 Goro Katsuumi
Goro Katsuumi Ippei Shimizu
Ippei Shimizu Yohko Yoshida
Yohko Yoshida Tohru Minamino
Tohru Minamino- 1Department of Cardiovascular Biology and Medicine, Niigata University Graduate School of Medical and Dental Sciences, Niigata, Japan
- 2Division of Molecular Aging and Cell Biology, Niigata University Graduate School of Medical and Dental Sciences, Niigata, Japan
In mammals, aging is associated with accumulation of senescent cells. Stresses such as telomere shortening and reactive oxygen species induce “cellular senescence”, which is characterized by growth arrest and alteration of the gene expression profile. Chronological aging is associated with development of age-related diseases, including heart failure, diabetes, and atherosclerotic disease, and studies have shown that accumulation of senescent cells has a causative role in the pathology of these age-related disorders. Endothelial cell senescence has been reported to develop in heart failure and promotes pathologic changes in the failing heart. Senescent endothelial cells and vascular smooth muscle cells are found in atherosclerotic plaque, and studies indicate that these cells are involved in progression of plaque. Diabetes is also linked to accumulation of senescent vascular endothelial cells, while endothelial cell senescence per se induces systemic glucose intolerance by inhibiting skeletal muscle metabolism. A close connection between derangement of systemic metabolism and cellular senescence is also well recognized. Aging is a complex phenomenon, and there is no simple approach to understanding the whole process. However, there is accumulating evidence that cellular senescence has a central role in the development and progression of various undesirable aspects of aging. Suppression of cellular senescence or elimination of senescent cells reverses phenotypic changes of aging in several models, and proof-of-concept has been established that inhibiting accumulation of senescent cells could become a next generation therapy for age-related disorders. It is clear that cellular senescence drives various pathological changes associated with aging. Accordingly, further investigation into the role of this biological process in age-related disorders and discovery of senolytic compounds are important fields for future exploration.
Introduction
In aging societies, the discrepancy between the total lifespan and the healthy lifespan is becoming a major problem. Chronological aging is associated with a higher prevalence of age-related diseases, including heart failure, diabetes, and atherosclerotic disorders with or without various comorbidities, resulting in impairment of the quality of life by limitation of normal activities. Thus, aging is associated with several undesirable processes. The mechanisms of aging and age-associated disorders are complex, and thus cannot be comprehended by a simple approach. However, recent studies have indicated a pivotal role of cellular senescence in the progression of age-related disorders (1–5). Back in the 1960s, Hayflick et al. demonstrated that fibroblasts have limited potential to replicate (6), indicating that aging also occurs at the cellular level. Such aging of cells is currently described as “cellular senescence”. Senescent cells become enlarged and flattened. In association with proliferative arrest, alterations of gene expression by these cells lead to secretion of pro-inflammatory molecules (7). This is known as the senescence-associated secretory phenotype, and it results in chronic sterile inflammation that promotes tissue remodeling. Senescent cells have been found in various organs of animal models, as well as in elderly humans and persons with age-related disorders. There is evidence that cellular senescence in the vasculature, termed “vascular senescence”, is crucially involved in the pathogenesis of cardiovascular and metabolic disorders. Vascular senescence has been reported to promote atherosclerosis (8), systolic cardiac dysfunction (9, 10), and systemic metabolic dysfunction (11). In this review, we delineate the role of cellular senescence in the diseases associated with aging, focusing on vascular senescence in cardiovascular and metabolic disorders, and discuss the potential usefulness of therapies targeting senescent cells.
Role of Cellular Senescence in Aging and Age-Related Diseases
Diverse deleterious changes of cells and tissues accumulate with the progression of aging, leading to a decline of physiological activity and an increased risk of death. Organ function deteriorates with chronological aging, and this biological process is characterized by cellular senescence, changes of intercellular communication, mitochondrial dysfunction, and deregulation of nutrient sensing (1). It is well known that DNA damage, telomere shortening, oncogenic stress, and exposure to high levels of reactive oxygen species (ROS) all occur with chronological aging, and signaling via the p53 pathway has been reported to increase under these conditions (12). The p53 protein is a transcriptional factor with a crucial role in maintenance of genomic stability that mediates the coordination of DNA repair, cell cycle regulation, apoptosis, and cellular senescence. Because p53 is involved in suppression of tumorigenesis, it has been described as the “guardian of the genome” (13). In addition to its role in the repair of DNA damage and maintenance of genomic stability, studies have indicated that p53 contributes to a broad spectrum of biological processes, such as cell metabolism, autophagy, antioxidant defenses, and angiogenesis (13–15). Accordingly, p53 signaling is thought to have a central role in cellular senescence. Somatic cells have a finite lifespan and eventually enter a state of irreversible growth arrest termed “replicative senescence.” Telomeres are repetitive nucleotide sequences located at the terminals of mammalian chromosomes that undergo incomplete replication during cell division, resulting in telomere shortening. Because telomeres are essential for chromosomal stability and DNA replication, DNA damage is recognized when telomere shortening exceeds the physiological range and this triggers cellular senescence, mainly via the p53 or p16 signaling pathways. “Stress-induced premature senescence” is another type of cellular senescence that is triggered by various stress signals, including DNA damage induced by oxidative stress or irradiation, constitutive activation of mitogenic stimuli, oncogenic activation, and metabolic stress. It is also mediated via the p53 or p16 signaling pathways. Preference for one pathway over the other depends on the cell type and also varies among species (16, 17). In humans, telomere dysfunction activates either p53 or p16 signaling, while only p53 signaling is activated in rodents (18). It is generally accepted that p53 signaling is primarily activated by DNA damage and telomere dysfunction, while p16 signaling is primarily linked to mitogenic stress, chromatin disruption, and general cellular stress (16, 17, 19) (Figure 1).
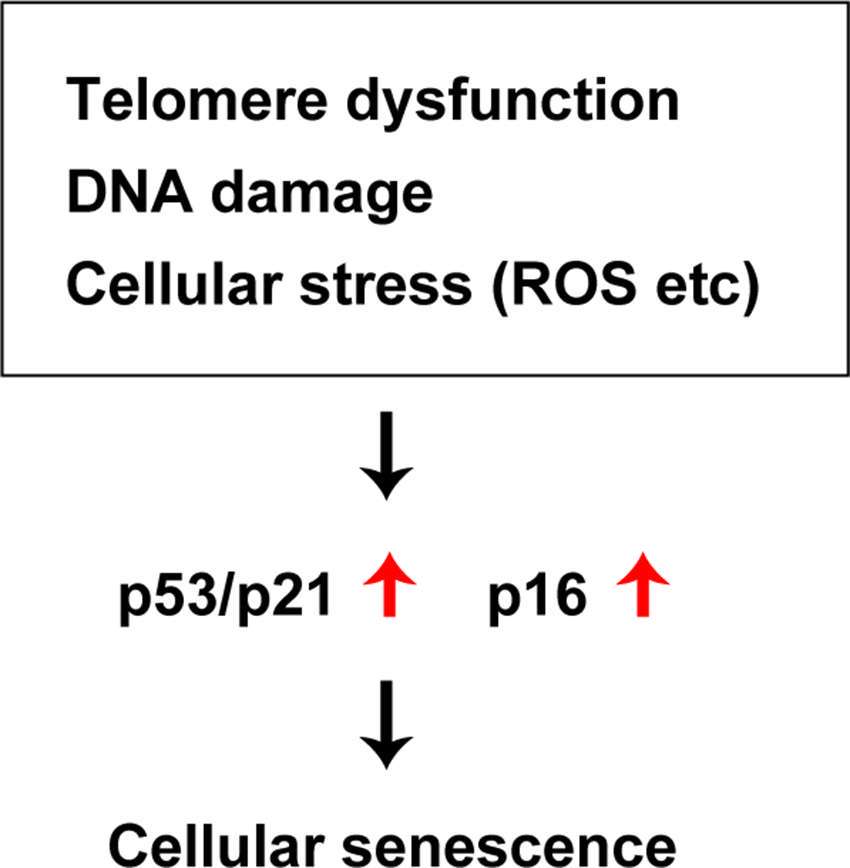
Figure 1. Common pathways of cellular senescence. Telomere dysfunction, DNA damage, cellular stress (ROS etc) up-regutate p53/p21, p16 signal and induces cellular senescence.
It was reported that p53 is increased in the failing heart, in aged vessels, and in the visceral fat of patients with obesity or heart failure. Studies have indicated a pathological role of p53-induced cellular senescence in aging and age-related disorders, including heart failure, atherosclerotic disease, obesity, and diabetes (10, 11, 15, 20–24). However, there is controversy about the role of p53 in aging and age-related diseases (3, 25, 26). In some settings, p53 signaling has been shown to have a beneficial effect by suppression of aging. Matheau and colleagues reported that Trp53/Cdkn2a transgenic mice were resistant to carcinogenesis and had a longer median lifespan (27). It was also reported that Trp53 transgenic (“Super p53”) mice displayed resistance to carcinogenesis without any signs of premature aging, and these mice showed normal glucose tolerance on a standard diet (28–30). Furthermore, Baker et al. found that loss of p53 or p21 accelerated cellular senescence in the adipose tissue and skeletal muscle of BubR1 progeroid mice (31). They also reported that p19Arf, acting upstream of p53, suppressed senescence and aging in the same progeroid model (32). Taken together, these various reports suggest that the p53/p21 signaling pathways regulate cellular senescence in a context-dependent manner.
Interestingly, it was recently reported that elimination of senescent cells by genetic manipulation inhibited age-related degenerative changes in several organs of mice, such as the heart and kidneys (33). Other studies have identified several pharmacological agents that selectively damage and remove senescent cells, and these compounds have been described as “senolytic agents”. For example, an inhibitor of anti-apoptotic proteins (ABT263) depletes senescent bone marrow hematopoietic stem cells and senescent muscle cells in a chronological aging model, leading to rejuvenation of these tissues (34). The gene expression profile of senescent cells is shifted toward a pro-inflammatory phenotype associated with the secretion of biologically active molecules (senescence-associated secretory phenotype). In vitro studies have shown that exposure of young fibroblasts to senescent fibroblast promotes senescence of the young cells via a gap junction-mediated process, which has been described as the “bystander effect” (35). Studies have shown that senescent cells damage their local environment and promote tissue remodeling in age-related disorders, suggesting that inhibition of cellular senescence and/or elimination of senescent cells could be potential next generation therapies for diseases associated with aging.
