 Jacob T. Mey
Jacob T. Mey Jacob M. Haus
Jacob M. Haus- 1Department of Pathobiology, Cleveland Clinic, Cleveland, OH, United States
- 2School of Kinesiology, University of Michigan, Ann Arbor, MI, United States
Glyoxalase-1 (GLO1) is a ubiquitously expressed cytosolic protein which plays a role in the natural maintenance of cellular health and is abundantly expressed in human skeletal muscle. A consequence of reduced GLO1 protein expression is cellular dicarbonyl stress, which is elevated in obesity, insulin resistance and type 2 diabetes (T2DM). Both in vitro and pre-clinical models suggest dicarbonyl stress per se induces insulin resistance and is prevented by GLO1 overexpression, implicating a potential role for GLO1 therapy in insulin resistance and type 2 diabetes (T2DM). Recent work has identified the therapeutic potential of novel natural agents as a GLO1 inducer, which resulted in improved whole-body metabolism in obese adults. Given skeletal muscle is a major contributor to whole-body glucose, lipid, and protein metabolism, such GLO1 inducers may act, in part, through mechanisms in skeletal muscle. Currently, investigations examining the specificity of dicarbonyl stress and GLO1 biology in human skeletal muscle are lacking. Recent work from our lab indicates that dysregulation of GLO1 in skeletal muscle may underlie human insulin resistance and that exercise training may impart therapeutic benefits. This minireview will summarize the existing human literature examining skeletal muscle GLO1 and highlight the emerging therapeutic concepts for GLO1 gain-of-function in conditions such as insulin resistance and cardiometabolic disease.
Introduction
Despite decades of coordinated efforts, T2DM remains a serious public health issue. In the United States alone, over 30 million adults are suffering from T2DM (1) and ~86 million are pre-diabetic and without intervention, may develop overt T2DM as well (2). T2DM is preceded by progressive skeletal muscle insulin resistance (3). Although the etiology and pathology of skeletal muscle insulin resistance and progression to T2DM remain multifactorial, emerging research implicates skeletal muscle dicarbonyl stress as a causative factor. Dicarbonyl stress is described as abnormally high concentrations of methylglyoxal (MG; a highly reactive dicarbonyl) which is elevated in diabetes (4). Methylglyoxal (MG) is a potent intracellular glycating agent that forms advanced glycation endproducts (AGEs). Formed spontaneously from 3-carbon glycolytic intermediates, MG rapidly glycates proteins and nucleotides, damage mitochondria and directly increase reactive oxygen species (ROS) production; thus, inducing a pro-oxidant state and senescent-like condition (4). Drug therapies developed to combat dicarbonyl stress that function as “MG scavengers” have had little clinical success; alternate therapies are being developed to utilize the natural defense against dicarbonyl stress, the glyoxalase enzymatic defense system.
The glyoxalase enzymatic defense system prevents dicarbonyl stress and is controlled by the protein expression and enzymatic activity of glyoxalase 1 (GLO1). GLO1 is a ubiquitously expressed enzyme that directly detoxifies MG, thus, mitigating dicarbonyl stress. However, under pathological conditions like insulin resistance and T2DM, GLO1 is reduced while MG generation is increased, creating an environment in which dicarbonyl stress persists. These phenomenon are well known to contribute to complications associated with diabetes such as nephropathy (5), neuropathy (6), and retinopathy (7). To date, much of the MG and GLO1 research has been directed at insulin-independent tissues (kidney, nervous, retina, respectively) whereas these tissues do not require insulin action for glucose disposal through facilitated glucose uptake via GLUT4 vesicles. However, recent evidence suggests the dysregulation of the MG-GLO1 axis extends to the highly metabolic skeletal muscle tissue and may play a causative role in the development of insulin resistance and overt T2DM.
We have recently published human data describing a dysregulation of dicarbonyl stress concomitant with reduced GLO1 protein expression in skeletal muscle of individuals with T2DM (8). Further, in vitro, pre-clinical evidence shows GLO1 overexpression protects against insulin resistance and T2DM. Clinically, two promising human therapies have arisen: nutraceutical GLO1 inducer therapy and aerobic exercise training. This minireview will summarize the physiologic impact of dicarbonyl stress and GLO1 in skeletal muscle metabolism and highlight the emerging therapeutic concepts for combating dicarbonyl stress.
Importance of Skeletal Muscle
Skeletal muscles are one of the most metabolically important tissues in the body. Striated muscle tissue accounts for >33% total body mass (9), >40% total body protein (10) and plays a major role in glucose disposal, accounting for >80% of insulin stimulated glucose uptake (11). Skeletal muscle insulin resistance is defined as reduced glucose uptake in response to insulin and is implicated in the pathogenesis of many diseases, most prominently T2DM.
Although multiple etiologies of T2DM exist, including ß-cell dysfunction, dysregulated fatty acid metabolism, and hepatic insulin resistance (12), the primary defect remains skeletal muscle insulin resistance (13–15), often as a consequence of a sedentary lifestyle combined with excessive kilocalorie consumption (16). Hyperinsulinemic-euglycemic clamp data has shown that individuals with T2DM have a >50% reduction in insulin stimulated glucose disposal compared to healthy individuals (17), primarily due to reductions in skeletal muscle glucose uptake.
Despite reductions in glucose disposal, even insulin-resistant skeletal muscle is exposed to a significant amount of glucose flux. For example, glucose disposal rates of insulin-resistant individuals measured during the hyperinsulinemic-euglycemic clamp, although reduced compared to insulin-sensitive individuals, remain at ~2–3 mg of glucose/kg body weight/minute (18). Improper handling of this glucose flux can have deleterious effects (19) which occur prior to the onset of overt T2DM (20); for example, the generation of methylglyoxal (MG) and dicarbonyl stress.
MG and Dicarbonyl Stress in Skeletal Muscle and Insulin Resistance
Methylglyoxal is a 3-carbon, highly reactive α-oxoaldehyde, or “dicarbonyl” that modifies proteins via covalent bonding. MG-modification may induce protein changes including, loss of side chain charge, alter structure and function (21, 22), signal proteolytic degradation (23) or AGE formation (24). MG-modifications are commonly directed at arginine residues and form methylglyoxal-derived hydroimidazolone adducts (MG-H1) (25). MG-H1 adducts are particularly deleterious due to the high probability of arginine residues to be located at functional sites of proteins (26). Proteins with altered function after MG-modification are known as the dicarbonyl proteome, which is an area of active research investigation. MG and MG-H1 are elevated in plasma and tissues of individuals with T2DM and are well-known to contribute to diabetic complications such as diabetic nephropathy (5), neuropathy (6), retinopathy (7), and early cardiovascular disease (27–29).
