 Saumik Biswas
Saumik Biswas Anu Alice Thomas
Anu Alice Thomas Subrata Chakrabarti
Subrata Chakrabarti- Department of Pathology and Laboratory Medicine, Western University, London, ON, Canada
Long non-coding RNAs (lncRNAs) are critical regulators in a multitude of biological processes. Recent evidences demonstrate potential pathogenetic implications of lncRNAs in diabetic cardiomyopathy (DCM); however, the majority of lncRNAs have not been comprehensively characterized. While the precise molecular mechanisms underlying the functions of lncRNAs remain to be deciphered in DCM, emerging data in other pathophysiological conditions suggests that lncRNAs can have versatile features such as genomic imprinting, acting as guides for certain histone-modifying complexes, serving as scaffolds for specific molecules, or acting as molecular sponges. In an effort to better understand these features of lncRNAs in the context of DCM, our review will first summarize some of the key molecular alterations that occur during fibrosis in the diabetic heart (extracellular proteins and endothelial-to-mesenchymal transitioning), followed by a review of the current knowledge on the crosstalk between lncRNAs and major epigenetic mechanisms (histone methylation, histone acetylation, DNA methylation, and microRNAs) within this fibrotic process.
Introduction
With the incidence of diabetic cardiomyopathy (DCM) increasing at an alarming rate, the need for broadening the therapeutic scope for disease management becomes fundamental. In order to develop new therapeutic agents to successfully impede the progression of DCM, a thorough understanding of the complex pathogenetic mechanisms implicated in DCM progression is an absolute requirement. There are currently several known metabolic pathway alterations that occur in a hyperglycemic environment; however, recent advances in genomic technology have identified that a significant number of epigenetic alterations contribute to the development and progression of DCM. Long non-coding RNAs (lncRNAs), which are involved in altering gene expression without modifying the underlying nucleotide composition of the genome, are beginning to emerge as key epigenetic regulators in various diseases (1). Despite possessing limited protein-coding potential and being greater than 200 nucleotides in length, lncRNAs can alter the dynamic configuration of the chromatin by interacting with several enzymes that facilitate chromatin remodeling and gene regulation (2). In the context of DCM, numerous lncRNAs have been identified to be aberrantly expressed in diabetic cardiac tissues (3); however, the majority of lncRNAs have not been comprehensively characterized (4). To fill some of the gap in knowledge, this review will first provide the necessary background behind DCM, extracellular matrix (ECM) proteins, and fibrosis and then discuss the postulated roles of lncRNAs in major epigenetic modifications during DCM fibrosis.
Diabetic Cardiomyopathy (DCM)
Cardiovascular complications are responsible for the majority of diabetes-related morbidity and mortality (5). Such complications include atherosclerosis, autonomic neuropathy and diabetic cardiomyopathy (DCM). The latter leads to an increased risk for the development of structural and functional changes in the myocardium independent of coronary artery disease and hypertension (6). Rubler and colleagues first described this ventricular dysfunction occurring in diabetic patients in 1972 (7). Alterations in myocardial structure, calcium signalling and metabolism are primary signs that precede accelerated left ventricular hypertrophy and increase susceptibility to ischemic injury and overall heart failure (HF) (8). The myocardial tissue undergoes structural and functional modifications after diabetes, which is induced by hyperglycemia, hyperlipidemia and insulin resistance (9). A histological trademark of DCM is interstitial and perivascular fibrosis, characterized by increased deposition of collagen accompanied by crosslinking of these collagen fibers contributing to reduced ventricular compliance (7, 10, 11). Myocardial fibrosis is accompanied by an increase in left ventricle (LV) mass, also known as left ventricle hypertrophy (LVH). LVH has been linked to elevated markers of systemic inflammation such as fibrinogen, C-reactive protein and microalbuminuria (12). These structural changes are accompanied by functional alterations, namely diastolic and systolic dysfunction. Diastolic dysfunction is defined as defective ventricular relaxation leading to pressure increase and impairment in blood filling during diastole (9). During systolic dysfunction, the myocardium fails to eject adequate blood volume and is observed at later stages in DCM after diastolic dysfunction has been established in patients (13, 14). Early identification of these abnormalities is important to provide appropriate treatment and prevent advancement to HF.
Multiple mechanisms have been proposed to explain the pathogenesis of DCM. These include autonomic dysfunction, defects in lipid metabolism, abnormalities in ion homeostasis, alterations in structural proteins, increased oxidative stress, interstitial fibrosis and alterations in myocardial substrates and energy metabolism (15–20). As compensation for glucose assimilation, fatty acid transporters are increased to generate ATP through fatty acid (FA) degradation (19). However, excess FAs accumulate in the cytosol and cause lipotoxicity through the generation of diacylglycerol and ROS. Hyperglycemia also triggers ROS and advanced glycation end-product (AGE) production, culminating in cardiac glucotoxicity. Hence, the lack of fuel and the presence of lipotoxicity and glucotoxicity trigger cardiac inflammation, fibrosis and contractile dysfunction (21). In response, RAS and TGF-β systems that mediate cytokine/chemokine responses are significantly enhanced (22).
Currently, there is no single therapy for treating DCM. Treatment options revolve around dietary glycemic control, direct and indirect regulators of fatty acid metabolism, and inhibitors of factors that trigger heart failure symptoms (13, 23). Medical advancements and lifestyle interventions have contributed to reduction in cardiovascular mortality in diabetic patients. However, epidemiological studies show higher incidence of diabetic cardiomyopathy despite adjustments for hypertension, microvascular diseases, hypercholesterolemia, body mass index and other factors (24–26). Therefore, further insights into the pathological mechanisms behind the advancement of DCM are warranted and the characterization of these processes may open novel avenues for targeted therapies. Since increased extracellular matrix (ECM) protein deposition or cardiac fibrosis is a key event of DCM, we chose to focus on this area.
Extracellular Matrix (ECM) Proteins
Structural and functional alterations in the vasculature arise in the presence of chronic diabetes. Among these alterations, modifications to the extracellular matrix (ECM) and basement membrane (BM) are the structural hallmarks in target organs of diabetic complications (27). First recorded by Siperstein and colleagues in 1968, the disturbances of ECM are directly linked functional loss in target organs (28). ECM encompasses an insoluble network of collagens, fibronectin, elastins, structural glycoproteins, proteoglycan hyaluronans and integrins that provide cells with mechanical support and mediate multifarious interactions between other cells or the ECM of vascular tissues (29). The cardiac ECM consists of fibrillar collagen localized within myocardial interstitium and non-fibrillary collagen, as well as fibronectin and laminin in the myocyte basement membrane (30, 31). In the context of DCM, chronic hyperglycemic environments can initiate a cascade of signals that disrupt the balance between synthesis and breakdown of ECM components (shown in Figure 1); which, ultimately contributes to the development of LVH and hypertension-induced diastolic dysfunction (32–37). Prior to the development of cardiac dysfunction, excessive ECM protein deposition occurs in the heart muscles, accompanied by abnormal proliferation of cardiac fibroblasts, and this phenomenon is known as cardiac fibrosis (38). Cardiac fibrosis plays a critical role in the development of DCM and TGF-ß is one of the most studied mediators of this phenomenon (39, 40). Numerous studies have reported high-glucose mediated elevation in the transcription of TGF-ß genes, thereby increasing the levels of the protein and its downstream signaling (41, 42). In addition to its role in the canonical SMAD signaling pathway, TGF-ß is also the key stimulator of three known MAP pathways: ERK, JNK and p38 pathways (43–45). Evidently, TGF-ß is the chief cytokine in the regulation of ECM protein synthesis and is responsible for stimulating the production of its components including proteoglycans, fibronectin and collagen, while blocking matrix degradation (46). Moreover, in addition to TGF-ß, hyperglycemia-induced damage of endothelial cells (ECs) can activate vascular endothelial growth factor (VEGF)-mediated angiogenic responses that may further contribute to increased basement membrane (BM) thickening and ECM protein deposition (47–49).
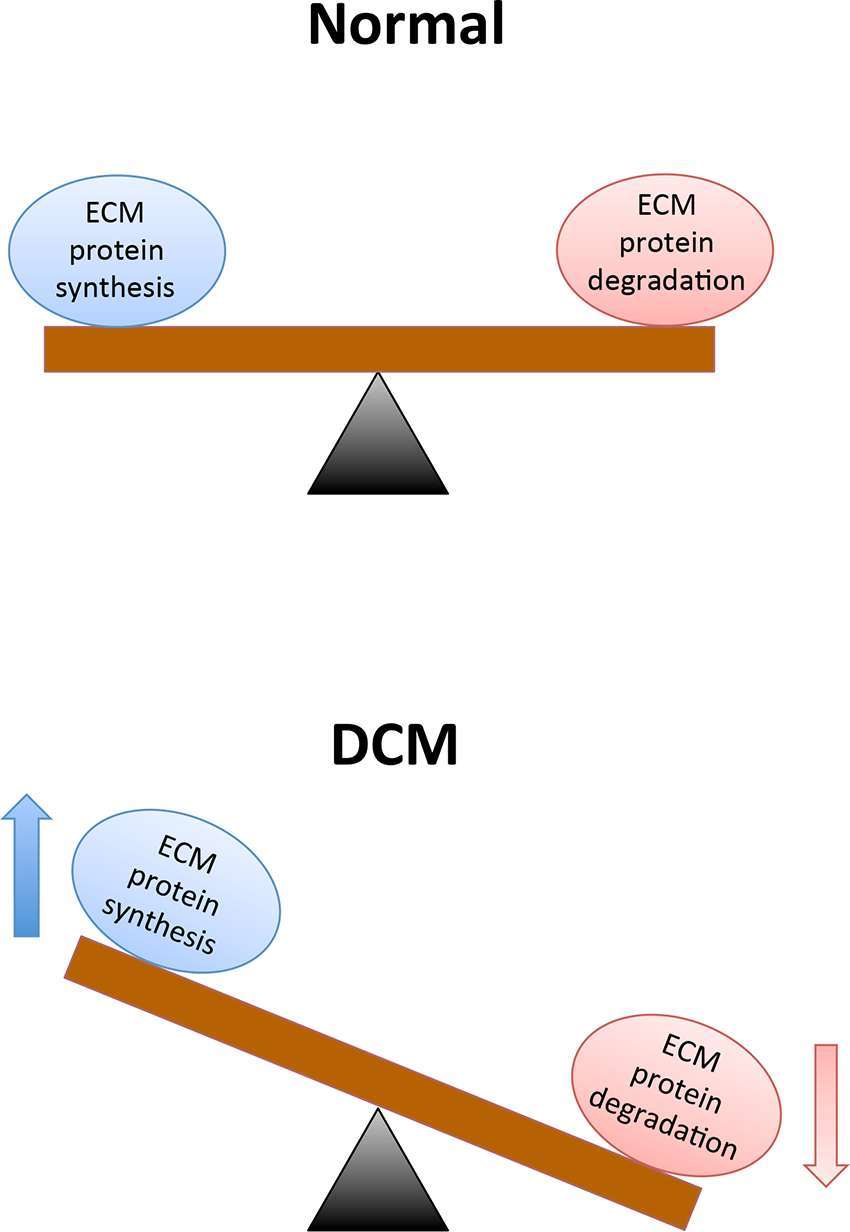
Figure 1. A chronic hyperglycemic environment causes excessive extracellular matrix protein deposition, which subsequently contributes to cardiac fibrosis and cardiac dysfunction. Legend: ECM = Extracellular matrix, and DCM = Diabetic cardiomyopathy.
