 Anindita Das
Anindita Das Arun Samidurai
Arun Samidurai Fadi N. Salloum
Fadi N. Salloum- Pauley Heart Center, Division of Cardiology, Department of Internal Medicine, Virginia Commonwealth University, Richmond, VA, United States
After being long considered as “junk” in the human genome, non-coding RNAs (ncRNAs) currently represent one of the newest frontiers in cardiovascular disease (CVD) since they have emerged in recent years as potential therapeutic targets. Different types of ncRNAs exist, including small ncRNAs that have fewer than 200 nucleotides, which are mostly known as microRNAs (miRNAs), and long ncRNAs that have more than 200 nucleotides. Recent discoveries on the role of ncRNAs in epigenetic and transcriptional regulation, atherosclerosis, myocardial ischemia/reperfusion (I/R) injury and infarction (MI), adverse cardiac remodeling and hypertrophy, insulin resistance, and diabetic cardiomyopathy prompted vast interest in exploring candidate ncRNAs for utilization as potential therapeutic targets and/or diagnostic/prognostic biomarkers in CVDs. This review will discuss our current knowledge concerning the roles of different types of ncRNAs in cardiovascular health and disease and provide some insight on the cardioprotective signaling pathways elicited by the non-coding genome. We will highlight important basic and clinical breakthroughs that support employing ncRNAs for treatment or early diagnosis of a variety of CVDs, and also depict the most relevant limitations that challenge this novel therapeutic approach.
Introduction
The discovery of the new class of ribonucleic acids (RNAs), namely non-coding RNAs (ncRNAs), revolutionized our knowledge about the epigenetic, post-transcriptional, and post-translational modification of gene expression in the regulation of tissue homeostasis in health and disease. Recent advances in the field of genomics enabled with technologies like next generation sequencing (NGS), ChIP RNA Seq, and transcriptome analysis have offered new perspectives and completely changed our understanding on small ncRNA molecules, once considered as “junk DNA.” Nearly 99% of the genome consists of non-coding DNA, whereas only 1% codes for functional proteins, which reflects the complexity and importance of ncRNAs in controlling gene expression. Regulatory ncRNAs such as microRNAs (miRNAs) and long non-coding RNAs (lncRNAs) have drastically impacted research in multiple fields, including cardiovascular diseases (1–3) (CVD), diabetes (4, 5), and cancer (6, 7). The epigenetic regulation of these ncRNAs, like miRs, plays a very significant role both in the early stage of development and during the pathogenesis of heart disease (3, 8, 9).
Cardiovascular disease (CVD) remains the leading cause of mortality and morbidity worldwide (10, 11). CVD is a broad term used to describe abnormalities affecting the heart and its associated circulatory system, such as atherosclerosis, myocardial ischemia/reperfusion (I/R) injury and infarction (MI), hypertension, and arrhythmias. The occurrence of risk factors, such as diabetes, obesity and advanced age leads to substantial complications of CVD ultimately leading to heart failure (HF). Although current management has improved survival in patients with CVD, such therapies do not fully address the underlying cause and, as a result, HF progresses. This highlights an urgent need to develop novel diagnostic and therapeutic approaches to alleviate symptoms, improve cardiac function and quality of life, slow disease progression, and reduce mortality in patients with CVD and HF (12, 13). Emerging concepts based on genomic information have redesigned diagnostic and treatment strategies, enabling early detection of abnormalities and offering hope for more effective treatment options. This review aims to provide the readers with an updated summary on the role of ncRNAs in cardiovascular physiology and pathophysiology, with emphasis on MI, atherosclerosis, diabetic cardiomyopathy, and HF. We will also review the potential utility of ncRNAs as a therapeutic option and describe some of the current limitations.
Non-coding RNAs: Classification and Mechanisms
Non-coding RNAs are classified into several types based on their length and mechanism of gene regulation. This includes miRNAs (<25 nt), lncRNA (>200 nt), piRNAs (RNA-protein complexes), and siRNAs (double-stranded RNA, ~20–25 nt), which are among the most thoroughly investigated ncRNAs. Even though regulation of gene expression is the primary function of these ncRNAs, they each achieve this goal via different methods.
miRNA Biosynthesis and Function
MicroRNAs (miRNAs) are small ncRNAs that are approximately 18–25 nt in length and regulate gene expression by binding to the 3′UTR of mRNA, leading to either degradation or translational suppression of its target mRNA (14, 15). The first miRNA was discovered in 1993 in a study examining developmental regulatory genes in Caenorhabditis elegans (14). miRNAs are among the most abundantly occurring ncRNAs that are widely distributed in several tissues and are conserved among species. There are more than 2,500 miRNAs reported in the human genome (miRBase) (16) that are known to specifically regulate gene expression. Almost all miRNAs are transcribed from either introns or intergenic region, by the enzyme RNA polymerase II or in some cases RNA polymerase III into a hairpin precursor molecule called primary-miRNA. These primary-miRNAs (100 nt) undergo maturation process by the enzyme Drosha, which cleaves them to Pre-miRNAs (~70 nt) (17). Pre-miRNAs are later exported to the cytoplasm by Exportin-5. Once out of the nucleus, the pre-miRNA further undergoes cleavage to a mature double-stranded miRNA of 22 nt by the enzyme RNase III (18). The active strand of the mature miRNA binds to RNA-induced silencing complex (RISC) and interferes in the transactivation-responsive RNA-binding protein (TRBP) and Argonaute 2 (Ago2) and inhibits the 3′UTR of the target mRNA through base-pair interactions (19) and negatively regulates its expression. These steps are collectively illustrated in Figure 1. Each miRNA can have multiple targets and is primarily based on the presence of complementary binding sequence in the mRNA. The extent of sequence complementarity between the miRNA and mRNA determines whether the target mRNA is destined for complete degradation or translational inhibition. Nucleotide sequence (2–7) at the 5′ end of miRNA, which forms the seed region, is critical for the formation miRNA-mRNA binding complex. Also, miRNAs can be generated from both guide as well as passenger strands of the DNA and are denoted by 3-p or 5-p suffix. The opposite strand, often called the passenger strand due to its relatively lower levels in the steady state, is denoted with an asterisk (*) and is normally degraded. In some cases, miRNA generated from both strands are viable and are incorporated in RISC complex and become functional miRNAs that target different mRNAs (20, 21). The loci of miRNA are located at various genomic contexts and while the majority of them are found in the intronic region, they can also be encoded at coding transcripts and even in the exonic regions (22). Often, several miRNAs that belong to the same cluster are co-transcribed simultaneously, but may have their own individual function after undergoing a separate post-transcriptional regulation (23, 24). Apart from the aforementioned classical canonical miRNA biogenesis pathway, miRNA synthesis may also follow a non-canonical pathway to maturation (25, 26), where short introns are processed through splicing independent of Drosha/DGCR8 processing known as mirtron pathway (27–29). Several miRNAs such as endogenous short hairpin RNAs (30), small nucleolar RNAs (snoRNAs) (31), and tRNAs (31, 32) belong to this class of miRNAs. Some mirtrons are processed through simtron route, where Drosha is required but does not necessitate Drosha's binding partner DGCR8 or endonuclease (33). Both simtrons and mirtrons are capable of silencing target transcripts and are associated with the RISC complex as evidenced by their interaction with Argonaute proteins. Regardless of the differences in various miRNA biogenesis pathways, they all result in functional mature miRNAs. The existence of several mechanisms in the biogenesis of miRNAs further reflects the complexity of RNA processing.
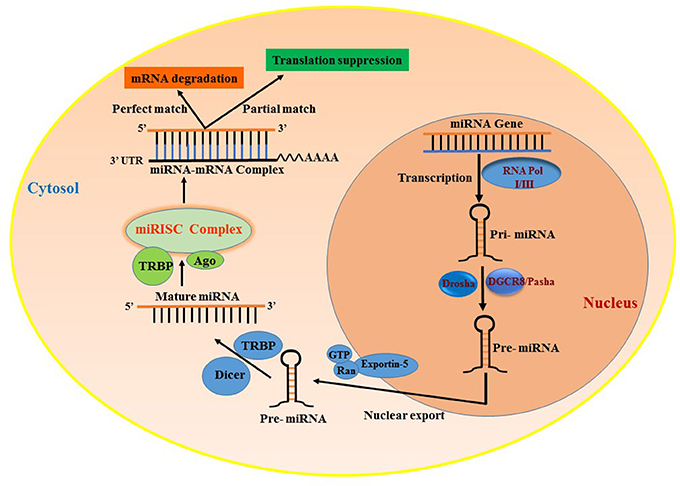
Figure 1. Biogenesis of miRNAs and their mode of transcript suppression. RNA-Pol, RNA polymerase; Drosha; RISC, RNA induced silencing complex; DGCR8, diGeorge syndrome critical region-8; GTP, guanosine triphosphate; Ras, RAs-related nuclear protein; TRBP, human immunodeficiency virus trans-activating response RNA-binding protein; Ago2, Argonaute 2.
Long Non-coding RNA
Long non-coding RNAs (LncRNAs) represent a highly diverse group of regulatory ncRNAs with respect to their characteristics, localization, and mode of action. The lncRNAs are longer than 200 nucleotides in size and are regulators of gene expression both at the transcriptional as well as post-transcriptional levels (34). They are synthesized by RNA polymerase II as co-factors along with gene activation and contain poly-A tail end and 5′capping. They function as cis and trans acting elements for protein-coding DNA sequences and therefore are powerful epigenetic mediators (35, 36). Due to this functionality, they can act both as negative or positive effectors of gene expression. LncRNAs are synthesized similar to regular mRNA transcripts but lack a defined open reading frame (ORF). They also contain their own promoter elements and can be transcribed as part of the gene. Interestingly, they can also be regulated by miRNAs (37) which adds another layer of transcriptional regulation. LncRNAs contain complementary sequences to miRNAs and can act as miRNA sponge/decoy. Briefly, lncRNAs are classified based on their location in the genome, their length, proximity to protein-coding genes, association with DNA elements, mechanisms of action, and sub-cellular localization (nucleus or cytoplasm). They are broadly classified based on their genomic loci and function (34) (1) Sense lncRNAs are synthesized from exons of protein-coding genes utilizing the same promoter region of the gene (38, 39), (2) Antisense lncRNAs are aberrant transcripts synthesized from the opposite strand of protein-coding region (40, 41), (3) Intronic lncRNAs are generated from an intron of a protein-coding gene (39, 42), (4) Intergenic lncRNAs, also referred to as large intergenic (or intervening) non-coding RNAs (lincRNAs), are encoded between protein-coding genes and are transcribed independently (43, 44), (5) Enhancer lncRNAs, also known as enhancer RNA (eRNA), are synthesized from the transcription binding regions i.e., activator/repressor elements of a protein-coding gene (45), (6) Circular forms are RNAs where the 3′-5′ ends are covalently enclosed to create a circular loop derived from splicing of a protein-coding gene (46, 47), and (7) Bidirectional transcripts are transcribed from the same promoter as coding genes, but in the opposite direction (48). The classification and function of lncRNAs are illustrated in Figure 2.
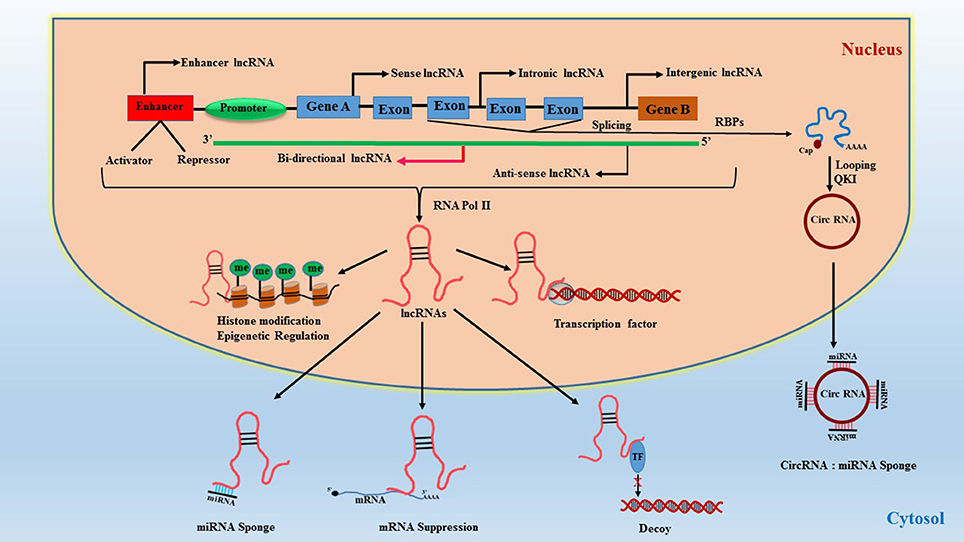
Figure 2. Classification of lncRNAs and their function. RBPs, RNA-binding proteins; QKI, quaking protein; Circ-RNA, circular RNA; TF, transcription factor.
A plethora of lncRNAs have been identified, but their function and regulation are not yet completely understood (49, 50). Nevertheless, several studies report a role for lncRNA in organ development and differentiation, and also in human diseases (51–53) especially in CVD (13, 54). Apart from direct gene silencing, lncRNAs regulate histones and influence epigenetics through modulation of DNA methylation at CpG dinucleotides, which is critical for the repression of genes (55).
siRNA
Discovered in 1999 (56), small interfering RNA or short interfering RNA (siRNA) is one of the most extensively exploited ncRNA in RNA interference therapies. siRNAs are closely related to miRNAs in terms of size and biogenesis, but slightly differ in their mechanism of RNA silencing. Unlike miRNAs, which are single-stranded RNA, siRNAs are double-stranded RNA (dsRNA) and are approximately 20–24-bp in size with a well-defined structure (57). siRNAs bind their target with 100% complementarity in the sequence and typically cleave mRNA before entering the translation process. Therefore, they are very highly specific in annealing to their target. They are also processed in a much similar fashion to miRNAs, synthesized by RNA pol III, cleaved by the enzyme Dicer and induce mRNA degradation via RISC formation (58–60). Due to their stability and the convenience of generating synthetic dsRNA that can be easily introduced exogenously into cells, siRNAs are widely used in gene therapy to silence mRNA transcripts.
Piwi-Interacting RNA (piRNA)
First identified in 1997 (61–64), piRNAs are ncRNAs that are altogether different from miRNAs and lncRNAs. piRNAs are mostly found in the genome as clusters and range in size from 25 to 30 nucleotides (65, 66). piRNAs interact with piwi (P-element induced wimpy testis) proteins of Argonaute family, thereby leading to the formation of silencing ribonucleoprotein complex, which recognizes and silences the complementary sequence (67, 68). The piRNA/PIWI complex primarily functions as epigenetic silencer by targeting transposable elements (TEs) in both germline and gonadal somatic cells and it regulates the process of transcription itself rather than transcripts (69).
The mechanism of piRNA biogenesis and function is not yet completely clear and rather very complex. However, reports suggest that piRNAs regulate mobile sequences in the genome by canonically involving endonucleolytic cleavage of the target sequence after complementary base-pair recognition through piRISC (piRNA-induced silencing complex) (70), as illustrated in Figure 3. There are more than 50,000 piRNA sequences identified in the murine genome, but inconsistencies in the sequence homology between species makes it difficult to determine their function. Research to elucidate the function of piRNAs is still in its early stages. Nevertheless, piRNAs are widely accepted to play a strong role in epigenetics via regulation of TEs. Since TEs are important for genetic diversity and genome instability, any abnormalities in TEs can lead to gene deregulation, chromosome rearrangement and gene mutations causing cancer and genetic diseases (71, 72).
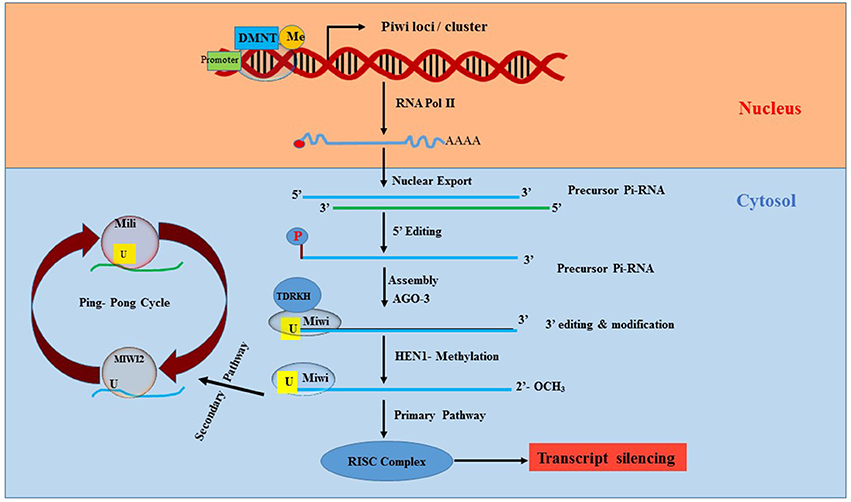
Figure 3. Synthesis and processing of Pi-RNA. Piwi, P-element Induced WImpy testis in Drosophila; piRNA, Piwi protein interaction RNA; DNMT, DNA methyltransferase; AGO3, Argonaute 3; HEN1, HUA ENHANCER 1; RISC, RNA induced silencing complex; Miwi, Mouse homolog of PIWI; TDRKH, Tudor domain to arginine methylated Miwi.
MicroRNAs and Cardiovascular Diseases
The most extensively studied ncRNAs are miRNAs, which are abundantly present in many cardiac cell types including fibroblasts, endothelial cells (ECs), and cardiomyocytes. They play a significant role in several cellular processes like proliferation, apoptosis, autophagy, and cell metabolism. Dysregulation of individual or cluster of miRNAs is linked to the pathogenesis of heart diseases and its risk factors such as diabetes, hypertension, atherosclerosis, myocardial I/R injury, and HF (8, 73). The role of miRNAs in several CVD has been well-established by taking advantage of genetically modified animal models and in vitro cell lines, and utilizing miRNA mimics and inhibitors (antagomiRs). Moreover, the aberrant expression of miRNAs and subsequent impact on cellular signaling pathways are well-documented in the literature (74, 75).
Adverse Cardiac Remodeling and Heart Failure
Cardiac remodeling is a progressive reactive phenomenon to myocardial injury that involves cellular, molecular and interstitial changes that manifest physiologically and ultimately lead to HF. Significant dysregulation of miRNA expression has been implicated during cardiac hypertrophy and HF (8, 73) (Figure 4). Preliminary evidence for the role of miRNA in myocardial development was reported from studies using Drosophila, where miRNA-1 (miR-1) was identified to regulate differentiation of cardiac and somatic muscle progenitors through Notch 1 receptor (76). After aortic constriction-induced hypertrophy in a mouse model, the muscle-specific miR-1 was significantly downregulated, plausibly through a serum response factor (SRF)-dependent mechanism (73). Overexpression of miR-1 inhibited its in silico-predicted, growth-related targets, including Ras GTPase-activating protein (RasGAP), cyclin-dependent kinase 9 (Cdk9), fibronectin, and Ras homolog enriched in brain (Rheb), in addition to protein synthesis and cell size. In this context, more supportive evidence was reported using cardiac muscle-specific targeted deletion of miR-1-2 in mouse, demonstrating a role of miR-1 in cardiac morphogenesis and cell-cycle control (77). Two mature miRNAs, miR-1 and miR-133, are derived from the same miRNA polycistron and transcribed together during development, but have distinct roles in modulating skeletal muscle proliferation and differentiation in cultured myoblasts (78). miR-1 promotes myogenesis by targeting histone deacetylase 4 (HDAC4), a transcriptional repressor of muscle gene expression, whereas miR-133 enhances myoblast proliferation by repressing SRF, a positive regulator of cardiac growth and HF. Among the miRs that were down-regulated during cardiac hypertrophy, both miR-1 and miR-133 have been prominently repressed in the left ventricle and atria in murine models as well as human subjects with cardiac hypertrophy (79). Overexpression of miR-133 or miR-1 inhibited cardiac hypertrophy. This notion was also confirmed in studies using antagomiR-133 showing sustained cardiac hypertrophy via RhoA, a GDP-GTP exchange protein target of miR-133. In contrast, another study demonstrated that overexpression of miR-1 in mouse cardiac progenitors has a negative effect on proliferation, where it targets the Hand transcription factor Hand2, which is involved in myocyte expansion (80).
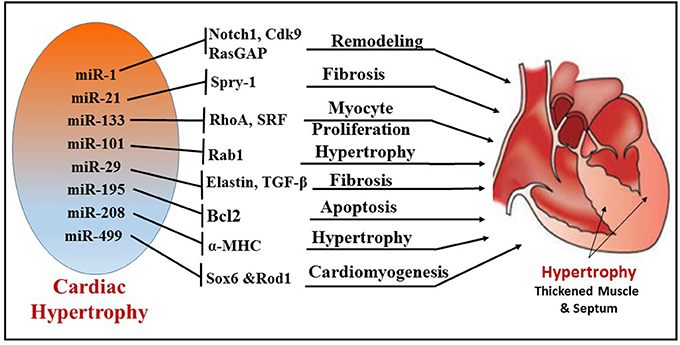
Figure 4. Illustration depicting the different miRNAs and their targets, which regulate corresponding cellular functions, like fibrosis, myocyte proliferation, apoptosis, cardiomyogenesis, and remodeling during cardiac hypertrophy.
A novel antifibrotic miRNA, miR-101, was found to be frequently downregulated in hypertrophic and post-infarcted hearts (81). MiR-101 plays a significant role in hypertrophy by regulating ras-related protein-1A (Rab1A), a member of the Rab family of small GTPases and an important regulator of cardiac hypertrophy (82). Expression of miR-101 in cardiomyocytes was downregulated in both the transverse abdominal aortic constriction rat model and angiotensin II (AngII)-induced hypertrophy. In addition, overexpression of miR-101 significantly suppressed AngII-induced cardiac hypertrophy by targeting Rab1A. In contrast, the inhibition of miR-101 expression promoted cardiac hypertrophy. The expression of miR-29 cluster (miR-29a, 29b, and 29c) inhibits the expression of targets involved in the extracellular matrix production and fibrosis (83). Moreover, the miR-29 family also controls pro-fibrotic genes such as elastin. Recent evidence also suggests that TGF-β signaling, an important regulator of fibrogenesis and collagen deposition, is regulated by several miRs including miR-29b, miR-26, miR-101a, and miR-24 miR101a (81, 84–86).
Overexpression of specific miRNAs evokes morphological changes in cardiomyocytes, which subsequently leads to ventricular hypertrophy and HF in humans (87). miRNA expression in idiopathic end-stage failing human hearts showed increased expression of miR-23a, miR-24, miR-125b, miR-195, miR-199a, and miR-214 (8). In addition, cardiac overexpression of miR-195 resulted in pathological cardiac remodeling and HF in transgenic mice. Other miRNAs have also been identified as important players in cardiac hypertrophy. For instance, cardiac-specific miR-208 encoded by an intron of the α-MHC (myosin heavy chain) gene is required for cardiomyocyte hypertrophy, fibrosis, and expression of β-MHC in response to stress and hypothyroidism (88). Moreover, the overexpression of miR-212 and miR-132 directly targets the anti-hypertrophic and pro-autophagic FoxO3 transcription factor and leads to cardiac hypertrophy and HF by inducing pro-hypertrophic calcineurin/NFAT signaling (89).
Several expression profile studies identified one of the most abundantly expressed miRNAs, namely miR-21, in murine and human hypertrophic and failing hearts. MiR-21 plays a crucial role in cardiac fibrosis and hypertrophy (90). Multiple studies reported that increased expression of miR-21 in fibroblasts of the failing heart induces the extent of interstitial fibrosis and cardiac hypertrophy by augmenting ERK–MAP kinase activity via inhibition of sprouty homolog 1 (Spry1) (91) or enhancing matrix metalloproteinase-2 (MMP-2) via PTEN (phosphatase and tensin homolog) pathway (92). Recently, miRNAs have emerged as regulators of intercellular communication in cardiac tissue (93). Bang et al. identified a high abundance of miR-21-3p (miR-21*) in cardiac fibroblast-derived exosomes as a paracrine signaling mediator that promotes cardiomyocyte hypertrophy by targeting sorbin and SH3 domain-containing protein 2 (SORBS2), PDZ and LIM domain 5 (PDLIM5). On the other hand, an antihypertrophic effect of miR-21 was also reported in transverse aortic constriction (TAC) and Ang II-treated mice (94). Overexpression of miR-21-3p suppressed TAC- and Ang II-induced cardiac hypertrophy by targeting histone deacetylase-8 (HDAC8) and modulating p-AKT/p-GSK3β pathway.
Apart from the direct involvement of certain miRNAs in hypertrophy and fibrosis, several other miRNAs were shown to be pro-hypertrophic. By targeting p53-induced nuclear protein (Tp53inp1), miR-155 regulates hypertrophy and cardiac remodeling (95). It was shown that miR-499 expression was upregulated in pressure overload-induced murine cardiac hypertrophy. This finding also correlated with increased expression of miR-499 in human failing and hypertrophied heart (94, 96, 97). Interestingly, miR-499 was responsible for the differentiation of cardiac stem cells (CSCs) into mature functional cardiomyocytes. To this end, overexpression of miR-499 in human cardiac stem cells (hCSCs) enhanced cardiomyogenesis by suppressing its target Sox6 and Rod1 (98).