Biological Markers of Cellular Senescence
Biological markers reflecting direct evidence of cellular senescence have not yet been identified, but several markers are used to indirectly detect senescent cells, among which senescence-associated beta-galactosidase (SA-β-gal) activity is the most common. Lysosomal beta-galactosidase activity is normally detected at a low pH (usually around pH 4), but becomes detectable at a higher pH (pH 6) in senescent cells due to marked expansion of the lysosomal compartment (36). Other established markers of cellular senescence include high expression of p53, p16, p21, p38 mitogen-activated protein kinase (p38MAPK) and γH2AX, reflecting the activation of DNA damage responses (4, 37–40). In addition, high mobility group A (HMGA) proteins or heterochromatin markers, including HP1 and tri-methylated lysine 9 histone H3 (H3K9me3), are recognized as molecular markers of senescence-associated heterochromatin foci and are considered to indicate cellular senescence (40).
Cardiac Aging Predisposes to Heart Failure
Heart failure has a high prevalence among the elderly (41). The prognosis of severe heart failure is still unacceptably poor, and there is an urgent need to find better therapies for this condition. Age-related heart failure develops in persons without established risk factors, such as hypertension, obesity, diabetes, or atherosclerotic diseases (42, 43). Heart failure without systolic dysfunction is classified as heart failure with a preserved ejection fraction (HFpEF), and occurs in approximately half of all patients with heart failure. HFpEF is prevalent among the elderly and lack of specific therapy for this type of heart failure is a major clinical problem. The mechanism of HFpEF is still not fully understood, although there is evidence of cardiac endothelial cell remodeling being involved in its onset and progression (44). It was also reported that coronary microvascular endothelial inflammation is critically involved in the pathology of HFpEF (45), while a recent study indicated a causative role of senescent signaling in this disorder (46). Thus, the physiological aging process seems to increase susceptibility to the onset of heart failure, considering that the prevalence of heart failure increases with age. Various studies have indicated that cellular senescence is critically involved in the pathology of heart failure, as described below.
Vascular Senescence and Heart Failure
Endothelial Cell Senescence
Although the role of cellular senescence in the failing heart is still not fully understood, a number of studies have suggested a pathological influence on heart failure. The cardiac level of p53 is increased in a murine model of left ventricular pressure overload, leading to suppression of myocardial angiogenesis that results in capillary rarefaction, tissue hypoxia, and cardiac dysfunction (15). Chronic sterile inflammation develops in the failing heart, and it is now well accepted that such inflammation is one of the mechanisms underlying cardiac remodeling (47). It was recently demonstrated that activation of p53 signaling in vascular endothelial cells induces cardiac inflammation and remodeling in a murine model of left ventricular (LV) pressure overload (10). Expression of p53 by capillary endothelial cells in the left ventricle increases in response to LV pressure overload, leading to elevated expression of intercellular adhesion molecule (ICAM)−1 by these cells that promotes infiltration of macrophages and cardiac inflammation. Conversely, depletion of p53 from capillary endothelial cells results in suppression of ICAM-1 expression and cardiac inflammation with improvement of cardiac dysfunction. Activation of the sympathetic nervous system occurs in heart failure and is associated with a poor prognosis (48). It was reported that the sympathetic nervous system/ROS axis increases p53 expression by endothelial cells in a murine model of LV pressure overload (10). In another study, depletion of p53 from endothelial cells improved capillary rarefaction and cardiac function, while suppressing cardiac fibrosis and remodeling (9). These findings indicate that endothelial p53 signaling suppresses angiogenesis, thereby promoting capillary rarefaction in the failing heart. Inhibition of p53 in endothelial cells could potentially become a next generation therapy for patients with heart failure and a reduced ejection fraction. As mentioned above, about half of all heart failure patients have HFpEF with a preserved ejection fraction. There are several established risk factors for HFpEF, including overweight/obesity, hypertension, diabetes, and aging. Cardiomyocyte hypertrophy and interstitial fibrosis develop in patients with HFpEF, leading to incomplete myocardial relaxation and increased wall stiffness (49–51). It is generally accepted that coronary microvascular inflammation is central to the pathogenesis of HFpEF (45), and it was recently demonstrated that endothelial cell senescence also makes a contribution. When mice with accelerated senescence were fed a high-fat, high-salt diet, both endothelial cell senescence and inflammation increased in cardiac tissue, along with the typical hemodynamic and structural changes of HFpEF (46). Considering that cellular senescence induces vascular dysfunction and inflammation, it seems reasonable that it would also promote pathologic changes of HFpEF. Accordingly, suppression of endothelial cell senescence may be a therapeutic option for this currently untreatable disorder (Figure 2).
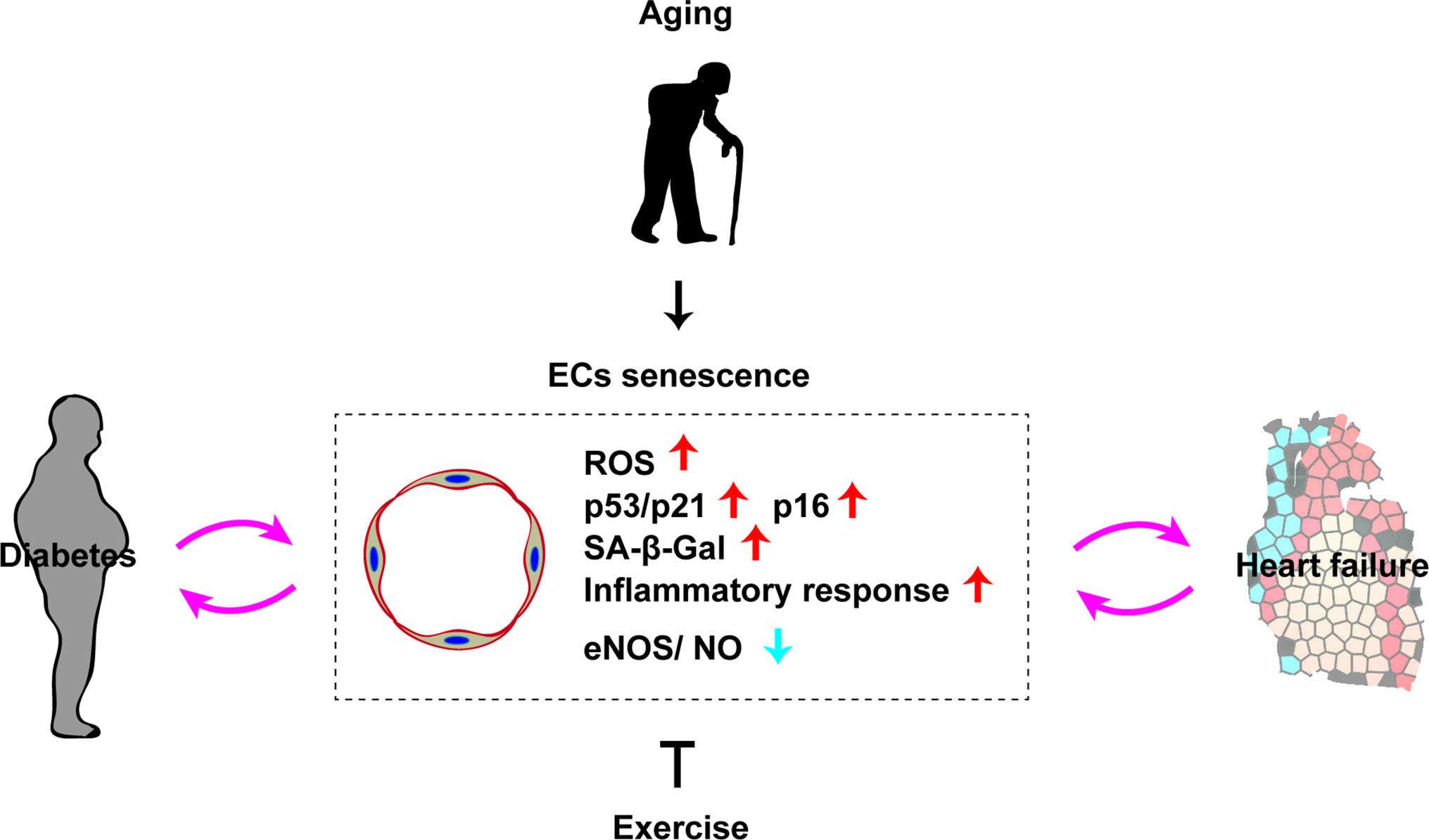
Figure 2. Role of endothelial senescence in cardio-metabolic disease. Chronological aging induces vascular endothelial cell senescence (EC senescence as characterized with an increase of ROS, p53/p21, p16, SA-β-Gal, inflammatory response, and reduced eNOS/NO level). EC senescence has a pivotal role in the progression of diabetes and heart failure. Exercise has a potential to suppress ECs senescence.
Vascular Aging Predisposes to Atherosclerotic Diseases
Structural and Functional Changes of Aging Arteries
Coronary artery disease and stroke are associated with arterial dysfunction. Arterial remodeling occurs with aging, even in the absence of cardiovascular disease and cardiovascular risk factors. While aging is a physiological process and not a pathological condition, studies indicate that aging per se is linked with vascular remodeling that predisposes to cardiovascular disease. Aged arteries are characterized by an increase of the intima/media thickness ratio, which was reported to increase by 2- to 3-fold from 20 to 90 years of age (52, 53). Vascular smooth muscle cells switch from the “contractile” to “synthetic” phenotype with aging and this change contributes to intimal thickening, which is associated with increased arterial permeability and leads to development of atherosclerotic disease. The arterial media also becomes thicker with aging and its cellularity decreases simultaneously (54). Moreover, the length and circumference of the aorta increase with aging (55), and these structural changes reflect increased collagen production and a corresponding decline of the elastin content (56). In association with such changes, aged vessels show reduced compliance, reduced elasticity/distensibility, and increased stiffness, resulting in a higher systolic blood pressure and lower diastolic pressure (57). Medial calcification is another characteristic of aged vessels. In association with other age-related disorders like hypertension, dyslipidemia, and diabetes, such vascular remodeling increases susceptibility to atherosclerotic vascular diseases. In elderly patients, atherosclerotic plaques tend to become larger and vascular stenosis becomes more severe over time. Aged rabbits fed a high fat diet developed more severe atherosclerotic lesions compared to young animals on the same diet. Therefore, it is well accepted that aging per se promotes the pathogenesis of atherosclerotic disorders, and studies have suggested that cellular senescence has a critical role in this process.
