 Anja Karlstaedt
Anja Karlstaedt Walter Schiffer
Walter Schiffer Heinrich Taegtmeyer
Heinrich Taegtmeyer- Division of Cardiology, Department of Internal Medicine, McGovern Medical School, University of Texas Health Science Center at Houston, Houston, TX, United States
Recent advances in cancer cell metabolism provide unprecedented opportunities for a new understanding of heart metabolism and may offer new approaches for the treatment of heart failure. Key questions driving the cancer field to understand how tumor cells reprogram metabolism and to benefit tumorigenesis are also applicable to the heart. Recent experimental and conceptual advances in cancer cell metabolism provide the cardiovascular field with the unique opportunity to target metabolism. This review compares cancer cell metabolism and cardiac metabolism with an emphasis on strategies of cellular adaptation, and how to exploit metabolic changes for therapeutic benefit.
Introduction
The intermediary metabolism of substrates defines every living cell, including heart and cancer cells. Metabolism in mammalian cells has four specific functions: (a) to provide chemical energy from nutrients, (b) to convert exogenous nutrients into macromolecules or building blocks of cell components, (c) to assemble building blocks into proteins, nucleic acid, lipids, and other cell components, and (d) to synthesize or degrade biomolecules required in specialized functions of cells (1). Although intermediary metabolism involves a complex network of pathways, the function of metabolism is remarkably similar in living cells. Recent advances in mass-spectrometry-based proteomics, metabolomics and flux analysis facilitate a more precise dissection of the pathways involved in myocardial dysfunction on a molecular level (2–6). Eventually these tools may lead to actionable and individualized therapies for heart failure patients.
Cardiac metabolism is a dynamic process that adapts to stress by altering its activity to maintain cardiac contraction, thereby ensuring cell survival in the near term. Some metabolic responses shift cardiac metabolism toward an energetically unfavorable state, and turn an initial adaptation into maladaptation, which leads to further disease progression (7–9). Precisely how metabolism affects structural remodeling in the heart, how metabolic activities regulate this transformation, and how metabolic changes can be targeted for therapeutic strategies are key questions under investigation. This review compares cancer cell metabolism and cardiac metabolism with an emphasis on strategies of cellular adaptation, and how to exploit metabolic changes for therapeutic benefit. Here, we have only provided a brief overview on common concepts in the metabolism of heart disease and cancer due to the breadth of literature available in both fields. In cancer cells, the principle applies that alteration in metabolic activities supports the acquisition of biosynthetic material and maintenance of cell proliferation. In other words: reprogrammed metabolism is a hallmark of cancer (10–12). Similarly, alterations in cardiac metabolism contribute to disease progression and severity during cardiac hypertrophy, atrophy, and heart failure (8).
Metabolic adaptation in the heart supports contractile function by maintaining ATP provision. While cancer cells optimize metabolic flux to maximize cell proliferation and growth at pathological levels, the heart tries to optimize the use of energy-providing substrates to ensure cardiac contraction and cell survival. Both cancer cells and the heart show a high metabolic turnover to rapidly adapt themselves to changes in the chemical composition of their exogenous nutrients. The question arises: how do cancer cells successfully improve their cellular fitness by coopting metabolic machinery, while the heart fails to do so under long-term stress? Is it time to seriously rethink cardiac metabolism? To address this question, we will discuss modulation of energy substrate metabolism, biosynthesis of macromolecules, and redox balance in both cancer and the heart below. Regardless of whether specific metabolic activities provide benefits or liabilities to cancer cells or the heart, the rationale is that these activities may be exploited as therapeutic targets. For example, pharmacologic inhibition of fatty acid oxidation by agonists of the peroxisome proliferator-activated receptor (PPAR) ligand-activated nuclear hormone receptors decreases myocardial fatty acid oxidation; which, in turn, promotes increased glucose uptake and oxidation, and improves contractile function (13, 14). There is a strong precedent for using pharmacologic modification of metabolic pathways to improve our understanding and treatment heart diseases and cancer. We postulate that the analysis of metabolic patterns common to human cancers and the failing heart may also provide important insights into the relationships between energy substrates, and lead to metabolic targets in the heart.
Comparing Metabolic Pathways in Cancer and the Heart
We begin our comparison by considering how cancer cells and cardiomyocytes employ pathways that catalyze the degradation of nutrients and the recovery of part of their chemical energy as ATP. The metabolic control of enzyme-catalyzed reactions is tightly regulated in eukaryotic cells through spatial localization, cooperativity, allosteric interactions, substrate availability, expression, and post-translational modification of enzymes. In certain tumors, impairment of mitochondrial function by somatic mutations of Krebs cycle enzymes (e.g., succinate dehydrogenase and fumarate hydratase) leads to activation of glycolysis even in the presence of oxygen (15). Most of these tumors maintain their ability to provide ATP and thrive on glycolysis and glucose oxidation. This phenomenon has been first described by Otto Warburg, who discovered that ascites cancer cells from mice obtain approximately the same amount of ATP from fermentation as from respiration. In fact, limiting the complete oxidation of glucose by inhibiting pyruvate decarboxylation through modulation of pyruvate dehydrogenase activity in tumors fails to prevent tumorigenesis (16). Recent studies in cancer cell lines showed that a switch toward glycolysis is caused by impaired mitochondrial function and regulated by reductive carboxylation of glutamine (17–20). Upregulation of glycolysis allows cancer cells to satisfy their increased demand for biosynthetic intermediates that can be derived from glucose; hence increased glycolysis enables cell proliferation and growth. In the heart, plasma substrate composition and workload dictate nutrient utilization. Under normal physiologic conditions, the heart predominately oxidizes fatty acids (21, 22). However, this substrate preference can quickly shift toward carbohydrates or ketone bodies based on the availability of substrates, the workload, physical activity or periods of starvation. In fact, experimental studies of acutely stressed hearts (21, 23), ischemia and hypertrophy models of transverse aortic constriction (22, 24–26) reveal that glycolysis and glucose oxidation are preferred over fatty acid oxidation. However, this does not mean that the heart is not utilizing carbohydrates under normal physiologic conditions. Both ex vivo and in silico studies (21, 23, 27) showed that simultaneous oxidation of long-chain fatty acids and glucose allow most efficient ATP provision in the heart during physiologic workload. Degradation of glucose through glycolysis does not only ensure ATP provision, but also provides intermediates for other important pathways, in particular the pentose phosphate pathway and serine synthesis. In tumors and the heart, the glycolytic intermediate glyceraldehyde 3-phosphate is required for the generation of NADPH in the pentose phosphate pathway via glyceraldehyde 3-phosphate dehydrogenase (GAPDH), and anabolic precursors for pentoses and nucleotide synthesis. Flux through the pentose phosphate pathway is regulated by GAPDH, and thus changes in GAPDH activity may impact redox regulation and synthesis of nucleic acids, as well as aromatic amino acids. Recent studies show that tumors exhibiting the Warburg effect are also characterized by increased GAPDH activity (e.g., non-small lung cancers, colorectal cancers) (28, 29, 30, 31). Similarly, during myocardial infarction GAPDH activity increases in the heart and later decreases again during disease progression (32). This change in activity is potentially caused by post-ischemic myocardial reperfusion and may be linked to the production of reactive oxygen species, which have been shown to reduce GAPDH activity in the heart (28, 33). The central role of GAPDH in nucleotide synthesis and generation of reducing equivalents make it critical for survival of cells during stress, and therefore make it a potential target for pharmacologic strategies.
Fatty Acid Metabolism
Fatty acids are important metabolic building blocks for membranes to generate acetyl-CoA for post-translational protein modifications [e.g., histone acetylations; (34)], to provide reducing equivalents in the form of NADH and FADH2, and to provide ATP through β-oxidation. De novo fatty acid synthesis includes several key regulatory enzymes: ATP citrate lyase (ACL) which generates acetyl-CoA from citrate; acetyl-CoA carboxylase which catalyzes the irreversible carboxylation of acetyl-CoA from malonyl-CoA, and fatty acid synthase (FASN) which catalyzes the sequential addition of carbon-units to assemble long-chain fatty acids. In most tissues, including the heart, FASN expression and de novo fatty acid synthesis is relatively low, indicating that most cells preferentially take up exogenous or dietary lipids from the blood to provide energy and macromolecule biosynthesis. However, proliferating cells, like several human cancers, have been shown to up-regulate FASN expression (35) and take up free fatty acids to generate phospholipids (36–40). For example, KRAS-driven tumors (e.g., NSCLCs and ovarian cancer) increase fatty acid uptake and oxidation, thereby decreasing the need for de novo synthesis. Increased fatty acid oxidation may be driven by increased activation of AMP-activated protein kinase (AMPK) by reduced [ATP]:[AMP] ratio in RAS-mutant cells. Several studies in cancer cell lines and other mammalian tissues showed that ATP and AMP availability regulate the activation of AMPK and the mechanistic target of rapamycin (mTOR), which, in turn, regulate fatty acid metabolism on the molecular level [reviewed by Laplante et al. (41)]. These multiple levels of regulation enable tumors to optimize nutrient utilization and biomass synthesis.
Fatty acid oxidation is a major ATP source for the heart, and depends on cardiac energy demand, oxygen supply and free fatty acid supply from the blood. One of the hallmarks of metabolic perturbations during the development of cardiac hypertrophy and heart failure is decreased use of fatty acids. This metabolic pattern has been observed in both animal and human studies and has been compared to the metabolism in fetal hearts (42–45). Both the fetal and failing heart are characterized by a repression of various genes encoding rate-limiting enzymes of the fatty acid oxidation pathway [e.g., carnithine palmitoyl transferase 1 (CPT1), medium chain acyl-CoA dehydrogenase, and acetyl-CoA carboxylase] (43, 44) and their upstream regulators, including PPARα (46, 47). Downregulation of these genes is not fully understood. However, recent experimental evidence supports the hypothesis that fatty acid oxidation is less efficient (in terms of ATP per oxygen molecule consumed) during mitochondrial dysfunction and limited oxygen availability during ischemic heart disease (48). Therefore, in the short-term this metabolic reprogramming ensures energy provision and cardiac contractile function. In the long-term, reduction of fatty acid oxidation may cause an imbalance between the increased energy demand and simultaneously increased fatty acid availability during heart failure. Several studies have argued that a mismatch between fatty acid uptake and oxidation leads to an accumulation of acetyl-derivatives of CoA and acetyl-CoA as well as carnitine (49), which contributes to cell death and cardiac remodeling (50). In this way, lipotoxicity may contribute to cardiac dysfunction.
