-200103000-00005
18. Gasiorowski A, Dutkiewicz J. Comprehensive rehabilitation in chronic heart failure. Ann Agric Environ Med. (2013) 20:606–12.
19. Heinonen I, Kalliokoski KK, Hannukainen JC, Duncker DJ, Nuutila P, Knuuti J. Organ-specific physiological responses to acute physical exercise and long-term training in humans. Physiology (2014) 29:421–36. doi: 10.1152/physiol.00067.2013
20. Galen Green RM. A Translation of Galen's Hygiene (De sanitate tuenda). Thomas: Springfield, Ill (1951).
21. Vega RB, Konhilas JP, Kelly DP, Leinwand LA. Molecular Mechanisms Underlying Cardiac Adaptation to Exercise. Cell Metab. (2017) 25:1012–26. doi: 10.1016/j.cmet.2017.04.025
22. Lerchenmuller C, Rosenzweig A. Mechanisms of exercise-induced cardiac growth. Drug Discov Today (2014) 19:1003–9. doi: 10.1016/j.drudis.2014.03.010
23. Haas TL, Nwadozi E. Regulation of skeletal muscle capillary growth in exercise and disease. Appl Physiol Nutr Metab. (2015) 40:1221–32. doi: 10.1139/apnm-2015-0336
24. Olver TD, Ferguson BS, Laughlin MH. molecular mechanisms for exercise training-induced changes in vascular structure and function: skeletal muscle, cardiac muscle, and the brain. Progressmolec Biol Trans Sci. (2015) 135:227–57 doi: 10.1016/bs.pmbts.2015.07.017
25. Laughlin MH, Bowles DK, Duncker DJ. The coronary circulation in exercise training. Am J Physiol Heart Circ Physiol. (2012) 302:H10–23 doi: 10.1152/ajpheart.00574.2011
26. Henschen S. Skidlauf und Skidwettlauf. Eine medizinische Sportstudie. Mitt Med Klin Upsala. Jena. Fischer Verlag. (1899) 2:15.
27. Darling EA. The effects of training. A study of the Harvard University crews. Boston Med Surg J. (1899) 141:205–9. doi: 10.1056/NEJM189909071411001
28. Beckner GL, Winsor T. Cardiovascular adaptations to prolonged physical effort. Circulation (1954) 9:835–46. doi: 10.1161/01.CIR.9.6.835
29. Roskamm H, Reindell H, Musshoff K, Koenig K. [Relations between heart size and physical efficiency in male and female athletes in comparison with normal male and female subjects. III]. Arch Kreislaufforsch (1961) 35:67–102 doi: 10.1007/BF02119723
30. Reindell H, Roskamm H, Steim H. [The heart and blood circulation in athletes]. Med Welt. (1960) 31:1557–63.
31. Bulychev VV, Khmelevskii VA, IuV R. [Roentgenological and Instrumental Examination of the Heart in Athletes]. Klin Med (Mosk). (1965) 43:108–14.
32. Weiner RB, Baggish AL. Exercise-induced cardiac remodeling. Prog Cardiovasc Dis. (2012) 54:380–6. doi: 10.1016/j.pcad.2012.01.006
33. Adolph Fick (1829-1901) mathematician physicist physiologist. JAMA (1967) 202:1100–1. doi: 10.1001/jama.1967.03130250082020
34. Rowell LB, Murray JA, Brengelmann GL, Kraning KK II Human cardiovascular adjustments to rapid changes in skin temperature during exercise. Circ Res. (1969) 24:711–24. doi: 10.1161/01.RES.24.5.711
35. Rowell LB. Human Circulation: Regulation During Physical Stress. New York, NY: Oxford University Press (1986).
36. Coyle EF, Gonzalez-Alonso J. Cardiovascular drift during prolonged exercise: new perspectives. Exerc Sport Sci Rev. (2001) 29:88–92.
37. Rowland T. Echocardiography and circulatory response to progressive endurance exercise. Sports Med. (2008) 38:541–51. doi: 10.2165/00007256-200838070-00002
38. Plowman SA, Smith DL. Exercise Physiology for Health, Fitness, and Performance. Philadelphia, PA: Wolters Kluwer (2017).
39. Clausen JP. Circulatory adjustments to dynamic exercise and effect of physical training in normal subjects and in patients with coronary artery disease. Prog Cardiovasc Dis. (1976) 18:459–95. doi: 10.1016/0033-0620(76)90012-8
40. Bezucha GR, Lenser MC, Hanson PG, Nagle FJ. Comparison of hemodynamic responses to static and dynamic exercise. J Appl Physiol Respir Environ Exerc Physiol. (1982) 53:1589–93. doi: 10.1152/jappl.1982.53.6.1589
41. Hill DW, Butler SD. Haemodynamic responses to weightlifting exercise. Sports Med. (1991) 12:1–7. doi: 10.2165/00007256-199112010-00001
42. MacDougall JD, Tuxen D, Sale DG, Moroz JR, Sutton JR. Arterial blood pressure response to heavy resistance exercise. J Appl Physiol. (1985) 58:785–90. doi: 10.1152/jappl.1985.58.3.785
43. MacDougall JD, McKelvie RS, Moroz DE, Sale DG, McCartney N, Buick F. Factors affecting blood pressure during heavy weight lifting and static contractions. J Appl Physiol. (1985) 73:1590–7. doi: 10.1152/jappl.1992.73.4.1590
44. DeMaria AN, Neumann A, Lee G, Fowler W, Mason DT. Alterations in ventricular mass and performance induced by exercise training in man evaluated by echocardiography. Circulation (1978) 57:237–44. doi: 10.1161/01.CIR.57.2.237
45. Maillet M, van Berlo JH, Molkentin JD. Molecular basis of physiological heart growth: fundamental concepts and new players. Nat Rev Mol Cell Biol. (2013) 14:38–48. doi: 10.1038/nrm3495
46. Maron BJ, Pelliccia A, Spataro A, Granata M. Reduction in left ventricular wall thickness after deconditioning in highly trained Olympic athletes. Br Heart J. (1993) 69:125–8. doi: 10.1136/hrt.69.2.125
47. Olah A, Kellermayer D, Matyas C, Nemeth BT, Lux A, Szabo L, et al. Complete reversion of cardiac functional adaptation induced by exercise training. Med Sci Sports Exerc. (2017) 49:420–9. doi: 10.1249/MSS.0000000000001127
48. Waring CD, Henning BJ, Smith AJ, Nadal-Ginard B, Torella D, Ellison GM. Cardiac adaptations from 4 weeks of intensity-controlled vigorous exercise are lost after a similar period of detraining. Physiol Rep. (2015) 3:e12302. doi: 10.14814/phy2.12302
49. Morganroth J, Maron BJ, Henry WL, Epstein SE. Comparative left ventricular dimensions in trained athletes. Ann Intern Med. (1975) 82:521–4. doi: 10.7326/0003-4819-82-4-521
50. Spence AL, Naylor LH, Carter HH, Buck CL, Dembo L, Murray CP, et al. A prospective randomised longitudinal MRI study of left ventricular adaptation to endurance and resistance exercise training in humans. J Physiol. (2011) 589:5443–52. doi: 10.1113/jphysiol.2011.217125
51. Mihl C, Dassen WR, Kuipers H. Cardiac remodelling: concentric versus eccentric hypertrophy in strength and endurance athletes. Neth Heart J. (2008) 16:129–33. doi: 10.1007/BF03086131
52. Arbab-Zadeh A, Perhonen M, Howden E, Peshock RM, Zhang R, Adams-Huet B, et al. Cardiac remodeling in response to 1 year of intensive endurance training. Circulation (2014) 130:2152–61. doi: 10.1161/CIRCULATIONAHA.114.010775
53. Weiner RB, DeLuca JR, Wang F, Lin J, Wasfy MM, Berkstresser B, et al. Exercise-induced left ventricular remodeling among competitive athletes: a phasic phenomenon. Circ Cardiovasc Imaging (2015) 8:e003651. doi: 10.1161/CIRCIMAGING.115.003651
54. Utomi V, Oxborough D, Whyte GP, Somauroo J, Sharma S, Shave R, et al. Systematic review and meta-analysis of training mode, imaging modality and body size influences on the morphology and function of the male athlete's heart. Heart (2013) 99:1727–33. doi: 10.1136/heartjnl-2012-303465
55. Pluim BM, Zwinderman AH, van der Laarse A, van der Wall EE. The athlete's heart. A meta-analysis of cardiac structure and function. Circulation (2000) 101:336–44. doi: 10.1161/01.CIR.101.3.336
56. Baggish AL, Yared K, Weiner RB, Wang F, Demes R, Picard MH, et al. Differences in cardiac parameters among elite rowers and subelite rowers. Med Sci Sports Exerc. (2010) 42:1215–20. doi: 10.1249/MSS.0b013e3181c81604