Vascular Senescence in Arterial Diseases
ROS and chronic low-grade sterile inflammation are two major contributors to the progression of age-related vascular dysfunction. Senescent cells accumulate in the arteries with aging irrespective of whether or not a person has age-related vascular disorders (58–61). Along with aging, vascular tissues of rodents and humans show elevation of the levels of p16, p21, phosphorylated p38, and double-stranded DNA breaks, in association with high SA-β Gal activity (62–65). It was reported that expression of p53 and p21 is increased in the arteries of elderly persons, together with structural breakdown of telomeres known as telomere uncapping (61). Both telomere length and telomerase activity were found to be reduced in endothelial progenitor cells from patients with coronary heart disease (66). In patients with chronic heart failure, telomere attrition was identified in circulating leukocytes (67). Moreover, the leucocyte telomere length displays an inverse association with the risk of coronary heart disease independently of conventional vascular risk factors (68). Accordingly, it is generally accepted that telomere length and telomerase activity are involved in human cardiovascular disease (69). Interestingly, senescent cells are increased in the coronary arteries of patients with ischemic heart disease, but not in the internal mammary arteries (58). Endothelial cells and vascular smooth muscle cells (VSMCs) from patients with abdominal aortic aneurysm (AAA) have the phenotypic features commonly observed in senescent cells (60). Hypertension is an established risk factor for atherosclerotic diseases, and it was reported that binding of p53 to the p21 promoter is increased in the arteries of hypertensive patients. While telomere length is comparable between patients with hypertension and controls, telomere uncapping is 2-fold higher in hypertensive patients (70). A murine model of genomic instability demonstrated senescence of endothelial cells and VSMCs in the aorta, along with impaired vasodilation, increased vascular stiffness, and hypertension (71). In hypertensive rats treated with deoxycorticosterone acetate and salt, overexpression of p16 was detected in the coronary arteries (72). Aortic p16 expression was elevated in another model of hypertension (mice administered an endothelial nitric oxide synthase inhibitor) (73), indicating the existence of a vicious circle between cellular senescence and hypertension. Thus, studies have shown that senescent cells accumulate in the vessels of patients with atherosclerosis, hypertension, aneurysms, diabetes, and intimal hyperplasia (Figure 3), so the role of endothelial cell, VSMC and immune cell senescence in arterial diseases is discussed next.
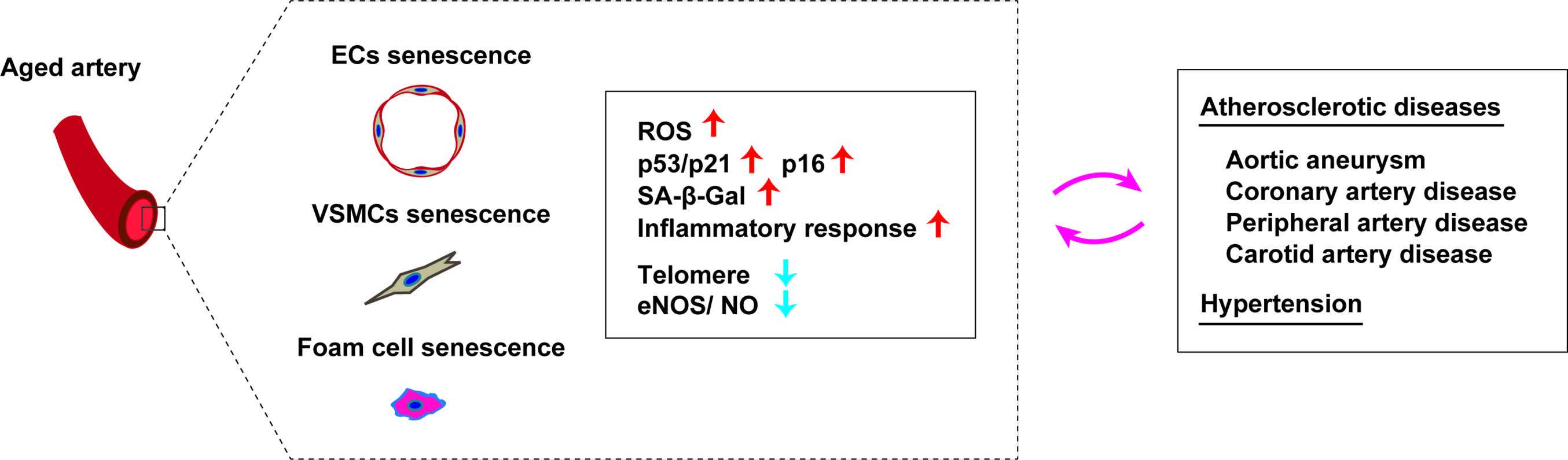
Figure 3. Cellular senescence in aged arteries. Aged arteries are characterized by accumulation of senescent vascular endothelial cells (EC), senescent vascular smooth muscle cells (VSMCs), and senescent foam cells. Accumulation of these senescent cells are associated with an increase of ROS, p53/p21, p16, SA-β-Gal, inflammatory response, telomere attrition and reduced eNOS/NO level). Senescence of these cells promotes pathological changes in atherosclerotic diseases and also has a role in development of hypertension.
Endothelial Cell Senescence in Arterial Diseases
Endothelial cells are critically important for maintaining vascular homeostasis and are involved in various biological functions, including angiogenesis, blood pressure regulation, coagulation, and systemic metabolism. Aged endothelial cells develop a dysfunctional phenotype that is characterized by reduced proliferation and migration, decreased expression of angiogenic molecules, and low production of nitric oxide (NO), which is synthetized by NO synthase (NOS) and mediates vasodilatation. In dysfunctional endothelial cells, NO production is generally reduced due to low NOS activity. This change is associated with impairment of endothelium-dependent dilatation (EDD), which is reported to predict future cardiovascular events. Dysfunctional endothelial cells develop pro-oxidant, pro-inflammatory, vasoconstrictor, and prothrombotic properties, and studies indicate that cellular senescence has a pathological role in such phenotypic aging.
Senescent endothelial cells have been found in atherosclerotic plaque (58). An autopsy study of patients with ischemic heart disease revealed that SA-β-gal activity is increased in the coronary arteries, but not in the internal mammary arteries. In the coronary arteries, SA-β-gal activity is high in cells located on the luminal surface (probably endothelial cells). ICAM-1 is also increased in senescent human aortic endothelial cells, while both endothelial nitric oxide synthase (eNOS) and NO activity are reduced in these cells compared to young cells. Importantly, these pathological phenotypic changes induced by replicative senescence were suppressed by activation of telomerase reverse transcriptase in aged human aortic endothelial cells, indicating that telomere shortening induces endothelial cell senescence and has pathological consequences in atherosclerotic diseases (58). In patients with AAA, telomeres are significantly shorter and oxidative DNA damage is more severe in endothelial cells from the aneurysmal region (60). In atherosclerotic mice, disturbance of flow in the ascending aorta and aortic arch promotes endothelial cell senescence, and in vitro studies have indicated that aberrant flow is a signal inducing cellular senescence (74). One of the problems related to an increase of senescent cells is development of the senescence-associated secretory phenotype, which is characterized by production of pro-inflammatory cytokines with a causal role in tissue remodeling (7). In human arterial endothelial cells with replicative senescence, levels of H2O2 and O2– are high and NO production is reduced. High ROS levels in senescent endothelial cells are thought to accelerate senescence. Aging is reported to be linked with increased circulating levels of pro-inflammatory cytokines, such as interleukin-6, tumor necrosis factor alpha, and monocyte chemoattractant protein-1 (75). It is highly possible that accumulation of senescent endothelial cells in the arteries of elderly persons induces chronic sterile inflammation and vascular remodeling, increasing susceptibility to atherosclerotic diseases. Controversy exists as to whether physical activity is associated with telomere length, since physical activity is positively correlated with telomere length in some studies, but not in other studies (76). It is generally accepted that physical activity improves vascular structure and function in humans and rodents. Bioavailability of NO declines with aging in association with elevation of ROS levels, while these changes are ameliorated or reversed by physical activity. Recently, it was shown that older individuals who performed exercise had lower levels of p53, p21, and p16 in endothelial cells from the brachial arteries and antecubital veins compared to sedentary older individuals, indicating that physical activity suppresses senescence of human vascular cells (77). Thus, there is evidence that physical activity improves vascular function, but further studies are needed to identify the detailed molecular mechanisms involved (Figure 2).
Vascular Smooth Muscle Cell Senescence in Arterial Diseases
Functional changes of VSMCs occur with aging, partly due to deregulation of TGF-β signaling, and these cells undergo transformation from a “contractile” to a “synthetic” phenotype. It was reported that aged SMCs show enhancement of inducible NOS (iNOS) activity, as well as higher expression of ICAM-1 and angiotensinogen in response to stress (78, 79). Intimal thickening develops with aging, partly due to increased production of collagen and a corresponding decrease of elastin (56). Generally, intimal thickening is widespread and concentric in the aorta, while it is eccentric in the coronary arteries. Intimal thickening is a preatherosclerotic lesion because topographic correspondence has been demonstrated between the sites of intimal thickening and atherosclerotic plaques. Telomeres are shorter in cells from atherosclerotic plaque in the human aorta compared to normal vessels. Telomeres are also shorter in VSMCs from the fibrous cap of atheroma compared to VSMCs from the normal vascular media, and these cells are positive for SA-β-gal staining along with elevated p16 and p21 expression. It was reported that oxidative stress induces DNA damage in VSMCs and suppresses telomerase activity, leading to telomere shortening and cellular senescence that contribute to acceleration of atherosclerotic disorders (80). In addition, senescence of VSMCs was reported in apolipoprotein E null mice treated with angiotensin II (81). Smooth muscle 22α is an actin-binding protein that is known as a marker of smooth muscle cell senescence. Recently, it was shown that smooth muscle 22α promotes angiotensin II-induced cellular senescence by suppressing Mdm-2-mediated degradation of p53 (82). In addition, Cafueri et al. reported that VSMCs from the aneurysms of AAA patients display telomere attrition and marked oxidative DNA damage (60). Senescent VSMCs have been identified in atherosclerotic lesions of patients with coronary artery disease, AAA, and peripheral artery disease (58). It was also reported that 18% of VSMCs from carotid artery plaques are positive for p16 and p21, in association with detectable SA-β-gal activity (80). Furthermore, SA-β-gal positive VSMCs in carotid plaques express interleukin-6, suggesting that senescent VSMCs have a senescence-associated secretory profile and a causative role in the progression of atherosclerotic disorders (83). Senescent VSMCs show dysregulated production of pro-inflammatory cytokines, growth factors, and extracellular matrix modifiers, accelerating the process of vascular remodeling. Some inducers of senescence are common to VSMCs and vascular endothelial cells, such as ROS and angiotensin II (84, 85), while hypoxic stress was reported to inhibit senescence by promoting telomerase activity (86). It was recently found that senescent VSMCs in atherosclerotic plaque display loss of telomeric repeat-binding factor-2 (TRF2), a protein localized in the telomeres. TRF2 overexpression reduces DNA damage, accelerates DNA repair, and suppresses cellular senescence in vitro, while introduction of TRF2 into loss of function mutants results in the opposite phenotype. Studies using transgenic mice have shown that VSMC-specific loss of TRF2 function increases atherosclerosis and necrotic core formation in vivo, while these pathological changes are suppressed in mice with VSMC-specific gain of TRF2 function (87). These results clearly indicate that inhibition of VSMC senescence is extremely important for suppressing the progression of atherosclerotic diseases.