MicroRNAs in Myocardial Ischemia/Reperfusion Injury and Infarction
Ischemia/reperfusion (I/R) injury is a major cause of necrotic, apoptotic and autophagic cardiomyocyte death, all of which are highly regulated by miRNAs (Figure 5). Some of the prominent regulators of cardiomyocyte death are miR-1(99) miR-15b (100), miR-21 (101), miR-30b (102), miR-34a (103, 104), and miR-497 (105). Almost all these miRNAs target the anti-apoptotic gene BCL2 and negatively regulate them. MiR-1 was found to be markedly up-regulated during I/R injury and its expression level was inversely correlated to Bcl-2 expression in cardiomyocytes (99). In line with this finding, using miR-1 transgenic mice, Pan et al. revealed that miR-1 exacerbated cardiac I/R injury whereas knockdown of miR-1 with LNA-antimiR-1 alleviated cardiac I/R injury (99, 106). The same study also showed that inhibition of miR-1 can reduce apoptosis via regulating protein kinase C (PKC) and HSP60. A recent study also determined that myocardial I/R injury causes induction of miR-1 expression and subsequent downregulation of Bcl-2, which were reversed with hydrogen sulfide treatment resulting in attenuation of cardiomyocyte apoptosis (107). Functional studies indicate contrasting roles of miR-1 and miR-133 in the regulation of stress-induced cardiomyocyte survival, with a pro-apoptotic role of miR-1 and anti-apoptotic role of miR-133 (108). Post-MI, increased miR-1 represses multiple anti-apoptotic genes (i.e., Hsp60, Hsp70, IGF-1, and Bcl-2); whereas miR-133 negatively regulates a pro-apoptotic gene (i.e., Caspase-9) (99, 108, 109). Specifically, cardiac expression of miR-133 in patients who died following MI was significantly reduced in the infarcted areas of the heart compared to healthy adult hearts who died from non-cardiac causes (110). On the contrary, down-regulation of both miR-1 and miR-133 were reported in rat hearts after 30 min of coronary artery occlusion and 180 min reperfusion (111).
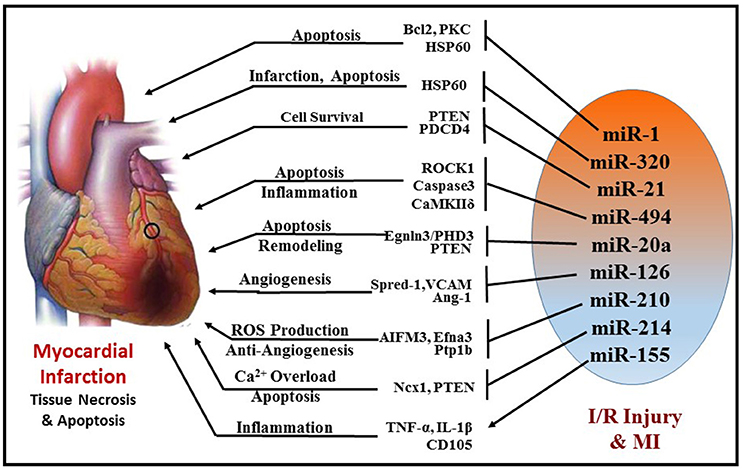
Figure 5. Schematic representation of miRNAs and their targets that are involved in cellular survival, apoptosis, angiogenesis, fibrosis, and inflammation during MI.
Another miRNA, miR-320, was shown to be differentially regulated in murine hearts after I/R injury both in vivo and ex vivo. Transgenic mice with cardiac-specific overexpression of miR-320 exhibited increased cardiomyocyte apoptosis and MI following I/R injury relative to the wild-type controls (112). Simultaneously, knockdown of endogenous miR-320 with antagomir-320 reduced infarct size and cardiomyocyte death. Using luciferase/GFP reporter assay, HSP20 (small heat-shock protein) was proven to be a target of miR-320. HSP20 plays a major role in cardioprotection against I/R injury by developing an adaptive response (113).
MiR-494 has also attracted considerable attention in the recent years. It is downregulated in failing human hearts and animal models of cardiac ischemia/hypertrophy. Transgenic mice with cardiac-specific overexpression of miR-494 displayed remarkable protection against myocardial I/R injury by reducing apoptosis and infarct size (114). Similarly, overexpression of miR-494 in cultured adult cardiomyocytes demonstrated inhibition of caspase-3 activity and reduced cell death upon simulated I/R. Furthermore, in vivo silencing of miR-494 aggravated I/R injury in mice. In this study, miR-494 was shown to target both pro-apoptotic (PTEN, ROCK1, and CaMKIIδ) as well as anti-apoptotic proteins (FGFR2 and LIF), but ultimately led to protection against myocardial I/R injury by activating AKT signaling.
miR-21 has also been shown to play a crucial role in attenuation of I/R injury by inducing several pro-survival signaling pathways in cardiomyocytes and targeting several pro-apoptotic genes, i.e., programmed cell death 4 (PDCD4), PTEN, and Fas ligand (FasL) (115–117). PTEN is essential for the activation of pro-survival AKT kinase pathway (118, 119) and inhibition of PTEN is known to limit infarct size. Transgenic mice with cardiac-specific over-expression of miR-21 exhibited suppression of ischemia-induced up-regulation of PTEN and FasL expression, increase in phospho-AKT, which collectively resulted in attenuation of infarct size and subsequent HF. Furthermore, ischemic pre-conditioning was shown to induce miR-21 in the mouse heart, which may mediate its cardioprotective effects against I/R injury (120, 121). Overexpression of miR-21 in rat hearts reduced myocardial infarct size with improved left ventricular remodeling 2 weeks after acute MI (122). Interestingly, induction of miR-21 by hydrogen sulfide was also proven to be beneficial in protecting the heart against MI and inflammasome activation (123). The protective effects of hydrogen sulfide were absent in cardiomyocytes treated with antagomiR-21 and in miR-21 knockout mice. Recently, miR-21 was also found to be pivotal in isoflurane-induced protection of cardiomyocytes against hypoxia/reoxygenation injury (124, 125). The beneficial effects of isoflurane against myocardial I/R injury were lost in miR-21 KO. This study demonstrates that Akt/NOS/mPTP pathway is involved in miR-21-mediated protective effect of isoflurane. Collectively, these studies indicate that miR-21 is induced in cardiomyocytes in the early phase of MI and contributes to myocardial protection. However, in the late phase of MI, induction of miR-21 predominantly in fibroblasts was shown to cause fibrosis and cardiac remodeling (125).
Another interesting player in myocardial I/R injury is miR-17-92 cluster consisting of family members, miR-17, miR-18a, miR-19a, miR-20a, miR-19b-1, and miR-92a, which is indispensable for cell proliferation and normal cardiac development (126, 127). Several studies have demonstrated that miR-17-92 is vital during cardiac morphogenesis and controls proliferation by targeting PTEN (128–130). Upregulation of miR-20a in neonatal rat cardiomyocytes following hypoxia/reoxygenation inhibits apoptosis, while its targeted knockdown induces cardiomyocyte apoptosis (131). The anti-apoptotic effect of miR-20a is mediated through targeted suppression of the pro-apoptotic factor Egnln3/PHD3 (prolyl hydroxylase 3). Moreover, cardiac-specific overexpression of miR-17–92 cluster also alleviates MI-induced injury and improves cardiac function in mice (128). A recent study revealed that miR-17-3p contributes to exercise-induced cardiac growth and protects against adverse ventricular remodeling after cardiac I/R injury (128, 132).
On the contrary, this cluster was also indicated to negatively affect angiogenesis (133) and the use of antagomiR-92a enhanced angiogenesis, improved left ventricular function, attenuated myocardial infarct size, and reduced apoptosis (134). This conflicting observation was in part due to the ablation of multiple miRNA members of the same cluster, as it is possible that members of the same cluster may have independent/conflicting functions by targeting different genes.
MiR-126 has been implicated as a protective miRNA, which is highly expressed in the heart. It functions as a promotor of new blood vessel formation by enhancing proangiogenic factor vascular endothelial growth factor (VEGF), inhibiting Sprouty-related protein-1 (Spred-1) and vascular cell adhesion molecule 1 (VCAM-1) and Angiopoietin-1 (Ang-1) (135–138). Consistent with this finding, Wang et al. demonstrated that targeted deletion of miR-126 in mice resulted in defective cardiac neovascularization with impairment of EC proliferation, migration, and angiogenesis following MI (135). Consistently, antogomiR-mediated silencing of EC specific miR-126 impaired angiogenesis following hindlimb ischemia (139). Moreover, miR-126 was also found in endothelial apoptotic bodies and was shown to mediate chemokine factor CXCL12 production leading to apoptosis during I/R injury. Similar to miR-126, miR-210 also promotes angiogenesis since its overexpression under normoxic conditions increased EC tubulogenesis and migration, whereas miR-210 inhibition in the presence of hypoxia decreased capillary-like formation, EC migration, survival, and induced apoptosis (140). Ephrin-A3 plays a crucial role in the development of the cardiovascular system and also in vascular remodeling (141). In response to hypoxia, miR-210 directly inhibits Ephrin-A3, which leads to stimulation of capillary-like formation and angiogenic response to ischemia (140). MiR-210 is also upregulated in hypoxic cardiomyocytes through Akt- and p53-dependent pathways and exerts cytoprotective effects by potentially reducing mitochondrial ROS production (142). Hu et al. demonstrated that induction of miR-210 rescues cardiac function after MI by upregulation of angiogenesis and inhibition of cellular apoptosis in the heart (143). Myocardial I/R injury is accompanied by mitochondrial calcium (Ca2+) overload, which contributes to mitochondrial dysfunction and cardiomyocyte death (144).
MiR-214 is yet another cardioprotective mediator against excessive Ca2+ overload in response to I/R injury. It targets sodium/calcium exchanger 1 (Ncx1)—a key regulator of Ca2+ influx, and influences several downstream effectors of Ca2+ signaling and cell death (145). MiR-214 protects the heart against I/R injury by inhibiting Ca2+ overload and cardiomyocyte death in response to I/R injury through its repression of NCX1, CaMKIIδ, CypD, and BIM. The beneficial role of miR-214 against I/R injury was further supported by reports demonstrating that genetic deletion of miR-214 in mice resulted in loss of calcium homeostasis, cardiac contractility, increased apoptosis and excessive fibrosis post-I/R (145). Alternatively, miR-214 can also inhibit PTEN and thus can regulate PI3-AKT mechanism during myocardial IR injury (146).
Recently, an interesting study showed miR-155 exacerbates I/R injury by enhancing the inflammation process in human muscle tissue (147). Data from this study showed that upregulation of miR-155 aggravates inflammatory response, leukocyte infiltration as well as cell death via induction of TNF-α, IL-1β, CD105, and Caspase3. Moreover, experiments conducted in miR-155 knockout mice displayed decreased inflammation upon I/R injury by regulation of suppressor of cytokine signaling 1 (SOCS-1) in a ROS-dependent manner (147).
Rane et al. reported that miR-199a is acutely down-regulated in cardiomyocytes during hypoxic conditions, which is obligatory for the rapid upregulation of its target, hypoxia-inducible factor HIF-1α, and hypoxia-induced apoptosis (148). Downregulation of miR-199 also induces hypoxia-induced pro-apoptotic genes like caspase −3,−6,−9, and−12 and FasL, AIF, and Bnip1. Replenishing miR-199a during hypoxia inhibits HIF-1α expression and reduces apoptosis. The same study also identified Sirt1 as another direct target of miR-199, which is responsible for downregulating prolyl hydroxylase 2 (PHD2)—required for stabilization of HIF-1α.
MicroRNAs and Atherosclerosis
Atherosclerosis is a progressive disease of the coronary arteries caused by plaque formation and lipid accumulation, accompanied by inflammation in the interior wall of blood vessels (149). The narrowing of the artery can limit or block coronary blood flow and lead to MI and related complications. miRNAs are important regulators of pathophysiological processes involved in the development of atherosclerosis such as cellular adhesion, proliferation, lipid uptake, and efflux, and recruitment of inflammatory mediators (Figure 6). The liver plays a central role in lipoprotein metabolism and several hepatic-enriched miRNAs have been identified to have significant impact on lipid homeostasis (150). Among these, hepatic-miR-122 was the first to be identified as a crucial regulator of cholesterol and fatty acid synthesis, and thus lipoprotein homeostasis (151, 152). A number of other miRNAs have been implicated in cholesterol efflux to apoA1, including miR-33 (153–155), miR-758 (156), miR-26 (157), miR-106 (158), and miR-144 (159). miR-33 is one of the most extensively investigated miRNAs and it represses multiple genes involved in cellular cholesterol trafficking (150). MiR-33a/b is embedded within introns of the SREBP (sterol regulatory element-binding protein) genes, the key transcription regulators of many cholesterogenic and lipogenic genes (160). In concert with the transcription of SREBP, miR-33 inhibits cellular cholesterol efflux by targeting ATP-binding cassette transporter A1 (ABCA1) and ABCG1 genes (161). Studies using ApoE/miR-33 double knockout mice demonstrated reduced atherosclerotic plaque with significant increase in HDL levels and enhanced cholesterol efflux via ABCA1 and ABCG1 (162). Interestingly, antagonism of miR-33 in Ldlr−/− mouse models of atherosclerosis impeded the progression of atherosclerosis (163) and also regressed established atherosclerosis (155). Anti-atherosclerotic effects of anti-miR-33 therapy have been attributed to increasing circulating levels of HDL or improving macrophage cholesterol efflux via ABCA1 and ABCG1, two of the well-established targets of miR-33 (155, 162, 164). Macrophage-specific loss of miR-33 was determined to impede atherosclerotic plaque formation by reducing inflammation and lipid accumulation in Ldlr−/− mice under hyperlipidemic conditions (165). Alternatively, whole body loss of miR-33 in Ldlr−/− mice resulted in increased body weight, impaired insulin sensitivity, and a pro-atherogenic lipid profile without significant changes in the plaque size. Humans possess another isoform of miR-33, namely miR-33b. However, bone marrow transplants from miR-33b-KI mice into the Ldlr−/− background did not show any impact on plaque formation or lipid accumulation (165). Recently, miR-302a and miR-26 have also been reported to be involved in cholesterol transport and efflux by targeting ABCA1 (157, 166).
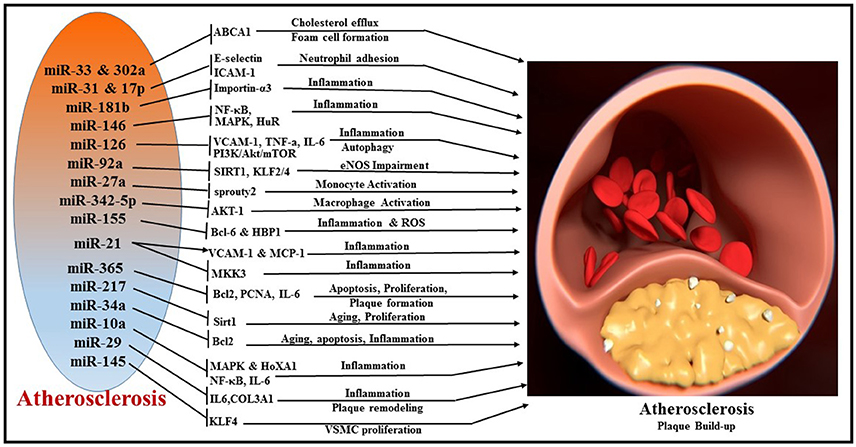
Figure 6. Overview of miRNAs and their respective target genes regulating multiple cellular processes, like cholesterol efflux, foam cell formation, neutrophil adhesion, inflammation, autophagy, monocyte activation, macrophage activation, apoptosis, cellular proliferation, and plaque remodeling during the pathogenesis of atherosclerosis.
Inflammatory activation of ECs promotes atherosclerosis by recruitment of leukocytes (167). Vascular adhesion molecule (VCAM)-1, intracellular adhesion molecule (ICAM)-1, and E-selectin are important players in the leukocyte recruitment to the vessel wall, which eventually leads to plaque formation (168). In human ECs, E-selectin and ICAM-1 are direct targets of pro-inflammatory cytokine TNFα-induced miR-31 and miR-17-3p, respectively, regulating neutrophil adhesion to ECs (169). MiR-181b regulates NF-κB-mediated EC activation, vascular inflammation as well as atherosclerosis via repression of importin-α3, a protein that is required for nuclear translocation of NF-κB (170, 171). MiR-146 maintains vascular homeostasis with repression of the pro-inflammatory signaling pathways, i.e., NF-κB pathway as well as the MAP kinase pathway and downstream early growth response (EGR) transcription factors through regulation of IL-1β signaling pathway adaptor proteins (i.e., TRAF6, IRAK1/2) (172). In addition, miR-146 modulates post-transcriptional pro-inflammatory pathways via targeting the RNA binding protein HuR (human antigen R), which promotes endothelial activation by antagonizing eNOS (endothelial nitric oxide synthase) expression (172).
A wealth of evidence suggests the involvement of miR-126 in the inflammatory responses associated with atherosclerosis (173, 174). MiR-126 suppresses VCAM-1 expression and limits leukocyte adherence to ECs and regulates vascular inflammation (138). Overexpression of miR-126 poses beneficial effects by decreasing the pro-inflammatory cytokine expression (TNF-α, IL-6) and reducing the accumulation of macrophages in atherosclerotic lesions by inhibiting MAPK pathway proteins (p38, ERK1/2, and JNK) (174). Another interesting study showed that overexpression of miR-126 prevented against ox-LDL-induced injury in HUVECs through restoring autophagy flux via repressing PI3K/Akt/mTOR pathway. This observation was supported by decreased LC3-II, Beclin 1, and p62 expression profiles that were induced by ox-LDL.
HUVECs treated with ox-LDL exhibited a robust increase in the expression level of miR-365 and downregulation of its target Bcl-2 (175). However, inhibition of miR-365 attenuated ox-LDL-induced EC apoptosis by restoring the expression of Bcl-2 (175). In contrast to this finding, miR-365 levels were downregulated in plaques (vs. healthy adjacent tissue) and in monocytes of coronary atherosclerosis (AS) patients compared to control subjects (176). Interestingly, the levels of IL-6 (direct target of miR-365), in both plaques and monocytes correlated with the expression level of miR-365 (176), suggesting a role for miR-365 in the pathogenesis of AS. MiR-365 was also reported to reduce proliferation of vascular smooth muscle cells (VSMCs) by targeting cell cycle-specific cyclin D1 (CD-1) both in vitro and in balloon injury-induced carotid artery proliferation model in rat (177). In specific, transfection of miR-365 mimics in VSMCs blunted PDGF (platelet-derived growth factor) or ANG-II-induced cell proliferation by decreasing the level of Proliferating Cell Nuclear Antigen (PCNA) through targeting CD-1 (177).
Recent studies reported that miR-92a is induced by oxidative stress in ECs (178) and is also involved in the development of atherosclerosis (179). MiR-92a targets the 3′ untranslated region of mRNAs encoding sirtuin 1 (SIRT1), Krüppel-like factor 2 (KLF2), and KLF4, and impairs eNOS-driven NO bioavailability (180). Specific in vivo inhibition of miR-92a expression in mice was shown to reduce endothelial inflammation and limit the development of atherosclerosis (179).
Macrophage foam cell formation, a hallmark of atherosclerosis, was determined to be regulated by miR-27a, which can activate CD14, CD68 expression, and CD206 and DC-SIGN, a marker of M2 and secretion of IL-10. Experiments using monocytes indicated that overexpression of miR-27a increased IL-10 secretion by activating ERK signaling pathway (181). MicroRNA expression profile reveals that macrophage-derived miR-342-5p and miR-155 are selectively upregulated in early atherosclerotic lesions in ApoE-knockout mice (182). This study indicates that miR-342-5p promotes atherosclerosis and induces the pro-inflammatory activation of macrophages by suppressing Akt1-mediated inhibition of miR-155 expression. In turn, miR-155 also promotes atherosclerosis by directly repressing the expression of BCL6 (B-cell leukemia/lymphoma 6), a transcription factor that attenuates pro-inflammatory NF-κB signaling (183). Systemic delivery of antagomiR-155 diminishes lipid-loading in macrophages and reduces atherosclerotic plaques in ApoE knockout mice (184). Ectopic overexpression and knockdown of miR-155 identified that HMG box-transcription protein 1 (HBP1) is a novel target of miR-155. miR-155, by direct repression of HBP1 expression, promoted lipid uptake and ROS production of macrophages to enhance foam cell formation (184). Furthermore, miR-155 also directly inhibits SOCS1 expression and induces p-STAT3 and PDCD4, which leads to production of inflammation mediators in macrophages to promote atherosclerotic plaque formation (185).
Aging is one of the major risk factors for type 2 diabetes mellitus and its associated endothelial dysfunction and atherogenesis (186). Interestingly, endothelial senescence seems to be dependent on the age-progressive increase in miR-217 (187). Upregulation of miR-217 was shown to negatively regulate Silent information regulator 1 (Sirt1) in human atherosclerotic plaques (187). This study reported a fascinating role linking miR-217 to aging ECs. Data showed a progressive upregulation of miR-217 during aging in cell lines including young human umbilical vein endothelial cells (HUVECs), human aortic ECs and human coronary artery ECs (187). Sirt1, a master regulator of aging was identified as a direct target of miR-217 and was shown to decline over age in correlation with potentiation of miR-217. Conversely, inhibition of miR-217 restored the levels of Sirt1 and modulated forkhead box O1 (FoxO1) and enhanced angiogenesis (187). Similarly, increased miR-34a in concert with suppression of SIRT1 expression were also reported in aged mouse aortas and in replicative senescent human aortic smooth muscle cells (HASMCs) (188). In addition, overexpression of miR-34a increased several pro-inflammatory factors like IL1β, IL8, IL6, and Mcp-1 in both endothelial and VSMCs (188). miR-34a was also found to be upregulated in HFD-induced ApoE−/− mice and ox-LDL-treated HAECs (189). Inhibition of miR-34a decreased atherosclerotic lesions and reduced EC apoptosis in HFD-induced ApoE−/− mice through suppression of its target Bcl-2 (189). Moreover, anti-miR-34a released the cell cycle arrest at the G1 phase induced by ox-LDL treatment of HAECs, suggesting that miR-34a promoted cell proliferation (189). Taken together, these reported findings imply that miR-34a regulates growth and apoptosis in ECs and plays an important role in atherosclerosis.
MiR-21 has received significant attention with respect to its role in CVDs because it was shown to be up-regulated in the arteries of patients with atherosclerosis (190). In early stages of atherosclerosis, miR-21 exhibits pro-inflammatory effect in ECs via activation of pro-inflammatory protein VCAM-1 and MCP-1 (monocyte chemotactic protein-1 (191). However, in later stages of the pathological process, it can facilitate suppression of inflammation via induction of eNOS with enhanced production of athero-protective NO, which suppresses activation and adhesion of monocytes and expression of pro-inflammatory cytokines (192). To this end, numerous studies identified the controversial role of miR-21, as a pro- or anti-atherogenic miRNA (193–195). A recent study revealed that miR-21 expression increased in macrophages and decreased in serum of patients with non-calcified coronary plaque. miR-21 participates in plaque instability by inducing the expression and secretion of pro-MMP-9 and active-MMP-9 in human macrophages via targeting gene RECK (Reversion-inducing cysteine-rich protein with Kazal motifs) (196). A report also suggested that miR-21 is the most abundantly expressed miR in macrophages and its absence leads to atherosclerosis in Ldlr−/− mice fed with western diet (197). Initial data from RNA sequencing using bone marrow-derived macrophages (BMDMs) identified rich expression of miR-21 in macrophages (197). Further experiments showed that Ldlr−/− mice that received BM from miR-21-deficient mice developed larger lesions than mice transplanted with wild type BM. In this context, mitogen-activated protein kinase kinase 3 (MKK3), a target gene of miR-21, was significantly increased in macrophages derived from miR-21−/− mice, which resulted in the activation of the p38 MAP Kinase-C/EBP homologous protein (p38-CHOP) and c-Jun N-terminal kinase (JNK) signaling pathways (197). The study also revealed that the absence of miR-21 reduced expression of the ATP-binding cassette transporter G1 (ABCG1), thus promoting the development of foam cell formation (197).
Shear stress plays an important role in the induction of inflammation in ECs and contributes to the severity of atherosclerosis by increasing proinflammatory factors in plaque regions (198–200). In this regard, miR-663 was reported to regulate shear stress in ECs (201). miRNA microarray analysis using HUVECs identified an upregulation of miR-663 upon exposure to oscillatory shear stress (OS) for 24 h (201). Antagonism of miR-663 using miR-663-locked nucleic acids (LNAs) blocked OS and TNF-α induced monocyte adhesion (201). This study identified 35 potential miR-663 targets including inflammatory genes (BMP, IL-6, and PCK) and transcription factors (FOSB, CEBPB, DDIT3, ATF3, and MYCN) that are differentially regulated with OS HUVECs (201). These observations suggest that miRNAs, including miR-663, are sensitive to shear stress and can play a delicate role in regulating inflammation and plaque formation during atherosclerosis. Similarly, studies using inner aortic arch of pig suggest that miR-10a expression was significantly reduced in the athero-susceptible regions (202). Overexpression of miR-10a inhibited the expression of VCAM-1 and E-selectin as well as phosphorylation of IκBα and NF-κB signaling in human aortic ECs. Experiments using knock-in and knockdown of miR-10a suggested that miR-10a regulates proinflammatory endothelial phenotypes in athero-susceptible regions both in vivo and in ECs by targeting NF-κB, MAPK and Homeobox A1 (HOXA1) genes (202). Importantly, a recent study reported lower expression of miR-10a and simultaneous higher expression of IL-6 and TNF-α in peripheral blood mononuclear cells (PBMCs) of patients with coronary artery disease (CAD) compared to control subjects (203).
Apart from mechanical shear stress, abnormal remodeling of plaques also increases its susceptibility to rupture. To this end, miR-29 was shown to impose positive remodeling of plaque and thus reducing the risk of plaque lesion (204). Interestingly, administration of LNA-miR-29 biweekly for 14 weeks reduced atherosclerotic lesion size in APOE−/− mice fed with high fat diet (204). Further, LNA-miR-29 increased fibrous cap thickness and SMA staining and reduced necrotic zones in lesions. Mechanistically, LNA-miR-29 increased collagen COL1A and COL3A1 (targets of miR-29) only in the risk-prone plaque region and stabilized them without inducing systemic fibrosis (204). Similarly, overexpression of miR-145 in APOE−/− mice before the onset of western diet for 12 weeks displayed reduced plaque formation (205). Specifically, VSMC-targeted expression of miR-145 resulted in plaque stability and decreased macrophage infiltration (205). Furthermore, overexpression of miR-145 resulted in a reduction in KLF4 levels with a concomitant increase in myocardin expression to promote a contractile phenotype of VSMC (205).
MicroRNAs in Diabetes and Insulin Signaling
Diabetes is a major risk factor for CVD and is characterized by elevated blood glucose, insulin resistance/deficiency and metabolic abnormalities. Several miRNAs were identified to play a role in diabetes by regulating insulin signaling and glucose metabolism (Figure 7). Some of the prominent players are miR-34a (206), miR-204 (207), miR-103/107 (208), miR-134 (209), miR-130a (210), miR-155 (211), miR-21 (212), miR-320 (213), and miR-27b (214).