Endothelial-Mesenchymal Transition (EndMT)
The entire circulatory system is lined by ECs, forming a boundary between circulating blood in the lumen and all vessel walls from the heart to small capillaries (50). ECs allow the blood to be pumped further by reducing turbulence of flow and are unambiguously known to play a critical role in maintaining overall homeostasis (50, 51). The endothelium secretes a number of factors that regulate coagulation, fibrinolysis, platelet aggregation and vascular tone (51). Hyperglycemia causes the endothelium to be exposed to a range of negative intracellular occurrences that promote endothelial dysfunction, where the endothelium loses its physiological properties (51). Fibrosis, or accumulation of fibrous connective tissue, and excess ECM in and around inflamed or damaged tissue that culminates in organ failure or death is a characteristic feature of endothelial dysfunction (52–55). Fibrosis involves proliferation of local fibroblasts and their differentiation into myofibroblasts (55). Myofibroblasts, in comparison to fibroblasts, have elevated α-smooth muscle actin (α-SMA) and an upregulated production of ECM proteins like type IV collagens (55, 56). Initially, the origin of these myofibroblasts were thought to be from local proliferating resident fibroblasts in response to factors like TGF-ß, but subsequent research suggests other cellular sources such as ECs that can form myofibroblasts (57, 58). ECs can adopt a mesenchymal phenotype and express markers characteristic of myofibroblast differentiation, which can include α-SMA, smooth muscle 22α (SM22α), vimentin, fibroblast-specific protein 1 (FSP1) and ECM proteins like fibronectin (FN) and collagen. As well, EC markers such as vascular endothelial cadherin (VE-Cadherin) and cluster of differentiation 31 (CD-31) are downregulated in these mesenchymal-like ECs (58–60). ECs undergo this phenomenon known as endothelial-mesenchymal transition (EndMT) to gain an altered differentiated phenotype and obtain invasive and migratory abilities in order to affect pathological processes in different ways (shown in Figure 2) (50). EndMT can be defined as loss of cell adhesion and actin reorganization to convert apical-basal polarity to front-end/back-end polarity resulting in change from compact, well-structured cobblestone-like shape to less organized spindle-shaped cells (60, 61). The major regulatory cytokines that stimulate EndMT are the TGF-ß superfamily of proteins including TGF-ß1 and TGF-ß2 (58, 61–63). Importance of this major regulator in the activation of EndMT has been described previously (64). TGF promotes EndMT through Smad-dependent and Smad-independent pathways like protein kinase C (PKC) (62, 65). Moreover, several studies have shown that inhibition at TGF-signaling at different stages have reduced EndMT and fibrosis in animal models (62, 64, 66). TGF-signaling play important roles in myofibroblast differentiation and ECM alterations, which favor myofibroblast transdifferentiation through altered responses to mechanical stress or transduction of growth factor signals (58). TGF is responsible for the induction of cardiac fibroblast transdifferentiation to myofibroblasts and promotes cardiac fibrosis by inducing ECM protein synthesis and reducing collagenase expression (67, 68). In general, TGF-signaling and EndMT are major contributors to generation of myofibroblasts that are key role players in the development of fibrotic diseases such as DCM.
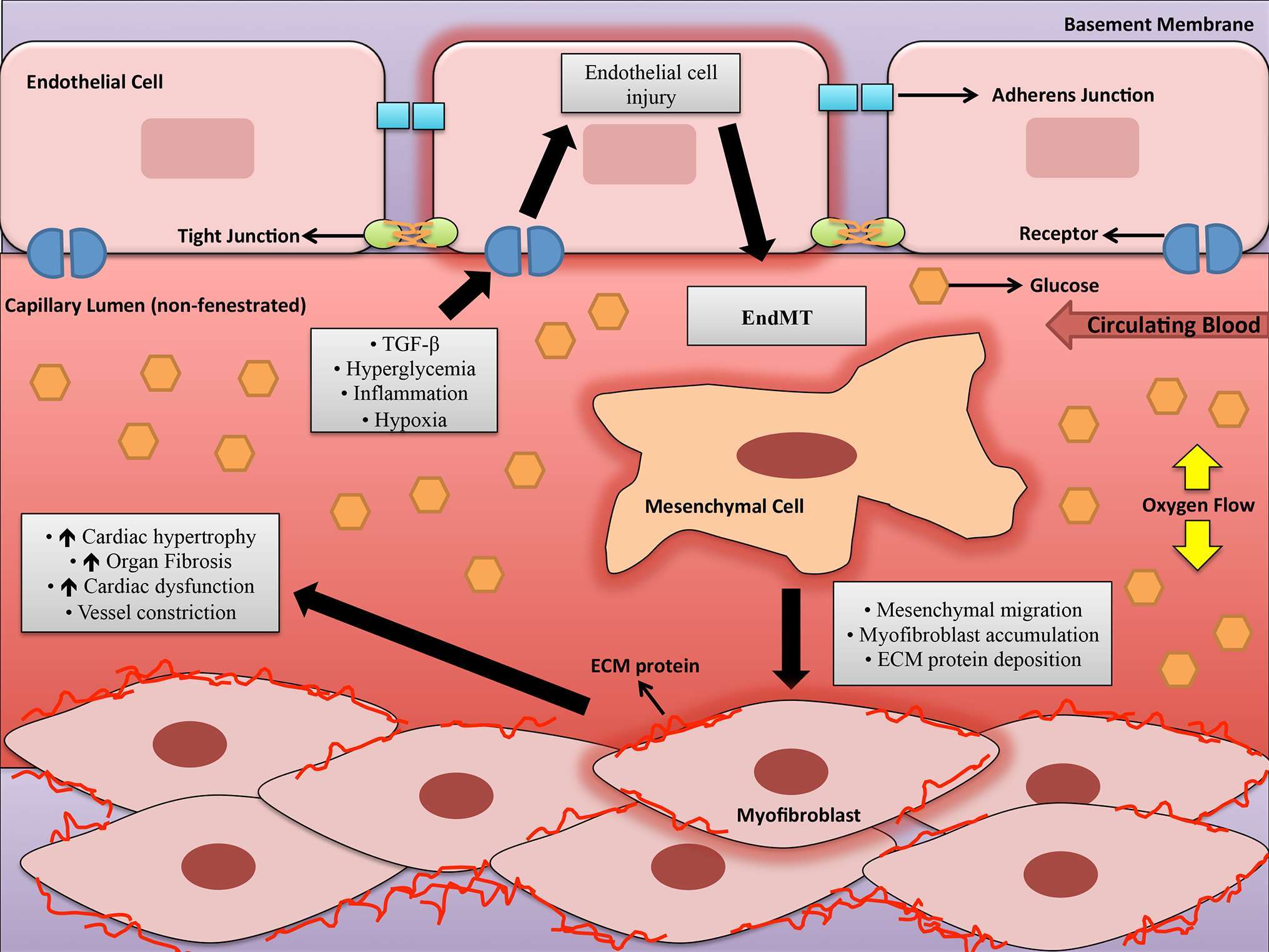
Figure 2. A schematic depicting some of the factors involved in endothelial-to-mesenchymal transition in the capillary lumen during diabetic cardiomyopathy. Legend: ECM = Extracellular matrix, EndMT = Endothelial to-mesenchymal transition, and TGF-β=Transforming growth factor-beta.
The Emergence of Epigenetics
Genetic factors are thought to be important for the development of diabetes and its various complications. In addition to genetics, environmental factors such as sedentary lifestyle, age and obesity are important for elevating the risk for the disease. In fact, there is considerable evidence indicating that the interaction between genes and the environment can influence an individual’s susceptibility to develop a chronic complication (69–71). The environmental factors can alter signaling pathways and alter gene expression through epigenetic modifications. Epigenetics can be defined as the study of heritable changes in gene expression that does not involve changes to the underlying DNA sequence. This represents a change in phenotype without a change in genotype that affects how cells read genes (72). The chromosomal DNA is tightly packaged into chromatin, and its status between transcriptionally “active” (euchromatin) and “inactive” (hetero-chromatin) in response to extracellular signals is a key aspect that can govern the expression of genes (73–75). The chromatin is composed of subunits known as nucleosomes. Each nucleosome has an octamer of histones, with two copies of histone proteins (H2A, H2B, H3 and H4), wrapped by 147 base pairs of chromosomal DNA. Post-translational modifications of these histones (PTHMs) alter their interactions with DNA and represent one of the key epigenetic regulations (76, 77). Epigenetic regulations can lead to non-heritable or heritable effects. Cells are able to respond fast to changing factors in the environment when effects are non-heritable (78). Heritable or long-term epigenetic effects occur in response to long acting stimuli and can be transmitted to memory of the offspring cells (79, 80). PTHMs, along with DNA methylation, microRNAs (miRNAs) and lncRNAs regulate chromosomal function and gene expression.
Interplay of lncRNAs and Other Epigenetic Mechanisms in Cardiac Fibrosis
With the emergence of genome-wide association studies (GWAS) in the early to late 2000s, nearly 88% of trait/disease-associated single nucleotide polymorphisms were identified to reside in non-protein coding regions (81); which, suggests that alterations in lncRNAs may be implicated in the genetic susceptibility of DCM.
LncRNAs
LncRNAs are defined as transcripts that are greater than 200 nucleotides in length and do not possess protein-coding potential (82). Based on their genomic localization, lncRNAs can be categorized as sense, anti-sense, bidirectional, enhancer, intronic, and intergenic lncRNAs (82, 83). Moreover, studies within the last decade, have provided unique insights behind the involvement of lncRNAs in a number of biological processes that include genomic imprinting (epigenetic regulation), enhancer activation, scaffold and guide for transcription and epigenetic factors, molecular sponges, and cell-cycle control (4, 82–87). In fact, the localization of the lncRNA transcript can govern its functional capabilities. For example, nuclear lncRNAs can influence gene activation by interacting with chromatin-remodelling complexes that initiates histone or DNA modifications (4, 82–86). Whereas, cytoplasmic lncRNAs are capable of secluding miRNAs (miRs) to indirectly impact protein expressions (4, 82–86).
LncRNAs and Their Implications in the Heart
Following the findings from GWAS, recent transcriptomic analyses revealed that lncRNA expression profiles differed considerably between failing and non-failing human hearts (88). In fact, out of the 18,480 total lncRNAs detected in the human heart, nearly 1249 lncRNAs (from ischemic and non-ischemic origins) were differentially expressed with heart failure (88). These findings, along with transcriptomic profiling from other studies (89, 90), opened the door for lncRNA research in cardiac pathologies. To date, there has been a multitude of lncRNAs identified in cardiovascular complications (summarized in Table 1); however, their exact mechanisms in cardiac fibrosis still require further characterization. Nevertheless, emerging studies are beginning to provide insight into some of the roles of lncRNAs in influencing other epigenetic mechanisms such as DNA methylation, histone methylation, histone acetylation and miRs in fibrosis. We will discuss some of the specific lncRNAs and their roles in DCM. However, it is to be noted that lncRNAs can interact and regulate multiple other epigenetic mechanisms such as methylation, acetylation, and miR alterations (shown in Figure 3). A concerted effort of all these pathways ultimately regulates gene expression and increased ECM production. Outlined below is a discussion of these mechanisms and their interactions with lncRNAs.