57. Naylor LH, Arnolda LF, Deague JA, Playford D, Maurogiovanni A, O'Driscoll G, et al. Reduced ventricular flow propagation velocity in elite athletes is augmented with the resumption of exercise training. J Physiol. (2005) 563:957–63. doi: 10.1113/jphysiol.2004.078360
58. Caso P, D'Andrea A, Galderisi M, Liccardo B, Severino S, De Simone L, et al. Pulsed Doppler tissue imaging in endurance athletes: relation between left ventricular preload and myocardial regional diastolic function. Am J Cardiol. (2000) 85:1131–6. doi: 10.1016/S0002-9149(00)00709-8
59. Prasad A, Popovic ZB, Arbab-Zadeh A, Fu Q, Palmer D, Dijk E, et al. The effects of aging and physical activity on Doppler measures of diastolic function. Am J Cardiol. (2007) 99:1629–36. doi: 10.1016/j.amjcard.2007.01.050
60. D'Andrea A, Cocchia R, Riegler L, Scarafile R, Salerno G, Gravino R, et al. Left ventricular myocardial velocities and deformation indexes in top-level athletes. J Am Soc Echocardiogr. (2010) 23:1281–8. doi: 10.1016/j.echo.2010.09.020
61. Baggish AL, Wang F, Weiner RB, Elinoff JM, Tournoux F, Boland A, et al. Training-specific changes in cardiac structure and function: a prospective and longitudinal assessment of competitive athletes. J Appl Physiol. (2008) 104:1121–8. doi: 10.1152/japplphysiol.01170.2007
62. Bar-Shlomo BZ, Druck MN, Morch JE, Jablonsky G, Hilton JD, Feiglin DH, et al. Left ventricular function in trained and untrained healthy subjects. Circulation (1982) 65:484–8. doi: 10.1161/01.CIR.65.3.484
63. Bekaert I, Pannier JL, Van de Weghe C, Van Durme JP, Clement DL, Pannier R. Non-invasive evaluation of cardiac function in professional cyclists. Br Heart J. (1981) 45:213–8. doi: 10.1136/hrt.45.2.213
64. Douglas PS, O'Toole ML, Hiller WD, Reichek N. Left ventricular structure and function by echocardiography in ultraendurance athletes. Am J Cardiol. (1986) 58:805–9. doi: 10.1016/0002-9149(86)90358-9
65. Gilbert CA, Nutter DO, Felner JM, Perkins JV, Heymsfield SB, Schlant RC. Echocardiographic study of cardiac dimensions and function in the endurance-trained athlete. Am J Cardiol. (1977) 40:528–33. doi: 10.1016/0002-9149(77)90067-4
66. Baggish AL, Yared K, Wang F, Weiner RB, Hutter AM Jr, Picard MH, et al. The impact of endurance exercise training on left ventricular systolic mechanics. Am J Physiol Heart Circ Physiol. (2008) 295:H1109–16. doi: 10.1152/ajpheart.00395.2008
67. Weiner RB, Hutter AM Jr, Wang F, Kim J, Weyman AE, Wood MJ, et al. The impact of endurance exercise training on left ventricular torsion. JACC Cardiovasc Imaging (2010) 3:1001–9. doi: 10.1016/j.jcmg.2010.08.003
68. Simsek Z, Hakan Tas M, Degirmenci H, Gokhan Yazici A, Ipek E, Duman H, et al. Speckle tracking echocardiographic analysis of left ventricular systolic and diastolic functions of young elite athletes with eccentric and concentric type of cardiac remodeling. Echocardiography (2013) 30:1202–8. doi: 10.1111/echo.12263
69. Heal JM, Brightman A. Exercise and circulating hematopoietic progenitor cells (CFU-GM) in humans. Transfusion (1987) 27:155–8. doi: 10.1046/j.1537-2995.1987.27287150188.x
70. Rehman J, Li J, Parvathaneni L, Karlsson G, Panchal VR, Temm CJ, et al. Exercise acutely increases circulating endothelial progenitor cells and monocyte-/macrophage-derived angiogenic cells. J Am Coll Cardiol. (2004) 43:2314–8. doi: 10.1016/j.jacc.2004.02.049
71. Walther C, Gaede L, Adams V, Gelbrich G, Leichtle A, Erbs S, et al. Effect of increased exercise in school children on physical fitness and endothelial progenitor cells: a prospective randomized trial. Circulation (2009) 120:2251–9. doi: 10.1161/CIRCULATIONAHA.109.865808
72. Bonsignore MR, Morici G, Santoro A, Pagano M, Cascio L, Bonanno A, et al. Circulating hematopoietic progenitor cells in runners. J Appl Physiol. (2002) 93:1691–7. doi: 10.1152/japplphysiol.00376.2002
73. Thijssen DH, Torella D, Hopman MT, Ellison GM. The role of endothelial progenitor and cardiac stem cells in the cardiovascular adaptations to age and exercise. Front Biosci. (2009) 14:4685–702. doi: 10.2741/3560
74. Brehm M, Picard F, Ebner P, Turan G, Bolke E, Kostering M, et al. Effects of exercise training on mobilization and functional activity of blood-derived progenitor cells in patients with acute myocardial infarction. Eur J Med Res. (2009) 14:393–405. doi: 10.1186/2047-783X-14-9-393
75. Van Craenenbroeck EM, Hoymans VY, Beckers PJ, Possemiers NM, Wuyts K, Paelinck BP, et al. Exercise training improves function of circulating angiogenic cells in patients with chronic heart failure. Basic Res Cardiol. (2010) 105:665–76. doi: 10.1007/s00395-010-0105-4
76. Xiao J, Xu T, Li J, Lv D, Chen P, Zhou Q, et al. Exercise-induced physiological hypertrophy initiates activation of cardiac progenitor cells. Int J Clin Exp Pathol. (2014) 7:663–9.
77. Leite CF, Lopes CS, Alves AC, Fuzaro CS, Silva MV, Oliveira LF, et al. Endogenous resident c-Kit cardiac stem cells increase in mice with an exercise-induced, physiologically hypertrophied heart. Stem Cell Res. (2015) 15:151–64. doi: 10.1016/j.scr.2015.05.011
78. Waring CD, Vicinanza C, Papalamprou A, Smith AJ, Purushothaman S, Goldspink DF, et al. The adult heart responds to increased workload with physiologic hypertrophy, cardiac stem cell activation, and new myocyte formation. Eur Heart J. (2014) 35:2722–31. doi: 10.1093/eurheartj/ehs338
79. Kolwicz SC, MacDonnell SM, Renna BF, Reger PO, Seqqat R, Rafiq K, et al. Left ventricular remodeling with exercise in hypertension. Am J Physiol Heart Circ Physiol. (2009) 297:H1361–8. doi: 10.1152/ajpheart.01253.2008
80. Mitchell A, Fujisawa T, Newby D, Mills N, Cruden NL. Vascular injury and repair: a potential target for cell therapies. Future Cardiol. (2015) 11:45–60. doi: 10.2217/fca.14.77
81. Broughton KM, Wang BJ, Firouzi F, Khalafalla F, Dimmeler S, Fernandez-Aviles F, et al. Mechanisms of Cardiac Repair and Regeneration. Circ Res. (2018) 122:1151–63. doi: 10.1161/CIRCRESAHA.117.312586
82. Wysoczynski M, Dassanayaka S, Zafir A, Ghafghazi S, Long BW, Noble C, et al. A new method to stabilize c-kit expression in reparative cardiac mesenchymal cells. Front Cell Dev Biol. (2016) 4:78. doi: 10.3389/fcell.2016.00078
83. Ross MD, Wekesa AL, Phelan JP, Harrison M. Resistance exercise increases endothelial progenitor cells and angiogenic factors. Med Sci Sports Exerc. (2014) 46:16–23. doi: 10.1249/MSS.0b013e3182a142da
84. Ellison GM, Waring CD, Vicinanza C, Torella D. Physiological cardiac remodelling in response to endurance exercise training: cellular and molecular mechanisms. Heart (2012) 98:5–10. doi: 10.1136/heartjnl-2011-300639
85. Rakobowchuk M, Harris E, Taylor A, Baliga V, Cubbon RM, Rossiter HB, et al. Heavy and moderate interval exercise training alters low-flow-mediated constriction but does not increase circulating progenitor cells in healthy humans. Exp Physiol. (2012) 97:375–85. doi: 10.1113/expphysiol.2011.062836
86. Tsai HH, Lin CP, Lin YH, Hsu CC, Wang JS. High-intensity Interval training enhances mobilization/functionality of endothelial progenitor cells and depressed shedding of vascular endothelial cells undergoing hypoxia. Eur J Appl Physiol. (2016) 116:2375–88. doi: 10.1007/s00421-016-3490-z
87. Ribeiro F, Ribeiro IP, Goncalves AC, Alves AJ, Melo E, Fernandes R, et al. Effects of resistance exercise on endothelial progenitor cell mobilization in women. Sci Rep. (2017) 7:17880. doi: 10.1038/s41598-017-18156-6