Ketone Bodies
In contrast to fatty acid and glucose metabolism, ketone body metabolism has been less investigated in both tumor metabolism and heart failure. Ketone bodies (e.g., acetate, acetoacetate, and beta-hydroxybutyrate) are released by the liver during a wide range of physiologic states; including fasting, starvation, low carbohydrate diets, the neonatal period, post-exercise, pregnancy and diabetes. In extrahepatic tissue (e.g., the heart, brain and skeletal muscle) ketone bodies play an important role for energy provision, post-translational modifications, as signaling mediators, and as modulators of inflammation and oxidative stress. Depending on the tumor type, ketone bodies can either support or diminish cancer cell progression. On the one hand, recent studies showed that ketone bodies support tumor progression and growth in breast cancer and glioblastoma by providing acetyl-CoA for de novo lipid synthesis, which is associated with shorter patient survival and increased metastasis of the tumor (51–53). On the other hand, ketogenic diets have been used in animal models and human studies, with potential benefits depending on the tumor location, type and time of diet initiation (54, 55). For example, ketone bodies inhibited growth, proliferation and glycolysis in pancreatic cancer and metastatic glioblastoma cell models and reduced in vivo tumor size and attenuated tumor-associated muscle loss (56). Similarly, recent studies indicate that altered cardiac ketone body metabolism contributes to the progression of heart failure (57, 58). These studies provide evidence that ketone body oxidation is increased in the failing heart. However, several questions remain unanswered, including which mechanisms are involved in upregulating ketone body utilization, whether increased use of ketone bodies is adaptive or maladaptive; whether normalization of ketone body oxidation is beneficial or detrimental for the non-failing and failing heart; and whether increased NAD+ levels during ketogenic diets may improve cardiovascular function. Additional work is needed to answer these questions, which will help understanding the role of ketone body metabolism in the pathogenesis of heart failure and evaluating potential risks during cancer treatments.
Amino Acid Metabolism
Compared to fatty acids and glucose, amino acids are not predominately used as substrates for energy provision. In general, amino acids are mostly used to provide substrates for protein synthesis or anaplerosis, and function as signaling molecules. For example, aspartate and leucine levels are sensed by mTOR complex 1 at the lysosomal membrane and promote activation of mTOR signaling (59–61). In cancer cells, the contribution of glutamine metabolism to energy provision and tumor progression has been widely studied (17, 20). Reductive carboxylation of glutamine is a common metabolic strategy, which enables cancer cells with somatic mutations of mitochondrial enzymes to maintain growth (e.g., de novo lipid synthesis). Furthermore, reductive glutamine carboxylation allows cancer cells to regenerate NADH and NADPH via malate dehydrogenase and/or isocitrate dehydrogenase. In the heart, amino acids predominately serve as fuels for protein synthesis and contribute only in a limited way to ATP provision. Under normal physiologic conditions, amidation of glutamate to glutamine occurs in the heart (62), but glutamine has only a marginal anaplerotic potential and may play a larger role in posttranslational modifications of proteins (e.g., β-linked N-acetylglucosamine) (63). During myocardial anoxia and ischemia, amino acids are used as anaplerotic substrates in the Krebs cycle (64, 65). The question remains whether glutamine plays a similar role in redox regulation in the heart as has been shown in cancer. Metabolomic analysis of failing mouse hearts and human plasma from heart failure patients showed that amino acid levels were increased (66–68). These changes suggest an association between amino acid levels and the progression of heart failure and have been attributed to increased protein breakdown in skeletal muscle. During heart failure, skeletal muscle serves as an additional amino acid source for the heart (69–71). Moreover, several studies suggest that amino acid supplementation helps to increase cardiac function in heart failure. For example, branched chain alpha-keto acids are elevated in hearts of heart failure patients, indicating that breakdown of branched chain amino acids (BCAA) is impaired. However, pharmacologic activation of BCAA catabolism and BCAA supplementation increased BCAA oxidation and improved cardiac contractility in heart failure patients and animal models (67, 72, 73). Cachexia is an independent risk factor for mortality during heart failure (74). With this in mind, improving cardiac amino acid oxidation and protein synthesis in skeletal muscle may protect the heart.
Biosynthesis and Turnover of Macromolecules
The biosynthesis of macromolecules is an essential aspect of metabolism in all living cells, as it ensures cellular homeostasis and is essential for cell proliferation and growth. Macromolecules are large molecules, commonly created by polymerization of a smaller subunit. In cells, these smaller subunits are nutrients (e.g., glucose, amino acids, fatty acids), which are converted into biosynthetic precursors through key pathways of intermediary metabolism, including glycolysis, the Krebs cycle, phospholipid pathways, and amino acid synthesis. These biosynthetic precursors then form the three most important biopolymers in the cell: proteins, lipids, and nucleic acids.
Protein biosynthesis is highly regulated in every living cell and requires sufficient supply of essential and non-essential amino acids. A complex network of growth factors, transporters, metabolic intermediates, and cofactors regulate activity of the mTOR signaling system, which is central for the activation of protein synthesis. Somatic mutations of either TSC1 or TSC2 genes causes the formation of hamartomas - a discovery that provided the first molecular link between mTOR and tumorigenesis (75). Phosphorylation and inhibition of TSC2 by AKT promotes activation of mTORC1, which is a common feature of oncogenic deregulation in cancer and may result from PTEN deletion, PIK3CA activating mutations or BCR-ABL translocation (76). Proliferating cancer cells further optimize uptake of amino acids and synthesis of non-essential amino acids through transamination of glutamate. For example, excess glutamine can be exported in exchange for leucine or other essential amino acids, which, in turn, ensures mTORC1 activation (77). At the same time, glutamate uptake and glutaminase activity are stimulated by mTORC1. This bidirectional regulation of mTORC1 activity and the glutamine pool further facilitates protein synthesis in cancer cells.
Protein synthesis and degradation in the heart are highly dynamic processes which are regulated by amino acid availability (78–81), regulation of specific mRNAs, oxygen supply and energy demand. Morgan et al. showed in perfused working hearts that the rate of protein synthesis could be increased by 40% when amino acids levels were increased by five-fold from normal plasma levels (80, 81). These early studies also showed that leucine primarily stimulates protein synthesis. However, overall, the net amount of protein synthesis in the adult heart is low compared to proliferating cells like tumors even when considering substantial increases in protein synthesis during physiologic as well as pathologic hypertrophy of the heart. Cardiac hypertrophy is mediated by protein phosphatases and kinases, such as MAPKs, Janus kinases (JAKs), and the PI3K/PDK/Akt pathway (82, 83). mTOR can be activated by the PI3K/PDK/Akt pathway. Chronic upregulation of mTORC1 is associated with increased cardiac hypertrophy (both physiologic and pathologic) (84, 85). Data from human subjects as well as animal studies show that decreased oxygen in the heart increases glutamine uptake and alanine release into the blood (86, 87). These effects are not seen until oxygen supply is decreased to less than 5% of normal oxygen concentrations, indicating that amino acids are necessary to provide protein precursors during stress (87). Cardiac remodeling is associated with increased glutamine deamination (glutaminolysis) (88). Thus, cardiac glutamine metabolism may enable mTORC1-mediated activation of protein synthesis in a way that mirrors cancer cells.
When nutrients are scarce, two main degradative pathways, autophagy and the ubiquitin proteasome system, enable cells to degrade macromolecules and replenish metabolic intermediates. Autophagy allows cells to maintain homeostasis by delivering protein aggregates and damaged organelles to the lysosome for degradation (89, 90). The formation of autophagosomes is controlled by specific yeast Atg-related proteins, which are tightly regulated by intracellular and extracellular nutrient availability and energy homeostasis of the cell (91–96). Autophagy functions as a cellular stress response that can increase the supply of amino acids by scavenging proteins. However, this contribution is unable to change net protein synthesis or increase nitrogen balance. Therefore, upregulation of glutamine uptake increases the cellular glutamate pool which is required for the synthesis of non-essential amino acids, and thus supports protein synthesis. Tumor cells use extracellular proteins as additional nitrogen source through micropinocytosis. Glutamine metabolism in cancer cells is highly dependent on the tumor type, oncogenic drivers, and the tumor microenvironment (97). Hypoxic tumor regions most distant from nutrient supply upregulate autophagy and sustain mitochondrial glutamine metabolism. Several studies have shown that tumor cells rely on extracellular amino acid supply to sustain cell growth (98). Therefore tumors may employ autophagy, or increase extracellular substrate uptake to buffet growth, while non-cancerous cells rely on autophagy alone during times of stress.
In the heart, autophagy recycles organelles and maintains supply of energy providing substrates during periods of reduced extracellular supply (e.g., starvation), oxygen deprivation (e.g., ischemia), or hemodynamic stress (e.g., valvular heart disease or systemic hypertension) (99–107). Tissue-specific deletion of Atg5 in heart causes cardiac hypertrophy and contractile dysfunction, indicating that autophagy activation under physiologic conditions is required to maintain cardiomyocytes size and cardiac structure and function (108). Upregulation of autophagy in failing hearts is currently considered an adaptive response to protect cells from stress. However, the role of autophagy in regulating amino acid metabolism in the heart remains unknown, and it remains unclear whether prolonged upregulation of autophagy is beneficial or detrimental in cardiovascular diseases.
De novo fatty acid synthesis is required in mammalian cells for membrane biosynthesis, lipidation reactions, signaling pathways and the formation of lipid rafts. Fatty acid synthesis depends on cytosolic acetyl-CoA levels and reducing equivalents in the form of cytosolic NADPH, which is provided through glycolysis via the pentose phosphate pathway. This link between carbohydrate, redox and fatty acid metabolism has been widely studied. Tumor cells in culture use glucose as a source for acetyl-CoA and fatty acid synthesis (19, 109). Most tumor cells also use glutamine, acetate or leucine degradation to enable lipogenesis when glucose availability is reduced, during hypoxia and when mitochondrial function is impaired (17, 34, 110–112). In contrast, the contribution of de novo fatty acid synthesis to the lipid pool in the heart is thought to be minimal under physiologic conditions and mostly limited to the synthesis of acetyl-CoA (113–115). The heart is capable taking up complex lipids through lipoprotein particles delivered by the liver (116, 117). Furthermore, glucose is used for glycerol synthesis and storage of fatty acids in the form of triacylglycerides. On the molecular level, Li et al. (118) showed that mTOR complex 1 directly affects de novo lipid synthesis through insulin-dependent activation and phosphorylation of S6 kinase (S6K), which then upregulates the sterol regulatory element binding protein 1 (SREBP1) [reviewed by Laplante et al. (41)]. Several questions remain unanswered regarding the metabolic regulation of (i) phospholipid synthesis/turnover, (ii) storage lipids accumulation/utilization, and (iii) cholesterol homeostasis. Recent advances in lipidomics and nutrient flux analysis will help further our understanding of these interconnected processes.