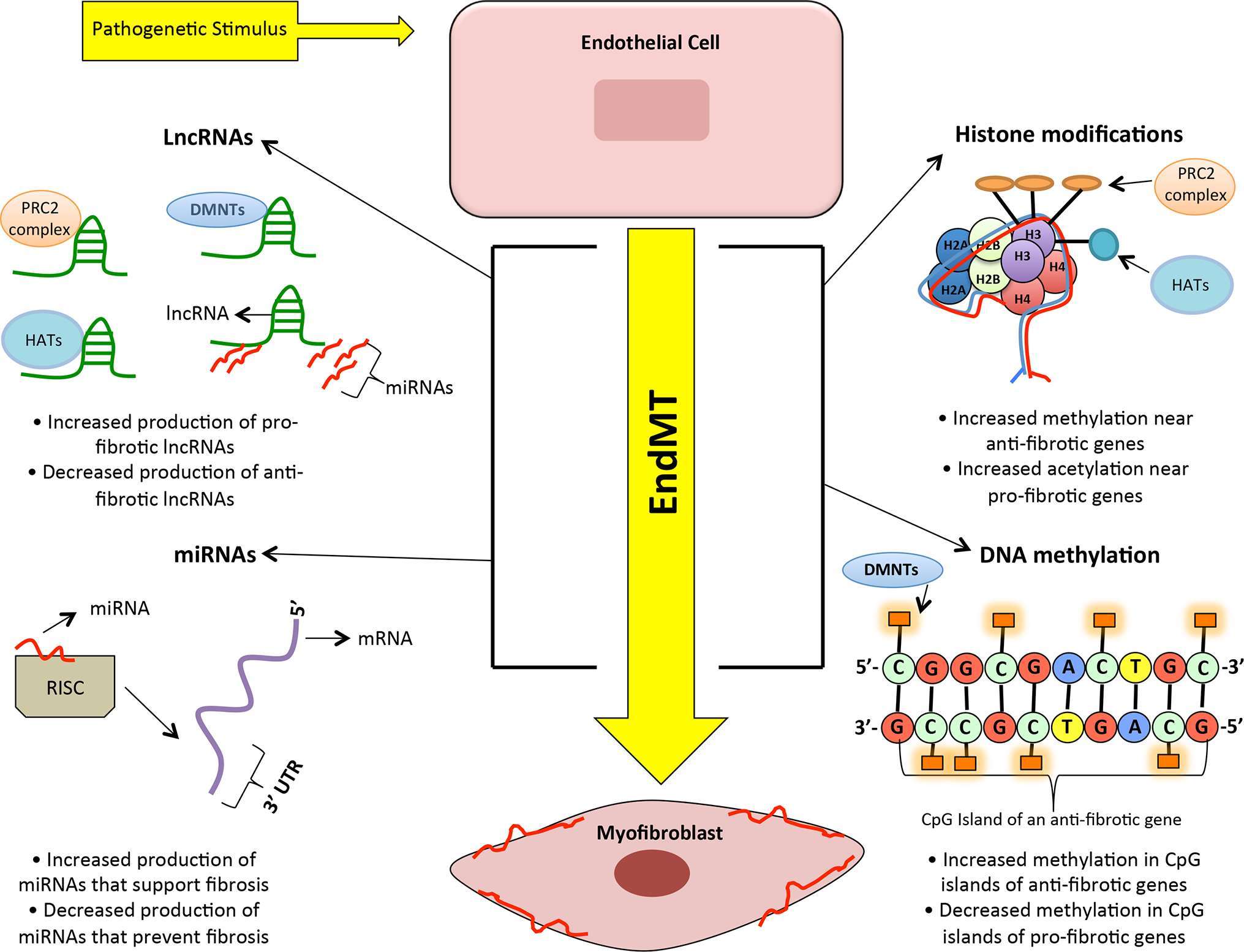
Figure 3. A simplified visual representing the interplay of many epigenetic processes that enhance endothelial-to-mesenchymal transition in diabetic cardiomyopathy. Legend: PRC2 complex = Polycomb repressive complex 2, HDAC = Histone acetyltransferases, DMNTs = DNA methyltransferases, LncRNA = Long non-coding RNAs, miRNA = microRNA, 3’ UTR = three prime untranslated region, mRNA = messenger RNA, and EndMT = endothelial to-mesenchymal transition.
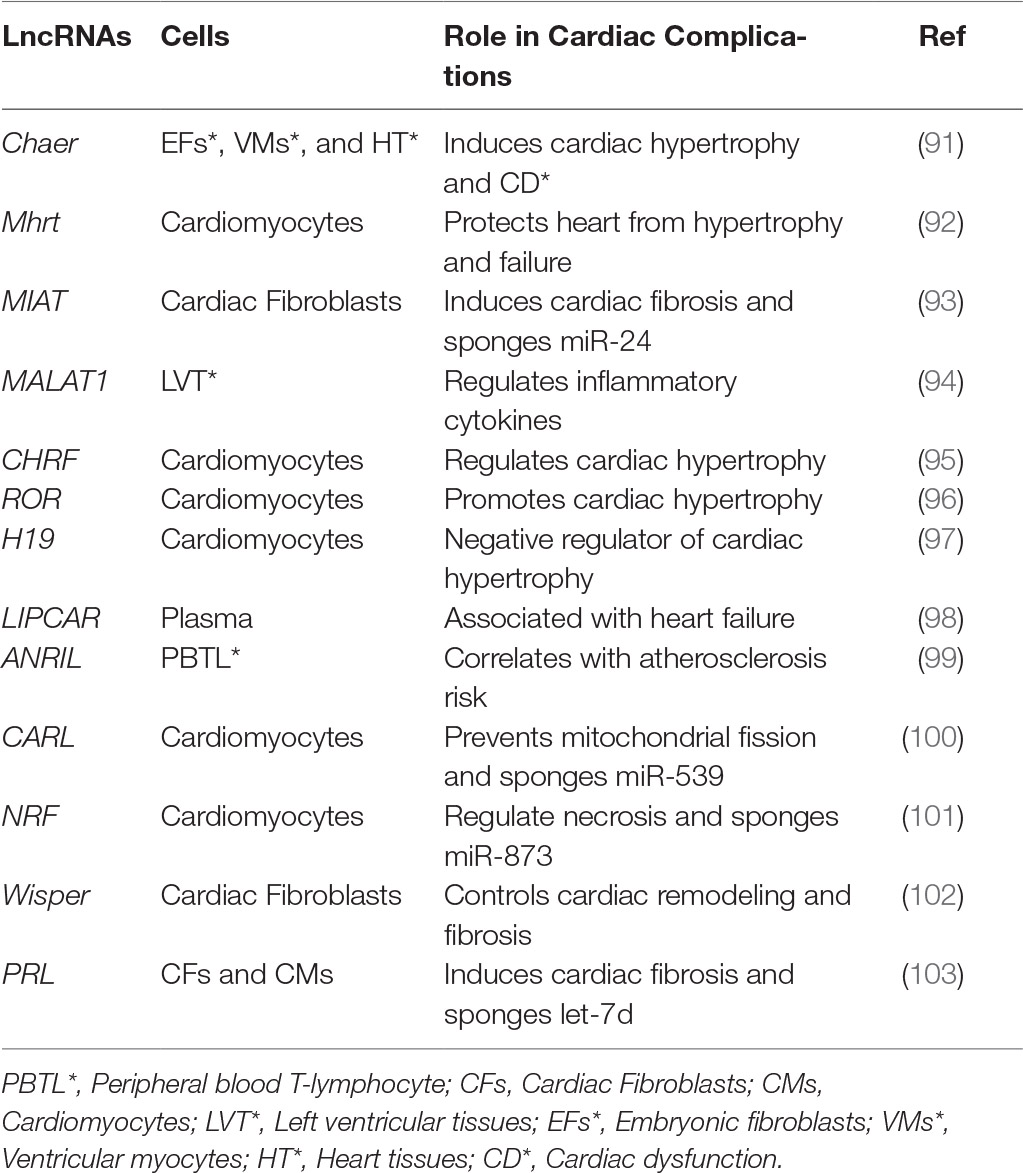
Table 1. List of the prominent lncRNAs implicated in cardiac complications.
DNA Methylation
DNA methylation is a critical epigenetic mechanism that involves the interaction between DNA methyltransferases (DMNTs) and DNA demethylases. Typically, gene silencing, via the elevated activity of DMNTs, is associated with aberrant methylation patterns of CpG dinucleotide clusters (CpG islands) in certain genes (104). In the context of cardiac fibrosis, Watson and colleagues have previously demonstrated in vitro that global DNA hypermethylation and elevated activities of DMNTs (DNMT1 and DMNT3B) were associated with hypoxic and pro-fibrotic human cardiac fibroblasts (105). In fact, silencing DMNT3B by small interfering RNAs (siRNAs) or administering the pan-DMNT inhibitor, 5-aza-2’-deoxycytidine, resulted in significant downregulations of α-SMA, collagen 1, and subsequently suppressed the pro-fibrotic effects of TGF-β (105). On the other hand, Pan et al. specifically analyzed CpG sites in the collagen type 1 alpha 1 chain (COL1A1) promoter of rat cardiac fibroblasts following TGF-β treatment and observed significant reductions in DNA methylation and DMNT activity (106). They conclude that TGF-β is capable of promoting collagen type I expression through inhibition of DMNTs in the COL1A1 promoter of cardiac fibroblasts. Moreover, Xu et al. have previously demonstrated that TGF-β also evokes aberrant methylation patterns in the promoter of the Ras-GTPase RASAL1, which in turn hinders the expression of RASAL1 (107). The subsequent reduction in RASAL1 expression allows for heightened Ras-GTP activity, which enhances EndMT and contributes to cardiac fibrosis (107). Xu and colleagues have also documented similar observations in fibrotic cardiac tissues from patients and mice (107, 108). Overall, these findings suggest that pathological stimuli can impact overall DNA methylation activity and ultimately influence the activation of pro-fibrotic genes in cardiac fibrosis.
Although the link between DNA methyltransferases and lncRNAs in cardiac fibrosis has not been made clear, previous evidence in lung fibrosis suggests that miRs (a group of small non-coding RNAs; sncRNAs) exhibit a complex regulatory relationship with DMNTs (109). In fact, Dakhlallah and colleagues revealed that several miRNAs from the miR-17 ~92 cluster targeted DNMT-1 expression and this ultimately produced a negative feedback loop (109). They also identified reduced miR-17 ~92 expressions, and increased DNMT-1 expression and promoter methylation of miR-17 ~92 in fibrotic lung tissues. Furthermore, when looking at lncRNAs, there are several evidences in neural differentiation, skeletal myoblast differentiation, colon cancer, and somatic cell reprogramming that document the capability of lncRNAs to modulate DNA methylation through direct or indirect interactions with various DMNTs (110–113). Whether these mechanistic actions of lncRNAs are present during cardiac fibrosis, requires further exploration.
Histone Methylation
Histone methylation involves the transfer of methyl groups from the methyl donor S-adenosyl methionine (SAM) to amino acid residues (lysine, arginine and histidine) by histone methyltransferases (114, 115). Methylation at the arginine and lysine residues leads to either activation or repression of transcription. Arginine residue methylation results in only activation, while methylation at lysine can lead to activation or repression of transcription. Generally, H3K4 (histone 3 methylated at lysine 4), H3K36 and H3K79 are associated with transcriptional activation and H3K9, H3K27 and H4K20 are seen in repressed regions(76). To add to the complexity of this regulation, lysine residues can be mono-methylated (m1), di-methylated (m2) or tri-methylated (m3). Furthermore, the specific actions of methyltransferases and demethylases controls chromatin accessibility to transcriptional enzymes and therefore influences protein expression. The arginine and lysine methyltransferase enzymes are part of three protein families: the protein arginine methyltransferase family, the SET-domain-containing protein family of lysine methyltransferases and the non-SET-domain DOTI-like protein (116). A recent study has demonstrated that human ECs transiently exposed to high glucose induced transcriptional activation of NFκB (p65), followed by the overexpression of inflammatory factors. These alterations were attributed to Set7-dependent monomethylation of H3K4 (117, 118). Reversal of oxidative stress through overexpression of ROS-scavenger enzymes prevented both NF-κB activation and vascular inflammation. Hence, oxidative stress plays a critical role in chromatin remodelling and gene alterations during diabetes (117, 118). Furthermore, evidences suggest involvement of important histone modifications in gene regulation that are associated with the pathogenesis of DCM. For example, the H3K79 methyltransferase DOT1L is downregulated in DCM patient samples and may regulate pathologic cardiac remodelling that ultimately contributes to eccentric hypertrophy and reactive fibrosis (119). Our lab has also described regulatory roles of tri-methylation at lysine27 (H3K27me3) catalyzed by H3K27me3 transferase EZH2 (PRC2 component) in diabetic complications (120). EZH2 is part of the polycomb repressive complex 2 (PRC2), which is a multimeric complex known to negatively regulate expression of genes including miRs. Other components of PRC2 are EED, SUZ12 and RpAp46/48 (121). This complex has been widely studied in tumor biology, where increased EZH2 promoted VEGF stimulation and subsequent angiogenesis by inhibitory methylation of anti-angiogenic factor vasohibin1 (vash1) (122). Additionally, Zhu et al. have demonstrated that miR-214–3 p is capable of targeting and reducing the expressions of EZH1 and EZH2, which subsequently attenuated the expressions of Col1a1 and Col3a1 (extracellular matrix genes in cardiac fibrosis) in mice myofibroblasts (123). On the other hand, the enforced expressions of EZH1 and EZH2 significantly contributed to elevated levels of Col1a1 and Col3a1 in myofibroblasts (123). Accumulating evidences in renal and hepatic fibrosis suggests that EZH2 plays a critical role in fibrotic development through the downregulation of PPAR-γ, Smad7 and PTEN to further enhance pathways implicated in profibrotic signalling (124, 125).