88. Laufs U, Urhausen A, Werner N, Scharhag J, Heitz A, Kissner G, et al. Running exercise of different duration and intensity: effect on endothelial progenitor cells in healthy subjects. Eur J Cardiovasc Prev Rehabil. (2005) 12:407–14. doi: 10.1097/01.hjr.0000174823.87269.2e
89. White FC, Bloor CM, McKirnan MD, Carroll SM. Exercise training in swine promotes growth of arteriolar bed and capillary angiogenesis in heart. J Appl Physiol. (1998) 85:1160–8. doi: 10.1152/jappl.1998.85.3.1160
90. Brown MD. Exercise and coronary vascular remodelling in the healthy heart. Exp Physiol. (2003) 88:645–58. doi: 10.1113/eph8802618
91. Burelle Y, Wambolt RB, Grist M, Parsons HL, Chow JC, Antler C, et al. Regular exercise is associated with a protective metabolic phenotype in the rat heart. Am J Physiol Heart Circ Physiol. (2004) 287:H1055–3. doi: 10.1152/ajpheart.00925.2003
92. Calvert JW, Condit ME, Aragon JP, Nicholson CK, Moody BF, Hood RL, et al. Exercise protects against myocardial ischemia-reperfusion injury via stimulation of beta(3)-adrenergic receptors and increased nitric oxide signaling: role of nitrite and nitrosothiols. Circ Res. (2011) 108:1448–58. doi: 10.1161/CIRCRESAHA.111.241117
93. Vujic A, Lerchenmuller C, Wu TD, Guillermier C, Rabolli CP, Gonzalez E, et al. Exercise induces new cardiomyocyte generation in the adult mammalian heart. Nat Commun. (2018) 9:1659. doi: 10.1038/s41467-018-04083-1
94. Booth FW, Roberts CK, Thyfault JP, Ruegsegger GN, Toedebusch RG. Role of inactivity in chronic diseases: evolutionary insight and pathophysiological mechanisms. Physiol Rev. (2017) 97:1351–402. doi: 10.1152/physrev.00019.2016
95. Eijsvogels TM, Fernandez AB, Thompson PD. Are there deleterious cardiac effects of acute and chronic endurance exercise? Physiol Rev. (2016) 96:99–125. doi: 10.1152/physrev.00029.2014
96. Meyer J, Morrison J, Zuniga J. the benefits and risks of crossfit: a systematic review. Workplace Health Saf. (2017) 65:612–8. doi: 10.1177/2165079916685568
97. Hoffman MD, Wegelin JA. The Western States 100-Mile Endurance Run: participation and performance trends. Med Sci Sports Exerc. (2009) 41:2191–8. doi: 10.1249/MSS.0b013e3181a8d553
98. Knechtle B, Knechtle P, Lepers R. Participation and performance trends in ultra-triathlons from 1985 to 2009. Scand J Med Sci Sports (2011) 21:e82–90 doi: 10.1111/j.1600-0838.2010.01160.x
99. D'Andrea A, La Gerche A, Golia E, Teske AJ, Bossone E, Russo MG, et al. Right heart structural and functional remodeling in athletes. Echocardiography (2015) 32(Suppl. 1):S11–22. doi: 10.1111/echo.12226
100. Maron BJ, Thompson PD, Ackerman MJ, Balady G, Berger S, Cohen D, et al. Recommendations and considerations related to preparticipation screening for cardiovascular abnormalities in competitive athletes: 2007 update: a scientific statement from the American heart association council on nutrition, physical activity, and metabolism: endorsed by the american college of cardiology foundation. Circulation (2007) 115:1643–455. doi: 10.1161/CIRCULATIONAHA.107.181423
101. Thompson PD, Franklin BA, Balady GJ, Blair SN, Corrado D, Estes NA III, et al. Exercise and acute cardiovascular events placing the risks into perspective: a scientific statement from the American heart association council on nutrition, physical activity, and metabolism and the council on clinical cardiology. Circulation (2007) 115:2358–68. doi: 10.1161/CIRCULATIONAHA.107.181485
102. Siscovick DS, Weiss NS, Fletcher RH, Lasky T. The incidence of primary cardiac arrest during vigorous exercise. New Engl J Med. (1984) 311:874–7. doi: 10.1056/NEJM198410043111402
103. Thompson PD, Funk EJ, Carleton RA, Sturner WQ. Incidence of death during jogging in Rhode Island from 1975 through 1980. JAMA (1982) 247:2535–8. doi: 10.1001/jama.1982.03320430039028
104. Dawson E, George K, Shave R, Whyte G, Ball D. Does the human heart fatigue subsequent to prolonged exercise? Sports Med. (2003) 33:365–80. doi: 10.2165/00007256-200333050-00003
105. Middleton N, Shave R, George K, Whyte G, Hart E, Atkinson G. (2006) Left ventricular function immediately following prolonged exercise: a meta-analysis. Med Sci Sports Exerc. 38:681–7. doi: 10.1249/01.mss.0000210203.10200.12
106. McGavock JM, Warburton DE, Taylor D, Welsh RC, Quinney HA, Haykowsky MJ. The effects of prolonged strenuous exercise on left ventricular function: a brief review. Heart Lung (2002) 31:279–92; quiz 293–274. doi: 10.1067/mhl.2002.126106
107. Thygesen K, Alpert JS, Jaffe AS, Simoons ML, Chaitman BR, White HD, et al. Third universal definition of myocardial infarction. Circulation (2012) 126:2020–35. doi: 10.1161/CIR.0b013e31826e1058
108. Eijsvogels TM, Hoogerwerf MD, Oudegeest-Sander MH, Hopman MT, Thijssen DH. The impact of exercise intensity on cardiac troponin I release. Int J Cardiol. (2014) 171:e3–4. doi: 10.1016/j.ijcard.2013.11.050
109. Ohba H, Takada H, Musha H, Nagashima J, Mori N, Awaya T, et al. Effects of prolonged strenuous exercise on plasma levels of atrial natriuretic peptide and brain natriuretic peptide in healthy men. Am Heart J. (2001) 141:751–8. doi: 10.1067/mhj.2001.114371
110. Scharhag J, Urhausen A, Herrmann M, Schneider G, Kramann B, Herrmann W, et al. No difference in N-terminal pro-brain natriuretic peptide (NT-proBNP) concentrations between endurance athletes with athlete's heart and healthy untrained controls. Heart (2004) 90:1055–16. doi: 10.1136/hrt.2003.020420
111. Neely JR, Morgan HE. Relationship between carbohydrate and lipid metabolism and the energy balance of heart muscle. Annu Rev Physiol. (1974) 36:413–59. doi: 10.1146/annurev.ph.36.030174.002213
112. Opie LH. Metabolism of the heart in health and disease. I. Am Heart J. (1968) 76:685–98. doi: 10.1016/0002-8703(68)90168-3
113. Opie LH. Metabolism of the heart in health and disease. II. Am Heart J. (1969) 77:100–122 contd
114. Doenst T, Nguyen TD, Abel ED. Cardiac metabolism in heart failure: implications beyond ATP production. Circ Res. (2013) 113:709–24. doi: 10.1161/CIRCRESAHA.113.300376
115. Opie LH. Heart Physiology: From Cell to Circulation. Philadelphia, PA: Lippincott Williams & Wilkins (2004).
116. Gibbs CL. Cardiac energetics. Physiol Rev. (1978) 58:174–254 doi: 10.1152/physrev.1978.58.1.174
117. Suga H. Ventricular energetics. Physiol Rev. (1990) 70:247–77 doi: 10.1152/physrev.1990.70.2.247
118. Gibb AA, Hill BG. Metabolic coordination of physiological and pathological cardiac remodeling. Circ Res. (2018) 123:107–28. doi: 10.1161/CIRCRESAHA.118.312017
119. Taegtmeyer H, Young ME, Lopaschuk GD, Abel ED, Brunengraber H, Darley-Usmar V, et al. Assessing cardiac metabolism: a scientific statement from the american heart association. Circ Res. (2016) 118:1659–701. doi: 10.1161/RES.0000000000000097
120. Taegtmeyer H, Lam T, Davogustto G. Cardiac Metabolism in perspective. Compr Physiol. (2016) 6:1675–99. doi: 10.1002/cphy.c150056
121. Wisneski JA, Gertz EW, Neese RA, Mayr M. Myocardial metabolism of free fatty acids. Studies with 14C-labeled substrates in humans. J Clin Invest. (1987) 79:359–66. doi: 10.1172/JCI112820
122. Saddik M, Lopaschuk GD. Myocardial triglyceride turnover and contribution to energy substrate utilization in isolated working rat hearts. J Biol Chem. (1991) 266:8162–70.