In addition to proteins and lipids, all living cells also rely on the synthesis of nucleic acids (e.g., RNA and DNA) from purines and pyrimidines. Therefore, it is not surprising that nucleotide analogs and antifolates targeting nucleotide biosynthesis have formed an integral part of cancer chemotherapeutic regimens. De novo synthesis of purines and pyrimidines requires non-essential amino acids and methyl groups donated from the one-carbon/folate pool. Aspartate and glutamine are required to synthesize the pyrimidine ring. Precursors for nucleotide synthesis are provided by central metabolic pathways including glycolysis, PPP, the serine-glycine pathways, the Krebs cycle and glutamine amidotransferase reactions. The metabolic energy required to enable nucleotide synthesis is substantial and proliferating cells have developed strategies to optimize flux into pathways providing precursors for nucleotides [reviewed by Lane et al. (119)]. The non-essential amino acid, aspartate, is a critical precursor, and most cells generate aspartate through deamination from glutamine rather than through uptake from the blood (120, 121). For many tumors, the rates of aspartate and folate synthesis limit proliferation and growth (122–124). In the heart, aspartate and other amino acids are preferentially used as anaplerotic substrates to provide ATP during ischemia reperfusion injury and cardiac hypertrophy (86, 87, 125). The purine nucleotide cycle in the heart provides fumarate from aspartate to replenish Krebs cycle intermediates (126). Recent reports indicate that purine and pyrimidine metabolism is potentially regulated by signaling pathways, including mTORC1 pathway (127). Depletion of purines, but not pyrimidines, is associated with mTORC1 inhibition, suggesting that in addition to leucine and aspartate, purines may play a role in regulating mTORC1 activity. Additional work is needed to determine how aspartate metabolism, the purine nucleotide cycle and other aspects of de novo nucleotide synthesis are regulated in the diseased heart to support protein synthesis as well as energy provision. In all, nucleic acid synthesis represents a rate-limiting step for the growth of tumors, which divert metabolic substrates to maintain cellular proliferation. Conversely, nucleotide metabolism in the heart helps to fuel the Krebs cycle, as there is currently no experimental evidence that its growth depends on nucleotide availability. However, more research may uncover a connection between growth and nucleotide synthesis in the heart.
Targeting Metabolism in the Failing Heart
Despite striking similarities in the metabolism of the failing heart and cancer cells, there are fundamental differences that need to be considered for the development of pharmacologic treatments. Cancer treatments targeting metabolism have the goal to limit or prevent tumor growth and induce cell death. Interventions targeting cardiac metabolism during heart failure aim to reverse structural remodeling and improve cardiac function. Are there therapies that may target metabolism to reverse heart failure? Are there strategies that both protect the heart and target the cancer? Common pharmacologic strategies for both heart failure and cancer are summarized in Table 1.
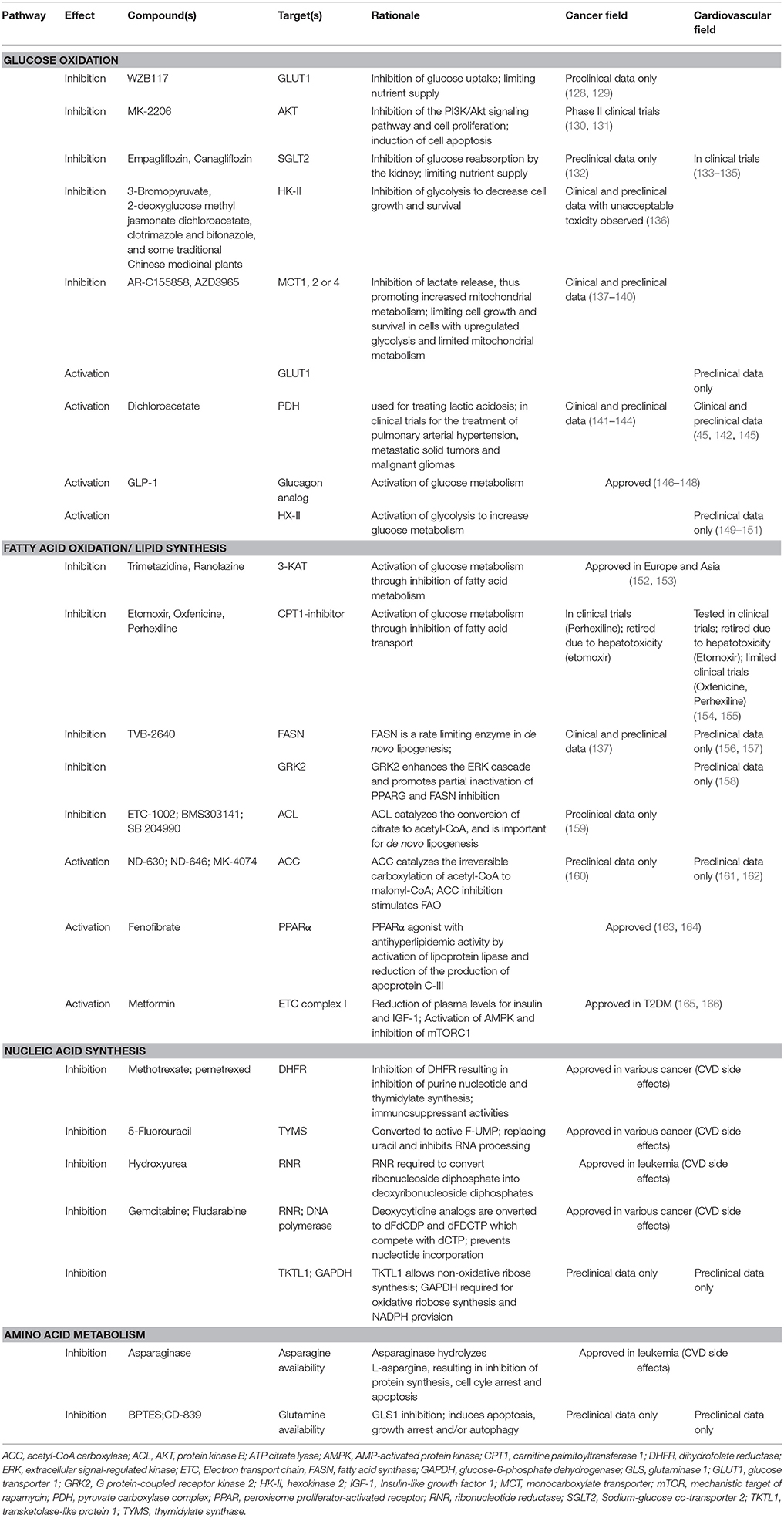
Table 1. Strategies to target metabolic enzymes for treatment of heart failure and cancer.
Glucose metabolism is an attractive target for the treatment of cancer and heart failure, because many solid tumors, as well as the failing heart, upregulate glucose utilization (167, 168). Glucose transporter 1 (GLUT1), a uniporter protein that facilitates glucose transport across the plasma membrane in mammalian cells, is a target for treatment of both cancer and heart failure (Figure 1). Genetic or pharmacologic inhibition of GLUT1 in lung and breast cancer diminishes tumor growth without systemic toxicity (Table 1). In the heart, the opposite approach has been studied. Cardiac-specific overexpression of GLUT1 has been shown to prevent cardiac hypertrophy in a transgenic mouse model subjected to pressure overload by transaortic constriction (169). Intriguingly, inhibition of Sodium glucose co-transporter 2 (SGLT2) in the kidney has proven to be an effective strategy in the treatment of type 2 diabetes with beneficial effects on the heart and at the same time implicated as a potential target in pancreatic and prostate cancers. Thus, depending on the disease progression and tumor type, inhibition of glucose transport may prevent cardiac hypertrophy and reduce cancer growth.
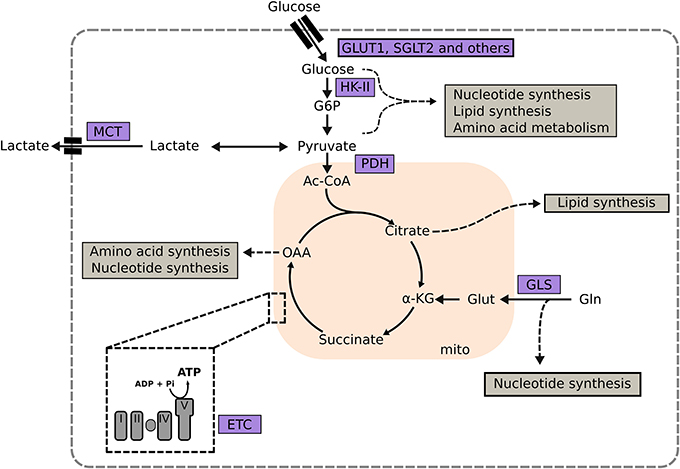
Figure 1. Targeting metabolic enzymes as a strategy in heart failure and cancer. Central metabolic pathways and the involvement of key metabolic enzymes in the synthesis of macromolecules are depicted (shown in gray boxes). α-KG, α-ketoglutarate; G6P, glucose-6-phosphate; GLS, glutaminase; GLUT1, glucose transporter type 1; Glut, glutamate; Gln, glutamine; HK-II, hexokinase II; I, complex I; III, complex III; IV, complex IV; MCT, monocarboxylate transporter; OAA, oxaloacetate; PC, pyruvate dehydrogenase complex; PDK, pyruvate dehydrogenase kinase; Succ, succinate V, complex V.
Another potential therapeutic target is Hexokinase II (HK-II; Figure 1 and Table 1), which catalyzes the phosphorylation of glucose to glucose 6-phosphate as the first rate limiting of glycolysis. HK-II binds and inactivates mTORC1 during glucose deprivation, which in turn activates autophagy. Under normal physiologic conditions, mammalian cells predominately express HK-I. Many tumors, including gliomas and NSCLCs, overexpress HK-II making it an attractive metabolic target for pharmacologic inhibitors that disrupt the binding between HK-II and mitochondria. However, HK-II inhibitors showed unacceptable systemic toxicity, e.g., development of cardiac cell death, in clinical and preclinical trials when used at high dosage (170, 171). These observations are supported by heterozygotic HK-II-knockout mouse models, which display increased cardiac susceptibility to ischemia and reperfusion injury, and increased hypertrophy and fibrosis in response to pressure overload (149). In contrast, overexpression of HK II in the heart attenuates cardiac hypertrophy by increasing flux through glycolysis and pentose phosphate pathway (150, 151, 172).
Another potential metabolic target is the PDH complex, which catalyzes the decarboxylation of the glycolytic product pyruvate to acetyl-CoA (Figure 1). The transcription factors c-MYC and HIF induce HK II and pyruvate dehydrogenase kinase (PDK) in a subset of lymphoma, which, in turn, decreases PDH activity. Pharmacologic activation of PDC by the PDK inhibitor dichloroacetate is currently in clinical trials for the treatment of pulmonary hypertension, as well as solid metastatic tumors, and gliomas. The rationale behind this strategy is to promote a tighter coupling between glucose uptake and oxidation. In the heart the premise is to increase ATP provision by increasing complete oxidation of glucose; while in cancer cells that are relying on glycolysis due to mitochondrial dysfunction, dichloroacetate potentially decreases tumor growth, and progression.