Within the last decade, various cancer- and inflammatory-based studies have identified several lncRNAs that bind to chromatin modification complexes, such as PRC2, to regulate gene expression (126–128). In the context of the heart, the lncRNA Fendrr is known to bind to both PRC2 and TrxG/MLL complexes in order to facilitate proper cardiac development (129). With this in mind, studies depicting the relationship between lncRNAs and chromatin-remodelling complexes in facilitating cardiac fibrosis are slowly beginning to emerge. For example, the lncRNA Chaer (cardiac-hypertrophy-associated epigenetic regulator) directly interacts with EZH2 to alter PRC2 targeting, which ultimately inhibits H3K27me3 at the promoter regions of genes implicated in cardiac hypertrophy (130). Klattenhoff and colleagues demonstrate that the lncRNA Bvht interacts with SUZ12 of PRC2 to mediate epigenetic regulation of cardiovascular lineage commitment (131). While, Mhrt is a cardioprotective lncRNA that is capable of protecting the heart from hypertrophy and failure by sequestering a stress-induced chromatin-remodelling factor (Brg1)— this factor is then unable to activate aberrant gene expressions (92). Moreover, in the context of lung cancer and epithelial-mesenchymal transition, Terashima and colleagues have shown through chromatin immunoprecipitation that the lncRNA MEG3 can transcriptionally repress miR-200 family genes by recruiting EZH2, and histone H3 methylation to these specific regulatory regions (132). Whether similar mechanisms exist in EndMT during DCM still remains elusive; however, previous researches have already documented unique lncRNAs in cardiovascular complications: MIAT (93), MALAT1 (94), CHRF (133), ROR (96), H19 (97), LIPCAR (98) and ANRIL (99). Using this information, identifying the chromatin-lncRNA interactions will provide additional mechanistic/functional insight behind these unique lncRNAs in cardiac fibrosis. We have previously shown that ANRIL regulates glucose-mediated upregulation of VEGF through its interactions with EZH2 and p300 (a histone acetyltransferase) in glucose-treated ECs (134). Nevertheless, of note, EZH2 can also act as a platform for the recruitment of DMNTs and directly control DNA methylation (135)—which alludes to the importance of understanding epigenetic mechanisms in its entirety.
Histone Acetylation
Histone acetylation involves either the addition or removal of acetyl groups to lysine residues, which is facilitated by histone acetyltransferases (HATs) and histone deacetylases (HDACs), respectively (136, 137). The interplay between HATs and HDACs can govern gene regulation in which elevated acetylation at specific lysine residues (lysines 9, 14, 18 and 56 in histone H3 and lysines 5, 8, 13 and 16 in histone H4) allows for chromatin relaxation and heightened transcription factor recruitment—contributing to gene activation (70, 138). As well, HATs and HDACs can also directly modify regulatory proteins and transcription factors (138). Previous work by our lab demonstrated that the transcriptional coactivator and HAT, p300 was markedly expressed in the diabetic animal heart and an increase in p300 activity led to an upregulation of VEGF, endothelin-1 (ET-1), and FN—molecules that are implicated in DCM (139, 140). In fact, silencing p300 prevented the diabetes-induced expression of VEGF, ET-1, and FN (139, 140). Our findings also demonstrated that p300 had an increased binding to ET-1 and FN promoters in large vessel ECs cultured with high glucose, which was also associated with augmented histone acetylation, H2AX phosphorylation, activation of several transcription factors, and mRNA expressions of ECM proteins and vasoactive factors (139). Similarly, Ghosh and colleagues have further indicated that p300 is an essential coactivator of pro-fibrotic signalling and is dramatically elevated during cardiac EndMT (141). Moreover, in the context of histone deacetylases, silent mating type information regulation 2 homolog (SIRTs) are important NAD-dependent deacetylases that are involved in a number of cellular processes such as apoptosis, and fatty acid and glucose metabolism (142–144). In vitro and in vivo findings from our lab have previously demonstrated that SIRT1 (a type III HDAC) activity is significantly reduced under chronic hyperglycemic environments in ECs and the subsequent reduction of SIRT1 drove the formation of reactive oxygen species (ROS), which is mediated by FOXO1(forkhead box protein O1) acetylation through elevated p300 activity (145). SIRT1 is also capable of regulating TGF-β1 and ET-1 expressions by p300, while SIRT1 overexpression can prevent diabetes-induced FN upregulation (146). Rizk and colleagues have also shown similar findings where elevating SIRT1 levels, with the administration of L-arginine, significantly prevented diabetes-induced myocardial fibrosis in male Wistar rats (146). Along with their histopathological findings, Rizk and colleagues observed a reduction in the expression of cardiac fibrotic markers (FN, TGF-β, brain naturetic peptide, and connective tissue growth factor) following SIRT1 upregulation (146). Other studies are also in agreement that SIRT1 activation protects against cardiovascular damage (147–149).
Although lncRNAs such as ANRIL (134) and Khps1 (150) have been shown to interact with p300 in microvascular ECs and cancer cell lines, respectively, this form of interaction has not yet been documented in cardiac fibrosis. However, our lab has previously identified that miR-200b (a sncRNA) is capable of mediating EndMT and VEGF through direct targeting of p300 in heart and retinal tissues of diabetic mice and rats (151–153). Additional work by Shehadeh and colleagues have reported the ability of miR-20a to bind to the 3’ untranslated region (UTR) of p300 and directly repress the expression of p300—subsequently reducing pro-angiogenic gene expressions implicated in cardiac hypertrophy (154). As for interactions between SIRTs and lncRNAs, recent evidence suggests that the Sirt1 antisense (AS) lncRNA can inhibit muscle formation by activating Sirt1 and impeding the function of miR-34a by competitively interacting with the 3’ UTR of the Sirt1 transcript (130). On a similar note, several miRs that are capable of modulating SIRT1 activity have been documented previously (155). Nevertheless, further follow-up is required for the role of Sirt1 AS lncRNA in cardiac fibrosis.
miRNAs (MiRs)
As previously mentioned, miRs belong to the class of small non-coding RNAs that are ~20–25 nucleotides in size (156, 157). MiRs are synthesized by RNA polymerase II, processed to precursors in the nucleus by RNAse III Drosha and DiGeorge syndrome critical region 8 (DGCR8) and then exported into cytoplasm by exportin 5 (157, 158). Dicer further processes miRs in the cytoplasm into functionally active miRNAs (159). From there, with the help of argonaute proteins, active miRNAs are incorporated into the RNA-induced silencing complex (RISC) (157–159). Following the complete formation of RISC, miRs are then capable of binding to complementary sequences within the 3’ UTR of targeted mRNAs, which subjects these transcripts to degradation or repressed translation—ultimately, inhibiting protein expression (157–159). From our lab, we have previously reported hyperglycemia-induced alterations of miRs in ECs and in numerous tissues impacted by chronic diabetes: miR-1, miR-133a, miR-146a, miR-195, miR-200b and miR-320 (120, 152, 153, 160–164). Earlier findings from our previous reports also demonstrate a novel regulation mechanism between PRC2 and miRNAs through histone methylation in diabetic complications (120, 153). As well, we show that the repression of miRs-146a and 200b plays an integral role in enhancing glucose-induced EndMT (152, 162). Moreover, within the last decade, a multitude of studies have emerged that investigate the regulatory capabilities of miRNAs in diabetes and its complications (165–167), and explore the changes of miRNAs upon various treatments (168–170). In the context of the heart, miRs-1, 22, 29, 31 101, 133, and 489 are few examples of the many miRs that may be functioning as inhibitors of cardiac fibrosis and hypertrophy (133, 171–175). Whereas, miRs- 21, 34, 132, 199b, 208a and 212 have been reported to enhance cardiac complications (87, 176–180).
One of the important functions of lncRNAs is to act as a molecular sponge to certain miRs, which can ultimately hinder the expression of these small non-coding molecules (181). For instance, the cardiac-apoptosis related lncRNA (CARL) has been shown to block the actions of miR-539 by acting as an endogenous sponge that consequently augments the expression of PHB2 (a miR-539 target) and prevents cardiomyocyte apoptosis and mitochondrial fission (133). Similarly, the lncRNA NRF (necrosis-related factor) can regulate necrosis in cardiomyocytes by sponging miR-873, which is an important miR implicated in the translational repression of RIPK1/RIPK3 (130). Furthermore, in cardiac fibroblasts, MIAT (myocardial infarction associated transcript) can block the actions of miR-24 (a critical regulator in TGF-β1 activation) by its sponging capabilities, and induce cardiac fibrosis (182). The findings from the study by Qu and colleagues identified and characterized MIAT as the first profibrotic lncRNA involved in cardiac fibrosis. Moreover, a recent study by Liang et al. has demonstrated that the pro-fibrotic lncRNA (PFL) can act as a competitive endogenous RNA for miR let-7d (103). Specifically, the overexpression of PFL stimulated proliferation, fibroblast-myofibroblast transition and fibrogenesis in mice cardiac fibroblasts by reducing the activity and expression of let-7D (103). Micheletti and colleagues have also recently shown that the lncRNA Wisper can regulate cardiac remodeling and fibrosis; however, whether Wisper can interact with miRs remains to be determined (102). Additional characterization of other profibrotic lncRNAs will provide significant insight behind the pathogenetic mechanisms of cardiac pathologies. Therefore, establishing a database with the fibrotic capabilities of previously documented lncRNAs in other diseases could help achieve this. For example, in the context of liver fibrosis, Hotair has been shown to act as an endogenous sponge for miR-148b to facilitate DMNT1 expression (183). Further follow-up on the sponging capabilities of Hotair in a cardiac-specific context may provide important information behind the precise nature of these regulatory mechanisms behind fibrosis and other cardiac complications.
Future Direction
Our review summarized the key epigenetic mechanisms implicated in DCM fibrosis; as well, we have provided unique insights behind lncRNAs and their impact on DNA methylation, histone modifications, and miRs. Although direct evidence for the mechanisms of lncRNAs is still limited in the context of cardiac fibrosis, emerging studies are beginning to provide further understanding of the regulatory capabilities of lncRNAs during DCM. Moreover, the critical implications of lncRNAs in a multitude of biological processes make it a valuable target for therapeutic applications, which necessitates the need for additional research behind lncRNA and epigenetic protein interactions. Therefore, understanding these epigenetic mechanisms for lncRNAs in cardiac fibrosis will not only provide critical information behind the intricate gene network, but it will also facilitate the development of better-targeted treatment strategies that take into account the regulatory gene network in its entirety. We hope that our review will allow for critical discussions and further experimentation that will extend the current findings of these epigenetic mechanisms on lncRNAs during cardiac fibrosis in DCM.
Data statement
The figures and table presented in this review were drawn by Saumik Biswas and Subrata Chakrabarti. We provide permission to Frontiers to publish our illustration in all formats (i.e. print and digital).
Author Contributions
Writing of manuscript: SB, AT, SC. Figure drawings: SB, SC.