123. Lopaschuk GD, Ussher JR, Folmes CD, Jaswal JS, Stanley WC. Myocardial fatty acid metabolism in health and disease. Physiol Rev. (2010) 90:207–58. doi: 10.1152/physrev.00015.2009
124. Carley AN, Lewandowski ED. Triacylglycerol turnover in the failing heart. Biochim Biophys Acta (2016) 1861:1492–9. doi: 10.1016/j.bbalip.2016.03.012
125. Lopaschuk GD. Metabolic modulators in heart disease: past, present, and future. Can J Cardiol. (2017) 33:838–49. doi: 10.1016/j.cjca.2016.12.013
126. Taha M, Lopaschuk GD. Alterations in energy metabolism in cardiomyopathies. Ann Med. (2007) 39:594–607. doi: 10.1080/07853890701618305
127. Lopaschuk GD, Saddik M. The relative contribution of glucose and fatty acids to ATP production in hearts reperfused following ischemia. Mol Cell Biochem. (1992) 116:111–6. doi: 10.1007/BF01270577
128. Lopaschuk GD, Belke DD, Gamble J, Itoi T, Schonekess BO. Regulation of fatty acid oxidation in the mammalian heart in health and disease. Biochim Biophys Acta (1994) 1213:263–76. doi: 10.1016/0005-2760(94)00082-4
129. Stanley WC, Chandler MP. Energy metabolism in the normal and failing heart: potential for therapeutic interventions. Heart Fail Rev. (2002) 7:115–30. doi: 10.1023/A:1015320423577
130. Goodwin GW, Ahmad F, Doenst T, Taegtmeyer H. Energy provision from glycogen, glucose, and fatty acids on adrenergic stimulation of isolated working rat hearts. Am J Physiol. (1998) 274:H1239–47. doi: 10.1152/ajpheart.1998.274.4.H1239
131. Taegtmeyer H, Golfman L, Sharma S, Razeghi P, van Arsdall M. Linking gene expression to function: metabolic flexibility in the normal and diseased heart. Ann N Y Acad Sci. (2004) 1015:202–13. doi: 10.1196/annals.1302.017
132. Borst P, Loos JA, Christ EJ, Slater EC. Uncoupling activity of long-chain fatty acids. Biochim Biophys Acta (1962) 62:509–18. doi: 10.1016/0006-3002(62)90232-9
133. Bian F, Kasumov T, Thomas KR, Jobbins KA, David F, Minkler PE, et al. Peroxisomal and mitochondrial oxidation of fatty acids in the heart, assessed from the 13C labeling of malonyl-CoA and the acetyl moiety of citrate. J Biol Chem. (2005) 280:9265–71. doi: 10.1074/jbc.M412850200
134. Peng GS, Chen YC, Tsao TP, Wang MF, Yin SJ. Pharmacokinetic and pharmacodynamic basis for partial protection against alcoholism in Asians, heterozygous for the variant ALDH2*2 gene allele. Pharmacogenet Genomics (2007) 17:845–55. doi: 10.1097/FPC.0b013e3282609e67
135. Nuutinen H, Lindros K, Hekali P, Salaspuro M. Elevated blood acetate as indicator of fast ethanol elimination in chronic alcoholics. Alcohol (1985) 2:623–6. doi: 10.1016/0741-8329(85)90090-4
136. Pachinger O, Mao J, Fauvel JM, Bing RJ. Mitochondrial function and excitation-contraction coupling in the development of alcoholic cardiomyopathy. Recent Adv Stud Cardiac Struct Metab. (1975) 5:423–9.
137. Wu S, White R, Wikman-Coffelt J, Sievers R, Wendland M, Garrett J, et al. The preventive effect of verapamil on ethanol-induced cardiac depression: phosphorus-31 nuclear magnetic resonance and high-pressure liquid chromatographic studies of hamsters. Circulation (1987) 75:1058–64. doi: 10.1161/01.CIR.75.5.1058
138. Hu C, Ge F, Hyodo E, Arai K, Iwata S, Lobdell Ht, et al. Chronic ethanol consumption increases cardiomyocyte fatty acid uptake and decreases ventricular contractile function in C57BL/6J mice. J Mol Cell Cardiol. (2013) 59:30–40. doi: 10.1016/j.yjmcc.2013.02.005
139. Regan TJ, Khan MI, Ettinger PO, Haider B, Lyons MM, Oldewurtel HA. Myocardial function and lipid metabolism in the chronic alcoholic animal. J Clin Invest. (1974) 54:740–52. doi: 10.1172/JCI107812
140. Wisneski JA, Gertz EW, Neese RA, Gruenke LD, Morris DL, Craig JC. Metabolic fate of extracted glucose in normal human myocardium. J Clin Invest. (1985) 76:1819–27. doi: 10.1172/JCI112174
141. Gertz EW, Wisneski JA, Stanley WC, Neese RA. Myocardial substrate utilization during exercise in humans. Dual carbon-labeled carbohydrate isotope experiments. J Clin Invest. (1988) 82:2017–25. doi: 10.1172/JCI113822
142. Willebrands AF, van der Veen KJ. Influence of substrate on oxygen consumption of isolated perfused rat heart. (1967) Am J Physiol. 212:1529–35. doi: 10.1152/ajplegacy.1967.212.6.1529
143. Russell RR III, Cline GW, Guthrie PH, Goodwin GW, Shulman GI, Taegtmeyer H. Regulation of exogenous and endogenous glucose metabolism by insulin and acetoacetate in the isolated working rat heart. A three tracer study of glycolysis, glycogen metabolism, and glucose oxidation. J Clin Invest. (1997) 100:2892–9. doi: 10.1172/JCI119838
144. Goodwin GW, Cohen DM, Taegtmeyer H. [5-3H]glucose overestimates glycolytic flux in isolated working rat heart: role of the pentose phosphate pathway. Am J Physiol Endocrinol Metab. (2001) 280:E502–8. doi: 10.1152/ajpendo.2001.280.3.E502
145. Gibb AA, Lorkiewicz PK, Zheng YT, Zhang X, Bhatnagar A, Jones SP, et al. Integration of flux measurements to resolve changes in anabolic and catabolic metabolism in cardiac myocytes. Biochem J. (2017) 474:2785–801. doi: 10.1042/BCJ20170474.
146. Gertz EW, Wisneski JA, Neese R, Bristow JD, Searle GL, Hanlon JT. Myocardial lactate metabolism: evidence of lactate release during net chemical extraction in man. Circulation (1981) 63:1273–9. doi: 10.1161/01.CIR.63.6.1273
147. Lassers BW, Wahlqvist ML, Kaijser L, Carlson LA. Effect of nicotinic acid on myocardial metabolism in man at rest and during exercise. J Appl Physiol. (1972) 33:72–80. doi: 10.1152/jappl.1972.33.1.72
148. Wisneski JA, Gertz EW, Neese RA, Gruenke LD, Craig JC. Dual carbon-labeled isotope experiments using D-[6-14C] glucose and L-[1,2,3-13C3] lactate: a new approach for investigating human myocardial metabolism during ischemia. J Am Coll Cardiol. (1985) 5:1138–46. doi: 10.1016/S0735-1097(85)80016-4
149. Kaijser L, Berglund B. Myocardial lactate extraction and release at rest and during heavy exercise in healthy men. Acta Physiol Scand. (1992) 144:39–45. doi: 10.1111/j.1748-1716.1992.tb09265.x
150. Schonekess BO. Competition between lactate and fatty acids as sources of ATP in the isolated working rat heart. J Mol Cell Cardiol. (1997) 29:2725–33. doi: 10.1006/jmcc.1997.0504
151. Drake AJ, Haines JR, Noble MI. Preferential uptake of lactate by the normal myocardium in dogs. Cardiovasc Res. (1980) 14:65–72. doi: 10.1093/cvr/14.2.65
152. Bedi KC Jr, Snyder NW, Brandimarto J, Aziz M, Mesaros C, Worth AJ, et al. Evidence for intramyocardial disruption of lipid metabolism and increased myocardial ketone utilization in advanced human heart failure. Circulation (2016) 133:706–16. doi: 10.1161/CIRCULATIONAHA.115.017545
153. Aubert G, Martin OJ, Horton JL, Lai L, Vega RB, Leone TC, et al. The Failing heart relies on ketone bodies as a fuel. Circulation (2016) 133:698–705. doi: 10.1161/CIRCULATIONAHA.115.017355
154. Uchihashi M, Hoshino A, Okawa Y, Ariyoshi M, Kaimoto S, Tateishi S, et al. Cardiac-specific Bdh1 overexpression ameliorates oxidative stress and cardiac remodeling in pressure overload-induced heart failure. Circ Heart Fail. (2017) 10:e004417. doi: 10.1161/CIRCHEARTFAILURE.117.004417
155. Barnes RH, Mackay EM, Moe GK, Visscher MB. The utilization of beta-hydroxybutyric acid bo the isolated mammalian heart and lungs. Am J Physiol. (1938) 123:272–9.
