 Maan A. Awad
Maan A. Awad Sarah R. Aldosari
Sarah R. Aldosari M. Ruhul Abid
M. Ruhul Abid- Division of Cardiothoracic Surgery, Department of Surgery, Cardiovascular Research Center, Rhode Island Hospital, Brown University Alpert Medical School, Providence, RI, United States
Cardiovascular diseases (CVD) are one of the prime causes of mortality worldwide. Experimental animal models have become a valuable tool to investigate and further advance our knowledge on etiology, pathophysiology and intervention. They also provide a great opportunity to understand the contribution of different genes and effector molecules in the pathogenesis and development of diseases at the sub-cellular levels. High levels of reactive oxygen species (ROS) have been associated with the progression of CVD such as ischemic heart disease (IHD), myocardial infarction, hypertension, atherosclerosis, aortic aneurysm, aortic dissection and others. On the contrary, low levels of antioxidants were associated with exacerbated cardiovascular event. Major focus of this review is on vascular pathogenesis that leads to CVD, with special emphasis on the roles of oxidant/antioxidant enzymes in health and disease progression in vascular cells including vascular endothelium. The major oxidant enzymes that have been implicated with the progression of CVD include NADPH Oxidase, nitric oxide synthase, monoamine oxidase, and xanthine oxidoreductase. The major antioxidant enzymes that have been attributed to normalizing the levels of oxidative stress include superoxide dismutases, catalase and glutathione peroxidases (GPx), and thioredoxin. Cardiovascular phenotypes of major oxidants and antioxidants knockout and transgenic animal models are discussed here.
Introduction
Cardiovascular diseases (CVD) are the leading cause of morbidity and mortality in the world. Reactive oxygen species (ROS) are believed to play major roles in the pathogenesis and development of CVD including ischemic heart disease and myocardial infarction. This review will focus on vascular pathogenesis leading to the development of CVD. Major emphasis will be given to the roles of oxidant/antioxidant enzymes in the vascular cells including vascular endothelium. The knockout and transgenic animal models of major oxidant/antioxidant enzymes affecting ROS levels in the vasculature and CVD progression are excellent experimental tools to further our understanding of CVD.
Studies have suggested a pivotal role of ROS in various physiological conditions as well as in a wide range of diseases including CVD (1–5). Oxidative stress results from a disrupted balance between ROS production and activity of antioxidant enzymes. Several enzymes, including superoxide dismutase (SOD), glutathione peroxidases (GPx) and thioredoxin act as scavengers to reduce the levels of ROS. In contrast, oxidases including nicotinic adenine dinucleotide phosphate (NADPH) oxidase, nitric oxide synthase (NOS), monoxides and xanthine oxidoreductase (XO) increase oxidant levels in the cardiovascular system (CVS). Several studies have illustrated the roles of ROS in CVD using animal models.
In the current review, we will present several transgenic and knockout animal models for genes encoding major oxidant and antioxidant enzymes and their cardiovascular phenotypes. We will discuss specific phenotypic changes associated with the manipulated expression of these genes in the vascular cells. For the convenience of the readership, we have included two tables, one with the major oxidant and antioxidant knockout (KO) models and the resulting cardiovascular phenotypes (Table 1), and the other with transgenic animal models and their cardiovascular phenotypes (Table 2).
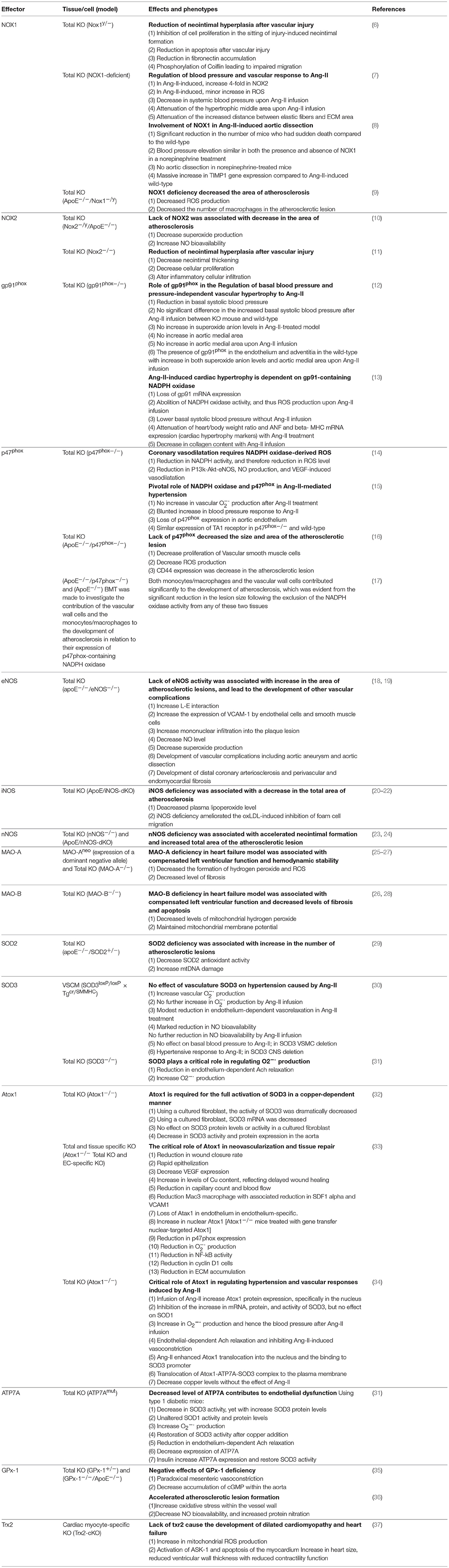
Table 1. Knockout oxidant and antioxidant models and their effects on cardiovascular system.
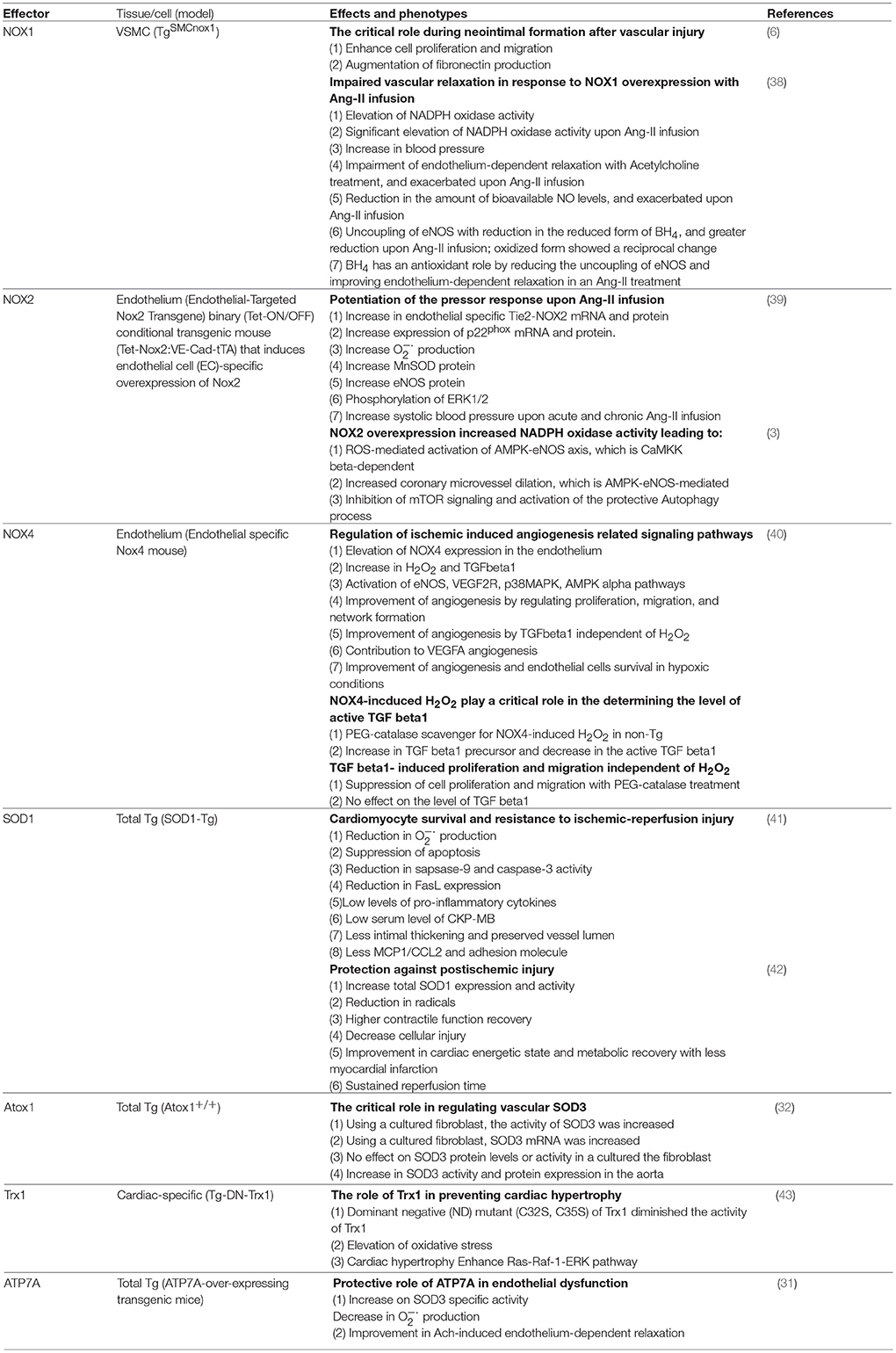
Table 2. Transgenic oxidant and antioxidant animals and their effects on cardiovascular system.
Oxidants
There is compelling evidence that indicates involvement of ROS molecules in CVD and their progression (5). In the heart and vasculature, ROS are synthesized from different locations and enzymes including NADPH oxidases, XO, cytochrome P450 monooxygenase, monoamine oxidase (MAO) and from the uncoupling of NOS. Under normal conditions, some of these enzymes such as NADPH oxidase produce physiologic amounts of ROS that are involved in multiple signaling pathways (3, 5, 14). While other enzymes such as MAO generate ROS as byproducts during metabolism of other molecules (25, 28, 44–46). In disease conditions, the activity of most of these enzymes is upregulated, generating increasing amounts of ROS that are thought to be involved in the pathogenesis and progression of the underlying conditions. In this section, we will be addressing some of the transgenic and KO animal models that were used to demonstrate the roles and activities of the major ROS-generating enzymes in CVD.
NADPH Oxidase
NADPH oxidases have different isoforms, among them, NOX1, NOX2 (gp91phox), NOX4, and NOX5 are the major contributor in the vasculature (5). NADPH oxidase is a complex containing membrane bound and cytosolic subunits. The membrane bound subunits are NOX2 (gp91phox) and p22phox. The cytoplasmic subunits are p47phox, p67phox, and Rac1-containing GTPase (5). This multi-subunit enzyme complex is triggered by different stimuli including stress, hypoxia, hormones, or cytokines (47). Activation or inhibition of one or more of the regulatory subunits modulates ROS levels in CVS which in turn induces changes in sub-cellular signal transduction and determines CVD phenotype.
