 Alexia Hulin
Alexia Hulin Alexandre Hego
Alexandre Hego Patrizio Lancellotti
Patrizio Lancellotti Cécile Oury
Cécile Oury- 1GIGA Cardiovascular Sciences, Laboratory of Thrombosis and Hemostasis and Valvular Heart Disease, University of Liège, CHU Sart Tilman, Liège, Belgium
- 2GIGA Cardiovascular Sciences, Department of Cardiology, University of Liège Hospital, Heart Valve Clinic, CHU Sart Tilman, Liège, Belgium
- 3Gruppo Villa Maria Care and Research, Anthea Hospital, Bari, Italy
Calcific Aortic Valve Disease (CAVD) is the most common heart valve disease and its incidence is expected to rise with aging population. No medical treatment so far has shown slowing progression of CAVD progression. Surgery remains to this day the only way to treat it. Effective drug therapy can only be achieved through a better insight into the pathogenic mechanisms underlying CAVD. The cellular and molecular events leading to leaflets calcification are complex. Upon endothelium cell damage, oxidized LDLs trigger a proinflammatory response disrupting healthy cross-talk between valve endothelial and interstitial cells. Therefore, valve interstitial cells transform into osteoblasts and mineralize the leaflets. Studies have investigated signaling pathways driving and connecting lipid metabolism, inflammation and osteogenesis. This review draws a summary of the recent advances and discusses their exploitation as promising therapeutic targets to treat CAVD and reduce valve replacement.
Introduction
Over the course of an average day, aortic valve (AoV) leaflets open and close 100,000 times allowing unidirectionality blood flow from the left ventricle to the systemic circulation. The proper function of AoV is achieved by thin leaflets composed of three distinct layers of extracellular matrix (ECM), rich in fibrillar collagen, glycosaminoglycans (GAGs) and elastin. Calcific Aortic Valve Disease (CAVD) appears first as AoV sclerosis developing into AoV stenosis (1, 2). Macroscopically, leaflets are thickened and progressively calcified resulting into stiff leaflets with restricted movement.
CAVD is one of the most common heart valve disease and its prevalence increases with aging (3). Nowadays, in western countries, 2.8% of the general population aged over 75 years is affected with moderate to severe aortic stenosis (3, 4). With life expectancy increasing, prevalence of heart valve disease is expecting to rise. Nevertheless, due to a lack of drug treatment (5), surgery remains the only way to treat it through surgical valve replacement or transcatheter aortic valve implantation.
The seeking of therapeutic targets relies on mechanistic understanding of CAVD. Due to its association with aging, CAVD used to be considered as a passive disease, but is now established that CAVD is an active cellular-driven regulated process (6). Heart valve homeostasis is tightly controlled by valve interstitial cells (VICs) embedded in ECM, valve endothelial cells (VECs) covering the leaflet, and circulant and resident immune cells. When CAVD develops, lipid deposition, inflammation and angiogenesis occur while VICs are entering an osteogenic program as a response to exposure to risk factors including age, congenital heart defect, male gender, tobacco use, diabetes, hypertension, obesity and dyslipidemia (7–9). As a result, homeostasis is disrupted, ECM is remodeled, and formation of calcium nodules occurs. Although mechanisms leading to CAVD are still unclear, studies on diseased human aortic valves and animal models of CAVD, reviewed by Sider et al. (10), have provided valuable insights into cellular components and signaling pathways involved in the pathogenesis. This review will summarize the current findings with emphasis on valuable therapeutic candidates.
CAVD: Multi-Step Process with Endothelium Damage as Starting Point
Endothelium dysfunction is an early feature of CAVD (11, 12) and likely the result of altered blood shear stress (13). There is indeed a spatial correlation between the calcific lesions, located almost exclusively on the aortic side of AoV leaflet, and the local hemodynamic environment (14–16).The hypothesis of hemodynamic onset is reinforced by the predisposition and accelerated progression of CAVD in patients with bicuspid aortic valve (17) that display different blood flow patterns than observed with tricuspid AoV (18, 19). Endothelium damage favors lipid deposit followed by infiltration of inflammatory cells, two hallmarks of early AoV lesions (20). Therefore, lipids and cytokines will influence neighbored VECs and VICs to promote activation of VICs, ECM remodeling and mineralization of AoV leaflets (Figure 1).
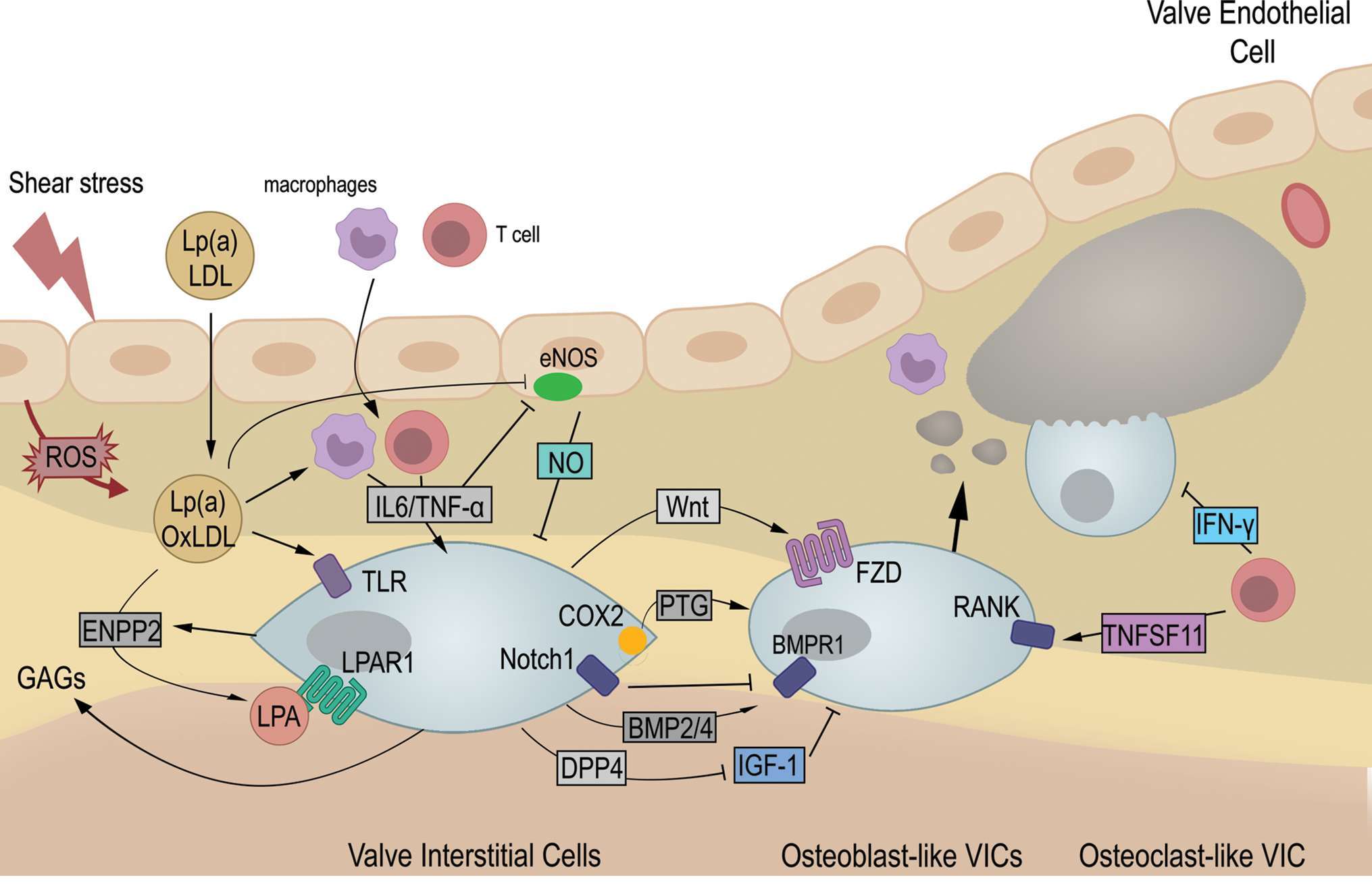