156. Williamson JR, Krebs HA. Acetoacetate as fuel of respiration in the perfused rat heart. Biochem J. (1961) 80:540–7. doi: 10.1042/bj0800540
157. Hall LM. Preferential oxidation of acetoacetate by the perfused heart. Biochem Biophys Res Commun. (1961) 6:177–9. doi: 10.1016/0006-291X(61)90124-3
158. Bassenge E, Wendt VE, Schollmeyer P, Bluemchen G, Gudbjarnason S, Bing RJ. Effect of ketone bodies on cardiac metabolism. Am J Physiol. (1965) 208:162–8. doi: 10.1152/ajplegacy.1965.208.1.162
159. Little JR, Goto M, Spitzer JJ. Effect of ketones on metabolism of FFA by dog myocardium and skeletal muscle in vivo. Am J Physiol. (1970) 219:1458–63. doi: 10.1152/ajplegacy.1970.219.5.1458
160. Taegtmeyer H, Hems R, Krebs HA. Utilization of energy-providing substrates in the isolated working rat heart. Biochem J. (1980) 186:701–11. doi: 10.1042/bj1860701
161. Taegtmeyer H. On the inability of ketone bodies to serve as the only energy providing substrate for rat heart at physiological work load. Basic Res Cardiol. (1983) 78:435–50. doi: 10.1007/BF02070167
162. Russell RR III, Taegtmeyer H. Changes in citric acid cycle flux and anaplerosis antedate the functional decline in isolated rat hearts utilizing acetoacetate. J Clin Invest. (1991) 87:384–90. doi: 10.1172/JCI115008
163. Sato K, Kashiwaya Y, Keon CA, Tsuchiya N, King MT, Radda GK, et al. Insulin, ketone bodies, and mitochondrial energy transduction. FASEB J. (1995) 9:651–8. doi: 10.1096/fasebj.9.8.7768357
164. Cahill GF Jr, Veech RL. Ketoacids? Good medicine? Trans Am Clin Climatol Assoc. (2003) 114:149–161; discussion 162–143.
165. Veech RL. The therapeutic implications of ketone bodies: the effects of ketone bodies in pathological conditions: ketosis, ketogenic diet, redox states, insulin resistance, and mitochondrial metabolism. Prostaglandins Leukot Essent Fatty Acids (2004) 70:309–9. doi: 10.1016/j.plefa.2003.09.007
166. Taegtmeyer H. Failing heart and starving brain: ketone bodies to the rescue. Circulation (2016) 134:265–6. doi: 10.1161/CIRCULATIONAHA.116.022141
167. Murray AJ, Knight NS, Cole MA, Cochlin LE, Carter E, Tchabanenko K, et al. Novel ketone diet enhances physical and cognitive performance. FASEB J. (2016) 30:4021–32. doi: 10.1096/fj.201600773R
168. Evans M, Cogan KE, Egan B. Metabolism of ketone bodies during exercise and training: physiological basis for exogenous supplementation. J Physiol. (2017) 595:2857–71. doi: 10.1113/JP273185
169. Schwartz RG, Barrett EJ, Francis CK, Jacob R, Zaret BL. Regulation of myocardial amino acid balance in the conscious dog. J Clin Invest. (1985) 75:1204–11. doi: 10.1172/JCI111817
170. Verrey F. System L: heteromeric exchangers of large, neutral amino acids involved in directional transport. Pflugers Archiv Eur J Physiol. (2003) 445:529–33. doi: 10.1007/s00424-002-0973-z
171. Nicklin P, Bergman P, Zhang B, Triantafellow E, Wang H, Nyfeler B, et al. Bidirectional transport of amino acids regulates mTOR and autophagy. Cell (2009) 136:521–34. doi: 10.1016/j.cell.2008.11.044
172. Broer S, Broer A. Amino acid homeostasis and signalling in mammalian cells and organisms. Biochem J. (2017) 474:1935–63. doi: 10.1042/BCJ20160822
173. Ichihara K, Neely JR, Siehl DL, Morgan HE. Utilization of leucine by working rat heart. Am J Physiol. (1980) 239:E430–6. doi: 10.1152/ajpendo.1980.239.6.E430
174. Huang Y, Zhou M, Sun H, Wang Y. Branched-chain amino acid metabolism in heart disease: an epiphenomenon or a real culprit? Cardiovasc Res. (2011) 90:220–3. doi: 10.1093/cvr/cvr070
175. Brosnan JT, Brosnan ME. Branched-chain amino acids: enzyme and substrate regulation. J Nutr. (2006) 136:207S–11S. doi: 10.1093/jn/136.1.207S
176. Sciarretta S, Volpe M, Sadoshima J. Mammalian target of rapamycin signaling in cardiac physiology and disease. Circ Res. (2014) 114:549–64. doi: 10.1161/CIRCRESAHA.114.302022
177. Sansbury BE, DeMartino AM, Xie Z, Brooks AC, Brainard RE, Watson LJ, et al. Metabolomic analysis of pressure-overloaded and infarcted mouse hearts. Circ Heart Fail. (2014) 7:634–42. doi: 10.1161/CIRCHEARTFAILURE.114.001151
178. Wang W, Zhang F, Xia Y, Zhao S, Yan W, Wang H, et al. Defective branched chain amino acid catabolism contributes to cardiac dysfunction and remodeling following myocardial infarction. Am J Physiol Heart Circ Physiol. (2016) 311:H1160–9. doi: 10.1152/ajpheart.00114.2016
179. Sun H, Olson KC, Gao C, Prosdocimo DA, Zhou M, Wang Z, et al. Catabolic Defect of branched-chain amino acids promotes heart failure. Circulation (2016) 133:2038–49. doi: 10.1161/CIRCULATIONAHA.115.020226
180. Shao D, Villet O, Zhang Z, Choi SW, Yan J, Ritterhoff J, et al. Glucose promotes cell growth by suppressing branched-chain amino acid degradation. Nat Commun. (2018) 9:2935. doi: 10.1038/s41467-018-05362-7
181. Jackson RH, Singer TP. Inactivation of the 2-ketoglutarate and pyruvate dehydrogenase complexes of beef heart by branched chain keto acids. J Biol Chem. (1983) 258:1857–65.
182. Williamson JR, Walajtys-Rode E, Coll KE. Effects of branched chain alpha-ketoacids on the metabolism of isolated rat liver cells. I. Regulation of branched chain alpha-ketoacid metabolism. J Biol Chem. (1979) 254:11511–20.