NADPH Oxidase 1
NADPH Oxidase 1 (NOX1) is an enzyme expressed in CVS (9, 48–50). Several researchers have investigated specific role of NOX1 and the extent to which NOX1 contributes to cardiovascular pathophysiology. For example, NOX1 has been recently shown to be involved in Angiotensin-II (Ang-II) induced vascular responses and in CVD (7, 8, 51) (Tables 1, 2).
Ang-II induced-vascular responses
Ang-II plays a role in activating NADPH oxidases enzymes, especially NOX1 and NOX2, resulting in increase in ROS level (8, 38, 51). Ang-II is a critical component of renin-angiotensin system, which plays an important role in renal and cardiovascular pathophysiology (52). In CVS, Ang-II is involved in several functions such as: (i) potent vasoconstriction (ii) elevation of blood pressure (iii) increase in vascular smooth muscle cell (VSMC) proliferation and hyperplasia in the aorta (iv) participation in the process of oxidants formation resulting in vascular damage, and accumulation of extracellular matrix metalloproteinases (MMP), and (v) aortic aneurysm and dissection (8, 52–55).
Using different models of transgenic and knockout animals, NOX1 has been shown to play critical roles in cardiovascular pathophysiology, especially in augmenting and maintaining Ang-II responses (Tables 1, 2). In TgSMCnox1 mice treated with Ang-II, blood pressure was elevated. One possible mechanism is reduction in nitric oxide (NO) bioavailability due to increase quenching of NO by ROS, hence impaired endothelial-dependent vessel relaxation (Table 2) (38, 56). This notion was further supported by the findings where a decrease in systolic blood pressure was observed in NOX1−/− mice treated with Ang-II (Table 1) (7). Taken together, NOX1 appears to have a critical role in modulating Ang-II-induced vascular response and hypertension. In NOX1−/− mice, both aortic medial hypertrophy and extracellular matrix accumulation were attenuated, suggesting a crucial role of NOX1 in VSMC proliferation and MMP synthesis (7, 8).
Neointimal injury
VSMC is the main element in neointimal injury. The abnormal growth of neointimal tissue in the blood vessels contributes to the development of vascular occlusive diseases (6, 9). Following an injury or endothelial breach of continuity, VSMCs migrate to the damaged area and proliferate. Platelet-derived growth factor (PDGF) and reduction in NO have been believed to play critical roles in this process. ROS production from the injured tissue has also been considered a major player in neointimal growth (6). As mentioned previously, NOX1 in VSMC is one of the main sources of ROS (6, 9). In NOX−/− mice, cell proliferation in the setting of an injury-induced neointimal formation was inhibited. Interestingly, whereas apoptosis, fibronectin accumulation and migration of VSMC were reduced in the KO model (6), cell proliferation, migration, and fibronectin production were augmented in NOX1 overexpressing TgSMCnox1 mice (9). These findings suggest that NOX1 is essential for neointimal formation. Intriguingly, NOX1 seems to have a role in regulating matrix components as fibronectin production is reduced when NOX1 was knocked out, and augmented when it was expressed. Furthermore, NOX1 was shown to be involved in VSMC migration through coiflin. In NOX1−/−, coiflin was phosphorylated and Serine/threonine-protein kinase (PAK1) levels were reduced, explaining coiflin's involvement in the migration process (6).
Aortic dissection and aneurysm
Another important cardiovascular pathology is formation of aneurysm and aortic dissection. In order to understand its pathobiology, NOX1-deficient mice were treated with Ang-II and were shown to have reduction in aortic dissection and aneurysm (57). In this model, inhibition of MMP was observed (8) and the number of sudden death of mice was significantly reduced. Together, these findings suggested that NOX1 was involved in Ang-II induced aortic aneurysm and dissection.
Atherosclerosis
Atherosclerosis is a condition characterized by plaque formation in the medium and large blood vessels and is considered as one of the primary causes of CVD (58). In ApoE−/− and NOX1−/− animals, a reduction in the atherosclerotic area and the macrophages within the plaque were observed, suggesting a crucial for NOX1-induced ROS formation in the pathogenic process of ApoE-mediated atherosclerosis (6).
NADPH Oxidase 2
NOX2 (gp91phox) is a major membrane-bound subunit of the NADPH oxidase enzyme. Like NOX4, NOX2 localization is not limited to the cellular membrane, it is found in different subcellular compartments such as the perinuclear membrane and the endoplasmic reticulum (ER) (5). NOX2 has been implicated in the pathogenesis of CVD including atherosclerosis (10, 11). However, recent studies have shown beneficial effects for the endothelial cell-specific overexpression of NOX2 on endothelial function and proliferation (3, 4).
Using NOX2 deficient (NOX2−/−) animal model, intimal thickening following arterial injury was found to be reduced by 34% compared to the wild-type (WT) counterpart (11). In this study, progressive thickening of the medial arterial layer was observed up to 28 days after the injury in both NOX2 deficient and wild-type mice. Whereas the intima/media ratio (I:M ratio) increased from 0.24 at day 7 to 0.97 at the 28th day from the injury in wild-type mice, the I:M ratio was 40% lower by day 28 in the NOX2 deficient mice compared to WT (11). These findings suggest an important role for NOX2-containing NADPH oxidase in inducing intimal thickening and cellular proliferation after vascular injury. Furthermore, altered leukocyte infiltration was observed in NOX2−/− mice suggesting that NOX2 plays critical roles in the accumulation of inflammatory cells within the vessel wall in response to injury (11).
In another study, [(ApoE−/−)/(Nox2y/−)] double knockout mice were used to examine the effects of NOX2-containing NADPH oxidase on the development of atherosclerosis in an atherogenic, ApoE-deficient mice (10) (Table 1). Although NOX2 knockout in [(ApoE−/−)/(Nox2y/−)] mice had minimal effect on the plasma-lipid levels compared to (ApoE−/−) mice, the area of atherosclerotic lesions in the aorta was 50% less in [(ApoE−/−)/(Nox2y/−)] compared to (ApoE−/−) mice (10). The decrease in the atherosclerotic plaque area was associated with decreased ROS levels. NO bioavailability was improved, and superoxide level was reduced by 75% in [(ApoE−/−)/(Nox2−/−)] mice compared to (ApoE−/−) animals (10). These results suggest that NOX2-containing NADPH oxidase enzyme-induced ROS production and resulting effects on NO bioavailability within the vessel wall has significant effect on the atherosclerotic plaque size (10).
To examine the effect of NOX2-containing NADPH oxidase on the vascular oxidative stress and response to hemodynamic alterations, Tie2-NOX2 transgenic mouse model was developed (39) (Table 2). The transgenic mouse showed an overall increase in NADPH activity resulting in increased production (39). With the increased amount of , there was a compensatory upregulation of Manganese-dependent Superoxide Dismutase (MnSOD) (SOD2) and eNOS protein followed by activation of the downstream signaling pathway ERK1/2 (39). Interestingly, the authors reported a lack of effect of NOX2-Tg on the basal blood pressure, however, it had greatly potentiated the pressor response upon acute and chronic Ang-II infusion (39). Together, these findings suggest an important role in modulation of the hemodynamic response to Ang-II by NOX2-containing NADPH oxidase (39).
Increased levels of ROS have been found to be associated with several cardiac diseases (3, 4). However, several clinical trials using global antioxidants have failed to improve and rather sometimes have worsen the outcome of CVD (3–5). In addition, decreased endothelial ROS level resulted in PI3K-Akt-eNOS inhibition and impairment of endothelium-dependent coronary vasodilation (3). These paradoxical findings suggest that ROS may also have positive effects on endothelial cells (EC). To examine the hypothesis, a novel binary (Tet-ON/OFF) conditional transgenic mouse (Tet-Nox2:VE-Cad-tTA), which induces endothelium-specific generation of Nox2-dependent NADPH oxidase-derived ROS was developed (3, 4). Isolated coronary microvessels from Tet-OFF mice showed significant increase in coronary vasodilation, upon stimulation by VEGF or Acetylcholine compared to Tet-ON vessels (5). The increased coronary vasodilation in Tet-OFF coronary microvessels was found to be the result of ROS-mediated activation of the AMPK-eNOS axis, which resulted in increased levels of NO in vascular endothelium. ROS-induced activation of AMPK showed inhibitory effect on mTOR signaling and subsequent activation of the protective autophagy process resulting in enhanced survival of Tet-OFF mouse heart endothelial cells (MHEC) (3).
To examine whether the duration of exposure to high levels of ROS dictates the ultimate effect on the vascular endothelium, Tet-ON and Tet-OFF animals were divided into short-term (8 weeks) and long-term (20 weeks) groups. Interestingly, the coronary microvessels isolated from animals that are 8 weeks Tet-OFF showed 22% increase in vasodilation, whereas those isolated from animals that were 20 weeks Tet-OFF showed 44% reduction in vasodilation in response to Acetylcholine and VEGF, compared to Tet-ON animals (4). MHEC from animals that were Tet-OFF for 20 weeks showed 2-fold increase in the level of peroxynitrite compared to the short-term exposure group (4). This finding indicates that long-term exposure to higher levels of ROS resulted in superoxide and NO interaction, leading to the formation of peroxynitrite and impairment of the ROS-activated AMPK-eNOS-mediated vasodilation. On the contrary, short-term exposure to ROS in Tet-OFF animals increased EC proliferation and endothelial sprouting from the aorta, while long-term exposure inhibited EC proliferation and aortic sprouting (4). The long-term exposure to high levels of ROS in Tet-OFF animals resulted in increased levels of mitochondrial ROS (mtROS). This led to mitochondrial damage by altering the mitochondrial membrane potential and significant reduction in the mitochondrial DNA (4). Increase in mtROS in short-term Tet-OFF animals was counteracted by the increased expression and activity of SOD2 enzyme. SOD2 was found to undergo nitration modification when exposed to long-term ROS increase, leading to the enzyme's inhibition (4). Together, these results demonstrated that duration of ROS exposure on EC is critical in determining phenotypic effects of ROS on blood vessels.
NADPH Oxidase 4
NADPH Oxidase 4 (NOX4) enzyme is expressed in endothelial as well as vascular smooth muscle cell (59). NOX4 can generate ROS, specifically H2O2, without requiring activating cytosolic factors (5, 59). Several studies have implicated role of H2O2 in cardiovascular cell survival.
Animal models with NOX4 transgene have been shown to have higher levels of H2O2 resulting in enhanced activation of eNOS, VEGFR2, p38MAPK, and AMPK-alpha pathways (40, 60, 61). These signaling pathways are critical in inducing angiogenesis by regulating proliferation, migration, and tubular network formation by ECs (40, 60). NOX4−/− mice demonstrated reduction in hemoxygenase-1 (HO-1) accompanied by an increase in apoptosis and increase in E-Cadherin expression (61). These finding suggested an important role for NOX4 in regulating HO enzyme and thus heme degradation.