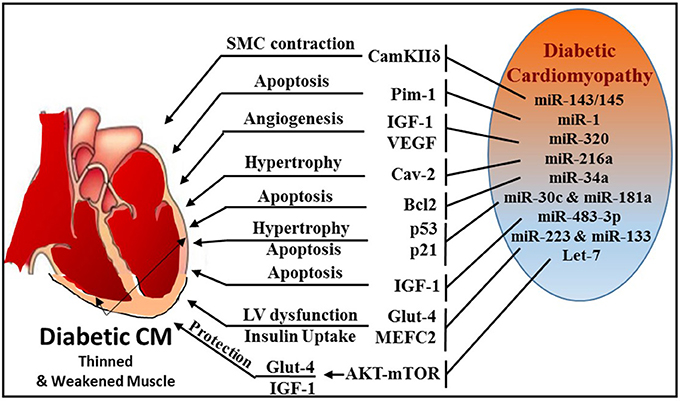
Figure 7. Schematic diagram illustrating multiple miRNAs and their targets, which mediate various cellular events, like SMC contraction, apoptosis, angiogenesis, hypertrophy, and glucose homeostasis during diabetic cardiomyopathy.
Formation of Advanced Glycation Products (AGE) that leads excessive reactive oxygen species (ROS) generation is a major mechanism of diabetes-related complications (215, 216). AGE plays an important role in the activation of PKC/Rho pathway induced by hyperglycemia (217, 218). Using Akita type 1 diabetic mice and miR-143/145 cluster knockout mice, Hien et al. established a pivotal link between hyperglycemia and smooth muscle cell (SMC) contractility (219). This study demonstrated that inhibition of PKC/Rho/MRTF (myosin phosphatase-targeting protein) signaling and genetic knockdown of miR-143/145 cluster reduced glucose-induced contractile gene expression.
Zampetaki et al. revealed that reduced miR-126 expression levels are responsible for impaired vascular repair capacities in diabetes (220). Patients with type 2 diabetes exhibited significant reduction in the level of vesicular miR-126, while non-vesicle-associated miR-126 was unchanged. Jansen et al. also reported decreased miR-126 in circulating micro-particles from patients with stable CAD and diabetes (221).
miR-1 was also found to play an important role in diabetes by directly targeting insulin-like growth factor-1 and its receptor (222) or signaling cascades related to IGF pathway (223). MiR-1 increases from the early to the late phases of diabetic cardiomyopathy, which leads to consequential cardiomyocyte apoptosis through targeting Pim-1 (proviral integration site for Moloney murine leukemia virus-1) (224). Intriguingly, blocking miR-1-dependent inhibition of Pim-1 using anti-miR-1 resulted in elevation of phosphorylated Akt and abrogation of diabetic-induced cardiac apoptosis.
Wang et al. reported that miR-320 was directly involved in regulation of insulin-like growth factor-1 in T2D rats and led to impaired angiogenesis in diabetes (213). The study also revealed that in diabetic myocardial microvascular endothelial cells (MMVECs), miR-320 is potentially targeting multiple angiogenesis-related genes, including Flk-1 (fetal liver kinase), VEGF, IGF-1 (insulin growth factor-1), IGF-1R (IGF-1 receptor), and FGFs (fibroblast growth factors). A notable finding was reported by Trajkovski et al. showing upregulation of miR-103/107 in obese mice. Adenovirus-mediated overexpression of miR-103/107 impaired glucose homeostasis in liver and fat, whereas antagomir-based inhibition of miR-103/107 increased insulin sensitivity and glucose uptake. The study also implicated caveolin-1 as a functional target gene of miR-103/107. Whether such mechanism exists in diabetes-related cardiovascular complications remains to be answered. Another miR, namely miR-216a, also targets caveolin-2, a scaffolding protein and substrate of the insulin receptor that helps recruit IRS-1 to the insulin receptor and propagate insulin signaling (225). Loss of caveolin expression results in the activation of a program of progressive hypertrophy in cardiac myocytes, and its deletion results in severe cardiomyopathy (226). Greco et al. revealed that miR-216a is induced in failing hearts of patients with and without diabetes, and this induction is negatively correlated with left ventricular ejection fraction (225).
The expression of miR-34a was found to be highly induced in H9c2 cells subjected to high glucose and concurrently, Bcl-2 expression was markedly reduced (227). In vivo experiments using miR-34a mimic prevented cardiac recovery post-MI in neonatal hearts whereas inhibition of miR-34a in adult hearts improved cardiac repair (228). Many key proteins involved in apoptosis and necrosis, such as Bcl-2, CD-1, and SIRT1 are also regulated by miR-34a (228). He et al. identified that members of the miR-34 family are direct transcriptional targets of p53 (229) and p53 is stimulated in high glucose cultured cardiomyocytes with induction of miR-34, which was also associated with a marked diminution of pro-survival SIRT1 (230). Therefore, the induction of miR-34 in diabetic heart may promote cardiomyocyte apoptosis in diabetic patients. Furthermore, miR-30 and miR-181a have also been recently shown to regulate p53 expression in cardiomyocytes (231, 232). Raut et al. revealed that myocardial expression of p53 and p21 genes is increased with simultaneous reduction in miR-30c and miR-181a in hearts of diabetic patients, rats with diabetic cardiomyopathy and in high glucose-treated cardiomyocytes (233).
MiR-483-3p is a critical regulator of heart development and a prognostic factor for heart disease (234). Induction of diabetes using streptozotocin in miR-483 transgenic mouse model increased cardiomyocyte apoptosis by silencing insulin growth factor 1 (IGF-1) (235). IGF-1 is known regulator of Myocyte enhance factor 2C (MEF2C), which plays an active role in diabetes-provoked cardiac hypertrophy (236). In this regard, it was observed that downregulation of miR-133a in diabetic cardiomyopathy resulted in an upregulation of serum and glucocorticoid regulated kinase 1 (SGK1) and IGFR1 (237). This in turn leads to the activation of MEF2C and p300 gene, paving the way for myocyte hypertrophy. On the contrary, miR-133 was also shown to target MEF2C and suppress it expression thereby blocking hypertrophy (237, 238). Downregulation of miR-133a is associated with induction of cardiac autophagy in diabetic patients with HF (177). MiR-133a improves the contractility of diabetic hearts by targeting tyrosine aminotransferase which leads to induction of norepinephrine biosynthesis, and consequently, activating β-adrenergic receptor (175).
Glucose transporter 4 (Glut4) is a major regulator that facilitates entry of glucose into cells and miR-223 was reported to control the expression of Glut4 gene in cardiomyocytes. A broad miRNA analysis study using left ventricular biopsies from patients with or without type 2 diabetes who presented with left ventricular dysfunction revealed that miR-223 was robustly upregulated in patient with diabetes, whereas Glut4 expression was low (239). Moreover, Horie et al. found that miR-133 overexpression lowered Glut4 levels by targeting KLF15 (Kruppel-like factor) and reduced insulin-induced glucose uptake in cardiomyocytes (240).
Overexpression of let-7 has been reported to mediate insulin resistance and impair glucose metabolism in high fat diet-induced diabetic mice (241), whereas the inhibition of let-7 resulted in improvement of glucose metabolism and insulin sensitivity (242). Recently Li et al. established that inhibition of the let-7 family improves glucose uptake and insulin resistance in streptozotocin-induced diabetic rats and confers cardioprotection against I/R injury through Akt and mTOR pathways (243). The study also determined that inhibition of let-7 enhanced the expression of IGF-1R, InsR (insulin receptor), and Glut4 in diabetic hearts.
Zheng et al. demonstrated that miR-195 is induced in streptozotocin-induced type 1 and db/db type 2 diabetic mouse hearts with reduction of its target proteins (B cell leukemia/lymphoma 2, Bcl-2 and sirtuin 1, Sirt1) (244). They also indicated that upregulation of miR-195 in diabetic hearts is associated with oxidative stress, apoptosis, myocardial hypertrophy, and dysfunction as well as reduction in coronary blood flow. Additionally, silencing of miR-195 reduced myocardial hypertrophy and apoptosis, increased myocardial capillary density and improved coronary blood flow and myocardial function in diabetic mice.
ncRNA as Predictors and Prognostic Tools in CVD
The identification of circulating miRNAs in blood and other liquid samples has immediately gained the attention of clinical research for their potential utility as biomarkers. Perhaps one of the most highly focused areas of research in ncRNAs is miRNAs as predictors and prognostic tools for human diseases (Table 1). These miRNAs are quite stable and withstand degradation in the blood stream largely due their association with proteins (266), apoptotic bodies (267), microvesicles (268), and exosomes (269). The feasibility of obtaining serum samples from human patients accelerated the research in this field and therefore several miRNAs were identified as biomarkers for various diseases such as cancer (270, 271), diabetes (272, 273), and CVD (274, 275).
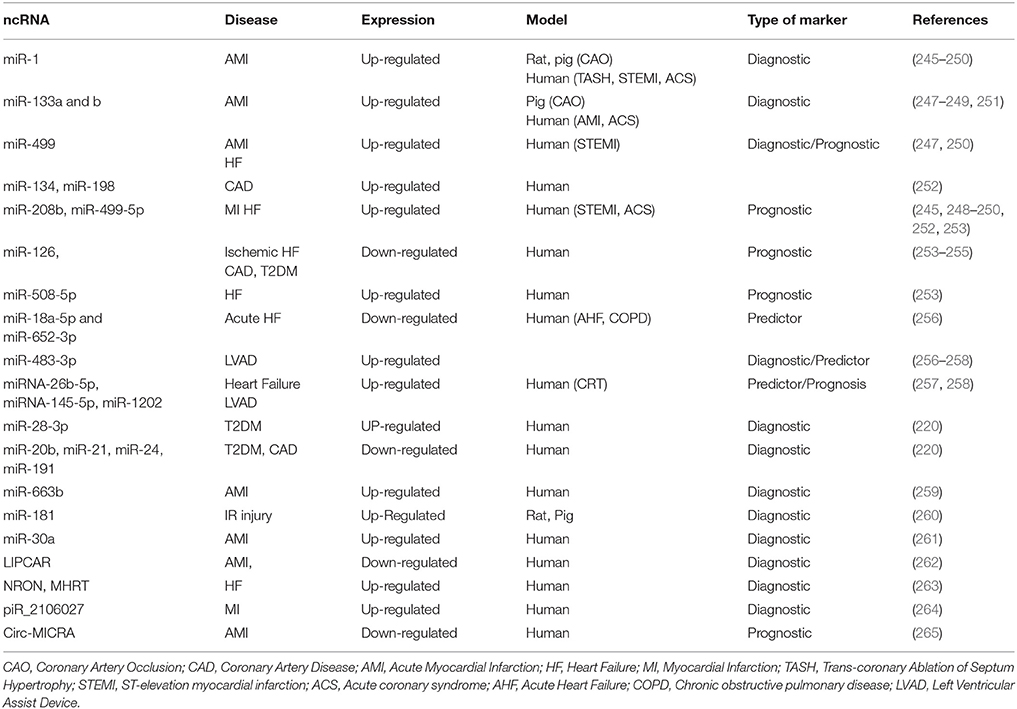
Table 1. Non-coding RNAs as biomarkers in cardiovascular diseases.
In patients with acute MI, miR-1, miR-133a, miR-499, and miR-208a have consistently been reported to be elevated in plasma (245, 246, 276). Clinicians are in pursuit of a reliable miRNA marker, similar to cardiac troponin, to evaluate the extent of MI injury. Numerous clinical studies indicated that miR-1 is markedly increased in the blood of patients with acute MI (245–247, 276). Similar to miR-1, miR-133 was also increased in plasma after coronary artery ligation in rats (245). Interestingly. miR-133 was also elevated in plasma of acute MI patients and positively correlated with cardiac troponin levels (251). A recent study conducted in patients with hypertrophic obstructive cardiomyopathy undergoing trans-coronary ablation of septal hypertrophy (TASH) procedure to identify time-dependent release of acute miRNAs that may be specific to cardiac tissue as an indicator of cardiomyocyte necrosis. The study showed that miR-1, miR-133a, and miR-208a increased continuously during the first 4 h post-MI (248). Interestingly the plasma concentration of miR-1 significantly increased (>3-fold) as early as 15 min after MI and reached peak level (>60-fold) after 75 min. A similar trend was also observed for miR-133a. In line with this finding, a comparative study between human and murine circulating miRNAs determined that the concentration human of miR-1, miR-133a, and miR-133b peaked even before cardiac troponin T post-MI, whereas in mice undergoing permanent coronary artery occlusion, miR-499 appeared to be a more sensitive marker of acute MI (247). Besides serving as diagnostic markers, miRs were also identified as predictors of disease prognosis. In a large cohort study involving 444 patients with acute coronary syndrome (ACS), the expression of miR-1, miR-133a, miR-133b, and miR-208b, were higher compared to patients with unstable angina (249). These patients were monitored for 6 months and the study concluded that miR-133a and miR-208b levels were significantly associated with the risk of mortality (249). In another interesting study involving 424 patients suspected for MI during a 30-day follow-up period, elevated plasma levels of miR-208b and miR-499-5p were strongly associated with increased risk of mortality or HF (250). Another study also suggested that the high expression level of a cluster of three miRs, including miR-134, miR-198, and miR-370, can also be used to distinguish between with CAD patients and healthy subjects (252).
Several reports suggest that miRNA expression can indicate the response to therapy. In this regard, miR-126 and miR-508-5p served as independent prognostic factors of chronic HF secondary to ischemic cardiomyopathy or non-ischemic cardiomyopathy (253). Recently, a study in patients with acute HF revealed that declining levels of circulating miR-18a-5p and miR-652-3p are associated with increasing acuity of HF (256). Interestingly, miRNAs may also serve as effective biomarkers in distinguishing responders and non-responders who received left ventricular assist device (LVAD) and cardiac resynchronization therapy (CRT) (257, 258) procedures. Patients who received LVAD had a consistently high expression of miR-483-3p, whereas levels of miR-1202 were able to identify responders vs. non-responders. The study also showed that patients with higher expression of miRNA-26b-5p, miRNA-145-5p, miRNA-92a-3p, miRNA-30e-5p, and miRNA-29a-3p responded well to CRT (257, 258).
Several studies have been conducted to examine whether the levels of circulating miRs can assist in outcome prediction of diabetic patients with impaired glucose metabolism and high-risk cardiovascular complications (220, 221, 254). A cohort study involving 80 patients with type 2 diabetes showed upregulation of miR-28-3p and downregulation of miR-20b, miR-21, miR-24, miR-15a, miR-126, miR-191, miR-197, miR-223, miR-320, and miR-486 (220). The study also used Lep(ob) mice to show a decline in miR-126 content of endothelial apoptotic bodies upon exposure to high glucose concentrations. Recently, it was demonstrated that expression levels of circulating miR-126 were decreased in the blood samples of type 2 diabetic patients with or without CAD (254). It has also been suggested that miR-126 correlates negatively with LDL in diabetic patients with CAD (254, 255). Circulating levels of miR-663b, was shown as a reliable marker for atherosclerosis related acute myocardial infarction (AMI) (259).
MicroRNAs and Exosomes
Recently researchers have shown interest in miRNAs packed in exosomes, largely due to their role in cell-cell communication (277) and ease of delivery into cells. Exosomes are small transport vehicles that measures 40–100 nm in diameter and are secreted membrane vesicles that originate from intracellular endosomes (278–280). Exosome-mediated cellular communication has been shown to play an important role in MI (281, 282). Transport of miRNA via exosomes can act as a potential mechanism for molecular cross-talk of combined gene and cell therapy in ischemic heart disease (283). Ibrahim et al. established that cardiosphere-derived cell exosomes (CDCexo) contain a distinctive complement of miRNAs, with particular enrichment of miR-146a (284). MiR-146 in exosomes plays a key role in mediating the beneficial effects of CDCexo in infarcted heart, but alone does not suffice to confer comprehensive therapeutic benefit. Circulating levels of miR-663b were shown to serve as a reliable marker for atherosclerosis related to AMI (259).
Recently de Couto et al. proposed that CDCexo contains several miRNAs, including miR-146a, miR-181b, and miR-126 and when delivered at reperfusion limits infarct size in a pig model of MI (260). They also showed that miR-181b within CDCexo is a critical mediator of macrophage polarization in vitro and cardioprotection in vivo. Using EC-specific miR-126 knockout mice, Chen et al. showed a brain-to-heart communication through miR-126 in cerebral artery occlusion model (285). Interestingly, studies conducted in rat showed that miR-17-92 cluster enriched exosome delivery restores function after stroke via targeting PTEN-PI3K pathway (286).
Yang et al. detected that miR-30a is highly enriched in exosomes from the serum of acute MI patients in vivo and also in culture medium of cardiomyocytes after hypoxic stimulation in vitro (261). The study showed that hypoxia inducible factor (HIF)-1α controls miR-30a, which is efficiently transferred via exosomes between cardiomyocytes after hypoxia. Exosomes released from hypoxic cardiomyocytes inhibit autophagy by transferring miR-30a in a paracrine manner.
In addition, specific lipids found on lipoproteins, such as phosphatidylcholine (PC), have been shown to form stable ternary complexes with RNAs (287). In addition to exosomes, lipoproteins—especially high-density lipoprotein (HDL)—play a critical role as carriers of miRNAs in cardiometabolic disorders (288). HDL transports endogenous miRNAs and delivers them to recipient cells with functional gene regulatory consequences (289). Cellular export of miRNAs to HDL is regulated by neutral sphingomyelinase. Moreover, mouse models of hypercholesterolemia and dyslipidemia exhibit a significantly distinct HDL-miRNA profile compared to healthy subjects, indicating that miRNA cargo of HDL may be involved in the atherosclerotic disease and cardiometabolic disorders (289). HDL-bound miRNAs may also be used as biomarkers in cardiometabolic disorders (290). Several angiogenesis and inflammation-associated miRNAs, including miR-92a, miR-126, miR-150, miR-378, and miR-486 were also found in circulating HDL of patients with CAD (288, 291). A recent study revealed that normalization of miRNAs with HDL level shows significant decrease in cardio-enriched miRNAs (particularly miR-1, 133, and 499) in diabetic patients undergoing coronary artery bypass graft (CABG) surgery for ischemic heart disease (292). The study proposed the need to normalize miRNA levels with HDL to increase its sensitivity as a diagnostic biomarker.
Overall, accumulating evidence implicates multiple circulating miRNAs as potential diagnostic tools as well as prognostic biomarkers of CVD. Therefore, multicenter and large cohort studies should be carefully designed to further identify and confirm specific circulating miRNAs as novel biomarkers for early diagnosis and/or prognosis of CVD in patients.
LncRNA in Cardiovascular Diseases
LncRNAs regulate various biological processes, including cell proliferation, differentiation and apoptosis (293, 294), and are aberrantly expressed in several pathological conditions such as CVD (2), diabetes mellitus (5, 295), and cancer (296, 297). Moreover, the expression of lncRNAs is predominantly unique to tissues and cell types (49, 297) and lncRNAs are therefore relatively precise in their functionality. LncRNAs act as powerful epigenetic modulators and also play an important role in heart development. Global transcriptome analyses identified deregulation of thousands of novel lncRNAs during cardiac development and pathology, but only a few have been well-characterized (298–302) (Table 2). Perhaps, one of the earliest known lncRNA is myosin heavy chain-associated RNA transcript (Mhrt/myheart), which plays a role in cardiomyocyte proliferation (304). Mhrt is inhibited by Brg1-HDAC-PARP—a chromatin repressor, which governs the transition of alpha-MHC to beta-MHC. Pathological stress activates Brg1, represses Mhrt and results in cardiac hypertrophy in adults (304).
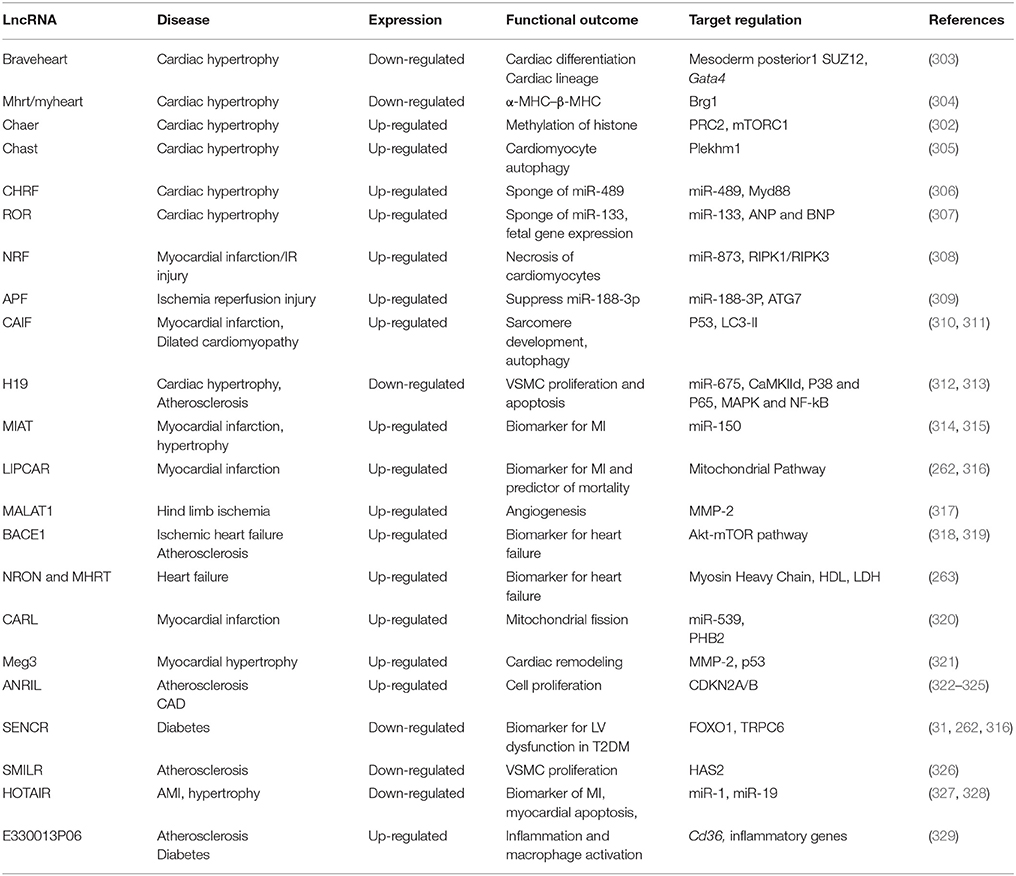
Table 2. Long non-coding RNAs and their function in the cardiovascular system.
Recent pioneering work has identified a significant role for Braveheart (BVHT) lncRNA in cardiac lineage (303). It was observed that Bvht directs mesoderm toward cardiac fate via mesoderm posterior 1 (MesP1), a mediator of cardiovascular progenitors. The study also showed that Bvht interacts with SUZ12, a core component of polycomb-repressive complex 2 (PRC2), suggesting an epigenetic regulation of chromatin. Moreover, several important transcription factors necessary for the commitment of cardiac lineage such as MesP1, Gata4, Hand1, Hand2, Nkx2.5, and Tbx5 are activated by Bvht.
LncRNAs are also being recognized as rigorous regulators of DNA methylation through interactions with DNA methyltransferases (DNMTs) and thereby act as epigenetic modulators both in normal and in pathological conditions (330). Evidence also suggests that cardiac-enriched lncRNAs, such as Cardiac Hypertrophy-Associated Epigenetic Regulator (Chaer), can block the methylation of histone H3 lysine 27 by interacting with polycomb repressor complex 2 (PRC2) and contribute to cardiac hypertrophy (302). This Chaer-PRC2 interaction is transiently enhanced at the onset of hypertrophic stress in a mammalian target of rapamycin complex 1 (mTORC1)-dependent manner, and is prerequisite for epigenetic reprogramming and induction of hypertrophic genes. Both genetic and siRNA-mediated inactivation of Chaer significantly attenuate cardiac hypertrophy and pathological progression. Cardiac Hypertrophy-Associated Transcript (Chast) is another novel cardiomyocyte-specific lncRNA known to be upregulated in TAC-induced hypertrophy in mice and aortic stenosis patients (305). It was shown that Chast impedes the expression of Pleckstrin homology domain-containing protein family member 1 (Plekhm1) as a cis regulatory element, which hampers cardiomyocyte autophagy and promotes hypertrophy.
Interestingly, lncRNAs can also interact with miRNAs and act as a decoy to regulate gene expression. Cardiac Hypertrophy-Related Factor (CHRF), another lncRNA, is able to directly bind to miR-489 and regulate the expression of MyD88 (myeloid differentiation primary response gene 88, as a miR-489 target) and consequent cardiac hypertrophy (306). MyD88 knockout mice and transgenic miR-489 overexpressing mice are resistant to hypertrophic stimuli with AngII treatment. Mechanistically it was shown that CHRF binds to miR-489 and acts as an endogenous sponge of miR-489 to downregulate its expression. Another lncRNA, ROR, was identified to be upregulated in hypertrophic mouse heart and cardiomyocytes (307). The pro-hypertrophic effect of lncRNA-ROR is mediated via repressing the expression and function of miR-133, overexpression of which attenuates lncRNA-ROR and expression of fetal genes (ANP and BNP). As evident from recent findings, gene regulation and the interplay among networks of ncRNAs is increasingly complex and yet precise. One such example is a three-way interaction between lncRNA H19, miR-675, and CaMKIId that was established in a mouse model of phenylephrine-induced hypertrophy (312). The study showed that overexpression of H19 attenuates cardiomyocyte hypertrophy in response to phenylephrine, whereas knock-down of H19 exacerbates it. Furthermore, in vivo silencing of miR-675 in a pressure overload-induced mouse model of HF increases cardiac CaMKIIδ expression and aggravates cardiac hypertrophy. Moreover, H19 was shown to be elevated in serum of patients with atherosclerosis as well as in atherosclerotic plaques of ApoE-knockout mice treated with high-fat diet (313). The study also demonstrated that overexpression of H19 enhances the expression of P38 and P65 and increased proliferation while reducing apoptosis in VSMC and HUVECs). The data suggested that H19 may regulate MAPK and NF-kB in atherosclerosis. Human MI-associated transcript (MIAT) was identified as a novel lncRNA associated with increased risk of MI (314). Vausort et al, reported that MIAT is significantly elevated in whole blood from patients with non-STsegment-elevation MI (NSTEMI) compared to STEMI patients, suggesting that MIAT may be associated with chronic cardiomyopathy. A recent study also revealed that MIAT is significantly increased in Ang II-induced cardiac hypertrophy in mice and in H9c2 cells with reduction of miR-150 (315). Accordingly, the study suggested that MIAT acts as sponge to inhibit miR-150 expression and enhanced cardiac hypertrophy.