183. Li T, Zhang Z, Kolwicz SC Jr, Abell L, Roe ND, et al. Defective branched-chain amino acid catabolism disrupts glucose metabolism and sensitizes the heart to ischemia-reperfusion injury. Cell Metab. (2017) 25:374–85. doi: 10.1016/j.cmet.2016.11.005
184. Xia Y, Wen HY, Young ME, Guthrie PH, Taegtmeyer H, Kellems RE. Mammalian target of rapamycin and protein kinase A signaling mediate the cardiac transcriptional response to glutamine. J Biol Chem. (2003) 278:13143–50. doi: 10.1074/jbc.M208500200
185. Khogali SE, Harper AA, Lyall JA, Rennie MJ. Effects of L-glutamine on post-ischaemic cardiac function: protection and rescue. J Mol Cell Cardiol. (1998) 30:819–27. doi: 10.1006/jmcc.1998.0647
186. Khogali SE, Pringle SD, Weryk BV, Rennie MJ. Is glutamine beneficial in ischemic heart disease? Nutrition (2002) 18:123–6. doi: 10.1016/S0899-9007(01)00768-7
187. Wischmeyer PE, Vanden Hoek TL, Li C, Shao Z, Ren H, Riehm J, et al. Glutamine preserves cardiomyocyte viability and enhances recovery of contractile function after ischemia-reperfusion injury. JPEN J Parenter Enteral Nutr. (2003) 27:116–22. doi: 10.1177/0148607103027002116
188. Wischmeyer PE, Jayakar D, Williams U, Singleton KD, Riehm J, Bacha EA, et al. Single dose of glutamine enhances myocardial tissue metabolism, glutathione content, and improves myocardial function after ischemia-reperfusion injury. JPEN J Parenter Enteral Nutr. (2003) 27:396–403. doi: 10.1177/0148607103027006396
189. Salabei JK, Lorkiewicz PK, Holden CR, Li Q, Hong KU, Bolli R, et al. Glutamine regulates cardiac progenitor cell metabolism and proliferation. Stem Cells (2015) 33:2613–27. doi: 10.1002/stem.2047
190. Moncada S, Higgs EA, Colombo SL. Fulfilling the metabolic requirements for cell proliferation. Biochem J. (2012) 446:1–7. doi: 10.1042/BJ20120427
191. Curi R, Lagranha CJ, Doi SQ, Sellitti DF, Procopio J, Pithon-Curi TC, et al. Molecular mechanisms of glutamine action. J Cell Physiol. (2005) 204:392–401. doi: 10.1002/jcp.20339
192. Cohen DM, Guthrie PH, Gao X, Sakai R, Taegtmeyer H. Glutamine cycling in isolated working rat heart. Am J Physiol Endocrinol Metab. (2003) 285:E1312–6. doi: 10.1152/ajpendo.00539.2002
193. Lauzier B, Vaillant F, Merlen C, Gelinas R, Bouchard B, Rivard ME, et al. Metabolic effects of glutamine on the heart: anaplerosis versus the hexosamine biosynthetic pathway. J Mol Cell Cardiol. (2013) 55:92–100. doi: 10.1016/j.yjmcc.2012.11.008
194. Liu J, Marchase RB, Chatham JC. Glutamine-induced protection of isolated rat heart from ischemia/reperfusion injury is mediated via the hexosamine biosynthesis pathway and increased protein O-GlcNAc levels. J Mol Cell Cardiol. (2007) 42:177–85. doi: 10.1016/j.yjmcc.2006.09.015
195. Laczy B, Hill BG, Wang K, Paterson AJ, White CR, Xing D, et al. Protein O-GlcNAcylation: a new signaling paradigm for the cardiovascular system. Am J Physiol Heart Circ Physiol. (2009) 296:H13–28. doi: 10.1152/ajpheart.01056.2008
196. Gibala MJ, MacLean DA, Graham TE, Saltin B. Tricarboxylic acid cycle intermediate pool size and estimated cycle flux in human muscle during exercise. Am J Physiol. (1998) 275:E235–42. doi: 10.1152/ajpendo.1998.275.2.E235
197. Kemppainen J, Fujimoto T, Kalliokoski KK, Viljanen T, Nuutila P, Knuuti J. Myocardial and skeletal muscle glucose uptake during exercise in humans. J Physiol. (2002) 542:403–12. doi: 10.1113/jphysiol.2002.018135
198. Lassers BW, Kaijser L, Wahlqvist M, Carlson LA. Effect of prolonged exercise on myocardial metabolism in man. Br Heart J. (1971) 33:609.
199. Goodwin GW, Taegtmeyer H. Improved energy homeostasis of the heart in the metabolic state of exercise. Am J Physiol Heart Circ Physiol. (2000) 279:H1490–501. doi: 10.1152/ajpheart.2000.279.4.H1490
200. Goodwin GW, Taylor CS, Taegtmeyer H. Regulation of energy metabolism of the heart during acute increase in heart work. J Biol Chem. (1998) 273:29530–9. doi: 10.1074/jbc.273.45.29530
201. Rodahl K, Miller HI, Issekutz B Jr. Plasma free fatty acids in exercise. J Appl Physiol. (1964) 19:489–92. doi: 10.1152/jappl.1964.19.3.489
202. Lassers BW, Kaijser L, Carlson LA. Myocardial lipid and carbohydrate metabolism in healthy, fasting men at rest: studies during continuous infusion of 3 H-palmitate. Eur J Clin Invest. (1972) 2:348–58. doi: 10.1111/j.1365-2362.1972.tb00661.x
203. Bergman BC, Tsvetkova T, Lowes B, Wolfel EE. Myocardial FFA metabolism during rest and atrial pacing in humans. Am J Physiol Endocrinol Metab. (2009) 296:E358–66. doi: 10.1152/ajpendo.90747.2008
204. de Groot MJ, Coumans WA, Willemsen PH, van der Vusse GJ. Substrate-induced changes in the lipid content of ischemic and reperfused myocardium. Its relation to hemodynamic recovery. Circ Res. (1993) 72:176–86. doi: 10.1161/01.RES.72.1.176
205. Strom CC, Aplin M, Ploug T, Christoffersen TE, Langfort J, Viese M, et al. Expression profiling reveals differences in metabolic gene expression between exercise-induced cardiac effects and maladaptive cardiac hypertrophy. FEBS J. (2005) 272:2684–95. doi: 10.1111/j.1742-4658.2005.04684.x
206. Bonen A, Luiken JJ, Arumugam Y, Glatz JF, Tandon NN. Acute regulation of fatty acid uptake involves the cellular redistribution of fatty acid translocase. J Biol Chem. (2000) 275:14501–8. doi: 10.1074/jbc.275.19.14501
207. Jeppesen J, Albers P, Luiken JJ, Glatz JF, Kiens B. Contractions but not AICAR increase FABPpm content in rat muscle sarcolemma. Mol Cell Biochem. (2009) 326:45–53. doi: 10.1007/s11010-008-0006-0
208. Stanley WC. Myocardial lactate metabolism during exercise. Med Sci Sports Exerc. (1991) 23:920–4. doi: 10.1249/00005768-199108000-00006
209. Bertrand ME, Carre AG, Ginestet AP, Lefebvre JM, Desplanque LA, Lekieffre JP. Maximal exercise in normal subjects: changes in coronary sinus blood flow, contractility and myocardial extraction of FFA and lactate. Eur J Cardiol. (1977) 5:481–91.
211. Coyle EF. Physical activity as a metabolic stressor. Am J Clin Nutr. (2000) 72:512S–20S. doi: 10.1093/ajcn/72.2.512S
212. Crass MF III, Shipp JC, Pieper GM. Effects of catecholamines on myocardial endogenous substrates and contractility. Am J Physiol. (1975) 228:618–27. doi: 10.1152/ajplegacy.1975.228.2.618
213. Takala TE, Ruskoaho HJ, Hassinen IE. Transmural distribution of cardiac glucose uptake in rat during physical exercise. Am J Physiol. (1983) 244:H131–7. doi: 10.1152/ajpheart.1983.244.1.H131
214. Gibb AA, Epstein PN, Uchida S, Zheng Y, McNally LA, Obal D, et al. Exercise-induced changes in glucose metabolism promote physiological cardiac growth. Circulation (2017) 136:2144–57. doi: 10.1161/CIRCULATIONAHA.117.028274
215. Brookes PS, Taegtmeyer H. Metabolism: a direct link between cardiac structure and function. Circulation (2017) 136:2158–61. doi: 10.1161/CIRCULATIONAHA.117.031372
216. Riehle C, Wende AR, Zhu Y, Oliveira KJ, Pereira RO, Jaishy BP, et al. Insulin receptor substrates are essential for the bioenergetic and hypertrophic response of the heart to exercise training. Mol Cell Biol. (2014) 34:3450–60. doi: 10.1128/MCB.00426-14
217. Hafstad AD, Boardman NT, Lund J, Hagve M, Khalid AM, Wisloff U, et al. High intensity interval training alters substrate utilization and reduces oxygen consumption in the heart. J Appl Physiol. (2011) 111:1235–41. doi: 10.1152/japplphysiol.00594.2011
218. Chatham JC, Young ME. Regulation of myocardial metabolism by the cardiomyocyte circadian clock. J Mol Cell Cardiol. (2013) 55:139–46. doi: 10.1016/j.yjmcc.2012.06.016
219. Wende AR, Young ME, Chatham J, Zhang J, Rajasekaran NS, Darley-Usmar VM. Redox biology and the interface between bioenergetics, autophagy and circadian control of metabolism. Free Radic Biol Med. (2016) 100:94–107. doi: 10.1016/j.freeradbiomed.2016.05.022
220. Steffens S, Winter C, Schloss MJ, Hidalgo A, Weber C, Soehnlein O. Circadian control of inflammatory processes in atherosclerosis and its complications. Arterioscler Thromb Vasc Biol. (2017) 37:1022–8. doi: 10.1161/ATVBAHA.117.309374
221. Seo DY, Lee S, Kim N, Ko KS, Rhee BD, Park BJ, et al. Morning and evening exercise. Integr Med Res. (2013) 2:139–44. doi: 10.1016/j.imr.2013.10.003
222. Tahara Y, Aoyama S, Shibata S. The mammalian circadian clock and its entrainment by stress and exercise. J Physiol Sci. (2017) 67:1–10. doi: 10.1007/s12576-016-0450-7
223. Coronado M, Fajardo G, Nguyen K, Zhao M, Kooiker KB, Jung G, et al. Physiologic mitochondrial fragmentation is a normal cardiac adaptation to increased energy demand. Circ Res. (2017) 122:282–95. doi: 10.1161/CIRCRESAHA.117.310725.