P47phox
P47phox is one of the cytosolic subunits of the NADPH oxidase enzyme (17). p47phox has been shown to have a pivotal role in the development or progression of several diseases, for example, atherosclerosis and hypertension (15, 16, 62).
Using knockdown model, researchers demonstrated that p47phox is involved in cardiovascular pathology through multi-subunit NADPH oxidase enzyme complex (Table 1). In p47phox−/− animals, level of NADPH oxidase enzyme-derived ROS production was inhibited (14, 17). This resulted in the reduction of PI3K-Akt-eNOS, NO production, and VEGF-induced vasodilatation (14). In ApoE−/− and p47phox−/− model, there was a significant decrease in atherosclerotic lesion (16, 62). Additionally, it was also shown to be involved in neointimal hyperplasia (16, 17). These findings suggested that p47phox plays a crucial role in the development of atherosclerosis and progression of neointimal hyperplasia. Another study using KO model of p47phox−/− mice showed that there was a reduction in ROS formation as well as reduction in Ang-II-mediated increase in blood pressure, suggesting a pivotal role for NADPH-p47phox in mediating Ang-II-induced hypertension (15).
Xanthine Oxidoreductase
Purines catabolism is carried out by the enzyme XO, which degrades adenine to hypoxanthine and guanine to xanthine (63, 64). Hypoxanthine and xanthine are then oxidized by XO, catalyzing the oxidation of hypoxanthine to xanthine and subsequently to uric acid (65). Both steps require oxygen, forming the superoxide molecules, which are then converted to hydrogen peroxide (64, 65). In human, the inactive state of the enzyme made determination of the enzyme distribution difficult, yet it was identified in the liver, small intestine, and mammary glands. It has been identified in other mammalian tissues as well as in bovine and rat heart (66–68).
The byproducts (i.e., superoxide and hydrogen peroxide) of XO enzyme are believed to have detrimental effects on heart. XO has also been associated with endothelial dysfunction and was found to be elevated during ischemia-reperfusion (IR) injury (69). Using rat heart, researchers showed that there was an elevation of H2O2 levels and associated left ventricular dysfunction in IR injury (70). Rabbit heart with ischemia experienced less ventricular dysfunction when treated with allopurinol (XO inhibitor) compared to untreated heart (71). Additionally, in spontaneous hypertensive/heart failure rat model, chronic use of XO inhibitor induced reverse cardiac remodeling and preserved the structure and function of the heart (72), suggesting critical roles of XO in IR and cardiac remodeling. Several clinical trials are being conducted to determine the effect of XO inhibitor in patients with CVD (73–75). Precise mechanisms by which XO exerts its effects on the CVS and how XO inhibitor can attenuate the adverse effects remain yet to be elucidated.
Nitric Oxide Synthase
NO is one of the major effector molecules in cardiovascular physiology (76). In addition to its critical role in inducing endothelium-dependent vasorelaxation, it also inhibits platelet aggregation, vascular smooth muscle proliferation, and leukocyte adhesion (18, 76, 77). It is important to maintain an adequate level of NO for the integrity of the vasculature and prevention of atherosclerosis (18, 76). Hypertension, atherosclerosis and other CVD have been linked to altered NO bioavailability and abnormalities in NO signaling (76).
There are three isoforms of NOS which produce NO: endothelial NOS (eNOS), Inducible NOS (iNOS) and Neuronal NOS (nNOS). These enzymes are encoded by distinct genes, yet they share similar mechanisms and sites for cofactor binding (76). They all require transfer of electron from oxygen to catalyze the conversion of L-arginine to L-citrulline (76). Under normal conditions, eNOS is the major isoform expressed within the vasculature. However, all three isoforms have been found to be overexpressed in atherosclerotic lesions (19, 24, 78). Previous studies have shown that nNOS and eNOS have protective roles against artificially induced and spontaneous atherosclerosis (18, 19, 23, 24), while iNOS expression aggravated progression of atherosclerotic plaque (20).
Endothelial Nitric Oxide Synthase
eNOS enzyme is thought to have a protective role against the development of atherosclerotic lesions. Although eNOS has beneficial effects on CVS, uncoupling of eNOS results in ROS production instead of NO synthesis. Different studies have investigated the effects of eNOS deficiency in the context of atherosclerosis, using the atherogenic (ApoE−/−) mice as a model (19, 77). In one study, expression of VCAM-1 by endothelial and smooth muscle cells was reported to be increased in [(ApoE−/−)/(eNOS−/−)] compared to (ApoE−/−) mice, resulting in increased Leukocyte-Endothelial interaction. The mononuclear accumulation within the vessel wall was also increased in this animal (19). Interestingly, genetic deletion of eNOS in ApoE deficient mice resulted in significant reduction in superoxide level compared to wild-type mice, suggesting that eNOS contributed to the formation of superoxide in the atherogenic (ApoE−/−) mice (19). Additionally, inhibition of eNOS activity resulted in a significant reduction in superoxide level in (ApoE−/−) but not in wild-type or [(ApoE−/−)/(eNOS−/−)] mice, further confirming that the increased level of superoxide in (ApoE−/−) mice was the result of eNOS uncoupling (19).
Inducible Nitric Oxide Synthase
iNOS expression in the atherosclerotic lesion is limited to the mononuclear leukocytes and smooth muscle cells (20). Whereas the activities of eNOS and nNOS are regulated by the intracellular calcium concentration, iNOS activity is mostly regulated by cytokines and bacterial endotoxins released under pathological conditions. Using the hyperlipidemic ApoE deficient mice model, several studies have investigated the role of iNOS in the atherosclerotic plaque formation (20, 21, 23).
Although body weight and total cholesterol and triglyceride levels were not different between ApoE-KO and ApoE/iNOS-dKO mice fed on high fat diet, the total area of the atherosclerotic lesion was significantly decreased in ApoE/iNOS-dKO mice compared to their ApoE−/− controls (20, 22), suggesting a deleterious role for iNOS in CVD. The reduction in the area of atherosclerosis was found to increase over time, suggesting that protection from lesion formation increases over time in iNOS deficient mice (20). The decrease in atherosclerotic lesion formation seen in ApoE/iNOS-dKO mice was accompanied by decreased plasma levels of lipoperoxide, suggesting that iNOS-mediated oxidative stress was also decreased in this animal (20). Oxidized low density lipoprotein (oxLDL) was found to induce accumulation of the macrophage-derived foam cells within the atherosclerotic plaque by increasing iNOS-mediated oxidative stress (21). Treatment with oxLDL resulted in increased iNOS expression, peroxynitrite formation and significant decrease in cell migration, which was reversed upon co-administration of iNOS inhibitor (21), further supporting a role for iNOS in atherosclerosis.
Neuronal Nitric Oxide Synthase
In early and advanced atherosclerotic lesion, nNOS is expressed in endothelial cells, macrophages and smooth muscle cells (23). Several studies have demonstrated a protective role for nNOS in atherosclerosis (23, 24). Using a mouse model, in which unilateral carotid artery ligation was performed to induce vascular injury, it was shown that neointimal formation and vasoconstriction were significantly accelerated in nNOS-KO mice compared to their wild-type littermates (24). These results suggested a protective role for nNOS against the development of atherosclerosis.
The nNOS gene produces multiple mRNA splice variants, leading to the formation of four distinct proteins (24). In order to study the role and activity of nNOS in spontaneous atherosclerosis, nNOS deficient mice model (nNOS-KO) was combined with the atherogenic ApoE-KO mice model (23). Total lesion area was significantly increased in male ApoE/nNOS-dKO mice compared to their ApoE-KO controls at 14 weeks of western-type diet, while there was no difference in the lesion area of the female animals (23). However, the lesion area of female ApoE/nNOS-dKO mice was markedly increased after 24 weeks of western-type diet compared to ApoE-KO female mice, plausibly due to the reduced blood pressure observed in ApoE/nNOS-dKO female mice (23). These results suggested a vasculo-protective role for nNOS in atherosclerosis.
Monoamine Oxidase
MAO are enzymes located on the outer membrane of the mitochondria. These enzymes are responsible for the metabolism of catecholamines including epinephrine and norepinephrine, and other biogenic amines such as serotonin (25, 28, 44, 79). There are two types of MAOs, MAO-A, and MAO-B, which are different in their substrate and inhibitor sensitivity (25, 26, 28, 44, 79). MAO-A is the predominant isoform expressed in the cardiac tissue of humans and rats, while MAO-B is also abundant in the myocardium of mice and humans (26, 28, 79, 80). Under stressful conditions, such as in congestive heart failure, the sympathetic tone and circulating catecholamines are increased leading to upregulation of MAO activity due to the increased substrate availability (25, 28, 79). Metabolism of catecholamines and other biogenic amines by MAO enzymes results in the formation of hydrogen peroxide, aldehyde intermediates and ammonia as end products (28, 79). Due to their ability to generate increasing levels of ROS, MAOs have been considered as the major contributors to the oxidative damage in several CVD such as heart failure and IR injury (25, 28, 44–46).
Monoamine Oxidase A
Being a major source of hydrogen peroxide and ROS under stressful conditions, both genetic deficiency and pharmacological modulation of the enzymatic activity of MAO-A have been found to improve the outcome of different pathological conditions (25, 44). A study showed that treatment of rat cardiomyocytes with serotonin resulted in mitochondrial dysfunction and apoptosis (44). These deleterious effects were fully prevented upon treating the cells with Pargyline, a MAO inhibitor. This suggests that the serotonin-induced apoptosis and mitochondrial dysfunction are mediated by the accumulation of hydrogen peroxide generated by MAO-A. Moreover, the infarction size following an ischemic injury was significantly reduced in rats treated with MAO inhibitors compared to those treated with vehicle only (44).
Another study demonstrated that treatment of isolated cardiomyocytes with Norepinephrine (NE) resulted in increased MAO-A expression, ROS production and cellular hypertrophy (25). The concomitant administration of MAO inhibitor prevented NE-induced ROS production and cellular hypertrophy. Furthermore, the administration of MAO inhibitor to a heart failure mouse model enhanced preservation of the left ventricular function and decreased heart weight (25). The genetic deficiency of MAO-A due to the expression of a dominant negative allele (MAO-Aneo) was found to be associated with improved ventricular function and decreased level of fibrosis (25, 26). Similarly, (MAO-A−/−) mice were found to have compensated left ventricular function and hemodynamic stability despite the presence of hypertrophic myocardium and ventricular dilation (26, 27). These results suggest that MAO-A-mediated oxidative stress contributes to the impairment of the cardiac function in heart failure.