Similarly, another lncRNA, Necrosis-Related Factor (NRF) served as a sponge to reduce the expression of miR-873 (308). MiR-873 suppresses myocardial infarct size upon experimental I/R injury by reducing the translation of RIPK1/RIPK3 as well as RIPK1/RIPK3-mediated necrotic cell death in cardiomyocytes. Knockdown of NRF reduces necrosis and MI upon I/R injury. The transcription factor p53 has been identified as an activator of NRF expression and regulates cardiomyocyte necrosis and myocardial I/R injury through NRF-miR-873-RIPK1/RIPK3 pathway.
Autophagy is a clearance mechanism where degraded proteins or damaged organelles are removed from the system to alleviate cellular burden (331). A recent study revealed that among lncRNAs, only AK079427, named as autophagy promoting factor (APF) is significantly upregulated following I/R injury in mouse heart along with reduction in miR-188-3P (309). The study also shows that enforced expression of miR-188-3p in vivo attenuates autophagy and myocardial infarct size in response to I/R injury by targeting ATG7, an autophagy-regulating protein. Accordingly, the study indicates that I/R induces APF expression, which interacts with miR-188-3p and inhibits its repressing activity on its downstream target ATG7, and finally leads to autophagy. Similarly, cardiac autophagy inhibitory factor (CAIF), was reported to suppress cardiac autophagy and attenuate MI (310). CAIF directly interacts with p53 protein and prevents its binding to the promotor region of myocardin and abolishes its transcription. The loss of myocardin, in turn, decreases the accumulation of LC3-II and attenuates autophagy. However, reports also suggest that myocardin transcription factor in cardiomyocytes is required for healthy sarcomere development. Ablation of myocardin causes loss of cardiomyocytes due to increased apoptosis and results in dilated cardiomyopathy (311).
The homeostasis of mitochondrial dynamics in heart during I/R injury is critical. Mitochondrial fusion is able to inhibit apoptosis, while growing body of evidence indicates that abnormal mitochondrial fission could initiate cellular apoptosis in the pathogenesis of many diseases (320). Prohibitin complexes, PHB1 and PHB2, are present in the inner mitochondrial membrane and play an important role in mitochondrial fusion and fission (332). PHB1-overexpressing transgenic mice that are subjected to MI showed reduced mitochondrial fission and lesser myocardial infarct size (333). On the other hand, Wang et al. demonstrated that overexpression of PHB2 inhibits post-ischemic mitochondrial fission with reduction in myocardial apoptosis and MI (320). They also identified that PHB2 is negatively regulated by miR-539, which can affect mitochondrial fission and apoptosis. Intriguingly, the same study revealed that cardiac apoptosis-related lncRNA (CARL) acts as a sponge and negatively regulates miR-539 and enhances PHB2 expression to inhibit mitochondrial fission and myocardial apoptosis, consequently attenuating MI. Another mitochondrial lncRNA, LIPCAR/uc022bqs.1 (long intergenic non-coding RNA predicting cardiac remodeling), was found to be decreased early during MI, but upregulated during later stages. Circulating LIPCAR was used as a biomarker for MI and as a prognostic tool for cardiac remodeling (262, 316).
In a landmark study, Michalik et al. elucidated the role of metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) in ECs. Using genetic MALAT1 knockout mice the authors showed MALAT1 is necessary for proper development of blood vessels and regulates the angiogenic features of vascular cells (317). In contrast, suppression of maternally expressed 3 (Meg3) lncRNA resulted in enhanced expression of angiogenesis-promoting genes (321) and prevented cardiac fibrosis and remodeling by reducing MMP-2 (334).
BACE1 has recently been recognized to play an important role in neurodegenerative diseases like Alzheimer's disease via regulating beta-amyloid peptide (Aβ) production (335–337). Interestingly, a recent study showed that Beta-Site Amyloid Precursor Protein Cleaving Enzyme (BACE1) and BACE1-AS expression was upregulated in left ventricle biopsies from patients with non-end stage ischemic HF (338) and in animal models of ischemic HF (318). The report demonstrated that BACE1 increased the production of beta-amyloid peptide and decreased the number of ECs and cardiomyocytes by activation of apoptosis. In a separate study using high fat diet-induced obesity in mice, Kim et al. showed that BACE1 transcriptional activity was activated through Akt-mTOR signaling in response to palmitic acid treatment resulting in increased beta amyloid peptide accumulation in neuronal cells (339). Whether similar mechanism exists in cardiomyocytes needs further investigation. Moreover, BACE1 was also found to play a role in atherosclerosis (319) and MI-induced neuro-inflammation in brain (318). Another human lncRNA, ANRIL (antisense non-coding RNA in the INK4 locus) has been associated with a locus implicated in CVD (340). ANRIL was shown to be highly upregulated in atherosclerotic plaques in patients with genetic polymorphism in chromosome 9p21 locus, which overlaps with ANRIL coding region (322–324, 340). A recent study suggests that the higher expression level of ANRIL is associated with presence of CAD in diabetic patients and could be considered as a potential peripheral biomarker (325).
According to reports, the smooth muscle and EC-enriched migration/differentiation associated long non-coding RNA (SENCR) was downregulated in VSMCs of diabetic mice and enhanced VSMC proliferation and migration through induction of FOXO1 and short transient receptor potential Channel (TRPC6), a target of SENCR (31, 341). SENCR is also recognized as a strong circulating biomarker for left ventricular dysfunction in type 2 diabetes (316). In line with this, smooth muscle-induced lncRNA enhances replication (SMILR) was also shown to be highly upregulated in unstable atherosclerotic plaques and in plasma of patients with increased plasma C-reactive protein levels (326). Further, knockdown of SMILR in primary human saphenous vein-derived endothelial cells (HSVECs) treated with IL-6/PDGF reduced proliferation (326). Another interesting lncRNA, E330013P06, is upregulated in macrophages from db/db and diet-induced insulin-resistant type 2 diabetic mice and in monocytes of diabetic patients (329). It is also increased along with inflammatory genes in mouse macrophages treated with high glucose and palmitic acid. Overexpression of E330013P06 in macrophages induces the expression of pro-inflammatory and pro-atherogenic genes, which leads to enhanced inflammatory signals and foam cell formation. Silencing E330013P06 was shown to reverse the upregulation of inflammatory genes induced by diabetes (342). Another lncRNA, MeXis, was identified to play a role in macrophage cholesterol efflux and atheroegenesis (343). A causal link between liver X receptors (LXRs), sterol-activated nuclear receptors that regulate the expression of genes involved in cholesterol homeostasis, and MeXis was established by Sallam et al. (343). The study showed that MeXis and ABCA1 expression were induced by LXR in macrophages. MeXis knockout mice displayed decreased expression of Abca1 in heart and enhanced development of atherosclerosis. Induction of MeXis expression in response to activation of LXRs augmented Abca1 expression and macrophage cholesterol efflux.
A recent clinical study identified two cardiac-specific/relevant circulating lncRNAs, namely NRON (non-coding repressor of NFAT) and MHRT (myosin heavy-chain-associated RNA transcripts), which are significantly elevated in plasma of HF patients as compared to healthy participants (263). The study suggested that circulating levels of NRON and MHRT may be new independent predictors for HF. Spearman's rank correlation analysis showed that NRON is negatively correlated with HDL and positively correlated with LDH (lactate dehydrogenase), whereas MHRT is positively correlated with AST (Aspartate aminotransferase) and LDH.
Interestingly, HOX (Homeobox) antisense intergenic RNA (HOTAIR), a reportedly cardioprotective lncRNA, was reduced in AMI patients and its concentration was inversely correlated with cTnI and miR-1 levels (327). Moreover, adenovirus-mediated overexpression of HOTAIR reduced hypoxia-induced apoptosis in cultured cardiomyocytes with suppression of miR-1 expression (327). Reports in the literature also indicate that the expression of HOTAIR is dramatically down-regulated in both hypertrophied heart tissues and cultured cardiomyocytes treated with Ang-II, which correlated with increased cell surface area and up-regulated expression of ANP and BNP (328). Overexpression of HOTAIR reduces the expression of pro-hypertrophic markers like ANP, BNP, and β-MHC in response to Ang-II stimulation (328). The same study also suggests that HOTAIR serves as a miR-19 sponge and functionally interacts with it. Overexpression of HOTAIR inhibits miR-19 with subsequent retrieval of the expression of PTEN (a direct target of miR-19), which is involved in HOTAIR-mediated inhibition of cardiac hypertrophy.
Taken together, although much more is known about miRNAs than lncRNAs, there are exceedingly more lncRNAs (~30,000) compared to miRNAs (~2,000). More targeted research to provide comprehensive understanding of lncRNAs, coupled with improved delivery methods will further advance the field of lncRNAs and offer more insight into their involvement in CVD before they can be used as therapeutic targets.
Piwi-RNA
P-Element induced wimpy testis RNAs (PIWI-RNA, piRNA) are lncRNAs that interact with piwi protein family and act as RNA-guided gene regulatory elements (342, 344). They play an important role in epigenetic control of gene expression through DNA methylation, regulation of TEs, and maintenance of genome integrity (345–351). They are involved in the pathogenesis of various types of cancer (352–356) and are master regulators of inheritance. However, relatively much less is known about the functional role of piRNAs in the field of CVDs. piRNAs are generally known to interact with several TEs and silence them to maintain genome stability (63, 67). In this regard, piRNAs influence long interspersed nuclear elements (LINEs) in the heart and suppression of LINE-1 decreases ischemic damage through activation of the Akt/PKB signaling (357, 358). Similarly, global microarray analysis revealed differentially expressed piRNAs in cardiosphere and cardiosphere-derived cells. The study also reported that 181 piRNAs are upregulated and 129 are downregulated in cardiosphere-derived cells with respect to cardiac fibroblasts (357, 359). Interestingly all upregulated piRNAs targeted LINE elements and in particular, piRNAs DQ594975, DQ572313, DQ586118 activated pro-survival AKT signaling. The overall result indicated that piRNAs could play a functional role in cardiomyocyte proliferation and regeneration. Reports also speculate that piRNAs can regulate AKT signaling through interaction with PIWIL2 protein. One study showed that PIWIL2 protein is highly expressed in tumor cells. Experiments using constitutive expression of PIWIL2 in NIH-3T3 cells demonstrated that it inhibits apoptosis through activation of Stat3/Bcl-XL pathway (360). Since piwi-interacting RNAs predominantly regulate TEs, aberrant expression mostly results in gene mutations leading to cancer and genome instability in inherited genetic disorders. Nevertheless, research findings of piRNAs in heart disease, such as cardiac hypertrophy and other proliferation related abnormalities are emerging and can further enhance our understanding on the role of piRNAs in CVD. In a recent publication, Rajan et al. reported abundant expression of piRNAs in the cardiac system and altered expression profile during cardiac hypertrophy in both chronic swimming-induced hypertrophied rat heart and control rat heart in vivo, and alpha-2 macroglobulin-induced hypertrophied H9c2 cells and control H9c2 cells in vitro (264). The study identified 22 potential piRNAs showing differential expression during cardiac hypertrophy, which was validated by qPCR and by RNA immunoprecipitation using piwi antibodies for piR_2106027. Analyzing the expression of piR_2106027 in different patients with and without MI, Rajan et al. also revealed that piR_2106027 is significantly elevated in patients with MI. Interestingly, they also identified that the level of piR_2106027 correlates with cTnI levels as this piRNA was not elevated in cTnI-negative MI patients. Accordingly, piRNAs were suggested as vibrant diagnostic markers as well as potential therapeutic targets for CVD.
Circular RNA (circRNA)
Relatively new players in the world of ncRNAs, much less is known about the role of circular RNA (circRNA, a closed continuous loop of ncRNA with 3′ and 5′ ends joined together) in CVD. However, circRNAs have gained much attention recently due to their sustained stability (361) and recognized as novel biomarkers (362, 363). CircRNAs were identified as competing endogenous RNAs (ceRNAs) that sponge specific miRNAs by complementary base pairing (364). A recent publication reported that mice with overexpression of ARC (apoptosis repressor with caspase recruitment domain) exhibit reduced hypertrophic response and that ARC is a direct downstream target of miR-223 (365). A heart-related circRNA (HRCR) acts as an endogenous miR-223 sponge and inhibits miR-223 activity, which resulted in the increase of ARC expression and attenuation of cardiac hypertrophy and HF.
Studies using mouse models of MI and isolated cardiomyocytes subjected to hypoxia showed that cerebellar degeneration-related protein 1 transcript (Cdr1as) is highly upregulated with MI and overexpression of miR-7 rescues Cdr1as-induced cardiac apoptosis (366). It was also observed that overexpression of Cdr1as induces the transcript levels of PARP and SP1 and that Cdr1as functions as a miR-7a sponge in promoting MI-related injury. Similarly, cZNF292 was identified to play a role in endothelial function and was up-regulated upon hypoxic treatment. Inhibition of cZNF292 reduced angiogenic sprouting of ECs and its overexpression increased endothelial proliferation (367). Several circRNAs were implicated as novel biomarkers in MI. A recent study using blood samples from 472 patients with acute MI showed MICRA as a novel biomarker for risk classification of MI patients (265). The study analyzed the correlation between the expression of MICRA to patient group to reduced EF (rEF), mid-range EF (mrEF), and preserved EF (pEF). Interestingly the data suggest that MICRA could be a significant predictor of LV dysfunction in patients after acute MI. A recent clinical study identified another circRNA, hsa_circ_0124644, which was remarkably increased in the peripheral blood of 137 patients with CAD compared to control subjects, suggesting that it may be a promising diagnostic biomarker (363). Gupta et al. identified specific circRNAs derived from Ttn (Titin), Fhod3 (Formin homology 2 domain containing 3), and Strn3 (Striatin, calmodulin-binding protein 3), which are altered in the heart upon doxorubicin treatment (368). Knockdown of Ttn-derived circRNA was shown to enhance the susceptibility to doxorubicin, elucidating the functional role of circRNAs in doxorubicin-induced cardiotoxicity.
Therapeutic Approach: Current and Future Perspectives and Controversies
Several promising therapeutic outcomes in animal models and pre-clinical trials have encouraged pharmaceutical companies to conduct ncRNA-based clinical trials in patients with CVD and other diseases. MRG-201, an anti-fibrotic synthetic miRNA mimic (promiR) of miR-29b, is designed to decrease the expression of collagen and reduce fibrotic scar in diseases like idiopathic pulmonary fibrosis and recently completed its phase I clinical trial in healthy volunteers to evaluate its tolerability (Miragen Therapeutics, Inc.). Miragen also has several miRNA-based pipeline projects including MRG-110, which targets miR-92a to enhance the revascularization process in ischemic heart disease. MGN-1374 (miR-15 family) to treat MI and MGN-9103 (miR-208) for the treatment of obesity and diabetes are also among the novel potential therapeutic candidates developed by Miragen. LNA-antimiR-34a, which targets Notch1 and Sema-4b (104, 369), is also being considered for the treatment of MI by miRNA Therapeutics, Inc. Several other miRNA-based therapies that are currently in clinical trials can be found on the National Institutes of Health (NIH) website (https://clinicaltrials.gov/). NcRNA-based therapies to treat CVD in clinical trials are very limited and are still in the developmental stage. However, RNA-based clinical trials, especially with siRNA and miRNA to treat devastating diseases like cancer (370–372) have shown promising results and are therefore encouraging to extend similar approaches to treat CVDs.
Although the path to ncRNA-based therapy looks promising, it is also fraught with limitations. The fact that a single miRNA can target several mRNAs, which is considered an advantage to exert broad effects on multiple pathological pathways can also be viewed as a major limitation that may evoke undesired responses. Moreover, a critical issue with ncRNA-based therapy is delivery of miRNA mimics/antagomiRs to the target organ while withstanding degradation in the blood stream. Researchers have tried several approaches to circumvent this problem such as adhesive hydrogel patch delivery (373), nanoparticle-mediated delivery (371, 374), exosome-mediated delivery (375, 376), and antibody-fused nanoparticles (377). Important considerations to improve miRNA based therapy include enhancement of target affinity, stability, specificity and ADME (absorption, distribution, metabolism, and excretion) (378). Several chemically modified oligonucleotides either antagonizing or mimicking miRNAs have been efficiently used in preclinical models (379–381). These synthetic oligonucleotides possess better stability, absorption in the cell and offer greater affinity toward their target (382, 383). The efficacy of these novel oligonucleotide-based drugs has accelerated clinical trials to target miRNA-based gene therapy. Several classes of oligonucleotide drugs with unique modifications in their structure have enhanced their pharmacokinetics, metabolism, stability, resistance against nuclease activity in the blood stream and allowed for better bioavailability (380, 384).
Since exonucleases cleave the phosphodiester bond, a substitution of phosphodiester group with phosphorothioate in the nucleotide backbone and replacing the ribose sugar moieties with 2′-O-methyl or other 2′s confer substantial resistance toward nuclease activity (381, 385, 386). Antisense oligonucleotides (ASO) are first generation miRNA inhibitors, with increased oligonucleotide stability but less binding affinity toward target RNA due to the presence of phosphorothioate. However, this obstacle was overcome with the use of 2′-O-methyl (2′-OMe) modification of RNA (2′-OMe-RNA), which possesses both improved binding affinity and nuclease resistance (387–389). Among the ASOs, 2′-O-methoxyethyl (2′-MOE) is the most successful and widely used oligonucleotide inhibitor, with increased nuclease resistance and substantially higher binding affinity than its predecessors (390, 391). Several other modifications, such as 2′-fluoro (2′F), which further increases the affinity of oligonucleotides also proved effective (392, 393). Locked nucleic acid (LNA) is another modified RNA oligonucleotide that has an additional bond between the 2′ oxygen and 4′ carbon that was shown to have increased binding affinity for its target (394–396). LNA-based miRNA inhibition has the advantage over other chemically modified oligonucleotides by interfering in the alternative gene splicing (397) and is also widely used in pharmaceutical industry (398). Indeed, the first miRNA-targeted drug tested for safety in phase II clinical trial of HCV infection was based on LNA chemistry (399). Several other chemically modified ASOs, such as aptamers, single-stranded oligonucleotides that bind to the protein and disrupt the protein-protein interaction (400), and LNA modified with constrained ethyl' (cEt) (401) and Ribonuclease H1-dependent LNA-Gapmers (402) have also been investigated with moderate success. All these methodologies have been used with considerable success in animal models thus far and are promising for further development. Novel delivery methods using AAV vectors guided with cardiac specific promoters such as α-MHC or troponin can also be considered when translating these findings to human subjects. Emerging revolutionary genome editing techniques like Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR-CAS9) are already considered for clinical trials to correct genetic disorders like β-thalassemia (372), cystic fibrosis transmembrane conductor receptor (CFTR) mutation (373), and Duchenne muscular dystrophy (374) (DMD). CTX001 is a new initiative by European Union and United States Food and Drug Administration (FDA) for CRISPR-based treatment of β-thalassemia, which is expected to start in 2018. Exploiting CRISPR/CAS9 technology to edit miRNA and other ncRNAs is also a novel option and demonstrated to be viable for stable suppression of miRNAs (403). Nevertheless, the scientific community should approach these new treatment strategies with extreme caution to avoid off-target effects.
In spite of its therapeutic potential, miRNA-based gene therapy is also challenged with controversies. One such example is miR-21, where antagomir-mediated inhibition of miR-21 was protective against pressure overload-induced cardiac stress (91), yet genetic ablation or LNA-based inhibition of miR-21 in mice failed to protect against TAC (404). These discrepancies between studies caution us to consider the efficiency of inhibition when using different types of inhibitors, such as LNA- or cholesterol-based antagomir. In addition, the non-specificity of blocking multiple miRNAs that have similar complementary sequences in the seed region may contribute to the functional outcome. Genetic knockout and antagomir-based inhibition of miR-143/145 demonstrated different response to stress in VSMCs (405, 406). Ablation of miR-143/145 blocked the induction of VSMC proliferation and developed neointimal lesions (405, 406). In striking contrast, miR143/145 knockout mice displayed reduced neointima formation in response to carotid artery ligation (407). However, these studies investigated different targets of miR-143/145 that are involved in cytoskeletal regulation (405–407). Therefore, these controversial outcomes, either due to the redundancy of miRNA targets or the types of methods used for miRNA inhibition, warrant further in-depth studies.
Conclusion
The robust development and the application of ncRNAs in myriad fields has opened up venues for many novel research areas such as precision and personalized medicine and may offer a unique class of biomarkers. The powerful role of ncRNAs in defining cell fate both in normal and pathological states has provided an opportunity to device new therapeutic strategies to combat CVD. Recent preclinical and clinical studies suggest that ncRNA-based gene therapy is not too far from clinical trials. However, despite the recent surge in the field of ncRNAs, our understanding about the function of these small molecules remains rather limited. Future studies are warranted to shed new light on the involvement of ncRNAs in CVD so we may be able to utilize them as biomarkers and/or target them to carefully regulate cell function for treatment of CVD and its risk factors.
Author Contributions
AS, AD, and FS drafted, edited, and approved the manuscript and figures.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by grants from the National Institutes of Health (HL134366) to AD and (HL133167 and AG053654) to FS.
