 Takuro Numaga-Tomita1,2
Takuro Numaga-Tomita1,2 Sayaka Oda1,2
Sayaka Oda1,2 Tsukasa Shimauchi1,3
Tsukasa Shimauchi1,3 Akiyuki Nishimura1,2
Akiyuki Nishimura1,2 Supachoke Mangmool4
Supachoke Mangmool4 Motohiro Nishida1,2,3,5*
Motohiro Nishida1,2,3,5*
- 1Division of Cardiocirculatory Signaling, Okazaki Institute for Integrative Bioscience, National Institute for Physiological Sciences, National Institutes of Natural Sciences, Okazaki, Japan
- 2Department of Physiological Sciences, Graduate University for Advanced Studies (SOKENDAI), Okazaki, Japan
- 3Department of Translational Pharmaceutical Sciences, Graduate School of Pharmaceutical Sciences, Kyushu University, Fukuoka, Japan
- 4Faculty of Pharmacy, Department of Pharmacology, Mahidol University, Bangkok, Thailand
- 5Precursory Research for Embryonic Science and Technology, Japan Science and Technology Agency, Kawaguchi, Japan
Cardiac stiffness, caused by interstitial fibrosis due to deposition of extracellular matrix proteins, is thought as a major clinical outcome of heart failure with preserved ejection fraction (HFpEF). Canonical transient receptor potential (TRPC) subfamily proteins are components of Ca2+-permeable non-selective cation channels activated by receptor stimulation and mechanical stress, and have been attracted attention as a key mediator of maladaptive cardiac remodeling. How TRPC-mediated local Ca2+ influx encodes a specific signal to induce maladaptive cardiac remodeling has been long obscure, but our recent studies suggest a pathophysiological significance of channel activity-independent function of TRPC proteins for amplifying redox signaling in heart. This review introduces the current understanding of the physiological and pathophysiological roles of TRPCs, especially focuses on the role of TRPC3 as a positive regulator of reactive oxygen species (PRROS) in heart. We have revealed that TRPC3 stabilizes NADPH oxidase 2 (Nox2), a membrane-bound reactive oxygen species (ROS)-generating enzyme, by forming stable protein complex with Nox2, which leads to amplification of mechanical stress-induced ROS signaling in cardiomyocytes, resulting in induction of fibrotic responses in cardiomyocytes and cardiac fibroblasts. Thus, the TRPC3 function as PRROS will offer a new therapeutic strategy for the prevention or treatment of HFpEF.
Introduction
The physiological and pathophysiological significance of Ca2+ influx across the plasma membrane in cardiomyocytes has been discussed for a long time, but how the heart decodes a specific Ca2+ influx as pathological signal under the background of rhythmic Ca2+ entry is obscure. There are two major roles of Ca2+ influx in cardiomyocytes: one is to mediate “excitation–contraction (E–C) coupling,” where a local Ca2+ influx through voltage-dependent L-type Ca2+ channels activated by membrane depolarization (i.e., excitation) induces substantial Ca2+ release from sarcoplasmic reticulum (SR), which leads to rhythmic myocardial contraction by increasing myosin ATPase activity through Ca2+/troponin C-dependent structural changes of actin-tropomyosin filaments, and the other is to mediate “excitation–transcription (E–T) coupling,” where a local Ca2+ influx evoked by neurohumoral excitation and/or hemodynamic load through activation of voltage-independent (or mechano-activated) cation channels induces hypertrophic gene expressions through activating Ca2+-dependent transcriptional factors, such as nuclear factor of activated T cells (NFAT) and myocyte enhancer factor (MEF). Transient receptor potential (TRP) proteins, especially canonical TRP subfamily [canonical transient receptor potential (TRPC)] members, have been suggested to function as receptor-activated cation channels (RACCs) regulating E–T coupling in the heart (1). We have also reported that diacylglycerol-activated TRPC3 and TRPC6 heteromultimer channels (TRPC3/6) act as a key mediator of pathological hypertrophy in receptor-stimulated rat cardiomyocytes (2, 3) and pressure-overloaded mouse hearts (4), while our recent studies using TRPC3/6-deficient mice have revealed that TRPC3 specifically mediates pressure overload-induced maladaptive cardiac fibrosis, independently of TRPC6 channels (5, 6). This review focuses on the putative molecular mechanism underlying TRPC3-mediated maladaptive cardiac fibrosis in rodent hearts and discusses its therapeutic possibilities.
TRPC Channels and Their Physiological Functions
The trp gene was first identified in 1989 as a causative gene mutant of phototransduction in Drosophila (7). Twenty-eight mammalian TRP homologs have been identified and these are subdivided into six related protein subfamilies based on their genetic and functional similarities: TRPC (canonical), TRPV (vanilloid), TRPM (melastatin), TRPP (polycystin), TRPML (mucolipin), TRPA (ankyrin). TRP proteins commonly possess structural 6 transmembrane domains and preserved 25 amino acid sequence called “TRP domain.” The TRPC family proteins, composed of seven mammalian homologs (TRPC1–TRPC7), are believed as molecular candidates of RACCs (8). TRPC4 and TRPC5 share an about 65% amino acid homology in their group, while TRPC3, TRPC6, and TRPC7 show the best homology covering ~75% of amino acid sequence (9). TRPC1 shares lower sequence homology compared to other TRPC members. TRPC1 is first suggested as a candidate subunit of store-operated Ca2+ channels (SOCCs) (10–13). TRPC1 contributes to coordination with elementary Ca2+ signaling events though promoting functional coupling between the endoplasmic reticulum (ER) and the plasma membrane in receptor-induced Ca2+ signaling (14). TRPC1 also functions as stretch-activated cation channels in mammalian cells (15). Thus, TRPC proteins have two important roles: one is to act as a critical component of stretch-activated or store-operated Ca2+ (SOC)-permeable channels and the other is to act as a signaling platform to amplify receptor-activated Ca2+ signaling via interacting with intracellular signaling molecules (16).
TRPC are generally known to be activated downstream of phospholipase C (PLC)-coupled receptors, such as G-protein-coupled receptors (GPCRs) and receptor tyrosine kinases (16). TRPC proteins comprise non-selective cation channels by forming homo- or hetero-tetramer complex. Due to their universal activation mechanism in many cell types, TRPC channels play important roles in basic cellular responses, including proliferation, differentiation, and death in response to various environmental stimuli. TRPC channels are also linked to physical stimulations such as mechanical stretch, hypoxia, and oxidative stress (17). TRPC1 and TRPC6 are suggested as a component of the tarantula toxin-sensitive mechanosensitive cation channel (15, 18). In fact, inhibition or deletion of TRPC6 has been reported to blunt the chronic mechanical stress-induced muscular contraction in mouse myocytes with Duchenne muscular dystrophy (19). In addition, intracellular lipid mediators, such as diacylglycerol and 20-HETE, also mediate activation of TRPC6 induced by oxidative stress (20) and mechanical stretch (21). Considering the role of TRPC3/6 heterotetramer channels in cardiac hypertrophy, TRPC6 protein signaling complex, including TRPC1 and TRPC3, may function as mechano-activated cation channels in the cardiovascular system.