Immune Cell Senescence in Arterial Diseases
In human vascular cells, telomere length shows a strong inverse correlation with aging, and telomere shortening is linked with cardiovascular disease and diabetes (88, 89). Elderly individuals with short telomeres in leukocyte DNA are reported to have a higher mortality rate, partly attributable to increased death from heart disease (90). Monocytes from patients with atherosclerosis exhibit increased production of ROS and various pro-inflammatory cytokines, including MCP-1, IL-6, IL-1β, and TNF-α (91). It was recently demonstrated that senescent intimal foam cells accumulate in atherosclerotic lesions and act as the key drivers of atheroma formation. Importantly, specific deletion of these senescent cells by genetic or pharmacological approaches has been shown to reverse atherosclerosis in mice (92). According to another report, cellular senescence mediated by p16INK4a promotes pro-inflammatory phenotypic changes in macrophages (93). These studies indicate that leukocyte senescence is involved in the progression of atherosclerotic plaque, suggesting that suppression of leukocyte senescence may be an important approach for combating atherosclerotic diseases.
Vascular Senescence in Metabolic Syndrome
Chronic sterile inflammation of visceral fat develops in patients with heart failure, obesity, and/or diabetes, and is well accepted to have a central role in inducing systemic insulin resistance and progression of metabolic disorders. It was previously reported that p53 induces inflammation of visceral adipose tissue in murine models of obesity or heart failure, which is involved in the progression of these age-related diseases (21, 24, 94). Occurrence of cellular senescence in visceral fat was reported to result in deterioration of systemic metabolic health. Capillaries have a crucial role in metabolically active organs, including skeletal muscle and brown adipose tissue. Capillarization of skeletal muscle was reported to increase in older adults after exercise training, and this increase of capillaries leads to enhancement of systemic insulin sensitivity (95). It was recently demonstrated that vascular endothelial cell senescence induces systemic metabolic dysfunction (11) (Figure 2). Conversely, it has been well documented that obesity is associated with vascular senescence. In metabolically unhealthy persons with obesity and/or diabetes, insulin/insulin receptor/insulin receptor substrate/phosphoinositide 3-kinase/Akt signaling is down-regulated in vascular cells, while insulin receptor/son of sevenless/growth factor receptor bound protein/mitogen-activated protein kinase (MAPK) signaling is enhanced. This is known as “selective insulin resistance”, and it mediates pro-atherosclerotic responses by activation of MAPK signaling. It was reported that activation of MAPK signaling by hyperinsulinemia and selective insulin resistance induces vascular remodeling through vasoconstriction, proliferation, and vascular cell migration (96). Various studies have indicated the existence of a vicious circle between vascular senescence and metabolic syndrome, as discussed below.
Endothelial Cell Senescence in Metabolic Syndrome
Capillary network formation is critically important for morphogenesis and maintenance of homeostasis, while vascular dysfunction induces organ malfunction and systemic metabolic disorders (97, 98). In animal studies, diabetes has been shown to induce vascular cell senescence. For example, endothelial cell senescence develops in the aortas of Zucker diabetic rats and hyperglycemic mice or rats (99–101). These reports indicate that hyperglycemia induces cellular senescence, while there is also evidence that cellular senescence per se promotes systemic metabolic dysfunction. It is widely accepted that skeletal muscle contributes to glucose disposal, so maintaining skeletal muscle homeostasis is crucial for systemic metabolic health. Metabolic stress induces accumulation of lipids and chronic sterile inflammation in skeletal muscle, contributing to development of systemic insulin resistance (102, 103). It was recently demonstrated that endothelial cell senescence suppresses skeletal muscle metabolism, leading to systemic glucose intolerance. Metabolic stress induced by dietary obesity increases p53 expression in the vascular endothelium (11), while endothelial cell-specific depletion of p53 reduces both visceral and subcutaneous fat volumes and improves systemic glucose intolerance. It is generally accepted that eNOS has a protective role in the cardiovascular system, which is mainly mediated by production of NO. It was reported that eNOS up-regulates skeletal muscle expression of peroxisome proliferator-activated receptor-γ coactivator-1α, a master regulator of mitochondrial biogenesis and cell metabolism, while this up-regulation is suppressed by p53. Down-regulation of p53 expression in vascular endothelial cells promotes glucose uptake by skeletal muscle through up-regulation of glucose transporter-1 expression in endothelial cells, and contributes to better systemic metabolic health. These findings indicate that suppression of endothelial cell senescence is important for maintenance of systemic metabolic health (11).
Potential Next Generation Therapies Targeting Senescent Cells for Cardiovascular and Metabolic Disorders
It was recently established that selective depletion of senescent cells (“senolysis”) reverses phenotypic changes of aging (33, 34, 104–106). Accumulation of senescent cells promotes chronic sterile inflammation in the visceral adipose tissue of patients with obesity and elderly persons. In a murine model of premature aging, elimination of p16-positive senescent cells contributed to suppression of the aging phenotype in several organs, including epididymal/inguinal white adipose tissue, the heart, and the kidney (33). Several agents causing selective depletion of senescent cells (senolytic activity) have been identified. It was reported that an anticancer agent (ABT263) has a senolytic effect by selectively removing p16-positive senescent cells from the bone marrow via apoptosis, leading to rejuvenation of hematopoietic stem cells during aging (34). In addition, Xu et al. showed that depletion of senescent cells in aged mice preserved adipogenesis and increased insulin sensitivity (104). Zhu et al. showed that administration of another senolytic therapy (dasatinib + quercetin: D + Q) significantly improved systolic cardiac function and reduced the left ventricular end-systolic dimension in 24-month-old mice (105). Treatment with D + Q also improved vasomotor function in aged mice, as well as reducing aortic calcification and osteogenic signaling in hypercholesterolemic mice (107). Regarding genetic approaches, elimination of p16-positive senescent cells from plaques suppressed pathologic changes in low-density lipoprotein receptor-deficient mice (92). Senescent VSMCs show hypermetabolic changes, with increased glycolysis and oxygen consumption. Administration of 2-deoxyglucose causes greater depletion of senescent VSMCs than control VSMCs in vitro, but clinical trials investigating the anticancer effect of 2-deoxyglucose have identified the issue of toxicity (108). Depletion of specific components to alter cell metabolism has now attracted attention in the field of anticancer therapy, including the search for senolytic agents targeting cell metabolism. There is evidence that elimination of senescent cells by administration of senolytic agents has the potential to become a next generation therapy for cardiovascular disorders (109, 110). Suppression of cellular senescence is another possibility. Sirtuin1 (SIRT1) is one of the most promising molecules to be studied in relation to suppression of aging. Activation of SIRT1-signaling was reported to prolong the lifespan of rodents, while overexpression of SIRT1 in VSMCs or vascular endothelial cells suppresses senescence and extends the survival of these cells. Resveratrol activates SIRT1, and administration of resveratrol was reported to prevent arterial wall inflammation and elevation of the pulse wave velocity by dietary obesity (111). Moreover, activation of SIRT1 attenuates arterial stiffness and hypertension in Klotho-haplodeficient mice (112). SIRT1 expression and activity are decreased in the VSMCs of patients with AAA, together with vascular cell senescence and elevated p21 expression, while SIRT1 inhibits p21-induced cellular senescence and contributes to suppression of vascular inflammation (113). Another study showed that calorie restriction up-regulates SIRT1 expression in vascular smooth muscle cells, and reduced the incidence of AAA (114). These results indicate that activation of SIRT1 in VSMCs may potentially prevent the progression of AAA (113, 114). Nicotinamide adenine dinucleotide (NAD) is a coenzyme involved in cell metabolism, redox reactions, and DNA repair, and it is well known to suppress aging (115). Nicotinamide phosphoribosyltransferase (Nampt) is the rate-limiting enzyme for conversion of nicotinamide to nicotinamide mononucleotide, enabling subsequent biosynthesis of NAD+. Nampt overexpression was reported to suppress senescence of VSMCs through a process mediated by SIRT1 signaling (116). In patients with aortic dilation, an inverse relationship between Nampt expression by VSMCs and the diameter of the ascending aorta was recently identified, and the authors concluded that NAD+ biosynthesis in the aortic media is important for protection against DNA damage and premature VSMC senescence (117). Systemic metabolic dysfunction is reported to induce cellular senescence in endothelial cells. Aged mice with systemic glucose intolerance and hyperinsulinemia show elevation of aortic NADPH oxidase-2 (Nox2) expression, while in vitro glucose and insulin challenge increases Nox2 and ROS levels in coronary microvascular endothelial cells, promoting cellular senescence along with elevation of p53 (118). Furthermore, hyperlipidemia associated with aging enhances mitochondrial oxidative stress and induces plaque instability in ApoE−/− mice (119). It was previously reported that Akt, which acts downstream of insulin signaling, negatively regulates the lifespan of human endothelial cells via p53/p21 signaling (120). Dietary intake of rapamycin (an inhibitor of mTOR, a molecule that acts downstream of the insulin signaling pathway) was shown to reverse age-related vascular dysfunction and oxidative stress, in association with reduced arterial expression of the senescence marker p19 (121). In the LEADER trial, a glucagon-like peptide 1 analogue (liraglutide) reduced the death rate from cardiovascular disease in patients with type 2 diabetes, as well as decreasing nonfatal myocardial infarction and nonfatal stroke (122). Another glucagon-like peptide 1 analogue (exenatide) showed beneficial vascular effects, partly via enhancing adiponectin production, and suppressed oxidative stress and inflammation in the vascular plaques of ApoE−/− mice (123). Taken together, these studies indicate that, in addition to use for the inhibition of systemic metabolic disorders, suppression of cellular senescence and/or elimination of senescent cells could become next generation therapies for cardiovascular disorders.