References
1. Yang Y, Cai Y, Wu G, Chen X, Liu Y, Wang X, et al. Plasma long non-coding RNA, CoroMarker, a novel biomarker for diagnosis of coronary artery disease. Clin Sci. (2015) 129:675–85. doi: 10.1042/CS20150121
2. Wu G, Cai J, Han Y, Chen J, Huang ZP, Chen C, et al. LincRNA-P21 regulates neointima formation, vascular smooth muscle cell proliferation, apoptosis, and atherosclerosis by enhancing P53 activity. Circulation (2014) 130:1452–65. doi: 10.1161/CIRCULATIONAHA.114.011675
3. Thum T, Galuppo P, Wolf C, Fiedler J, Kneitz S, van Laake LW, et al. MicroRNAs in the human heart: a clue to fetal gene reprogramming in heart failure. Circulation (2007) 116:258–67. doi: 10.1161/CIRCULATIONAHA.107.687947
4. Fan B, Luk AOY, Chan JCN, Ma RCW. MicroRNA and diabetic complications: a clinical perspective. Antioxid Redox Signal. (2017). doi: 10.1089/ars.2017.7318
5. Wang M, Wang S, Yao D, Yan Q, Lu W. A novel long non-coding RNA CYP4B1-PS1-001 regulates proliferation and fibrosis in diabetic nephropathy. Mol Cell Endocrinol. (2016) 426:136–45. doi: 10.1016/j.mce.2016.02.020
6. Hayes J, Peruzzi PP, Lawler S. MicroRNAs in cancer: biomarkers, functions and therapy. Trends Mol Med. (2014) 20:460–9. doi: 10.1016/j.molmed.2014.06.005
7. Huarte M. The emerging role of LncRNAs in cancer. Nat Med. (2015) 21:1253–61. doi: 10.1038/nm.3981
8. van Rooj E, Sutherland LB, Liu N, Williams AH, McAnally J, Gerard RD, et al. A signature pattern of stress-responsive microRNAs that can evoke cardiac hypertrophy and heart failure. Proc Natl Acad Sci USA. (2006) 103:18255–60. doi: 10.1073/pnas.0608791103
9. Ikeda S, Kong SW, Lu J, Bisping E, Zhang H, Allen PD, et al. Altered microRNA expression in human heart disease. Physiol Genomics (2007) 31:367–73. doi: 10.1152/physiolgenomics.00144.2007
10. Benjamin EJ, Blaha MJ, Chiuve SE, Cushman M, Das SR, Deo R, et al. Heart disease and stroke statistics-2017 update: a report from the american heart association. Circulation (2017) 135:e146–e603. doi: 10.1161/CIR.0000000000000485
11. Roger VL. Epidemiology of heart failure. Circ Res. (2013) 113:646–59. doi: 10.1161/CIRCRESAHA.113.300268
12. Napoli C, Grimaldi V, De Pascale MR, Sommese L, Infante T, Soricelli A. Novel epigenetic-based therapies useful in cardiovascular medicine. World J Cardiol. (2016) 8:211–9. doi: 10.4330/wjc.v8.i2.211
13. Bar C, Chatterjee S, Thum T. Long noncoding RNAs in cardiovascular pathology, diagnosis, and therapy. Circulation (2016) 134:1484–99. doi: 10.1161/CIRCULATIONAHA.116.023686
14. Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs With antisense complementarity to lin-14. Cell (1993) 75:843–54. doi: 10.1016/0092-8674(93)90529-Y
15. Rodriguez A, Griffiths-Jones S, Ashurst JL, Bradley A. Identification of mammalian microRNA host genes and transcription units. Genome Res. (2004) 14:1902–10. doi: 10.1101/gr.2722704
16. Kozomara A, Griffiths-Jones S. MiRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. (2014) 42:D68–73. doi: 10.1093/nar/gkt1181
17. Landthaler M, Yalcin A, Tuschl T. The human diGeorge syndrome critical region gene 8 and Its D. melanogaster homolog are required for miRNA biogenesis. Curr Biol. (2004) 14:2162–7. doi: 10.1016/j.cub.2004.11.001
18. Hutvagner G, McLachlan J, Pasquinelli AE, Balint E, Tuschl T, Zamore PD. A cellular function for the RNA-interference enzyme dicer in the maturation of the let-7 small temporal RNA. Science (2001) 293:834–8. doi: 10.1126/science.1062961
19. Filipowicz W, Bhattacharyya SN, Sonenberg N. Mechanisms of Post-transcriptional regulation by microRNAs: are the answers in sight? Nat Rev Genet. (2008) 9:102–14. doi: 10.1038/nrg2290
20. Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell (2009) 136:215–33. doi: 10.1016/j.cell.2009.01.002
22. Lee Y, Jeon K, Lee JT, Kim S, Kim VN. MicroRNA maturation: stepwise processing and subcellular localization. EMBO J. (2002) 21:4663–70. doi: 10.1093/emboj/cdf476
23. Ozsolak F, Poling LL, Wang Z, Liu H, Liu XS, Roeder RG, et al. Chromatin structure analyses identify miRNA promoters. Genes Dev. (2008) 22:3172–83. doi: 10.1101/gad.1706508
24. Monteys AM, Spengler RM, Wan J, Tecedor L, Lennox KA, Xing Y, et al. Structure and activity of putative intronic miRNA promoters. RNA (2010) 16:495–505. doi: 10.1261/rna.1731910
25. Berezikov E, van Tetering G, Verheul M, van de Belt J, van Laake L, Vos J, et al. Many novel mammalian microRNA candidates identified by extensive cloning and RAKE analysis. Genome Res. (2006) 16:1289–8. doi: 10.1101/gr.5159906
26. Miyoshi K, Miyoshi T, Siomi H. Many ways to generate microRNA-like small RNAs: non-canonical pathways for microRNA production. Mol Genet Genomics (2010) 284:95–103. doi: 10.1007/s00438-010-0556-1
27. Berezikov E, Chung WJ, Willis J, Cuppen E, Lai EC. Mammalian mirtron genes. Mol Cell. (2007) 28:328–36. doi: 10.1016/j.molcel.2007.09.028
28. Babiarz JE, Ruby JG, Wang Y, Bartel DP, Blelloch R. Mouse ES cells express endogenous ShRNAs, siRNAs, and other microprocessor-independent, dicer-dependent small RNAs. Genes Dev. (2008) 22:2773–85. doi: 10.1101/gad.1705308
29. Sibley CR, Seow Y, Saayman S, Dijkstra KK, El Andaloussi S, Weinberg MS, et al. The biogenesis and characterization of mammalian microRNAs of mirtron origin. Nucl Acids Res. (2012) 40:438–48. doi: 10.1093/nar/gkr722
30. Martinez J, Patkaniowska A, Urlaub H, Luhrmann R, Tuschl T. Single-stranded antisense siRNAs guide target RNA cleavage in RNAi. Cell (2002) 110:563–74. doi: 10.1016/S0092-8674(02)00908-X
31. Zou ZQ, Xu J, Li L, Han YS. Down-regulation of SENCR promotes smooth muscle cells proliferation and migration in Db/Db mice through up-regulation of FoxO1 and TRPC6. Biomed Pharmacother. (2015) 74:35–41. doi: 10.1016/j.biopha.2015.06.009
32. Cole C, Sobala A, Lu C, Thatcher SR, Bowman A, Brown JW, et al. Filtering of deep sequencing data reveals the existence of abundant dicer-dependent small RNAs derived from tRNAs. RNA (2009) 15:2147–60. doi: 10.1261/rna.1738409
33. Havens MA, Reich AA, Duelli DM, Hastings ML. Biogenesis of mammalian microRNAs by a non-canonical processing pathway. Nucleic Acids Res. (2012) 40:4626–40. doi: 10.1093/nar/gks026
34. Kapranov P, Cheng J, Dike S, Nix DA, Duttagupta R, Willingham AT, et al. RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science (2007) 316:1484–8. doi: 10.1126/science.1138341
35. Mao YS, Sunwoo H, Zhang B, Spector DL. Direct Visualization of the co-transcriptional assembly of a nuclear body by noncoding RNAs. Nat Cell Biol. (2011) 13:95–101. doi: 10.1038/ncb2140
36. Affymetrix/Cold Spring Harbor Laboratory ENCODE Transcriptome Project. Post-transcriptional processing generates a diversity of 5′-modified long and short RNAs. Nature (2009) 457:1028–32. doi: 10.1038/nature07759
37. Xue Z, Hennelly S, Doyle B, Gulati AA, Novikova IV, Sanbonmatsu KY, et al. A G-rich motif in the LncRNA braveheart interacts with a zinc-finger transcription factor to specify the cardiovascular lineage. Mol Cell. (2016) 64:37–50. doi: 10.1016/j.molcel.2016.08.010
38. Guttman M, Garber M, Levin JZ, Donaghey J, Robinson J, Adiconis X, et al. Ab initio reconstruction of cell type-specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincRNAs. Nat Biotechnol. (2010) 28:503–10. doi: 10.1038/nbt.1633
39. Kampa D, Cheng J, Kapranov P, Yamanaka M, Brubaker S, Cawley S, et al. Novel RNAs identified from an in-depth analysis of the transcriptome of human chromosomes 21 and 22. Genome Res. (2004) 14:331–42. doi: 10.1101/gr.2094104
40. Derrien T, Johnson R, Bussotti G, Tanzer A, Djebali S, Tilgner H, et al. The GENCODE V7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. (2012) 22:1775–89. doi: 10.1101/gr.132159.111
41. Katayama S, Tomaru Y, Kasukawa T, Waki K, Nakanishi M, Nakamura M, et al. Antisense transcription in the mammalian transcriptome. Science (2005) 309:1564–6. doi: 10.1126/science.1112009
42. Birney E, Stamatoyannopoulos JA, Dutta A, Guigo R, Gingeras TR, Margulies EH, et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature (2007) 447:799–816. doi: 10.1038/nature05874
43. Hangauer MJ, Vaughn IW, McManus MT. Pervasive transcription of the human genome produces thousands of previously unidentified long intergenic noncoding RNAs. PLoS Genet. (2013) 9:e1003569. doi: 10.1371/journal.pgen.1003569
44. Ulitsky I, Bartel DP. LincRNAs: genomics, evolution, and mechanisms. Cell (2013) 154:26–46. doi: 10.1016/j.cell.2013.06.020
45. Kim TK, Hemberg M, Gray JM, Costa AM, Bear DM, Wu J, et al. Widespread transcription at neuronal activity-regulated enhancers. Nature (2010) 465:182–7. doi: 10.1038/nature09033
46. Surono A, Takeshima Y, Wibawa T, Ikezawa M, Nonaka I, Matsuo M. Circular dystrophin RNAs consisting of exons that were skipped by alternative splicing. Hum Mol Genet. (1999) 8:493–500. doi: 10.1093/hmg/8.3.493
47. Cocquerelle C, Mascrez B, Hetuin D, Bailleul B. Mis-splicing yields circular RNA molecules. FASEB J. (1993) 7:155–60. doi: 10.1096/fasebj.7.1.7678559
48. Taft RJ, Pheasant M, Mattick JS. The relationship between non-protein-coding DNA and eukaryotic complexity. Bioessays (2007) 29:288–99. doi: 10.1002/bies.20544
49. Cabili MN, Trapnell C, Goff L, Koziol M, Tazon-Vega B, Regev A, et al. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. (2011) 25:1915–27. doi: 10.1101/gad.17446611
50. Cheng J, Kapranov P, Drenkow J, Dike S, Brubaker S, Patel S, et al. Transcriptional maps of 10 human chromosomes at 5-nucleotide resolution. Science (2005) 308:1149–54. doi: 10.1126/science.1108625
51. Wilusz JE, Sunwoo H, Spector DL. Long noncoding RNAs: functional surprises from the RNA world. Genes Dev. (2009) 23:1494–504. doi: 10.1101/gad.1800909
52. Taft RJ, Pang KC, Mercer TR, Dinger M, Mattick JS. Non-coding RNAs: regulators of disease. J Pathol. (2010) 220:126–39. doi: 10.1002/path.2638
53. Uchida S, Dimmeler S. Long noncoding RNAs in cardiovascular diseases. Circ Res. (2015) 116:737–50. doi: 10.1161/CIRCRESAHA.116.302521
54. Archer K, Broskova Z, Bayoumi AS, Teoh JP, Davila A, Tang Y, et al. Long non-coding RNAs as master regulators in cardiovascular diseases. Int J Mol Sci. (2015) 16:23651–67. doi: 10.3390/ijms161023651
55. Guttman M, Amit I, Garber M, French C, Lin MF, Feldser D, et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature (2009) 458:223–7. doi: 10.1038/nature07672
56. Hamilton AJ, Baulcombe DC. A species of small antisense RNA in posttranscriptional gene silencing in plants. Science (1999) 286:950–2. doi: 10.1126/science.286.5441.950
57. Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature (2001) 411:494–8. doi: 10.1038/35078107
58. Carthew RW, Sontheimer EJ. Origins and mechanisms of miRNAs and siRNAs. Cell (2009) 136:642–55. doi: 10.1016/j.cell.2009.01.035
59. May RC, Plasterk RH. RNA Interference spreading in C. elegans. Methods Enzymol. (2005) 392:308–15. doi: 10.1016/S0076-6879(04)92018-6
60. Ruby JG, Jan C, Player C, Axtell MJ, Lee W, Nusbaum C, et al. Large-scale sequencing reveals 21U-RNAs and additional microRNAs and endogenous siRNAs in C. elegans. Cell (2006) 127:1193–207. doi: 10.1016/j.cell.2006.10.040
61. Carmell MA, Xuan Z, Zhang MQ, Hannon GJ. The argonaute family: tentacles that reach into RNAi, developmental control, stem cell maintenance, and tumorigenesis. Genes Dev. (2002) 16:2733–42. doi: 10.1101/gad.1026102
62. Lin H, Spradling AC. A novel group of pumilio mutations affects the asymmetric division of germline stem cells in the Drosophila ovary. Development (1997) 124:2463–76.
63. Aravin A, Gaidatzis D, Pfeffer S, Lagos-Quintana M, Landgraf P, Iovino N, Morris P, et al. A novel class of small RNAs bind to MILI protein in mouse testes. Nature (2006) 442:203–7. doi: 10.1038/nature04916
64. Lau NC, Seto AG, Kim J, Kuramochi-Miyagawa S, Nakano T, Bartel DP, et al. Characterization of the piRNA complex from rat testes. Science (2006) 313:363–7. doi: 10.1126/science.1130164
65. O'Donnell KA, Boeke JD. Mighty piwis defend the germline against genome intruders. Cell (2007) 129:37–44. doi: 10.1016/j.cell.2007.03.028
66. Seto AG, Kingston RE, Lau NC. The coming of age for piwi proteins. Mol Cell (2007) 26:603–9. doi: 10.1016/j.molcel.2007.05.021
67. Siomi MC, Sato K, Pezic D, Aravin AA. PIWI-interacting small RNAs: the vanguard of genome defence. Nat Rev Mol Cell Biol. (2011) 12:246–58. doi: 10.1038/nrm3089
68. Ku HY, Lin H. PIWI proteins and their interactors in piRNA biogenesis, germline development and gene expression. Natl Sci Rev. (2014) 1:205–18. doi: 10.1093/nsr/nwu014
69. Huang XA, Yin H, Sweeney S, Raha D, Snyder M, Lin H. A major epigenetic programming mechanism guided by piRNAs. Dev Cell. (2013) 24:502–16. doi: 10.1016/j.devcel.2013.01.023
70. Das PP, Bagijn MP, Goldstein LD, Woolford JR, Lehrbach NJ, Sapetschnig A, et al. Piwi and piRNAs act upstream of an endogenous siRNA pathway to suppress Tc3 transposon mobility in the Caenorhabditis elegans germline. Mol Cell. (2008) 31:79–90. doi: 10.1016/j.molcel.2008.06.003
71. Cordaux R, Batzer MA. The impact of retrotransposons on human genome evolution. Nat Rev Genet. (2009) 10:691–703. doi: 10.1038/nrg2640
72. Kazazian HH Jr. Mobile elements: drivers of genome evolution. Science (2004) 303:1626–32. doi: 10.1126/science.1089670
73. Sayed D, Hong C, Chen IY, Lypowy J, Abdellatif M. MicroRNAs play an essential role in the development of cardiac hypertrophy. Circ Res. (2007) 100:416–24. doi: 10.1161/01.RES.0000257913.42552.23
74. Barolo S, Posakony JW. Three habits of highly effective signaling pathways: principles of transcriptional control by developmental cell signaling. Genes Dev. (2002) 16:1167–81. doi: 10.1101/gad.976502
75. Hagen JW, Lai EC. MicroRNA control of cell-cell signaling during development and disease. Cell Cycle (2008) 7:2327–32. doi: 10.4161/cc.6447
76. Kwon C, Han Z, Olson EN, Srivastava D. MicroRNA1 influences cardiac differentiation in drosophila and regulates notch signaling. Proc Natl Acad Sci USA. (2005) 102:18986–91. doi: 10.1073/pnas.0509535102
77. Zhao Y, Ransom JF, Li A, Vedantham V, von Drehle M, Muth AN, et al. Dysregulation of cardiogenesis, cardiac conduction, and cell cycle in mice lacking miRNA-1-2. Cell (2007) 129:303–17. doi: 10.1016/j.cell.2007.03.030
78. Chen JF, Mandel EM, Thomson JM, Wu Q, Callis TE, Hammond SM, et al. The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and differentiation. Nat Genet. (2006) 38:228–33. doi: 10.1038/ng1725
79. Care A, Catalucci D, Felicetti F, Bonci D, Addario A, Gallo P, et al. MicroRNA-133 controls cardiac hypertrophy. Nat Med. (2007) 13:613–8. doi: 10.1038/nm1582
80. Zhao Y, Samal E, Srivastava D. Serum response factor regulates a muscle-specific microRNA that targets Hand2 during cardiogenesis. Nature (2005) 436:214–20. doi: 10.1038/nature03817
81. Pan Z, Sun X, Shan H, Wang N, Wang J, Ren J, et al. MicroRNA-101 inhibited postinfarct cardiac fibrosis and improved left ventricular compliance via the FBJ osteosarcoma oncogene/transforming growth factor-β1 pathway. Circulation (2012) 126:840–50. doi: 10.1161/CIRCULATIONAHA.112.094524
82. Wei L, Yuan M, Zhou R, Bai Q, Zhang W, Zhang M, et al. MicroRNA-101 inhibits rat cardiac hypertrophy by targeting Rab1a. J Cardiovasc Pharmacol. (2015) 65:357–63. doi: 10.1097/FJC.0000000000000203
83. van Rooij E, Sutherland LB, Thatcher JE, DiMaio JM, Naseem RH, Marshall WS, et al. Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc Natl Acad Sci USA. (2008) 105:13027–32. doi: 10.1073/pnas.0805038105
84. Wang J, Huang W, Xu R, Nie Y, Cao X, Meng J, et al. MicroRNA-24 regulates cardiac fibrosis after myocardial infarction. J Cell Mol Med. (2012) 16:2150–60. doi: 10.1111/j.1582-4934.2012.01523.x
85. Duisters RF, Tijsen AJ, Schroen B, Leenders JJ, Lentink V, van der Made I, et al. MiR-133 and MiR-30 regulate connective tissue growth factor: implications for a role of microRNAs in myocardial matrix remodeling. Circ Res. (2009) 104:170–8, 6p following 178. doi: 10.1161/CIRCRESAHA.108.182535
86. Zhang Y, Huang XR, Wei LH, Chung AC, Yu CM, Lan HY. miR-29b as a therapeutic agent for angiotensin II-induced cardiac fibrosis by targeting TGF-β/Smad3 signaling. Mol Ther. (2014) 22:974–85. doi: 10.1038/mt.2014.25
87. Arad M, Seidman JG, Seidman CE. Phenotypic diversity in hypertrophic cardiomyopathy. Hum Mol Genet. (2002) 11:2499–506. doi: 10.1093/hmg/11.20.2499
88. van Rooij E, Sutherland LB, Qi X, Richardson JA, Hill J, Olson EN. Control of stress-dependent cardiac growth and gene expression by a microRNA. Science (2007) 316:575–9. doi: 10.1126/science.1139089
89. Ucar A, Gupta SK, Fiedler J, Erikci E, Kardasinski M, Batkai S, et al. The miRNA-212/132 family regulates both cardiac hypertrophy and cardiomyocyte autophagy. Nat Commun. (2012) 3:1078. doi: 10.1038/ncomms2090
90. Duygu B, Da Costa Martins PA. MiR-21: a star player in cardiac hypertrophy. Cardiovasc Res. (2015) 105:235–7. doi: 10.1093/cvr/cvv026
91. Thum T, Gross C, Fiedler J, Fischer T, Kissler S, Bussen M, et al. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature (2008) 456:980–4. doi: 10.1038/nature07511
92. Roy S, Khanna S, Hussain SR, Biswas S, Azad A, Rink C, et al. MicroRNA expression in response to murine myocardial infarction: miR-21 regulates fibroblast metalloprotease-2 via phosphatase and tensin homologue. Cardiovasc Res. (2009) 82:21–9. doi: 10.1093/cvr/cvp015
93. Bang C, Batkai S, Dangwal S, Gupta SK, Foinquinos A, Holzmann A, et al. Cardiac fibroblast-derived microRNA passenger strand-enriched exosomes mediate cardiomyocyte hypertrophy. J Clin Invest. (2014) 124:2136–46. doi: 10.1172/JCI70577
94. Yan M, Chen C, Gong W, Yin Z, Zhou L, Chaugai S, et al. MiR-21-3p regulates cardiac hypertrophic response by targeting histone deacetylase-8. Cardiovasc Res. (2015) 105:340–352. doi: 10.1093/cvr/cvu254
95. He W, Huang H, Xie Q, Wang Z, Fan Y, Kong B, et al. MiR-155 Knockout in fibroblasts improves cardiac remodeling by targeting tumor protein P53-inducible nuclear protein 1. J Cardiovasc Pharmacol Ther. (2016) 21:423–35. doi: 10.1177/1074248415616188
96. Matkovich SJ, Hu Y, Eschenbacher WH, Dorn LE, Dorn GW. Direct and indirect involvement of microRNA-499 in clinical and experimental cardiomyopathy. Circ Res. (2012) 111:521–31. doi: 10.1161/CIRCRESAHA.112.265736
97. Zhang L, Chen X, Su T, Li H, Huang Q, Wu D, et al. Circulating miR-499 are novel and sensitive biomarker of acute myocardial infarction. J Thorac Dis. (2015) 7:303–8. doi: 10.1016/j.jacc.2015.06.318
98. Hosoda T, Zheng H, Cabral-da-Silva M, Sanada F, Ide-Iwata N, Ogorek B, et al. Human cardiac stem cell differentiation is regulated by a mircrine mechanism. Circulation (2011) 123:1287–96. doi: 10.1161/CIRCULATIONAHA.110.982918
99. Tang Y, Zheng J, Sun Y, Wu Z, Liu Z, Huang G. MicroRNA-1 regulates cardiomyocyte apoptosis by targeting Bcl-2. Int Heart J. (2009) 50:377–87. doi: 10.1536/ihj.50.377
100. Liu L, Zhang G, Liang Z, Liu X, Li T, Fan J, et al. MicroRNA-15b enhances hypoxia/reoxygenation-induced apoptosis of cardiomyocytes via a mitochondrial apoptotic pathway. Apoptosis (2014) 19:19–29. doi: 10.1007/s10495-013-0899-2
101. Cheng Y, Zhang C. MicroRNA-21 in cardiovascular disease. J Cardiovasc Transl Res. (2010) 3:251–5. doi: 10.1007/s12265-010-9169-7
102. Wei C, Li L, Gupta S. NF-κB-mediated miR-30b regulation in cardiomyocytes cell death by targeting Bcl-2. Mol Cell Biochem. (2014) 387:135–41. doi: 10.1007/s11010-013-1878-1
103. Fu BC, Lang JL, Zhang DY, Sun L, Chen W, Liu W, et al. Suppression of miR-34a expression in the myocardium protects against ischemia-reperfusion injury through SIRT1 protective pathway. Stem Cells Dev. (2017) 26:1270–82. doi: 10.1089/scd.2017.0062
104. Bernardo BC, Gao XM, Winbanks CE, Boey EJ, Tham YK, Kiriazis H, et al. Therapeutic inhibition of the miR-34 family attenuates pathological cardiac remodeling and improves heart function. Proc Natl Acad Sci USA. (2012) 109:17615–20. doi: 10.1073/pnas.1206432109
105. Li X, Zeng Z, Li Q, Xu Q, Xie J, Hao H, et al. Inhibition of microRNA-497 ameliorates anoxia/reoxygenation injury in cardiomyocytes by suppressing cell apoptosis and enhancing autophagy. Oncotarget (2015) 6:18829–44. doi: 10.18632/oncotarget.4774
106. Pan Z, Sun X, Ren J, Li X, Gao X, Lu C, et al. MiR-1 exacerbates cardiac ischemia-reperfusion injury in mouse models. PLoS ONE (2012) 7:e50515. doi: 10.1371/journal.pone.0050515
107. Kang B, Hong J, Xiao J, Zhu X, Ni X, Zhang Y, et al. Involvement of miR-1 in the protective effect of hydrogen sulfide against cardiomyocyte apoptosis induced by ischemia/reperfusion. Mol Biol Rep. (2014) 41:6845–53. doi: 10.1007/s11033-014-3570-2
108. Zhu H, Fan GC. Role of microRNAs in the reperfused myocardium towards post-infarct remodelling. Cardiovasc Res. (2012) 94:284–92. doi: 10.1093/cvr/cvr291
109. Yu XY, Song YH, Geng YJ, Lin QX, Shan ZX, Lin SG, et al. Glucose induces apoptosis of cardiomyocytes via microRNA-1 and IGF-1. Biochem Biophys Res Commun. (2008) 376:548–52. doi: 10.1016/j.bbrc.2008.09.025
110. Bostjancic E, Zidar N, Stajer D, Glavac D. MicroRNAs miR-1, miR-133a, miR-133b and miR-208 are dysregulated in human myocardial infarction. Cardiology (2010) 115:163–9. doi: 10.1159/000268088
111. He B, Xiao J, Ren AJ, Zhang YF, Zhang H, Chen M, et al. Role of miR-1 and mir-133a in myocardial ischemic postconditioning. J Biomed Sci. (2011) 18:22. doi: 10.1186/1423-0127-18-22
112. Ren XP, Wu J, Wang X, Sartor MA, Jones K, Qian J, et al. MicroRNA-320 is involved in the regulation of cardiac ischemia/reperfusion injury by targeting heat-shock protein 20. Circulation (2009) 119:2357–66. doi: 10.1161/CIRCULATIONAHA.108.814145
113. Fan GC, Ren X, Qian J, Yuan Q, Nicolaou P, Wang Y, et al. Novel cardioprotective role of a small heat-shock protein, Hsp20, against ischemia/reperfusion injury. Circulation (2005) 111:1792–99. doi: 10.1161/01.CIR.0000160851.41872.C6
114. Wang X, Zhang X, Ren XP, Chen J, Liu H, Yang J, et al. MicroRNA-494 targeting both proapoptotic and antiapoptotic proteins protects against ischemia/reperfusion-induced cardiac injury. Circulation (2010) 122:1308–18. doi: 10.1161/CIRCULATIONAHA.110.964684
115. Cheng Y, Liu X, Zhang S, Lin Y, Yang J, Zhang C. MicroRNA-21 protects against the H(2)O(2)-induced injury on cardiac myocytes via its target gene PDCD4. J Mol Cell Cardiol. (2009) 47:5–14. doi: 10.1016/j.yjmcc.2009.01.008
116. Cheng Y, Zhu P, Yang J, Liu X, Dong S, Wang X, et al. Ischaemic preconditioning-regulated miR-21 protects heart against ischaemia/reperfusion injury via anti-apoptosis through its target PDCD4. Cardiovasc Res. (2010) 87:431–9. doi: 10.1093/cvr/cvq082