Regulation of DAG-Activated TRPC3/C6 Channel Activities
TRPC3/C6/C7 subfamilies are directly activated by diacylglycerol (22, 23). TRPC3 and TRPC6 are mainly expressed in central nervous system, but the physiological significances of both channels have been emerged from vascular physiology. TRPC6 channel is activated downstream of α-adrenergic receptor and mediates cation influx, which evokes membrane depolarization and activation of voltage-dependent Ca2+ channel to induce smooth muscle contraction in rat portal vein (24). Following this prominent work, other researches including ours demonstrated that TRPC3/C6 channels function to depolarize the plasma membrane in response to vasoconstrictive GPCR agonists (25, 26). In addition, there are several reports demonstrated the physiological importance of these channels in non-excitable cells. In these cellular contexts, TRPC channels mainly function as Ca2+ influx channels. However, because the number and conductance of endogenously expressed TRPC channels seem to be very small, TRPC-mediated Ca2+ influx is considered to be involved in local Ca2+ signaling rather than global intracellular Ca2+ mobilization. In fact, TRPC3-mediated local Ca2+ influx is specifically and efficiently transduced to downstream signaling pathways in B lymphocytes (27, 28). TRPC3 is found to interact with several signaling molecules, such as PLC, protein kinase C (PKC), receptor for activated C-kinase-1, inositol 1,4,5-trisphosphate receptor, and calmodulin (27–31). These interactions may be critical for the diversity of downstream signaling pathways induced by TRPC3-mediated local Ca2+ influx, since local Ca2+ per se is highly mobile and easily buffered by buffering proteins in the cytosol.
TRPC3/6 channel activities are negatively regulated by Ser/Thr phosphorylation of TRPC3/6 proteins via PKC, protein kinase A (PKA), and protein kinase G (PKG). PKG is reported to phosphorylate human TRPC3 at Thr-11 and Ser-263, and human TRPC6 at Thr-70 and Ser-322 (32). Nitric oxide (NO), atrial natriuretic peptide, and inhibition of phosphodiesterase 5 can activate PKG. The PKG-dependent negative regulation of TRPC6 channel activity by NO is physiologically important in endothelium-dependent vasodilation (33). PKA and PKG recognize a similar substrate sequence, and PKA-dependent phosphorylation of rodent TRPC6 at Thr-69 is found to participate in endothelium-independent vasodilation (26). Increased PKG activity is also reported to suppress Ca2+/calcineurin-dependent cardiac hypertrophy induced by agonist stimulation and pressure overload, and blockade of PKG phosphorylation by TRPC6 mutagenesis canceled the PKG-dependent anti-hypertrophic action (34). By contrast, reduction of cGMP/PKG signaling by guanylate cyclase-A gene deletion is reported to develop spontaneous cardiac hypertrophy through TRPC3/6 channel activation (35). In fact, this hypertrophic phenotype was attenuated by the treatment with pyrazole-2, an inhibitor of TRPC1-7 channels.
TRPC3/6 Channels in Cardiac Remodeling
The heart can adapt itself to various environmental stresses by flexibly changing its structure and morphology. Physiological stimuli, such as physical exercise or pregnancy, induce cardiac hypertrophy to adapt the increases of oxygen and nutrition demands, which is fully reversible. By contrast, pathological conditions also induce cardiac hypertrophy, which is followed by interstitial fibrosis and eventual left ventricular dilation and dysfunction (36). These physiological and pathological cardiac remodelings are chronic tissue responses accompanied with gene expression. Several pieces of evidence indicate the involvement of TRPC channels in the cardiac remodeling processes. Intracellular Ca2+ increase and subsequent NFAT activation are the best known pathway that mediates pathological cardiac hypertrophy (37). Thus, TRPC channels were identified as Ca2+ permeable channels to activate calcineurin/NFAT pathway. However, now TRPC channels are thought to be not only a cation channel but also a scaffold or membrane anchor to organize downstream signaling complex and participate in pathological cardiac remodeling (Tables 1 and 2). Recently, we have revealed that TRPC3 channel functions as a mediator linking Ca2+ signaling and reactive oxygen species (ROS) production which exacerbates pathological cardiac remodeling (5, 6).
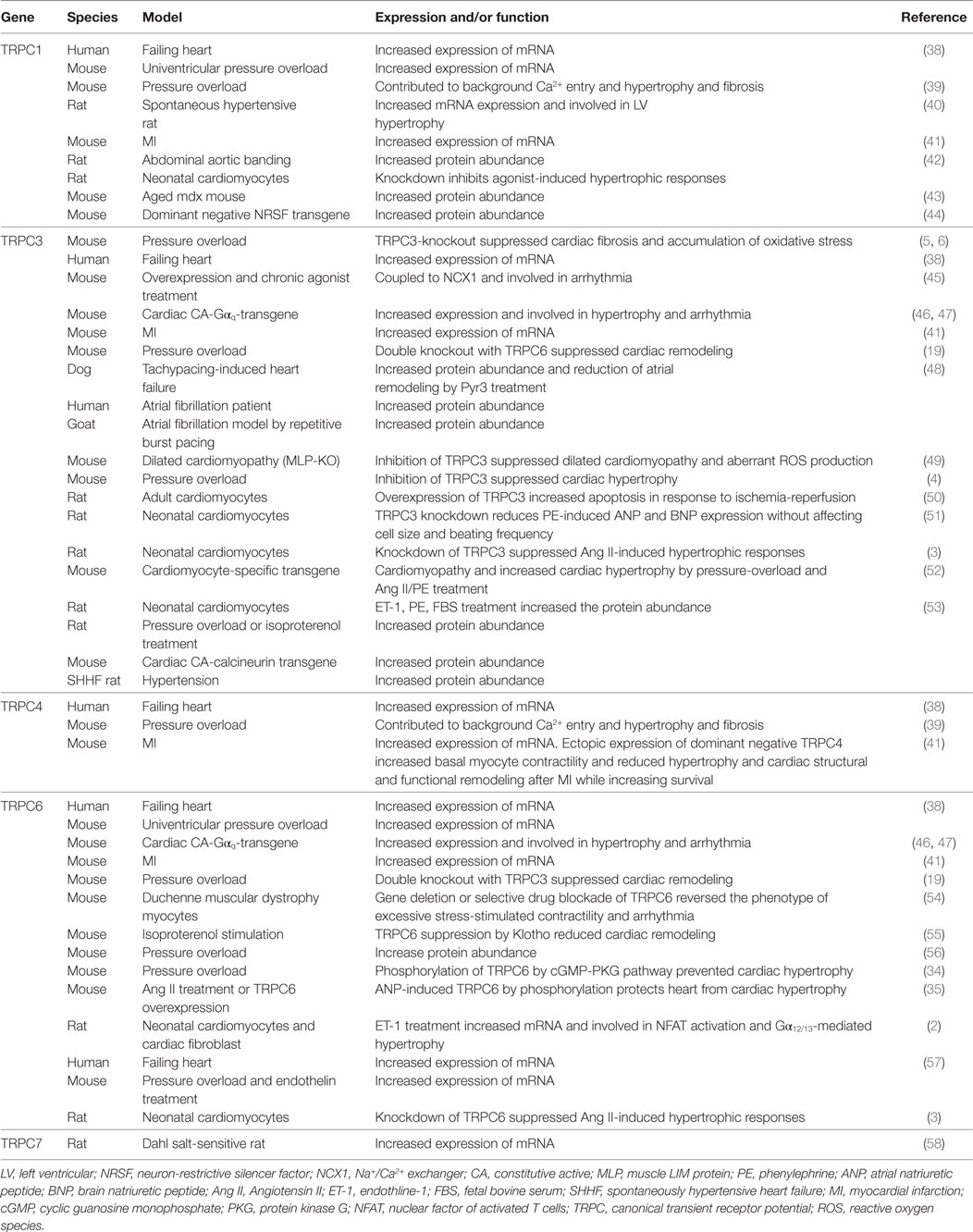
Table 1. Involvement of TRPC channels in cardiomyopathy.