Figure 1. Upon valve endothelium damage, low-density-lipoprotein (LDL) and lipoprotein a [Lp(a)] accumulate. Oxidation of LDL (oxLDL) trigger infiltration of macrophages and T cells that express pro-inflammatory cytokines among which IL-6 and TNF-α. Proinflammatory cytokines impairs protective role of valve endothelial by inhibition of endothelial nitric oxide synthase (eNOS) and production of nitric oxide (NO). Therefore, oxidative stress (ROS) increases and contributes to enhance oxLDL. Concomitantly, valve interstitial cells (VICs) get activated by cytokines and oxLDL directly, or indirectly through autotaxin (ENPP2) and lysophosphatidyl acid (LPA). Therefore, VICs enter an osteogenic differentiation leading to calcific deposit and nodule formation. Activated VICs secrete glycosaminoglycans (GAGs), favoring further accumulation of oxLDL. Increased cyclooxygenase 2 (COX2) and its product prostaglandin (PTG), Wnt and BMP signaling have been shown to drive osteogenic differentiation while inhibition of IGF-1 signaling by dipeptidyl peptidase-4 (DPP4) contributes as well to pathogenesis. Also, Notch signaling, induced by NO, repress osteogenic differentiation. Finally, T cells favor osteogenesis and osteoclast formation by production of TNFSF11 but secrete Interferon-γ (IFN-γ) which limits calcium resorption. Altogether, aortic valve leaflets gets remodeled and stiffen leading to aortic valve stenosis. ROS: Reactive Oxygen Species. LPAR1: Lysosphosphatidyl Acid Receptor 1. TLR: Toll-like receptor. IL6: interleukin 6. TNF-α: Tumor necrosis factor- α. BMP: Bone Morphogenetic Protein. Morphogen. BMPR1: BMP Receptor 1. Fzd: Frizzled. RANK: Receptor Activator of Nuclear Factor kB. TNFSF11: RANK ligand.
Oxidized LDLs Mediate Inflammation and Mineralization
The importance of dyslipidemia in CAVD was confirmed by prevalence of CAVD in familial hypercholesterolemia caused by mutation of LDL receptor (Ldlr) and leading to abnormal circulating level of LDL (21–23). Hypercholesterolemia induced in animal models by genetic mutation (Ldlr−/−, ApoE−/−, ApoB100/100) and/or combined with enriched diet further indicate that increased lipid deposits precede the emergence of inflammatory and calcification processes (21, 24). Due to association between lipid and CAVD, clinical trial using lipid level lowering drug have been carried out, but it has shown negative results in regard with reducing CAVD (5, 25–27). One of the reasons might be that statins are ineffective to reduce Lp(a) level (28, 29) . Lp(a) consists of low density lipoprotein (LDL)-like particle in which apoliprotein(a) is covalently linked to apoliprotein B100 (30). Histopathologic studies demonstrated accumulation of apoliproteins and lipid in early stages of CAVD (31). Genome wide association study further described a SNP in LPA gene that was strongly associated with CAVD. Individuals with that SNP had higher Lp(a) plasma level and higher risk of aortic valve stenosis (32–34).
Altogether, Lp(a) appears genuinely to mediate the onset of CAVD. Deciphering the pathogenic mechanisms linking Lp(a) to CAVD has been recently acknowledged as a priority (35). Several studies highlighted a link between lipid metabolism and calcification through oxidation of LDLs. Lp(a) is a carrier of oxidized phospholipids (OxPLs), used by Lp(a)-associated phospholipase A2 (Lp-PLA2), to generate lysophosphatidyl choline (LPC), all highly expressed in human CAVD (36, 37). LPC is then transformed into lysophosphatidyl acid (LPA) by ectonucleotide pyrophosphatase/phosphodiesterase 2 (ENPP2), secreted by stimulated VICs (38). LPA is also produced during non- oxidative transformation of LDLs. Therefore, LPA activates VICs through enzymatic LPAR1/RhoA/NF-κb signaling, and mediates mineralization through BMP2 expression (38, 39). The requirement for RhoA to promote calcific nodule was also illustrated in vitro (40). The signaling pathway is confirmed with decreased AoV mineralization when using Ki16425, an inhibitor of LPAR1, in Ldlr−/−, ApoB100/100 mice fed with high fat and high sucrose diet (39). It is important to mention that changes in the ECM, with accumulation of glycosaminoglycans, precede and favor oxLDL retention (24, 41, 42).
The findings indicate that lowering Lp(a), OxLDL or targeting LPAR1 are attractive options and might be used to prevent the onset of CAVD. Multiple treatment options are currently suggested to decrease Lp(a). IONIS-APO(a)Rx, and IONIS-APO(a)-LRx, antisense oligonucleotide targeting Apo(a) mRNA have been shown to lower Lp(a) level (43). Targeting Proprotein Convertase Subtilisin/Kexin type 9 (PCSK9), a hepatic protease that promotes LDLR destruction, might be a way to decrease LDL and oxidative products. This might be achieved with monoclonal antibodies, Alirocumab and Evolocumab (44), or by using Inclisiran, a small RNAi targeting PCSK9 (45, 46).
Inflammation Contributes to Calcification
Inflammation occurs after endothelium activation and lipid deposition. Microarray analysis of human CAVD (47) and Rapacz familial hypercholesterolemia swine, an established model of human FH (21) shows upregulation of inflammation-related genes and chemokines. Histological studies present inflammatory cells, composed of macrophages, B and T cells found near osteoblast-like cells and calcified area in human CAVD (20, 48, 49). PET imaging using 18-Flurodexoxyglucose uptake (18F-FDG) to monitor inflammation reports higher 18F-FDG uptake in patients with AoV sclerosis and stenosis and a raise of the activity as the disease gets more severe(50).
Besides activation of endothelial cells (11, 12), OxLDLs trigger proinflammatory cytokines expression and promotes infiltration of immune cells into AoV leaflets (42, 51, 52). In diseased AoV, higher oxLDL content correlates with higher amounts of inflammatory cells (53) . During inflammation, immune cells secrete inflammatory cytokines including IL-2 (54), IL-1β (55), TNF-α (56, 57) , IL6 (58) and MMPs (55, 59) than stimulates VICs, ECM remodeling and promote the expression of genes involved in osteogenesis (52). Altogether, data support that CAVD is an inflammatory disease, and inflammation may drive calcification.