117. Sayed D, He M, Hong C, Gao S, Rane S, Yang Z, et al. MicroRNA-21 is a downstream effector of AKT that mediates its antiapoptotic effects via suppression of Fas ligand. J Biol Chem. (2010) 285:20281–90. doi: 10.1074/jbc.M110.109207
118. Keyes KT, Xu J, Long B, Zhang C, Hu Z, Ye Y. Pharmacological inhibition of PTEN limits myocardial infarct size and improves left ventricular function postinfarction. Am J Physiol Heart Circ Physiol. (2010) 298:H1198–208. doi: 10.1152/ajpheart.00915.2009
119. Mocanu MM, Yellon DM. PTEN, the Achilles' heel of myocardial ischaemia/reperfusion injury? Br J Pharmacol. (2007) 150:833–8. doi: 10.1038/sj.bjp.0707155
120. Yin C, Salloum FN, Kukreja RC. A novel role of microRNA in late preconditioning: upregulation of endothelial nitric oxide synthase and heat shock protein 70. Circ Res. (2009) 104:572–5. doi: 10.1161/CIRCRESAHA.108.193250
121. Yin C, Wang X, Kukreja RC. Endogenous microRNAs induced by heat-shock reduce myocardial infarction following ischemia-reperfusion in mice. FEBS Lett. (2008) 582:4137–42. doi: 10.1016/j.febslet.2008.11.014
122. Dong S, Cheng Y, Yang J, Li J, Liu X, Wang X, et al. MicroRNA expression signature and the role of microRNA-21 in the early phase of acute myocardial infarction. J Biol Chem. (2009) 284:29514–25. doi: 10.1074/jbc.M109.027896
123. Toldo S, Das A, Mezzaroma E, Chau VQ, Marchetti C, Durrant D, et al. Induction of microRNA-21 with exogenous hydrogen sulfide attenuates myocardial ischemic and inflammatory injury in mice. Circ Cardiovasc Genet. (2014) 7:311–20. doi: 10.1161/CIRCGENETICS.113.000381
124. Olson JM, Yan Y, Bai X, Ge ZD, Liang M, Kriegel AJ, et al. Up-regulation of microRNA-21 mediates isoflurane-induced protection of cardiomyocytes. Anesthesiology (2015) 122:795–805. doi: 10.1097/ALN.0000000000000567
125. Qiao S, Olson JM, Paterson M, Yan Y, Zaja I, Liu Y, et al. MicroRNA-21 mediates isoflurane-induced cardioprotection against ischemia-reperfusion injury via Akt/nitric oxide synthase/mitochondrial permeability transition pore pathway. Anesthesiology (2015) 123:786–798. doi: 10.1097/ALN.0000000000000807
126. Ventura A, Young AG, Winslow MM, Lintault L, Meissner A, Erkeland SJ, et al. Targeted deletion reveals essential and overlapping functions of the miR-17 through 92 family of miRNA clusters. Cell (2008) 132:875–86. doi: 10.1016/j.cell.2008.02.019
127. Wang J, Greene SB, Bonilla-Claudio M, Tao Y, Zhang J, Bai Y, et al. Bmp signaling regulates myocardial differentiation from cardiac progenitors through a microRNA-mediated mechanism. Dev Cell. (2010) 19:903–12. doi: 10.1016/j.devcel.2010.10.022
128. Chen J, Huang ZP, Seok HY, Ding J, Kataoka M, Zhang Z, et al. Mir-17-92 cluster is required for and sufficient to induce cardiomyocyte proliferation in postnatal and adult hearts. Circ Res. (2013) 112:1557–66. doi: 10.1161/CIRCRESAHA.112.300658
129. Mogilyansky E, Rigoutsos I. The miR-17/92 cluster: a comprehensive update on its genomics, genetics, functions and increasingly important and numerous roles in health and disease. Cell Death Differ. (2013) 20:1603–14. doi: 10.1038/cdd.2013.125
130. Foshay KM, Gallicano GI. MiR-17 Family miRNAs are expressed during early mammalian development and regulate stem cell differentiation. Dev Biol. (2009) 326:431–43. doi: 10.1016/j.ydbio.2008.11.016
131. Frank D, Gantenberg J, Boomgaarden I, Kuhn C, Will R, Jarr KU, et al. MicroRNA-20a inhibits stress-induced cardiomyocyte apoptosis involving its novel target Egln3/PHD3. J Mol Cell Cardiol. (2012) 52:711–7. doi: 10.1016/j.yjmcc.2011.12.001
132. Shi J, Bei Y, Kong X, Liu X, Lei Z, Xu T, et al. MiR-17-3p contributes to exercise-induced cardiac growth and protects against myocardial ischemia-reperfusion injury. Theranostics (2017) 7:664–76. doi: 10.7150/thno.15162
133. Doebele C, Bonauer A, Fischer A, Scholz A, Reiss Y, Urbich C, et al. Members of the MicroRNA-17-92 Cluster exhibit a cell-intrinsic antiangiogenic function in endothelial cells. Blood (2010) 115:4944–50. doi: 10.1182/blood-2010-01-264812
134. Bonauer A, Carmona G, Iwasaki M, Mione M, Koyanagi M, Fischer A, et al. MicroRNA-92a controls angiogenesis and functional recovery of ischemic tissues in mice. Science (2009) 324:1710–3. doi: 10.1126/science.1174381
135. Wang S, Aurora AB, Johnson BA, Qi X, McAnally J, Hill JA, et al. The endothelial-specific microRNA miR-126 governs vascular integrity and angiogenesis. Dev. Cell. (2008) 15:261–71. doi: 10.1016/j.devcel.2008.07.002
136. Fish JE, Santoro MM, Morton SU, Yu S, Yeh RF, Wythe JD, et al. MiR-126 regulates angiogenic signaling and vascular integrity. Dev Cell. (2008) 15:272–84. doi: 10.1016/j.devcel.2008.07.008
137. Sessa R, Seano G, di Blasio L, Gagliardi PA, Isella C, Medico E, et al. The miR-126 regulates angiopoietin-1 signaling and vessel maturation by targeting p85β. Biochim Biophys Acta (2012) 1823:1925–35. doi: 10.1016/j.bbamcr.2012.07.011
138. Harris TA, Yamakuchi M, Ferlito M, Mendell JT, Lowenstein CJ. MicroRNA-126 regulates endothelial expression of vascular cell adhesion molecule 1. Proc Natl Acad Sci USA. (2008) 105:1516–21. doi: 10.1073/pnas.0707493105
139. van Solingen C, Seghers L, Bijkerk R, Duijs JM, Roeten MK, van Oeveren-Rietdijk AM, et al. Antagomir-mediated silencing of endothelial cell specific microRNA-126 impairs ischemia-induced angiogenesis. J Cell Mol Med. (2009) 13:1577–85. doi: 10.1111/j.1582-4934.2008.00613.x
140. Fasanaro P, D'Alessandra Y, Di Stefano V, Melchionna R, Romani S, Pompilio G, et al. MicroRNA-210 modulates endothelial cell response to hypoxia and inhibits the receptor tyrosine kinase ligand ephrin-A3. J Biol Chem. (2008) 283:15878–83. doi: 10.1074/jbc.M800731200
141. Kuijper S, Turner CJ, Adams RH. Regulation of angiogenesis by Eph-ephrin interactions. Trends Cardiovasc Med. (2007) 17:145–51. doi: 10.1016/j.tcm.2007.03.003
142. Mutharasan RK, Nagpal V, Ichikawa Y, Ardehali H. microRNA-210 is upregulated in hypoxic cardiomyocytes through Akt- and p53-dependent pathways and exerts cytoprotective effects. Am J Physiol Heart Circ Physiol. (2011) 301:H1519–30. doi: 10.1152/ajpheart.01080.2010
143. Hu S, Huang M, Li Z, Jia F, Ghosh Z, Lijkwan MA, et al. MicroRNA-210 as a novel therapy for treatment of ischemic heart disease. Circulation (2010) 122:S124–31. doi: 10.1161/CIRCULATIONAHA.109.928424
144. Frey N, McKinsey TA, Olson EN. Decoding calcium signals involved in cardiac growth and function. Nat Med. (2000) 6:1221–27. doi: 10.1038/81321
145. Aurora AB, Mahmoud AI, Luo X, Johnson BA, van Rooij E, Matsuzaki S, et al. MicroRNA-214 protects the mouse heart from ischemic injury by controlling Ca(2)(+) overload and cell death. J Clin Invest. (2012) 122:1222–32. doi: 10.1172/JCI59327
146. Wang X, Ha T, Hu Y, Lu C, Liu L, Zhang X, et al. MicroRNA-214 protects against hypoxia/reoxygenation induced cell damage and myocardial ischemia/reperfusion injury via suppression of PTEN and Bim1 expression. Oncotarget (2016) 7:86926–36. doi: 10.18632/oncotarget.13494
147. Eisenhardt SU, Weiss JB, Smolka C, Maxeiner J, Pankratz F, Bemtgen X, et al. MicroRNA-155 aggravates ischemia-reperfusion injury by modulation of inflammatory cell recruitment and the respiratory oxidative burst. Basic Res Cardiol. (2015) 110:32. doi: 10.1007/s00395-015-0490-9
148. Rane S, He M, Sayed D, Vashistha H, Malhotra A, Sadoshima J, et al. Downregulation of miR-199a derepresses hypoxia-inducible factor-1alpha and Sirtuin 1 and recapitulates hypoxia preconditioning in cardiac myocytes. Circ Res. (2009) 104:879–86. doi: 10.1161/CIRCRESAHA.108.193102
149. Tabas I, Garcia-Cardena G, Owens GK. Recent insights into the cellular biology of atherosclerosis. J Cell Biol. (2015) 209:13–22. doi: 10.1083/jcb.201412052
150. Moore KJ, Rayner KJ, Suarez Y, Fernandez-Hernando C. The role of microRNAs in cholesterol efflux and hepatic lipid metabolism. Annu Rev Nutr. (2011) 31:49–63. doi: 10.1146/annurev-nutr-081810-160756
151. Elmen J, Lindow M, Schutz S, Lawrence M, Petri A, Obad S, et al. LNA-mediated microRNA silencing in non-human primates. Nature (2008) 452:896–9. doi: 10.1038/nature06783
152. Elmen J, Lindow M, Silahtaroglu A, Bak M, Christensen M, Lind-Thomsen A, et al. Antagonism of microRNA-122 in mice by systemically administered LNA-antimiR leads to up-regulation of a large set of predicted target mRNAs in the liver. Nucleic Acids Res. (2008) 36:1153–62. doi: 10.1093/nar/gkm1113
153. Gerin I, Clerbaux LA, Haumont O, Lanthier N, Das AK, Burant CF, et al. Expression of miR-33 from an SREBP2 intron inhibits cholesterol export and fatty acid oxidation. J Biol Chem. (2010) 285:33652–61. doi: 10.1074/jbc.M110.152090
154. Horie T, Ono K, Horiguchi M, Nishi H, Nakamura T, Nagao K, et al. MicroRNA-33 encoded by an intron of sterol regulatory element-binding protein 2 (Srebp2) regulates HDL in vivo. Proc Natl Acad Sci USA. (2010) 107:17321–6. doi: 10.1073/pnas.1008499107
155. Rayner KJ, Sheedy FJ, Esau CC, Hussain FN, Temel RE, Parathath S, et al. Antagonism of miR-33 in mice promotes reverse cholesterol transport and regression of atherosclerosis. J Clin Invest. (2011) 121:2921–31. doi: 10.1172/JCI57275
156. Ramirez CM, Davalos A, Goedeke L, Salerno AG, Warrier N, Cirera-Salinas D, et al. MicroRNA-758 regulates cholesterol efflux through posttranscriptional repression of ATP-binding cassette transporter A1. Arterioscler Thromb Vasc Biol. (2011) 31:2707–14. doi: 10.1161/ATVBAHA.111.232066
157. Sun D, Zhang J, Xie J, Wei W, Chen M, Zhao X. MiR-26 controls LXR-dependent cholesterol efflux by targeting ABCA1 and ARL7. FEBS Lett. (2012) 586:1472–9. doi: 10.1016/j.febslet.2012.03.068
158. Kim J, Yoon H, Ramirez CM, Lee SM, Hoe HS, Fernandez-Hernando C, et al. MiR-106b impairs cholesterol efflux and increases Aβ levels by repressing ABCA1 expression. Exp Neurol. (2012) 235:476–83. doi: 10.1016/j.expneurol.2011.11.010
159. de Aguiar Vallim TQ, Tarling EJ, Kim T, Civelek M, Baldan A, Esau C, et al. MicroRNA-144 regulates hepatic ATP binding cassette transporter A1 and plasma high-density lipoprotein after activation of the nuclear receptor farnesoid X receptor. Circ Res. (2013) 112:1602–12. doi: 10.1161/CIRCRESAHA.112.300648
160. Najafi-Shoushtari SH, Kristo F, Li Y, Shioda T, Cohen DE, Gerszten RE, et al. MicroRNA-33 and the SREBP host genes cooperate to control cholesterol homeostasis. Science (2010) 328:1566–9. doi: 10.1126/science.1189123
161. Marquart TJ, Allen RM, Ory DS, Baldan A. miR-33 links SREBP-2 induction to repression of sterol transporters. Proc Natl Acad Sci USA. (2010) 107:12228–32. doi: 10.1073/pnas.1005191107
162. Horie T, Baba O, Kuwabara Y, Chujo Y, Watanabe S, Kinoshita M, et al. MicroRNA-33 deficiency reduces the progression of atherosclerotic plaque in ApoE−/− mice. J Am Heart Assoc. (2012) 1:e003376. doi: 10.1161/JAHA.112.003376
163. Rotllan N, Ramirez CM, Aryal B, Esau CC, Fernandez-Hernando C. Therapeutic silencing of microRNA-33 inhibits the progression of atherosclerosis in Ldlr−/− mice–brief report. Arterioscler Thromb Vasc Biol. (2013) 33:1973–7. doi: 10.1161/ATVBAHA.113.301732
164. Rayner KJ, Suarez Y, Davalos A, Parathath S, Fitzgerald ML, Tamehiro N, et al. MiR-33 contributes to the regulation of cholesterol homeostasis. Science (2010) 328:1570–3. doi: 10.1126/science.1189862
165. Price NL, Rotllan N, Canfran-Duque A, Zhang X, Pati P, Arias N, et al. Genetic dissection of the impact of miR-33a and miR-33b during the progression of atherosclerosis. Cell Rep. (2017) 21:1317–30. doi: 10.1016/j.celrep.2017.10.023
166. Meiler S, Baumer Y, Toulmin E, Seng K, Boisvert WA. MicroRNA 302a is a novel modulator of cholesterol homeostasis and atherosclerosis. Arterioscler Thromb Vasc Biol. (2015) 35:323–31. doi: 10.1161/ATVBAHA.114.304878
167. Cuhlmann S, Van der Heiden K, Saliba D, Tremoleda JL, Khalil M, Zakkar M, et al. Disturbed blood flow induces relA expression via c-Jun N-terminal kinase 1: a novel mode of NF-κB regulation that promotes arterial inflammation. Circ Res. (2011) 108:950–9. doi: 10.1161/CIRCRESAHA.110.233841
168. Braun M, Pietsch P, Schror K, Baumann G, Felix SB. Cellular adhesion molecules on vascular smooth muscle cells. Cardiovasc Res. (1999) 41:395–401. doi: 10.1016/S0008-6363(98)00302-2
169. Suarez Y, Wang C, Manes TD, Pober JS. Cutting edge: TNF-induced microRNAs regulate TNF-induced expression of E-selectin and intercellular adhesion molecule-1 on human endothelial cells: feedback control of inflammation. J Immunol. (2010) 184:21–5. doi: 10.4049/jimmunol.0902369
170. Sun X, He S, Wara AKM, Icli B, Shvartz E, Tesmenitsky Y, et al. Systemic delivery of microRNA-181b inhibits nuclear factor-κB activation, vascular inflammation, and atherosclerosis in apolipoprotein E-Deficient mice. Circ Res. (2014) 114:32–40. doi: 10.1161/CIRCRESAHA.113.302089
171. Sun X, Icli B, Wara AK, Belkin N, He S, Kobzik L, et al. MicroRNA-181b regulates NF-κB-mediated vascular inflammation. J Clin Invest. (2012) 122:1973–90. doi: 10.1172/JCI61495
172. Cheng HS, Sivachandran N, Lau A, Boudreau E, Zhao JL, Baltimore D, et al. MicroRNA-146 represses endothelial activation by inhibiting pro-inflammatory pathways. EMBO Mol Med. (2013) 5:1017–34. doi: 10.1002/emmm.201202318
173. Schober A, Nazari-Jahantigh M, Wei Y, Bidzhekov K, Gremse F, Grommes J, et al. MicroRNA-126-5p promotes endothelial proliferation and limits atherosclerosis by suppressing Dlk1. Nat Med. (2014) 20:368–76. doi: 10.1038/nm.3487
174. Hao XZ, Fan HM. Identification of miRNAs as atherosclerosis biomarkers and functional role of miR-126 in atherosclerosis progression through MAPK signalling pathway. Eur Rev Med Pharmacol Sci. (2017) 21:2725–33.
175. Qin B, Xiao B, Liang D, Xia J, Li Y, Yang H. MicroRNAs expression in Ox-LDL treated HUVECs: MiR-365 modulates apoptosis and Bcl-2 expression. Biochem Biophys Res Commun. (2011) 410:127–33. doi: 10.1016/j.bbrc.2011.05.118
176. Lin B, Feng DG, Wang F, Wang JX, Xu CG, Zhao H, et al. MiR-365 Participates in coronary atherosclerosis through regulating IL-6. Eur Rev Med Pharmacol Sci. (2016) 20:5186–92.
177. Kim MH, Ham O, Lee SY, Choi E, Lee CY, Park JH, et al. MicroRNA-365 inhibits the proliferation of vascular smooth muscle cells by targeting cyclin D1. J Cell Biochem. (2014) 115:1752–61. doi: 10.1002/jcb.24841
178. Chen Z, Wen L, Martin M, Hsu CY, Fang L, Lin FM, et al. Oxidative stress activates endothelial innate immunity via sterol regulatory element binding protein 2 (SREBP2) transactivation of microRNA-92a. Circulation (2015) 131:805–14. doi: 10.1161/CIRCULATIONAHA.114.013675
179. Loyer X, Potteaux S, Vion AC, Guerin CL, Boulkroun S, Rautou PE, et al. Inhibition of microRNA-92a prevents endothelial dysfunction and atherosclerosis in mice. Circ Res. (2014) 114:434–43. doi: 10.1161/CIRCRESAHA.114.302213
180. Fang Y, Davies PF. Site-specific microRNA-92a regulation of Kruppel-like factors 4 and 2 in atherosusceptible endothelium. Arterioscler Thromb Vasc Biol. (2012) 32:979–87. doi: 10.1161/ATVBAHA.111.244053
181. Saha B, Bruneau JC, Kodys K, Szabo G. Alcohol-induced miR-27a regulates differentiation and M2 macrophage polarization of normal human monocytes. J Immunol. (2015) 194:3079–87. doi: 10.4049/jimmunol.1402190
182. Wei Y, Nazari-Jahantigh M, Chan L, Zhu M, Heyll K, Corbalan-Campos J, et al. The microRNA-342-5p fosters inflammatory macrophage activation through an Akt1- and microRNA-155-dependent pathway during atherosclerosis. Circulation (2013) 127:1609–19. doi: 10.1161/CIRCULATIONAHA.112.000736
183. Nazari-Jahantigh M, Wei Y, Noels H, Akhtar S, Zhou Z, Koenen RR, et al. MicroRNA-155 promotes atherosclerosis by repressing Bcl6 in macrophages. J Clin Invest. (2012) 122:4190–202. doi: 10.1172/JCI61716
184. Tian FJ, An LN, Wang GK, Zhu JQ, Li Q, Zhang YY, et al. Elevated microRNA-155 promotes foam cell formation by targeting HBP1 in atherogenesis. Cardiovasc Res. (2014) 103:100–10. doi: 10.1093/cvr/cvu070
185. Ye J, Guo R, Shi Y, Qi F, Guo C, Yang L. miR-155 regulated inflammation response by the SOCS1-STAT3-PDCD4 axis in atherogenesis. Mediators Inflamm. (2016) 2016:8060182. doi: 10.1155/2016/8060182
186. Minamino T, Komuro I. Vascular cell senescence: contribution to atherosclerosis. Circ Res. (2007) 100:15–26. doi: 10.1161/01.RES.0000256837.40544.4a
187. Menghini R, Casagrande V, Cardellini M, Martelli E, Terrinoni A, Amati F, et al. MicroRNA 217 modulates endothelial cell senescence via silent information regulator 1. Circulation (2009) 120:1524–32. doi: 10.1161/CIRCULATIONAHA.109.864629
188. Badi I, Burba I, Ruggeri C, Zeni F, Bertolotti M, Scopece A, et al. MicroRNA-34a induces vascular smooth muscle cells senescence by SIRT1 downregulation and promotes the expression of age-associated pro-inflammatory secretory factors. J Gerontol A Biol Sci Med Sci. (2015) 70:1304–11. doi: 10.1093/gerona/glu180
189. Su G, Sun G, Liu H, Shu L, Liang Z. Downregulation of miR-34a promotes endothelial cell growth and suppresses apoptosis in atherosclerosis by regulating Bcl-2. Heart Vessels (2018). doi: 10.1007/s00380-018-1169-6
190. Wang M, Li W, Chang GQ, Ye CS, Ou JS, Li XX, et al. MicroRNA-21 regulates vascular smooth muscle cell function via targeting tropomyosin 1 in arteriosclerosis obliterans of lower extremities. Arterioscler Thromb Vasc Biol. (2011) 31:2044–53. doi: 10.1161/ATVBAHA.111.229559
191. Zhou J, Wang KC, Wu W, Subramaniam S, Shyy JY, Chiu JJ, et al. MicroRNA-21 targets peroxisome proliferators-activated receptor-alpha in an autoregulatory loop to modulate flow-induced endothelial inflammation. Proc Natl Acad Sci USA. (2011) 108:10355–60. doi: 10.1073/pnas.1107052108
192. Weber M, Baker MB, Moore JP, Searles CD. MiR-21 is induced in endothelial cells by shear stress and modulates apoptosis and ENOS activity. Biochem Biophys Res Commun. (2010) 393:643–8. doi: 10.1016/j.bbrc.2010.02.045
193. Kumar S, Kim CW, Simmons RD, Jo H. Role of flow-sensitive microRNAs in endothelial dysfunction and atherosclerosis: mechanosensitive athero-miRs. Arterioscler Thromb Vasc Biol. (2014) 34:2206–16. doi: 10.1161/ATVBAHA.114.303425
194. Koroleva IA, Nazarenko MS, Kucher AN. Role of microRNA in development of instability of atherosclerotic plaques. Biochemistry (Mosc.) (2017) 82:1380–90. doi: 10.1134/S0006297917110165
195. Andreou I, Sun X, Stone PH, Edelman ER, Feinberg MW. miRNAs in atherosclerotic plaque initiation, progression, and rupture. Trends Mol Med. (2015) 21:307–18. doi: 10.1016/j.molmed.2015.02.003
196. Fan X, Wang E, Wang X, Cong X, Chen X. MicroRNA-21 is a unique signature associated with coronary plaque instability in humans by regulating matrix metalloproteinase-9 via reversion-inducing cysteine-rich protein with kazal motifs. Exp Mol Pathol. (2014) 96:242–9. doi: 10.1016/j.yexmp.2014.02.009
197. Canfran-Duque A, Rotllan N, Zhang X, Fernandez-Fuertes M, Ramirez-Hidalgo C, Araldi E, et al. Macrophage deficiency of miR-21 promotes apoptosis, plaque necrosis, and vascular inflammation during atherogenesis. EMBO Mol Med. (2017) 9:1244–62. doi: 10.15252/emmm.201607492
198. Davies PF. Hemodynamic shear stress and the endothelium in cardiovascular pathophysiology. Nat Clin Pract Cardiovasc Med. (2009) 6:16–26. doi: 10.1038/ncpcardio1397
199. Cheng C, Tempel D, van Haperen R, de Boer HC, Segers D, Huisman M, et al. Shear stress-induced changes in atherosclerotic plaque composition are modulated by chemokines. J Clin Invest. (2007) 117:616–26. doi: 10.1172/JCI28180
200. Heo KS, Fujiwara K, Abe J. Shear stress and atherosclerosis. Mol Cell. (2014) 37:435–40. doi: 10.14348/molcells.2014.0078
201. Ni CW, Qiu H, Jo H. MicroRNA-663 upregulated by oscillatory shear stress plays a role in inflammatory response of endothelial cells. Am J Physiol Heart Circ Physiol. (2011) 300:H1762–9. doi: 10.1152/ajpheart.00829.2010
202. Fang Y, Shi C, Manduchi E, Civelek M, Davies PF. MicroRNA-10a regulation of proinflammatory phenotype in athero-susceptible endothelium in vivo and in vitro. Proc Natl Acad Sci USA. (2010) 107:13450–5. doi: 10.1073/pnas.1002120107
203. Moradi N, Fadaei R, Ahmadi R, Kazemian E, Fallah S. Lower expression of miR-10a in coronary artery disease and its association with pro/anti-inflammatory cytokines. Clin Lab. (2018) 64:847–54. doi: 10.7754/Clin.Lab.2018.171222
204. Ulrich V, Rotllan N, Araldi E, Luciano A, Skroblin P, Abonnenc M, et al. Chronic miR-29 antagonism promotes favorable plaque remodeling in atherosclerotic mice. EMBO Mol Med. (2016) 8:643–53. doi: 10.15252/emmm.201506031
205. Lovren F, Pan Y, Quan A, Singh KK, Shukla PC, Gupta N, et al. MicroRNA-145 targeted therapy reduces atherosclerosis. Circulation (2012) 126:S81–90. doi: 10.1161/CIRCULATIONAHA.111.084186
206. Choi SE, Fu T, Seok S, Kim DH, Yu E, Lee KW, et al. Elevated microRNA-34a in obesity reduces NAD+ levels and SIRT1 activity by directly targeting NAMPT. Aging Cell. (2013) 12:1062–72. doi: 10.1111/acel.12135
207. Xu G, Chen J, Jing G, Shalev A. Thioredoxin-interacting protein regulates insulin transcription through microRNA-204. Nat Med. (2013) 19:1141–6. doi: 10.1038/nm.3287
208. Trajkovski M, Hausser J, Soutschek J, Bhat B, Akin A, Zavolan M, et al. MicroRNAs 103 and 107 regulate insulin sensitivity. Nature (2011) 474:649–53. doi: 10.1038/nature10112
209. Wang KJ, Zhao X, Liu YZ, Zeng QT, Mao XB, Li SN, et al. Circulating miR-19b-3p, miR-134-5p and miR-186-5p are promising novel biomarkers for early diagnosis of acute myocardial infarction. Cell Physiol Biochem. (2016) 38:1015–29. doi: 10.1159/000443053
210. Xiao F, Yu J, Liu B, Guo Y, Li K, Deng J, et al. A novel function of microRNA 130a-3p in hepatic insulin sensitivity and liver steatosis. Diabetes (2014) 63:2631–42. doi: 10.2337/db13-1689
211. Kishore R, Verma SK, Mackie AR, Vaughan EE, Abramova TV, Aiko I, et al. Bone marrow progenitor cell therapy-mediated paracrine regulation of cardiac miRNA-155 modulates fibrotic response in diabetic hearts. PLoS ONE (2013) 8:e60161. doi: 10.1371/journal.pone.0060161
212. Ruan Q, Wang T, Kameswaran V, Wei Q, Johnson DS, Matschinsky F, et al. The microRNA-21-PDCD4 axis prevents type 1 diabetes by blocking pancreatic beta cell death. Proc Natl Acad Sci USA. (2011) 108:12030–5. doi: 10.1073/pnas.1101450108
213. Wang XH, Qian RZ, Zhang W, Chen SF, Jin HM, Hu RM. MicroRNA-320 expression in myocardial microvascular endothelial cells and its relationship with insulin-like growth factor-1 in type 2 diabetic rats. Clin Exp Pharmacol Physiol. (2009) 36:181–8. doi: 10.1111/j.1440-1681.2008.05057.x
214. Wang JM, Tao J, Chen DD, Cai JJ, Irani K, Wang Q, et al. MicroRNA miR-27b rescues bone marrow-derived angiogenic cell function and accelerates wound healing in type 2 diabetes mellitus. Arterioscler Thromb Vasc Biol. (2014) 34:99–109. doi: 10.1161/ATVBAHA.113.302104