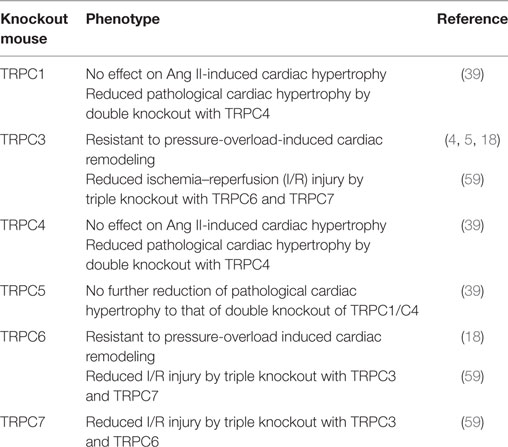
Table 2. Cardiac phenotype of canonical transient receptor potential (TRPC) knockout mice.
Canonical transient receptor potential channels were historically presumed to be the molecular entity of SOCCs. Now Stromal interacting molecule 1 and Orai1 channel are identified as a molecular entity of SOCCs. SOC entry is known to be critical for activation of NFAT, which is one of the main transcription factors in cardiac hypertrophy. Therefore, several papers addressed the involvement of TRPC channels in cardiac hypertrophy. Nakayama et al. first demonstrated the involvement of TRPC3 in cardiac hypertrophy (52). It has been known that Ca2+ influx and subsequent activation of NFAT play critical roles in cardiac hypertrophy (35, 36). Their group produced transgenic mice overexpressing TRPC3 specifically in cardiomyocytes (37). Those mice showed elevated SOC entry and basal NFAT activity, and eventually exhibited cardiomyopathy. This prominent study clearly indicated that TRPC3 expression per se evokes cardiac hypertrophy. Consistent with this report, TRPC3 protein abundance is increased in rodent hypertrophic cardiomyocytes (53). Neurohumoral factor-induced cardiac hypertrophy was also mediated by the increase of TRPC3/C6 expression (3). Ectopic expression of TRPC6 in cardiomyocytes also promoted the induction of pathological cardiac remodeling (57). Consistent with the data obtained from TRPC3/C6 ectopic expression model, cardiomyocyte-specific overexpression of dominant negative mutants of TRPC3 or TRPC6 [N-terminal fragment of TRPC3 or pore-dead mutant (L678-W680 replaced to three alanine residues) of TRPC6] suppressed both neurohumoral factor-induced and pressure-overload-induced cardiac hypertrophy and dysfunction (60). The involvement of TRPC3 was also demonstrated in cardiac remodeling by myocardial infarction (MI) and arrhythmia (41, 48, 50, 61). These reports strongly suggest that TRPC3/C6 channels are prominent molecules mediating cardiac remodeling induced by exposure to several stresses. Recently, we and others reported the effect of TRPC3/C6 genetic deletion on pressure-overload-induced cardiac dysfunction (5, 6, 19). Seo et al. reported that TRPC3/C6 double knockout mice, but not single knockout mice, were resistant to pressure-overload-induced cardiac remodeling (19). However, TRPC3 single deletion was sufficient to suppress cardiac remodeling in response to pressure overload in our study (5, 6). This discrepancy can be partially explained by the difference of mouse strains. In our study, we used 129/Sv mouse and Seo et al. used the mouse backcrossed with C57BL/6 mice. It has been reported that the responses of the heart to pressure overload differ among mouse strains (62). Interestingly, while cardiac hypertrophy was not affected by TRPC3 deletion, cardiac fibrosis was diminished in TRPC3-deficient mice in response to pressure overload (5, 6).
ROS in Cardiac Physiology and Pathophysiology
Production of ROS is observed in most of the pathophysiological conditions of the heart, which exacerbate cardiac remodeling and dysfunction. ROS are generated from both defect of mitochondrial respiratory chains and NADPH oxidase (Nox) activation. Among seven members of Nox proteins, NADPH oxidase 2 (Nox2) and Nox4 are predominantly expressed in the heart. In resting conditions, Nox2 only interacted with p22phox subunit, which is crucial for the expression of Nox2 by preventing proteasomal degradation. Upon cellular activation, other cytoplasmic subunits p67phox, p40phox, p47phox, and small G protein Rac1 are recruited and activate Nox2 protein (Figure 1). Among the cytoplasmic subunits, p47phox mainly regulates Nox2 complex formation. To form complex, phosphorylation of p47phox is necessary. Phosphorylation of p47phox is reported to be mediated by PKC, mitogen-activated protein kinases (MAPKs), and p21-activated kinase (63). Nox2 is located in the membrane of the T-tubules in close apposition to the junctional SR (64). The involvement of Nox in cardiac pathophysiology was demonstrated in myocardial ischemia, pressure-overload and chemical toxicity (65–67). However, Nox plays a critical role in cardiac physiology. During regular heartbeat, diastole is very important regarding intracellular Ca2+ homeostasis. Diastolic LV filling causes stretch of cardiomyocytes, which evokes mechano-signal transduction. Prosser et al. demonstrated that mechanical stretch of cardiomyocytes during diastole evokes ROS production via Nox2 activation in microtubule-dependent manner (68). Those ROS oxidize ryanodine receptors in junctional SR, which sensitizes ryanodine receptors to Ca2+ and thereby increases Ca2+ release in coming systolic contraction.