Monoamine Oxidase B
Until recently, most studies focused on addressing the role of MAO-A in heart diseases because it was believed to be the predominant isoform expressed in the cardiac tissue of humans and rats (26, 28, 79, 80). As stated earlier, MAO-B is also abundantly present in the human heart, and there are several labs currently studying MAO-B to elucidate the role of MAO-B in CVD. To that end, using MAO-B deficient mice (MAO-B−/−), trans-aortic constriction was performed to induce heart failure. Whereas both (MAO-B−/−) and wild-Type (WT) mice were found to have ventricular hypertrophy following the trans-aortic constriction, only (MAO-B−/−) mice maintained compensated left ventricular function (28). Additionally, level of dopamine was greatly reduced in the heart tissue of WT mice in spite of the unchanged MAO-B protein expression (26, 28). Together, these findings suggested that activity of the enzyme was upregulated upon exposure to higher levels of the substrate. Fibrosis and apoptosis were markedly increased in WT mice compared to their (MAO-B−/−) counterparts (26, 28). Cardiomyocytes of WT mice with dopamine showed significant increase in mitochondrial hydrogen peroxide levels and significant drop in the mitochondrial membrane potential. These effects were ameliorated upon co-incubation with a MAO-B inhibitor and were completely absent in MAO-B deficient mice (28). Together, these findings suggested that MAO-B plays a critical role in inducing mitochondrial oxidative stress and dysfunction in CVD.
Antioxidants
Antioxidants include both enzymatic and non-enzymatic antioxidants that work as a line of defense in protecting the tissues from the oxidative damages. Thus, antioxidants are critical for the maintenance of homeostatic balance of oxidants at the tissue levels. When levels of ROS exceed physiological limit and the antioxidants become overwhelmed, the resulting impaired balance affects cellular functions (81, 82). There are several important intracellular enzymatic antioxidants that include SODs, glutathione peroxidase, and thioredoxin (82). To conceptualize the importance of antioxidants and equivalent molecules in CVD, transgenic and knockout models have been utilized to elucidate the functional roles of each of these proteins.
Superoxide Dismutase
SODs are copper-containing and zinc-containing enzymes (32). They convert to H2O2, minimizing the damaging effects of superoxide. There are three isoforms: (i) cytoplasmic CuZnSOD (SOD1) (ii) mitochondrial MnSOD (SOD2), and (iii) extracellular SOD (SOD3) (40).
Superoxide Dismutase 1
Superoxide dismutase 1 (SOD1) is a cytoplasmic zinc- and copper-containing enzyme that catalyzes the conversion of to H2O2 intracellularly (83). Subsequently, H2O2 is reduced to water and molecular oxygen by glutathione peroxidase or catalase (42, 84). Several studies have examined the role of SOD1 in IR injury and whether the overexpression of SOD1 is detrimental or beneficial to cells under the pathophysiological conditions with higher oxidative stress (42, 84, 85).
Ischemia-reperfusion injury
IR injury is one of the major causes of pathologies seen in the heart. It has been also linked as one of the prime complications of peri-transplant injury during heart transplantation (41). Ischemia denotes to the sudden cessation of blood supply to the organ, resulting in cellular damage (86). SOD1 was shown to protect from IR injury (41, 42, 84). In animal models overexpressing SOD1, the levels of ROS, specifically , generated in post-ischemic tissues were significantly diminished compared to WT animals (42, 84). One of these models further showed improvement in the myocardial energetic state, metabolic recovery and reduction in myocardial infarction size (42). In another model with SOD1 overexpression, inflammatory cytokines, adhesion molecules, apoptotic proteins including caspase-9, caspase-3, and FasL were all reduced (41, 84). These results implicated SOD1 in reducing cardiomyocyte apoptosis and inflammation (42, 84). Taken together, all of the above findings suggest a critical role for SOD1 in minimizing IR injury in post-ischemia and post-transplantation settings (41, 42, 84). Other studies have thoroughly investigated the effects of exogenous administration of SOD1 on ischemic tissues, which was associated with dose-dependent toxic effect. This necessitates further studies aimed at examining whether SOD1 protect or potentiate adverse cellular injury in CVD (87).
Superoxide Dismutase 2
SOD2 constitutes the major mitochondrial antioxidant defense system against superoxide radicals produced by the leakage of electron transport chain (88). Like other SOD enzymes, SOD2 catalyzes the conversion of superoxide to hydrogen peroxide. Deficiency in SOD2 is associated with oxidative mitochondrial damage, which is mediated primarily by superoxide. Whereas homozygous SOD2 deficiency (SOD2−/−) is not compatible with life and produces lethal mice phenotypes (88), heterozygous deficiency (SOD2−/+) has been associated with increased mitochondrial damage in the heart (89).
Mitochondrial DNA (mtDNA) damage is an indicator of ROS-mediated cellular damage (29). A study has addressed the question whether mtDNA damage plays a causal role in atherosclerosis or atherosclerosis results from the oxidative damage happening at the site of the lesion. Researchers used single KO (ApoE−/−) and double KO [(ApoE−/−)/(SOD2+/−)] mice models for this study (29). Results showed that aortic tissue from (ApoE−/−) mice had significant increase in mtDNA damage during the earliest stages of the atherosclerotic lesion development (29). [(ApoE−/−)/(SOD2−/+)] mice showed significant increase in the number of atherosclerotic lesions compared to their (ApoE−/−) counterparts (29). Interestingly, [(ApoE+/+)/(SOD2+/−)] mice did not develop atherosclerotic lesions at all, suggesting that the phenotype observed in [(ApoE−/−)/(SOD2+/−)] mice resulted from the combined effect of having an atherogenic environment and increased mitochondrial ROS levels (29).
Superoxide Dismutase 3
Superoxide Dismutase 3 (SOD3) is a major extracellular antioxidant enzyme. It is synthesized inside the cell and anchored in the extracellular matrix and also on endothelial surface (31, 90–93). This enzyme acts as a first line of defense against ROS because of its extracellular location. Similar to other SODs, it catalyzes the conversion of to H2O2. The regulation of redox balance highlights the critical role of SOD3 in the pathogenesis of CVD.
SOD3 requires a catalytic copper for its full activity and since the intracellular copper levels are restricted, a soluble copper carrier is required to deliver the copper to this extracellular enzyme. This is achieved by the antioxidant-1 copper chaperone (Atox1) that interacts with ATP7A. Atox1-ATP7A pathway helps in the activation of the secretory pathway for copper as well as regulates the level of intracellular copper by excreting excess amount (83, 94, 95). Thus, ATP7A delivers copper to SOD3 to scavenge and increase bioavailability of nitric oxide to preserve the endothelial functions (31).
In SOD3−/− mice, increase in ROS levels were reported [80.87] (Table 1). Increase in ROS was also observed in Atox1−/− and ATP7Amut mice, further supporting the physiological link of Atox1- ATP7A-SOD3 (30, 31, 83). In the knockdown animal model of SOD3 or ATP7A, a reduction in endothelium-dependent acetylcholine-mediated vasorelaxation, as a consequence of increased ROS production, was reported (31). SOD3loxP/loxP × Tgcr/SMMHC animals treated with Ang-II showed a marked reduction in nitric oxide bioavailability (30, 96). However, in a VSMC-specific SOD3 KO model, there was no effect of lack of SOD activity on Ang-II-induced hypertension (96), suggesting SOD3 plays a major role specifically in endothelial NO homeostasis by modulating ROS levels.
Since Atox1 plays a role in the regulation of SOD3, Atox1−/− mice were used to see the endothelial cell specific role during wound healing. Atox1−/− mice showed delay in wound healing which could be explained by the loss of copper sensor function implicated by increase copper content. These animals also showed a reduction in the number of capillaries, blood flow, Mac3+ macrophage, stromal cell-derived factor 1 (SDF1) alpha, and vascular cell adhesion molecule 1 (VCAM-1). These mediators are critical for neovascularization and tissue repair (34). Interestingly, Atox1−/− demonstrated reduction in p47phox expression, NF-kB activity and cyclin D levels. Together, these findings suggested a role for Atox1 in the downstream regulation of NF-kB via p47phox, and in cell proliferation via cyclin D, resulting in delay in wound healing.
Glutathione Peroxidase
Glutathione and GPx enzymes are amongst the most important antioxidant systems in mammalian cells (36). They play crucial roles in the defense against oxidative stress. The most abundant cellular form of GPx is GPx-1 [591]. GPx-1 converts hydrogen peroxide to water and molecular oxygen, and converts lipid peroxides to their respective alcohols using glutathione as a reducing agent (33, 36). Lipid peroxides can react with NO forming lipid peroxynitrite, which results in decreased NO bioavailability (33). Deficiency in GPx-1 has been linked to the development of endothelial dysfunction, inflammation and neointimal formation, suggesting a quenching role of ROS against NO (33, 35). GPx-1 deficiency was found to have deleterious effects also in the context of atherosclerosis (36).
GPx-1 and glutathione have been shown to have vasodilatory effects on the vasculature (33). Studies showed that infusion of wild-type mice with bradykinin or b-methacholine lead to dose-dependent vasodilation of the mesenteric arterioles in vivo. On the contrary, superfusion of the GPx-1 deficient mice with the same vasodilators resulted in paradoxical vasoconstriction of the mesenteric arterioles (33). Interestingly, superfusion of both animal types with the NO donor, sodium nitroprusside (SNP), resulted in dose-dependent vasodilation, suggesting that the lack of GPx-1 was associated with decreased NO bioavailability (33). Moreover, upon stimulation with Bradykinin, aortic tissue from GPx-1 deficient mice showed decreased accumulation of cGMP compared to wild-type aortic tissue, suggesting Bradykinin's involvement in the activation of NO-cGMP signaling cascade (33).
The double knockout [(ApoE−/−)/(GPx-1−/−)] mice developed more aggressive atherosclerosis compared to their (ApoE−/−) controls (36). There was marked increase in ROS levels within the vessel wall, mostly in the endothelium of the double-knockout animals compared to the controls (36). A decrease in NO bioavailability was also associated with increased protein nitration within the atherosclerotic lesions of [(ApoE−/−)/(GPx-1−/−)] compared to (ApoE−/−) mice (36). These findings suggest that GPx-1 deficiency is associated with accelerated atherosclerotic lesion formation and extensive oxidative stress within the plaque formation (36).
Thioredoxin
Thioredoxin (Trx) is a small ubiquitin enzyme that serves as a reducing agent. Trx has been shown to be efficient in keeping a reduced state intracellularly (97–99). Trx maintains cellular functions via different mechanisms; primarily, it operates by reducing ROS and thereby protecting the cells from deleterious effects. It also plays a role in regulating apoptosis and inflammatory response (100, 101). Trx is active in the reduced form and operates by scavenging ROS and inhibiting apoptosis through inhibitory binding to apoptosis signal-regulating kinase-1 (ASK-1) (98, 100). Trx enzyme requires Thioredoxin Reductase (TrxR) to regenerate the reduced form of Trx at the expense of NADPH molecule (102). There are at least three isoforms of Trx in mammals: cytosolic/nuclear thioredoxin 1 (Trx1), mitochondrial thioredoxin 2 (Trx2), and sperm thioredoxin (SpTrx) (100–102). In the following sections, we will only discuss Trx1 and Trx2, and their roles in cardiac hypertrophy, IR injury, and heart failure.