The majority of MG formation is spontaneously produced in vivo during glycolysis from the 3-carbon glycolytic intermediates, dihydroxy-acetone-phosphate (DHAP) and glyceraldehyde-3-phosphate (G3P). Spontaneous MG formation from DHAP and G3P occurs at a constant rate of ~0.05–0.1% of glycolytic flux (30). In T2DM, a dysregulation of glycolysis leads to a buildup of 3-carbon intermediates and subsequent increased generation of MG (28). Alternatively, MG can form from lipid peroxidation mechanisms, known to be exacerbated with oxidative stress, a common characteristic of insulin resistance and T2DM (13). MG induces pathways known to contribute to insulin resistance including: (1) oxidative stress caused by damage to mitochondria (31) and mitochondrial DNA (32), (2) generation of AGEs [for more information on the independent effects of AGEs in diabetes and diabetic complications, the reader is directed to a recent review by Brings et al. (33)] and (3) inflammation mediated through the Receptor for Advanced Glycation Endproducts (RAGE) signaling (34–36). RAGE is highly expressed in skeletal muscle (37, 38) and upon binding of a RAGE-ligands (i.e., MG-H1) a well characterized inflammatory signaling cascade ensues (39–41). Further, in vitro experiments in L6 myotubes treated with MG or MG-modified proteins, inhibit insulin-stimulated glucose uptake via impaired phosphorylation of phosphatidylinositol-4,5-bisphosphate 3-kinase (P13K) and extracellular-signal-regulated kinase 1 and 2 (ERK1/2) (42–44). This induction of insulin resistance was independent of MG-generated oxidative stress and likely due to direct binding of MG to the important insulin signaling protein, IRS-1.
The Glyoxalase Enzymatic Defense System
The biological natural defense against dicarbonyl stress is the glyoxalase enzymatic defense system which detoxifies MG by converting it to a stable byproduct (D-Lactate). This enzymatic system is controlled by the protein expression and enzymatic activity of GLO1. GLO1 is ubiquitously expressed in all cells and catalyzes the first step in the glutathione-dependent sequestration of MG at an astounding (>99%) efficiency (45). For further information on the glyoxalase detoxification mechanism, we refer the reader to literature and reviews by Thornalley et al. (46–49). GLO1 is essential to protecting cells from dicarbonyl stress and is highly expressed in skeletal muscle (50–55).
Human, animal and in vitro studies describe GLO1 as the primary regulator of dicarbonyl stress in tissues (48), including skeletal muscle (56). The importance of GLO1 in preventing dicarbonyl stress in human physiology is evidenced by its ubiquitously expressed nature, as it is present in the cytosol of all cells, and its abundance, remaining in the top 13% of human proteins with a concentration of ~0.2 μg of GLO1 per 1 milligram of human tissue (57). Additionally, a rare mutation in human GLO1 gene that produces a non-functional GLO1 protein is embryonically lethal due to an inability to prevent dicarbonyl stress (58). Gene deletion of GLO1 is also embryonically lethal in mice (59) whereas in Drosophila, GLO1 knockout animals displayed increased MG and recapitulated the progression of T2DM with obesity, insulin resistance and hyperglycemia (60). Mechanistic evidence from in vitro studies using the GLO1 inhibitor, Staltil, resulted in loss of GLO1 function and MG accumulation in a variety of tissues including skeletal muscle (61, 62). Further, silencing of GLO1 also increases MG and contributes to the development of multiple disease conditions (63).
In metabolically healthy muscle cells, transient increases in MG stimulate GLO1 protein expression resulting in efficient detoxification of MG despite increased MG flux (64). However, in metabolically compromised muscle (as seen in insulin resistance and T2DM), reduced efficiency of the glyoxalase system leads to MG accumulation (8, 61, 62). This increase in intracellular MG results in inhibition of insulin signaling (42–44, 65), damage to mitochondria (31) and mitochondrial DNA (32) increased ROS production (35), inflammation related to the accumulation of MG-H1 (34) and structural changes to skeletal muscle proteins (56). The causes of reduced GLO1 in skeletal muscle is largely unknown however, we recently described concomitant reductions in GLO1 and NRF2 protein expression, the transcriptional regulator of GLO1 in T2DM subjects (8). NRF2 is highly sensitive to REDOX perturbations and hypoxia suggesting that adequate tissue blood flow may also play a role in GLO1 biology. Recent murine data has also provided evidence for reduced GLO1 protein expression with diet induced obesity (66).
In contrast to the theory that MG is causative to skeletal muscle insulin resistance, Gawlowski et al. (67) recorded increased glucose uptake in L6 myoblasts with siRNA GLO1 knockdown and reported altered GLUT4 trafficking mediated by MG accumulation. This discrepancy is likely due, in part, to MG-mediated oxidative stress. MG is known to increase mitochondrial reactive oxygen species (ROS) generation, which increases glucose uptake and stimulates GLUT4 translocation in muscle cells (68, 69). The increased glucose uptake observed by Gawlowski et al. may be an artifact of mitochondrial ROS rather than from the MG accumulation per se, which would agree with the findings of others (42–44). Future studies should continue to differentiate the effects of MG-mediated ROS production and independent effects of MG.
Although GLO1 is highly expressed in skeletal muscle tissue in humans (50–55), information on GLO1 protein expression and activity as it pertains to insulin resistance and T2DM is lacking. To date, there are limited investigations in the area of GLO1 and skeletal muscle. Table 1 highlights these studies and their major findings. In muscular dystrophy [a disease characterized by skeletal muscle insulin resistance (70)] GLO1 protein was reduced. Similarly, observations from our lab show GLO1 protein expression and enzyme activity is reduced in obese insulin resistant individuals and GLO1 activity was strongly correlated with insulin sensitivity, and VO2max but inversely correlated with chronological age and percentage body fat (73). Moreover, we recently reported marked reduction in skeletal muscle GLO1 protein expression in individuals with T2DM compared to lean healthy control subjects (8). Of note, shotgun proteomic approaches have yielded conflicting reports with regard to GLO1 (53–55, 72). Despite the current state of the literature, we cannot differentiate independent effects of obesity, insulin resistance or clinical T2DM on GLO1 protein expression or enzymatic activity; nor can it be determined if reductions in GLO1 are causative or a consequence of metabolic pathologies. Future studies should utilize clinical-translational approaches to better elucidate the role of skeletal muscle GLO1 protein expression and enzymatic activity in metabolic pathologies.
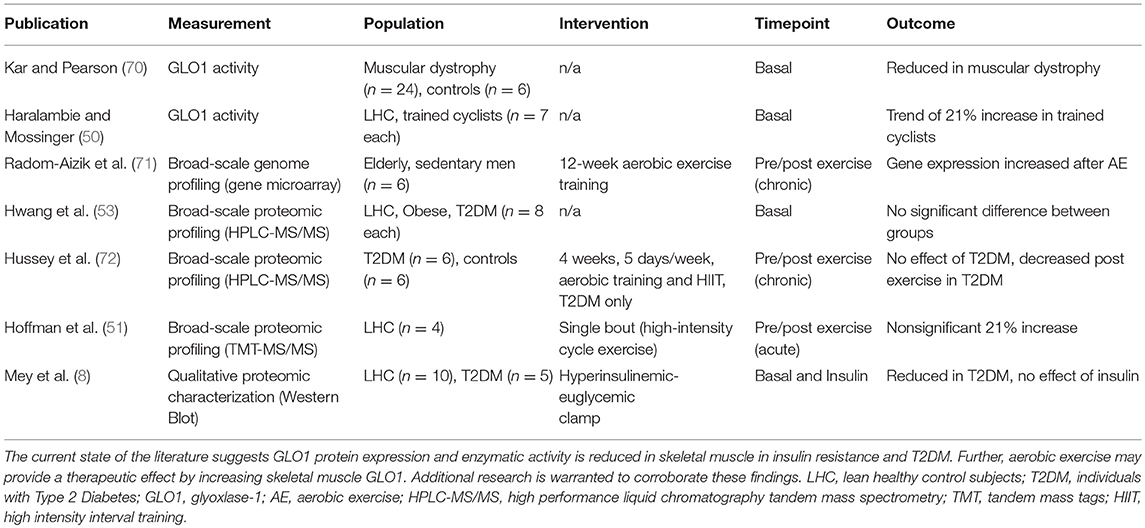
Table 1. Studies Investigating Glo1 In Human Skeletal Muscle.