Conclusion and Future Directions
This review outlined the pathological role of vascular senescence in cardiovascular disease and metabolic disease. Both capillaries and arteries are critically important for delivery of nutrients and oxygen to the organs/tissues for maintenance of physiological function. Vascular endothelial cells and VSMCs have a crucial role in vascular homeostasis. Senescence of vascular cells promotes the development of age-related disorders, including heart failure, diabetes, and atherosclerotic diseases, while suppression of vascular cell senescence ameliorates phenotypic features of aging in various models. Recent findings have indicated that specific depletion of senescent cells reverses age-related changes. Considering that suppression of cellular senescence is associated with a risk of tumorigenesis, specific depletion of senescent cells may be a more promising approach to the treatment of age-related diseases. An issue that remains to be explored is the potential side effects of such treatment. For example, Demaria and colleagues found that genetic removal of senescent cells delayed wound healing in mice (124). We also need to identify the best senolytic agents, and optimize the dosage and administration and combinations for treatment of various conditions. Potential gender differences are another important research topic. Although the biological networks contributing to maintenance of homeostasis are extremely complex, it seems reasonable to explore senolytic agents that can act on specific cellular components or tissues. Several clinical trials of senolytic agents are currently ongoing. Survivors of hematopoietic stem cell transplantation are prone to premature aging, and one pilot clinical study is designed to test whether D + Q can suppress aging in these patients (Clinical Trials. Gov Identifier: NCT02652052). Another clinical trial is testing whether D + Q reduces pro-inflammatory cells obtained by skin biopsy in patients with idiopathic pulmonary fibrosis (Clinical Trials. Gov Identifier: NCT02874989). Furthermore, a clinical trial is ongoing to determine whether D + Q can reduce the senescent cell burden and frailty in patients with chronic kidney disease, as well as improving the function of adipose tissue-derived mesenchymal stem cells (Clinical Trials. Gov Identifier: NCT02848131). So far, only D + Q has been assessed in the clinical setting, and none of the current clinical trials are testing whether senolytic agents can inhibit cardiovascular disorders. However, depletion of senescent cells was demonstrated to suppress pathological progression of atherosclerotic plaque in rodents, suggesting that senolytic agents could become a next generation therapy for cardiovascular disorders (Table 1).
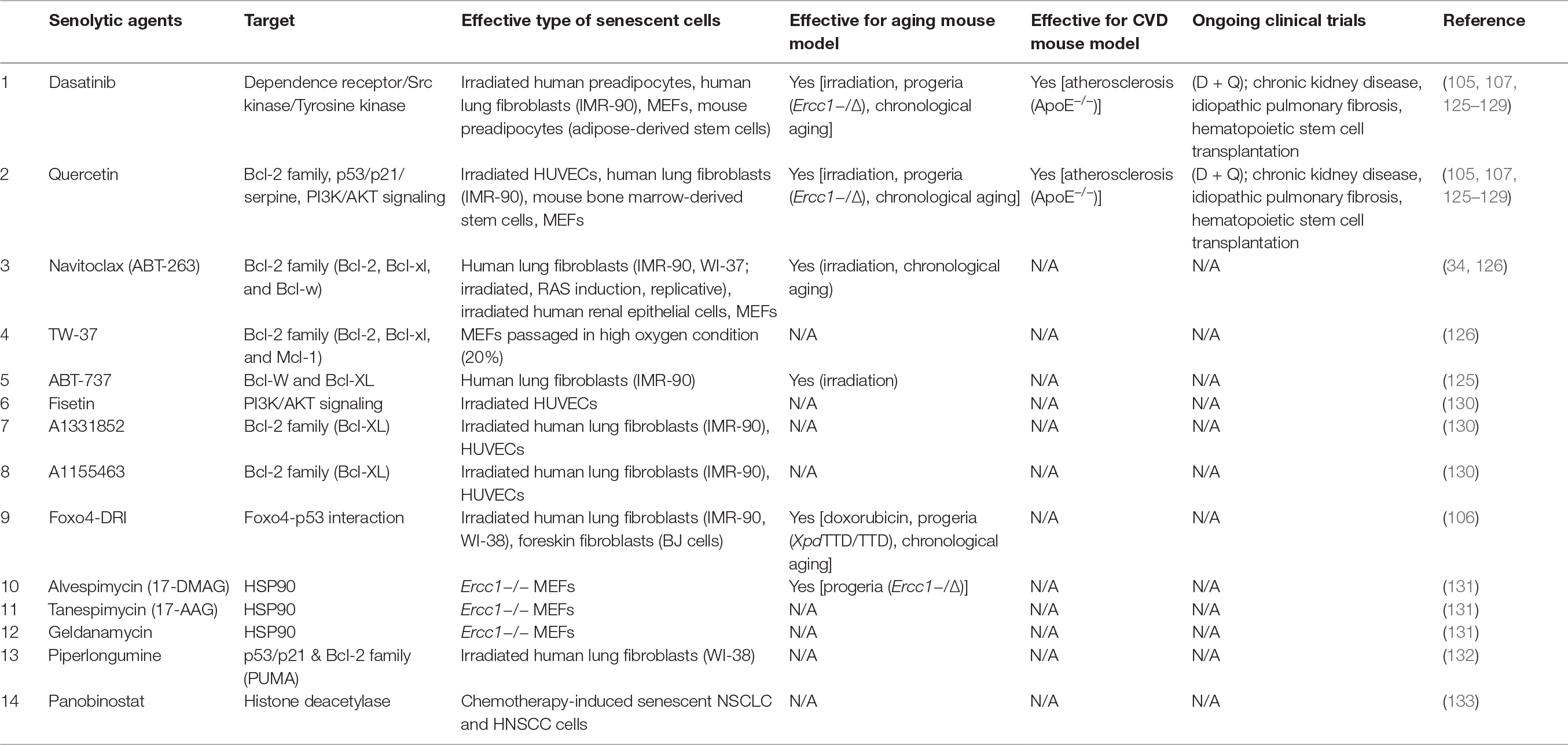
Table 1. Senolytic agents and their molecular targets
Author Contributions
GK, IS, YY, TM wrote the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by a Grant-in-Aid for Scientific Research (Grant number 17H04172, 25870127), a Grant-in-Aid for Scientific Research on Innovative Areas (Grant number 26115008), and a Grant-in-Aid for Exploratory Research from the Ministry of Education, Culture, Sports, Science and Technology (MEXT, Grant number 15K15306) of Japan and grants from the Takeda Medical Research Foundation, the Japan Foundation for Applied Enzymology, the Takeda Science Foundation, the SENSHIN Medical Research Foundation, the Terumo Foundation, the Manpei Suzuki Diabetes Foundation, the Naito Foundation, and the NOVARITIS foundation (to T.M.).
References
1. López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell (2013) 153(6):1194–217. doi: 10.1016/j.cell.2013.05.039
2. de Cabo R, Carmona-Gutierrez D, Bernier M, Hall MN, Madeo F. The search for antiaging interventions: from elixirs to fasting regimens. Cell (2014) 157(7):1515–26. doi: 10.1016/j.cell.2014.05.031
3. Shimizu I, Yoshida Y, Suda M, Minamino T. DNA damage response and metabolic disease. Cell Metab (2014) 20(6):967–77. doi: 10.1016/j.cmet.2014.10.008
4. van Deursen JM. The role of senescent cells in ageing. Nature (2014) 509(7501):439–46. doi: 10.1038/nature13193
5. Childs BG, Durik M, Baker DJ, van Deursen JM. Cellular senescence in aging and age-related disease: from mechanisms to therapy. Nat Med (2015) 21(12):1424–35. doi: 10.1038/nm.4000
6. Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res (1961) 25:585–621. doi: 10.1016/0014-4827(61)90192-6
7. Tchkonia T, Zhu Y, van Deursen J, Campisi J, Kirkland JL. Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J Clin Invest (2013) 123(3):966–72. doi: 10.1172/JCI64098
8. Wang JC, Bennett M. Aging and atherosclerosis: mechanisms, functional consequences, and potential therapeutics for cellular senescence. Circ Res (2012) 111(2):245–59. doi: 10.1161/CIRCRESAHA.111.261388
9. Gogiraju R, Xu X, Bochenek ML, Steinbrecher JH, Lehnart SE, Wenzel P, et al. Endothelial p53 deletion improves angiogenesis and prevents cardiac fibrosis and heart failure induced by pressure overload in mice. J Am Heart Assoc (2015) 4(2):e001770. doi: 10.1161/JAHA.115.001770
10. Yoshida Y, Shimizu I, Katsuumi G, Jiao S, Suda M, Hayashi Y, et al. p53-Induced inflammation exacerbates cardiac dysfunction during pressure overload. J Mol Cell Cardiol (2015) 85:183–98. doi: 10.1016/j.yjmcc.2015.06.001
11. Yokoyama M, Okada S, Nakagomi A, Moriya J, Shimizu I, Nojima A, et al. Inhibition of endothelial p53 improves metabolic abnormalities related to dietary obesity. Cell Rep (2014) 7(5):1691–703. doi: 10.1016/j.celrep.2014.04.046
12. Stewart SA, Weinberg RA. Telomeres: cancer to human aging. Annu Rev Cell Dev Biol (2006) 22:531–57. doi: 10.1146/annurev.cellbio.22.010305.104518
13. Lane DP. Cancer. p53, guardian of the genome. Nature (1992) 358(6381):15–16. doi: 10.1038/358015a0
14. Green DR, Chipuk JE. p53 and metabolism: Inside the TIGAR. Cell (2006) 126(1):30–2. doi: 10.1016/j.cell.2006.06.032
15. Sano M, Minamino T, Toko H, Miyauchi H, Orimo M, Qin Y, et al. p53-induced inhibition of Hif-1 causes cardiac dysfunction during pressure overload. Nature (2007) 446(7134):444–8. doi: 10.1038/nature05602
16. Beauséjour CM, Krtolica A, Galimi F, Narita M, Lowe SW, Yaswen P, et al. Reversal of human cellular senescence: roles of the p53 and p16 pathways. Embo J (2003) 22(16):4212–22. doi: 10.1093/emboj/cdg417
17. Campisi J. Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell (2005) 120(4):513–22. doi: 10.1016/j.cell.2005.02.003
18. Jacobs JJ, de Lange T. Significant role for p16INK4a in p53-independent telomere-directed senescence. Curr Biol (2004) 14(24):2302–8. doi: 10.1016/j.cub.2004.12.025
19. Donato AJ, Morgan RG, Walker AE, Lesniewski LA. Cellular and molecular biology of aging endothelial cells. J Mol Cell Cardiol (2015) 89(Pt B):122–35. doi: 10.1016/j.yjmcc.2015.01.021
20. Varela I, Cadiñanos J, Pendás AM, Gutiérrez-Fernández A, Folgueras AR, Sánchez LM, et al. Accelerated ageing in mice deficient in Zmpste24 protease is linked to p53 signalling activation. Nature (2005) 437(7058):564–8. doi: 10.1038/nature04019
21. Minamino T, Orimo M, Shimizu I, Kunieda T, Yokoyama M, Ito T, et al. A crucial role for adipose tissue p53 in the regulation of insulin resistance. Nat Med (2009) 15(9):1082–7. doi: 10.1038/nm.2014
22. Sahin E, Colla S, Liesa M, Moslehi J, Müller FL, Guo M, et al. Telomere dysfunction induces metabolic and mitochondrial compromise. Nature (2011) 470(7334):359–65. doi: 10.1038/nature09787
23. Shimizu I, Yoshida Y, Katsuno T, Tateno K, Okada S, Moriya J, et al. p53-induced adipose tissue inflammation is critically involved in the development of insulin resistance in heart failure. Cell Metab (2012) 15(1):787–787. doi: 10.1016/j.cmet.2012.04.014
24. Shimizu I, Yoshida Y, Moriya J, Nojima A, Uemura A, Kobayashi Y, et al. Semaphorin3E-induced inflammation contributes to insulin resistance in dietary obesity. Cell Metab (2013) 18(4):491–504. doi: 10.1016/j.cmet.2013.09.001
25. Vousden KH, Prives C. Blinded by the light: the growing complexity of p53. Cell (2009) 137(3):413–31. doi: 10.1016/j.cell.2009.04.037
26. Sahin E, Depinho RA. Axis of ageing: telomeres, p53 and mitochondria. Nat Rev Mol Cell Biol (2012) 13(6):397–404. doi: 10.1038/nrm3352