Fatty acid and mitochondrial metabolism have also emerged as targets for treatment of heart failure and cancer (173). The rationale in cancer treatment is to limit tumor proliferation by inhibiting de novo lipogenesis or stimulating fatty acid oxidation. Intriguingly, pharmacologic strategies when targeting fatty acid metabolism have been similar in heart disease and cancer. Modulation of fatty oxidation by selective inhibition of 3-ketoacyl coenzyme-A thiolase (3-KAT) and CPT1 have been either approved (e.g., 3-KAT inhibitors like trimetazidin) or tested in clinical trials for both heart failure and cancer (Table 1). However, application of CPT-1 inhibitors is limited due to hepatotoxicity and other severe side effects. Other approaches focus on limiting de novo lipid synthesis by inhibiting FASN or ATP citrate lyase (ACL). FASN is the rate limiting enzyme for de novo lipogenesis, while ACL catalyzes the conversion of glucose-derived citrate to acetyl-CoA and regulates cytosolic acetyl-CoA levels. Similarly, pharmacologic inhibition of GRK2 partially inactivates PPARγ and inhibits FASN through mitogen-activated protein kinases (MAPK) (158). Ongoing clinical trials indicate the efficacy for FASN inhibition in cancer. Similar trials in heart failure have not been successful. Another common metabolic target that may be employed is the electron transport chain (ETC), and specifically metformin, which has been increasingly used as an anti-cancer agent (174–177). By inhibiting ETC complex I (Figure 1), metformin decreases mitochondrial ATP provision (120, 178). In cancer cells, this inhibition increases the reliance on glycolysis for ATP provision, and makes cancer cells vulnerable when glucose availability is limited. Additionally, metformin reduces plasma levels of insulin and insulin-like growth factor 1 (IGF-1), which further constricts glucose availability to glycolysis-dependent cancer cells.
A further potential treatment strategy is targeting nucleic acid synthesis and amino acid metabolism. Among the various pharmacologic agents targeting nucleic acid synthesis that are available for cancer therapy, almost all have been reported to have cardiovascular side effects. Glutaminase inhibitors offer a potential way to inhibit mitochondrial amino acid metabolism (Figure 1), to induce apoptosis, growth arrest and autophagy. Certain tumors (e.g., NSCLCs and pancreatic tumors) show increased uptake and utilization of glutamine to support macromolecule synthesis and ATP provision (18, 179–181). Prolonged activation of autophagy may be involved with disease progression and decreased cardiac contractility (182, 183); thus, glutaminase inhibitors can reduce tumor burden and potentially improve cardiac function during advanced stages of heart failure.
Outlook and Conclusions
We presented several common metabolic strategies that both cancer cells and cardiomyocytes employ to optimize nutrient flux and cell growth. Metabolic reprogramming is a hallmark of both heart failure and malignant cells, which provides them with the ability to survive and sustain stress. Recent progress in molecular techniques (e.g., CRISPR/Cas9) and metabolic flux analysis using stable isotope labeling improved our understanding of mechanisms, biological consequences, and vulnerabilities associated with metabolic reprogramming in heart disease and cancer. Somatic mutations in metabolic reprogramming predominately stems from redirections of metabolic intermediates and increased ATP demand in the context of decreased cardiac contractility. Intermediary metabolites serve as signals that activate signaling pathways, modulate posttranslational modifications of proteins and alter gene expression. Examination of these relationships has inspired pharmacologic strategies that aim to either correct or enhance metabolic vulnerabilities in cancers and the failing heart. In cancer, potential pharmacologic targets manifest in pathways that regulate energy homeostasis and macromolecule biosynthesis. In the heart, similar strategies are often accompanied by severe side effects and increased cell death. Developing rational therapeutic strategies for both cancer and cardiovascular diseases will be aided by integrating findings on a systems level from pre-clinical and clinical studies. Little is known about the metabolic interaction between tumors and the heart. However, recent studies show that oncometabolic dysregulation can promote cardiac dysfunction (27). Despite the vast metabolic differences and functions of cancer cells and the heart, their common metabolic requirements present opportunities to find intersections for new therapies. Recent experimental and conceptual advances in cancer cell metabolism [reviewed by Vander Heiden et al. (184)] provide the cardiovascular field with the unique opportunity to target metabolism. This strategy holds the potential for new therapies to combat heart failure, as well as chemotherapies that may protect the heart as much as they subvert cancer.
Author Contributions
AK drafted and wrote the manuscript. WS edited the manuscript, critically evaluated and searched the literature. HT provided guidance and edited the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer XP and handling Editor declared their shared affiliation.
Acknowledgments
Funding was from the American Heart Association (17POST33660221 to AK), Cancer Prevention and Research Institute of Texas (RP170619 to HT) and NIH/NHLBI (2R01-HL-061483-15 to HT).
References
1. Lehninger AL, Nelson DL, Cox MM. Lehninger Principles of Biochemistry. New York, NY: Freeman WH (2013).
2. Sheng S, Chen D, Van Eyk JE. Multidimensional liquid chromatography separation of intact proteins by chromatographic focusing and reversed phase of the human serum proteome: optimization and protein database. Mol Cell Proteomics (2006) 5:26–34. doi: 10.1074/mcp.T500019-MCP200
3. Agnetti G, Kaludercic N, Kane LA, Elliott ST, Guo Y, Chakir K, et al. Modulation of mitochondrial proteome and improved mitochondrial function by biventricular pacing of dyssynchronous failing hearts. Circ Cardiovasc Genet. (2010) 3:78–87. doi: 10.1161/CIRCGENETICS.109.871236
4. Chouchani ET, Pell VR, Gaude E, Aksentijevic D, Sundier SY, Robb EL, et al. and Murphy MP. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature (2014) 515:431–5. doi: 10.1038/nature13909
5. Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, et al. Proteomics. Tissue-based map of the human proteome. Science (2015) 347:1260419. doi: 10.1126/science.1260419
6. Brown JM, Hazen SL. Seeking a unique lipid signature predicting cardiovascular disease risk. Circulation (2014) 129:1799–803. doi: 10.1161/CIRCULATIONAHA.114.009224
7. Essop MF, Opie LH. Metabolic therapy for heart failure. Eur Heart J. (2004) 25:1765–8. doi: 10.1016/j.ehj.2004.08.019
8. Lopaschuk GD, Ussher JR, Folmes CD, Jaswal JS, Stanley WC. Myocardial fatty acid metabolism in health and disease. Physiol Rev. (2010) 90:207–58. doi: 10.1152/physrev.00015.2009
9. Stanley WC, Lopaschuk GD, Hall JL, McCormack JG. Regulation of myocardial carbohydrate metabolism under normal and ischaemic conditions. Potential for pharmacological interventions. Cardiovasc Res. (1997) 33:243–57. doi: 10.1016/S0008-6363(96)00245-3
10. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
11. Pavlova NN, Thompson CB. The emerging hallmarks of cancer metabolism. Cell Metab. (2016) 23:27–47. doi: 10.1016/j.cmet.2015.12.006
12. DeBerardinis RJ, Chandel NS. Fundamentals of cancer metabolism. Sci Adv. (2016) 2:e1600200. doi: 10.1126/sciadv.1600200
13. Sidell RJ, Cole MA, Draper NJ, Desrois M, Buckingham RE, Clarke K. Thiazolidinedione treatment normalizes insulin resistance and ischemic injury in the zucker Fatty rat heart. Diabetes (2002) 51:1110–7. doi: 10.2337/diabetes.51.4.1110
14. Son NH, Park TS, Yamashita H, Yokoyama M, Huggins LA, Okajima K, et al. Cardiomyocyte expression of PPARgamma leads to cardiac dysfunction in mice. J Clin Invest. (2007) 117:2791–801. doi: 10.1172/JCI30335
15. Larman TC, DePalma SR, Hadjipanayis AG, Protopopov A, Zhang J, Gabriel SB, et al. Spectrum of somatic mitochondrial mutations in five cancers. Proc Natl Acad Sci USA. (2012) 109:14087–91. doi: 10.1073/pnas.1211502109
16. Christen S, Lorendeau D, Schmieder R, Broekaert D, Metzger K, Veys K, et al. Breast cancer-derived lung metastases show increased pyruvate carboxylase-dependent anaplerosis. Cell Rep. (2016) 17:837–48. doi: 10.1016/j.celrep.2016.09.042
17. Mullen AR, Wheaton WW, Jin ES, Chen PH, Sullivan LB, Cheng T, et al. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature (2011) 481:385–8. doi: 10.1038/nature10642
18. Jiang L, Shestov AA, Swain P, Yang C, Parker SJ, Wang QA, et al. Reductive carboxylation supports redox homeostasis during anchorage-independent growth. Nature (2016) 532:255–8. doi: 10.1038/nature17393
19. DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, et al. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci USA. (2007) 104:19345–50. doi: 10.1073/pnas.0709747104
20. Gaude E, Schmidt C, Gammage PA, Dugourd A, Blacker T, Chew SP, et al. NADH shuttling couples cytosolic reductive carboxylation of glutamine with glycolysis in cells with mitochondrial dysfunction. Mol Cell (2018) 69:581–593.e7. doi: 10.1016/j.molcel.2018.01.034
21. Goodwin GW, Ahmad F, Doenst T, Taegtmeyer H. Energy provision from glycogen, glucose, and fatty acids on adrenergic stimulation of isolated working rat hearts. Am J Physiol. (1998) 274:H1239–47. doi: 10.1152/ajpheart.1998.274.4.H1239
22. Allard MF, Schonekess BO, Henning SL, English DR, Lopaschuk GD. Contribution of oxidative metabolism and glycolysis to ATP production in hypertrophied hearts. Am J Physiol. (1994) 267:H742–50. doi: 10.1152/ajpheart.1994.267.2.H742
23. Goodwin GW, Taylor CS, Taegtmeyer H. Regulation of energy metabolism of the heart during acute increase in heart work. J Biol Chem. (1998) 273:29530–9. doi: 10.1074/jbc.273.45.29530
24. Akki A, Smith K, Seymour AM. Compensated cardiac hypertrophy is characterised by a decline in palmitate oxidation. Mol Cell Biochem. (2008) 311:215–24. doi: 10.1007/s11010-008-9711-y
25. Lagerstrom CF, Walker WE, Taegtmeyer H. Failure of glycogen depletion to improve left ventricular function of the rabbit heart after hypothermic ischemic arrest. Circ Res. (1988) 63:81–6. doi: 10.1161/01.RES.63.1.81
26. Schneider CA, Taegtmeyer H. Fasting in vivo delays myocardial cell damage after brief periods of ischemia in the isolated working rat heart. Circ Res. (1991) 68:1045–50. doi: 10.1161/01.RES.68.4.1045
27. Karlstaedt A, Zhang X, Vitrac H, Harmancey R, Vasquez H, Wang JH, et al. Oncometabolite d-2-hydroxyglutarate impairs α-ketoglutarate dehydrogenase and contractile function in rodent heart. Proc Natl Acad Sci USA. (2016) 113:10436–41. doi: 10.1073/pnas.1601650113
28. Yogalingam G, Hwang S, Ferreira JC, Mochly-Rosen D. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) phosphorylation by protein kinase Cdelta (PKCdelta) inhibits mitochondria elimination by lysosomal-like structures following ischemia and reoxygenation-induced injury. J Biol Chem. (2013) 288:18947–60. doi: 10.1074/jbc.M113.466870
29. Tang Z, Yuan S, Hu Y, Zhang H, Wu W, Zeng Z, et al. Over-expression of GAPDH in human colorectal carcinoma as a preferred target of 3-bromopyruvate propyl ester. J Bioenerg Biomembr. (2012) 44:117–25. doi: 10.1007/s10863-012-9420-9
30. Puzone R, Savarino G, Salvi S, Dal Bello MG, Barletta G, Genova C, et al. Glyceraldehyde-3-phosphate dehydrogenase gene over expression correlates with poor prognosis in non small cell lung cancer patients. Mol Cancer (2013) 12:97. doi: 10.1186/1476-4598-12-97
31. Liu DW, Chen ST, Liu HP. Choice of endogenous control for gene expression in nonsmall cell lung cancer. Eur Respir J. (2005) 26:1002–8. doi: 10.1183/09031936.05.00050205
32. Aksentijevic D, Lygate CA, Makinen K, Zervou S, Sebag-Montefiore L, Medway D, et al. High-energy phosphotransfer in the failing mouse heart: role of adenylate kinase and glycolytic enzymes. Eur J Heart Fail. (2010) 12:1282–9. doi: 10.1093/eurjhf/hfq174
33. Knight RJ, Kofoed KF, Schelbert HR, Buxton DB. Inhibition of glyceraldehyde-3-phosphate dehydrogenase in post-ischaemic myocardium. Cardiovasc Res. (1996) 32:1016–23. doi: 10.1016/S0008-6363(96)00137-X
34. Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, Thompson CB. ATP-citrate lyase links cellular metabolism to histone acetylation. Science (2009) 324:1076–80. doi: 10.1126/science.1164097
35. Furuta E, Pai SK, Zhan R, Bandyopadhyay S, Watabe M, Mo YY, et al. Fatty acid synthase gene is up-regulated by hypoxia via activation of Akt and sterol regulatory element binding protein-1. Cancer Res. (2008) 68:1003–11. doi: 10.1158/0008-5472.CAN-07-2489
36. Deberardinis RJ, Lum JJ, Thompson CB. Phosphatidylinositol 3-kinase-dependent modulation of carnitine palmitoyltransferase 1A expression regulates lipid metabolism during hematopoietic cell growth. J Biol Chem. (2006) 281:37372–80. doi: 10.1074/jbc.M608372200
37. Spector AA, Steinberg D. The effect of fatty acid structure on utilization by Ehrlich ascites tumor cells. Cancer Res. (1967) 27:1587–94.