Although inflammation precedes ECM remodeling and calcification, inflammation over the course of the disease has not been fully explored yet. Similarly, immune cells display a broad heterogeneity with specific function. Thorough characterization of macrophages, T cells or B cells is now just starting to be done in the context of CAVD. M1 macrophage subset have recently be found to be the predominant macrophage subset in CAVD, promoting osteogenic differentiation of VICs through TNF-α and IL-6 secretion (58, 60). T cells are also reported surrounding calcified area. T cells favor calcification through cytokine TNF-α and TNFSF11 expression (56, 61, 62). Increased T cells in diseased AoV is likely the result of increased circulating CD8 +T cells (63). Activated T cells infiltrate the leaflets and surround calcified area and display high level of inflammatory cytokine IFN-γ (62). Although TNFSF11 promotes osteoclast activity, aberrant IFN-γ level impairs calcium resorption by valve osteoclast. Therefore, calcium accumulates in the leaflets and facilitates nodule formations (62). A similar study indicates that macrophages surrounding calcium deposits in human atherosclerotic are defective and unable to resorb calcification (64). Such role of macrophage in CAVD have not been explored yet. Circulating Tregs are also measured in patients with CAVD and associate with disease progression (65). Although dendritic cells are found abundantly in heart valve and accumulate in AoV stenosis, their contribution to CAVD is still unknown (51, 66).
Deeper understanding of regulation, timing and functional role of immune cells in CAVD will bring valuable information to determine how targeting inflammation might help preventing pathogenesis.
VECs Are Natural Inhibitors of Calcification, Through NO Release, but Activators Through Oxidative Stress
Inflammatory cytokines, TNF-α and IL-6, induce valve endothelial-to-mesenchymal (EMT) transformation through Akt/NF-κb signaling and reduce endothelial nitric-oxide synthase (eNOS) expression (67). Although some markers of EMT are measured in human calcified aortic valves (67), studies have still to address if EMT contribute to pathogenesis of CAVD.
VECs have the particularity to display side-specific heterogeneity. Endothelium on the aortic side displays an antioxidative and anti-inflammatory phenotype defined by its RNA expression profile (15). Thus, aortic side of AoV demonstrates protection against repetitive insult in normal AoV. As consequence, VECs are releasing nitric-oxide (NO), a natural inhibitor of pathogenic differentiation of VICs into myofibroblast andosteoblasts (68). Increased NO release has been shown to inhibit calcific nodule formation in vitro (69) and in vivo with atorvastatin treatment (70). On the opposite, in CAVD, altered mechanical stimulus, oxLDLs or TNF-α impair eNOS expression (68, 71, 72). Concomitantly, uncoupling of NO synthase leads to increased production of superoxide and oxidative stress which drives calcification (73).The critical role of endothelium and eNOS was further illustrated through modulation of a multifunctional enzyme dipeptidyl peptidase-4 (DPP4) and insulin growth factor-1 (IGF-1). Upon NO depletion, DPP4 increases in human VICs and limits IGF-1 signaling leading to enhanced calcification. Treatment of rabbit and mouse model of CAVD with Sitagliptin, a selective DPP4 inhibitor, was protective against AoV calcification (74). Similarly, the protective role of VECs is illustrated by TGF-β1 expression that translocates Sox9 into VICs nucleus and prevent calcific nodule formation (75, 76). Therefore, enhancing protective role of VECs, during early phase of disease, must be exploited. Notably, increasing NO production with statins or using DPP4 inhibitor ,broadly used as hypoglycemic drugs for treatment of type 2 diabetes mellitus, might mitigate CAVD.
VICs Differentiate Into Osteoblast-Like Cells and Mineralize the Leaflets
Histological studies report the formation of bone nodules in stenotic CAVD resulting from deposition of calcium in the form of hydroxyapatite in the valve leaflet (49). Once heart valve development is complete, VICs become quiescent, but in disease get activated and turn into active phenotype. In response to pathological stimuli, VICs differentiate into osteoblast-like cells with abnormal expression of typical bone genes, including Runx2, Alkaline Phosphatase (ALP), Osteopontin (SPP1), Osteocalcin (BGLAP) (47) resulting in calcified ECM. Apart from promoting inflammation, OxLDLs and Lp(a) can also directly activate VICs through LPAR1 (38, 39) and TLR activation (52, 77–79), This interaction contributes to trigger GAG accumulation, in a positive feedback loop, and upregulates osteogenic gene expression through BMP2 and IL6 expression (38, 42, 80).
Different molecular mechanisms are involved in VICs osteogenic differentiation and shared with bone formation (81, 82). Stimulation of VICs culture with OxLDLs and hypercholesterolemia animal model have been used to investigate signaling pathway underlying osteogenic differentiation. Also studies in klotho null mice have been useful to investigate AoV calcification with minimal inflammation (83).BMP2, along with osteogenic gene expression, are the usual markers measured to assess VICs osteogenic differentiation. BMP signaling is increased in human CAVD illustrated by increased BMP2, BMP4 ligands and phosphorylation of Smad1/5/8 (82, 84, 85). Downregulation of Smad6, an inhibitor of BMP signaling, enhance BMP signaling (84, 86). Inhibition of osteogenic gene expression and calcific nodule formation by targeting Alk3, BMP receptor type-1A, strongly indicate that LDN-193189, a small molecule inhibitor of BMP signaling, should be used to prevent calcification in late stage of CAVD (85).
Mutation in Notch1 and its association with BAV and AoV calcification highlighted the role of Notch signaling in CAVD (87). Later, studies confirms that Notch signaling represses osteogenic gene expression (88, 89) and is regulated by NO released by endothelial cells (90). Decreased Notch signaling is not just observed in patients with mutated Notch1 but also in patients with idiopathic CAVD where increased long non-coding RNA H19, resulting from hypomethylation, prevents Notch1 expression (91).The role of prostaglandins has been illustrated in osteogenesis (92, 93), but only recently in CAVD. Prostaglandins are synthesized by COX2, an enzyme highly expressed by VICs in CAVD (94). Pharmacological inhibition of COX2 activity with Celecoxib, a nonsteroidal anti-inflammatory (NSAID) drugs, is sufficient to reduces AoV calcification in Klotho null mice (94). Celecoxib is clinically used to treat joint and/or muscle pain(95) but was associated with increased cardiovascular risk (96). Cardiovascular safety of celecoxib is nowadays controversial (97) as recent report indicate that cardiovascular risk associated with moderate doses of celecoxib is not greater than associated with non-selective-NSAID ibuprofen (98). Additional research must evaluate the effectiveness of COX2 inhibitor in human CAVD.
Non-canonical Wnt5b and Wnt11 ligands are found elevated in macrophages of human calcified AoV. Moreover, the ligands stimulate VICs, apoptosis and calcium deposits (99). Abundant expression of Fzd receptors and co-receptors Lrp5/6 also suggest the involvement of canonical Wnt/β-catenin signaling in CAVD (81, 100). In vitro, Wnt treatment of VICs inhibit chondrogenic differentiation and promote osteogenic gene expression (101, 102) while Lrp5/6 is required to promote calcification in hypercholesterolemia mouse model (103). In Axin2 KO mice, increased canonical Wnt/β-catenin signaling promotes ECM remodeling and BMP signaling but fails to calcify AoV (104). The findings illustrate that Wnt signaling is required but might not be sufficient to promote end-stage calcification. These data illustrate the importance to further study the role of Wnt signaling in CAVD as specific inhibitors are being tested (105).