27. Matheu A, Maraver A, Klatt P, Flores I, Garcia-Cao I, Borras C, et al. Delayed ageing through damage protection by the Arf/p53 pathway. Nature (2007) 448(7151):375–9. doi: 10.1038/nature05949
28. García-Cao I, García-Cao M, Martín-Caballero J, Criado LM, Klatt P, Flores JM, et al. "Super p53" mice exhibit enhanced DNA damage response, are tumor resistant and age normally. Embo J (2002) 21(22):6225–35. doi: 10.1093/emboj/cdf595
29. Tomás-Loba A, Flores I, Fernández-Marcos PJ, Cayuela ML, Maraver A, Tejera A, et al. Telomerase reverse transcriptase delays aging in cancer-resistant mice. Cell (2008) 135(4):609–22. doi: 10.1016/j.cell.2008.09.034
30. Franck D, Tracy L, Armata HL, Delaney CL, Jung DY, Ko HJ, et al. Glucose tolerance in mice is linked to the dose of the p53 transactivation domain. Endocr Res (2013) 38(3):139–50. doi: 10.3109/07435800.2012.735735
31. Baker DJ, Weaver RL, van Deursen JM. p21 both attenuates and drives senescence and aging in BubR1 progeroid mice. Cell Rep (2013) 3(4):1164–74. doi: 10.1016/j.celrep.2013.03.028
32. Baker DJ, Perez-Terzic C, Jin F, Pitel KS, Niederlander NJ, Jeganathan K. Opposing roles for p16(Ink4a) and p19(Arf) in senescence and ageing caused by BubR1 insufficiency (vol 10, pg 825, 2008). Nat Cell Biol (2012) 14(6):649.
33. Baker DJ, Childs BG, Durik M, Wijers ME, Sieben CJ, Zhong J, et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature (2016) 530(7589):184–9. doi: 10.1038/nature16932
34. Chang J, Wang Y, Shao L, Laberge RM, Demaria M, Campisi J, et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat. Med. (2016) 22(1):78–83. doi: 10.1038/nm.4010
35. Nelson G, Wordsworth J, Wang C, Jurk D, Lawless C, Martin-Ruiz C, et al. A senescent cell bystander effect: senescence-induced senescence. Aging Cell (2012) 11(2):345–9. doi: 10.1111/j.1474-9726.2012.00795.x
36. Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci USA (1995) 92(20):9363–7. doi: 10.1073/pnas.92.20.9363
37. Iwasa H, Han J, Ishikawa F. Mitogen-activated protein kinase p38 defines the common senescence-signalling pathway. Genes Cells (2003) 8(2):131–44. doi: 10.1046/j.1365-2443.2003.00620.x
38. Minamino T, Komuro I. Vascular cell senescence: contribution to atherosclerosis. Circ Res (2007) 100(1):15–26. doi: 10.1161/01.RES.0000256837.40544.4a
39. Muñoz-Espín D, Serrano M. Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol (2014) 15(7):482–96. doi: 10.1038/nrm3823
40. Salama R, Sadaie M, Hoare M, Narita M. Cellular senescence and its effector programs. Genes Dev (2014) 28(2):99–114. doi: 10.1101/gad.235184.113
41. Borlaug BA. The pathophysiology of heart failure with preserved ejection fraction. Nat Rev Cardiol (2014) 11(9):507–15. doi: 10.1038/nrcardio.2014.83
42. Lakatta EG. Cardiovascular regulatory mechanisms in advanced age. Physiol Rev (1993) 73(2):413–67. doi: 10.1152/physrev.1993.73.2.413
43. Shih H, Lee B, Lee RJ, Boyle AJ. The aging heart and post-infarction left ventricular remodeling. J Am Coll Cardiol (2011) 57(1):9–17. doi: 10.1016/j.jacc.2010.08.623
44. Mohammed SF, Redfield MM. Response to letters regarding article, "coronary microvascular rarefaction and myocardial fibrosis in heart failure with preserved ejection fraction". Circulation (2015) 132(16):e206. doi: 10.1161/CIRCULATIONAHA.115.017050
45. Paulus WJ, Tschöpe C. A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J Am Coll Cardiol (2013) 62(4):263–71. doi: 10.1016/j.jacc.2013.02.092
46. Gevaert AB, Shakeri H, Leloup AJ, van Hove CE, de Meyer GRY, Vrints CJ, et al. Endothelial senescence contributes to heart failure with preserved ejection fraction in an aging mouse model. Circ Heart Fail (2017) 10(6):e003806. doi: 10.1161/CIRCHEARTFAILURE.116.003806
47. Frieler RA, Mortensen RM. Immune cell and other noncardiomyocyte regulation of cardiac hypertrophy and remodeling. Circulation (2015) 131(11):1019–30. doi: 10.1161/CIRCULATIONAHA.114.008788
48. Cohn JN, Levine TB, Olivari MT, Garberg V, Lura D, Francis GS, et al. Plasma norepinephrine as a guide to prognosis in patients with chronic congestive heart failure. N Engl J Med (1984) 311(13):819–23. doi: 10.1056/NEJM198409273111303
49. van Heerebeek L, Borbély A, Niessen HW, Bronzwaer JG, van der Velden J, Stienen GJ, et al. Myocardial structure and function differ in systolic and diastolic heart failure. Circulation (2006) 113(16):1966–73. doi: 10.1161/CIRCULATIONAHA.105.587519
50. Kasner M, Westermann D, Lopez B, Gaub R, Escher F, Kühl U, et al. Diastolic tissue Doppler indexes correlate with the degree of collagen expression and cross-linking in heart failure and normal ejection fraction. J Am Coll Cardiol (2011) 57(8):977–85. doi: 10.1016/j.jacc.2010.10.024
51. Selby DE, Palmer BM, Lewinter MM, Meyer M. Tachycardia-induced diastolic dysfunction and resting tone in myocardium from patients with a normal ejection fraction. J Am Coll Cardiol (2011) 58(2):147–54. doi: 10.1016/j.jacc.2010.10.069
52. Lakatta EG, Levy D. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: Part I: aging arteries: a "set up" for vascular disease. Circulation (2003) 107(1):139–46. doi: 10.1161/01.CIR.0000048892.83521.58
53. Lakatta EG. The reality of aging viewed from the arterial wall. Artery Res (2013) 7(2):73–80. doi: 10.1016/j.artres.2013.01.003
54. Spina M, Garbisa S, Hinnie J, Hunter JC, Serafini-Fracassini A. Age-related changes in composition and mechanical properties of the tunica media of the upper thoracic human aorta. Arteriosclerosis (1983) 3(1):64–76. doi: 10.1161/01.ATV.3.1.64
55. Gerstenblith G, Frederiksen J, Yin FC, Fortuin NJ, Lakatta EG, Weisfeldt ML. Echocardiographic assessment of a normal adult aging population. Circulation (1977) 56(2):273–8. doi: 10.1161/01.CIR.56.2.273
56. Mauriello A, Orlandi A, Palmieri G, Spagnoli LG, Oberholzer M, Christen H. Age-related modification of average volume and anisotropy of vascular smooth muscle cells. Pathol Res Pract (1992) 188(4-5):630–6. doi: 10.1016/S0344-0338(11)80070-1
57. Harvey A, Montezano AC, Touyz RM. Vascular biology of ageing-Implications in hypertension. J Mol Cell Cardiol (2015) 83:112–21. doi: 10.1016/j.yjmcc.2015.04.011
58. Minamino T, Miyauchi H, Yoshida T, Ishida Y, Yoshida H, Komuro I. Endothelial cell senescence in human atherosclerosis: role of telomere in endothelial dysfunction. Circulation (2002) 105(13):1541–4. doi: 10.1161/01.CIR.0000013836.85741.17
59. Marchand A, Atassi F, Gaaya A, Leprince P, Le Feuvre C, Soubrier F, et al. The Wnt/beta-catenin pathway is activated during advanced arterial aging in humans. Aging Cell (2011) 10(2):220–32. doi: 10.1111/j.1474-9726.2010.00661.x
60. Cafueri G, Parodi F, Pistorio A, Bertolotto M, Ventura F, Gambini C, et al. Endothelial and smooth muscle cells from abdominal aortic aneurysm have increased oxidative stress and telomere attrition. PLoS ONE (2012) 7(4):e35312. doi: 10.1371/journal.pone.0035312
61. Morgan RG, Ives SJ, Lesniewski LA, Cawthon RM, Andtbacka RH, Noyes RD, et al. Age-related telomere uncapping is associated with cellular senescence and inflammation independent of telomere shortening in human arteries. Am J Physiol Heart Circ Physiol (2013) 305(2):H251–58. doi: 10.1152/ajpheart.00197.2013
62. Melk A, Schmidt BM, Takeuchi O, Sawitzki B, Rayner DC, Halloran PF. Expression of p16INK4a and other cell cycle regulator and senescence associated genes in aging human kidney. Kidney Int. (2004) 65(2):510–20. doi: 10.1111/j.1523-1755.2004.00438.x
63. Yang D, Mccrann DJ, Nguyen H, St Hilaire C, Depinho RA, Jones MR, et al. Increased polyploidy in aortic vascular smooth muscle cells during aging is marked by cellular senescence. Aging Cell (2007) 6(2):257–60. doi: 10.1111/j.1474-9726.2007.00274.x
64. Rajapakse AG, Yepuri G, Carvas JM, Stein S, Matter CM, Scerri I, et al. Hyperactive S6K1 mediates oxidative stress and endothelial dysfunction in aging: inhibition by resveratrol. PLoS ONE (2011) 6(4):e19237. doi: 10.1371/journal.pone.0019237
65. Lin JR, Shen WL, Yan C, Gao PJ. Downregulation of dynamin-related protein 1 contributes to impaired autophagic flux and angiogenic function in senescent endothelial cells. Arterioscler Thromb Vasc Biol (2015) 35(6):1413–22. doi: 10.1161/ATVBAHA.115.305706