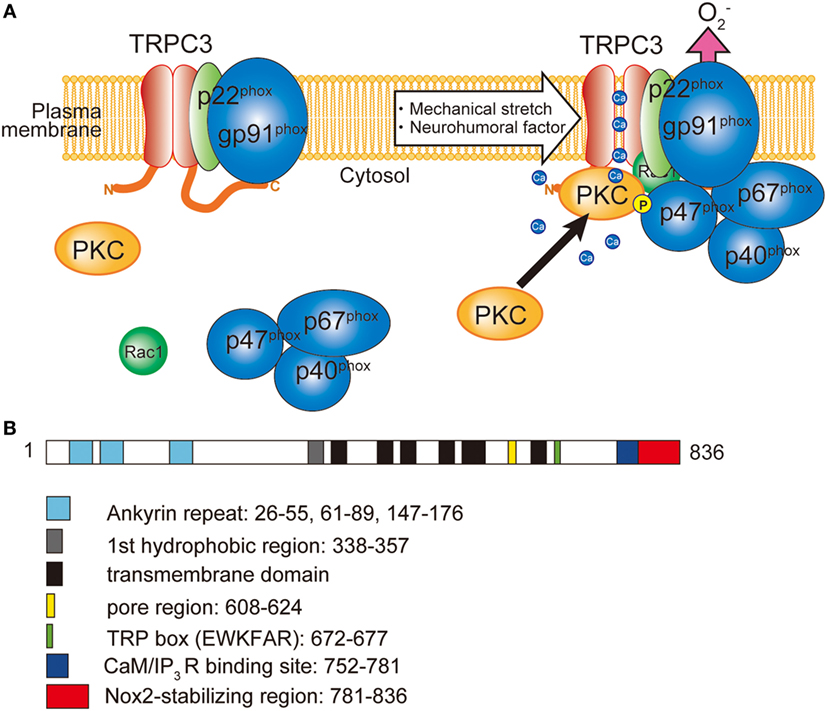
Figure 1. Involvement of TRPC3 in the activation of NADPH oxidase 2 (Nox2). (A) TRPC3-mediated Ca2+ influx recruits and activates protein kinase C (PKC) which phosphorylates p47phox and evokes Nox2 enzymatic activation. (B) Schematic illustration of the domain structure of TRPC3. TRPC3 interacts with Nox2 through the C-terminal region. Numbers represent the positions of amino acids from first methionine.
In pathological situations, involvement of cardiac hypertrophy is manifested. Nox activity is increased in the end-stage failing human heart and that it is likely to be an important source of increased cardiac ROS in human chronic heart failure (69). Bendall et al. first reported that Ang II-induced cardiac hypertrophy was blunted by deletion of gp91phox subunit in mice (65). Nox2 mediates Ang II-induced cardiac hypertrophy by modulating Akt and Wnt signaling (70, 71). However, pressure-overload-induced cardiac hypertrophy was not affected by deletion of gp91phox (72, 73). Besides the cardiac hypertrophy, interstitial fibrosis is manifested in heart failure observed in the elderly population, and patients with HFpEF caused by hypertensive heart disease, aortic valve stenosis, and hypertrophic cardiomyopathy (74). Nox2 was important for transforming growth factor β-induced cardiac fibrosis in hypertensive rat (75). gp91phox knockout mice also showed vulnerability to MI. In contrast to the different involvement of cardiac hypertrophy induced by neurohumoral factors versus pressure overload, interstitial fibrosis in response to above factors were abolished in either Nox2 or Rac1-deficient mice (65, 72, 76–78).
Different from tunable Nox2, Nox4 is regulated only by its expression. Nox4 also requires p22phox. Therefore, Nox4 is likely to contribute to basal ROS production. It has been demonstrated that Nox4 localizes in intracellular membrane especially perinuclear location associated with SR or mitochondria. Downregulation of Nox4, the major Nox isoform presents during early stages of differentiation, suppressed cardiogenesis. This was rescued by a pulse of low concentrations of hydrogen peroxide (H2O2) 4 days before spontaneous beating appears. The mechanisms of ROS-dependent signaling included p38 MAPK activation and nuclear translocation of the cardiac transcription factor MEF2C (79). Cardiomyocyte-specific knockout of Nox4 reportedly suppressed pressure-overload-induced cardiac hypertrophy, fibrosis and dysfunction (80). However, null knockout of Nox4 mice showed opposite phenotype as exaggeration of contractile dysfunction, hypertrophy, and cardiac dilatation (81). Cardiomyocyte-specific overexpression of Nox4 counteracted cardiac dysfunction by increasing angiogenic activity in cardiomyocytes, suggesting that increases of Nox4 expression is an adaptive response against chronic heart stress (81). Low tonic production of H2O2 by Nox4 in endothelial cells has a vasoprotective role by increasing antioxidant systems such as heme oxygenase-1 and NO synthases (82, 83). Therefore, Nox4 seemingly plays a protective role in cardiovascular homeostasis, in contrast to Nox2. Although expression of Nox1 is relatively low in heart compared to Nox2 and Nox4, sepsis-induced myocardial cell death and ROS production were significantly suppressed in Nox1-deficient mice (84).
Coupling of Nox Proteins and TRPC Channels
Besides the activation mechanism of Nox2 mentioned earlier, Nox2 requires extracellular Ca2+ influx to be activated (85–87). In neutrophil-like cell line HL-60, TRPC3, and TRPC6 are critical Ca2+ channel for the activation of Nox2 (88). In these cells, GPCR activation induced large increase of intracellular Ca2+ concentration and removal or pharmacological blocking attenuated Nox2 activation. Therefore, TRPC channels function as a provider of Ca2+ for the enzymatic activation. Kitajima et al. reported that TRPC3 functions not only Ca2+ channel but also protein stabilizer by physical interaction (Figures 1 and 2). Previous work demonstrated that interaction with p22phox is critical for Nox2 stabilization. Recently, an ER resident membrane protein competes with p22phox to interact with Nox2. By releasing from p22phox and proceeding to proteasomal degradation, the protein termed negative regulator of ROS facilitates degradation of Nox2 to reduce basal expression (89). By contrast, increased stability of Nox2 by TRPC3 is not simple facilitation of Nox2-p22phox interaction. In fact, p22phox by itself could interact with TRPC3 and be stabilized by the interaction (Figure 2). In pressure-overloaded heart, Nox2 expression was significantly increased, which was completely abolished in TRPC3-deficient mouse hearts. In addition, TRPC3 silencing reduced basal expression of Nox2 in rat neonatal cardiomyocytes (NRCMs), although there was only slight reduction of basal Nox2 expression in normal hearts of TRPC3 knockout mouse compared to those of wild type. In both experimental samples, there were no differences regarding Nox2 mRNA levels. Furthermore, the reduction of Nox2 in TRPC3-silenced NRCMs was mostly rescued by proteasome inhibitor, indicating that TRPC3 increases Nox2 protein abundance by protecting from proteasome-dependent degradation (Figure 2).
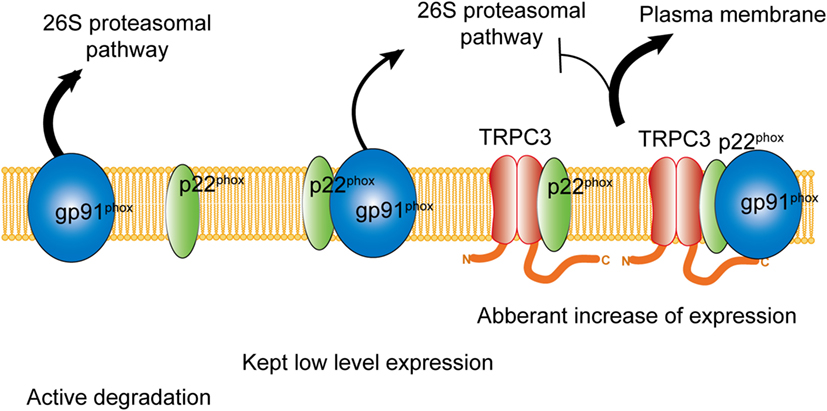
Figure 2. Physical interaction with TRPC3 prevents NADPH oxidase 2 (Nox2) from proteasome-dependent downregulation. In physiological condition, level of Nox2 expression is kept low by proteasomal degradation. Without interaction with p22phox, actively gp91phox is degraded. By physical interaction with TRPC3, both gp91phox and p22phox are protected from proteasomal degradation, which leads to excess expression of Nox2 enzyme on the plasma membrane.