VIC osteogenic differentiation has been one of the most studied process in CAVD due to available cell culture model. However, VIC remains a poorly defined cell type. Heterogeneity of VIC population is underappreciated during heart valve homeostasis and disease. Being able to define which cell type is activated and/or differentiated across disease is a major goal in order to present innovative therapeutic options.
Conclusions
CAVD is a complex multi-step event that involves numerous biological processes from lipid accumulation, inflammation to osteogenesis. Understanding the underlying molecular and cellular processes is crucial in the establishment of therapeutic targets. Clinical, histological and animal model studies have allowed better characterization of the disease and show the importance of cross-talk between lipids, immune cells, VECs and VICs. As a result, putative molecular targets with available treatments (Table 1) emerge for each stage of CAVD. Giving the multifactorial and complex interplay, timing and combination of therapy should be considered. In the context of appropriate therapeutic timing, accurate biomarkers should be defined. Similarly, thorough knowledge of the heterogeneity and function of valve cell subtype, over the course of the disease, may provide better targeting of the “diseased” cells. Overall, recent advances and future directions bring hope for the development of efficient drug treatment and for the reduction of valve replacement surgeries.
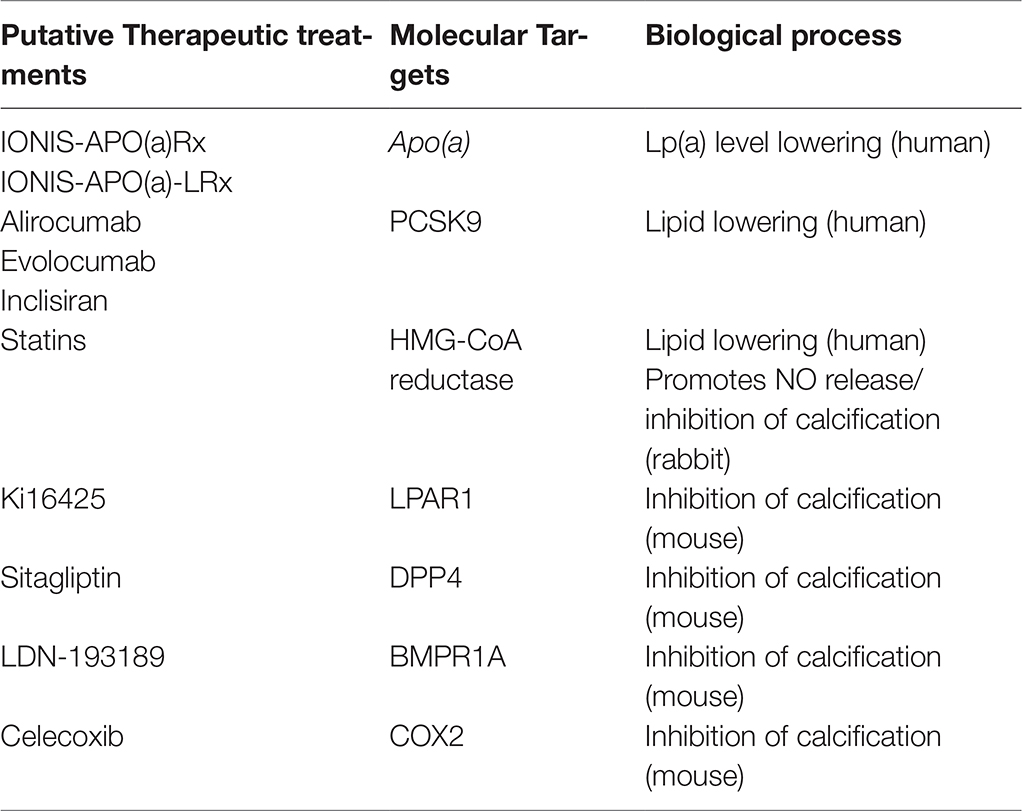
Table 1. Putative available therapeutic treatments and molecular targets that might affect the pathophysiology of CAVD. In brackets, species where the drug effect has been reported.
Author Contributions
AHu wrote the manuscript. AHe drafted the figure. PL provided intellectual contributions and edited the manuscript. CO drafted and revised the manuscript.
Funding
The authors received a grant from the European Regional Development Fund (Interreg V - Polyvalve).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
AHu is supported by an Interreg V grant. CO is Senior Research Associate at the Belgian Funds for Scientific Research (F.R.S.-FNRS, Belgium).
References
1. Lindman BR, Clavel MA, Mathieu P, Iung B, Lancellotti P, Otto CM, et al. Calcific aortic stenosis. Nat Rev Dis Primers (2016) 2:16006. doi: 10.1038/nrdp.2016.6
2. Yutzey KE, Demer LL, Body SC, Huggins GS, Towler DA, Giachelli CM, et al. Calcific aortic valve disease: a consensus summary from the alliance of Investigators on calcific aortic valve disease. Arterioscler Thromb Vasc Biol (2014) 34(11):2387–93. doi: 10.1161/ATVBAHA.114.302523
3. Nkomo VT, Gardin JM, Skelton TN, Gottdiener JS, Scott CG, Enriquez-Sarano M. Burden of valvular heart diseases: a population-based study. Lancet (2006) 368(9540):1005–11. doi: 10.1016/S0140-6736(06)69208-8
4. Benjamin EJ, Blaha MJ, Chiuve SE, Cushman M, Das SR, Deo R, et al. Heart disease and stroke statistics-2017 update: a report from the American heart association. Circulation (2017) 135(10):e146–603. doi: 10.1161/CIR.0000000000000485
5. Hutcheson JD, Aikawa E, Merryman WD. Potential drug targets for calcific aortic valve disease. Nat Rev Cardiol (2014) 11(4):218–31. doi: 10.1038/nrcardio.2014.1
6. Rajamannan NM, Evans FJ, Aikawa E, Grande-Allen KJ, Demer LL, Heistad DD, et al. Calcific aortic valve disease: not simply a degenerative process: a review and agenda for research from the national heart and lung and blood institute aortic stenosis working group. Executive summary: Calcific aortic valve disease-2011 update. Circulation (2011) 124(16):1783–91. doi: 10.1161/CIRCULATIONAHA.110.006767
7. Mosch J, Gleissner CA, Body S, Aikawa E. Histopathological assessment of calcification and inflammation of calcific aortic valves from patients with and without diabetes mellitus. Histol Histopathol (2017) 32(3):293–306. doi: 10.14670/HH-11-797
8. Stewart BF, Siscovick D, Lind BK, Gardin JM, Gottdiener JS, Smith VE, et al. Clinical factors associated with calcific aortic valve disease. Cardiovascular health study. J Am Coll Cardiol (1997) 29(3):630–4.