66. Satoh M, Ishikawa Y, Takahashi Y, Itoh T, Minami Y, Nakamura M. Association between oxidative DNA damage and telomere shortening in circulating endothelial progenitor cells obtained from metabolic syndrome patients with coronary artery disease. Atherosclerosis (2008) 198(2):347–53. doi: 10.1016/j.atherosclerosis.2007.09.040
67. van der Harst P, van der Steege G, de Boer RA, Voors AA, Hall AS, Mulder MJ, et al. Telomere length of circulating leukocytes is decreased in patients with chronic heart failure. J Am Coll Cardiol (2007) 49(13):1459–64. doi: 10.1016/j.jacc.2007.01.027
68. Haycock PC, Heydon EE, Kaptoge S, Butterworth AS, Thompson A, Willeit P. Leucocyte telomere length and risk of cardiovascular disease: systematic review and meta-analysis. BMJ (2014a) 349(3):g4227. doi: 10.1136/bmj.g4227
69. Salpea KD, Humphries SE. Telomere length in atherosclerosis and diabetes. Atherosclerosis (2010) 209(1):35–8. doi: 10.1016/j.atherosclerosis.2009.12.021
70. Morgan RG, Ives SJ, Walker AE, Cawthon RM, Andtbacka RH, Noyes D, et al. Role of arterial telomere dysfunction in hypertension: relative contributions of telomere shortening and telomere uncapping. J Hypertens (2014) 32(6):1293–9. doi: 10.1097/HJH.0000000000000157
71. Durik M, Kavousi M, van der Pluijm I, Isaacs A, Cheng C, Verdonk K, et al. Nucleotide excision DNA repair is associated with age-related vascular dysfunction. Circulation (2012) 126(4):468–478. doi: 10.1161/CIRCULATIONAHA.112.104380
72. Westhoff JH, Hilgers KF, Steinbach MP, Hartner A, Klanke B, Amann K, et al. Hypertension induces somatic cellular senescence in rats and humans by induction of cell cycle inhibitor p16INK4a. Hypertension (2008) 52(1):123–9. doi: 10.1161/HYPERTENSIONAHA.107.099432
73. Boe AE, Eren M, Murphy SB, Kamide CE, Ichimura A, Terry D, et al. Plasminogen activator inhibitor-1 antagonist TM5441 attenuates Nω-nitro-L-arginine methyl ester-induced hypertension and vascular senescence. Circulation (2013) 128(21):2318–24. doi: 10.1161/CIRCULATIONAHA.113.003192
74. Warboys CM, de Luca A, Amini N, Luong L, Duckles H, Hsiao S, et al. Disturbed flow promotes endothelial senescence via a p53-dependent pathway. Arterioscler Thromb Vasc Biol (2014) 34(5):985–95. doi: 10.1161/ATVBAHA.114.303415
75. Ungvari Z, Kaley G, de Cabo R, Sonntag WE, Csiszar A. Mechanisms of vascular aging: new perspectives. J Gerontol A Biol Sci Med Sci (2010) 65(10):1028–41. doi: 10.1093/gerona/glq113
76. Chilton W, O'Brien B, Charchar F. Telomeres, aging and exercise: guilty by association? Int J Mol Sci (2017) 18(12):2573. doi: 10.3390/ijms18122573
77. Rossman MJ, Kaplon RE, Hill SD, Mcnamara MN, Santos-Parker JR, Pierce GL, et al. Endothelial cell senescence with aging in healthy humans: prevention by habitual exercise and relation to vascular endothelial function. Am J Physiol Heart Circ Physiol (2017) 313(5):H890–95. doi: 10.1152/ajpheart.00416.2017
78. Faggiotto A, Ross R, Harker L. Studies of hypercholesterolemia in the nonhuman primate. I. Changes that lead to fatty streak formation. Arteriosclerosis (1984) 4(4):323–40. doi: 10.1161/01.ATV.4.4.323
79. Lakatta EG. So! What's aging? Is cardiovascular aging a disease? J Mol Cell Cardiol (2015) 83:1–13. doi: 10.1016/j.yjmcc.2015.04.005
80. Matthews C, Gorenne I, Scott S, Figg N, Kirkpatrick P, Ritchie A, et al. Vascular smooth muscle cells undergo telomere-based senescence in human atherosclerosis: effects of telomerase and oxidative stress. Circ Res (2006) 99(2):156–64. doi: 10.1161/01.RES.0000233315.38086.bc
81. Kunieda T, Minamino T, Nishi J, Tateno K, Oyama T, Katsuno T, et al. Angiotensin II induces premature senescence of vascular smooth muscle cells and accelerates the development of atherosclerosis via a p21-dependent pathway. Circulation (2006) 114(9):953–60. doi: 10.1161/CIRCULATIONAHA.106.626606
82. Miao SB, Xie XL, Yin YJ, Zhao LL, Zhang F, Shu YN, et al. Accumulation of smooth muscle 22α protein accelerates senescence of vascular smooth muscle cells via stabilization of p53 in vitro and in vivo. Arterioscler Thromb Vasc Biol (2017) 37(10):1849–59. doi: 10.1161/ATVBAHA.117.309378
83. Gardner SE, Humphry M, Bennett MR, Clarke MCH. Senescent vascular smooth muscle cells drive inflammation through an interleukin-1α-dependent senescence-associated secretory phenotype. Atherosclerosis (2016) 244:e5. doi: 10.1016/j.atherosclerosis.2015.10.065
84. Herbert KE, Mistry Y, Hastings R, Poolman T, Niklason L, Williams B. Angiotensin II-mediated oxidative DNA damage accelerates cellular senescence in cultured human vascular smooth muscle cells via telomere-dependent and independent pathways. Circ Res (2008) 102(2):201–8. doi: 10.1161/CIRCRESAHA.107.158626
85. Zhao W, Zheng XL, Peng DQ, Zhao SP. Myocyte enhancer factor 2A regulates hydrogen peroxide-induced senescence of vascular smooth muscle cells via microRNA-143. J Cell Physiol (2015) 230(9):2202–11. doi: 10.1002/jcp.24948
86. Minamino T, Mitsialis SA, Kourembanas S. Hypoxia extends the life span of vascular smooth muscle cells through telomerase activation. Mol Cell Biol (2001) 21(10):3336–42. doi: 10.1128/MCB.21.10.3336-3342.2001
87. Wang J, Uryga AK, Reinhold J, Figg N, Baker L, Finigan A, et al. Vascular smooth muscle cell senescence promotes atherosclerosis and features of plaque vulnerability. Circulation (2015) 132(20):1909–19. doi: 10.1161/CIRCULATIONAHA.115.016457
88. Haycock PC, Heydon EE, Kaptoge S, Butterworth AS, Thompson A, Willeit P. Leucocyte telomere length and risk of cardiovascular disease: systematic review and meta-analysis. BMJ (2014) 349:g4227. doi: 10.1136/bmj.g4227
89. D'Mello MJ, Ross SA, Briel M, Anand SS, Gerstein H, Paré G. Association between shortened leukocyte telomere length and cardiometabolic outcomes: systematic review and meta-analysis. Circ Cardiovasc Genet (2015) 8(1):82–90. doi: 10.1161/CIRCGENETICS.113.000485
90. Cawthon RM, Smith KR, O'Brien E, Sivatchenko A, Kerber RA. Association between telomere length in blood and mortality in people aged 60 years or older. Lancet (2003) 361(9355):393–5. doi: 10.1016/S0140-6736(03)12384-7
91. Calvert PA, Liew TV, Gorenne I, Clarke M, Costopoulos C, Obaid DR, et al. Leukocyte telomere length is associated with high-risk plaques on virtual histology intravascular ultrasound and increased proinflammatory activity. Arterioscler Thromb Vasc Biol (2011) 31(9):2157–64. doi: 10.1161/ATVBAHA.111.229237
92. Childs BG, Baker DJ, Wijshake T, Conover CA, Campisi J, van Deursen JM. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science (2016) 354(6311):472–7. doi: 10.1126/science.aaf6659
93. Cudejko C, Wouters K, Fuentes L, Hannou SA, Paquet C, Bantubungi K, et al. p16INK4a deficiency promotes IL-4-induced polarization and inhibits proinflammatory signaling in macrophages. Blood (2011) 118(9):2556–66. doi: 10.1182/blood-2010-10-313106
94. Shimizu I, Yoshida Y, Katsuno T, Tateno K, Okada S, Moriya J, et al. p53-induced adipose tissue inflammation is critically involved in the development of insulin resistance in heart failure. Cell Metab (2012) 15(1):51–64. doi: 10.1016/j.cmet.2011.12.006
95. Prior SJ, Goldberg AP, Ortmeyer HK, Chin ER, Chen D, Blumenthal JB, et al. Increased skeletal muscle capillarization independently enhances insulin sensitivity in older adults after exercise training and detraining. Diabetes (2015) 64(10):3386–95. doi: 10.2337/db14-1771
96. King GL, Park K, Li Q. Selective insulin resistance and the development of cardiovascular diseases in diabetes: the 2015 Edwin Bierman award lecture. Diabetes (2016) 65(6):1462–71. doi: 10.2337/db16-0152
97. Ferrara N. Vascular endothelial growth factor: basic science and clinical progress. Endocr Rev (2004) 25(4):581–611. doi: 10.1210/er.2003-0027
98. Shimizu I, Aprahamian T, Kikuchi R, Shimizu A, Papanicolaou KN, Maclauchlan S, et al. Vascular rarefaction mediates whitening of brown fat in obesity. J Clin Invest (2014) 124(5):2099–112. doi: 10.1172/JCI71643
99. Yokoi T, Fukuo K, Yasuda O, Hotta M, Miyazaki J, Takemura Y, et al. Apoptosis signal-regulating kinase 1 mediates cellular senescence induced by high glucose in endothelial cells. Diabetes (2006) 55(6):1660–5. doi: 10.2337/db05-1607