38. Spector AA. Effect of carnitine on free fatty acid utilization in Ehrlich ascites tumor cells. Arch Biochem Biophys. (1967) 122:55–61. doi: 10.1016/0003-9861(67)90123-3
40. Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer (2007) 7:763–77. doi: 10.1038/nrc2222
41. Laplante M, Sabatini DM. mTORC1 activates SREBP-1c and uncouples lipogenesis from gluconeogenesis. Proc Natl Acad Sci USA. (2010) 107:3281–2. doi: 10.1073/pnas.1000323107
42. Yazaki Y, Isobe M, Takahashi W, Kitabayashi H, Nishiyama O, Sekiguchi M, et al. Assessment of myocardial fatty acid metabolic abnormalities in patients with idiopathic dilated cardiomyopathy using 123I BMIPP SPECT: correlation with clinicopathological findings and clinical course. Heart (1999) 81:153–9. doi: 10.1136/hrt.81.2.153
43. Razeghi P, Sharma S, Ying J, Li YP, Stepkowski S, Reid MB, et al. Atrophic remodeling of the heart in vivo simultaneously activates pathways of protein synthesis and degradation. Circulation (2003) 108:2536–41. doi: 10.1161/01.CIR.0000096481.45105.13
44. Sack MN, Disch DL, Rockman HA, Kelly DP. A role for Sp and nuclear receptor transcription factors in a cardiac hypertrophic growth program. Proc Natl Acad Sci USA. (1997) 94:6438–43. doi: 10.1073/pnas.94.12.6438
45. Kato T, Niizuma S, Inuzuka Y, Kawashima T, Okuda J, Tamaki Y, et al. Analysis of metabolic remodeling in compensated left ventricular hypertrophy and heart failure. Circ Heart Fail. (2010) 3:420–30. doi: 10.1161/CIRCHEARTFAILURE.109.888479
46. Kaimoto S, Hoshino A, Ariyoshi M, Okawa Y, Tateishi S, Ono K, et al. Activation of PPAR-α in the early stage of heart failure maintained myocardial function and energetics in pressure-overload heart failure. Am J Physiol Heart Circ Physiol. (2017) 312:H305–13. doi: 10.1152/ajpheart.00553.2016
47. Young ME, Laws FA, Goodwin GW, Taegtmeyer H. (2001). Reactivation of peroxisome proliferator-activated receptor alpha is associated with contractile dysfunction in hypertrophied rat heart. J Biol Chem. 276:44390–5. doi: 10.1074/jbc.M103826200
48. Karlstadt A, Fliegner D, Kararigas G, Ruderisch HS, Regitz-Zagrosek V, Holzhutter HG. CardioNet: a human metabolic network suited for the study of cardiomyocyte metabolism. BMC Syst Biol. (2012) 6:114. doi: 10.1186/1752-0509-6-114
49. Sharma S, Adrogue JV, Golfman L, Uray I, Lemm J, Youker K, et al. Intramyocardial lipid accumulation in the failing human heart resembles the lipotoxic rat heart. FASEB J. (2004) 18:1692–1700. doi: 10.1096/fj.04-2263com
50. Beer M, Seyfarth T, Sandstede J, Landschutz W, Lipke C, Kostler H, et al. Absolute concentrations of high-energy phosphate metabolites in normal, hypertrophied, and failing human myocardium measured noninvasively with (31)P-SLOOP magnetic resonance spectroscopy. J Am Coll Cardiol. (2002) 40:1267–1274. doi: 10.1016/S0735-1097(02)02160-5
51. Mashimo T, Pichumani K, Vemireddy V, Hatanpaa KJ, Singh DK, Sirasanagandla S, et al. Acetate is a bioenergetic substrate for human glioblastoma and brain metastases. Cell (2014) 159:1603–14. doi: 10.1016/j.cell.2014.11.025
52. Salem AF, Howell A, Sartini M, Sotgia F, Lisanti MP. Downregulation of stromal BRCA1 drives breast cancer tumor growth via upregulation of HIF-1alpha, autophagy and ketone body production. Cell Cycle (2012) 11:4167–73. doi: 10.4161/cc.22316
53. Martinez-Outschoorn UE, Lin Z, Whitaker-Menezes D, Howell A, Sotgia F, Lisanti MP. Ketone body utilization drives tumor growth and metastasis. Cell Cycle (2012) 11:3964–71. doi: 10.4161/cc.22137
54. Klement RJ, Sweeney RA. Impact of a ketogenic diet intervention during radiotherapy on body composition: I. Initial clinical experience with six prospectively studied patients. BMC Res Notes (2016) 9:143. doi: 10.1186/s13104-016-1959-9
55. Woolf EC, Syed N, Scheck AC. Tumor metabolism, the ketogenic diet and beta-hydroxybutyrate: novel approaches to adjuvant brain tumor therapy. Front Mol Neurosci. (2016) 9:122. doi: 10.3389/fnmol.2016.00122
56. Shukla SK, Gebregiworgis T, Purohit V, Chaika NV, Gunda V, Radhakrishnan P, et al. Metabolic reprogramming induced by ketone bodies diminishes pancreatic cancer cachexia. Cancer Metab. (2014) 2:18. doi: 10.1186/2049-3002-2-18
57. Aubert G, Martin OJ, Horton JL, Lai L, Vega RB, Leone TC, et al. The failing heart relies on ketone bodies as a fuel. Circulation (2016) 133:698–705. doi: 10.1161/CIRCULATIONAHA.115.017355
58. Bedi KC Jr, Snyder NW, Brandimarto J, Aziz M, Mesaros C, Worth AJ, et al. Evidence for intramyocardial disruption of lipid metabolism and increased myocardial ketone utilization in advanced human heart failure. Circulation (2016) 133:706–16. doi: 10.1161/CIRCULATIONAHA.115.017545
59. Wolfson RL, Chantranupong L, Saxton RA, Shen K, Scaria SM, Cantor JR, et al. Sestrin2 is a leucine sensor for the mTORC1 pathway. Science (2016) 351:43–8. doi: 10.1126/science.aab2674
60. Saxton RA, Knockenhauer KE, Wolfson RL, Chantranupong L, Pacold ME, Wang T, et al. Structural basis for leucine sensing by the Sestrin2-mTORC1 pathway. Science (2016) 351:53–8. doi: 10.1126/science.aad2087
61. Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell (2012) 149:274–93. doi: 10.1016/j.cell.2012.03.017
62. Cohen DM, Guthrie PH, Gao X, Sakai R, Taegtmeyer H. Glutamine cycling in isolated working rat heart. Am J Physiol Endocrinol Metab. (2003) 285:E1312–6. doi: 10.1152/ajpendo.00539.2002
63. Lauzier B, Vaillant F, Merlen C, Gelinas R, Bouchard B, Rivard ME, et al. Metabolic effects of glutamine on the heart: anaplerosis versus the hexosamine biosynthetic pathway. J Mol Cell Cardiol. (2013) 55:92–100. doi: 10.1016/j.yjmcc.2012.11.008
64. Taegtmeyer H. Metabolic responses to cardiac hypoxia. Increased production of succinate by rabbit papillary muscles. Circ Res. (1978) 43:808–15. doi: 10.1161/01.RES.43.5.808
65. Wischmeyer PE, Vanden Hoek TL, Li C, Shao Z, Ren H, Riehm J, et al. Glutamine preserves cardiomyocyte viability and enhances recovery of contractile function after ischemia-reperfusion injury. J Parenter Enteral Nutr. (2003) 27:116–22. doi: 10.1177/0148607103027002116
66. Pasini E, Aquilani R, Dioguardi FS, D'Antona G, Gheorghiade M, Taegtmeyer H. Hypercatabolic syndrome: molecular basis and effects of nutritional supplements with amino acids. Am J Cardiol. (2008) 101:11E−15E. doi: 10.1016/j.amjcard.2008.02.074
67. Sun H, Olson KC, Gao C, Prosdocimo DA, Zhou M, Wang Z, et al. Catabolic defect of branched-chain amino acids promotes heart failure. Circulation (2016) 133:2038–49. doi: 10.1161/CIRCULATIONAHA.115.020226
68. Hakuno D, Hamba Y, Toya T, Adachi T. Plasma amino acid profiling identifies specific amino acid associations with cardiovascular function in patients with systolic heart failure. PLoS ONE (2015) 10:e0117325. doi: 10.1371/journal.pone.0117325
69. Mangner N, Weikert B, Bowen TS, Sandri M, Hollriegel R, Erbs S, et al. Skeletal muscle alterations in chronic heart failure: differential effects on quadriceps and diaphragm. J Cachexia Sarcopenia Muscle (2015) 6:381–90. doi: 10.1002/jcsm.12034
70. Toth MJ, Gottlieb SS, Fisher ML, Poehlman ET. Skeletal muscle atrophy and peak oxygen consumption in heart failure. Am J Cardiol. (1997) 79:1267–9. doi: 10.1016/S0002-9149(97).00098-2
71. Morrison WL, Gibson JN, Rennie MJ. Skeletal muscle and whole body protein turnover in cardiac cachexia: influence of branched-chain amino acid administration. Eur J Clin Invest. (1988) 18:648–54. doi: 10.1111/j.1365-2362.1988.tb01282.x
72. Uchino Y, Watanabe M, Takata M, Amiya E, Tsushima K, Adachi T, et al. Effect of oral branched-chain amino acids on serum albumin concentration in heart failure patients with hypoalbuminemia: results of a preliminary study. Am J Cardiovasc Drugs (2018). doi: 10.1007/s40256-018-0269-0. [Epub ahead of print].