Funding
The Heart and Stroke Foundation of Ontario (G-16-00012556) and the Canadian Diabetes Association (OG-3-15-4890-SC) supported the work presented in this review.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would also like to acknowledge past and current members in the Chakrabarti lab for their contributions in the advancement of diabetes research.
References
1. Esteller M. Non-coding RNAs in human disease. Nat Rev Genet (2011) 12(12):861–74. doi: 10.1038/nrg3074
2. Rinn JL. lncRNAs: linking RNA to chromatin. Cold Spring Harb Perspect Biol (2014) 6(8):a018614. doi: 10.1101/cshperspect.a018614
3. Thum T, Condorelli G. Long noncoding RNAs and microRNAs in cardiovascular pathophysiology. Circ Res (2015) 116(4):751–62. doi: 10.1161/CIRCRESAHA.116.303549
4. Quinn JJ, Chang HY. Unique features of long non-coding RNA biogenesis and function. Nat Rev Genet (2016) 17(1):47–62. doi: 10.1038/nrg.2015.10
5. Winer N, Sowers JR. Epidemiology of diabetes. J Clin Pharmacol (2004) 44(4):397–405. doi: 10.1177/0091270004263017
6. Bugger H, Abel ED. Rodent models of diabetic cardiomyopathy. Dis Model Mech (2009) 2(9-10):454–66. doi: 10.1242/dmm.001941
7. Rubler S, Dlugash J, Yuceoglu YZ, Kumral T, Branwood AW, Grishman A. New type of cardiomyopathy associated with diabetic glomerulosclerosis. Am J Cardiol (1972) 30(6):595–602. doi: 10.1016/0002-9149(72)90595-4
8. Boudina S, Abel ED, Cardiomyopathy D. Causes and effects. Rev Endocr Metab Disord (2010) 11:31–9.
9. Lorenzo-Almorós A, Tuñón J, Orejas M, Cortés M, Egido J, Lorenzo Ó. Diagnostic approaches for diabetic cardiomyopathy. Cardiovasc Diabetol (2017) 16(1):1–14. doi: 10.1186/s12933-017-0506-x
10. Aneja A, Tang WH, Bansilal S, Garcia MJ, Farkouh ME. Diabetic cardiomyopathy: insights into pathogenesis, diagnostic challenges, and therapeutic options. Am J Med (2008) 121(9):748–57. doi: 10.1016/j.amjmed.2008.03.046
11. Goldin A, Beckman JA, Schmidt AM, Creager MA. Advanced glycation end products: sparking the development of diabetic vascular injury. Circulation (2006) 114(6):597–605. doi: 10.1161/CIRCULATIONAHA.106.621854
12. Palmieri V, Tracy RP, Roman MJ, Liu JE, Best LG, Bella JN, et al. Relation of left ventricular hypertrophy to inflammation and albuminuria in adults with type 2 diabetes: the strong heart study. Diabetes Care (2003) 26(10):2764–9. doi: 10.2337/diacare.26.10.2764
13. Hayat SA, Patel B, Khattar RS, Malik RA. Diabetic cardiomyopathy: mechanisms, diagnosis and treatment. Clin Sci (2004) 107(6):539–57. doi: 10.1042/CS20040057
14. Ernande L, Bergerot C, Rietzschel ER, de Buyzere ML, Thibault H, Pignonblanc PG, et al. Diastolic dysfunction in patients with type 2 diabetes mellitus: is it really the first marker of diabetic cardiomyopathy? J Am Soc Echocardiogr (2011) 24(11):1268–75. doi: 10.1016/j.echo.2011.07.017
15. Taegtmeyer H, Mcnulty P, Young ME. Adaptation and maladaptation of the heart in diabetes: Part I: general concepts. Circulation (2002) 105(14):1727–33. doi: 10.1161/01.CIR.0000012466.50373.E8
16. Wende AR, Abel ED. Lipotoxicity in the heart. Biochim Biophys Acta (2010) 1801(3):311–9. doi: 10.1016/j.bbalip.2009.09.023
17. Spector KS. Diabetic cardiomyopathy. Clin Cardiol (1998) 21(12):885–7. doi: 10.1002/clc.4960211205
18. Tziakas DN, Chalikias GK, Kaski JC. Epidemiology of the diabetic heart. Coron Artery Dis (2005) 16(Suppl 1):S3–10. doi: 10.1097/00019501-200511001-00002
19. Cai L, Wang Y, Zhou G, Chen T, Song Y, Li X, et al. Attenuation by metallothionein of early cardiac cell death via suppression of mitochondrial oxidative stress results in a prevention of diabetic cardiomyopathy. J Am Coll Cardiol (2006) 48(8):1688–97. doi: 10.1016/j.jacc.2006.07.022
20. Lopaschuk GD. Metabolic abnormalities in the diabetic heart. Heart Fail Rev (2002) 7(2):149–59. doi: 10.1023/A:1015328625394
21. Frustaci A, Kajstura J, Chimenti C, Jakoniuk I, Leri A, Maseri A, et al. Myocardial cell death in human diabetes. Circ Res (2000) 87(12):1123–32. doi: 10.1161/01.RES.87.12.1123
22. Bugger H, Abel ED. Molecular mechanisms of diabetic cardiomyopathy. Diabetologia (2014) 57(4):660–71. doi: 10.1007/s00125-014-3171-6
23. Bayeva M, Sawicki KT, Ardehali H. Taking diabetes to heart-deregulation of myocardial lipid metabolism in diabetic cardiomyopathy. J Am Heart Assoc (2013) 2(6):e000433. doi: 10.1161/JAHA.113.000433
24. Kannel WB, Hjortland M, Castelli WP. Role of diabetes in congestive heart failure: the Framingham study. Am J Cardiol (1974) 34(1):29–34. doi: 10.1016/0002-9149(74)90089-7
25. Devereux RB, Roman MJ, Paranicas M, O'Grady MJ, Lee ET, Welty TK, et al. Impact of diabetes on cardiac structure and function: the strong heart study. Circulation (2000) 101(19):2271–6. doi: 10.1161/01.CIR.101.19.2271
26. Bertoni AG, Tsai A, Kasper EK, Brancati FL. Diabetes and idiopathic cardiomyopathy: a nationwide case-control study. Diabetes Care (2003) 26(10):2791–5. doi: 10.2337/diacare.26.10.2791
27. Brownlee M, Spiro RG. Biochemistry of the basement membrane in diabetes mellitus. Adv Exp Med Biol (1979) 124:141–56.
28. Siperstein MD, Unger RH, Madison LL. Studies of muscle capillary basement membranes in normal subjects, diabetic, and prediabetic patients. J. Clin. Invest. (1968) 47(9):1973–99. doi: 10.1172/JCI105886
29. Hayden MR, Tyagi SC. The central role of vascular extracellular matrix and basement membrane remodeling in metabolic syndrome and type 2 diabetes: the matrix preloaded. Cardiovasc Diabetol (2005) 4(1):1–20. doi: 10.1186/1475-2840-4-1
30. Eghbali M, Weber KT. Collagen and the myocardium: fibrillar structure, biosynthesis and degradation in relation to hypertrophy and its regression. Mol Cell Biochem (1990) 96(1):1–14. doi: 10.1007/BF00228448
31. Bishop JE, Laurent GJ. Collagen turnover and its regulation in the normal and hypertrophying heart. Eur Heart J (1995) 16(suppl C):38–44. doi: 10.1093/eurheartj/16.suppl_C.38
32. Regan TJ, Wu CF, Yeh CK, Oldewurtel HA, Haider B. Myocardial composition and function in diabetes. The effects of chronic insulin use. Circ Res (1981) 49(6):1268–77. doi: 10.1161/01.RES.49.6.1268
33. Shapiro LM, Leatherdale BA, Mackinnon J, Fletcher RF. Left ventricular function in diabetes mellitus. II: Relation between clinical features and left ventricular function. Br Heart J (1981) 45(2):129–32. doi: 10.1136/hrt.45.2.129
34. Nishikawa T, Giardino I, Edelstein D, Brownlee M. Changes in diabetic retinal matrix protein mRNA levels in a common transgenic mouse strain. Curr Eye Res (2000) 21(1):581–7. doi: 10.1076/0271-3683(200007)2111-ZFT581
35. Roy S, Maiello M, Lorenzi M. Increased expression of basement membrane collagen in human diabetic retinopathy. J Clin Invest (1994) 93(1):438–42. doi: 10.1172/JCI116979
36. Spirin KS, Saghizadeh M, Lewin SL, Zardi L, Kenney MC, Ljubimov AV. Basement membrane and growth factor gene expression in normal and diabetic human retinas. Curr Eye Res (1999) 18(6):490–9. doi: 10.1076/ceyr.18.6.490.5267
37. Arden GB, Sidman RL, Arap W, Schlingemann RO. Spare the rod and spoil the eye. Br J Ophthalmol (2005) 89(6):764–9. doi: 10.1136/bjo.2004.062547
38. Yue Y, Meng K, Pu Y, Zhang X. Transforming growth factor beta (TGF-β) mediates cardiac fibrosis and induces diabetic cardiomyopathy. Diabetes Res Clin Pract (2017) 133:124–30. doi: 10.1016/j.diabres.2017.08.018
39. Gao Y, Kang L, Li C, Wang X, Sun C, Li Q, et al. Resveratrol ameliorates diabetes-induced cardiac dysfunction through AT1R-ERK/p38 MAPK signaling pathway. Cardiovasc Toxicol (2016) 16(2):130–7. doi: 10.1007/s12012-015-9321-3
40. Suematsu Y, Miura S, Goto M, Matsuo Y, Arimura T, Kuwano T, et al. LCZ696, an angiotensin receptor-neprilysin inhibitor, improves cardiac function with the attenuation of fibrosis in heart failure with reduced ejection fraction in streptozotocin-induced diabetic mice. Eur J Heart Fail (2016) 18(4):386–93. doi: 10.1002/ejhf.474
41. Iwano M, Kubo A, Nishino T, Sato H, Nishioka H, Akai Y, et al. Quantification of glomerular TGF-beta 1 mRNA in patients with diabetes mellitus. Kidney Int (1996) 49(4):1120–6. doi: 10.1038/ki.1996.162
42. Dagher Z, Gerhardinger C, Vaz J, Goodridge M, Tecilazich F, Lorenzi M. The increased transforming growth factor-β signaling induced by diabetes protects retinal vessels. Am J Pathol (2017) 187(3):627–38. doi: 10.1016/j.ajpath.2016.11.007
43. Li ZL, Shi Y, Ding Y, Ran Y, Le G. Dietary oxidized tyrosine (O-Tyr) stimulates TGF-β1-induced extracellular matrix production via the JNK/p38 signaling pathway in rat kidneys. Amino Acids (2017) 49(2):241–60. doi: 10.1007/s00726-016-2353-6
44. Ferreira RR, de Souza EM, de Oliveira FL, Ferrão PM, Gomes LH, Mendonça-Lima L, et al. Proteins involved on TGF-β pathway are up-regulated during the acute phase of experimental Chagas disease. Immunobiology (2016) 221(5):587–94. doi: 10.1016/j.imbio.2016.01.009
45. Wojciechowski MC, Mahmutovic L, Shu DY, Lovicu FJ. ERK1/2 signaling is required for the initiation but not progression of TGFβ-induced lens epithelial to mesenchymal transition (EMT). Exp Eye Res (2017) 159:98–113. doi: 10.1016/j.exer.2017.03.012
46. Rivarola EW, Moyses-Neto M, Dantas M, da-Silva CG, Volpini R, Coimbra TM. Transforming growth factor beta activity in urine of patients with type 2 diabetes and diabetic nephropathy. Braz J Med Biol Res (1999) 32(12):1525–8. doi: 10.1590/S0100-879X1999001200011
47. Khan ZA, Chakrabarti S. Growth factors in proliferative diabetic retinopathy. Exp Diabesity Res (2003) 4(4):287–301. doi: 10.1155/EDR.2003.287
48. Kuiper EJ, Hughes JM, van Geest RJ, Vogels IM, Goldschmeding R, van Noorden CJ, et al. Effect of VEGF-A on expression of profibrotic growth factor and extracellular matrix genes in the retina. Invest Ophthalmol Vis Sci (2007) 48(9):4267–76. doi: 10.1167/iovs.06-0804
49. Branton MH, Kopp JB. TGF-β and fibrosis. Microbes and Infection (1999) 1(15):1349–65. doi: 10.1016/S1286-4579(99)00250-6
50. Lin F, Wang N, Zhang TC. The role of endothelial-mesenchymal transition in development and pathological process. IUBMB Life (2012) 64(9):717–23. doi: 10.1002/iub.1059