215. Aragno M, Mastrocola R, Medana C, Catalano MG, Vercellinatto I, Danni O, et al. Oxidative stress-dependent impairment of cardiac-specific transcription factors in experimental diabetes. Endocrinology (2006) 147:5967–74. doi: 10.1210/en.2006-0728
216. Basta G, Schmidt AM, De Caterina R. Advanced glycation end products and vascular inflammation: implications for accelerated atherosclerosis in diabetes. Cardiovasc Res. (2004) 63:582–92. doi: 10.1016/j.cardiores.2004.05.001
217. Thallas-Bonke V, Lindschau C, Rizkalla B, Bach LA, Boner G, Meier M, et al. Attenuation of extracellular matrix accumulation in diabetic nephropathy by the advanced glycation end product cross-link breaker ALT-711 via a protein kinase C-α-dependent pathway. Diabetes (2004) 53:2921–30. doi: 10.2337/diabetes.53.11.2921
218. Bu DX, Rai V, Shen X, Rosario R, Lu Y, D'Agati V, et al. Activation of the ROCK1 branch of the transforming growth factor-β pathway contributes to RAGE-dependent acceleration of atherosclerosis in diabetic ApoE-null mice. Circ Res. (2010) 106:1040–51. doi: 10.1161/CIRCRESAHA.109.201103
219. Hien TT, Turczynska KM, Dahan D, Ekman M, Grossi M, Sjogren J, et al. Elevated glucose levels promote contractile and cytoskeletal gene expression in vascular smooth muscle via Rho/protein kinase C and actin polymerization. J Biol Chem. (2016) 291:3552–68. doi: 10.1074/jbc.M115.654384
220. Zampetaki A, Kiechl S, Drozdov I, Willeit P, Mayr U, Prokopi M, et al. Plasma microRNA profiling reveals loss of endothelial miR-126 and other microRNAs in type 2 diabetes. Circ Res. (2010) 107:810–7. doi: 10.1161/CIRCRESAHA.110.226357
221. Jansen F, Yang X, Hoelscher M, Cattelan A, Schmitz T, Proebsting S, et al. Endothelial microparticle-mediated transfer of microRNA-126 promotes vascular endothelial cell repair via SPRED1 and is abrogated in glucose-damaged endothelial microparticles. Circulation (2013) 128:2026–38. doi: 10.1161/CIRCULATIONAHA.113.001720
222. Elia L, Contu R, Quintavalle M, Varrone F, Chimenti C, Russo MA, et al. Reciprocal regulation of microRNA-1 and insulin-like growth factor-1 signal transduction cascade in cardiac and skeletal muscle in physiological and pathological conditions. Circulation (2009) 120:2377–385. doi: 10.1161/CIRCULATIONAHA.109.879429
223. Varrone F, Gargano B, Carullo P, Di Silvestre D, De Palma A, Grasso L, et al. The circulating level of FABP3 is an indirect biomarker of microRNA-1. J Am Coll Cardiol. (2013) 61:88–95. doi: 10.1016/j.jacc.2012.08.1003
224. Katare R, Caporali A, Zentilin L, Avolio E, Sala-Newby G, Oikawa A, et al. Intravenous gene therapy with PIM-1 via a cardiotropic viral vector halts the progression of diabetic cardiomyopathy through promotion of prosurvival signaling. Circ Res. (2011) 108:1238–51. doi: 10.1161/CIRCRESAHA.110.239111
225. Greco S, Fasanaro P, Castelvecchio S, D'Alessandra Y, Arcelli D, Di Donato M, et al. MicroRNA dysregulation in diabetic ischemic heart failure patients. Diabetes (2012) 61:1633–41. doi: 10.2337/db11-0952
226. Park DS, Woodman SE, Schubert W, Cohen AW, Frank PG, Chandra M, et al. Caveolin-1/3 double-knockout mice are viable, but lack both muscle and non-muscle caveolae, and develop a severe cardiomyopathic phenotype. Am J Pathol. (2002) 160:2207–17. doi: 10.1016/S0002-9440(10)61168-6
227. Zhao F, Li B, Wei YZ, Zhou B, Wang H, Chen M, et al. MicroRNA-34a regulates high glucose-induced apoptosis in H9c2 cardiomyocytes. J Huazhong Univ Sci Technolog Med Sci. (2013) 33:834–9. doi: 10.1007/s11596-013-1207-7
228. Yang Y, Cheng HW, Qiu Y, Dupee D, Noonan M, Lin YD, et al. MicroRNA-34a plays a key role in cardiac repair and regeneration following myocardial infarction. Circ Res. (2015) 117:450–9. doi: 10.1161/CIRCRESAHA.117.305962
229. He L, He X, Lim LP, de Stanchina E, Xuan Z, Liang Y, et al. A microRNA component of the p53 tumour suppressor network. Nature (2007) 447:1130–4. doi: 10.1038/nature05939
230. Fomison-Nurse I, Saw EEL, Gandhi S, Munasinghe PE, Van Hout I, Williams MJA, et al. Diabetes induces the activation of pro-ageing miR-34a in the heart, but has differential effects on cardiomyocytes and cardiac progenitor cells. Cell Death Differ. (2018). doi: 10.1038/s41418-017-0047-6
231. Li J, Donath S, Li Y, Qin D, Prabhakar BS, Li P. MiR-30 regulates mitochondrial fission through targeting P53 and the dynamin-related protein-1 pathway. PLoS Genet. (2010) 6:e1000795. doi: 10.1371/journal.pgen.1000795
232. Forini F, Kusmic C, Nicolini G, Mariani L, Zucchi R, Matteucci M, et al. Triiodothyronine prevents cardiac ischemia/reperfusion mitochondrial impairment and cell loss by regulating miR30a/P53 axis. Endocrinology (2014) 155:4581–90. doi: 10.1210/en.2014-1106
233. Raut SK, Singh GB, Rastogi B, Saikia UN, Mittal A, Dogra N, et al. MiR-30c and MiR-181a Synergistically modulate P53-P21 pathway in diabetes induced cardiac hypertrophy. Mol Cell Biochem. (2016) 417:191–203. doi: 10.1007/s11010-016-2729-7
234. Qiao Y, Ma N, Wang X, Hui Y, Li F, Xiang Y, et al. MiR-483-5p controls angiogenesis in vitro and targets serum response factor. FEBS Lett. (2011) 585:3095–100. doi: 10.1016/j.febslet.2011.08.039
235. Qiao Y, Zhao Y, Liu Y, Ma N, Wang C, Zou J, et al. MiR-483-3p regulates hyperglycaemia-induced cardiomyocyte apoptosis in transgenic mice. Biochem Biophys Res Commun. (2016) 477:541–7. doi: 10.1016/j.bbrc.2016.06.051
236. Munoz JP, Collao A, Chiong M, Maldonado C, Adasme T, Carrasco L, et al. The transcription factor MEF2C mediates cardiomyocyte hypertrophy induced by IGF-1 signaling. Biochem Biophys Res Commun. (2009) 388:155–60. doi: 10.1016/j.bbrc.2009.07.147
237. Ruiz MA, Chakrabarti S. MicroRNAs: the underlying mediators of pathogenetic processes in vascular complications of diabetes. Can J Diabetes (2013) 37:339–44. doi: 10.1016/j.jcjd.2013.07.003
238. Feng B, Chen S, George B, Feng Q, Chakrabarti S. MiR133a regulates cardiomyocyte hypertrophy in diabetes. Diabetes Metab Res Rev. (2010) 26:40–9. doi: 10.1002/dmrr.1054
239. Lu H, Buchan RJ, Cook SA. MicroRNA-223 regulates glut4 expression and cardiomyocyte glucose metabolism. Cardiovasc Res. (2010) 86:410–20. doi: 10.1093/cvr/cvq010
240. Horie T, Ono K, Nishi H, Iwanaga Y, Nagao K, Kinoshita M, et al. MicroRNA-133 regulates the expression of GLUT4 by targeting KLF15 and is involved in metabolic control in cardiac myocytes. Biochem Biophys Res Commun. (2009) 389:315–20. doi: 10.1016/j.bbrc.2009.08.136
241. Zhu H, Shyh-Chang N, Segre AV, Shinoda G, Shah SP, Einhorn WS, et al. The Lin28/let-7 axis regulates glucose metabolism. Cell (2011) 147:81–94. doi: 10.1016/j.cell.2011.08.033
242. Zhu H, Shah S, Shyh-Chang N, Shinoda G, Einhorn WS, Viswanathan SR, et al. Lin28a transgenic mice manifest size and puberty phenotypes identified in human genetic association studies. Nat Genet. (2010) 42:626–30. doi: 10.1038/ng.593
243. Li J, Ren Y, Shi E, Tan Z, Xiong J, Yan L, et al. Inhibition of the Let-7 Family microRNAs induces cardioprotection against ischemia-reperfusion injury in diabetic rats. Ann Thorac Surg. (2016) 102:829–35. doi: 10.1016/j.athoracsur.2016.02.016
244. Zheng D, Ma J, Yu Y, Li M, Ni R, Wang G, et al. Silencing of miR-195 reduces diabetic cardiomyopathy in C57BL/6 mice. Diabetologia (2015) 58:1949–58. doi: 10.1007/s00125-015-3622-8
245. Wang GK, Zhu JQ, Zhang JT, Li Q, Li Y, He J, et al. Circulating microRNA: a novel potential biomarker for early diagnosis of acute myocardial infarction in humans. Eur Heart J. (2010) 31:659–66. doi: 10.1093/eurheartj/ehq013
246. Gidlof O, Andersson P, van der Pals J, Gotberg M, Erlinge D. Cardiospecific microRNA plasma levels correlate with troponin and cardiac function in patients with ST elevation myocardial infarction, are selectively dependent on renal elimination, and can be detected in urine samples. Cardiology (2011) 118:217–26. doi: 10.1159/000328869
247. D'Alessandra Y, Devanna P, Limana F, Straino S, Di Carlo A, Brambilla PG, et al. Circulating microRNAs are new and sensitive biomarkers of myocardial infarction. Eur Heart J. (2010) 31:2765–73. doi: 10.1093/eurheartj/ehq167
248. Liebetrau C, Mollmann H, Dorr O, Szardien S, Troidl C, Willmer M, et al. Release kinetics of circulating muscle-enriched microRNAs in patients undergoing transcoronary ablation of septal hypertrophy. J Am Coll Cardiol. (2013) 62:992–8. doi: 10.1016/j.jacc.2013.05.025
249. Widera C, Gupta SK, Lorenzen JM, Bang C, Bauersachs J, Bethmann K, et al. Diagnostic and prognostic impact of six circulating microRNAs in acute coronary syndrome. J Mol Cell Cardiol. (2011) 51:872–5. doi: 10.1016/j.yjmcc.2011.07.011
250. Gidlof O, Smith JG, Miyazu K, Gilje P, Spencer A, Blomquist S, et al. Circulating cardio-enriched microRNAs are associated with long-term prognosis following myocardial infarction. BMC Cardiovasc Disord. (2013) 13:12. doi: 10.1186/1471-2261-13-12
251. Wang F, Long G, Zhao C, Li H, Chaugai S, Wang Y, et al. Plasma microRNA-133a is a new marker for both acute myocardial infarction and underlying coronary artery stenosis. J Transl Med. (2013) 11:222. doi: 10.1186/1479-5876-11-222
252. Hoekstra M, van der Lans CA, Halvorsen B, Gullestad L, Kuiper J, Aukrust P, et al. The peripheral blood mononuclear cell microRNA signature of coronary artery disease. Biochem Biophys Res Commun. (2010) 394:792–7. doi: 10.1016/j.bbrc.2010.03.075
253. Qiang L, Hong L, Ningfu W, Huaihong C, Jing W. Expression of miR-126 and miR-508-5p in endothelial progenitor cells is associated with the prognosis of chronic heart failure patients. Int J Cardiol. (2013) 168:2082–8. doi: 10.1016/j.ijcard.2013.01.160
254. Al-Kafaji G, Al-Mahroos G, Abdulla Al-Muhtaresh H, Sabry MA, Abdul RR, Salem AH. Circulating endothelium-enriched microRNA-126 as a potential biomarker for coronary artery disease in type 2 diabetes mellitus patients. Biomarkers (2017) 22:268–78. doi: 10.1080/1354750X.2016.1204004
255. Zernecke A, Bidzhekov K, Noels H, Shagdarsuren E, Gan L, Denecke B, et al. Delivery of microRNA-126 by apoptotic bodies induces CXCL12-dependent vascular protection. Sci Signal (2009) 2:ra81. doi: 10.1126/scisignal.2000610
256. Ovchinnikova ES, Schmitter D, Vegter EL, Ter Maaten JM, Valente MA, Liu LC, et al. Signature of circulating microRNAs in patients with acute heart failure. Eur J Heart Fail. (2016) 18:414–23. doi: 10.1002/ejhf.332
257. Morley-Smith AC, Mills A, Jacobs S, Meyns B, Rega F, Simon AR, et al. Circulating microRNAs for predicting and monitoring response to mechanical circulatory support from a left ventricular assist device. Eur J Heart Fail. (2014) 16:871–9. doi: 10.1002/ejhf.116
258. Marfella R, Di Filippo C, Potenza N, Sardu C, Rizzo MR, Siniscalchi M, et al. Circulating microRNA changes in heart failure patients treated with cardiac resynchronization therapy: responders vs. non-responders. Eur J Heart Fail. (2013) 15:1277–88. doi: 10.1093/eurjhf/hft088
259. Peng L, Chun-guang Q, Bei-fang L, Xue-zhi D, Zi-hao W, Yun-fu L, et al. Clinical impact of circulating miR-133, miR-1291 and miR-663b in plasma of patients with acute myocardial infarction. Diagn Pathol. (2014) 9:89. doi: 10.1186/1746-1596-9-89
260. de Couto G, Gallet R, Cambier L, Jaghatspanyan E, Makkar N, Dawkins JF, et al. Exosomal microRNA transfer into macrophages mediates cellular postconditioning. Circulation (2017) 136:200–14. doi: 10.1161/CIRCULATIONAHA.116.024590
261. Yang Y, Li Y, Chen X, Cheng X, Liao Y, Yu X. Exosomal transfer of miR-30a between cardiomyocytes regulates autophagy after hypoxia. J Mol Med (Berl.) (2016) 94:711–24. doi: 10.1007/s00109-016-1387-2
262. Kumarswamy R, Bauters C, Volkmann I, Maury F, Fetisch J, Holzmann A, et al. Circulating long noncoding RNA, LIPCAR, predicts survival in patients with heart failure. Circ Res. (2014) 114:1569–75. doi: 10.1161/CIRCRESAHA.114.303915
263. Xuan L, Sun L, Zhang Y, Huang Y, Hou Y, Li Q, et al. Circulating long non-coding RNAs NRON and MHRT as novel predictive biomarkers of heart failure. J Cell Mol Med. (2017) 21:1803–14. doi: 10.1111/jcmm.13101
264. Rajan KS, Velmurugan G, Gopal P, Ramprasath T, Babu DD, Krithika S, et al. Abundant and altered expression of PIWI-interacting RNAs during cardiac hypertrophy. Heart Lung Circ. (2016) 25:1013–20. doi: 10.1016/j.hlc.2016.02.015
265. Salgado-Somoza A, Zhang L, Vausort M, Devaux Y. The circular RNA MICRA for risk stratification after myocardial infarction. Int J Cardiol Heart Vasc. (2017) 17:33–6. doi: 10.1016/j.ijcha.2017.11.001
266. Wang K, Zhang S, Weber J, Baxter D, Galas DJ. Export of microRNAs and microRNA-protective protein by mammalian cells. Nucleic Acids Res. (2010) 38:7248–59. doi: 10.1093/nar/gkq601
267. Xu L, Yang BF, Ai J. MicroRNA transport: a new way in cell communication. J Cell Physiol. (2013) 228:1713–9. doi: 10.1002/jcp.24344
268. Hunter MP, Ismail N, Zhang X, Aguda BD, Lee EJ, Yu L, et al. Detection of microRNA expression in human peripheral blood microvesicles. PLoS ONE (2008) 3:e3694. doi: 10.1371/journal.pone.0003694
269. Gibbings DJ, Ciaudo C, Erhardt M, Voinnet O. Multivesicular bodies associate with components of miRNA effector complexes and modulate miRNA activity. Nat Cell Biol. (2009) 11:1143–9. doi: 10.1038/ncb1929
270. Lawrie CH, Gal S, Dunlop HM, Pushkaran B, Liggins AP, Pulford K, et al. Detection of elevated levels of tumour-associated microRNAs in serum of patients with diffuse large B-cell lymphoma. Br J Haematol. (2008) 141:672–5. doi: 10.1111/j.1365-2141.2008.07077.x
271. Mitchell PS, Parkin RK, Kroh EM, Fritz BR, Wyman SK, Pogosova-Agadjanyan EL, et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proc Natl Acad Sci USA. (2008) 105:10513–8. doi: 10.1073/pnas.0804549105
272. Willeit P, Skroblin P, Moschen AR, Yin X, Kaudewitz D, Zampetaki A, et al. Circulating microRNA-122 is associated with the risk of new-onset metabolic syndrome and type 2 diabetes. Diabetes (2017) 66:347–57. doi: 10.2337/db16-0731
273. Zhu H, Leung SW. Identification of microRNA biomarkers in type 2 diabetes: a meta-analysis of controlled profiling studies. Diabetologia (2015) 58:900–11. doi: 10.1007/s00125-015-3510-2
274. Jones Buie JN, Goodwin AJ, Cook JA, Halushka PV, Fan H. The role of miRNAs in cardiovascular disease risk factors. Atherosclerosis (2016) 254:271–81. doi: 10.1016/j.atherosclerosis.2016.09.067
275. Viereck J, Thum T. Circulating noncoding RNAs as biomarkers of cardiovascular disease and injury. Circ Res. (2017) 120:381–99. doi: 10.1161/CIRCRESAHA.116.308434
276. Ai J, Zhang R, Li Y, Pu J, Lu Y, Jiao J, et al. Circulating microRNA-1 as a potential novel biomarker for acute myocardial infarction. Biochem Biophys Res Commun. (2010) 391:73–7. doi: 10.1016/j.bbrc.2009.11.005
277. Hergenreider E, Heydt S, Treguer K, Boettger T, Horrevoets AJ, Zeiher AM, et al. Atheroprotective communication between endothelial cells and smooth muscle cells through miRNAs. Nat Cell Biol. (2012) 14:249–56. doi: 10.1038/ncb2441
278. Raposo G, Stoorvogel W. Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol. (2013) 200:373–83. doi: 10.1083/jcb.201211138
279. Thery C. Exosomes: secreted vesicles and intercellular communications. F1000 Biol Rep. (2011) 3:15. doi: 10.3410/B3-15
280. Yang M, Chen J, Su F, Yu B, Su F, Lin L, et al. Microvesicles secreted by macrophages shuttle invasion-potentiating microRNAs into breast cancer cells. Mol Cancer (2011) 10:117. doi: 10.1186/1476-4598-10-117
281. Yuan MJ, Maghsoudi T, Wang T. Exosomes mediate the intercellular communication after myocardial infarction. Int J Med Sci. (2016) 13:113–16. doi: 10.7150/ijms.14112
282. Li Y, Shen Z, Yu XY. Transport of microRNAs via exosomes. Nat Rev Cardiol. (2015) 12:198. doi: 10.1038/nrcardio.2014.207-c1
283. Ong SG, Lee WH, Huang M, Dey D, Kodo K, Sanchez-Freire V, et al. Cross talk of combined gene and cell therapy in ischemic heart disease: role of exosomal microRNA transfer. Circulation (2014) 130:S60–9. doi: 10.1161/CIRCULATIONAHA.113.007917
284. Ibrahim AG, Cheng K, Marban E. Exosomes as critical agents of cardiac regeneration triggered by cell therapy. Stem Cell Reports (2014) 2:606–19. doi: 10.1016/j.stemcr.2014.04.006
285. Chen J, Cui C, Yang X, Xu J, Venkat P, Zacharek A, et al. MiR-126 affects brain-heart interaction after cerebral ischemic stroke. Transl Stroke Res. (2017) 8:374–85. doi: 10.1007/s12975-017-0520-z
286. Xin H, Katakowski M, Wang F, Qian JY, Liu XS, Ali MM, et al. MicroRNA cluster miR-17-92 cluster in exosomes enhance neuroplasticity and functional recovery after stroke in rats. Stroke (2017) 48:747–53. doi: 10.1161/STROKEAHA.116.015204
287. Janas T, Janas T, Yarus M. Specific RNA binding to ordered phospholipid bilayers. Nucleic Acids Res. (2006) 34:2128–136. doi: 10.1093/nar/gkl220
288. Wagner J, Riwanto M, Besler C, Knau A, Fichtlscherer S, Roxe T, et al. Characterization of levels and cellular transfer of circulating lipoprotein-bound microRNAs. Arterioscler Thromb Vasc Biol. (2013) 33:1392–400. doi: 10.1161/ATVBAHA.112.300741
289. Vickers KC, Palmisano BT, Shoucri BM, Shamburek RD, Remaley AT. MicroRNAs are transported in plasma and delivered to recipient cells by high-density lipoproteins. Nat Cell Biol. (2011) 13:423–33. doi: 10.1038/ncb2210
290. Canfran-Duque A, Lin CS, Goedeke L, Suarez Y, Fernandez-Hernando C. Micro-RNAs and high-density lipoprotein metabolism. Arterioscler Thromb Vasc Biol. (2016) 36:1076–84. doi: 10.1161/ATVBAHA.116.307028
291. Niculescu LS, Simionescu N, Sanda GM, Carnuta MG, Stancu CS, Popescu AC, et al. MiR-486 and miR-92a identified in circulating HDL discriminate between stable and vulnerable coronary artery disease patients. PLoS ONE (2015) 10:e0140958. doi: 10.1371/journal.pone.0140958
292. Ram TP, Fomison-Nurse I, Gandhi S, Coffey S, Saxena P, Galvin I, et al. The diagnostic sensitivity of circulating cardio-enriched microRNAs is increased after normalization of high-density lipoprotein levels. Int J Cardiol. (2017) 236:498–500. doi: 10.1016/j.ijcard.2017.01.119
293. Di Gesualdo F, Capaccioli S, Lulli M. A pathophysiological view of the long non-coding RNA world. Oncotarget (2014) 5:10976–96. doi: 10.18632/oncotarget.2770
294. Orom UA, Derrien T, Beringer M, Gumireddy K, Gardini A, Bussotti G, et al. Long noncoding RNAs with enhancer-like function in human cells. Cell (2010) 143:46–58. doi: 10.1016/j.cell.2010.09.001
295. Kaur S, Mirza AH, Brorsson CA, Floyel T, Storling J, Mortensen HB, et al. The genetic and regulatory architecture of ERBB3-type 1 diabetes susceptibility locus. Mol Cell Endocrinol. (2016) 419:83–91. doi: 10.1016/j.mce.2015.10.002
296. Jin Y, Cui Z, Li X, Jin X, Peng J. Upregulation of long non-coding RNA PlncRNA-1 promotes proliferation and induces epithelial-mesenchymal transition in prostate cancer. Oncotarget (2017) 8:26090–9. doi: 10.18632/oncotarget.15318
297. He X, Zheng Y, Zhang Y, Gan Y, Zhou Y, Liang H, et al. Long non-coding RNA AK058003, as a precursor of MiR-15a, interacts with HuR to inhibit the expression of γ-synuclein in hepatocellular carcinoma cells. Oncotarget (2017) 8:9451–65. doi: 10.18632/oncotarget.14276
298. Matkovich SJ, Edwards JR, Grossenheider TC, de Guzman SC, Dorn GW. Epigenetic coordination of embryonic heart transcription by dynamically regulated long noncoding RNAs. Proc Natl Acad Sci USA. (2014) 111:12264–9. doi: 10.1073/pnas.1410622111
299. Ounzain S, Micheletti R, Beckmann T, Schroen B, Alexanian M, Pezzuto I, et al. Genome-wide profiling of the cardiac transcriptome after myocardial infarction identifies novel heart-specific long non-coding RNAs. Eur Heart J. (2015) 36:353–68a. doi: 10.1093/eurheartj/ehu180
300. Grote P, Wittler L, Hendrix D, Koch F, Wahrisch S, Beisaw A, et al. The tissue-specific lncRNA Fendrr is an essential regulator of heart and body wall development in the mouse. Dev Cell. (2013) 24:206–14. doi: 10.1016/j.devcel.2012.12.012
301. Lee JH, Gao C, Peng G, Greer C, Ren S, Wang Y, et al. Analysis of transcriptome complexity through RNA sequencing in normal and failing murine hearts. Circ Res. (2011) 109:1332–41. doi: 10.1161/CIRCRESAHA.111.249433
302. Wang Z, Zhang XJ, Ji YX, Zhang P, Deng KQ, Gong J, et al. The long noncoding RNA chaer defines an epigenetic checkpoint in cardiac hypertrophy. Nat Med. (2016) 22:1131–9. doi: 10.1038/nm.4179
303. Klattenhoff CA, Scheuermann JC, Surface LE, Bradley RK, Fields PA, Steinhauser ML, et al. Braveheart, a long noncoding RNA required for cardiovascular lineage commitment. Cell (2013) 152:570–83. doi: 10.1016/j.cell.2013.01.003
304. Han P, Li W, Lin CH, Yang J, Shang C, Nuernberg ST, et al. A long noncoding RNA protects the heart from pathological hypertrophy. Nature (2014) 514:102–6. doi: 10.1038/nature13596
305. Viereck J, Kumarswamy R, Foinquinos A, Xiao K, Avramopoulos P, Kunz M, et al. Long noncoding RNA chast promotes cardiac remodeling. Sci Transl Med. (2016) 8:326ra22. doi: 10.1126/scitranslmed.aaf1475
306. Wang K, Liu F, Zhou LY, Long B, Yuan SM, Wang Y, et al. The long noncoding RNA CHRF regulates cardiac hypertrophy by targeting miR-489. Circ Res. (2014) 114:1377–88. doi: 10.1161/CIRCRESAHA.114.302476
307. Jiang F, Zhou X, Huang J. Long non-coding RNA-ROR mediates the reprogramming in cardiac hypertrophy. PLoS ONE (2016) 11:e0152767. doi: 10.1371/journal.pone.0152767
308. Wang K, Liu F, Liu CY, An T, Zhang J, Zhou LY, et al. The long noncoding RNA NRF regulates programmed necrosis and myocardial injury during ischemia and reperfusion by targeting miR-873. Cell Death Differ. (2016) 23:1394–405. doi: 10.1038/cdd.2016.28
309. Wang K, Liu CY, Zhou LY, Wang JX, Wang M, Zhao B, et al. APF lncRNA regulates autophagy and myocardial infarction by targeting miR-188-3p. Nat Commun. (2015) 6:6779. doi: 10.1038/ncomms7779
310. Liu CY, Zhang YH, Li RB, Zhou LY, An T, Zhang RC, et al. LncRNA CAIF inhibits autophagy and attenuates myocardial infarction by blocking p53-mediated myocardin transcription. Nat Commun. (2018) 9:29. doi: 10.1038/s41467-017-02280-y
311. Huang J, Min LM, Cheng L, Yuan LJ, Zhu X, Stout AL, et al. Myocardin is required for cardiomyocyte survival and maintenance of heart function. Proc Natl Acad Sci USA. (2009) 106:18734–9. doi: 10.1073/pnas.0910749106
312. Liu L, An X, Li Z, Song Y, Li L, Zuo S, et al. The H19 long noncoding RNA is a novel negative regulator of cardiomyocyte hypertrophy. Cardiovasc Res. (2016) 111:56–65. doi: 10.1093/cvr/cvw078
313. Pan JX. LncRNA H19 promotes atherosclerosis by regulating MAPK and NF-KB signaling pathway. Eur Rev Med Pharmacol Sci. (2017) 21:322–8.