9. Testuz A, Nguyen V, Mathieu T, Kerneis C, Arangalage D, Kubota N, et al. Influence of metabolic syndrome and diabetes on progression of calcific aortic valve stenosis. Int J Cardiol (2017) 244:248–53. doi: 10.1016/j.ijcard.2017.06.104
10. Sider KL, Blaser MC, Simmons CA. Animal models of calcific aortic valve disease. Int J Inflam (2011) 2011:1–18. doi: 10.4061/2011/364310
11. Ghaisas NK, Foley JB, O'Briain DS, Crean P, Kelleher D, Walsh M. Adhesion molecules in nonrheumatic aortic valve disease: endothelial expression, serum levels and effects of valve replacement. J Am Coll Cardiol (2000) 36(7):2257–62. doi: 10.1016/S0735-1097(00)00998-0
12. Poggianti E, Venneri L, Chubuchny V, Jambrik Z, Baroncini LA, Picano E. Aortic valve sclerosis is associated with systemic endothelial dysfunction. J Am Coll Cardiol (2003) 41(1):136–41. doi: 10.1016/S0735-1097(02)02622-0
13. Sucosky P, Balachandran K, Elhammali A, Jo H, Yoganathan AP. Altered shear stress stimulates upregulation of endothelial VCAM-1 and ICAM-1 in a BMP-4- and TGF-beta1-dependent pathway. Arterioscler Thromb Vasc Biol (2009) 29(2):254–60. doi: 10.1161/ATVBAHA.108.176347
14. Gould ST, Srigunapalan S, Simmons CA, Anseth KS. Hemodynamic and cellular response feedback in calcific aortic valve disease. Circ Res (2013) 113(2):186–97. doi: 10.1161/CIRCRESAHA.112.300154
15. Simmons CA, Grant GR, Manduchi E, Davies PF. Spatial heterogeneity of endothelial phenotypes correlates with side-specific vulnerability to calcification in normal porcine aortic valves. Circ Res (2005) 96(7):792–9. doi: 10.1161/01.RES.0000161998.92009.64
16. Yap CH, Saikrishnan N, Yoganathan AP. Experimental measurement of dynamic fluid shear stress on the ventricular surface of the aortic valve leaflet. Biomech Model Mechanobiol (2012) 11(1-2):231–44. doi: 10.1007/s10237-011-0306-2
17. Roberts WC, Ko JM. Frequency by decades of unicuspid, bicuspid, and tricuspid aortic valves in adults having isolated aortic valve replacement for aortic stenosis, with or without associated aortic regurgitation. Circulation (2005) 111(7):920–5. doi: 10.1161/01.CIR.0000155623.48408.C5
18. Meierhofer C, Schneider EP, Lyko C, Hutter A, Martinoff S, Markl M, et al. Wall shear stress and flow patterns in the ascending aorta in patients with bicuspid aortic valves differ significantly from tricuspid aortic valves: a prospective study. Eur Heart J Cardiovasc Imaging (2013) 14(8):797–804. doi: 10.1093/ehjci/jes273
19. Saikrishnan N, Mirabella L, Yoganathan AP. Bicuspid aortic valves are associated with increased wall and turbulence shear stress levels compared to trileaflet aortic valves. Biomech Model Mechanobiol (2015) 14(3):577–88. doi: 10.1007/s10237-014-0623-3
20. Otto CM, Kuusisto J, Reichenbach DD, Gown AM, O'Brien KD. Characterization of the early lesion of 'degenerative' valvular aortic stenosis. Histological and immunohistochemical studies. Circulation (1994) 90(2):844–53. doi: 10.1161/01.CIR.90.2.844
21. Porras AM, Shanmuganayagam D, Meudt JJ, Krueger CG, Hacker TA, Rahko PS, et al. Development of aortic valve disease in familial hypercholesterolemic swine: implications for elucidating disease etiology. J Am Heart Assoc (2015) 4(10):e002254. doi: 10.1161/JAHA.115.002254
22. Rajamannan NM. The role of Lrp5/6 in cardiac valve disease: experimental hypercholesterolemia in the ApoE-/- /Lrp5-/- mice. J Cell Biochem (2011a) 112(10):2987–91. doi: 10.1002/jcb.23221
23. Varghese MJ. Familial hypercholesterolemia: a review. Ann Pediatr Cardiol (2014) 7(2):107. doi: 10.4103/0974-2069.132478
24. Sider KL, Zhu C, Kwong AV, Mirzaei Z, de Langé CF, Simmons CA. Evaluation of a porcine model of early aortic valve sclerosis. Cardiovasc Pathol (2014) 23(5):289–97. doi: 10.1016/j.carpath.2014.05.004
25. Chan KL, Teo K, Dumesnil JG, Ni A,Tam J ASTRONOMER Investigators. Effect of Lipid lowering with rosuvastatin on progression of aortic stenosis: results of the aortic stenosis progression observation: measuring effects of rosuvastatin (ASTRONOMER) trial. Circulation (2010) 121(2):306–14. doi: 10.1161/CIRCULATIONAHA.109.900027
26. Cowell SJ, Newby DE, Prescott RJ, Bloomfield P, Reid J, Northridge DB, et al. A randomized trial of intensive lipid-lowering therapy in calcific aortic stenosis. N Engl J Med (2005) 352(23):2389–97. doi: 10.1056/NEJMoa043876
27. Rossebø AB, Pedersen TR, Boman K, Brudi P, Chambers JB, Egstrup K, et al. Intensive lipid lowering with simvastatin and ezetimibe in aortic stenosis. N Engl J Med (2008) 359(13):1343–56. doi: 10.1056/NEJMoa0804602
28. Kostner GM, Gavish D, Leopold B, Bolzano K, Weintraub MS, Breslow JL. HMG CoA reductase inhibitors lower LDL cholesterol without reducing Lp(a) levels. Circulation (1989) 80(5):1313–9. doi: 10.1161/01.CIR.80.5.1313
29. Peeters FECM, Meex SJR, Dweck MR, Aikawa E, Crijns HJGM, Schurgers LJ, et al. Calcific aortic valve stenosis: hard disease in the heart. Eur Heart J (2017) 131. doi: 10.1093/eurheartj/ehx653
30. Rogers MA, Aikawa E. A Not-So-Little Role for Lipoprotein(a) in the Development of Calcific Aortic Valve Disease. Circulation (2015) 132(8):621–3. doi: 10.1161/CIRCULATIONAHA.115.018139
31. O'Brien KD, Reichenbach DD, Marcovina SM, Kuusisto J, Alpers CE, Otto CM. Apolipoproteins B, (a), and E accumulate in the morphologically early lesion of 'degenerative' valvular aortic stenosis. Arterioscler Thromb Vasc Biol (1996) 16(4):523–32. doi: 10.1161/01.ATV.16.4.523
32. Arsenault BJ, Boekholdt SM, Dubé MP, Rhéaume E, Wareham NJ, Khaw KT, et al. Lipoprotein(a) levels, genotype, and incident aortic valve stenosis: a prospective Mendelian randomization study and replication in a case-control cohort. Circ Cardiovasc Genet (2014) 7(3):304–10. doi: 10.1161/CIRCGENETICS.113.000400
33. Kamstrup PR, Tybjærg-Hansen A, Nordestgaard BG. Elevated lipoprotein(a) and risk of aortic valve stenosis in the general population. J Am Coll Cardiol (2014) 63(5):470–7. doi: 10.1016/j.jacc.2013.09.038
34. Thanassoulis G, Campbell CY, Owens DS, Smith JG, Smith AV, Peloso GM, et al. Genetic associations with valvular calcification and aortic stenosis. N Engl J Med (2013) 368(6):503–12. doi: 10.1056/NEJMoa1109034
35. Tsimikas S, Fazio S, Ferdinand KC, Ginsberg HN, Koschinsky ML, Marcovina SM, et al. NHLBI working group recommendations to reduce lipoprotein(a)-mediated risk of cardiovascular disease and aortic stenosis. J Am Coll Cardiol (2018) 71(2):177–92. doi: 10.1016/j.jacc.2017.11.014