51. Hadi HA, Suwaidi JA. Endothelial dysfunction in diabetes mellitus. Vasc Health Risk Manag (2007) 3(6):853–76.
52. Karsdal MA, Manon-Jensen T, Genovese F, Kristensen JH, Nielsen MJ, Sand JM, et al. Novel insights into the function and dynamics of extracellular matrix in liver fibrosis. Am J Physiol Gastrointest Liver Physiol (2015) 308(10):G807–30. doi: 10.1152/ajpgi.00447.2014
53. Rockey DC, Bell PD, Hill JA. Fibrosis--a common pathway to organ injury and failure. N Engl J Med (2015) 372(12):1138–49. doi: 10.1056/NEJMra1300575
54. Wynn TA. Common and unique mechanisms regulate fibrosis in various fibroproliferative diseases. J Clin Invest (2007) 117(3):524–9. doi: 10.1172/JCI31487
55. Ho YY, Lagares D, Tager AM, Kapoor M. Fibrosis--a lethal component of systemic sclerosis. Nat Rev Rheumatol (2014) 10(7):390–402. doi: 10.1038/nrrheum.2014.53
56. Hinz B, Phan SH, Thannickal VJ, Prunotto M, Desmoulière A, Varga J, et al. Recent developments in myofibroblast biology: paradigms for connective tissue remodeling. Am J Pathol (2012) 180(4):1340–55. doi: 10.1016/j.ajpath.2012.02.004
57. Postlethwaite AE, Shigemitsu H, Kanangat S. Cellular origins of fibroblasts: possible implications for organ fibrosis in systemic sclerosis. Curr Opin Rheumatol (2004) 16(6):733–8. doi: 10.1097/01.bor.0000139310.77347.9c
58. Pardali E, Sanchez-Duffhues G, Gomez-Puerto M, Ten Dijke P. TGF-β-Induced Endothelial-Mesenchymal Transition in Fibrotic Diseases. Int J Mol Sci (2017) 18(10):2157–22. doi: 10.3390/ijms18102157
59. Piera-Velazquez S, Li Z, Jimenez SA. Role of endothelial-mesenchymal transition (EndoMT) in the pathogenesis of fibrotic disorders. Am J Pathol (2011) 179(3):1074–80. doi: 10.1016/j.ajpath.2011.06.001
60. Sánchez-Duffhues G, García de Vinuesa A, Ten Dijke P. Endothelial-to-mesenchymal transition in cardiovascular diseases: Developmental signaling pathways gone awry. Dev Dyn (2018) 247(3):1–17. doi: 10.1002/dvdy.24589
61. Medici D. Endothelial-mesenchymal transition in regenerative medicine. Stem Cells Int (2016) 2016(10):1–7. doi: 10.1155/2016/6962801
62. Medici D, Potenta S, Kalluri R. Transforming growth factor-β2 promotes Snail-mediated endothelial-mesenchymal transition through convergence of Smad-dependent and Smad-independent signalling. Biochem J (2011) 437(3):515–20. doi: 10.1042/BJ20101500
63. Milani S, Herbst H, Schuppan D, Stein H, Surrenti C. Transforming growth factors beta 1 and beta 2 are differentially expressed in fibrotic liver disease. Am J Pathol (1991) 139(6):1221–9.
64. Cooley BC, Nevado J, Mellad J, Yang D, Hilaire CS, Negro A, et al. TGF-β signaling mediates endothelial-to-mesenchymal transition (EndMT) during vein graft remodeling. Sci Transl Med (2014) 6(227):227ra34. doi: 10.1126/scitranslmed.3006927
65. Li Z, Jimenez SA. Protein kinase Cδ and c-Abl kinase are required for transforming growth factor β induction of endothelial-mesenchymal transition in vitro. Arthritis Rheum (2011) 63(8):2473–83. doi: 10.1002/art.30317
66. Kokudo T, Suzuki Y, Yoshimatsu Y, Yamazaki T, Watabe T, Miyazono K. Snail is required for TGFbeta-induced endothelial-mesenchymal transition of embryonic stem cell-derived endothelial cells. J Cell Sci (2008) 121(Pt 20):3317–24. doi: 10.1242/jcs.028282
67. Dobaczewski M, Bujak M, Li N, Gonzalez-Quesada C, Mendoza LH, Wang XF, et al. Smad3 signaling critically regulates fibroblast phenotype and function in healing myocardial infarction. Circ Res (2010) 107(3):418–28. doi: 10.1161/CIRCRESAHA.109.216101
68. Bujak M, Ren G, Kweon HJ, Dobaczewski M, Reddy A, Taffet G, et al. Essential role of smad3 in infarct healing and in the pathogenesis of cardiac remodeling. Circulation (2007) 116(19):2127–38. doi: 10.1161/CIRCULATIONAHA.107.704197
69. Keating ST, El-Osta A. Epigenetic changes in diabetes. Clin. Genet. (2013) 84(1):1–10. doi: 10.1111/cge.12121
70. Pirola L, Balcerczyk A, Okabe J, El-Osta A. Epigenetic phenomena linked to diabetic complications. Nat Rev Endocrinol (2010) 6(12):665–75. doi: 10.1038/nrendo.2010.188
71. Ling C, Groop L. Epigenetics: a molecular link between environmental factors and type 2 diabetes. Diabetes (2009) 58(12):2718–25. doi: 10.2337/db09-1003
72. Goldberg AD, Allis CD, Bernstein E. Epigenetics: a landscape takes shape. Cell (2007) 128(4):635–8. doi: 10.1016/j.cell.2007.02.006
73. James LI, Frye SV. Targeting chromatin readers. Clin Pharmacol Ther (2013) 93(4):312–4. doi: 10.1038/clpt.2013.6
74. Grewal SI, Jia S. Heterochromatin revisited. Nat Rev Genet (2007) 8(1):35–46. doi: 10.1038/nrg2008
75. Bush KM, Yuen BT, Barrilleaux BL, Riggs JW, O'Geen H, Cotterman RF, et al. Endogenous mammalian histone H3.3 exhibits chromatin-related functions during development. Epigenetics Chromatin (2013) 6(1):7–16. doi: 10.1186/1756-8935-6-7
76. Kouzarides T. Chromatin modifications and their function. Cell (2007) 128(4):693–705. doi: 10.1016/j.cell.2007.02.005
77. Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet (2012) 13(7):484–92. doi: 10.1038/nrg3230
78. Karaguzel G, Ozer S, Akcurin S, Turkkahraman D, Bircan I. Type 1 diabetes-related epidemiological, clinical and laboratory findings. An evaluation with special regard to autoimmunity in children. Saudi Med J (2007) 28(4):584–9.
79. Fetita LS, Sobngwi E, Serradas P, Calvo F, Gautier JF. Consequences of fetal exposure to maternal diabetes in offspring. J Clin Endocrinol Metab (2006) 91(10):3718–24. doi: 10.1210/jc.2006-0624
80. Allis CD, Jenuwein T, Reinberg D. Epigenetics. Vol. 502. Cold Spring Habor: Inglis, John (2007). p. 1–14.
81. Hindorff LA, Sethupathy P, Junkins HA, Ramos EM, Mehta JP, Collins FS, et al. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci USA (2009) 106(23):9362–7. doi: 10.1073/pnas.0903103106
82. Rinn JL, Chang HY. Genome regulation by long noncoding RNAs. Annu Rev Biochem (2012) 81(1):145–66. doi: 10.1146/annurev-biochem-051410-092902
83. Devaux Y, Zangrando J, Schroen B, Creemers EE, Pedrazzini T, Chang CP, et al. Long noncoding RNAs in cardiac development and ageing. Nat Rev Cardiol (2015) 12(7):415–25. doi: 10.1038/nrcardio.2015.55
84. Mercer TR, Dinger ME, Mattick JS. Long non-coding RNAs: insights into functions. Nat Rev Genet (2009) 10(3):155–9. doi: 10.1038/nrg2521
85. Wapinski O, Chang HY. Long noncoding RNAs and human disease. Trends Cell Biol (2011) 21(6):354–61. doi: 10.1016/j.tcb.2011.04.001
86. Kung JT, Colognori D, Lee JT. Long noncoding RNAs: past, present, and future. Genetics (2013) 193(3):651–69. doi: 10.1534/genetics.112.146704
87. Ni YG, Berenji K, Wang N, Oh M, Sachan N, Dey A, et al. Foxo transcription factors blunt cardiac hypertrophy by inhibiting calcineurin signaling. Circulation (2006) 114(11):1159–68. doi: 10.1161/CIRCULATIONAHA.106.637124
88. Yang KC, Yamada KA, Patel AY, Topkara VK, George I, Cheema FH, et al. Deep RNA sequencing reveals dynamic regulation of myocardial noncoding RNAs in failing human heart and remodeling with mechanical circulatory support. Circulation (2014) 129(9):1009–21. doi: 10.1161/CIRCULATIONAHA.113.003863
89. Ounzain S, Micheletti R, Beckmann T, Schroen B, Alexanian M, Pezzuto I, et al. Genome-wide profiling of the cardiac transcriptome after myocardial infarction identifies novel heart-specific long non-coding RNAs. Eur Heart J (2015) 36(6):353–68. doi: 10.1093/eurheartj/ehu180
90. Zangrando J, Zhang L, Vausort M, Maskali F, Marie PY, Wagner DR, et al. Identification of candidate long non-coding RNAs in response to myocardial infarction. BMC Genomics (2014) 15(1):460. doi: 10.1186/1471-2164-15-460
91. Wang Z, Zhang XJ, Ji YX, Zhang P, Deng KQ, Gong J, et al. The long noncoding RNA Chaer defines an epigenetic checkpoint in cardiac hypertrophy. Nat Med (2016) 22(10):1131–9. doi: 10.1038/nm.4179
92. Han P, Li W, Lin CH, Yang J, Shang C, Nuernberg ST, et al. A long noncoding RNA protects the heart from pathological hypertrophy. Nature (2014) 514(7520):102–6. doi: 10.1038/nature13596
93. Ishii N, Ozaki K, Sato H, Mizuno H, Saito S, Takahashi A, et al. Identification of a novel non-coding RNA, MIAT, that confers risk of myocardial infarction. J Hum Genet (2006) 51(12):1087–99. doi: 10.1007/s10038-006-0070-9
94. Gordon AD, Biswas S, Feng B, Chakrabarti S. MALAT1: A regulator of inflammatory cytokines in diabetic complications. Endocrinol Diab Metab (2018) 65:e00010. doi: 10.1002/edm2.10
95. Wang K, Liu F, Zhou LY, Long B, Yuan SM, Wang Y, et al. The long noncoding RNA CHRF regulates cardiac hypertrophy by targeting miR-489. Circ Res (2014) 114(9):1377–88. doi: 10.1161/CIRCRESAHA.114.302476
96. Jiang F, Zhou X, Huang J. Long non-coding RNA-ROR mediates the reprogramming in cardiac hypertrophy. PLoS ONE (2016) 11(4):e0152767. doi: 10.1371/journal.pone.0152767
97. Liu L, An X, Li Z, Song Y, Li L, Zuo S, et al. The H19 long noncoding RNA is a novel negative regulator of cardiomyocyte hypertrophy. Cardiovasc Res (2016) 111(1):56–65. doi: 10.1093/cvr/cvw078
98. Kumarswamy R, Bauters C, Volkmann I, Maury F, Fetisch J, Holzmann A, et al. Circulating long noncoding RNA, LIPCAR, predicts survival in patients with heart failure. Circ Res (2014) 114(10):1569–75. doi: 10.1161/CIRCRESAHA.114.303915