73. Lombardi C, Carubelli V, Lazzarini V, Vizzardi E, Quinzani F, Guidetti F, et al. Effects of oral amino Acid supplements on functional capacity in patients with chronic heart failure. Clin Med Insights Cardiol. (2014) 8:39–44. doi: 10.4137/CMC.S14016
74. Anker SD, Ponikowski P, Varney S, Chua TP, Clark AL, Webb-Peploe KM, et al. Wasting as independent risk factor for mortality in chronic heart failure. Lancet (1997) 349:1050–3. doi: 10.1016/S0140-6736(96)07015-8
75. Crino PB, Nathanson KL, Henske EP. The tuberous sclerosis complex. N Engl J Med. (2006) 355:1345–56. doi: 10.1056/NEJMra055323
76. Guertin DA, Sabatini DM. An expanding role for mTOR in cancer. Trends Mol Med. (2005) 11:353–61. doi: 10.1016/j.molmed.2005.06.007
77. Nicklin P, Bergman P, Zhang B, Triantafellow E, Wang H, Nyfeler B, et al. Bidirectional transport of amino acids regulates mTOR and autophagy. Cell (2009) 136:521–34. doi: 10.1016/j.cell.2008.11.044
78. Noall MW, Riggs TR, Walker LM, Christensen HN. Endocrine control of amino acid transfer; distribution of an unmetabolizable amino acid. Science (1957) 126:1002–5. doi: 10.1126/science.126.3281.1002
79. Kipnis DM, Noall MW. Stimulation of amino acid transport by insulin in the isolated rat diaphragm. Biochim Biophys Acta (1958) 28:226–7. doi: 10.1016/0006-3002(58)90466-9
80. Morgan HE, Jefferson LS, Wolpert EB, Rannels DE. Regulation of protein synthesis in heart muscle. II. Effect of amino acid levels and insulin on ribosomal aggregation. J Biol Chem. (1971) 246:2163–70.
81. Morgan HE, Earl DC, Broadus A, Wolpert EB, Giger KE, Jefferson LS. Regulation of protein synthesis in heart muscle. I. Effect of amino acid levels on protein synthesis. J Biol Chem. (1971) 246:2152–62.
82. Wilkins BJ, Dai YS, Bueno OF, Parsons SA, Xu J, Plank DM, et al. Calcineurin/NFAT coupling participates in pathological, but not physiological, cardiac hypertrophy. Circ Res. (2004) 94:110–8. doi: 10.1161/01.RES.0000109415.17511.18
83. Frey N, Frank D, Lippl S, Kuhn C, Kogler H, Barrientos T, et al. Calsarcin-2 deficiency increases exercise capacity in mice through calcineurin/NFAT activation. J Clin Invest. (2008) 118:3598–608. doi: 10.1172/JCI36277
84. Hartupee J, Szalai GD, Wang W, Ma X, Diwan A, Mann DL. Impaired protein quality control during left ventricular remodeling in mice with cardiac restricted overexpression of tumor necrosis factor. Circ Heart Fail. (2017) 10:e004252. doi: 10.1161/CIRCHEARTFAILURE.117.004252
85. Sen S, Kundu BK, Wu HC, Hashmi SS, Guthrie P, Locke LW, et al. Glucose regulation of load-induced mTOR signaling and ER stress in mammalian heart. J Am Heart Assoc. (2013) 2:e004796. doi: 10.1161/JAHA.113.004796
86. Mudge GH, Jr., Mills RM Jr., Taegtmeyer H, Gorlin R, et al. Alterations of myocardial amino acid metabolism in chronic ischemic heart disease. J Clin Invest. (1976) 58:1185–92. doi: 10.1172/JCI108571
87. Taegtmeyer H, Peterson MB, Ragavan VV, Ferguson AG, Lesch M. De novo alanine synthesis in isolated oxygen-deprived rabbit myocardium. J Biol Chem. (1977) 252:5010–8.
88. Piao L, Fang YH, Parikh K, Ryan JJ, Toth PT, Archer SL. Cardiac glutaminolysis: a maladaptive cancer metabolism pathway in the right ventricle in pulmonary hypertension. J Mol Med. (2013) 91:1185–97. doi: 10.1007/s00109-013-1064-7
89. Drews O, Taegtmeyer H. Targeting the ubiquitin-proteasome system in heart disease: the basis for new therapeutic strategies. Antioxid Redox Signal. (2014) 21:2322–43. doi: 10.1089/ars.2013.5823
90. Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy (2016) 12:1–222. doi: 10.1080/15548627.2015.1100356
91. Kabeya Y, Kamada Y, Baba M, Takikawa H, Sasaki M, Ohsumi Y. Atg17 functions in cooperation with Atg1 and Atg13 in yeast autophagy. Mol Biol Cell (2005) 16:2544–53. doi: 10.1091/mbc.e04-08-0669
92. Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol. (2005) 169:425–34. doi: 10.1083/jcb.200412022
93. Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol. (2011) 27:107–32. doi: 10.1146/annurev-cellbio-092910-154005
94. Obara K, Sekito T, Ohsumi Y. Assortment of phosphatidylinositol 3-kinase complexes–Atg14p directs association of complex I to the pre-autophagosomal structure in Saccharomyces cerevisiae. Mol Biol Cell (2006) 17:1527–39. doi: 10.1091/mbc.e05-09-0841
95. Suzuki K, Kubota Y, Sekito T, Ohsumi Y. Hierarchy of Atg proteins in pre-autophagosomal structure organization. Genes Cells (2007) 12:209–18. doi: 10.1111/j.1365-2443.2007.01050.x
96. Suzuki K, Noda T, Ohsumi Y. Interrelationships among Atg proteins during autophagy in Saccharomyces cerevisiae. Yeast (2004) 21:1057–65. doi: 10.1002/yea.1152
97. Cluntun AA, Lukey MJ, Cerione RA, Locasale JW. Glutamine metabolism in cancer: understanding the heterogeneity. Trends Cancer (2017) 3:169–180. doi: 10.1016/j.trecan.2017.01.005
98. Seo JW, Choi J, Lee SY, Sung S, Yoo HJ, Kang MJ, et al. Autophagy is required for PDAC glutamine metabolism. Sci Rep. (2016) 6:37594. doi: 10.1038/srep37594
99. Andres AM, Tucker KC, Thomas A, Taylor DJ, Sengstock D, Jahania SM, et al. Mitophagy and mitochondrial biogenesis in atrial tissue of patients undergoing heart surgery with cardiopulmonary bypass. JCI Insight (2017) 2:e89303. doi: 10.1172/jci.insight.89303
100. Gustafsson AB, Gottlieb RA. Recycle or die: the role of autophagy in cardioprotection. J Mol Cell Cardiol. (2008) 44:654–61. doi: 10.1016/j.yjmcc.2008.01.010
101. Hernandez G, Thornton C, Stotland A, Lui D, Sin J, Ramil J, et al. MitoTimer: a novel tool for monitoring mitochondrial turnover. Autophagy (2013) 9:1852–61. doi: 10.4161/auto.26501
102. Stotland A, Gottlieb RA. alpha-MHC MitoTimer mouse: In vivo mitochondrial turnover model reveals remarkable mitochondrial heterogeneity in the heart. J Mol Cell Cardiol. (2016) 90:53–8. doi: 10.1016/j.yjmcc.2015.11.032
103. Hammerling BC, Najor RH, Cortez MQ, Shires SE, Leon LJ, Gonzalez ER, et al. A Rab5 endosomal pathway mediates Parkin-dependent mitochondrial clearance. Nat Commun. (2017) 8:14050. doi: 10.1038/ncomms14050
104. Moyzis AG, Sadoshima J, Gustafsson AB. Mending a broken heart: the role of mitophagy in cardioprotection. Am J Physiol Heart Circ Physiol. (2015) 308:H183–92. doi: 10.1152/ajpheart.00708.2014
105. Saito T, Sadoshima J. Unexpected functional consequences of the loss of the autophagy-related conjugation system. Circ Res. (2017) 120:610–612. doi: 10.1161/CIRCRESAHA.117.310450
106. Shirakabe A, Ikeda Y, Sciarretta S, Zablocki DK, Sadoshima J. Aging and autophagy in the heart. Circ Res. (2016) 118:1563–76. doi: 10.1161/CIRCRESAHA.116.307474
107. Shirakabe A, Zhai P, Ikeda Y, Saito T, Maejima Y, Hsu CP, et al. Drp1-dependent mitochondrial autophagy plays a protective role against pressure overload-induced mitochondrial dysfunction and heart failure. Circulation (2016) 133:1249–63. doi: 10.1161/CIRCULATIONAHA.115.020502
108. Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M, et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med. (2007) 13:619–24. doi: 10.1038/nm1574
109. Yoo H, Stephanopoulos G, Kelleher JK. Quantifying carbon sources for de novo lipogenesis in wild-type and IRS-1 knockout brown adipocytes. J Lipid Res. (2004) 45:1324–32. doi: 10.1194/jlr.M400031-JLR200
110. Schug ZT, Peck B, Jones DT, Zhang Q, Grosskurth S, Alam IS, et al. Acetyl-CoA synthetase 2 promotes acetate utilization and maintains cancer cell growth under metabolic stress. Cancer Cell (2015) 27:57–71. doi: 10.1016/j.ccell.2014.12.002
111. Metallo CM, Gameiro PA, Bell EL, Mattaini KR, Yang J, Hiller K, et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature (2011) 481:380–4. doi: 10.1038/nature10602
112. Wise DR, Ward PS, Shay JE, Cross JR, Gruber JJ, Sachdeva UM, et al. Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of alpha-ketoglutarate to citrate to support cell growth and viability. Proc Natl Acad Sci USA. (2011) 108:19611–6. doi: 10.1073/pnas.1117773108
113. Bianchi A, Evans JL, Iverson AJ, Nordlund AC, Watts TD, Witters LA. Identification of an isozymic form of acetyl-CoA carboxylase. J Biol Chem. (1990) 265:1502–9.