36. Lehti S, Käkelä R, Hörkkö S, Kummu O, Helske-Suihko S, Kupari M, et al. Modified lipoprotein-derived lipid particles accumulate in human stenotic aortic valves. PLoS ONE (2013) 8(6):e65810. doi: 10.1371/journal.pone.0065810
37. Mahmut A, Boulanger MC, El Husseini D, Fournier D, Bouchareb R, Després JP, et al. Elevated expression of lipoprotein-associated phospholipase A2 in calcific aortic valve disease: implications for valve mineralization. J Am Coll Cardiol (2014) 63(5):460–9. doi: 10.1016/j.jacc.2013.05.105
38. Bouchareb R, Mahmut A, Nsaibia MJ, Boulanger MC, Dahou A, Lépine JL, et al. Autotaxin derived from lipoprotein(a) and valve interstitial cells promotes inflammation and mineralization of the aortic valve. Circulation (2015) 132(8):677–90. doi: 10.1161/CIRCULATIONAHA.115.016757
39. Nsaibia MJ, Boulanger MC, Bouchareb R, Mkannez G, Le Quang K, Hadji F, et al. OxLDL-derived lysophosphatidic acid promotes the progression of aortic valve stenosis through a LPAR1-RhoA-NF-κB pathway. Cardiovasc Res (2017) 113(11):1351–63. doi: 10.1093/cvr/cvx089
40. Farrar EJ, Pramil V, Richards JM, Mosher CZ, Butcher JT. Valve interstitial cell tensional homeostasis directs calcification and extracellular matrix remodeling processes via RhoA signaling. Biomaterials (2016) 105:25–37. doi: 10.1016/j.biomaterials.2016.07.034
41. Osman N, Grande-Allen KJ, Ballinger ML, Getachew R, Marasco S, O'Brien KD, et al. Smad2-dependent glycosaminoglycan elongation in aortic valve interstitial cells enhances binding of LDL to proteoglycans. Cardiovasc Pathol (2013) 22(2):146–55. doi: 10.1016/j.carpath.2012.07.002
42. Porras AM, Westlund JA, Evans AD, Masters KS. Creation of disease-inspired biomaterial environments to mimic pathological events in early calcific aortic valve disease. Proc Natl Acad Sci USA (2018) 115(3):E363–71. doi: 10.1073/pnas.1704637115
43. Viney NJ, van Capelleveen JC, Geary RS, Xia S, Tami JA, Yu RZ, et al. Antisense oligonucleotides targeting apolipoprotein(a) in people with raised lipoprotein(a): two randomised, double-blind, placebo-controlled, dose-ranging trials. Lancet (2016) 388(10057):2239–53. doi: 10.1016/S0140-6736(16)31009-1
44. Karatasakis A, Danek BA, Karacsonyi J, Rangan BV, Roesle MK, Knickelbine T, et al. Effect of PCSK9 inhibitors on clinical outcomes in patients with hypercholesterolemia: a meta-analysis of 35 randomized controlled trials. J Am Heart Assoc (2017) 6(12):e006910. doi: 10.1161/JAHA.117.006910
45. Fitzgerald K, White S, Borodovsky A, Bettencourt BR, Strahs A, Clausen V, et al. A highly durable RNAi therapeutic inhibitor of PCSK9. N Engl J Med (2017) 376(1):41–51. doi: 10.1056/NEJMoa1609243
46. Ray KK, Landmesser U, Leiter LA, Kallend D, Dufour R, Karakas M, et al. Inclisiran in patients at high cardiovascular risk with elevated LDL cholesterol. N Engl J Med (2017) 376(15):1430–40. doi: 10.1056/NEJMoa1615758
47. Bossé Y, Miqdad A, Fournier D, Pépin A, Pibarot P, Mathieu P. Refining molecular pathways leading to calcific aortic valve stenosis by studying gene expression profile of normal and calcified stenotic human aortic valves. Circ Cardiovasc Genet (2009) 2(5):489 doi: 10.1161/CIRCGENETICS.108.820795
48. Coté N, Mahmut A, Bosse Y, Couture C, Pagé S, Trahan S, et al. Inflammation is associated with the remodeling of calcific aortic valve disease. Inflammation (2013) 36(3):573–81. doi: 10.1007/s10753-012-9579-6
49. Mohler ER, Gannon F, Reynolds C, Zimmerman R, Keane MG, Kaplan FS. Bone formation and inflammation in cardiac valves. Circulation (2001) 103(11):1522–8. doi: 10.1161/01.CIR.103.11.1522
50. Dweck MR, Jones C, Joshi NV, Fletcher AM, Richardson H, White A, et al. Assessment of valvular calcification and inflammation by positron emission tomography in patients with aortic stenosis. Circulation (2012) 125(1):76–86. doi: 10.1161/CIRCULATIONAHA.111.051052
51. Lee SH, Choi JH. Involvement of immune cell network in aortic valve stenosis: communication between valvular interstitial cells and immune cells. Immune Netw (2016) 16(1):26–32. doi: 10.4110/in.2016.16.1.26
52. Mathieu P, Bouchareb R, Boulanger MC. Innate and adaptive immunity in calcific aortic valve disease. J Immunol Res (2015) 2015:1–11. doi: 10.1155/2015/851945
53. Côté C, Pibarot P, Després JP, Mohty D, Cartier A, Arsenault BJ, et al. Association between circulating oxidised low-density lipoprotein and fibrocalcific remodelling of the aortic valve in aortic stenosis. Heart (2008) 94(9):1175–80. doi: 10.1136/hrt.2007.125740
54. Olsson M, Dalsgaard CJ, Haegerstrand A, Rosenqvist M, Rydén L, Nilsson J. Accumulation of T lymphocytes and expression of interleukin-2 receptors in nonrheumatic stenotic aortic valves. J Am Coll Cardiol (1994) 23(5):1162–70. doi: 10.1016/0735-1097(94)90606-8
55. Kaden JJ, Dempfle CE, Grobholz R, Tran HT, Kiliç R, Sarikoç A, et al. Interleukin-1 beta promotes matrix metalloproteinase expression and cell proliferation in calcific aortic valve stenosis. Atherosclerosis (2003) 170(2):205–11. doi: 10.1016/S0021-9150(03)00284-3
56. Isoda K, Matsuki T, Kondo H, Iwakura Y, Ohsuzu F. Deficiency of interleukin-1 receptor antagonist induces aortic valve disease in BALB/c mice. Arterioscler Thromb Vasc Biol (2010) 30(4):708–15. doi: 10.1161/ATVBAHA.109.201749
57. Kaden JJ, Kiliç R, Sarikoç A, Hagl S, Lang S, Hoffmann U, et al. Tumor necrosis factor alpha promotes an osteoblast-like phenotype in human aortic valve myofibroblasts: a potential regulatory mechanism of valvular calcification. Int J Mol Med (2005) 16(5):869–72. doi: 10.3892/ijmm.16.5.869
58. Li G, Qiao W, Zhang W, Li F, Shi J, Dong N. The shift of macrophages toward M1 phenotype promotes aortic valvular calcification. J Thorac Cardiovasc Surg (2017) 153(6):1318–27. doi: 10.1016/j.jtcvs.2017.01.052
59. Edep ME, Shirani J, Wolf P, Brown DL. Matrix metalloproteinase expression in nonrheumatic aortic stenosis. Cardiovasc. Pathol. (2000) 9(5):281–6. doi: 10.1016/S1054-8807(00)00043-0
60. Li XF, Wang Y, Zheng DD, Xu HX, Wang T, Pan M, et al. M1 macrophages promote aortic valve calcification mediated by microRNA-214/TWIST1 pathway in valvular interstitial cells. Am J Transl Res (2016) 8(12):5773–83.