Thioredoxin-1
Human Trx1 is a ubiquitous cytosolic protein, which can translocate to the nucleus or exit from the cell to the extracellular environment (103). It harbors two cysteines in the catalytic site (Cys-32 and Cys-35) and three additional non-catalytic cysteines (Cys-62, Cys-69, and Cys-73) (100). Under oxidative conditions, the cysteines in the catalytic site (Cys-32 and Cys-35) react with the oxidized substrate, generating a reduced substrate and an oxidized Trx1. The non-catalytic cysteines may suppress the enzymatic activity of Trx1 as Cys-62 and Cys-69 attenuate accessibility of TrxR to Trx1 by forming a disulfide bond, and Cys-73 can react with glutathione to form a mixed disulfide moiety (glutathionylation) (104, 105). Additionally, Cys-69 may also preserve the redox activity of Trx1 via nitrosylation (106).
Cardiac remodeling
The term “Remodeling” was first used to describe the replacement of the infarcted tissue with scars in myocardial infarction. Nowadays, the term is used more broadly to include adaptive and pathological changes of the heart after an injury (107). ROS have been shown to influence the progression of this process from the development of hypertrophy to compensated hypertrophy and subsequently to heart failure (107). Since ROS can induce hypertrophy, antioxidants such as Trx1 should logically suppress cardiac hypertrophy. In transgenic mouse overexpressing Trx1, myocyte hypertrophy was suppressed by Trx1-mediated inhibition of Ras-MAPK signaling pathway (43). Using adult rat ventricular myocyte, Trx1 was shown to inhibit activation of Ras by reducing thiol in Ras (108).
Another phenomenon that occurs in response to cardiac ischemia is the ischemic preconditioning. It has been described as a transient ischemia inducing a powerful protective mechanism against the longstanding ischemic insult in IR injury (109). It has been reported that ischemic preconditioning requires ROS production, which in turn induces protein kinase C (100, 110). Using ex vivo rat heart that overexpresses Trx1, it was demonstrated that Trx1 is essential for the cardiac ischemic preconditioning (111).
Prolonged ischemic injury increases ROS levels, resulting in the imbalance between the levels of ROS and NO, and consequently inactivation of Trx1 through post-translational modifications (106). Increased levels of ROS induce both apoptosis and fibrosis of cardiomyocytes (112, 113). As mentioned earlier, Trx1 exerts anti-apoptotic effect through ASK-1 and Akt signaling pathways (98, 100, 114). Trx1 activates Akt which in turn inhibits Bad (pro-apoptotic protein) and procaspase-9 (115). Together, Trx1 prevents pathological cardiac remodeling through intracellular signaling molecules that inhibits apoptosis and cell death. Further experiments are ongoing in several labs to elucidate the precise molecular mechanisms by which Trx1 modulate pathophysiology of CVD.
Thioredoxin-2
Trx2 is a mitochondrial protein, hence the name mitochondrial thioredoxin (100, 102). Like Trx1, it contains an active catalytic site but lacks the other non-catalytic cysteines (100). Due to high demand of ATP generation and oxygen consumption in the heart, mitochondria become susceptible to oxidative stress during highly active oxidative phosphorylation. Trx2 plays a critical role in protecting the mitochondria by respiration-dependent H2O2 removal in the organelle (116, 117). Mice with cardiac-specific ablation of Trx2 developed fatal cardiomyopathy (37). This was correlated with increased ASK-1 signaling and apoptosis of cardiomyocyte. These findings suggested that Trx2 helps preserve and maintain the integrity of the mitochondria in the heart with increased ROS levels (37, 118).
Limitations
The availability of animal models has undeniably advanced our current knowledge and offered novel insights into different aspects of CVD and interventions. Yet there are certain limitations of the studies that utilize animal models. A major limitation for using small animals like mouse is lack of the translational aspects of these studies and lack of reliability of the outcomes of such studies for clinical applications and interventions (119, 120). Additionally, the size of the animal restricts the amount of blood sampling and complicates vessel dissection (120). Other concerns include different etiology of diseases among different species, and sometimes the number of samples in a study can be limited due to imposed financial constraints (121, 122). Thereby, it is imperative to follow a rational choice of animal models, and to carefully interpret the outcomes of a study. Moreover, there exist some enzyme-specific limitations. In this review, any enzyme-related limitation has been mentioned under each corresponding enzyme to aid the understanding and the articulation of thoughts for the readership.
Concluding Remarks
CVD are the leading cause death in the world (3). Increased levels of ROS have been associated with the development and progression of CVD. Several groups of researchers hypothesized that halting ROS production and augmenting antioxidant levels can have a promising outcome in the treatment of CVD (3). On the contrary, the findings of more recent studies do not support this notion. In this review article, we have presented the studies that have taken objective approaches to examine these opposing hypotheses by utilizing transgenic and knockout models of oxidant producing and antioxidant enzymes and their effects on cardiovascular health and disease.
This review has systematically cataloged the phenotypic outcomes in transgenic and knockout models of the same or similar enzymes. The readership will be benefitted from the Tables of global/tissue-specific knockdown (Table 1) and transgenic (Table 2) animal models of oxidative and antioxidant genes.
Author Contributions
MA and SA wrote the article and prepared the Tables of the genetically altered animals. MRA conceptualized the topic theme, edited the article and the tables, and wrote the abstract and the concluding remarks.
Funding
This work was supported in part by the National Heart, Lung, and Blood Institute (NHLBI) and National Institute of General Medical Sciences (NIGMS) grant R01HL133624-01A1 (MRA) and American Heart Association Grant-in-Aid 14GRNT20460291 (MRA).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
Ang-II, angiotensin-II; Akt, protein kinase B; AMPK, 5′ AMP-activated protein kinase; Atox1, antioxidant-1 copper chaperone; ATP, adenosine tri-phosphate; CaMKKb, calcium/calmodulin-dependent protein kinase kinase; cGMP, cyclic guanosine monophosphate; CVD, cardiovascular disease, CVS, cardiovascular system; EC, endothelial cell; eNOS, endothelial nitric oxide synthase; GPx, glutathione peroxidase; HO-1, heme oxygenase; iNOS, inducible nitric oxide synthase; KO, knockout; MAO, monoamine oxidase; MAPK, mitogen-activated protein kinase; MHEC, mouse heart endothelial cell; MMP, metalloproteinase; mTOR, mammalian target of rapamycin; NADPH, nicotinic adenine dinucleotide phosphate oxidase; NF-kB, nuclear factor kappa-light-chain-enhancer of activated B cells; nNOS, neuronal nitric oxide synthase; NO, nitric oxide; NOS, nitric oxide synthase; PI3K, phosphatidylinositol 3-kinase; ROS, reactive oxygen species; SDF1, stromal cell-derived factor 1; SOD, superoxide dismutase; Tet, tetracycline; Tg, transgene; Trx, thioredoxin; TrxR, thioredoxin reductase; VCAM-1, vascular cell adhesion molecule 1; VEGFR2, vascular endothelial growth factor receptor 2; VSMC, vascular smooth muscle cell; WT, wild-type; XO, xanthine oxidoreductase; BMT, bone marrow transplant.
References
1. Ruiz-Lozano P, Rajan P. Stem cells as in vitro models of disease. Curr Stem Cell Res Ther. (2007) 2:280–92. doi: 10.2174/157488807782793772
2. Fearon IM, Gaça MD, Nordskog BK. In vitro models for assessing the potential cardiovascular disease risk associated with cigarette smoking. Toxicol In Vitro (2013) 27:513–22. doi: 10.1016/j.tiv.2012.08.018
3. Shafique E, Choy WC, Liu Y, Feng J, Arthur Lyra BC, Arafah M, et al. Oxidative stress improves coronary endothelial function through activation of the pro-survival kinase AMPK. Aging (2013) 5:515–30. doi: 10.18632/aging.100569
4. Shafique E, Torina A, Reichert K, Colantuono B, Nur N, Zeeshan K, et al. Mitochondrial redox plays a critical role in the paradoxical effects of NAPDH oxidase derived ROS on coronary endothelium. Cardiovasc Res. (2017) 113:234–46. doi: 10.1093/cvr/cvw249
5. Aldosari S, Awad M, Harrington E, Sellke F, Abid M. Subcellular reactive oxygen species (ROS) in cardiovascular pathophysiology. Antioxidants (2018) 7:14. doi: 10.3390/antiox7010014
6. Lee MY, Martin AS, Mehta PK, Dikalova AE, Garrido AM, Datla SR, et al. Mechanisms of vascular smooth muscle NADPH oxidase 1 (Nox1) contribution to injury-induced neointimal formation. Arterioscler Thromb Vasc Biol. (2009) 29:480–7. doi: 10.1161/ATVBAHA.108.181925
7. Gavazzi G, Banfi B, Deffert C, Fiette L, Schappi M, Herrmann F, et al. Decreased blood pressure in NOX1-deficient mice. FEBS Lett. (2006) 580:497–504. doi: 10.1016/j.febslet.2005.12.049
8. Gavazzi G, Deffert C, Trocme C, Schappi M, Herrmann FR, Krause KH. NOX1 deficiency protects from aortic dissection in response to angiotensin II. Hypertension (2007) 50:189–96. doi: 10.1161/HYPERTENSIONAHA.107.089706
9. Sheehan AL, Carrell S, Johnson B, Stanic B, Banfi B, Miller FJ. Role for Nox1 NADPH oxidase in atherosclerosis. Atherosclerosis (2011) 216:321–6. doi: 10.1016/j.atherosclerosis.2011.02.028