314. Ishii N, Ozaki K, Sato H, Mizuno H, Saito S, Takahashi A, et al. Identification of a novel non-coding RNA, MIAT, that confers risk of myocardial infarction. J Hum Genet. (2006) 51:1087–99. doi: 10.1007/s10038-006-0070-9
315. Zhu XH, Yuan YX, Rao SL, Wang P. LncRNA MIAT enhances cardiac hypertrophy partly through sponging MiR-150. Eur Rev Med Pharmacol Sci. (2016) 20:3653–60.
316. de Gonzalo-Calvo D, Kenneweg F, Bang C, Toro R, van der Meer RW, Rijzewijk LJ, et al. Circulating long-non coding RNAs as biomarkers of left ventricular diastolic function and remodelling in patients with well-controlled type 2 diabetes. Sci Rep. (2016) 6:37354. doi: 10.1038/srep37354
317. Michalik KM, You X, Manavski Y, Doddaballapur A, Zornig M, Braun T, et al. Long noncoding RNA MALAT1 regulates endothelial cell function and vessel growth. Circ Res. (2014) 114:1389–97. doi: 10.1161/CIRCRESAHA.114.303265
318. Nural-Guvener HF, Mutlu N, Gaballa MA. BACE1 levels are elevated in congestive heart failure. Neurosci Lett. (2013) 532:7–11. doi: 10.1016/j.neulet.2012.10.051
319. Kokjohn TA, Van Vickle GD, Maarouf CL, Kalback WM, Hunter JM, Daugs ID, et al. Chemical characterization of pro-inflammatory amyloid-β peptides in human atherosclerotic lesions and platelets. Biochim Biophys Acta (2011) 1812:1508–14. doi: 10.1016/j.bbadis.2011.07.004
320. Wang K, Long B, Zhou LY, Liu F, Zhou QY, Liu CY, et al. CARL lncRNA inhibits anoxia-induced mitochondrial fission and apoptosis in cardiomyocytes by impairing miR-539-dependent PHB2 downregulation. Nat Commun. (2014) 5:3596. doi: 10.1038/ncomms4596
321. Zhou Y, Zhang X, Klibanski A. MEG3 noncoding RNA: a tumor suppressor. J Mol Endocrinol. (2012) 48:R45–53. doi: 10.1530/JME-12-0008
322. Holdt LM, Beutner F, Scholz M, Gielen S, Gabel G, Bergert H, et al. ANRIL expression is associated with atherosclerosis risk at chromosome 9p21. Arterioscler Thromb Vasc Biol. (2010) 30:620–7. doi: 10.1161/ATVBAHA.109.196832
323. Jarinova O, Stewart AF, Roberts R, Wells G, Lau P, Naing T, et al. Functional analysis of the chromosome 9p21.3 coronary artery disease risk locus. Arterioscler Thromb Vasc Biol. (2009) 29:1671–7. doi: 10.1161/ATVBAHA.109.189522
324. Holdt LM, Teupser D. Recent studies of the human chromosome 9p21 locus, which is associated with atherosclerosis in human populations. Arterioscler Thromb Vasc Biol. (2012) 32:196–206. doi: 10.1161/ATVBAHA.111.232678
325. Rahimi E, Ahmadi A, Boroumand MA, Mohammad SB, Behmanesh M. Association of ANRIL expression with coronary artery disease in type 2 diabetic patients. Cell J. (2018) 20:41–5. doi: 10.22074/cellj.2018.4821
326. Ballantyne MD, Pinel K, Dakin R, Vesey AT, Diver L, Mackenzie R, et al. Smooth Muscle Enriched Long Noncoding RNA (SMILR) regulates cell proliferation. Circulation (2016) 133:2050–65. doi: 10.1161/CIRCULATIONAHA.115.021019
327. Gao L, Liu Y, Guo S, Yao R, Wu L, Xiao L, et al. Circulating long noncoding RNA HOTAIR is an essential mediator of acute myocardial infarction. Cell Physiol Biochem. (2017) 44:1497–508. doi: 10.1159/000485588
328. Lai Y, He S, Ma L, Lin H, Ren B, Ma J, et al. HOTAIR functions as a competing endogenous RNA to regulate PTEN expression by inhibiting miR-19 in cardiac hypertrophy. Mol Cell Biochem. (2017) 432:179–87. doi: 10.1007/s11010-017-3008-y
329. Reddy MA, Chen Z, Park JT, Wang M, Lanting L, Zhang Q, et al. Regulation of inflammatory phenotype in macrophages by a diabetes-induced long noncoding RNA. Diabetes (2014) 63:4249–61. doi: 10.2337/db14-0298
330. Wang L, Zhao Y, Bao X, Zhu X, Kwok YK, Sun K, et al. LncRNA Dum interacts with Dnmts to regulate Dppa2 expression during myogenic differentiation and muscle regeneration. Cell Res. (2015) 25:335–50. doi: 10.1038/cr.2015.21
331. Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science (2000) 290:1717–21. doi: 10.1126/science.290.5497.1717
332. Artal-Sanz M, Tavernarakis N. Prohibitin and mitochondrial biology. Trends Endocrinol Metab. (2009) 20:394–401. doi: 10.1016/j.tem.2009.04.004
333. Wang K, Liu CY, Zhang XJ, Feng C, Zhou LY, Zhao Y, et al. MiR-361-regulated prohibitin inhibits mitochondrial fission and apoptosis and protects heart from ischemia injury. Cell Death Differ. (2015) 22:1058–68. doi: 10.1038/cdd.2014.200
334. Piccoli MT, Gupta SK, Viereck J, Foinquinos A, Samolovac S, Kramer FL, et al. Inhibition of the cardiac fibroblast-enriched lncRNA Meg3 prevents cardiac fibrosis and diastolic dysfunction. Circ Res. (2017) 121:575–83. doi: 10.1161/CIRCRESAHA.117.310624
335. Riva P, Ratti A, Venturin M. The long non-coding RNAs in neurodegenerative diseases: novel mechanisms of pathogenesis. Curr Alzheimer Res. (2016) 13:1219–31. doi: 10.2174/1567205013666160622112234
336. Luo Q, Chen Y. Long noncoding RNAs and Alzheimer's disease. Clin Interv Aging (2016) 11:867–72. doi: 10.2147/CIA.S107037
337. Hessler S, Zheng F, Hartmann S, Rittger A, Lehnert S, Volkel M, et al. β-secretase BACE1 regulates hippocampal and reconstituted M-currents in a β-subunit-like fashion. J Neurosci. (2015) 35:3298–311. doi: 10.1523/JNEUROSCI.3127-14.2015
338. Greco S, Zaccagnini G, Fuschi P, Voellenkle C, Carrara M, Sadeghi I, et al. Increased BACE1-AS long noncoding RNA and β-amyloid levels in heart failure. Cardiovasc Res. (2017) 113:453–63. doi: 10.1093/cvr/cvx013
339. Kim JY, Lee HJ, Lee SJ, Jung YH, Yoo DY, Hwang IK, et al. Palmitic acid-BSA enhances amyloid-β production through GPR40-mediated dual pathways in neuronal cells: involvement of the Akt/mTOR/HIF-1α and Akt/NF-κB pathways. Sci Rep. (2017) 7:4335. doi: 10.1038/s41598-017-04175-w
340. Congrains A, Kamide K, Oguro R, Yasuda O, Miyata K, Yamamoto E, et al. Genetic variants at the 9p21 locus contribute to atherosclerosis through modulation of ANRIL and CDKN2A/B. Atherosclerosis (2012) 220:449–55. doi: 10.1016/j.atherosclerosis.2011.11.017
341. Bell RD, Long X, Lin M, Bergmann JH, Nanda V, Cowan SL, et al. Identification and Initial functional characterization of a human vascular cell-enriched long noncoding RNA. Arterioscler Thromb Vasc Biol. (2014) 34:1249–59. doi: 10.1161/ATVBAHA.114.303240
342. Ma JB, Yuan YR, Meister G, Pei Y, Tuschl T, Patel DJ. Structural basis for 5′-end-specific recognition of guide RNA by the A. fulgidus piwi protein. Nature (2005) 434:666–70. doi: 10.1038/nature03514
343. Sallam T, Jones M, Thomas BJ, Wu X, Gilliland T, Qian K, et al. Transcriptional regulation of macrophage cholesterol efflux and atherogenesis by a long noncoding RNA. Nat Med. (2018) 24:304–12. doi: 10.1038/nm.4479
344. Parker JS, Roe SM, Barford D. Structural insights into mRNA recognition from a PIWI domain-siRNA guide complex. Nature (2005) 434:663–6. doi: 10.1038/nature03462
345. Yin H, Lin H. An epigenetic activation role of Piwi and a Piwi-associated piRNA in Drosophila melanogaster. Nature (2007) 450:304–8. doi: 10.1038/nature06263
346. Teixeira FK, Okuniewska M, Malone CD, Coux RX, Rio DC, Lehmann R. piRNA-mediated regulation of transposon alternative splicing in the soma and germ line. Nature (2017) 552:268–72. doi: 10.1038/nature25018
347. Ernst C, Odom DT, Kutter C. The emergence of piRNAs against transposon invasion to preserve mammalian genome integrity. Nat Commun. (2017) 8:1411. doi: 10.1038/s41467-017-01049-7
348. Ferreira HJ, Heyn H, Garcia del Muro X, Vidal A, Larriba S, Munoz C, et al. Epigenetic loss of the PIWI/piRNA machinery in human testicular tumorigenesis. Epigenetics (2014) 9:113–18. doi: 10.4161/epi.27237
349. Siddiqi S, Matushansky I. Piwis and piwi-interacting RNAs in the epigenetics of cancer. J Cell Biochem. (2012) 113:373–80. doi: 10.1002/jcb.23363
350. Costa FF. Non-coding RNAs, epigenetics and complexity. Gene (2008) 410:9–17. doi: 10.1016/j.gene.2007.12.008
351. Klattenhoff C, Theurkauf W. Biogenesis and germline functions of piRNAs. Development (2008) 135:3–9. doi: 10.1242/dev.006486
352. Han YN, Li Y, Xia SQ, Zhang YY, Zheng JH, Li W. PIWI proteins and PIWI-interacting RNA: emerging roles in cancer. Cell Physiol Biochem. (2017) 44:1–20. doi: 10.1159/000484541
353. Pekarsky Y, Balatti V, Palamarchuk A, Rizzotto L, Veneziano D, Nigita G, et al. Dysregulation of a family of short noncoding RNAs, TsRNAs, in human cancer. Proc Natl Acad Sci USA. (2016) 113:5071–6. doi: 10.1073/pnas.1604266113
354. Fu A, Jacobs DI, Hoffman AE, Zheng T, Zhu Y. PIWI-interacting RNA 021285 is involved in breast tumorigenesis possibly by remodeling the cancer epigenome. Carcinogenesis (2015) 36:1094–102. doi: 10.1093/carcin/bgv105
355. Mei Y, Wang Y, Kumari P, Shetty AC, Clark D, Gable T, et al. A piRNA-like small RNA interacts with and modulates P-ERM proteins in human somatic cells. Nat Commun. (2015) 6:7316. doi: 10.1038/ncomms8316
356. Cheng J, Deng H, Xiao B, Zhou H, Zhou F, Shen Z, et al. PiR-823, a novel non-coding small RNA, demonstrates in vitro and in vivo tumor suppressive activity in human gastric cancer cells. Cancer Lett. (2012) 315:12–17. doi: 10.1016/j.canlet.2011.10.004
357. Duan L, Hu J, Xiong X, Liu Y, Wang J. The role of DNA methylation in coronary artery disease. Gene (2018) 646:91–7. doi: 10.1016/j.gene.2017.12.033
358. Lucchinetti E, Feng J, Silva R, Tolstonog GV, Schaub MC, Schumann GG, et al. Inhibition of LINE-1 expression in the heart decreases ischemic damage by activation of Akt/PKB signaling. Physiol Genomics (2006) 25:314–24. doi: 10.1152/physiolgenomics.00251.2005
359. Vella S, Gallo A, Lo NA, Galvagno D, Raffa GM, Pilato M, et al. PIWI-interacting RNA (piRNA) signatures in human cardiac progenitor cells. Int J Biochem Cell Biol. (2016) 76:1–11. doi: 10.1016/j.biocel.2016.04.012
360. Lee JH, Schutte D, Wulf G, Fuzesi L, Radzun HJ, Schweyer S, et al. Stem-cell protein Piwil2 is widely expressed in tumors and inhibits apoptosis through activation of Stat3/Bcl-XL pathway. Hum Mol Genet. (2006) 15:201–11. doi: 10.1093/hmg/ddi430
361. Wilusz JE, Sharp PA. Molecular biology. A circuitous route to noncoding RNA. Science (2013) 340:440–1. doi: 10.1126/science.1238522
362. Li Y, Zheng Q, Bao C, Li S, Guo W, Zhao J, et al. Circular rna is enriched and stable in exosomes: a promising biomarker for cancer diagnosis. Cell Res. (2015) 25:981–4. doi: 10.1038/cr.2015.82
363. Zhao Z, Li X, Gao C, Jian D, Hao P, Rao L, et al. Peripheral blood circular RNA Hsa_Circ_0124644 can be used as a diagnostic biomarker of coronary artery disease. Sci Rep. (2017) 7:39918. doi: 10.1038/srep39918
364. Hansen TB, Jensen TI, Clausen BH, Bramsen JB, Finsen B, Damgaard CK, et al. Natural RNA circles function as efficient microRNA sponges. Nature (2013) 495:384–8. doi: 10.1038/nature11993
365. Wang K, Long B, Liu F, Wang JX, Liu CY, Zhao B, et al. A circular RNA protects the heart from pathological hypertrophy and heart failure by targeting miR-223. Eur Heart J. (2016) 37:2602–11. doi: 10.1093/eurheartj/ehv713
366. Geng HH, Li R, Su YM, Xiao J, Pan M, Cai XX, et al. The circular RNA Cdr1as promotes myocardial infarction by mediating the regulation of miR-7a on its target genes expression. PLoS ONE (2016) 11:e0151753. doi: 10.1371/journal.pone.0151753
367. Boeckel JN, Jae N, Heumuller AW, Chen W, Boon RA, Stellos K, et al. Identification and characterization of hypoxia-regulated endothelial circular RNA. Circ Res. (2015) 117:884–90. doi: 10.1161/CIRCRESAHA.115.306319
368. Gupta SK, Garg A, Bar C, Chatterjee S, Foinquinos A, Milting H, et al. Quaking inhibits doxorubicin-mediated cardiotoxicity through regulation of cardiac circular RNA expression. Circ Res. (2018) 122:246–54. doi: 10.1161/CIRCRESAHA.117.311335
369. Ito Y, Inoue A, Seers T, Hato Y, Igarashi A, Toyama T, et al. Identification of targets of tumor suppressor microRNA-34a using a reporter library system. Proc Natl Acad Sci USA. (2017) 114:3927–32. doi: 10.1073/pnas.1620019114
370. Schultheis B, Strumberg D, Santel A, Vank C, Gebhardt F, Keil O, et al. First-in-human phase I study of the liposomal RNA interference therapeutic Atu027 in patients with advanced solid tumors. J Clin Oncol. (2014) 32:4141–8. doi: 10.1200/JCO.2013.55.0376
371. Davis ME, Zuckerman JE, Choi CH, Seligson D, Tolcher A, Alabi CA, et al. Evidence of RNAi in humans from systemically administered siRNA via targeted nanoparticles. Nature (2010) 464:1067–70. doi: 10.1038/nature08956
372. Tabernero J, Shapiro GI, LoRusso PM, Cervantes A, Schwartz GK, Weiss GJ, et al. First-in-humans trial of an RNA interference therapeutic targeting VEGF and KSP in Cancer patients with liver involvement. Cancer Discov. (2013) 3:406–17. doi: 10.1158/2159-8290.CD-12-0429
373. Gilam A, Conde J, Weissglas-Volkov D, Oliva N, Friedman E, Artzi N, et al. Local microRNA delivery targets palladin and prevents metastatic breast cancer. Nat Commun. (2016) 7:12868. doi: 10.1038/ncomms12868
374. Hong CA, Nam YS. Functional nanostructures for effective delivery of small interfering RNA therapeutics. Theranostics (2014) 4:1211–32. doi: 10.7150/thno.8491
375. Ohno S, Kuroda M. Exosome-mediated targeted delivery of miRNAs. Methods Mol Biol. (2016) 1448:261–70. doi: 10.1007/978-1-4939-3753-0_19
376. Zhang Y, Hu YW, Zheng L, Wang Q. Characteristics and roles of exosomes in cardiovascular disease. DNA Cell Biol. (2017) 36:202–11. doi: 10.1089/dna.2016.3496
377. Chen Y, Zhu X, Zhang X, Liu B, Huang L. Nanoparticles modified with tumor-targeting ScFv deliver siRNA and miRNA for cancer therapy. Mol Ther. (2010) 18:1650–6. doi: 10.1038/mt.2010.136
378. Hennessy EJ, Moore KJ. Using microRNA as an alternative treatment for hyperlipidemia and cardiovascular disease: cardio-miRs in the pipeline. J Cardiovasc Pharmacol. (2013) 62:247–54. doi: 10.1097/FJC.0b013e31829d48bf
379. Bumcrot D, Manoharan M, Koteliansky V, Sah DW. RNAi Therapeutics: a potential new class of pharmaceutical drugs. Nat Chem Biol. (2006) 2:711–9. doi: 10.1038/nchembio839
380. Lennox KA, Behlke MA. Chemical modification and design of anti-miRNA oligonucleotides. Gene Ther. (2011) 18:1111–20. doi: 10.1038/gt.2011.100
381. Krutzfeldt J, Rajewsky N, Braich R, Rajeev KG, Tuschl T, Manoharan M, et al. Silencing of micrornas in vivo with ‘antagomirs’. Nature (2005) 438:685–9. doi: 10.1038/nature04303
382. Kool ET. Preorganization of DNA: design principles for improving nucleic acid recognition by synthetic oligonucleotides. Chem Rev. (1997) 97:1473–88. doi: 10.1021/cr9603791
383. Owczarzy R, You Y, Groth CL, Tataurov AV. Stability and mismatch discrimination of locked nucleic acid-DNA duplexes. Biochemistry (2011) 50:9352–67. doi: 10.1021/bi200904e
384. Geary RS. Antisense oligonucleotide pharmacokinetics and metabolism. Expert Opin Drug Metab Toxicol. (2009) 5:381–91. doi: 10.1517/17425250902877680
385. Jensen TB, Langkjaer N, Wengel J. Unlocked Nucleic Acid (UNA) and UNA derivatives: thermal denaturation studies. Nucleic Acids Symp Ser (Oxf.) (2008) 2008:133–4. doi: 10.1093/nass/nrn068
386. Lennox KA, Behlke MA. A direct comparison of anti-microRNA oligonucleotide potency. Pharm Res. (2010) 27:1788–99. doi: 10.1007/s11095-010-0156-0
387. Manoharan M. 2′-Carbohydrate modifications in antisense oligonucleotide therapy: importance of conformation, configuration and conjugation. Biochim Biophys Acta (1999) 1489:117–30. doi: 10.1016/S0167-4781(99)00138-4
388. Mangos MM, Damha MJ. Flexible and frozen sugar-modified nucleic acids–modulation of biological activity through furanose ring dynamics in the antisense strand. Curr Top Med Chem. (2002) 2:1147–71. doi: 10.2174/1568026023393110
389. Egli M, Minasov G, Tereshko V, Pallan PS, Teplova M, Inamati GB, et al. Probing the Influence of stereoelectronic effects on the biophysical properties of oligonucleotides: comprehensive analysis of the RNA affinity, nuclease resistance, and crystal structure of ten 2′-O-ribonucleic acid modifications. Biochemistry (2005) 44:9045–57. doi: 10.1021/bi050574m
390. Crooke ST, Baker BF, Witztum JL, Kwoh TJ, Pham NC, Salgado N, et al. The effects of 2′-O-methoxyethyl containing antisense oligonucleotides on platelets in human clinical trials. Nucleic Acid Ther. (2017) 27:121–9. doi: 10.1089/nat.2016.0650
391. Crooke ST, Baker BF, Kwoh TJ, Cheng W, Schulz DJ, Xia S, et al. Integrated safety assessment of 2′-O-methoxyethyl chimeric antisense oligonucleotides in nonhuman primates and healthy human volunteers. Mol Ther. (2016) 24:1771–82. doi: 10.1038/mt.2016.136
392. Chorn G, Klein-McDowell M, Zhao L, Saunders MA, Flanagan WM, Willingham AT, et al. Single-stranded microRNA mimics. RNA (2012) 18:1796–804. doi: 10.1261/rna.031278.111
393. Matsui M, Prakash TP, Corey DR. Argonaute 2-dependent regulation of gene expression by single-stranded miRNA mimics. Mol Ther. (2016) 24:946–55. doi: 10.1038/mt.2016.39
394. Petersen M, Bondensgaard K, Wengel J, Jacobsen JP. Locked Nucleic Acid (LNA) recognition of RNA: NMR solution structures of LNA:RNA hybrids. J Am Chem Soc. (2002) 124:5974–82. doi: 10.1021/ja012288d
395. McTigue PM, Peterson RJ, Kahn JD. Sequence-dependent thermodynamic parameters for Locked Nucleic Acid (LNA)-DNA duplex formation. Biochemistry (2004) 43:5388–405. doi: 10.1021/bi035976d
396. Frieden M, Hansen HF, Koch T. Nuclease stability of LNA oligonucleotides and LNA-DNA chimeras. Nucleosides Nucleotides Nucleic Acids (2003) 22:1041–3. doi: 10.1081/NCN-120022731
397. Touznik A, Maruyama R, Hosoki K, Echigoya Y, Yokota T. LNA/DNA mixmer-based antisense oligonucleotides correct alternative splicing of the SMN2 gene and restore SMN protein expression in type 1 SMA fibroblasts. Sci Rep. (2017) 7:3672. doi: 10.1038/s41598-017-03850-2
398. Gebert LF, Rebhan MA, Crivelli SE, Denzler R, Stoffel M, Hall J. Miravirsen (SPC3649) Can inhibit the biogenesis of miR-122. Nucleic Acids Res. (2014) 42:609–21. doi: 10.1093/nar/gkt852
399. Janssen HL, Reesink HW, Lawitz EJ, Zeuzem S, Rodriguez-Torres M, Patel K, et al. Treatment of HCV infection by targeting microRNA. N Engl J Med. (2013) 368:1685–94. doi: 10.1056/NEJMoa1209026
400. Keefe AD, Pai S, Ellington A. Aptamers as therapeutics. Nat Rev Drug Discov. (2010) 9:537–50. doi: 10.1038/nrd3141
401. Watts JK. Locked nucleic acid: tighter is different. Chem Commun (Camb.) (2013) 49:5618–20. doi: 10.1039/c3cc40340h
402. Straarup EM, Fisker N, Hedtjarn M, Lindholm MW, Rosenbohm C, Aarup V, et al. Short locked nucleic acid antisense oligonucleotides potently reduce apolipoprotein B mRNA and serum cholesterol in mice and non-human primates. Nucleic Acids Res. (2010) 38:7100–11. doi: 10.1093/nar/gkq457
403. Chang H, Yi B, Ma R, Zhang X, Zhao H, Xi Y. CRISPR/Cas9, a novel genomic tool to knock down microRNA in vitro and in vivo. Sci Rep. (2016) 6:22312. doi: 10.1038/srep22312
404. Patrick DM, Montgomery RL, Qi X, Obad S, Kauppinen S, Hill JA, et al. Stress-dependent cardiac remodeling occurs in the absence of microRNA-21 in mice. J Clin Invest. (2010) 120:3912–6. doi: 10.1172/JCI43604
405. Cordes KR, Sheehy NT, White MP, Berry EC, Morton SU, Muth AN, et al. MiR-145 and miR-143 regulate smooth muscle cell fate and plasticity. Nature (2009) 460:705–10. doi: 10.1038/nature08195
406. Boettger T, Beetz N, Kostin S, Schneider J, Kruger M, Hein L, et al. Acquisition of the contractile phenotype by murine arterial smooth muscle cells depends on the mir143/145 gene cluster. J Clin Invest. (2009) 119:2634–47. doi: 10.1172/JCI38864
Keywords: long non-coding RNA, microRNA, myocardial infarction, hypertrophy, atherosclerosis, diabetic cardiomyopathy
Citation: Das A, Samidurai A and Salloum FN (2018) Deciphering Non-coding RNAs in Cardiovascular Health and Disease. Front. Cardiovasc. Med. 5:73. doi: 10.3389/fcvm.2018.00073
Received: 19 February 2018; Accepted: 29 May 2018;
Published: 02 July 2018.
Edited by:
Georges Nemer, American University of Beirut, LebanonReviewed by:
Noemi Rotllan Vila, Yale University, United StatesHector A. Cabrera-Fuentes, Justus Liebig Universität Gießen, Germany
Copyright © 2018 Das, Samidurai and Salloum. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fadi N. Salloum, ZmFkaS5zYWxsb3VtQHZjdWhlYWx0aC5vcmc=