114. Iverson AJ, Bianchi A, Nordlund AC, Witters LA. Immunological analysis of acetyl-CoA carboxylase mass, tissue distribution and subunit composition. Biochem J. (1990) 269:365–71. doi: 10.1042/bj2690365
115. Thampy KG. Formation of malonyl coenzyme A in rat heart. Identification and purification of an isozyme of A carboxylase from rat heart. J Biol Chem. (1989) 264:17631–4.
116. Ballard FB, Danforth WH, Naegle S, Bing RJ. Myocardial metabolism of fatty acids. J Clin Invest. (1960) 39:717–23. doi: 10.1172/JCI104088
117. Bharadwaj KG, Hiyama Y, Hu Y, Huggins LA, Ramakrishnan R, Abumrad NA, et al. Chylomicron- and VLDL-derived lipids enter the heart through different pathways: in vivo evidence for receptor- and non-receptor-mediated fatty acid uptake. J Biol Chem. (2010) 285:37976–86. doi: 10.1074/jbc.M110.174458
118. Li S, Brown MS, Goldstein JL. Bifurcation of insulin signaling pathway in rat liver: mTORC1 required for stimulation of lipogenesis, but not inhibition of gluconeogenesis. Proc Natl Acad Sci USA. (2010) 107:3441–6. doi: 10.1073/pnas.0914798107
119. Lane AN, Fan TW. Regulation of mammalian nucleotide metabolism and biosynthesis. Nucleic Acids Res. (2015) 43:2466–85. doi: 10.1093/nar/gkv047
120. Birsoy K, Wang T, Chen WW, Freinkman E, Abu-Remaileh M, Sabatini DM. An essential role of the mitochondrial electron transport chain in cell proliferation is to enable aspartate synthesis. Cell (2015) 162:540–51. doi: 10.1016/j.cell.2015.07.016
121. Sullivan LB, Gui DY, Hosios AM, Bush LN, Freinkman E, Vander Heiden MG. Supporting aspartate biosynthesis is an essential function of respiration in proliferating cells. Cell (2015) 162:552–63. doi: 10.1016/j.cell.2015.07.017
122. Bao XR, Ong SE, Goldberger O, Peng J, Sharma R, Thompson DA, et al. Mitochondrial dysfunction remodels one-carbon metabolism in human cells. Elife (2016) 5:e10575. doi: 10.7554/eLife.10575
123. Meiser J, Tumanov S, Maddocks O, Labuschagne CF, Athineos D, N., et al. Serine one-carbon catabolism with formate overflow. Sci Adv. (2016) 2:e1601273. doi: 10.1126/sciadv.1601273
124. Gui DY, Sullivan LB, Luengo A, Hosios AM, Bush LN, Gitego N, et al. Environment dictates dependence on mitochondrial complex I for NAD+ and aspartate production and determines cancer cell sensitivity to metformin. Cell Metab. (2016) 24:716–727. doi: 10.1016/j.cmet.2016.09.006
125. Peuhkurinen KJ, Takala TE, Nuutinen EM, Hassinen IE. Tricarboxylic acid cycle metabolites during ischemia in isolated perfused rat heart. Am J Physiol. (1983) 244:H281–8. doi: 10.1152/ajpheart.1983.244.2.H281
126. Taegtmeyer H. Is the purine nucleotide cycle important in heart muscle? Adv Myocardiol. (1985) 6:165–72.
127. Hoxhaj G, Hughes-Hallett J, Timson RC, Ilagan E, Yuan M, Asara JM, et al. The mTORC1 signaling network senses changes in cellular purine nucleotide levels. Cell Rep. (2017) 21:1331–46. doi: 10.1016/j.celrep.2017.10.029
128. Ojelabi OA, Lloyd KP, Simon AH, De Zutter JK, Carruthers A. WZB117 (2-Fluoro-6-(m-hydroxybenzoyloxy) Phenyl m-Hydroxybenzoate) Inhibits GLUT1-mediated sugar transport by binding reversibly at the exofacial sugar binding site. J Biol Chem. (2016) 291:26762–72. doi: 10.1074/jbc.M116.759175
129. Liu Y, Cao Y, Zhang W, Bergmeier S, Qian Y, Akbar H, et al. A small-molecule inhibitor of glucose transporter 1 downregulates glycolysis, induces cell-cycle arrest, and inhibits cancer cell growth in vitro and in vivo. Mol Cancer Ther. (2012) 11:1672–82. doi: 10.1158/1535-7163.MCT-12-0131
130. Oki Y, Fanale M, Romaguera J, Fayad L, Fowler N, Copeland A, et al. Phase II study of an AKT inhibitor MK2206 in patients with relapsed or refractory lymphoma. Br J Haematol. (2015) 171:463–70. doi: 10.1111/bjh.13603
131. Ramanathan RK, McDonough SL, Kennecke HF, Iqbal S, Baranda JC, Seery TE, et al. Phase 2 study of MK-2206, an allosteric inhibitor of AKT, as second-line therapy for advanced gastric and gastroesophageal junction cancer: a SWOG cooperative group trial (S1005). Cancer (2015) 121:2193–7. doi: 10.1002/cncr.29363
132. Scafoglio C, Hirayama BA, Kepe V, Liu J, Ghezzi C, Satyamurthy N, et al. Functional expression of sodium-glucose transporters in cancer. Proc Natl Acad Sci USA. (2015) 112:E4111–9. doi: 10.1073/pnas.1511698112
133. Fitchett D, Zinman B, Wanner C, Lachin JM, Hantel S, Salsali A, et al. Heart failure outcomes with empagliflozin in patients with type 2 diabetes at high cardiovascular risk: results of the EMPA-REG OUTCOME(R) trial. Eur Heart J. (2016) 37:1526–34. doi: 10.1093/eurheartj/ehv728
134. Neal B, Perkovic V, Mahaffey KW, de Zeeuw D, Fulcher G, Erondu N, et al. Canagliflozin and cardiovascular and renal events in type 2 diabetes. N Engl J Med. (2017) 377:644–57. doi: 10.1056/NEJMoa1611925
135. Ferrannini E, Baldi S, Frascerra S, Astiarraga B, Heise T, Bizzotto R, et al. Shift to fatty substrate utilization in response to sodium-glucose cotransporter 2 inhibition in subjects without diabetes and patients with type 2 diabetes. Diabetes (2016) 65:1190–5. doi: 10.2337/db15-1356
136. Raez LE, Papadopoulos K, Ricart AD, Chiorean EG, Dipaola RS, Stein MN, et al. A phase I dose-escalation trial of 2-deoxy-D-glucose alone or combined with docetaxel in patients with advanced solid tumors. Cancer Chemother Pharmacol. (2013) 71:523–30. doi: 10.1007/s00280-012-2045-1
137. O'Farrell M, Crowley R, Heuer T, Buckley D, Rubino CM, McCulloch W, et al. Biomarker and PK/PD analyses of first-in-class FASN inhibitor TVB-2640 in a first-in-human phase 1 study in solid tumor patients [abstract]. In: Proceedings of the 106th Annual Meeting of the American Association for Cancer Research. Philadelphia, PA: AACR (2015).