99. Burd CE, Jeck WR, Liu Y, Sanoff HK, Wang Z, Sharpless NE. Expression of linear and novel circular forms of an INK4/ARF-associated non-coding RNA correlates with atherosclerosis risk. PLoS Genet (2010) 6(12):e1001233. doi: 10.1371/journal.pgen.1001233
100. Wang K, Long B, Zhou LY, Liu F, Zhou QY, Liu CY, et al. CARL lncRNA inhibits anoxia-induced mitochondrial fission and apoptosis in cardiomyocytes by impairing miR-539-dependent PHB2 downregulation. Nat Commun (2014) 5:3596. doi: 10.1038/ncomms4596
101. Wang K, Liu F, Liu CY, An T, Zhang J, Zhou LY, et al. The long noncoding RNA NRF regulates programmed necrosis and myocardial injury during ischemia and reperfusion by targeting miR-873. Cell Death Differ (2016) 23(8):1394–405. doi: 10.1038/cdd.2016.28
102. Micheletti R, Plaisance I, Abraham BJ, Sarre A, Ting CC, Alexanian M, et al. The long noncoding RNA Wisper controls cardiac fibrosis and remodeling. Sci Transl Med (2017) 9(395):eaai9118. doi: 10.1126/scitranslmed.aai9118
103. Liang H, Pan Z, Zhao X, Liu L, Sun J, Su X, et al. LncRNA PFL contributes to cardiac fibrosis by acting as a competing endogenous RNA of let-7d. Theranostics (2018) 8(4):1180–94. doi: 10.7150/thno.20846
104. Deaton AM, Bird A. CpG islands and the regulation of transcription. Genes Dev (2011) 25(10):1010–22. doi: 10.1101/gad.2037511
105. Watson CJ, Collier P, Tea I, Neary R, Watson JA, Robinson C, et al. Hypoxia-induced epigenetic modifications are associated with cardiac tissue fibrosis and the development of a myofibroblast-like phenotype. Hum Mol Genet (2014) 23(8):2176–88. doi: 10.1093/hmg/ddt614
106. Pan X, Chen Z, Huang R, Yao Y, Ma G. Transforming growth factor β1 induces the expression of collagen type I by DNA methylation in cardiac fibroblasts. PLoS ONE (2013) 8(4):e60335. doi: 10.1371/journal.pone.0060335
107. Xu X, Tan X, Tampe B, Nyamsuren G, Liu X, Maier LS, et al. Epigenetic balance of aberrant Rasal1 promoter methylation and hydroxymethylation regulates cardiac fibrosis. Cardiovasc Res (2015) 105(3):279–91. doi: 10.1093/cvr/cvv015
108. Xu X, Tan X, Hulshoff MS, Wilhelmi T, Zeisberg M, Zeisberg EM. Hypoxia-induced endothelial-mesenchymal transition is associated with RASAL1 promoter hypermethylation in human coronary endothelial cells. FEBS Lett (2016) 590(8):1222–33. doi: 10.1002/1873-3468.12158
109. Dakhlallah D, Batte K, Wang Y, Cantemir-Stone CZ, Yan P, Nuovo G, et al. Epigenetic regulation of miR-17~92 contributes to the pathogenesis of pulmonary fibrosis. Am J Respir Crit Care Med (2013) 187(4):397–405. doi: 10.1164/rccm.201205-0888OC
110. Chalei V, Sansom SN, Kong L, Lee S, Montiel JF, Vance KW, et al. The long non-coding RNA Dali is an epigenetic regulator of neural differentiation. Elife (2014) 3:e04530. doi: 10.7554/eLife.04530
111. Bao X, Wu H, Zhu X, Guo X, Hutchins AP, Luo Z, et al. The p53-induced lincRNA-p21 derails somatic cell reprogramming by sustaining H3K9me3 and CpG methylation at pluripotency gene promoters. Cell Res (2015) 25(1):80–92. doi: 10.1038/cr.2014.165
112. Wang L, Zhao Y, Bao X, Zhu X, Kwok YK, Sun K, et al. LncRNA Dum interacts with Dnmts to regulate Dppa2 expression during myogenic differentiation and muscle regeneration. Cell Res (2015) 25(3):335–50. doi: 10.1038/cr.2015.21
113. Merry CR, Forrest ME, Sabers JN, Beard L, Gao XH, Hatzoglou M, et al. DNMT1-associated long non-coding RNAs regulate global gene expression and DNA methylation in colon cancer. Hum Mol Genet (2015) 24(21):6240–53. doi: 10.1093/hmg/ddv343
114. Grillo MA, Colombatto S. S-adenosylmethionine and protein methylation. Amino Acids (2005) 28(4):357–62. doi: 10.1007/s00726-005-0197-6
115. Byvoet P, Shepherd GR, Hardin JM, Noland BJ. The distribution and turnover of labeled methyl groups in histone fractions of cultured mammalian cells. Arch Biochem Biophys (1972) 148(2):558–67. doi: 10.1016/0003-9861(72)90174-9
116. Feng Q, Wang H, Ng HH, Erdjument-Bromage H, Tempst P, Struhl K, et al. Methylation of H3-lysine 79 is mediated by a new family of HMTases without a SET domain. Curr Biol (2002) 12(12):1052–8. doi: 10.1016/S0960-9822(02)00901-6
117. El-Osta A, Brasacchio D, Yao D, Pocai A, Jones PL, Roeder RG, et al. Transient high glucose causes persistent epigenetic changes and altered gene expression during subsequent normoglycemia. J. Exp. Med. (2008) 205(10):2409–17. doi: 10.1084/jem.20081188
118. Allan GM, Nouri F, Korownyk C, Kolber MR, Vandermeer B, Mccormack J. Agreement among cardiovascular disease risk calculators. Circulation (2013) 127(19):1948–56. doi: 10.1161/CIRCULATIONAHA.112.000412
119. Nguyen AT, Xiao B, Neppl RL, Kallin EM, Li J, Chen T, et al. DOT1L regulates dystrophin expression and is critical for cardiac function. Genes Dev (2011) 25(3):263–74. doi: 10.1101/gad.2018511
120. Ruiz MA, Feng B, Chakrabarti S. Polycomb repressive complex 2 regulates MiR-200b in retinal endothelial cells: potential relevance in diabetic retinopathy. PLoS ONE (2015) 10(4):e0123987. doi: 10.1371/journal.pone.0123987
121. di Croce L, Helin K. Transcriptional regulation by polycomb group proteins. Nat Struct Mol Biol (2013) 20(10):1147–55. doi: 10.1038/nsmb.2669
122. Chang CJ, Hung MC. The role of EZH2 in tumour progression. Br J Cancer (2012) 106(2):243–7. doi: 10.1038/bjc.2011.551
123. Zhu WS, Tang CM, Xiao Z, Zhu JN, Lin QX, Fu YH, et al. Targeting EZH1 and EZH2 contributes to the suppression of fibrosis-associated genes by miR-214-3p in cardiac myofibroblasts. Oncotarget (2016) 7(48):78331–42. doi: 10.18632/oncotarget.13048
124. Zhou X, Zang X, Ponnusamy M, Masucci MV, Tolbert E, Gong R, et al. Enhancer of zeste homolog 2 inhibition attenuates renal fibrosis by maintaining Smad7 and phosphatase and tensin homolog expression. J Am Soc Nephrol (2016) 27(7):2092–108. doi: 10.1681/ASN.2015040457
125. Atta H, El-Rehany M, Hammam O, Abdel-Ghany H, Ramzy M, Roderfeld M, et al. Mutant MMP-9 and HGF gene transfer enhance resolution of CCl4-induced liver fibrosis in rats: role of ASH1 and EZH2 methyltransferases repression. PLoS ONE (2014) 9(11):e112384. doi: 10.1371/journal.pone.0112384
126. Serresi M, Gargiulo G, Proost N, Siteur B, Cesaroni M, Koppens M, et al. Polycomb repressive complex 2 is a barrier to KRAS-driven inflammation and epithelial-mesenchymal transition in non-small-cell lung cancer. Cancer Cell (2016) 29(1):17–31. doi: 10.1016/j.ccell.2015.12.006
127. Lee ST, Li Z, Wu Z, Aau M, Guan P, Karuturi RK, et al. Context-specific regulation of NF-κB target gene expression by EZH2 in breast cancers. Mol Cell (2011) 43(5):798–810. doi: 10.1016/j.molcel.2011.08.011
128. Bayarsaihan D. Epigenetic mechanisms in inflammation. J Dent Res (2011) 90(1):9–17. doi: 10.1177/0022034510378683
129. Grote P, Wittler L, Hendrix D, Koch F, Währisch S, Beisaw A, et al. The tissue-specific lncRNA Fendrr is an essential regulator of heart and body wall development in the mouse. Dev Cell (2013) 24(2):206–14. doi: 10.1016/j.devcel.2012.12.012
130. Wang GQ, Wang Y, Xiong Y, Chen XC, Ma ML, Cai R, et al. Sirt1 AS lncRNA interacts with its mRNA to inhibit muscle formation by attenuating function of miR-34a. Sci Rep (2016) 6:21865. doi: 10.1038/srep21865
131. Klattenhoff CA, Scheuermann JC, Surface LE, Bradley RK, Fields PA, Steinhauser ML, et al. Braveheart, a long noncoding RNA required for cardiovascular lineage commitment. Cell (2013) 152(3):570–83. doi: 10.1016/j.cell.2013.01.003
132. Terashima M, Tange S, Ishimura A, Suzuki T. MEG3 long noncoding RNA contributes to the epigenetic regulation of epithelial-mesenchymal transition in lung cancer cell lines. J Biol Chem (2017) 292(1):82–99. doi: 10.1074/jbc.M116.750950
133. Wang K, Liu F, Zhou LY, Yuan SM, Wang Y, et al. The long noncoding RNA CHRF regulates cardiac hypertrophy by targeting miR-489. Circ Res (2014) 114(9):1377–88. doi: 10.1161/CIRCRESAHA.114.302476
134. Thomas AA, Feng B, Chakrabarti S. ANRIL: a regulator of VEGF in diabetic retinopathy. Invest Ophthalmol Vis Sci (2017) 58(1):470–80. doi: 10.1167/iovs.16-20569
135. Viré E, Brenner C, Deplus R, Blanchon L, Fraga M, Didelot C, et al. The Polycomb group protein EZH2 directly controls DNA methylation. Nature (2006) 439(7078):871–4. doi: 10.1038/nature04431
136. Hassig CA, Schreiber SL. Nuclear histone acetylases and deacetylases and transcriptional regulation: HATs off to HDACs. Curr Opin Chem Biol (1997) 1(3):300–8. doi: 10.1016/S1367-5931(97)80066-X
137. Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res (2011) 21(3):381–95. doi: 10.1038/cr.2011.22
138. Gray SG, de Meyts P. Role of histone and transcription factor acetylation in diabetes pathogenesis. Diabetes Metab Res Rev (2005) 21(5):416–33. doi: 10.1002/dmrr.559
139. Chen S, Feng B, George B, Chakrabarti R, Chen M, Chakrabarti S. Transcriptional coactivator p300 regulates glucose-induced gene expression in endothelial cells. Am J Physiol Endocrinol Metab (2010) 298(1):E127–37. doi: 10.1152/ajpendo.00432.2009
140. Kaur H, Chen S, Xin X, Chiu J, Khan ZA, Chakrabarti S. Diabetes-induced extracellular matrix protein expression is mediated by transcription coactivator p300. Diabetes (2006) 55(11):3104–11. doi: 10.2337/db06-0519
141. Ghosh AK, Nagpal V, Covington JW, Michaels MA, Vaughan DE. Molecular basis of cardiac endothelial-to-mesenchymal transition (EndMT): differential expression of microRNAs during EndMT. Cell Signal (2012) 24(5):1031–6. doi: 10.1016/j.cellsig.2011.12.024