In addition, there were reciprocal regulation between TRPC3 and Nox2, i.e., enhancement of Nox2 expression also increased TRPC3 expression and channel function (5). Similar regulation of TRPC channels by Nox protein has been reported. Nox4 expression is important for TRPC6 upregulation in podocytes (90–92). In these studies, TRPC6 was oxidized by ROS produced by Nox4 and its activation was facilitated. However, Nox2-dependent increase of TRPC3-mediated current was not affected by diphenyleneiodonium treatment. Therefore, the reciprocal regulation between TRPC3 and Nox2 also increased channel density on the plasma membrane reflecting the increase of gross expression of TRPC3 by co-expression with Nox2.
Proteomic analysis using RhoA (G17A)-agarose revealed that microtubule-associated Rho guanine nucleotide exchange factor, GEF-H1, was significantly associated with RhoA in TGFβ-stimulated cardiac fibroblasts (6). GEF-H1 is reportedly activated by microtubule depolymerization, and oxidative stress increases GEF-H1 activity through microtubule depolymerization-dependent manner (6). As inhibition of TRPC3 or Nox2 suppressed the mechanical stretch-induced RhoA activation in rat cardiomyocytes and the TGFβ-stimulated RhoA activation in rat cardiac fibroblasts, Nox2-derived ROS-mediated GEF-H1 activation may underlie the induction of fibrotic responses induced by mechanical stress in cardiomyocytes as well as TGFβ stimulation in cardiac fibroblasts.
The reciprocal positive regulation of TRPC3 and Nox2 caused aberrant increase of ROS production in mechanically stressed hearts, which lead to RhoA activation pathway in both cardiomyocytes and cardiac fibroblasts, resulting in eventual cardiac fibrosis (Figure 3). Interestingly, both TRPC3-deleted and Nox2-deleted mice suppressed only cardiac fibrosis in response to pressure overload (5, 73), while both hypertrophy and fibrosis were reduced in both mice chronically treated with Ang II (2, 63). These pieces of evidence indicate that TRPC3 and Nox2 have close association in pathological cardiac remodeling caused by various environmental stresses.
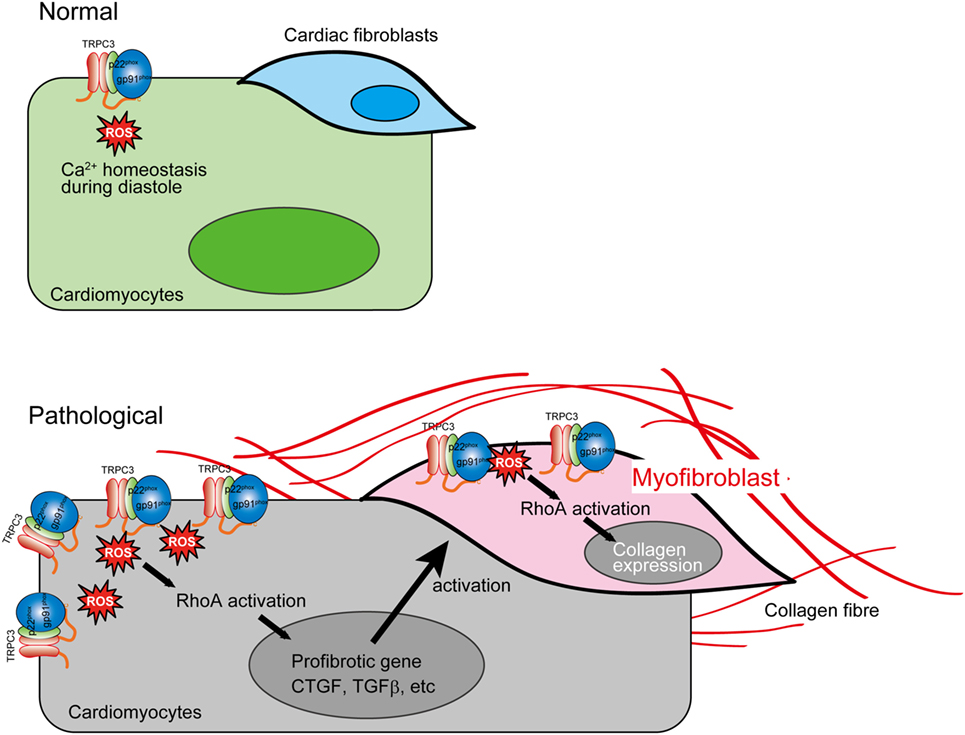
Figure 3. Aberrant reactive oxygen species (ROS) production by TRPC3–NADPH oxidase 2 (Nox2) coupling evokes cardiac fibrosis. In pathological conditions, TRPC3 protein abundance is increased, which leads to Nox2 protein stabilization. This positive regulation of Nox2 induces accumulation of excessive Nox2 complex on the plasma membrane. The ROS production mediated by TRPC3–Nox2 axis activates RhoA in both cardiomyocytes and cardiac fibroblast activated by mechanical stress and TGFβ, respectively, leading to cardiac fibrosis.
Therapeutic Insights
Cardiovascular disease is a leading cause of morbidity and mortality, accounting for more than a quarter of all deaths worldwide (45). Especially, heart failure is a final stage of all cardiovascular diseases, and the 5-year survival rate after diagnosis is less than 50% (93). Since accumulated oxidative stress is the major cause of heart failure, antioxidant agents have been paid attention to the novel therapeutics for heart failure. Based on the involvement of Nox2 in cardiac dysfunction as mentioned above, Nox2-targeted drugs seem to be promising. Several reports demonstrated that inhibitory action on Nox2 ameliorates cardiac dysfunction. Allicin protects against cardiac hypertrophy and fibrosis via attenuating ROS-dependent signaling pathways (94). Trimetazidine inhibits pressure overload-induced cardiac fibrosis (95). Nox inhibition ameliorates cardiac dysfunction in rabbits with heart failure by apocynin (96). However, most of Nox inhibitors are less selective among different Nox isoforms. As mentioned above, Nox2 and Nox4 play also critical role in cardiac physiology. Nox is also important for innate immunity. Therefore, complete and direct suppression of Nox enzyme need to be considered with caution. Seo et al. demonstrated that dual inhibitor of TRPC3/C6, GSK503A, could suppress cardiac fibrosis in pressure-overloaded rat hearts (19). In addition, chronic treatment of a relatively selective TRPC3 inhibitor, Pyr3 suppressed mouse cardiomyopathy in either genetic or pressure-overload mouse model of heart failure (4, 49). These reports strongly suggest that TRPC3 could be a potential pharamacological target. Although beneficial effects of Pyr3 on cardiac remodeling was initially caused by suppressing Ca2+ influx to activate Nox2 enzymatic activity, chronic treatment of Pyr3 indeed reduced Nox2 protein abundance in cardiomyocytes (5). Since chronic treatment of Pyr3 could interfere the physical interaction between TRPC3 and Nox2, Pyr3 could decrease Nox2 stability by disrupting the interaction with TRPC3. As mentioned above, various environmental stresses increase TRPC3 protein abundance in the heart, which concomitantly amplifies Nox2-mediated ROS production, and eventually evokes pathological cardiac remodeling. Therefore, any intervention that suppresses TRPC3–Nox2 interaction would be a novel therapeutic strategy. Kitajima et al. demonstrated that overexpression of C-terminal fragment of TRPC3 that is a critical region for the interaction with Nox2 in cardiomyocytes abrogated TRPC3 channel activity-dependent ROS production (Figures 1 and 2) (5).