61. Kaden JJ, Bickelhaupt S, Grobholz R, Haase KK, Sarikoç A, Kiliç R, et al. Receptor activator of nuclear factor kappaB ligand and osteoprotegerin regulate aortic valve calcification. J Mol Cell Cardiol (2004) 36(1):57–66. doi: 10.1016/j.yjmcc.2003.09.015
62. Nagy E, Lei Y, Martínez-Martínez E, Body SC, Schlotter F, Creager M, et al. Interferon-γ released by activated CD8+T lymphocytes impairs the calcium resorption potential of osteoclasts in calcified human aortic valves. Am J Pathol (2017) 187(6):1413–25. doi: 10.1016/j.ajpath.2017.02.012
63. Winchester R, Wiesendanger M, O'Brien W, Zhang HZ, Maurer MS, Gillam LD, et al. Circulating activated and effector memory T cells are associated with calcification and clonal expansions in bicuspid and tricuspid valves of calcific aortic stenosis. J Immunol (2011) 187(2):1006–14. doi: 10.4049/jimmunol.1003521
64. Chinetti-Gbaguidi G, Daoudi M, Rosa M, Vinod M, Louvet L, Copin C, et al. Human alternative macrophages populate calcified areas of atherosclerotic lesions and display impaired RANKL-induced osteoclastic bone resorption activity. Circ Res (2017) 121(1):19–30. doi: 10.1161/CIRCRESAHA.116.310262
65. Shimoni S, Bar I, Meledin V, Gandelman G, George J. Circulating regulatory T cells in patients with aortic valve stenosis: association with disease progression and aortic valve intervention. Int J Cardiol (2016) 218:181–7. doi: 10.1016/j.ijcard.2016.05.039
66. Choi JH, do Y, Cheong C, Koh H, Boscardin SB, Oh YS, et al. Identification of antigen-presenting dendritic cells in mouse aorta and cardiac valves. J Exp Med (2009) 206(3):497–505. doi: 10.1084/jem.20082129
67. Mahler GJ, Farrar EJ, Butcher JT. Inflammatory cytokines promote mesenchymal transformation in embryonic and adult valve endothelial cells. Arterioscler Thromb Vasc Biol (2013) 33(1):121–30. doi: 10.1161/ATVBAHA.112.300504
68. Richards J, El-Hamamsy I, Chen S, Sarang Z, Sarathchandra P, Yacoub MH, et al. Side-specific endothelial-dependent regulation of aortic valve calcification. Am J Pathol (2013) 182(5):1922–31. doi: 10.1016/j.ajpath.2013.01.037
69. Kennedy JA, Hua X, Mishra K, Murphy GA, Rosenkranz AC, Horowitz JD. Inhibition of calcifying nodule formation in cultured porcine aortic valve cells by nitric oxide donors. Eur J Pharmacol (2009) 602(1):28–35. doi: 10.1016/j.ejphar.2008.11.029
70. Rajamannan NM, Subramaniam M, Stock SR, Stone NJ, Springett M, Ignatiev KI, et al. Atorvastatin inhibits calcification and enhances nitric oxide synthase production in the hypercholesterolaemic aortic valve. Heart (2005) 91(6):806–10. doi: 10.1136/hrt.2003.029785
71. Davis ME, Grumbach IM, Fukai T, Cutchins A, Harrison DG. Shear stress regulates endothelial nitric-oxide synthase promoter activity through nuclear factor kappaB binding. J Biol Chem (2004) 279(1):163–8. doi: 10.1074/jbc.M307528200
72. Lubrano V, Vassalle C, Blandizzi C, del Tacca M, Palombo C, L'Abbate A, et al. The effect of lipoproteins on endothelial nitric oxide synthase is modulated by lipoperoxides. Eur J Clin Invest (2003) 33(2):117–25. doi: 10.1046/j.1365-2362.2003.01083.x
73. Miller JD, Chu Y, Brooks RM, Richenbacher WE, Peña-Silva R, Heistad DD. Dysregulation of antioxidant mechanisms contributes to increased oxidative stress in calcific aortic valvular stenosis in humans. J Am Coll Cardiol (2008) 52(10):843–50. doi: 10.1016/j.jacc.2008.05.043
74. Choi B, Lee S, Kim SM, Lee EJ, Lee SR, Kim DH, et al. Dipeptidyl peptidase-4 induces aortic valve calcifcation by inhibiting insulin-like growth factor-1 signaling in valvular interstitial cells. Circulation (2017) 135(20):1935–50. doi: 10.1161/CIRCULATIONAHA.116.024270
75. Huk DJ, Austin BF, Horne TE, Hinton RB, Ray WC, Heistad DD, et al. Valve endothelial cell-derived Tgfβ1 signaling promotes nuclear localization of Sox9 in interstitial cells associated with attenuated calcification. Arterioscler Thromb Vasc Biol (2016) 36(2):328–38. doi: 10.1161/ATVBAHA.115.306091
76. Peacock JD, Levay AK, Gillaspie DB, Tao G, Lincoln J. Reduced sox9 function promotes heart valve calcification phenotypes in vivo. Circ Res (2010) 106(4):712–9. doi: 10.1161/CIRCRESAHA.109.213702
77. Song R, Zeng Q, Ao L, Yu JA, Cleveland JC, Zhao KS, et al. Biglycan induces the expression of osteogenic factors in human aortic valve interstitial cells via Toll-like receptor-2. Arterioscler Thromb Vasc Biol (2012) 32(11):2711–20. doi: 10.1161/ATVBAHA.112.300116
78. Zeng Q, Song R, Fullerton DA, Ao L, Zhai Y, Li S, et al. Interleukin-37 suppresses the osteogenic responses of human aortic valve interstitial cells in vitro and alleviates valve lesions in mice. Proc Natl Acad Sci USA (2017) 114(7):1631–6. doi: 10.1073/pnas.1619667114
79. Zhan Q, Song R, Zeng Q, Yao Q, Ao L, Xu D, et al. Activation of TLR3 induces osteogenic responses in human aortic valve interstitial cells through the NF-κB and ERK1/2 pathways. Int J Biol Sci (2015) 11(4):482–93. doi: 10.7150/ijbs.10905
80. Zeng Q, Song R, Ao L, Xu D, Venardos N, Fullerton DA, et al. Augmented osteogenic responses in human aortic valve cells exposed to oxLDL and TLR4 agonist: a mechanistic role of Notch1 and NF-κB interaction. PLoS ONE (2014) 9(5):e95400. doi: 10.1371/journal.pone.0095400
81. Caira FC, Stock SR, Gleason TG, Mcgee EC, Huang J, Bonow RO, et al. Human degenerative valve disease is associated with up-regulation of low-density lipoprotein receptor-related protein 5 receptor-mediated bone formation. J Am Coll Cardiol (2006) 47(8):1707–12. doi: 10.1016/j.jacc.2006.02.040
82. Wirrig EE, Hinton RB, Yutzey KE. Differential expression of cartilage and bone-related proteins in pediatric and adult diseased aortic valves. J Mol Cell Cardiol (2011) 50(3):561–9. doi: 10.1016/j.yjmcc.2010.12.005
83. Cheek JD, Wirrig EE, Alfieri CM, James JF, Yutzey KE. Differential activation of valvulogenic, chondrogenic, and osteogenic pathways in mouse models of myxomatous and calcific aortic valve disease. J Mol Cell Cardiol (2012) 52(3):689–700. doi: 10.1016/j.yjmcc.2011.12.013
84. Ankeny RF, Thourani VH, Weiss D, Vega JD, Taylor WR, Nerem RM, et al. Preferential activation of SMAD1/5/8 on the fibrosa endothelium in calcified human aortic valves-association with low BMP antagonists and SMAD6. PLoS ONE (2011) 6(6):e20969. doi: 10.1371/journal.pone.0020969
85. Gomez-Stallons MV, Wirrig-Schwendeman EE, Hassel KR, Conway SJ,Yutzey KE Bone Morphogenetic Protein Signaling Is Required for Aortic Valve Calcification. Arterioscler. Thromb. Vasc. Biol. (2016) 36(7):1398–405.