100. Orimo M, Minamino T, Miyauchi H, Tateno K, Okada S, Moriya J, et al. Protective role of SIRT1 in diabetic vascular dysfunction. Arterioscler Thromb Vasc Biol (2009) 29(6):889–94. doi: 10.1161/ATVBAHA.109.185694
101. Hayashi T, Kotani H, Yamaguchi T, Taguchi K, Iida M, Ina K, et al. Endothelial cellular senescence is inhibited by liver X receptor activation with an additional mechanism for its atheroprotection in diabetes. Proc Natl Acad Sci USA (2014) 111(3):1168–73. doi: 10.1073/pnas.1322153111
102. Lumeng CN, Saltiel AR. Inflammatory links between obesity and metabolic disease. J Clin Invest (2011) 121(6):2111–7. doi: 10.1172/JCI57132
103. Pillon NJ, Bilan PJ, Fink LN, Klip A. Cross-talk between skeletal muscle and immune cells: muscle-derived mediators and metabolic implications. Am J Physiol Endocrinol Metab (2013) 304(5):E453–65. doi: 10.1152/ajpendo.00553.2012
104. Xu M, Palmer AK, Ding H, Weivoda MM, Pirtskhalava T, White TA, et al. Targeting senescent cells enhances adipogenesis and metabolic function in old age. Elife (2015) 4:e12997. doi: 10.7554/eLife.12997
105. Zhu Y, Tchkonia T, Pirtskhalava T, Gower AC, Ding H, Giorgadze N, et al. The Achilles' heel of senescent cells: from transcriptome to senolytic drugs. Aging Cell (2015) 14(4):644–58. doi: 10.1111/acel.12344
106. Baar MP, Brandt RMC, Putavet DA, Klein JDD, Derks KWJ, Bourgeois BRM, et al. Targeted apoptosis of senescent cells restores tissue homeostasis in response to chemotoxicity and aging. Cell (2017) 169(1):132–47. doi: 10.1016/j.cell.2017.02.031
107. Roos CM, Zhang B, Palmer AK, Ogrodnik MB, Pirtskhalava T, Thalji NM, et al. Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or atherosclerotic mice. Aging Cell (2016) 15(5):973–7. doi: 10.1111/acel.12458
108. Raez LE, Papadopoulos K, Ricart AD, Chiorean EG, Dipaola RS, Stein MN, et al. A phase I dose-escalation trial of 2-deoxy-D-glucose alone or combined with docetaxel in patients with advanced solid tumors. Cancer Chemother Pharmacol (2013) 71(2):523–30. doi: 10.1007/s00280-012-2045-1
109. Kirkland JL, Tchkonia T. Cellular Senescence: A Translational Perspective. EBioMedicine (2017) 21:21–8. doi: 10.1016/j.ebiom.2017.04.013
110. Kirkland JL, Tchkonia T, Zhu Y, Niedernhofer LJ, Robbins PD. The clinical potential of senolytic drugs. J Am Geriatr Soc (2017) 65(10):2297–301. doi: 10.1111/jgs.14969
111. Mattison JA, Wang M, Bernier M, Zhang J, Park SS, Maudsley S, et al. Resveratrol prevents high fat/sucrose diet-induced central arterial wall inflammation and stiffening in nonhuman primates. Cell Metab (2014) 20(1):183–90. doi: 10.1016/j.cmet.2014.04.018
112. Gao D, Zuo Z, Tian J, Ali Q, Lin Y, Lei H, et al. Activation of SIRT1 attenuates klotho deficiency-induced arterial stiffness and hypertension by enhancing AMP-activated protein kinase activity. Hypertension (2016) 68(5):1191–9. doi: 10.1161/HYPERTENSIONAHA.116.07709
113. Chen HZ, Wang F, Gao P, Pei JF, Liu Y, Xu TT, et al. Age-associated sirtuin 1 reduction in vascular smooth muscle links vascular senescence and inflammation to abdominal aortic aneurysm. Circ Res (2016) 119(10):1076–88. doi: 10.1161/CIRCRESAHA.116.308895
114. Liu Y, Wang TT, Zhang R, Fu WY, Wang X, Wang F, et al. Calorie restriction protects against experimental abdominal aortic aneurysms in mice. J Exp Med (2016) 213(11):2473–88. doi: 10.1084/jem.20151794
115. Imai S, Guarente L. NAD+ and sirtuins in aging and disease. Trends Cell Biol (2014) 24(8):464–71. doi: 10.1016/j.tcb.2014.04.002
116. van der Veer E, Ho C, O'Neil C, Barbosa N, Scott R, Cregan SP, et al. Extension of human cell lifespan by nicotinamide phosphoribosyltransferase. J Biol Chem (2007) 282(15):10841–5. doi: 10.1074/jbc.C700018200
117. Watson A, Nong Z, Yin H, O'Neil C, Fox S, Balint B, et al. Nicotinamide phosphoribosyltransferase in smooth muscle cells maintains genome integrity, resists aortic medial degeneration, and is suppressed in human thoracic aortic aneurysm disease. Circ Res (2017) 120(12):1889–902. doi: 10.1161/CIRCRESAHA.116.310022
118. Fan LM, Cahill-Smith S, Geng L, du J, Brooks G, Li JM. Aging-associated metabolic disorder induces Nox2 activation and oxidative damage of endothelial function. Free Radic Biol Med (2017) 108:940–51. doi: 10.1016/j.freeradbiomed.2017.05.008
119. Vendrov AE, Stevenson MD, Alahari S, Pan H, Wickline SA, Madamanchi NR, et al. Attenuated superoxide dismutase 2 activity induces atherosclerotic plaque instability during aging in hyperlipidemic mice. J Am Heart Assoc (2017) 6(11):e006775. doi: 10.1161/JAHA.117.006775
120. Miyauchi H, Minamino T, Tateno K, Kunieda T, Toko H, Komuro I. Akt negatively regulates the in vitro lifespan of human endothelial cells via a p53/p21-dependent pathway. Embo J (2004) 23(1):212–20. doi: 10.1038/sj.emboj.7600045
121. Lesniewski LA, Seals DR, Walker AE, Henson GD, Blimline MW, Trott DW, et al. Dietary rapamycin supplementation reverses age-related vascular dysfunction and oxidative stress, while modulating nutrient-sensing, cell cycle, and senescence pathways. Aging Cell (2017) 16(1):17–26. doi: 10.1111/acel.12524
122. Marso SP, Daniels GH, Brown-Frandsen K, Kristensen P, Mann JF, Nauck MA, et al. Liraglutide and cardiovascular outcomes in type 2 diabetes. N Engl J Med (2016) 375(4):311–22. doi: 10.1056/NEJMoa1603827
123. Yang G, Lei Y, Inoue A, Piao L, Hu L, Jiang H, et al. Exenatide mitigated diet-induced vascular aging and atherosclerotic plaque growth in ApoE-deficient mice under chronic stress. Atherosclerosis (2017) 264:1–10. doi: 10.1016/j.atherosclerosis.2017.07.014
124. Demaria M, Ohtani N, Youssef SA, Rodier F, Toussaint W, Mitchell JR, et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev Cell (2014) 31(6):722–33. doi: 10.1016/j.devcel.2014.11.012
125. Yosef R, Pilpel N, Tokarsky-Amiel R, Biran A, Ovadya Y, Cohen S, et al. Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL. Nat Commun (2016) 7:11190. doi: 10.1038/ncomms11190
126. Zhu Y, Tchkonia T, Fuhrmann-Stroissnigg H, Dai HM, Ling YY, Stout MB, et al. Identification of a novel senolytic agent, navitoclax, targeting the Bcl-2 family of anti-apoptotic factors. Aging Cell (2016) 15(3):428–35. doi: 10.1111/acel.12445
127. Farr JN, Xu M, Weivoda MM, Monroe DG, Fraser DG, Onken JL, et al. Targeting cellular senescence prevents age-related bone loss in mice. Nat Med (2017) 23(9):1072–9. doi: 10.1038/nm.4385
128. Ogrodnik M, Miwa S, Tchkonia T, Tiniakos D, Wilson CL, Lahat A, et al. Cellular senescence drives age-dependent hepatic steatosis. Nat Commun (2017) 8:15691. doi: 10.1038/ncomms15691
129. Schafer MJ, White TA, Iijima K, Haak AJ, Ligresti G, Atkinson EJ, et al. Cellular senescence mediates fibrotic pulmonary disease. Nat Commun (2017) 8:14532. doi: 10.1038/ncomms14532
130. Zhu Y, Doornebal EJ, Pirtskhalava T, Giorgadze N, Wentworth M, Fuhrmann-Stroissnigg H, et al. New agents that target senescent cells: the flavone, fisetin, and the BCL-XLinhibitors, A1331852 and A1155463. Aging (2017) 9(3):955–63. doi: 10.18632/aging.101202
131. Fuhrmann-Stroissnigg H, Ling YY, Zhao J, Mcgowan SJ, Zhu Y, Brooks RW, et al. Identification of HSP90 inhibitors as a novel class of senolytics. Nat Commun (2017) 8(1):422. doi: 10.1038/s41467-017-00314-z
132. Wang Y, Chang J, Liu X, Zhang X, Zhang S, Zhang X, et al. Discovery of piperlongumine as a potential novel lead for the development of senolytic agents. Aging (2016) 8(11):2915–26. doi: 10.18632/aging.101100
Keywords: cellular senescence, p53, atherosclerosis, heart failure, diabetes, senolysis
Citation: Katsuumi G, Shimizu I, Yoshida Y and Minamino T (2018). Vascular Senescence in Cardiovascular and Metabolic Diseases. Front. Cardiovasc. Med. 5:18. doi: 10.3389/fcvm.2018.00018
Received: 24 October 2017; Accepted: 21 February 2018;
Published: 05 March 2018
Edited by:
Masanori Aikawa, Harvard Medical School, United StatesReviewed by:
Andrea Caporali, University of Edinburgh, United KingdomHiroshi Kondoh, Kyoto University, Japan
Copyright © 2018 Katsuumi, Shimizu, Yoshida and Minamino. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tohru Minamino, dG1pbmFtaW5vQG1lZC5uaWlnYXRhLXUuYWMuanA=
†These authors have contributed equally to this work