Collectively, increasing GLO1 expression may play a protective role when MG generation becomes exacerbated. The protective role of GLO1 to attenuate dicarbonyl stress has potential in maintaining metabolic homeostasis in skeletal muscle. Further clarification of GLO1's role in metabolically compromised cells and elucidation of its regulatory proteins and their stimuli may lead to better avenues for T2DM treatment and prevention. A schematic summary of skeletal muscle MGGLO1 physiology in health and disease is presented in Figure 1.
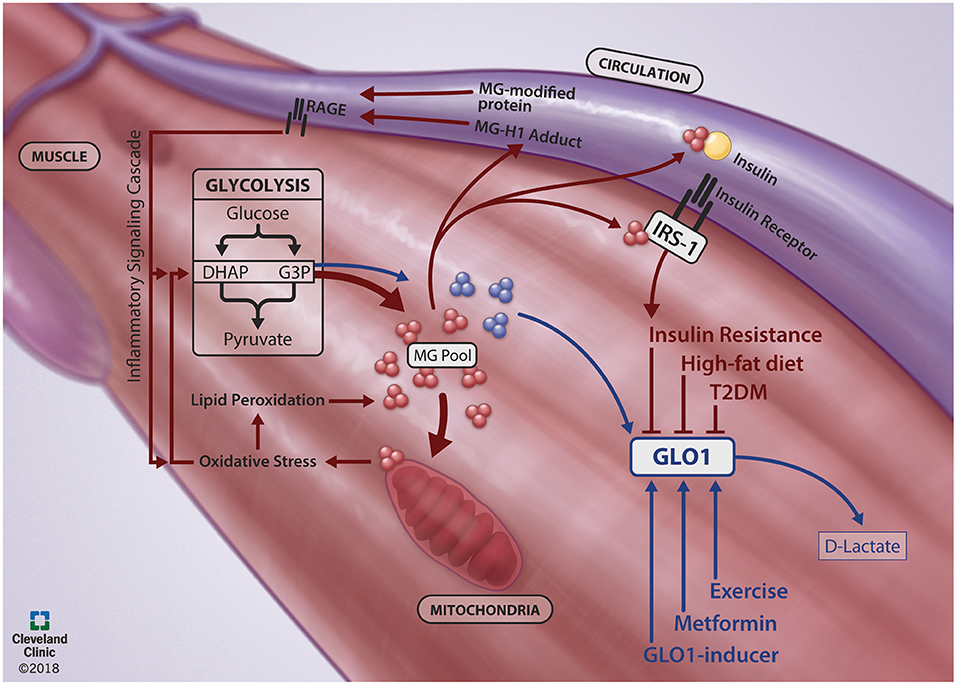
Figure 1. Skeletal muscle MG-GLO1 physiology in the context of health and disease. During healthy physiology (Blue Lines), methyglyoxal (MG) is generated in the skeletal muscle as a spontaneous byproduct from the 3-carbon intermediates (dihydroxyacetone phosphate, DHAP; glyceraldehyde-3-phosphate, G3P) of glycolysis. This MG is efficiently detoxified by glyoxalase-1 (GLO1) to D-lactate. However, GLO1 protein expression is reduced in metabolically impaired states, such as insulin resistance, type 2 diabetes mellitus (T2DM) and during high-fat diet feeding. In concert, MG is generated from glycolysis at an increased rate (Red Lines) and contributes to poor skeletal muscle metabolic health through multiple pathways. Excessive MG modifies mitochondria, contributing to oxidative stress, lipid peroxidation and further generation of MG. Excessive MG also directly inhibits insulin signaling by binding to IRS-1 within the skeletal muscle and by binding to circulating insulin. Further, excessive MG can bind to proteins, presenting in circulation as MG-H1 adducts or MG-modified proteins, which activate the RAGE (receptor for advanced glycation endproducts)-mediated inflammatory signaling cascade. Together, this creates a viscous cycle of MG generation. A potential route to combat dicarbonyl stress is through increasing GLO1 protein expression with targeted therapeutic strategies, such as exercise, metformin and GLO1-inducers. We thank Brandon Stelter and the Center for Medical Art and Photography at the Cleveland Clinic for generating the figure.
Therapeutic Strategies for the Prevention of Dicarbonyl Stress
Scavenging MG and MG-Modified Proteins
Drugs of the guanidine family are known to function as MG scavengers and bind to the highly reactive MG, preventing MG-induced protein modifications, like MG-H1, in plasma and tissues. The pharmacologic agent, Aminoguanidine, showed much promise, improving laboratory measures predictive of diabetic complications (74). However, lack of clinical outcomes (75) and serious side effects (76) limit its therapeutic application in diabetes. Metformin is the most commonly prescribed medication for T2DM and functions as an MG-scavenger due to its biguanidine structure. Conceptually, MG scavengers may be flawed for applications in skeletal muscle, as MG scavengers require an increase in stoichiometric proportion to MG production; this would require supraphysiologic concentrations of MG-scavengers to scale with the high glucose flux and MG generation within skeletal muscle.
GLO1 Inducer Therapy—Nutraceutical
GLO1 inducer therapy increases GLO1 protein expression by stimulating Nuclear factor-erythroid 2 p45 subunit-related factor 2 (NRF2). NRF2 is a transcription factor that promotes basal and inducible expression of GLO1 (77, 78) and is implicated in T2DM in both animals and humans (79–81). Recent human trials have shown the effectiveness of trans-resveratrol and hesperetin (tRES/HESP) to increase GLO1 protein expression and activity concomitant with reductions in plasma MG and MG-directed protein modifications (82) via NRF2 signaling (83). This recent work has achieved effective pharmacologic targeting of the NRF2-GLO1-MG axis in human plasma concomitant with clinically relevant improvements in whole body glucose metabolism. This profound effect on glucose control elicited by tRES/HESP therapy are suggestive of effects on highly metabolic tissues, like the liver and skeletal muscle. Future research should investigate the liver and skeletal muscle effects of tRES/HESP therapy on the tissue specific NRF2-GLO1-MG axis. For information on other GLO1 inducer formulations, the reader is directed to a recent review by Rabbani and Thornalley (84).
GLO1 Inducer Therapy—Aerobic Exercise
The relationship between physical activity and T2DM is well known. A sedentary lifestyle contributes to insulin resistance, while increased physical activity improves insulin sensitivity and whole-body glucose and lipid metabolism (85, 86). Chronic aerobic exercise has been shown to have robust benefits on insulin sensitivity, glucose disposal, glycogen content, fatty acid oxidation and metabolic flexibility (87–102). Both acute (103, 104) and chronic aerobic exercise (92, 99, 100) provide these benefits which are driven by adaptations in skeletal muscle (105, 106). Furthermore, individuals at risk for developing T2DM (obese, insulin-resistant individuals) can reduce the risk of progression to T2DM by over 50% with a lifestyle intervention that focuses on reducing calorie intake and increasing exercise (2). At this time, the signaling mechanisms that underline these metabolic improvements are not well understood but may involve regulation of GLO1 such as exercise-induced stimuli increase NRF2 (64), including oxidative stress (107) and AMPK signaling (108). It is reasonable to infer that NRF2 directs upregulation of skeletal muscle GLO1 gene in response to these or other exercise-mediated skeletal muscle stimuli.