10. Judkins CP, Diep H, Broughton BR, Mast AE, Hooker EU, Miller AA, et al. Direct evidence of a role for Nox2 in superoxide production, reduced nitric oxide bioavailability, and early atherosclerotic plaque formation in ApoE−/− mice. Am J Physiol Heart Circ Physiol. (2010) 298:H24–32. doi: 10.1152/ajpheart.00799.2009
11. Chen Z, Keaney JF, Schulz E, Levison B, Shan L, Sakuma M, et al. Decreased neointimal formation in Nox2-deficient mice reveals a direct role for NADPH oxidase in the response to arterial injury. Proc Natl Acad Sci. (2004) 101:13014–9. doi: 10.1073/pnas.0405389101
12. Wang HD, Xu S, Johns DG, Du Y, Quinn MT, Cayatte AJ, et al. Role of NADPH oxidase in the vascular hypertrophic and oxidative stress response to angiotensin II in mice. Circ Res. (2001) 88:947–53. doi: 10.1161/hh0901.089987
13. Bendall JK, Cave AC, Heymes C, Gall N, Shah AM. Pivotal role of a gp91phox-containing NADPH oxidase in angiotensin II-induced cardiac hypertrophy in mice. Circulation (2002) 105:293–6. doi: 10.1161/hc0302.103712
14. Feng J, Damrauer SM, Lee M, Sellke FW, Ferran C, Abid MR. Endothelium-dependent coronary vasodilatation requires NADPH oxidase-derived reactive oxygen species. Arterioscler Thromb Vasc Biol. (2010) 30:1703–10. doi: 10.1161/ATVBAHA.110.209726
15. Landmesser U, Cai H, Dikalov S, McCann L, Hwang J, Jo H, et al. Role of p47phox in vascular oxidative stress and hypertension caused by angiotensin II. Hypertension (2002) 40:511–15. doi: 10.1161/01.HYP.0000032100.23772.98
16. Vendrov AE, Madamanchi NR, Hakim ZS, Rojas M, Runge MS. Thrombin and NAD(P)H oxidase-mediated regulation of CD44 and BMP4-Id pathway in VSMC, restenosis, and atherosclerosis. Circ Res. (2006) 98:1254–63. doi: 10.1161/01.RES.0000221214.37803.79
17. Vendrov AE, Hakim ZS, Madamanchi NR, Rojas M, Madamanchi C, Runge MS. Atherosclerosis is attenuated by limiting superoxide generation in both macrophages and vessel wall cells. Arterioscler Thromb Vasc Biol. (2007) 27:2714–21. doi: 10.1161/ATVBAHA.107.152629
18. Kuhlencordt PJ, Gyurko R, Han F, Scherrer-Crosbie M, Aretz TH, Hajjar R, et al. Accelerated atherosclerosis, aortic aneurysm formation, and ischemic heart disease in apolipoprotein E/endothelial nitric oxide synthase double-knockout mice. Circulation (2001) 104:448–54. doi: 10.1161/hc2901.091399
19. Ponnuswamy P, Schröttle A, Ostermeier E, Grüner S, Huang PL, Ertl G, et al. ENOS protects from atherosclerosis despite relevant superoxide production by the enzyme in ApoE−/− mice. PLoS ONE (2012) 7:e30193. doi: 10.1371/journal.pone.0030193
20. Kuhlencordt PJ, Chen J, Han F, Astern J, Huang PL. Genetic deficiency of inducible nitric oxide synthase reduces atherosclerosis and lowers plasma lipid peroxides in apolipoprotein E-knockout mice. Circulation (2001) 103:3099–104. doi: 10.1161/01.CIR.103.25.3099
21. Huang H, Koelle P, Fendler M, Schröttle A, Czihal M, Hoffmann U, et al. Induction of inducible nitric oxide synthase (iNOS) expression by oxLDL inhibits macrophage derived foam cell migration. Atherosclerosis (2014) 235:213–22. doi: 10.1016/j.atherosclerosis.2014.04.020
22. Detmers PA, Hernandez M, Mudgett J, Hassing H, Burton C, Mundt S, et al. Deficiency in inducible nitric oxide synthase results in reduced atherosclerosis in apolipoprotein E-deficient mice. J Immunol. (2000) 165:3430–5. doi: 10.4049/jimmunol.165.6.3430
23. Kuhlencordt PJ, Hotten S, Schodel J, Rutzel S, Hu K, Widder J, et al. Atheroprotective effects of neuronal nitric oxide synthase in apolipoprotein e knockout mice. Arterioscler Thromb Vasc Biol. (2006) 26:1539–44. doi: 10.1161/01.ATV.0000223143.88128.19
24. Morishita T, Tsutsui M, Shimokawa H, Horiuchi M, Tanimoto A, Suda O, et al. Vasculoprotective roles of neuronal nitric oxide synthase. FASEB J. (2002) 16:1994–6. doi: 10.1096/fj.02-0155fje
25. Kaludercic N, Takimoto E, Nagayama T, Feng N, Lai EW, Bedja D, et al. Monoamine oxidase a-mediated enhanced catabolism of norepinephrine contributes to adverse remodeling and pump failure in hearts with pressure overload. Circ Res. (2010) 106:193–202. doi: 10.1161/CIRCRESAHA.109.198366
26. Kaludercic N, Mialet-Perez J, Paolocci N, Parini A, Di Lisa F. Monoamine oxidases as sources of oxidants in the heart. J Mol Cell Cardiol. (2014) 73:34–42. doi: 10.1016/j.yjmcc.2013.12.032
27. Lairez O, Calise D, Bianchi P, Ordener C, Spreux-Varoquaux O, Guilbeau-Frugier C, et al. Genetic deletion of MAO-A promotes serotonin-dependent ventricular hypertrophy by pressure overload. J Mol Cell Cardiol. (2009) 46:587–95. doi: 10.1016/j.yjmcc.2008.12.017
28. Kaludercic N, Carpi A, Nagayama T, Sivakumaran V, Zhu G, Lai EW, et al. Monoamine oxidase B prompts mitochondrial and cardiac dysfunction in pressure overloaded hearts. Antioxid Redox Signal. (2014) 20:267–80. doi: 10.1089/ars.2012.4616
29. Ballinger SW, Patterson C, Knight-Lozano CA, Burow DL, Conklin CA, Hu Z, et al. Mitochondrial integrity and function in atherogenesis. Circulation (2002) 106:544–9. doi: 10.1161/01.CIR.0000023921.93743.89
30. Ozumi K, Sudhahar V, Kim HW, Chen G-F, Kohno T, Finney L, et al. Role of copper transport protein antioxidant 1 in angiotensin II-induced hypertension: a key regulator of extracellular superoxide dismutase. Hypertension (2012) 60:476–86. doi: 10.1161/HYPERTENSIONAHA.111.189571
31. Sudhahar V, Urao N, Oshikawa J, McKinney RD, Llanos RM, Mercer JFB, et al. Copper transporter ATP7A protects against endothelial dysfunction in type 1 diabetic mice by regulating extracellular superoxide dismutase. Diabetes (2013) 62:3839–50. doi: 10.2337/db12-1228
32. Jeney V, Itoh S, Wendt M, Gradek Q, Ushio-Fukai M, Harrison DG, et al. Role of antioxidant-1 in extracellular superoxide dismutase function and expression. Circ Res. (2005) 96:723–9. doi: 10.1161/01.RES.0000162001.57896.66
33. Forgione MA, Weiss N, Heydrick S, Cap A, Klings ES, Bierl C, et al. Cellular glutathione peroxidase deficiency and endothelial dysfunction. Am J Physiol Heart Circ Physiol. (2002) 282:H1255–61. doi: 10.1152/ajpheart.00598.2001
34. Das A, Sudhahar V, Chen GF, Kim HW, Youn SW, Finney L, et al. Endothelial antioxidant-1: a key mediator of copper-dependent wound healing in vivo. Sci Rep. (2016) 6:33783. doi: 10.1038/srep33783
35. Forgione MA, Cap A, Liao R, Moldovan NI, Eberhardt RT, Lim CC, et al. Heterozygous cellular glutathione peroxidase deficiency in the mouse: abnormalities in vascular and cardiac function and structure. Circulation (2002) 106:1154–8. doi: 10.1161/01.CIR.0000026820.87824.6A
36. Torzewski M, Ochsenhirt V, Kleschyov AL, Oelze M, Daiber A, Li H, et al. Deficiency of glutathione peroxidase-1 accelerates the progression of atherosclerosis in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. (2007) 27:850–7. doi: 10.1161/01.ATV.0000258809.47285.07
37. Conrad M, Jakupoglu C, Moreno SG, Lippl S, Banjac A, Schneider M, et al. Essential role for mitochondrial thioredoxin reductase in hematopoiesis, heart development, and heart function. Mol Cell Biol. (2004) 24:9414–23. doi: 10.1128/MCB.24.21.9414-9423.2004
38. Dikalova AE, Góngora MC, Harrison DG, Lambeth JD, Dikalov S, Griendling KK, et al. Upregulation of Nox1 in vascular smooth muscle leads to impaired endothelium-dependent relaxation via enos uncoupling. AJP Heart Circ Physiol. (2010) 299:ajpheart. doi: 10.1152/ajpheart.00242.2010
39. Bendall JK, Rinze R, Adlam D, Tatham AL, De Bono J, Channon KM. Endothelial Nox2 overexpression potentiates vascular oxidative stress and hemodynamic response to angiotensin II: studies in endothelial-targeted Nox2 transgenic mice. Circ Res. (2007) 100:1016–25. doi: 10.1161/01.RES.0000263381.83835.7b
40. Chen L, Hou X, Xiao J, Kuroda J, Ago T, Sadoshima J, et al. Both hydrogen peroxide and transforming growth factor beta 1 contribute to endothelial Nox4 mediated angiogenesis in endothelial Nox4 transgenic mouse lines. Biochim Biophys Acta Mol Basis Dis. (2014) 1842:2489–99. doi: 10.1016/j.bbadis.2014.10.007
41. Gaudin PB, Rayburn BK, Hutchins GM, Kasper EK, Baughman KL, Goodman SN, et al. Peritransplant injury to the myocardium associated with the development of accelerated arteriosclerosis in heart transplant recipients. Am J Surg Pathol. (1994) 18:338–46. doi: 10.1097/00000478-199404000-00002
42. Wang P, Chen H, Qin H, Sankarapandi S, Becher MW, Wong PC, et al. Overexpression of human copper, zinc-superoxide dismutase (SOD1) prevents postischemic injury. Proc Natl Acad Sci USA. (1998) 95:4556–60. doi: 10.1073/pnas.95.8.4556
43. Yamamoto M, Yang G, Hong C, Liu J, Holle E, Yu X, et al. Inhibition of endogenous thioredoxin in the heart increases oxidative stress and cardiac hypertrophy. J Clin Invest. (2003) 112:1395–406. doi: 10.1172/JCI200317700
44. Bianchi P, Kunduzova O, Masini E, Cambon C, Bani D, Raimondi L, et al. Oxidative stress by monoamine oxidase mediates receptor-independent cardiomyocyte apoptosis by serotonin and postischemic myocardial injury. Circulation (2005) 112:3297–305. doi: 10.1161/CIRCULATIONAHA.104.528133
45. Villeneuve C, Guilbeau-Frugier C, Sicard P, Lairez O, Ordener C, Duparc T, et al. p53-PGC-1α Pathway mediates oxidative mitochondrial damage and cardiomyocyte necrosis induced by monoamine oxidase-A upregulation: role in chronic left ventricular dysfunction in mice. Antioxid Redox Signal. (2013) 18:5–18. doi: 10.1089/ars.2011.4373
46. Maurel A, Hernandez C, Kunduzova O, Bompart G, Cambon C, Parini A, et al. Age-dependent increase in hydrogen peroxide production by cardiac monoamine oxidase A in rats. Am J Physiol Heart Circ Physiol. (2003) 284:H1460–7. doi: 10.1152/ajpheart.00700.2002