224. Bezzerides VJ, Platt C, Lerchenmuller C, Paruchuri K, Oh NL, Xiao C, et al. CITED4 induces physiologic hypertrophy and promotes functional recovery after ischemic injury. JCI Insight (2016) 1:e85904. doi: 10.1172/jci.insight.85904
225. Bostrom P, Mann N, Wu J, Quintero PA, Plovie ER, Panakova D, et al. C/EBPbeta controls exercise-induced cardiac growth and protects against pathological cardiac remodeling. Cell (2010) 143:1072–83. doi: 10.1016/j.cell.2010.11.036
226. Salabei JK, Hill BG. Mitochondrial fission induced by platelet-derived growth factor regulates vascular smooth muscle cell bioenergetics and cell proliferation. Redox Biol. (2013) 1:542–51. doi: 10.1016/j.redox.2013.10.011
227. Buck MD, O'Sullivan D, Klein Geltink RI, Curtis JD, Chang CH, Sanin DE, et al. Mitochondrial dynamics controls T Cell fate through metabolic programming. Cell (2016) 166:63–76. doi: 10.1016/j.cell.2016.05.035
228. Shirihai OS, Song M, Dorn GW II. How mitochondrial dynamism orchestrates mitophagy. Circ Res. (2015) 116:1835–49. doi: 10.1161/CIRCRESAHA.116.306374
229. Vasquez-Trincado C, Garcia-Carvajal I, Pennanen C, Parra V, Hill JA, Rothermel BA, et al. Mitochondrial dynamics, mitophagy and cardiovascular disease. J Physiol. (2016) 594:509–25. doi: 10.1113/JP271301
230. Hill BG, Benavides GA, Lancaster JR Jr, Ballinger S, Dell'Italia L, Jianhua Z, et al. Integration of cellular bioenergetics with mitochondrial quality control and autophagy. Biol Chem. (2012) 393:1485–512. doi: 10.1515/hsz-2012-0198
231. Ogura Y, Iemitsu M, Naito H, Kakigi R, Kakehashi C, Maeda S, et al. Single bout of running exercise changes LC3-II expression in rat cardiac muscle. Biochem Biophys Res Commun. (2011) 414:756–60. doi: 10.1016/j.bbrc.2011.09.152
232. Lee Y, Kang EB, Kwon I, Cosio-Lima L, Cavnar P, Javan GT. Cardiac Kinetophagy coincides with activation of anabolic signaling. Med Sci Sports Exerc. (2016) 48:219–26. doi: 10.1249/MSS.0000000000000774
233. Noh J, Wende AR, Olsen CD, Kim B, Bevins J, Zhu Y, et al. Phosphoinositide dependent protein kinase 1 is required for exercise-induced cardiac hypertrophy but not the associated mitochondrial adaptations. J Mol Cell Cardiol. (2015) 89:297–305. doi: 10.1016/j.yjmcc.2015.10.015
234. Noor E, Eden E, Milo R, Alon U. Central carbon metabolism as a minimal biochemical walk between precursors for biomass and energy. Mol Cell (2010) 39:809–20. doi: 10.1016/j.molcel.2010.08.031
235. Lehninger AL, Nelson DL, Cox MM. (2000) Lehninger Principles of Biochemistry. New York, NY: Worth Publishers.
236. Yamamoto T, Takano N, Ishiwata K, Ohmura M, Nagahata Y, Matsuura T, et al. Reduced methylation of PFKFB3 in cancer cells shunts glucose towards the pentose phosphate pathway. Nat Commun. (2014) 5:3480. doi: 10.1038/ncomms4480
237. Yi W, Clark PM, Mason DE, Keenan MC, Hill C, Goddard WA III, et al. Phosphofructokinase 1 glycosylation regulates cell growth and metabolism. Science (2012) 337:975–80. doi: 10.1126/science.1222278
238. Boada J, Roig T, Perez X, Gamez A, Bartrons R, Cascante M, et al. Cells overexpressing fructose-2,6-bisphosphatase showed enhanced pentose phosphate pathway flux and resistance to oxidative stress. FEBS Lett. (2000) 480:261–4. doi: 10.1016/S0014-5793(00)01950-5
239. Blackmore PF, Shuman EA. Regulation of hepatic altro heptulose 1,7-bisphosphate levels and control of flux through the pentose pathway by fructose 2,6-bisphosphate. FEBS Lett. (1982) 142:255–9. doi: 10.1016/0014-5793(82)80147-6
240. Cortassa S, Caceres V, Bell LN, O'Rourke B, Paolocci N, Aon MA. From metabolomics to fluxomics: a computational procedure to translate metabolite profiles into metabolic fluxes. Biophys J. (2015) 108:163–72. doi: 10.1016/j.bpj.2014.11.1857
241. York JW, Penney DG, Oscai LB. Effects of physical training on several glycolytic enzymes in rat heart. Biochim Biophys Acta (1975) 381:22–7. doi: 10.1016/0304-4165(75)90185-3
242. Stuewe SR, Gwirtz PA, Agarwal N, Mallet RT. Exercise training enhances glycolytic and oxidative enzymes in canine ventricular myocardium. J Mol Cell Cardiol. (2000) 32:903–13. doi: 10.1006/jmcc.2000.1131
243. Belke DD. Swim-exercised mice show a decreased level of protein O-GlcNAcylation and expression of O-GlcNAc transferase in heart. J Appl Physiol. (2011) 111:157–62. doi: 10.1152/japplphysiol.00147.2011
244. Bennett CE, Johnsen VL, Shearer J, Belke DD. Exercise training mitigates aberrant cardiac protein O-GlcNAcylation in streptozotocin-induced diabetic mice. Life Sci. (2013) 92:657–63. doi: 10.1016/j.lfs.2012.09.007
245. Medford HM, Porter K, Marsh SA. Immediate effects of a single exercise bout on protein O-GlcNAcylation and chromatin regulation of cardiac hypertrophy. Am J Physiol Heart Circ Physiol. (2013) 305:H114–23. doi: 10.1152/ajpheart.00135.2013
246. Nelson BA, Robinson KA, Koning JS, Buse MG. Effects of exercise and feeding on the hexosamine biosynthetic pathway in rat skeletal muscle. Am J Physiol. (1997) 272:E848–55. doi: 10.1152/ajpendo.1997.272.5.E848
247. Jones SP, Zachara NE, Ngoh GA, Hill BG, Teshima Y, Bhatnagar A, et al. Cardioprotection by N-acetylglucosamine linkage to cellular proteins. Circulation (2008) 117:1172–82. doi: 10.1161/CIRCULATIONAHA.107.730515
248. Watson LJ, Long BW, DeMartino AM, Brittian KR, Readnower RD, Brainard RE, et al. Cardiomyocyte Ogt is essential for postnatal viability. Am J Physiol Heart Circ Physiol. (2014) 306:H142–53. doi: 10.1152/ajpheart.00438.2013
249. Zafir A, Readnower R, Long BW, McCracken J, Aird A, Alvarez A, et al. Protein O-GlcNAcylation is a novel cytoprotective signal in cardiac stem cells. Stem Cells (2013) 31:765–75. doi: 10.1002/stem.1325
250. Kalhan SC, Hanson RW. Resurgence of serine: an often neglected but indispensable amino Acid. J Biol Chem. (2012) 287:19786–91. doi: 10.1074/jbc.R112.357194
251. McMullen JR, Shioi T, Huang WY, Zhang L, Tarnavski O, Bisping E, et al. The insulin-like growth factor 1 receptor induces physiological heart growth via the phosphoinositide 3-kinase(p110alpha) pathway. J Biol Chem. (2004) 279:4782–93. doi: 10.1074/jbc.M310405200
252. Kim J, Wende AR, Sena S, Theobald HA, Soto J, Sloan C, et al. Insulin-like growth factor I receptor signaling is required for exercise-induced cardiac hypertrophy. Mol Endocrinol. (2008) 22:2531–43. doi: 10.1210/me.2008-0265
253. Cai MX, Shi XC, Chen T, Tan ZN, Lin QQ, Du SJ, et al. Exercise training activates neuregulin 1/ErbB signaling and promotes cardiac repair in a rat myocardial infarction model. Life Sci. (2016) 149:1–9. doi: 10.1016/j.lfs.2016.02.055
254. McMullen JR, Shioi T, Zhang L, Tarnavski O, Sherwood MC, Kang PM, et al. Phosphoinositide 3-kinase(p110alpha) plays a critical role for the induction of physiological, but not pathological, cardiac hypertrophy. Proc Natl Acad Sci USA. (2003) 100:12355–60. doi: 10.1073/pnas.1934654100
255. DeBosch B, Treskov I, Lupu TS, Weinheimer C, Kovacs A, Courtois M, et al. Akt1 is required for physiological cardiac growth. Circulation (2006) 113:2097–104. doi: 10.1161/CIRCULATIONAHA.105.595231
256. Baliga RR, Pimental DR, Zhao YY, Simmons WW, Marchionni MA, Sawyer DB, et al. NRG-1-induced cardiomyocyte hypertrophy. Role of PI-3-kinase, p70(S6K), and MEK-MAPK-RSK. Am J Physiol. (1999) 277:H2026–37.