Microarray studies with exercise have reported mixed results where one study found no differences in GLO1 when comparing three groups of young healthy men; untrained, aerobically trained and resistance trained (109). Yet another gene microarray showed skeletal muscle GLO1 mRNA increases with chronic aerobic exercise training in older men (71). Given the known relationship between increasing chronologic age and reductions in GLO1, it is reasonable to assume these older men had reduced GLO1 mRNA, however, there was no comparative lean healthy control group in this study. Together, this implicates a potential ceiling effect for aerobic exercise to increase GLO1 gene expression. Similar results were found measuring GLO1 enzymatic activity with exercise training. Haralambie et al. (50) performed a cross-sectional investigation of GLO1 activity in untrained and trained individuals, showing a trend for a 21% increase in GLO1 activity in the trained cohort. Importantly, the untrained cohort was young, and fit (VO2max: c.a. 46 ml/kg/min), and may have had optimal levels of GLO1, again providing evidence of a ceiling for GLO1.
Few studies in human skeletal muscle have investigated the effect of exercise on GLO1 protein expression (8, 51, 72, 82). Hoffman et al. showed acute exercise produces a non-significant 21% increase in GLO1 protein expression using large-scale proteomic profiling (51). Using a similar approach, Hussey et al. showed reduced GLO1 protein expression after 4 weeks of exercise training [3 days per week of aerobic training and 2 days per week of high-intensity interval training (HIIT)] in individuals with T2DM (72). The effect of long-term interventions or different training modalities (continuous aerobic vs. HIIT) is still under investigation. Additional research is needed to corroborate these findings that aerobic exercise confers a therapeutic benefit in individuals with reduced skeletal muscle GLO1 gene expression, protein expression, or enzymatic activity.
Other Considerations for Future Research
• Skeletal muscle GLO1 protein or gene expression data alone may not offer a complete picture without concomitant measures of GLO1 activity and dicarbonyl stress (i.e., MG-modified proteins and AGEs).
• Many MG-modifications on protein may be inert. Understanding which specific proteins acquire altered function and compose the skeletal muscle dicarbonyl proteome will advance the field greatly.
• Skeletal muscle GLO1 is reduced in mice fed a high-fat diet (66), but whether diet or obesity has independent effects on skeletal muscle GLO1 in humans has yet to be determined.
• Much is still unknown about GLO1 regulation. For example, GLO1 can undergo four unique post-translational modifications that inhibit GLO1 activity (110). Future research should aim to elucidate the independent importance of these modifications.
• With regard to exercise, dose response and time course studies in all modes of exercise (aerobic, resistance, interval, etc.) remain to be empirically tested.
Summary
Elevated dicarbonyl stress is characteristic of T2DM and associated with the development of diabetic complications. Recent research suggests dicarbonyl stress in skeletal muscle may play a causative role the development of insulin resistance and the onset of T2DM. The primary cellular defense against dicarbonyl stress is the glyoxalase enzymatic defense systems controlled by the protein expression and enzymatic activity of GLO1. Novel therapies targeted at inducing GLO1 improve whole body glucose control, indicative of effects in skeletal muscle, and may represent the next line of adjuvant therapy for T2DM. We have recently described aberrant dicarbonyl stress and GLO1 protein expression in the skeletal muscle of individuals with T2DM. Further, we postulate that aerobic exercise training will result in GLO1 gain-of-function through NRF2 mediated pathways. Additional research is warranted to determine the physiologic impact of targeting skeletal muscle GLO1 and dicarbonyl stress for the prevention and treatment of skeletal muscle insulin resistance and T2DM.
Author Contributions
JM conceptualized and wrote this work. JH conceptualized, wrote and edited this work, and provided funding for trainees. JH provided approval for publication of the content and agrees to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Funding
This work was supported by the American Diabetes Association Grant 1-14-JF-32 (to JH), National Institutes of Health Grants R01-DK-109948 and UL1-RR-029879 (to JH), and Sigma Xi Research Society Grant-in-Aid (to JM).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Solomon TP, Sistrun SN, Krishnan RK, Del Aguila LF, Marchetti CM, O'Carroll SM, et al. Exercise and diet enhance fat oxidation and reduce insulin resistance in older obese adults. J Appl Physiol. (2008)104:1313–9. doi: 10.1152/japplphysiol.00890.2007
2. McCarthy M. Nearly one in 10 US residents has diabetes, CDC reports. BMJ (2014) 348:g3962. doi: 10.1136/bmj.g3962
3. Petersen KF, Oral EA, Dufour S, Befroy D, Ariyan C, Yu C, et al. Leptin reverses insulin resistance and hepatic steatosis in patients with severe lipodystrophy. J Clin Invest. (2002)109:1345–50. doi: 10.1172/JCI0215001
4. Rabbani N, Thornalley PJ. Dicarbonyl stress in cell and tissue dysfunction contributing to ageing and disease. Biochem Biophys Res Commun. (2015)458:221–6. doi: 10.1016/j.bbrc.2015.01.140
5. Inagi R. RAGE and glyoxalase in kidney disease. Glycoconj J. (2016)33:619–26. doi: 10.1007/s10719-016-9689-8
6. Skapare E, Konrade I, Liepinsh E, Strele I, Makrecka M, Bierhaus A, et al. Association of reduced glyoxalase 1 activity and painful peripheral diabetic neuropathy in type 1 and 2 diabetes mellitus patients. J Diabetes Complications (2013) 27:262–7. doi: 10.1016/j.jdiacomp.2012.12.002
7. Berner AK, Brouwers O, Pringle R, Klaassen I, Colhoun L, McVicar C, et al. Protection against methylglyoxal-derived AGEs by regulation of glyoxalase 1 prevents retinal neuroglial and vasodegenerative pathology. Diabetologia (2012)55:845–54. doi: 10.1007/s00125-011-2393-0
8. Mey JT, Blackburn BK, Miranda ER, Chaves AB, Briller J, Bonini MG, et al. Dicarbonyl stress and glyoxalase enzymatic system regulation in human skeletal muscle. Am J Physiol Regul Integr Comp Physiol. (2017) 314:R181–90. doi: 10.1152/ajpregu.00159.2017
9. Kim J, Wang Z, Heymsfield SB, Baumgartner RN, Gallagher D. Total-body skeletal muscle mass: estimation by a new dual-energy X-ray absorptiometry method. Am J Clin Nutr. (2002)76:378–83. doi: 10.1093/ajcn/76.2.378
10. Lentner CC, Lentner C, Wink A editors. Geigy Scientific Tables. 8th, Rev. and Enl. Edn. West Caldwell, NJ: CIBA-Geigy (1981). p. c1981-4.