Conclusion
It will be no doubt that TRPC channels, especially TRPC3, play a key role in the development of maladaptive cardiac remodeling. Although how local background Ca2+ entry through TRPC channels specifically encodes signals for induction of hypertrophy has been long discussed, we proposed a new concept of physical interaction-dependent mechanism that TRPC3-mediated local Ca2+ influx is directly converted to amplification of Nox2-mediated ROS signaling by stabilizing Nox2 via physical interaction between TRPC3 and Nox2. The TRPC3–Nox2 complex-mediated ROS production leads to fibrotic responses in cardiomyocytes and cardiac fibroblasts through activation of ROS-sensitive GEF-H1 (5, 6). These observations will provide a new therapeutic strategy for the prevention and/or treatment of chronic heart failure. On the other hand, more detailed structure-based analyses must be required to understand how TRPC3 specifically stabilizes Nox2 and why closest analog TRPC6 is unable to stabilize Nox2, although the pharmacological significance of TRPC3–Nox2 complex formation through TRPC3 C-terminal region becomes relevant. It is also interesting whether post-translational modification of TRPC3/6 such as Ser/Thr phosphorylation affects the stability of TRPC3–Nox2 complex. These future studies will deepen the understanding of molecular mechanisms underlying regulation of cardiac plasticity by TRPC channels.
Author Contributions
TN-T, SO, TS, AN, and SM wrote the draft, and MN edited the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported in part by Grants-in-Aid for Scientific Research (16KT0013 and 16H05092 to MN and 17K15585 to TN-T), from the Ministry of Education, Culture, Sports, Science and Technology (MEXT). This work was also supported by PRESTO (No. JPMJPR1336), Japan Science and Technology Agency (JST) and Naito Memorial Foundation (to MN), and Salt Science Foundation (to TN-T).
References
1. Nishida M, Kurose H. Roles of TRP channels in the development of cardiac hypertrophy. Naunyn Schmiedebergs Arch Pharmacol (2008) 378(4):395–406. doi:10.1007/s00210-008-0321-8
2. Nishida M, Onohara N, Sato Y, Suda R, Ogushi M, Tanabe S, et al. Gα12/13-mediated up-regulation of TRPC6 negatively regulates endothelin-1-induced cardiac myofibroblast formation and collagen synthesis through nuclear factor of activated T cells activation. J Biol Chem (2007) 282(32):23117–28. doi:10.1074/jbc.M611780200
3. Onohara N, Nishida M, Inoue R, Kobayashi H, Sumimoto H, Sato Y, et al. TRPC3 and TRPC6 are essential for angiotensin II-induced cardiac hypertrophy. EMBO J (2006) 25(22):5305–16. doi:10.1038/sj.emboj.7601417
4. Kiyonaka S, Kato K, Nishida M, Mio K, Numaga T, Sawaguchi Y, et al. Selective and direct inhibition of TRPC3 channels underlies biological activities of a pyrazole compound. Proc Natl Acad Sci U S A (2009) 106(13):5400–5. doi:10.1073/pnas.0808793106
5. Kitajima N, Numaga-Tomita T, Watanabe M, Kuroda T, Nishimura A, Miyano K, et al. TRPC3 positively regulates reactive oxygen species driving maladaptive cardiac remodeling. Sci Rep (2016) 6:37001. doi:10.1038/srep37001
6. Numaga-Tomita T, Kitajima N, Kuroda T, Nishimura A, Miyano K, Yasuda S, et al. TRPC3-GEF-H1 axis mediates pressure overload-induced cardiac fibrosis. Sci Rep (2016) 6:39383. doi:10.1038/srep39383
7. Montell C, Rubin GM. Molecular characterization of the Drosophila trp locus: a putative integral membrane protein required for phototransduction. Neuron (1989) 2(4):1313–23. doi:10.1016/0896-6273(89)90069-X
8. Ramsey IS, Delling M, Clapham DE. An introduction to TRP channels. Annu Rev Physiol (2006) 68:619–47. doi:10.1146/annurev.physiol.68.040204.100431
9. Vazquez G, Wedel BJ, Aziz O, Trebak M, Putney JW Jr. The mammalian TRPC cation channels. Biochim Biophys Acta (2004) 1742(1–3):21–36. doi:10.1016/j.bbamcr.2004.08.015
10. Wes PD, Chevesich J, Jeromin A, Rosenberg C, Stetten G, Montell C. TRPC1, a human homolog of a Drosophila store-operated channel. Proc Natl Acad Sci U S A (1995) 92(21):9652–6. doi:10.1073/pnas.92.21.9652
11. Zitt C, Zobel A, Obukhov AG, Harteneck C, Kalkbrenner F, Luckhoff A, et al. Cloning and functional expression of a human Ca2+-permeable cation channel activated by calcium store depletion. Neuron (1996) 16(6):1189–96. doi:10.1016/S0896-6273(00)80145-2
12. Zhu X, Jiang M, Peyton M, Boulay G, Hurst R, Stefani E, et al. trp, a novel mammalian gene family essential for agonist-activated capacitative Ca2+ entry. Cell (1996) 85(5):661–71. doi:10.1016/S0092-8674(00)81233-7
13. Liu X, Cheng KT, Bandyopadhyay BC, Pani B, Dietrich A, Paria BC, et al. Attenuation of store-operated Ca2+ current impairs salivary gland fluid secretion in TRPC1(-/-) mice. Proc Natl Acad Sci U S A (2007) 104(44):17542–7. doi:10.1073/pnas.0701254104
14. Mori Y, Wakamori M, Miyakawa T, Hermosura M, Hara Y, Nishida M, et al. Transient receptor potential 1 regulates capacitative Ca2+ entry and Ca2+ release from endoplasmic reticulum in B lymphocytes. J Exp Med (2002) 195(6):673–81. doi:10.1084/jem.20011758
15. Maroto R, Raso A, Wood TG, Kurosky A, Martinac B, Hamill OP. TRPC1 forms the stretch-activated cation channel in vertebrate cells. Nat Cell Biol (2005) 7(2):179–85. doi:10.1038/ncb1218
16. Nishida M, Hara Y, Yoshida T, Inoue R, Mori Y. TRP channels: molecular diversity and physiological function. Microcirculation (2006) 13(7):535–50. doi:10.1080/10739680600885111
17. Poteser M, Graziani A, Rosker C, Eder P, Derler I, Kahr H, et al. TRPC3 and TRPC4 associate to form a redox-sensitive cation channel. Evidence for expression of native TRPC3-TRPC4 heteromeric channels in endothelial cells. J Biol Chem (2006) 281(19):13588–95. doi:10.1074/jbc.M512205200