257. Zhao YY, Sawyer DR, Baliga RR, Opel DJ, Han X, Marchionni MA, et al. Neuregulins promote survival and growth of cardiac myocytes. Persistence of ErbB2 and ErbB4 expression in neonatal and adult ventricular myocytes. J Biol Chem. v273:10261–9. doi: 10.1074/jbc.273.17.10261
258. Bersell K, Arab S, Haring B, Kuhn B. Neuregulin1/ErbB4 signaling induces cardiomyocyte proliferation and repair of heart injury. Cell (2009) 138:257–70. doi: 10.1016/j.cell.2009.04.060
259. Polizzotti BD, Ganapathy B, Walsh S, Choudhury S, Ammanamanchi N, Bennett DG, et al. Neuregulin stimulation of cardiomyocyte regeneration in mice and human myocardium reveals a therapeutic window. Sci Transl Med. (2015) 7:281ra245. doi: 10.1126/scitranslmed.aaa5171
260. D'Uva G, Aharonov A, Lauriola M, Kain D, Yahalom-Ronen Y, Carvalho S, et al. ERBB2 triggers mammalian heart regeneration by promoting cardiomyocyte dedifferentiation and proliferation. Nat Cell Biol. (2015) 17:627–38. doi: 10.1038/ncb3149
261. Williamson JR. Metabolic effects of epinephrine in the isolated, perfused rat heart. I. Dissociation of the glycogenolytic from the metabolic stimulatory effect. J Biol Chem. (1964) 239:2721–9.
262. Collins-Nakai RL, Noseworthy D, Lopaschuk GD. Epinephrine increases ATP production in hearts by preferentially increasing glucose metabolism. Am J Physiol. (1994) 267:H1862–71. doi: 10.1152/ajpheart.1994.267.5.H1862
263. Pozuelo Rubio M, Peggie M, Wong BH, Morrice N, MacKintosh C. 14-3-3s regulate fructose-2,6-bisphosphate levels by binding to PKB-phosphorylated cardiac fructose-2,6-bisphosphate kinase/phosphatase. EMBO J. (2003) 22:3514–23. doi: 10.1093/emboj/cdg363
264. Ren J, Samson WK, Sowers JR. Insulin-like growth factor I as a cardiac hormone: physiological and pathophysiological implications in heart disease. J Mol Cell Cardiol. (1999) 31:2049–61. doi: 10.1006/jmcc.1999.1036
265. Pentassuglia L, Heim P, Lebboukh S, Morandi C, Xu L, Brink M. Neuregulin-1beta promotes glucose uptake via PI3K/Akt in neonatal rat cardiomyocytes. Am J Physiol Endocrinol Metab. (2016) 310:E782–94. doi: 10.1152/ajpendo.00259.2015
266. Sen S, Kundu BK, Wu HC, Hashmi SS, Guthrie P, Locke LW, et al. Glucose regulation of load-induced mTOR signaling and ER stress in mammalian heart. J Am Heart Assoc. (2013) 2:e004796. doi: 10.1161/JAHA.113.004796
267. Kundu BK, Zhong M, Sen S, Davogustto G, Keller SR, Taegtmeyer H. Remodeling of glucose metabolism precedes pressure overload-induced left ventricular hypertrophy: review of a hypothesis. Cardiology (2015) 130:211–20. doi: 10.1159/000369782
268. Roberts DJ, Tan-Sah VP, Ding EY, Smith JM, Miyamoto S. Hexokinase-II positively regulates glucose starvation-induced autophagy through TORC1 inhibition. Mol Cell (2014) 53:521–33. doi: 10.1016/j.molcel.2013.12.019
269. Hurlimann HC, Laloo B, Simon-Kayser B, Saint-Marc C, Coulpier F, Lemoine S, et al. Physiological and toxic effects of purine intermediate 5-amino-4-imidazolecarboxamide ribonucleotide (AICAR) in yeast. J Biol Chem. (2011) 286:30994–1002. doi: 10.1074/jbc.M111.262659
270. Sullivan JE, Brocklehurst KJ, Marley AE, Carey F, Carling D, Beri RK. Inhibition of lipolysis and lipogenesis in isolated rat adipocytes with AICAR, a cell-permeable activator of AMP-activated protein kinase. FEBS Lett. (1994) 353:33–6. doi: 10.1016/0014-5793(94)01006-4
271. Chance B, Williams GR. Respiratory enzymes in oxidative phosphorylation. III. The steady state. J Biol Chem. (1955) 217:409–27.
272. Chance B, Williams GR, Holmes WF, Higgins J. Respiratory enzymes in oxidative phosphorylation. V. A mechanism for oxidative phosphorylation. J Biol Chem. (1955) 217:439–51.
273. Chance B, Williams GR. Respiratory enzymes in oxidative phosphorylation. VI. The effects of adenosine diphosphate on azide-treated mitochondria. J Biol Chem. (1956) 221:477–89.
274. Heinrich R, Rapoport TA. A linear steady-state treatment of enzymatic chains. Critique of the crossover theorem and a general procedure to identify interaction sites with an effector. Eur J Biochem. (1974) 42:97–105. doi: 10.1111/j.1432-1033.1974.tb03319.x
275. Foryst-Ludwig A, Kreissl MC, Benz V, Brix S, Smeir E, Ban Z, et al. Adipose tissue lipolysis promotes exercise-induced cardiac hypertrophy involving the lipokine C16:1n7-palmitoleate. J Biol Chem. (2015) 290:23603–15. doi: 10.1074/jbc.M115.645341
276. Riquelme CA, Magida JA, Harrison BC, Wall CE, Marr TG, Secor SM, et al. Fatty acids identified in the Burmese python promote beneficial cardiac growth. Science (2011) 334:528–31. doi: 10.1126/science.1210558
277. Papadopoulos NM, Bloor CM, Standefer JC. Effects of exercise and training on plasma lipids and lipoproteins in the rat. J Appl Physiol. (1969) 26:760–3. doi: 10.1152/jappl.1969.26.6.760
278. Monleon D, Garcia-Valles R, Morales JM, Brioche T, Olaso-Gonzalez G, Lopez-Grueso R, et al. Metabolomic analysis of long-term spontaneous exercise in mice suggests increased lipolysis and altered glucose metabolism when animals are at rest. J Appl Physiol. (2014) 117:1110–9. doi: 10.1152/japplphysiol.00585.2014
279. Husted AS, Trauelsen M, Rudenko O, Hjorth SA, Schwartz TW. GPCR-mediated signaling of metabolites. Cell Metab. (2017) 25:777–96. doi: 10.1016/j.cmet.2017.03.008
Keywords: exercise, heart, glucose, mitochondria, hypertrophy, cell signaling, metabokine, cardiomyopathy
Citation: Fulghum K and Hill BG (2018) Metabolic Mechanisms of Exercise-Induced Cardiac Remodeling. Front. Cardiovasc. Med. 5:127. doi: 10.3389/fcvm.2018.00127
Received: 15 June 2018; Accepted: 23 August 2018;
Published: 11 September 2018.
Edited by:
Umberto Campia, Brigham and Women's Hospital, Harvard Medical School, United StatesReviewed by:
Adam R. Wende, University of Alabama at Birmingham, United StatesJames C. Lo, Weill Cornell Medicine, Cornell University, United States
Copyright © 2018 Fulghum and Hill. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bradford G. Hill, YnJhZGZvcmQuaGlsbEBsb3Vpc3ZpbGxlLmVkdQ==