11. DeFronzo RA, Jacot E, Jequier E, Maeder E, Wahren J, Felber JP. The effect of insulin on the disposal of intravenous glucose : results from indirect calorimetry and hepatic and femoral venous catheterization. Diabetes (1981) 30:1000–7. doi: 10.2337/diab.30.12.1000
12. Stinkens R, Goossens GH, Jocken JW, Blaak EE. Targeting fatty acid metabolism to improve glucose metabolism. Obes Rev. (2015) 16:715–57. doi: 10.1111/obr.12298
13. Defronzo RA. Banting lecture. From the triumvirate to the ominous octet: a new paradigm for the treatment of type 2 diabetes mellitus. Diabetes (2009) 58:773–95. doi: 10.2337/db09-9028
14. DeFronzo RA. Glucose intolerance and aging: evidence for tissue insensitivity to insulin. Diabetes (1979) 28:1095–101. doi: 10.2337/diab.28.12.1095
15. DeFronzo RA, Tobin JD, Andres R. Glucose clamp technique: a method for quantifying insulin secretion and resistance. Am J Physiol. (1979) 237:E214–23. doi: 10.1152/ajpendo.1979.237.3.E214
16. DeFronzo RA. Pathogenesis of type 2 diabetes mellitus. Med Clin N Am. (2004) 88:787–835. doi: 10.1016/j.mcna.2004.04.013
17. Chavez AO, Kamath S, Jani R, Sharma LK, Monroy A, Abdul-Ghani MA, et al. Effect of short-term free fatty acids elevation on mitochondrial function in skeletal muscle of healthy individuals. J Clin Endocrinol Metab. (2010) 95:422–9. doi: 10.1210/jc.2009-1387
18. Williamson DL, Dungan CM, Mahmoud AM, Mey JT, Blackburn BK, Haus JM. Aberrant REDD1-mTORC1 responses to insulin in skeletal muscle from Type 2 diabetics. Am J Physiol Regul Integr Comp Physiol. (2015) 309:R855–63. doi: 10.1152/ajpregu.00285.2015
19. Kahn CR. Banting lecture. Insulin action, diabetogenes, and the cause of type II diabetes. Diabetes (1994) 43:1066–84. doi: 10.2337/diab.43.8.1066
20. DeFronzo RA. Lilly lecture 1987. The triumvirate: beta-cell, muscle, liver. A collusion responsible for NIDDM. Diabetes (1988) 37:667–87. doi: 10.2337/diab.37.6.667
21. DeGroot J, Verzijl N, Jacobs KM, Budde M, Bank RA, Bijlsma JW, et al. Accumulation of advanced glycation endproducts reduces chondrocyte-mediated extracellular matrix turnover in human articular cartilage. Osteoarthritis Cartilage (2001) 9:720–6. doi: 10.1053/joca.2001.0469
22. Ahmed N, Babaei-Jadidi R, Howell SK, Thornalley PJ, Beisswenger PJ. Glycated and oxidized protein degradation products are indicators of fasting and postprandial hyperglycemia in diabetes. Diabetes Care (2005) 28:2465–71. doi: 10.2337/diacare.28.10.2465
23. Vlassara H, Brownlee M, Cerami A. High-affinity-receptor-mediated uptake and degradation of glucose-modified proteins: a potential mechanism for the removal of senescent macromolecules. Proc Natl Acad Sci USA. (1985) 82:5588–92. doi: 10.1073/pnas.82.17.5588
24. Vlassara H, Striker GE. Advanced glycation endproducts in diabetes and diabetic complications. Endocrinol Metab Clin North Am. (2013) 42:697–719. doi: 10.1016/j.ecl.2013.07.005
25. Thornalley PJ. Measurement of protein glycation, glycated peptides, and glycation free adducts. Perit Dial Int. (2005) 25:522–33.
26. Gallet X, Charloteaux B, Thomas A, Brasseur R. A fast method to predict protein interaction sites from sequences. J Mol Biol. (2000) 302:917–26. doi: 10.1006/jmbi.2000.4092
27. Wang H, Liu J, Wu L. Methylglyoxal-induced mitochondrial dysfunction in vascular smooth muscle cells. Biochem. Pharmacol. (2009) 77:1709–16. doi: 10.1016/j.bcp.2009.02.024
28. Wang H, Meng QH, Gordon JR, Khandwala H, Wu L. Proinflammatory and proapoptotic effects of methylglyoxal on neutrophils from patients with type 2 diabetes mellitus. Clin. Biochem. (2007) 40:1232–9. doi: 10.1016/j.clinbiochem.2007.07.016
29. Wang X, Chang T, Jiang B, Desai K, Wu L. Attenuation of hypertension development by aminoguanidine in spontaneously hypertensive rats: role of methylglyoxal. Am J Hypertens. (2007) 20:629–36. doi: 10.1016/j.amjhyper.2006.12.003
30. Ellis KJ. Human body composition: in vivo methods. Physiol Rev. (2000) 80:649–80. doi: 10.1152/physrev.2000.80.2.649
31. Amicarelli F, Ragnelli AM, Aimola P, Bonfigli A, Colafarina S, Di Ilio C, et al. Age-dependent ultrastructural alterations and biochemical response of rat skeletal muscle after hypoxic or hyperoxic treatments. Biochim Biophys Acta (1999) 1453:105–14. doi: 10.1016/S0925-4439(98)00088-X
32. Blake R, Trounce IA. Mitochondrial dysfunction and complications associated with diabetes. Biochim Biophys Acta (2014) 1840:1404–12. doi: 10.1016/j.bbagen.2013.11.007
33. Brings S, Fleming T, Freichel M, Muckenthaler MU, Herzig S, Nawroth PP. Dicarbonyls and advanced glycation end-products in the development of diabetic complications and targets for intervention. Int J Mol Sci. (2017) 18:984. doi: 10.3390/ijms18050984
34. Brouwers O, de Vos-Houben JM, Niessen PM, Miyata T, van Nieuwenhoven F, Janssen BJ, et al. Mild oxidative damage in the diabetic rat heart is attenuated by glyoxalase-1 overexpression. Int J Mol Sci. (2013) 14:15724–39. doi: 10.3390/ijms140815724
35. Brouwers O, Niessen PM, Ferreira I, Miyata T, Scheffer PG, Teerlink T, et al. Overexpression of glyoxalase-I reduces hyperglycemia-induced levels of advanced glycation end products and oxidative stress in diabetic rats. J Biol Chem. (2011) 286:1374–80. doi: 10.1074/jbc.M110.144097
36. Brouwers O, Niessen PM, Haenen G, Miyata T, Brownlee M, Stehouwer CD, et al. Hyperglycaemia-induced impairment of endothelium-dependent vasorelaxation in rat mesenteric arteries is mediated by intracellular methylglyoxal levels in a pathway dependent on oxidative stress. Diabetologia (2010) 53:989–1000. doi: 10.1007/s00125-010-1677-0
37. Brett J, Schmidt AM, Yan SD, Zou YS, Weidman E, Pinsky D, et al. Survey of the distribution of a newly characterized receptor for advanced glycation end products in tissues. Am J Pathol. (1993) 143:1699–712.