86. Li X, Lim J, Lu J, Pedego TM, Demer L, Tintut Y. Protective Role of Smad6 in Inflammation-Induced Valvular Cell Calcification. J Cell Biochem (2015) 116(10):2354–64. doi: 10.1002/jcb.25186
87. Garg V, Muth AN, Ransom JF, Schluterman MK, Barnes R, King IN, et al. Mutations in NOTCH1 cause aortic valve disease. Nature (2005) 437(7056):270–4. doi: 10.1038/nature03940
88. Acharya A, Hans CP, Koenig SN, Nichols HA, Galindo CL, Garner HR, et al. Inhibitory role of Notch1 in calcific aortic valve disease. PLoS ONE (2011) 6(11):e27743. doi: 10.1371/journal.pone.0027743
89. Nigam V, Srivastava D. Notch1 represses osteogenic pathways in aortic valve cells. J Mol Cell Cardiol (2009) 47(6):828–34. doi: 10.1016/j.yjmcc.2009.08.008
90. Bosse K, Hans CP, Zhao N, Koenig SN, Huang N, Guggilam A, et al. Endothelial nitric oxide signaling regulates Notch1 in aortic valve disease. J Mol Cell Cardiol (2013) 60:27–35. doi: 10.1016/j.yjmcc.2013.04.001
91. Hadji F, Boulanger MC, Guay SP, Gaudreault N, Amellah S, Mkannez G, et al. Altered DNA methylation of long noncoding RNA H19 in calcific aortic valve disease promotes mineralization by silencing NOTCH1. Circulation (2016) 134(23):1848–62. doi: 10.1161/CIRCULATIONAHA.116.023116
92. Arikawa T, Omura K, Morita I. Regulation of bone morphogenetic protein-2 expression by endogenous prostaglandin E2 in human mesenchymal stem cells. J Cell Physiol (2004) 200(3):400–6. doi: 10.1002/jcp.20031
93. Zhang X, Schwarz EM, Young DA, Puzas JE, Rosier RN, O'Keefe RJ. Cyclooxygenase-2 regulates mesenchymal cell differentiation into the osteoblast lineage and is critically involved in bone repair. J Clin Invest (2002) 109(11):1405–15. doi: 10.1172/JCI0215681
94. Wirrig EE, Gomez MV, Hinton RB, Yutzey KE. COX2 inhibition reduces aortic valve calcification in vivo. Arterioscler Thromb Vasc Biol (2015) 35(4):938–47. doi: 10.1161/ATVBAHA.114.305159
95. Singh G, Fort JG, Goldstein JL, Levy RA, Hanrahan PS, Bello AE, et al. Celecoxib versus naproxen and diclofenac in osteoarthritis patients: SUCCESS-I Study. Am J Med (2006) 119(3):255–66. doi: 10.1016/j.amjmed.2005.09.054
96. Solomon SD, McMurray JJ, Pfeffer MA, Wittes J, Fowler R, Finn P, et al. Cardiovascular risk associated with celecoxib in a clinical trial for colorectal adenoma prevention. N Engl J Med (2005) 352(11):1071–80. doi: 10.1056/NEJMoa050405
97. Pepine CJ, Gurbel PA. Cardiovascular safety of NSAIDs: Additional insights after PRECISION and point of view. Clin Cardiol (2017) 40(12):1352–1356. doi: 10.1002/clc.22814
98. Nissen SE, Yeomans ND, Solomon DH, Lüscher TF, Libby P, Husni ME, et al. Cardiovascular safety of celecoxib, naproxen, or ibuprofen for arthritis. N Engl J Med (2016) 375(26):2519–29. doi: 10.1056/NEJMoa1611593
99. Albanese I, Yu B, Al-Kindi H, Barratt B, Ott L, Al-Refai M, et al. Role of noncanonical wnt signaling pathway in human aortic valve calcification. Arterioscler Thromb Vasc Biol (2017) 37(3):543–52. doi: 10.1161/ATVBAHA.116.308394
100. Siddique A, Yu B, Khan K, Buyting R, Al-Kindi H, Alaws H, et al. Expression of the Frizzled receptors and their co-receptors in calcified human aortic valves. Can J Physiol Pharmacol (2018) 96(2):208–14. doi: 10.1139/cjpp-2017-0577
101. Alfieri CM, Cheek J, Chakraborty S, Yutzey KE. Wnt signaling in heart valve development and osteogenic gene induction. Dev Biol (2010) 338(2):127–35. doi: 10.1016/j.ydbio.2009.11.030
102. Fang M, Alfieri CM, Hulin A, Conway SJ, Yutzey KE. Loss of β-catenin promotes chondrogenic differentiation of aortic valve interstitial cells. Arterioscler Thromb Vasc Biol (2014) 34(12):2601–8. doi: 10.1161/ATVBAHA.114.304579
103. Rajamannan NM. The role of Lrp5/6 in cardiac valve disease: LDL-density-pressure theory. J Cell Biochem (2011b) 112(9):2222–9. doi: 10.1002/jcb.23182
104. Hulin A, Moore V, James JM, Yutzey KE. Loss of Axin2 results in impaired heart valve maturation and subsequent myxomatous valve disease. Cardiovasc Res (2017) 113(1):40–51. doi: 10.1093/cvr/cvw229
Keywords: calcific aortic valve disease, calcification, inflammation, oxidative stress, lipids, signal transduction
Citation: Hulin A, Hego A, Lancellotti P and Oury C (2018). Advances in Pathophysiology of Calcific Aortic Valve Disease Propose Novel Molecular Therapeutic Targets. Front. Cardiovasc. Med. 5:21. doi: 10.3389/fcvm.2018.00021
Received: 31 January 2018; Accepted: 26 February 2018;
Published: 14 March 2018
Edited by:
Manvendra K. Singh, Duke Medical School, National University of Singapore, SingaporeReviewed by:
Chinmay M Trivedi, University of Massachusetts Medical School, United StatesKazufumi Nakamura, Okayama University, Japan
Copyright © 2018 Hulin, Hego, Lancellotti and Oury. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cécile Oury, Y2VjaWxlLm91cnlAdWxnLmFjLmJl