138. Doherty JR, Cleveland JL. Targeting lactate metabolism for cancer therapeutics. J Clin Invest. (2013) 123:3685–92. doi: 10.1172/JCI69741
139. Ovens MJ, Manoharan C, Wilson MC, Murray CM, Halestrap AP. The inhibition of monocarboxylate transporter 2 (MCT2) by AR-C155858 is modulated by the associated ancillary protein. Biochem J. (2010) 431:217–25. doi: 10.1042/BJ20100890
140. Ovens MJ, Davies AJ, Wilson MC, Murray CM, Halestrap AP. AR-C155858 is a potent inhibitor of monocarboxylate transporters MCT1 and MCT2 that binds to an intracellular site involving transmembrane helices 7-10. Biochem J. (2010) 425:523–30. doi: 10.1042/BJ20091515
141. Dunbar EM, Coats BS, Shroads AL, Langaee T, Lew A, Forder JR, et al. Phase 1 trial of dichloroacetate (DCA) in adults with recurrent malignant brain tumors. Invest New Drugs (2014) 32:452–64. doi: 10.1007/s10637-013-0047-4
142. McMurtry MS, Bonnet S, Wu X, Dyck JR, Haromy A, Hashimoto K, et al. Dichloroacetate prevents and reverses pulmonary hypertension by inducing pulmonary artery smooth muscle cell apoptosis. Circ Res. (2004) 95:830–40. doi: 10.1161/01.RES.0000145360.16770.9f
143. Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, et al. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell (2007) 11:37–51. doi: 10.1016/j.ccr.2006.10.020
144. Garon EB, Christofk HR, Hosmer W, Britten CD, Bahng A, Crabtree MJ, et al. Dichloroacetate should be considered with platinum-based chemotherapy in hypoxic tumors rather than as a single agent in advanced non-small cell lung cancer. J Cancer Res Clin Oncol. (2014) 140:443–52. doi: 10.1007/s00432-014-1583-9
145. Lewis JF, DaCosta M, Wargowich T, Stacpoole P. Effects of dichloroacetate in patients with congestive heart failure. Clin Cardiol. (1998) 21:888–92. doi: 10.1002/clc.4960211206
146. Margulies KB, McNulty SE, Cappola TP. Lack of benefit for liraglutide in heart failure-reply. JAMA (2016) 316:2429–30. doi: 10.1001/jama.2016.15394
147. Margulies KB, Hernandez AF, Redfield MM, Givertz MM, Oliveira GH, Cole R, et al. Effects of liraglutide on clinical stability among patients with advanced heart failure and reduced ejection fraction: a randomized clinical trial. JAMA (2016) 316:500–8. doi: 10.1001/jama.2016.10260
148. Margulies KB. Evolving challenges for targeting metabolic abnormalities in heart failure. JACC Heart Fail. (2016) 4:567–9. doi: 10.1016/j.jchf.2016.03.008
149. Wu R, Wyatt E, Chawla K, Tran M, Ghanefar M, Laakso M, et al. Hexokinase II knockdown results in exaggerated cardiac hypertrophy via increased ROS production. EMBO Mol Med. (2012) 4:633–46. doi: 10.1002/emmm.201200240
150. Roberts DJ, Tan-Sah VP, Ding EY, Smith JM, Miyamoto S. Hexokinase-II positively regulates glucose starvation-induced autophagy through TORC1 inhibition. Mol Cell (2014) 53:521–33. doi: 10.1016/j.molcel.2013.12.019
151. McCommis KS, Douglas DL, Krenz M, Baines CP. Cardiac-specific hexokinase 2 overexpression attenuates hypertrophy by increasing pentose phosphate pathway flux. J Am Heart Assoc. (2013) 2:e000355. doi: 10.1161/JAHA.113.000355
152. Kantor PF, Lucien A, Kozak R, Lopaschuk GD. The antianginal drug trimetazidine shifts cardiac energy metabolism from fatty acid oxidation to glucose oxidation by inhibiting mitochondrial long-chain 3-ketoacyl coenzyme A thiolase. Circ Res. (2000) 86:580–8. doi: 10.1161/01.RES.86.5.580
153. Gao D, Ning N, Niu X, Hao G, Meng Z. Trimetazidine: a meta-analysis of randomised controlled trials in heart failure. Heart (2011) 97:278–86. doi: 10.1136/hrt.2010.208751
154. Lee L, Campbell R, Scheuermann-Freestone M, Taylor R, Gunaruwan P, Williams L, et al. Metabolic modulation with perhexiline in chronic heart failure: a randomized, controlled trial of short-term use of a novel treatment. Circulation (2005) 112:3280–8. doi: 10.1161/CIRCULATIONAHA.105.551457
155. Cole PL, Beamer AD, McGowan N, Cantillon CO, Benfell K, Kelly RA, et al. Efficacy and safety of perhexiline maleate in refractory angina. A double-blind placebo-controlled clinical trial of a novel antianginal agent. Circulation (1990) 81:1260–70. doi: 10.1161/01.CIR.81.4.1260
156. Razani B, Zhang H, Schulze PC, Schilling JD, Verbsky J, Lodhi IJ, et al. Fatty acid synthase modulates homeostatic responses to myocardial stress. J Biol Chem. (2011) 286:30949–61. doi: 10.1074/jbc.M111.230508
157. Knobloch M, Braun SM, Zurkirchen L, von Schoultz C, Zamboni N, Arauzo-Bravo MJ, et al. Metabolic control of adult neural stem cell activity by Fasn-dependent lipogenesis. Nature (2013) 493:226–30. doi: 10.1038/nature11689
158. Abd Alla J, Graemer M, Fu X, Quitterer U. Inhibition of G-protein-coupled receptor kinase 2 prevents the dysfunctional cardiac substrate metabolism in fatty acid synthase transgenic mice. J Biol Chem. (2016) 291:2583–600. doi: 10.1074/jbc.M115.702688
159. Zhao S, Torres A, Henry RA, Trefely S, Wallace M, Lee JV, et al. ATP-citrate lyase controls a glucose-to-acetate metabolic switch. Cell Rep. (2016) 17:1037–52. doi: 10.1016/j.celrep.2016.09.069
160. Svensson RU, Parker SJ, Eichner LJ, Kolar MJ, Wallace M, Brun SN, et al. Inhibition of acetyl-CoA carboxylase suppresses fatty acid synthesis and tumor growth of non-small-cell lung cancer in preclinical models. Nat Med. (2016) 22:1108–19. doi: 10.1038/nm.4181
161. Harriman G, Greenwood J, Bhat S, Huang X, Wang R, Paul D, et al. Acetyl-CoA carboxylase inhibition by ND-630 reduces hepatic steatosis, improves insulin sensitivity, and modulates dyslipidemia in rats. Proc Natl Acad Sci USA. (2016) 113:E1796–805. doi: 10.1073/pnas.1520686113
162. Kim CW, Addy C, Kusunoki J, Anderson NN, Deja S, Fu X, et al. Acetyl CoA carboxylase inhibition reduces hepatic steatosis but elevates plasma triglycerides in mice and humans: a bedside to bench investigation. Cell Metab. (2017) 26:576. doi: 10.1016/j.cmet.2017.08.011
163. Colhoun H. After FIELD: should fibrates be used to prevent cardiovascular disease in diabetes? Lancet (2005) 366:1829–31. doi: 10.1016/S0140-6736(05)67668-4
164. Keech A, Simes RJ, Barter P, Best J, Scott R, Taskinen MR, et al. Effects of long-term fenofibrate therapy on cardiovascular events in 9795 people with type 2 diabetes mellitus (the FIELD study): randomised controlled trial. Lancet (2005) 366:1849–61. doi: 10.1016/S0140-6736(05)67667-2
165. Kalender A, Selvaraj A, Kim SY, Gulati P, Brule S, Viollet B, et al. Metformin, independent of AMPK, inhibits mTORC1 in a rag GTPase-dependent manner. Cell Metab. (2010) 11:390–401. doi: 10.1016/j.cmet.2010.03.014
166. Giovannucci E, Harlan DM, Archer MC, Bergenstal RM, Gapstur SM, Habel LA, et al. Diabetes and cancer: a consensus report. Diabetes Care (2010) 33:1674–85. doi: 10.2337/dc10-0666
167. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science (2009) 324:1029–33. doi: 10.1126/science.1160809
168. Taegtmeyer H, Stanley WC. Too much or not enough of a good thing? Cardiac glucolipotoxicity versus lipoprotection. J Mol Cell Cardiol. (2011) 50:2–5. doi: 10.1016/j.yjmcc.2010.09.014
169. Liao R, Jain M, Cui L, D'Agostino J, Aiello F, Luptak I, et al. Cardiac-specific overexpression of GLUT1 prevents the development of heart failure attributable to pressure overload in mice. Circulation (2002) 106:2125–31. doi: 10.1161/01.CIR.0000034049.61181.F3
170. Singh D, Banerji AK, Dwarakanath BS, Tripathi RP, Gupta JP, Mathew TL, et al. Optimizing cancer radiotherapy with 2-deoxy-d-glucose dose escalation studies in patients with glioblastoma multiforme. Strahlenther Onkol. (2005) 181:507–14. doi: 10.1007/s00066-005-1320-z
171. Mohanti BK, Rath GK, Anantha N, Kannan V, Das BS, Chandramouli BA, et al. Improving cancer radiotherapy with 2-deoxy-D-glucose: phase I/II clinical trials on human cerebral gliomas. Int J Radiat Oncol Biol Phys. (1996) 35:103–11. doi: 10.1016/S0360-3016(96)85017-6
172. Roberts DJ, Miyamoto S. Hexokinase II integrates energy metabolism and cellular protection: Akting on mitochondria and TORCing to autophagy. Cell Death Differ. (2015) 22:248–57. doi: 10.1038/cdd.2014.173
173. Samudio I, Harmancey R, Fiegl M, Kantarjian H, Konopleva M, Korchin B, et al. Pharmacologic inhibition of fatty acid oxidation sensitizes human leukemia cells to apoptosis induction. J Clin Invest. (2010) 120:142–56. doi: 10.1172/JCI38942
174. Dowling RJ, Niraula S, Stambolic V, Goodwin PJ. Metformin in cancer: translational challenges. J Mol Endocrinol. (2012) 48:R31–43. doi: 10.1530/JME-12-0007
175. Ota S, Horigome K, Ishii T, Nakai M, Hayashi K, Kawamura T, et al. Metformin suppresses glucose-6-phosphatase expression by a complex I inhibition and AMPK activation-independent mechanism. Biochem Biophys Res Commun. (2009) 388:311–6. doi: 10.1016/j.bbrc.2009.07.164
176. El-Mir MY, Nogueira V, Fontaine E, Averet N, Rigoulet M, Leverve X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J Biol Chem. (2000) 275:223–8. doi: 10.1074/jbc.275.1.223
177. Owen MR, Doran E, Halestrap AP. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem J. (2000) 348(Pt 3):607–14. doi: 10.1042/bj3480607
178. Wheaton WW, Weinberg SE, Hamanaka RB, Soberanes S, Sullivan LB, Anso E, et al. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. Elife (2014) 3:e02242. doi: 10.7554/eLife.02242
179. Kamphorst JJ, Nofal M, Commisso C, Hackett SR, Lu W, Grabocka E, et al. Human pancreatic cancer tumors are nutrient poor and tumor cells actively scavenge extracellular protein. Cancer Res. (2015) 75:544–53. doi: 10.1158/0008-5472.CAN-14-2211
180. Faubert B, Li KY, Cai L, Hensley CT, Kim J, Zacharias LG, et al. Lactate metabolism in human lung tumors. Cell (2017) 171:358–371.e9. doi: 10.1016/j.cell.2017.09.019
181. Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M, et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature (2013) 496:101–5. doi: 10.1038/nature12040
182. Zhu H, Tannous P, Johnstone JL, Kong Y, Shelton JM, Richardson JA, et al. Cardiac autophagy is a maladaptive response to hemodynamic stress. J Clin Invest. (2007) 117:1782–93. doi: 10.1172/JCI27523
183. Cao DJ, Wang ZV, Battiprolu PK, Jiang N, Morales CR, Kong Y, et al. Histone deacetylase (HDAC) inhibitors attenuate cardiac hypertrophy by suppressing autophagy. Proc Natl Acad Sci USA. (2011) 108:4123–8. doi: 10.1073/pnas.1015081108
Keywords: cardiac metabolism, cancer cell metabolism, heart failure, clinical trials as topic, targeted treatments, metabolic targets, systems biology, intermediary metabolism
Citation: Karlstaedt A, Schiffer W and Taegtmeyer H (2018) Actionable Metabolic Pathways in Heart Failure and Cancer—Lessons From Cancer Cell Metabolism. Front. Cardiovasc. Med. 5:71. doi: 10.3389/fcvm.2018.00071
Received: 19 December 2017; Accepted: 24 May 2018;
Published: 19 June 2018.
Edited by:
James Martin, Baylor College of Medicine, United StatesReviewed by:
Xinchun Pi, Baylor College of Medicine, United StatesTodd R. Heallen, Texas Heart Institute, United States
Sivareddy Kotla, University of Texas MD Anderson Cancer Center, United States
Copyright © 2018 Karlstaedt, Schiffer and Taegtmeyer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Heinrich Taegtmeyer, aGVpbnJpY2gudGFlZ3RtZXllckB1dGgudG1jLmVkdQ==