38. Lopez-Diez R, Shen X, Daffu G, Khursheed M, Hu J, Song F, et al. Ager deletion enhances ischemic muscle inflammation, angiogenesis, and blood flow recovery in diabetic mice. Arterioscler Thromb Vasc Biol. (2017) 37:1536–47. doi: 10.1161/ATVBAHA.117.309714
39. Cassese A, Esposito I, Fiory F, Barbagallo AP, Paturzo F, Mirra P, et al. In skeletal muscle advanced glycation end products (AGEs) inhibit insulin action and induce the formation of multimolecular complexes including the receptor for AGEs. J Biol Chem. (2008) 283:36088–99. doi: 10.1074/jbc.M801698200
40. Mahmoud AM, Szczurek MR, Blackburn BK, Mey JT, Chen Z, Robinson AT, et al. Hyperinsulinemia augments endothelin-1 protein expression and impairs vasodilation of human skeletal muscle arterioles. Physiol Rep. (2016) 4:e12895. doi: 10.14814/phy2.12895
41. Mahmoud AM, Brown MD, Phillips SA, Haus JM. Skeletal muscle vascular function: a counterbalance of insulin action. Microcirculation (2015) 22:327–47. doi: 10.1111/micc.12205
42. Miele C, Riboulet A, Maitan MA, Oriente F, Romano C, Formisano P, et al. Human glycated albumin affects glucose metabolism in L6 skeletal muscle cells by impairing insulin-induced insulin receptor substrate (IRS) signaling through a protein kinase C alpha-mediated mechanism. J Biol Chem. (2003) 278:47376–87. doi: 10.1074/jbc.M301088200
43. Riboulet-Chavey A, Pierron A, Durand I, Murdaca J, Giudicelli J, Van Obberghen E. Methylglyoxal impairs the insulin signaling pathways independently of the formation of intracellular reactive oxygen species. Diabetes (2006) 55:1289–99. doi: 10.2337/db05-0857
44. Pirola L, Bonnafous S, Johnston AM, Chaussade C, Portis F, Van Obberghen E. Phosphoinositide 3-kinase-mediated reduction of insulin receptor substrate-1/2 protein expression via different mechanisms contributes to the insulin-induced desensitization of its signaling pathways in L6 muscle cells. J Biol Chem. (2003) 278:15641–51. doi: 10.1074/jbc.M208984200
45. Shaheen F, Shmygol A, Rabbani N, Thornalley PJ. A fluorogenic assay for methylglyoxal. Biochem Soc Trans. (2014) 42:548–55. doi: 10.1042/BST20140028
46. Thornalley PJ. Protein and nucleotide damage by glyoxal and methylglyoxal in physiological systems–role in ageing and disease. Drug metabolism and drug interactions. (2008) 23:125–50. doi: 10.1515/DMDI.2008.23.1-2.125
47. Haik GM Jr, Lo TW, Thornalley PJ. Methylglyoxal concentration and glyoxalase activities in the human lens. Exp Eye Res. (1994) 59:497–500. doi: 10.1006/exer.1994.1135
48. Thornalley PJ. Glyoxalase I–structure, function and a critical role in the enzymatic defence against glycation. Biochem Soc Trans. (2003) 31(Pt 6):1343–8. doi: 10.1042/bst0311343
49. Thornalley PJ. Protecting the genome: defence against nucleotide glycation and emerging role of glyoxalase I overexpression in multidrug resistance in cancer chemotherapy. Biochem Soc Trans. (2003) 31(Pt 6):1372–7. doi: 10.1042/bst0311372
50. Haralambie G, Mossinger M. Metabolites of the aminoacetone pathway in blood after exercise. Metabolism (1980) 29:1258–61. doi: 10.1016/0026-0495(80)90155-9
51. Hoffman NJ, Parker BL, Chaudhuri R, Fisher-Wellman KH, Kleinert M, Humphrey SJ, et al. Global phosphoproteomic analysis of human skeletal muscle reveals a network of exercise-regulated kinases and AMPK substrates. Cell Metab. (2015) 22:922–35. doi: 10.1016/j.cmet.2015.09.001
52. Hojlund K, Yi Z, Hwang H, Bowen B, Lefort N, Flynn CR, et al. Characterization of the human skeletal muscle proteome by one-dimensional gel electrophoresis and HPLC-ESI-MS/MS. Mol Cell Proteomics (2008) 7:257–67. doi: 10.1074/mcp.M700304-MCP200
53. Hwang H, Bowen BP, Lefort N, Flynn CR, De Filippis EA, Roberts C, et al. Proteomics analysis of human skeletal muscle reveals novel abnormalities in obesity and type 2 diabetes. Diabetes (2010) 59:33–42. doi: 10.2337/db09-0214
54. Parker KC, Walsh RJ, Salajegheh M, Amato AA, Krastins B, Sarracino DA, Greenberg SA. Characterization of human skeletal muscle biopsy samples using shotgun proteomics. J Proteome Res. (2009) 8:3265–77. doi: 10.1021/pr800873q
55. Yi Z, Bowen BP, Hwang H, Jenkinson CP, Coletta DK, Lefort N, et al. Global relationship between the proteome and transcriptome of human skeletal muscle. J Proteome Res. (2008) 7:3230–41. doi: 10.1021/pr800064s
56. Stratmann B, Goldstein B, Thornalley PJ, Rabbani N, Tschoepe D. Intracellular accumulation of methylglyoxal by glyoxalase 1 knock down alters collagen homoeostasis in L6 myoblasts. Int J Mol Sci. (2017) 18:480. doi: 10.3390/ijms18030480
57. Thornalley PJ, Battah S, Ahmed N, Karachalias N, Agalou S, Babaei-Jadidi R, Dawnay A. Quantitative screening of advanced glycation endproducts in cellular and extracellular proteins by tandem mass spectrometry. Biochem J. (2003) 375:581–92. doi: 10.1042/BJ20030763
58. Xue M, Rabbani N, Thornalley PJ. Measurement of glyoxalase gene expression. Biochem Soc Trans. (2014) 42:495–9. doi: 10.1042/BST20140026
59. Arai M, Yuzawa H, Nohara I, Ohnishi T, Obata N, Iwayama Y, et al. Enhanced carbonyl stress in a subpopulation of schizophrenia. Arch Gen Psychiatry (2010) 67:589–97. doi: 10.1001/archgenpsychiatry.2010.62
60. Moraru A, Wiederstein J, Pfaff D, Fleming T, Miller AK, Nawroth P, et al. Elevated levels of the reactive metabolite methylglyoxal recapitulate progression of type 2 diabetes. Cell Metab. (2018) 27:926–34.e8. doi: 10.1016/j.cmet.2018.02.003