18. Spassova MA, Hewavitharana T, Xu W, Soboloff J, Gill DL. A common mechanism underlies stretch activation and receptor activation of TRPC6 channels. Proc Natl Acad Sci U S A (2006) 103(44):16586–91. doi:10.1073/pnas.0606894103
19. Seo K, Rainer PP, Shalkey Hahn V, Lee DI, Jo SH, Andersen A, et al. Combined TRPC3 and TRPC6 blockade by selective small-molecule or genetic deletion inhibits pathological cardiac hypertrophy. Proc Natl Acad Sci U S A (2014) 111(4):1551–6. doi:10.1073/pnas.1308963111
20. Weissmann N, Sydykov A, Kalwa H, Storch U, Fuchs B, Mederos y Schnitzler M, et al. Activation of TRPC6 channels is essential for lung ischaemia-reperfusion induced oedema in mice. Nat Commun (2012) 3:649. doi:10.1038/ncomms1660
21. Inoue R, Jensen LJ, Jian Z, Shi J, Hai L, Lurie AI, et al. Synergistic activation of vascular TRPC6 channel by receptor and mechanical stimulation via phospholipase C/diacylglycerol and phospholipase A2/omega-hydroxylase/20-HETE pathways. Circ Res (2009) 104(12):1399–409. doi:10.1161/CIRCRESAHA.108.193227
22. Hofmann T, Obukhov AG, Schaefer M, Harteneck C, Gudermann T, Schultz G. Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature (1999) 397(6716):259–63. doi:10.1038/16711
23. Trebak M, Vazquez G, Bird GS, Putney JW Jr. The TRPC3/6/7 subfamily of cation channels. Cell Calcium (2003) 33(5–6):451–61. doi:10.1016/S0143-4160(03)00056-3
24. Inoue R, Okada T, Onoue H, Hara Y, Shimizu S, Naitoh S, et al. The transient receptor potential protein homologue TRP6 is the essential component of vascular α1-adrenoceptor-activated Ca2+-permeable cation channel. Circ Res (2001) 88(3):325–32. doi:10.1161/01.RES.88.3.325
25. Nishida M, Watanabe K, Sato Y, Nakaya M, Kitajima N, Ide T, et al. Phosphorylation of TRPC6 channels at Thr69 is required for anti-hypertrophic effects of phosphodiesterase 5 inhibition. J Biol Chem (2010) 285(17):13244–53. doi:10.1074/jbc.M109.074104
26. Nishioka K, Nishida M, Ariyoshi M, Jian Z, Saiki S, Hirano M, et al. Cilostazol suppresses angiotensin II-induced vasoconstriction via protein kinase A-mediated phosphorylation of the transient receptor potential canonical 6 channel. Arterioscler Thromb Vasc Biol (2011) 31(10):2278–86. doi:10.1161/ATVBAHA.110.221010
27. Nishida M, Sugimoto K, Hara Y, Mori E, Morii T, Kurosaki T, et al. Amplification of receptor signalling by Ca2+ entry-mediated translocation and activation of PLCγ2 in B lymphocytes. EMBO J (2003) 22(18):4677–88. doi:10.1093/emboj/cdg457
28. Numaga T, Nishida M, Kiyonaka S, Kato K, Katano M, Mori E, et al. Ca2+ influx and protein scaffolding via TRPC3 sustain PKCβ and ERK activation in B cells. J Cell Sci (2010) 123(Pt 6):927–38. doi:10.1242/jcs.061051
29. Woodard GE, Lopez JJ, Jardin I, Salido GM, Rosado JA. TRPC3 regulates agonist-stimulated Ca2+ mobilization by mediating the interaction between type I inositol 1,4,5-trisphosphate receptor, RACK1, and Orai1. J Biol Chem (2010) 285(11):8045–53. doi:10.1074/jbc.M109.033605
30. Bandyopadhyay BC, Ong HL, Lockwich TP, Liu X, Paria BC, Singh BB, et al. TRPC3 controls agonist-stimulated intracellular Ca2+ release by mediating the interaction between inositol 1,4,5-trisphosphate receptor and RACK1. J Biol Chem (2008) 283(47):32821–30. doi:10.1074/jbc.M805382200
31. Tang J, Lin Y, Zhang Z, Tikunova S, Birnbaumer L, Zhu MX. Identification of common binding sites for calmodulin and inositol 1,4,5-trisphosphate receptors on the carboxyl termini of trp channels. J Biol Chem (2001) 276(24):21303–10. doi:10.1074/jbc.M102316200
32. Yao X. TRPC, cGMP-dependent protein kinases and cytosolic Ca2+. Handb Exp Pharmacol (2007) 179:527–40. doi:10.1007/978-3-540-34891-7_31
33. Takahashi S, Lin H, Geshi N, Mori Y, Kawarabayashi Y, Takami N, et al. Nitric oxide-cGMP-protein kinase G pathway negatively regulates vascular transient receptor potential channel TRPC6. J Physiol (2008) 586(17):4209–23. doi:10.1113/jphysiol.2008.156083
34. Koitabashi N, Aiba T, Hesketh GG, Rowell J, Zhang M, Takimoto E, et al. Cyclic GMP/PKG-dependent inhibition of TRPC6 channel activity and expression negatively regulates cardiomyocyte NFAT activation Novel mechanism of cardiac stress modulation by PDE5 inhibition. J Mol Cell Cardiol (2010) 48(4):713–24. doi:10.1016/j.yjmcc.2009.11.015
35. Kinoshita H, Kuwahara K, Nishida M, Jian Z, Rong X, Kiyonaka S, et al. Inhibition of TRPC6 channel activity contributes to the antihypertrophic effects of natriuretic peptides-guanylyl cyclase-A signaling in the heart. Circ Res (2010) 106(12):1849–60. doi:10.1161/CIRCRESAHA.109.208314
36. Hill JA, Olson EN. Cardiac plasticity. N Engl J Med (2008) 358(13):1370–80. doi:10.1056/NEJMra072139
37. Houser SR, Molkentin JD. Does contractile Ca2+ control calcineurin-NFAT signaling and pathological hypertrophy in cardiac myocytes? Sci Signal (2008) 1(25):e31. doi:10.1126/scisignal.125pe31
38. Morine KJ, Paruchuri V, Qiao X, Aronovitz M, Huggins GS, DeNofrio D, et al. Endoglin selectively modulates transient receptor potential channel expression in left and right heart failure. Cardiovasc Pathol (2016) 25(6):478–82. doi:10.1016/j.carpath.2016.08.004
39. Camacho Londono JE, Tian Q, Hammer K, Schroder L, Camacho Londono J, Reil JC, et al. A background Ca2+ entry pathway mediated by TRPC1/TRPC4 is critical for development of pathological cardiac remodelling. Eur Heart J (2015) 36(33):2257–66. doi:10.1093/eurheartj/ehv250