47. Lassègue B, Griendling KK. NADPH oxidases: functions and pathologies in the vasculature. Arterioscler Thromb Vasc Biol. (2010) 30:653–61. doi: 10.1161/ATVBAHA.108.181610
48. Bengtsson SHM, Gulluyan LM, Dusting GJ, Drummond GR. Novel isoforms of NADPH oxidase in vascular physiology and pathophysiology. Clin Exp Pharmacol Physiol. (2003) 30:849–54. doi: 10.1046/j.1440-1681.2003.03929.x
49. Szöcs K, Lassègue B, Sorescu D, Hilenski LL, Valppu L, Couse TL, et al. Upregulation of nox-based NAD(P)H oxidases in restenosis after carotid injury. Arterioscler Thromb Vasc Biol. (2002) 22:21–7. doi: 10.1161/hq0102.102189
50. Schröder K, Helmcke I, Palfi K, Krause K-H, Busse R, Brandes RP. Nox1 mediates basic fibroblast growth factor-induced migration of vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. (2007) 27:1736–43. doi: 10.1161/atvbaha.107.142117
51. Mollnau H, Wendt M, Szöcs K, Lassègue B, Schulz E, Oelze M, et al. Effects of angiotensin II infusion on the expression and function of NAD(P)H oxidase and components of nitric oxide/cGMP signaling. Circ Res. (2002) 90:E58–65. doi: 10.1161/01.RES.0000012569.55432.02
52. Sadoshima J, Izumo S. Molecular characterization of angiotensin II–induced hypertrophy of cardiac myocytes and hyperplasia of cardiac fibroblasts. Critical role of the AT1 receptor subtype. Circ Res. (1993) 73:413–23. doi: 10.1161/01.RES.73.3.413
53. Taubman MB. Angiotensin II: a vasoactive hormone with ever-increasing biological roles. Circ Res. (2003) 92:9–11. doi: 10.1161/01.res.0000052920.70316.ae
54. Giani JF, Shah KH, Khan Z, Bernstein EA, Shen XZ, McDonough AA, et al. The intrarenal generation of angiotensin II is required for experimental hypertension. Curr Opin Pharmacol. (2015) 21:73–81. doi: 10.1016/j.coph.2015.01.002
55. Ho Y-C, Wu M-L, Gung P-Y, Chen C-H, Kuo C-C, Yet S-F. Heme oxygenase-1 deficiency exacerbates angiotensin II-induced aortic aneurysm in mice. Oncotarget (2016) 7:67760–76. doi: 10.18632/oncotarget.11917
56. Paolocci N, Biondi R, Bettini M, Lee CI, Berlowitz CO, Rossi R, et al. Oxygen radical-mediated reduction in basal and agonist-evoked no release in isolated rat heart. J Mol Cell Cardiol. (2001) 33:671–9. doi: 10.1006/jmcc.2000.1334
57. Daugherty A, Cassis L. Angiotensin II-mediated development of vascular diseases. Trends Cardiovasc Med. (2004) 14:117–20. doi: 10.1016/j.tcm.2004.01.002
59. Ray R, Murdoch CE, Wang M, Santos CX, Zhang M, Alom-Ruiz S, et al. Endothelial Nox4 NADPH oxidase enhances vasodilatation and reduces blood pressure in vivo. Arterioscler Thromb Vasc Biol. (2011) 31:1368–76. doi: 10.1161/ATVBAHA.110.219238
60. Craige SM, Chen K, Pei Y, Li C, Huang X, Chen C, et al. NADPH oxidase 4 promotes endothelial angiogenesis through endothelial nitric oxide synthase activation. Circulation (2011) 124:731–40. doi: 10.1161/CIRCULATIONAHA.111.030775
61. Schröder K, Zhang M, Benkhoff S, Mieth A, Pliquett R, Kosowski J, et al. Nox4 is a protective reactive oxygen species generating vascular NADPH oxidase. Circ Res. (2012) 110:1217–25. doi: 10.1161/CIRCRESAHA.112.267054
62. Barry-Lane PA, Patterson C, Merwe MV, Hu Z, Holland SM, Yeh ET, et al. P47phox is required for atherosclerotic lesion progression in Apo−/− mice. J Clin Invest. (2001) 108:1513–22. doi: 10.1172/JCI11927.
63. Glantzounis GK, Tsimoyiannis EC, Kappas AM, Galaris DA. Uric acid and oxidative stress. Curr Pharm Des. (2005) 11:4145–51. doi: 10.2174/138161205774913255
64. Battelli MG, Bolognesi A, Polito L. Pathophysiology of circulating xanthine oxidoreductase: new emerging roles for a multi-tasking enzyme. Biochim Biophys Acta Mol Basis Dis. (2014) 1842:1502–17. doi: 10.1016/j.bbadis.2014.05.022
65. Kostić DA, Dimitrijević DS, Stojanović GS, Palić IR, Dordević AS, Ickovski JD. Xanthine oxidase: isolation, assays of activity, and inhibition. J Chem. (2015) 2015:294858. doi: 10.1155/2015/294858
66. Abadeh S, Case PC, Harrison R. Purification of xanthine oxidase from human heart. Biochem Soc Trans. (1993) 21:99S. doi: 10.1042/bst021099s
67. De Jong JW, Van der Meer P, Nieukoop AS, Huizer T, Stroeve RJ, Bos E. Xanthine oxidoreductase activity in perfused hearts of various species, including humans. Circ Res. (1990) 67:770–3. doi: 10.1161/01.RES.67.3.770
68. Berry CE, Hare JM. Xanthine oxidoreductase and cardiovascular disease: molecular mechanisms and pathophysiological implications. J Physiol. (2004) 555:589–606. doi: 10.1113/jphysiol.2003.055913
69. Landmesser U, Spiekermann S, Dikalov S, Tatge H, Wilke R, Kohler C, et al. Vascular oxidative stress and endothelial dysfunction in patients with chronic heart failure: role of xanthine-oxidase and extracellular superoxide dismutase. Circulation (2002) 106:3073–8. doi: 10.1161/01.CIR.0000041431.57222.AF
70. Brown JM, Terada LS, Grosso MA, Whitmann GJ, Velasco SE, Patt A, et al. Xanthine oxidase produces hydrogen peroxide which contributes to reperfusion injury of ischemic, isolated, perfused rat hearts. J Clin Invest. (1988) 81:1297–301. doi: 10.1172/JCI113448
71. Terada LS, Rubinstein JD, Lesnefsky EJ, Horwitz LD, Leff JA, Repine JE. Existence and participation of xanthine oxidase in reperfusion injury of ischemic rabbit myocardium. Am J Physiol. (1991) 260:H805–10. doi: 10.1152/ajpheart.1991.260.3.H805
72. Minhas KM, Saraiva RM, Schuleri KH, Lehrke S, Zheng M, Saliaris AP, et al. Xanthine oxidoreductase inhibition causes reverse remodeling in rats with dilated cardiomyopathy. Circ Res. (2006) 98:271–9. doi: 10.1161/01.RES.0000200181.59551.71
73. Gimpel JA, Lahpor JR, van der Molen AJ, Damen J, Hitchcock JF. Reduction of reperfusion injury of human myocardium by allopurinol: a clinical study. Free Radic Biol Med. (1995) 19:251–5.
74. Guan W, Osanai T, Kamada T, Hanada H, Ishizaka H, Onodera H, et al. Effect of allopurinol pretreatment on free radical generation after primary coronary angioplasty for acute myocardial infarction. J Cardiovasc Pharmacol. (2003) 41:699–705. doi: 10.1097/00005344-200305000-00005
75. Baldus S, Müllerleile K, Chumley P, Steven D, Rudolph V, Lund GK, et al. Inhibition of xanthine oxidase improves myocardial contractility in patients with ischemic cardiomyopathy. Free Radic Biol Med. (2006) 41:1282–8. doi: 10.1016/j.freeradbiomed.2006.07.010
76. Madamanchi NR, Runge MS. Redox signaling in cardiovascular health and disease. Free Radic Biol Med. (2013) 0:473–501. doi: 10.1016/j.freeradbiomed.2013.04.001
77. Gawaz M, Langer H, May AE. Platelets in inflammation and atherogenesis. J Clin Invest. (2005) 115:3378–84. doi: 10.1172/JCI27196
78. Wilcox JN, Subramanian RR, Sundell CL, Ross Tracey W, Pollock JS, Harrison DG, et al. Expression of multiple isoforms of nitric oxide synthase in normal and atherosclerotic vessels. Arterioscler Thromb Vasc Biol. (1997) 17:2479–88. doi: 10.1161/01.ATV.17.11.2479
79. Kaludercic N, Carpi A, Menabò R, Lisa FD, Paolocci N. Monoamine oxidases (MAO) in the pathogenesis of heart failure and ischemia/reperfusion injury. Biochim Biophys Acta Mol Cell Res. (2011) 1813:1323–32. doi: 10.1016/j.bbamcr.2010.09.010
80. Sivasubramaniam SD, Finch CC, Rodriguez MJ, Mahy N, Billett EE. A comparative study of the expression of monoamine oxidase-A and -B mRNA and protein in non-CNS human tissues. Cell Tissue Res. (2003) 313:291–300. doi: 10.1007/s00441-003-0765-6
81. Ighodaro O, Akinloye O. First line defence antioxidants-superoxide dismutase (SOD), catalase (CAT) and glutathione peroxidase (GPX): their fundamental role in the entire antioxidant defence grid. Alexandria J Med. (2017). doi: 10.1016/j.ajme.2017.09.001
82. Nimse SB, Pal D. Free radicals, natural antioxidants, and their reaction mechanisms. RSC Adv. (2015) 5:27986–8006. doi: 10.1039/C4RA13315C
83. Fridovich I. Superoxide radical and superoxide dismutases. Ann Rev Biochem. (1995) 64:97–112. doi: 10.1146/annurev.bi.64.070195.000525
84. Tanaka M, Mokhtari GK, Terry RD, Balsam LB, Lee K, Kofidis T, et al. Overexpression of human copper/zinc superoxide dismutase (SOD1) suppresses ischemia-reperfusion injury and subsequent development of graft coronary artery disease in murine cardiac grafts. Circulation (2004) 110:II200–6. doi: 10.1161/01.cir.0000138390.81640.54
85. Das DK, Maulik N. Antioxidant effectiveness in ischemia-reperfusion tissue injury. Methods Enzymol. (1994) 233:601–10. doi: 10.1016/S0076-6879(94)33063-8
86. Kalogeris T, Baines CP, Krenz M, Korthuis RJ. Cell biology of ischemia/reperfusion injury. Int Rev Cell Mol Biol. (2012) 298:229–317. doi: 10.1016/b978-0-12-394309-5.00006-7
87. Omar BA, Gad NM, Jordan MC, Striplin SP, Russell WJ, Downey JM, et al. Cardioprotection by Cu, Zn-superoxide dismutase is lost at high doses in the reoxygenated heart. Free Radic Biol Med. (1990) 9:465–71. doi: 10.1016/0891-5849(90)90123-Z
88. Hamilton RT. Mouse models of oxidative stress indicate a role for modulating healthy aging. J Clin Exp Pathol. (2012) 01:005. doi: 10.4172/2161-0681.S4-005
89. Van Remmen H, Williams MD, Guo Z, Estlack L, Yang H, Carlson EJ, et al. Knockout mice heterozygous for Sod2 show alterations in cardiac mitochondrial function and apoptosis. Am J Physiol Heart Circ Physiol. (2001) 281:H1422–32. doi: 10.1152/ajpheart.2001.281.3.H1422
90. Fukai T, Folz RJ, Landmesser U, Harrison DG. Extracellular superoxide dismutase and cardiovascular disease. Cardiovasc Res. (2002) 55:239–49. doi: 10.1016/S0008-6363(02)00328-0
91. Oury T, Day B, Crapo J. Extracellular superoxide dismutase: a regulator of nitric oxide bioavailability. Lab Invest. (1996) 75:617–36.