61. Allen RE, Lo TW, Thornalley PJ. A simplified method for the purification of human red blood cell glyoxalase. I. Characteristics, immunoblotting, and inhibitor studies. J Protein Chem. (1993) 12:111–9. doi: 10.1007/BF01026032
62. Piec I, Listrat A, Alliot J, Chambon C, Taylor RG, Bechet D. Differential proteome analysis of aging in rat skeletal muscle. FASEB J. (2005) 19:1143–5. doi: 10.1096/fj.04-3084fje
63. Rabbani N, Thornalley PJ. The critical role of methylglyoxal and glyoxalase 1 in diabetic nephropathy. Diabetes (2014) 63:50–2. doi: 10.2337/db13-1606
64. Xue M, Rabbani N, Momiji H, Imbasi P, Anwar MM, Kitteringham N, et al. Transcriptional control of glyoxalase 1 by Nrf2 provides a stress-responsive defence against dicarbonyl glycation. Biochem J. (2012) 443:213–22. doi: 10.1042/BJ20111648
65. Campbell AK, Matthews SB, Vassel N, Cox CD, Naseem R, Chaichi J, et al. Bacterial metabolic 'toxins': a new mechanism for lactose and food intolerance, and irritable bowel syndrome. Toxicology (2010) 278:268–76. doi: 10.1016/j.tox.2010.09.001
66. Arrabal S, Lucena MA, Canduela MJ, Ramos-Uriarte A, Rivera P, Serrano A, et al. Pharmacological blockade of cannabinoid cb1 receptors in diet-induced obesity regulates mitochondrial dihydrolipoamide dehydrogenase in muscle. PLoS ONE (2015) 10:e0145244. doi: 10.1371/journal.pone.0145244
67. Engelbrecht B, Mattern Y, Scheibler S, Tschoepe D, Gawlowski T, Stratmann B. Methylglyoxal impairs GLUT4 trafficking and leads to increased glucose uptake in L6 myoblasts. Horm Metab Res. (2014) 46:77–84. doi: 10.1055/s-0033-1357121
68. Higaki Y, Mikami T, Fujii N, Hirshman MF, Koyama K, Seino T, et al. Oxidative stress stimulates skeletal muscle glucose uptake through a phosphatidylinositol 3-kinase-dependent pathway. Am J Physiol Endocrinol Metab. (2008) 294:E889–97. doi: 10.1152/ajpendo.00150.2007
69. Toyoda T, Hayashi T, Miyamoto L, Yonemitsu S, Nakano M, Tanaka S, et al. Possible involvement of the alpha1 isoform of 5'AMP-activated protein kinase in oxidative stress-stimulated glucose transport in skeletal muscle. Am J Physiol Endocrinol Metab. (2004) 287:E166–73. doi: 10.1152/ajpendo.00487.2003
70. Kar NC, Pearson CM. Glyoxalase enzyme system in human muscular dystrophy. Clin Chim Acta (1975) 65:153–5. doi: 10.1016/0009-8981(75)90348-4
71. Radom-Aizik S, Hayek S, Shahar I, Rechavi G, Kaminski N, Ben-Dov I. Effects of aerobic training on gene expression in skeletal muscle of elderly men. Med Sci Sports Exerc. (2005) 37:1680–96. doi: 10.1249/01.mss.0000181838.96815.4d
72. Hussey SE, Sharoff CG, Garnham A, Yi Z, Bowen BP, Mandarino LJ, Hargreaves M. Effect of exercise on the skeletal muscle proteome in patients with type 2 diabetes. Med Sci Sports Exerc. (2013) 45:1069–76. doi: 10.1249/MSS.0b013e3182814917
73. Nadolski LE, Solomon TPJ, Fealy CE, Malin SK, Kirwan JP, Haus JM. Diminished glyoxalase I activity in skeletal muscle of older obese individuals is associated with determinants of metabolic health. Diabetes (2013) 62:A500-A.
74. Brownlee M, Vlassara H, Kooney A, Ulrich P, Cerami A. Aminoguanidine prevents diabetes-induced arterial wall protein cross-linking. Science (1986) 232:1629–32. doi: 10.1126/science.3487117
75. Bolton WK, Cattran DC, Williams ME, Adler SG, Appel GB, Cartwright K, et al. Randomized trial of an inhibitor of formation of advanced glycation end products in diabetic nephropathy. Am J Nephrol. (2004) 24:32–40. doi: 10.1159/000075627
76. Freedman BI, Wuerth JP, Cartwright K, Bain RP, Dippe S, Hershon K, et al. Design and baseline characteristics for the aminoguanidine clinical trial in overt type 2 diabetic nephropathy (ACTION II). Control Clin Trials (1999) 20:493–510. doi: 10.1016/S0197-2456(99)00024-0
77. Kwak MK, Wakabayashi N, Itoh K, Motohashi H, Yamamoto M, Kensler TW. Modulation of gene expression by cancer chemopreventive dithiolethiones through the Keap1-Nrf2 pathway. Identification of novel gene clusters for cell survival. J Biol Chem. (2003) 278:8135–45. doi: 10.1074/jbc.M211898200
78. Tanaka Y, Aleksunes LM, Yeager RL, Gyamfi MA, Esterly N, Guo GL, et al. NF-E2-related factor 2 inhibits lipid accumulation and oxidative stress in mice fed a high-fat diet. J Pharmacol Exp Ther. (2008) 325:655–64. doi: 10.1124/jpet.107.135822
79. Shin S, Wakabayashi J, Yates MS, Wakabayashi N, Dolan PM, Aja S, et al. Role of Nrf2 in prevention of high-fat diet-induced obesity by synthetic triterpenoid CDDO-imidazolide. Eur J Pharmacol. (2009) 620:138–44. doi: 10.1016/j.ejphar.2009.08.022
80. Lu MC, Ji JA, Jiang ZY, You QD. The Keap1-Nrf2-ARE pathway as a potential preventive and therapeutic target: an update. Med Res Rev. 36:924–63. (2016). doi: 10.1002/med.21396
81. Abed DA, Goldstein M, Albanyan H, Jin H, Hu L. Discovery of direct inhibitors of Keap1-Nrf2 protein-protein interaction as potential therapeutic and preventive agents. Acta Pharm Sin B. (2015) 5:285–99. doi: 10.1016/j.apsb.2015.05.008
82. Xue M, Weickert MO, Qureshi S, Kandala NB, Anwar A, Waldron M, et al. Improved glycemic control and vascular function in overweight and obese subjects by glyoxalase 1 inducer formulation. Diabetes (2016) 65:2282–94. doi: 10.2337/db16-0153
83. Xue M, Momiji H, Rabbani N, Bretschneider T, Rand DA, Thornalley PJ. Frequency modulated translocational oscillations of Nrf2, a transcription factor functioning like a wireless sensor. Biochem Soc Trans. (2015) 43:669–73. doi: 10.1042/BST20150060
84. Rabbani N, Thornalley PJ. Glyoxalase 1 modulation in obesity and diabetes. Antioxid Redox Signal. (2018). doi: 10.1089/ars.2017.7424. [Epub ahead of print].