142. Feng B, Ruiz MA, Chakrabarti S. Oxidative-stress-induced epigenetic changes in chronic diabetic complications. Can J Physiol Pharmacol (2013) 91(3):213–20. doi: 10.1139/cjpp-2012-0251
143. Rahman S, Islam R. Mammalian Sirt1: insights on its biological functions. Cell Commun Signal (2011) 9(1):11. doi: 10.1186/1478-811X-9-11
144. Sauve AA, Wolberger C, Schramm VL, Boeke JD. The biochemistry of sirtuins. Annu Rev Biochem (2006) 75:435–65. doi: 10.1146/annurev.biochem.74.082803.133500
145. Mortuza R, Feng B, Chakrabarti S. SIRT1 reduction causes renal and retinal injury in diabetes through endothelin 1 and transforming growth factor β1. J Cell Mol Med (2015) 19(8):1857–67. doi: 10.1111/jcmm.12557
146. Rizk SM, El-Maraghy SA, Nassar NN. A novel role for SIRT-1 in L-arginine protection against STZ induced myocardial fibrosis in rats. PLoS ONE (2014) 9(12):e114560. doi: 10.1371/journal.pone.0114560
147. Cappetta D, Esposito G, Piegari E, Russo R, Ciuffreda LP, Rivellino A, et al. SIRT1 activation attenuates diastolic dysfunction by reducing cardiac fibrosis in a model of anthracycline cardiomyopathy. Int J Cardiol (2016) 205:99–110. doi: 10.1016/j.ijcard.2015.12.008
148. Xiao J, Sheng X, Zhang X, Guo M, Ji X. Curcumin protects against myocardial infarction-induced cardiac fibrosis via SIRT1 activation in vivo and in vitro. Drug Des Devel Ther (2016) 10:1267–77. doi: 10.2147/DDDT.S104925
149. Ma S, Feng J, Zhang R, Chen J, Han D, Li X, et al. SIRT1 activation by resveratrol alleviates cardiac dysfunction via mitochondrial regulation in diabetic cardiomyopathy mice. Oxid Med Cell Longev (2017) 2017:1–15. doi: 10.1155/2017/4602715
150. Postepska-Igielska A, Giwojna A, Gasri-Plotnitsky L, Schmitt N, Dold A, Ginsberg D, et al. LncRNA Khps1 regulates expression of the proto-oncogene SPHK1 via triplex-mediated changes in chromatin structure. Mol Cell (2015) 60(4):626–36. doi: 10.1016/j.molcel.2015.10.001
151. Feng B, Cao Y, Chen S, Chu X, Chu Y, Chakrabarti S. miR-200b mediates endothelial-to-mesenchymal transition in diabetic cardiomyopathy. Diabetes (2016) 65(3):768–79. doi: 10.2337/db15-1033
152. Cao Y, Feng B, Chen S, Chu Y, Chakrabarti S. Mechanisms of endothelial to mesenchymal transition in the retina in diabetes. Invest Ophthalmol Vis Sci (2014) 55(11):7321–31. doi: 10.1167/iovs.14-15167
153. Mcarthur K, Feng B, Wu Y, Chen S, Chakrabarti S. MicroRNA-200b regulates vascular endothelial growth factor-mediated alterations in diabetic retinopathy. Diabetes (2011) 60(4):1314–23. doi: 10.2337/db10-1557
154. Shehadeh LA, Sharma S, Pessanha M, Wei JQ, Liu J, Yuan H, et al. MicroRNA-20a constrains p300-driven myocardial angiogenic transcription by direct targeting of p300. PLoS ONE (2013) 8(11):e79133. doi: 10.1371/journal.pone.0079133
155. Yamakuchi M. MicroRNA regulation of SIRT1. Front Physiol (2012) 3:68. doi: 10.3389/fphys.2012.00068
156. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell (2004) 116(2):281–97.
157. Wahid F, Shehzad A, Khan T, Kim YY. MicroRNAs: synthesis, mechanism, function, and recent clinical trials. Biochim Biophys Acta (2010) 1803(11):1231–43. doi: 10.1016/j.bbamcr.2010.06.013
158. Kim VN, Han J, Siomi MC. Biogenesis of small RNAs in animals. Nat Rev Mol Cell Biol (2009) 10(2):126–39. doi: 10.1038/nrm2632
159. Kim VN. MicroRNA biogenesis: coordinated cropping and dicing. Nat Rev Mol Cell Biol (2005) 6(5):376–85. doi: 10.1038/nrm1644
160. Feng B, Cao Y, Chen S, Ruiz M, Chakrabarti S. miRNA-1 regulates endothelin-1 in diabetes. Life Sci (2014) 98(1):18–23. doi: 10.1016/j.lfs.2013.12.199
161. Chen S, Puthanveetil P, Feng B, Matkovich SJ, Dorn GW, Chakrabarti S. Cardiac miR-133a overexpression prevents early cardiac fibrosis in diabetes. J Cell Mol Med (2014) 18(3):415–21. doi: 10.1111/jcmm.12218
162. Feng B, Chen S, Mcarthur K, Wu Y, Sen S, Ding Q, et al. miR-146a-Mediated extracellular matrix protein production in chronic diabetes complications. Diabetes (2011) 60(11):2975–84. doi: 10.2337/db11-0478
163. Mortuza R, Feng B, Chakrabarti S. miR-195 regulates SIRT1-mediated changes in diabetic retinopathy. Diabetologia (2014) 57(5):1037–46. doi: 10.1007/s00125-014-3197-9
164. Feng B, Chakrabarti S. miR-320 regulates glucose-induced gene expression in diabetes. ISRN Endocrinol (2012) 2012:1–6. doi: 10.5402/2012/549875
165. Asrih M, Steffens S. Emerging role of epigenetics and miRNA in diabetic cardiomyopathy. Cardiovasc Pathol (2013) 22(2):117–25. doi: 10.1016/j.carpath.2012.07.004
166. Guay C, Regazzi R. New emerging tasks for microRNAs in the control of β-cell activities. Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids (2016) 1861(12):2121–9. doi: 10.1016/j.bbalip.2016.05.003
167. Mastropasqua R, Toto L, Cipollone F, Santovito D, Carpineto P, Mastropasqua L. Role of microRNAs in the modulation of diabetic retinopathy. Prog Retin Eye Res (2014) 43:92–107. doi: 10.1016/j.preteyeres.2014.07.003
168. Ortega FJ, Mercader JM, Moreno-Navarrete JM, Rovira O, Guerra E, Esteve E, et al. Profiling of circulating microRNAs reveals common microRNAs linked to type 2 diabetes that change with insulin sensitization. Diabetes Care (2014) 37(5):1375–83. doi: 10.2337/dc13-1847
169. Santovito D, de Nardis V, Marcantonio P, Mandolini C, Paganelli C, Vitale E, et al. Plasma exosome microRNA profiling unravels a new potential modulator of adiponectin pathway in diabetes: effect of glycemic control. J Clin Endocrinol Metab (2014) 99(9):E1681–5. doi: 10.1210/jc.2013-3843
170. Nunez Lopez YO, Coen PM, Goodpaster BH, Seyhan AA. Gastric bypass surgery with exercise alters plasma microRNAs that predict improvements in cardiometabolic risk. Int J Obes (2017) 41(7):1121–30. doi: 10.1038/ijo.2017.84
171. Pan Z, Sun X, Shan H, Wang N, Wang J, Ren J, et al. MicroRNA-101 inhibited postinfarct cardiac fibrosis and improved left ventricular compliance via the FBJ osteosarcoma oncogene/transforming growth factor-β1 pathway. Circulation (2012) 126(7):840–50. doi: 10.1161/CIRCULATIONAHA.112.094524
172. Liu N, Olson EN. MicroRNA regulatory networks in cardiovascular development. Dev Cell (2010) 18(4):510–25. doi: 10.1016/j.devcel.2010.03.010
173. Gurha P, Abreu-Goodger C, Wang T, Ramirez MO, Drumond AL, van Dongen S, et al. Targeted deletion of microRNA-22 promotes stress-induced cardiac dilation and contractile dysfunction. Circulation (2012) 125(22):2751–61. doi: 10.1161/CIRCULATIONAHA.111.044354
174. van Rooij E, Sutherland LB, Thatcher JE, Dimaio JM, Naseem RH, Marshall WS, et al. Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc Natl Acad Sci USA (2008) 105(35):13027–32. doi: 10.1073/pnas.0805038105
175. Huang ZP, Chen J, Seok HY, Zhang Z, Kataoka M, Hu X, et al. MicroRNA-22 regulates cardiac hypertrophy and remodeling in response to stress. Circ Res (2013) 112(9):1234–43. doi: 10.1161/CIRCRESAHA.112.300682
176. van Rooij E, Sutherland LB, Qi X, Richardson JA, Hill J, Olson EN. Control of stress-dependent cardiac growth and gene expression by a microRNA. Science (2007) 316(5824):575–9. doi: 10.1126/science.1139089
177. Ucar A, Gupta SK, Fiedler J, Erikci E, Kardasinski M, Batkai S, et al. The miRNA-212/132 family regulates both cardiac hypertrophy and cardiomyocyte autophagy. Nat Commun (2012) 3:1078. doi: 10.1038/ncomms2090
178. da Costa Martins PA, Salic K, Gladka MM, Armand AS, Leptidis S, El Azzouzi H, et al. MicroRNA-199b targets the nuclear kinase Dyrk1a in an auto-amplification loop promoting calcineurin/NFAT signalling. Nat Cell Biol (2010) 12(12):1220–7. doi: 10.1038/ncb2126
179. Bernardo BC, Gao XM, Winbanks CE, Boey EJ, Tham YK, Kiriazis H, et al. Therapeutic inhibition of the miR-34 family attenuates pathological cardiac remodeling and improves heart function. Proc Natl Acad Sci USA (2012) 109(43):17615–20. doi: 10.1073/pnas.1206432109
180. Thum T, Gross C, Fiedler J, Fischer T, Kissler S, Bussen M, et al. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature (2008) 456(7224):980–4. doi: 10.1038/nature07511
181. Militello G, Weirick T, John D, Döring C, Dimmeler S, Uchida S. Screening and validation of lncRNAs and circRNAs as miRNA sponges. Brief Bioinformatics (2017) 18(5):780–8. doi: 10.1093/bib/bbw053
182. Qu X, du Y, Shu Y, Gao M, Sun F, Luo S, et al. MIAT is a pro-fibrotic long non-coding RNA governing cardiac fibrosis in post-infarct myocardium. Sci Rep (2017) 7:42657. doi: 10.1038/srep42657
Keywords: diabetic cardiomyopathy, epigenetics, long non-coding RNAs, cardiac fibrosis, EndMT
Citation: Biswas S, Thomas AA and Chakrabarti S (2018). LncRNAs: Proverbial Genomic “Junk” or Key Epigenetic Regulators During Cardiac Fibrosis in Diabetes? Front. Cardiovasc. Med. 5:28. doi: 10.3389/fcvm.2018.00028
Received: 01 February 2018; Accepted: 15 March 2018;
Published: 04 April 2018
Edited by:
Paras Kumar Mishra, University of Nebraska Medical Center, United StatesReviewed by:
Siva K. Panguluri, University of South Florida, United StatesDonato Santovito, Ludwig-Maximilians-University Munich, Germany
Copyright © 2018 Biswas, Thomas and Chakrabarti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Subrata Chakrabarti, c3VicmF0YS5jaGFrcmFiYXJ0aUBsaHNjLm9uLmNh
†These authors have contributed equally to this work