92. Fattman CL, Schaefer LM, Oury TD. Extracellular superoxide dismutase in biology and medicine. Free Radic Biol Med. (2003) 35:236–56. doi: 10.1016/S0891-5849(03)00275-2
93. Jung O, Marklund SL, Geiger H, Pedrazzini T, Busse R, Brandes RP. Extracellular superoxide dismutase is a major determinant of nitric oxide bioavailability: in vivo and ex vivo evidence from ecSOD-deficient mice. Circ Res. (2003) 93:622–9. doi: 10.1161/01.RES.0000092140.81594.A8
94. Itoh S, Ozumi K, Kim H, Nakagawa O, Mckinney R, Folz R, et al. Novel mechanism for regulation of extracellular SOD transcription and activity by copper: role of antioxidant-1. Free Radic Biol Med. (2009) 46:95–104. doi: 10.1016/j.freeradbiomed.2008.09.039
95. Qin Z, Itoh S, Jeney V, Ushio-Fukai M, Fukai T. Essential role for the Menkes ATPase in activation of extracellular superoxide dismutase: implication for vascular oxidative stress. FASEB J. (2006) 20:334–6. doi: 10.1096/fj.05-4564fje
96. Lob HE, Vinh A, Li L, Blinder Y, Offermanns S, Harrison DG. Role of vascular extracellular superoxide dismutase in hypertension. Hypertension (2011) 58:232–9. doi: 10.1161/HYPERTENSIONAHA.111.172718
97. Nakamura H, Nakamura K, Yodoi J. Redox regulation of cellular activation. Ann Rev Immunol. (1997) 15:351–69. doi: 10.1146/annurev.immunol.15.1.351
98. Nordberg J, Arnér ESJ. Reactive oxygen species, antioxidants, and the mammalian thioredoxin system. Free Radic Biol Med. (2001) 31:1287–312. doi: 10.1016/S0891-5849(01)00724-9
99. Watson WH, Yang X, Choi YE, Jones DP, Kehrer JP. Thioredoxin and its role in toxicology. Toxicol Sci. (2004) 78:3–14. doi: 10.1093/toxsci/kfh050
100. Ago T, Sadoshima J. Thioredoxin and ventricular remodeling. J Mol Cell Cardiol. (2006) 41:762–73. doi: 10.1016/j.yjmcc.2006.08.006
101. Schwertassek U, Weingarten L, Dick TP. Identification of redox-active cell-surface proteins by mechanism-based kinetic trapping. Science's STKE (2007) 2007:pl8. doi: 10.1126/stke.4172007pl8
102. Yoshioka J, Chutkow WA, Lee S, Kim JB, Yan J, Tian R, et al. Deletion of thioredoxin-interacting protein in mice impairs mitochondrial function but protects the myocardium from ischemia-reperfusion injury. J Clin Invest. (2012) 122:267–79. doi: 10.1172/JCI44927
103. Tanaka T, Nishiyama Y, Okada K, Hirota K, Matsui M, Yodoi J, et al. Induction and nuclear translocation of thioredoxin by oxidative damage in the mouse kidney: independence of tubular necrosis and sulfhydryl depletion. Lab Invest. (1997) 77:145–55.
104. Watson WH, Pohl J, Montfort WR, Stuchlik O, Reed MS, Powis G, et al. Redox potential of human thioredoxin 1 and identification of a second dithiol/disulfide motif. J Biol Chem. (2003) 278:33408–15. doi: 10.1074/jbc.m211107200
105. Casagrande S, Bonetto V, Fratelli M, Gianazza E, Eberini I, Massignan T, et al. Glutathionylation of human thioredoxin: a possible crosstalk between the glutathione and thioredoxin systems. Proc Natl Acad Sci. (2002) 99:9745–9. doi: 10.1073/pnas.152168599
106. Haendeler J, Hoffmann J, Tischler V, Berk BC, Zeiher AM, Dimmeler S. Redox regulatory and anti-apoptotic functions of thioredoxin depend on S-nitrosylation at cysteine 69. Nat Cell Biol. (2002) 4:743–9. doi: 10.1038/ncb851
107. Azevedo PS, Polegato BF, Minicucci MF, Paiva SAR, Zornoff LAM. Cardiac remodeling: concepts, clinical impact, pathophysiological mechanisms and pharmacologic treatment. Arq Bras Cardiol. (2015) 106:62–9. doi: 10.5935/abc.20160005
108. Kuster GM, Pimentel DR, Adachi T, Ido Y, Brenner DA, Cohen RA, et al. α-Adrenergic receptor-stimulated hypertrophy in adult rat ventricular myocytes is mediated via thioredoxin-1-sensitive oxidative modification of thiols on ras. Circulation (2005) 111:1192–8. doi: 10.1161/01.CIR.0000157148.59308.F5
109. Iliodromitis EK, Lazou A, Kremastinos DT. Ischemic preconditioning: protection against myocardial necrosis and apoptosis. Vasc Health Risk Manag. (2007) 3:629–37.
110. Gray MO, Zhou HZ, Schafhalter-Zoppoth I, Zhu P, Mochly-Rosen D, Messing RO. Preservation of base-line hemodynamic function and loss of inducible cardioprotection in adult mice lacking protein kinase Cε. J Biol Chem. (2004) 279:3596–604. doi: 10.1074/jbc.M311459200
111. Turoczi T, Chang VWH, Engelman RM, Maulik N, Ho YS, Das DK. Thioredoxin redox signaling in the ischemic heart: an insight with transgenic mice overexpressing Trx1. J Mol Cell Cardiol. (2003) 35:695–704. doi: 10.1016/S0022-2828(03)00117-2
112. Scarabelli TM, Gottlieb RA. Functional and clinical repercussions of myocyte apoptosis in the multifaceted damage by ischemia/reperfusion injury: old and new concepts after 10 years of contributions. Cell Death Differ. (2004) 11:S144–52. doi: 10.1038/sj.cdd.4401544
113. Chen K, Chen J, Li D, Zhang X, Mehta JL. Angiotensin II regulation of collagen type I expression in cardiac fibroblasts: modulation by PPAR-gamma ligand pioglitazone. Hypertension (2004) 44:655–61. doi: 10.1161/01.HYP.0000144400.49062.6b
114. Saitoh M, Nishitoh H, Fujii M, Takeda K, Tobiume K, Sawada Y, et al. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. (1998) 17:2596–606. doi: 10.1093/emboj/17.9.2596
115. Datta SR, Dudek H, Xu T, Masters S, Haian F, Gotoh Y, et al. Akt phosphorylation of BAD couples survival signals to the cell- intrinsic death machinery. Cell (1997) 91:231–41. doi: 10.1016/S0092-8674(00)80405-5
116. Drechsel DA, Patel M. Respiration-dependent H2O2 removal in brain mitochondria via the thioredoxin/peroxiredoxin system. J Biol Chem. (2010) 285:27850–8. doi: 10.1074/jbc.m110.101196
117. Stanley BA, Sivakumaran V, Shi S, McDonald I, Lloyd D, Watson WH, et al. Thioredoxin reductase-2 is essential for keeping low levels of H2O2 emission from isolated heart mitochondria. J Biol Chem. (2011) 286:33669–77. doi: 10.1074/jbc.M111.284612
118. Huang Q, Zhou HJ, Zhang H, Huang Y, Hinojosa-Kirschenbaum F, Fan P, et al. Thioredoxin-2 inhibits mitochondrial reactive oxygen species generation and apoptosis stress kinase-1 activity to maintain cardiac function. Circulation (2015) 131:1082–97. doi: 10.1161/CIRCULATIONAHA.114.012725
119. Camacho P, Fan H, Liu Z, He J-Q. Small mammalian animal models of heart disease. Am J Cardiovasc Dis. (2016) 6:70–80. doi: 10.3390/jcdd3040030
120. Milani-Nejad N, Janssen PML. Small and large animal models in cardiac contraction research: advantages and disadvantages. Pharmacol Ther. (2014) 141:235–49. doi: 10.1016/j.pharmthera.2013.10.007
121. Leong XF, Ng CY, Jaarin K. Animal models in cardiovascular research: hypertension and atherosclerosis. BioMed Res Int. (2015) 2015:528757. doi: 10.1155/2015/528757
122. Zaragoza C, Gomez-Guerrero C, Martin-Ventura JL, Blanco-Colio L, Lavin B, Mallavia B, et al. Animal models of cardiovascular diseases. J Biomed Biotechnol. (2011) 2011:497841. doi: 10.1155/2011/497841
Key Concepts
• Paradoxical effects of ROS: ROS may exert beneficial or detrimental effects on cardiovascular system depending on the levels, sub-cellular localization and duration 1281 of exposure to ROS.
• Regulatable oxidant and antioxidant enzymes: Genetically modified animals with on-off capability of the transgene.
• Tissue-specific: Tissue-specific (endothelium, cardiac, VSMC) knockout and transgene models
• Sub-cellular ROS: ROS in cytosol vs mitochondria vs. ER.
Keywords: reactive oxygen species, cardiovascular disease, coronary endothelium, animal model, oxidant enzymes, antioxidant enzymes
Citation: Awad MA, Aldosari SR and Abid MR (2018) Genetic Alterations in Oxidant and Anti-Oxidant Enzymes in the Vascular System. Front. Cardiovasc. Med. 5:107. doi: 10.3389/fcvm.2018.00107
Received: 07 May 2018; Accepted: 16 July 2018;
Published: 09 August 2018.
Edited by:
Jason Bazil, Michigan State University, United StatesReviewed by:
Sabzali Javadov, University of Puerto Rico, Puerto RicoNazareno Paolocci, Johns Hopkins University, United States
Copyright © 2018 Awad, Aldosari and Abid. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: M. Ruhul Abid, cnVodWxfYWJpZEBicm93bi5lZHU=