85. Hawley JA, Hargreaves M, Joyner MJ, Zierath JR. Integrative biology of exercise. Cell (2014) 159:738–49. doi: 10.1016/j.cell.2014.10.029
86. Pesta DH, Goncalves RLS, Madiraju AK, Strasser B, Sparks LM. Resistance training to improve type 2 diabetes: working toward a prescription for the future. Nutr Metab. (2017) 14:24. doi: 10.1186/s12986-017-0173-7
87. Haus JM, Solomon TP, Marchetti CM, Edmison JM, Gonzalez F, Kirwan JP. Free fatty acid-induced hepatic insulin resistance is attenuated following lifestyle intervention in obese individuals with impaired glucose tolerance. J Clin Endocrinol Metab. (2010) 95:323–7. doi: 10.1210/jc.2009-1101
88. Solomon TPJ, Haus JM, Kelly KR, Cook MD, Riccardi M, Rocco M, et al. Dietary glycemic index does not influence insulin sensitivity and substrate metabolism following short-term exercise training in older obese humans. Obesity (2008) 19:373–P. doi: 10.1097/SLA.0b013e31818b7595
89. Solomon TP, Haus JM, Kelly KR, Cook MD, Riccardi M, Rocco M, et al. Randomized trial on the effects of a 7-d low-glycemic diet and exercise intervention on insulin resistance in older obese humans. Am J Clin Nutr. (2009) 90:1222–9. doi: 10.3945/ajcn.2009.28293
90. Solomon TP, Haus JM, Marchetti CM, Stanley WC, Kirwan JP. Effects of exercise training and diet on lipid kinetics during free fatty acid-induced insulin resistance in older obese humans with impaired glucose tolerance. Am J Physiol Endocrinol Metab. (2009) 297:E552–9. doi: 10.1152/ajpendo.00220.2009
91. Haus JM, Solomon TP, Marchetti CM, O'Leary VB, Brooks LM, Gonzalez F, et al. Decreased visfatin after exercise training correlates with improved glucose tolerance. Med Sci Sports Exerc. (2009) 41:1255–60. doi: 10.1249/MSS.0b013e318195bad5
92. Solomon TP, Haus JM, Kelly KR, Cook MD, Filion J, Rocco M, et al. A low-glycemic index diet combined with exercise reduces insulin resistance, postprandial hyperinsulinemia, and glucose-dependent insulinotropic polypeptide responses in obese, prediabetic humans. Am J Clin Nutr. (2010) 92:1359–68. doi: 10.3945/ajcn.2010.29771
93. Solomon TP, Haus JM, Kelly KR, Rocco M, Kashyap SR, Kirwan JP. Improved pancreatic beta-cell function in type 2 diabetic patients after lifestyle-induced weight loss is related to glucose-dependent insulinotropic polypeptide. Diabetes Care (2010) 33:1561–6. doi: 10.2337/dc09-2021
94. Mulya A, Haus JM, Solomon TP, Kelly KR, Malin SK, Rocco M, Barkoukis H, Kirwan JP. Exercise training-induced improvement in skeletal muscle PGC-1α-mediated fat metabolism is independent of dietary glycemic index. Obesity (2017) 25:721–9. doi: 10.1002/oby.21799
95. Haus JM, Solomon TP, Lu L, Jesberger JA, Barkoukis H, Flask CA, et al. Intramyocellular lipid content and insulin sensitivity are increased following a short-term low-glycemic index diet and exercise intervention. Am J Physiol Endocrinol Metab. (2011) 301:E511–6. doi: 10.1152/ajpendo.00221.2011
96. Kelly KR, Blaszczak A, Haus JM, Patrick-Melin A, Fealy CE, Solomon TP, et al. A 7-d exercise program increases high-molecular weight adiponectin in obese adults. Med Sci Sports Exerc. (2012) 44:69–74. doi: 10.1249/MSS.0b013e318228bf85
97. Kelly KR, Haus JM, Solomon TP, Patrick-Melin AJ, Cook M, Rocco M, et al. A low-glycemic index diet and exercise intervention reduces TNF(alpha) in isolated mononuclear cells of older, obese adults. J Nutr. (2011) 141:1089–94. doi: 10.3945/jn.111.139964
98. Fealy CE, Haus JM, Solomon TP, Pagadala M, Flask CA, McCullough AJ, et al. Short-term exercise reduces markers of hepatocyte apoptosis in non-alcoholic fatty liver disease. J Appl Physiol. (2012). 13:1–6. doi: 10.1152/japplphysiol.00127.2012
99. Malin SK, Niemi N, Solomon TP, Haus JM, Kelly KR, Filion J, et al. Exercise training with weight loss and either a high- or low-glycemic index diet reduces metabolic syndrome severity in older adults. Ann Nutr Metab. (2012) 61:135–41. doi: 10.1159/000342084
100. Solomon TP, Malin SK, Karstoft K, Haus JM, Kirwan JP. The influence of hyperglycemia on the therapeutic effect of exercise on glycemic control in patients with type 2 diabetes mellitus. JAMA Intern Med. (2013) 173:1834–6. doi: 10.1001/jamainternmed.2013.7783
101. Phillips SA, Mahmoud AM, Brown MD, Haus JM. Exercise interventions and peripheral arterial function: implications for cardio-metabolic disease. Prog Cardiovasc Dis. (2015) 57:521–34. doi: 10.1016/j.pcad.2014.12.005
102. Kasumov T, Solomon TP, Hwang C, Huang H, Haus JM, Zhang R, et al. Improved insulin sensitivity after exercise training is linked to reduced plasma C14:0 ceramide in obesity and type 2 diabetes. Obesity (2015) 23:1414–21. doi: 10.1002/oby.21117
103. Devlin JT, Hirshman MF, Horton ED, Horton ES. Enhanced peripheral and spanchnic insulin sensitivity in NIDDM men after single bout of exercise. Diabetes (1987) 36:434–9. doi: 10.2337/diab.36.4.434
104. Schenk S, Horowitz JF. Acute exercise increases triglyceride synthesis in skeletal muscle and prevents fatty acid-induced insulin resistance. J Clin Invest. (2007) 117:1690–8. doi: 10.1172/JCI30566
105. Borghouts LB, Keizer HA. Exercise and insulin sensitivity: a review. Int J Sports Med. (2000) 21:1–12. doi: 10.1055/s-2000-8847
106. Perry CG, Lally J, Holloway GP, Heigenhauser GJ, Bonen A, Spriet LL. Repeated transient mRNA bursts precede increases in transcriptional and mitochondrial proteins during training in human skeletal muscle. J Physiol. (2010) 588(Pt 23):4795–810. doi: 10.1113/jphysiol.2010.199448
107. Done AJ, Traustadottir T. Nrf2 mediates redox adaptations to exercise. Redox Biol. (2016) 10:191–9. doi: 10.1016/j.redox.2016.10.003
108. Kjobsted R, Wojtaszewski JF, Treebak JT. Role of AMP-activated protein kinase for regulating post-exercise insulin sensitivity. EXS (2016) 107:81–126. doi: 10.1007/978-3-319-43589-3_5
109. Stepto NK, Coffey VG, Carey AL, Ponnampalam AP, Canny BJ, Powell D, et al. Global gene expression in skeletal muscle from well-trained strength and endurance athletes. Med Sci Sports Exerc. (2009) 41:546–65. doi: 10.1249/MSS.0b013e31818c6be9
Keywords: type 2 diabetes, insulin resistance, aerobic exercise, methylglyoxal, advanced glycation endproducts
Citation: Mey JT and Haus JM (2018) Dicarbonyl Stress and Glyoxalase-1 in Skeletal Muscle: Implications for Insulin Resistance and Type 2 Diabetes. Front. Cardiovasc. Med. 5:117. doi: 10.3389/fcvm.2018.00117
Received: 15 June 2018; Accepted: 09 August 2018;
Published: 10 September 2018.
Edited by:
Ichiro Sakuma, Hokko Memorial Hospital, JapanReviewed by:
Koichi Okita, Hokusho University, JapanArata Fukushima, Hokkaido University, Japan
Takashi Yokota, Hokkaido University, Japan
Copyright © 2018 Mey and Haus. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jacob M. Haus, am1oYXVzQHVtaWNoLmVkdQ==