40. Zou G, Hong H, Lin X, Shi X, Wu Y, Chen L. TRPC1, CaN and NFATC3 signaling pathway in the pathogenesis and progression of left ventricular hypertrophy in spontaneously hypertensive rats. Clin Exp Hypertens (2015) 37(3):223–34. doi:10.3109/10641963.2014.943405
41. Makarewich CA, Zhang H, Davis J, Correll RN, Trappanese DM, Hoffman NE, et al. Transient receptor potential channels contribute to pathological structural and functional remodeling after myocardial infarction. Circ Res (2014) 115(6):567–80. doi:10.1161/CIRCRESAHA.115.303831
42. Ohba T, Watanabe H, Murakami M, Takahashi Y, Iino K, Kuromitsu S, et al. Upregulation of TRPC1 in the development of cardiac hypertrophy. J Mol Cell Cardiol (2007) 42(3):498–507. doi:10.1016/j.yjmcc.2006.10.020
43. Williams IA, Allen DG. Intracellular calcium handling in ventricular myocytes from mdx mice. Am J Physiol Heart Circ Physiol (2007) 292(2):H846–55. doi:10.1152/ajpheart.00688.2006
44. Ohba T, Watanabe H, Takahashi Y, Suzuki T, Miyoshi I, Nakayama S, et al. Regulatory role of neuron-restrictive silencing factor in expression of TRPC1. Biochem Biophys Res Commun (2006) 351(3):764–70. doi:10.1016/j.bbrc.2006.10.107
45. Rowell J, Koitabashi N, Kass DA. TRP-ing up heart and vessels: canonical transient receptor potential channels and cardiovascular disease. J Cardiovasc Transl Res (2010) 3(5):516–24. doi:10.1007/s12265-010-9208-4
46. Hirose M, Takeishi Y, Niizeki T, Nakada T, Shimojo H, Kashihara T, et al. Diacylglycerol kinase zeta inhibits ventricular tachyarrhythmias in a mouse model of heart failure. Circ J (2011) 75(10):2333–42. doi:10.1253/circj.CJ-10-1213
47. Matsushita N, Kashihara T, Shimojo H, Suzuki S, Nakada T, Takeishi Y, et al. Cardiac overexpression of constitutively active Galpha q causes angiotensin II type1 receptor activation, leading to progressive heart failure and ventricular arrhythmias in transgenic mice. PLoS One (2014) 9(8):e106354. doi:10.1371/journal.pone.0106354
48. Harada M, Luo X, Qi XY, Tadevosyan A, Maguy A, Ordog B, et al. Transient receptor potential canonical-3 channel-dependent fibroblast regulation in atrial fibrillation. Circulation (2012) 126(17):2051–64. doi:10.1161/CIRCULATIONAHA.112.121830
49. Kitajima N, Watanabe K, Morimoto S, Sato Y, Kiyonaka S, Hoshijima M, et al. TRPC3-mediated Ca2+ influx contributes to Rac1-mediated production of reactive oxygen species in MLP-deficient mouse hearts. Biochem Biophys Res Commun (2011) 409(1):108–13. doi:10.1016/j.bbrc.2011.04.124
50. Shan D, Marchase RB, Chatham JC. Overexpression of TRPC3 increases apoptosis but not necrosis in response to ischemia-reperfusion in adult mouse cardiomyocytes. Am J Physiol Cell Physiol (2008) 294(3):C833–41. doi:10.1152/ajpcell.00313.2007
51. Brenner JS, Dolmetsch RE. TrpC3 regulates hypertrophy-associated gene expression without affecting myocyte beating or cell size. PLoS One (2007) 2(8):e802. doi:10.1371/journal.pone.0000802
52. Nakayama H, Wilkin BJ, Bodi I, Molkentin JD. Calcineurin-dependent cardiomyopathy is activated by TRPC in the adult mouse heart. FASEB J (2006) 20(10):1660–70. doi:10.1096/fj.05-5560com
53. Bush EW, Hood DB, Papst PJ, Chapo JA, Minobe W, Bristow MR, et al. Canonical transient receptor potential channels promote cardiomyocyte hypertrophy through activation of calcineurin signaling. J Biol Chem (2006) 281(44):33487–96. doi:10.1074/jbc.M605536200
54. Seo K, Rainer PP, Lee DI, Hao S, Bedja D, Birnbaumer L, et al. Hyperactive adverse mechanical stress responses in dystrophic heart are coupled to transient receptor potential canonical 6 and blocked by cGMP-protein kinase G modulation. Circ Res (2014) 114(5):823–32. doi:10.1161/CIRCRESAHA.114.302614
55. Xie J, Cha SK, An SW, Kuro OM, Birnbaumer L, Huang CL. Cardioprotection by Klotho through downregulation of TRPC6 channels in the mouse heart. Nat Commun (2012) 3:1238. doi:10.1038/ncomms2240
56. Niizeki T, Takeishi Y, Kitahara T, Arimoto T, Koyama Y, Goto K, et al. Diacylglycerol kinase zeta rescues Gαq-induced heart failure in transgenic mice. Circ J (2008) 72(2):309–17. doi:10.1253/circj.72.309
57. Kuwahara K, Wang Y, McAnally J, Richardson JA, Bassel-Duby R, Hill JA, et al. TRPC6 fulfills a calcineurin signaling circuit during pathologic cardiac remodeling. J Clin Invest (2006) 116(12):3114–26. doi:10.1172/JCI27702
58. Satoh S, Tanaka H, Ueda Y, Oyama J, Sugano M, Sumimoto H, et al. Transient receptor potential (TRP) protein 7 acts as a G protein-activated Ca2+ channel mediating angiotensin II-induced myocardial apoptosis. Mol Cell Biochem (2007) 294(1–2):205–15. doi:10.1007/s11010-006-9261-0
59. He X, Li S, Liu B, Susperreguy S, Formoso K, Yao J, et al. Major contribution of the 3/6/7 class of TRPC channels to myocardial ischemia/reperfusion and cellular hypoxia/reoxygenation injuries. Proc Natl Acad Sci U S A (2017) 114(23):E4582–91. doi:10.1073/pnas.1621384114
60. Wu X, Eder P, Chang B, Molkentin JD. TRPC channels are necessary mediators of pathologic cardiac hypertrophy. Proc Natl Acad Sci U S A (2010) 107(15):7000–5. doi:10.1073/pnas.1001825107
61. Dominguez-Rodriguez A, Ruiz-Hurtado G, Sabourin J, Gomez AM, Alvarez JL, Benitah JP. Proarrhythmic effect of sustained EPAC activation on TRPC3/4 in rat ventricular cardiomyocytes. J Mol Cell Cardiol (2015) 87:74–8. doi:10.1016/j.yjmcc.2015.07.002
62. Barrick CJ, Rojas M, Schoonhoven R, Smyth SS, Threadgill DW. Cardiac response to pressure overload in 129S1/SvImJ and C57BL/6J mice: temporal- and background-dependent development of concentric left ventricular hypertrophy. Am J Physiol Heart Circ Physiol (2007) 292(5):H2119–30. doi:10.1152/ajpheart.00816.2006
63. El-Benna J, Dang PM, Gougerot-Pocidalo MA, Marie JC, Braut-Boucher F. p47phox, the phagocyte NADPH oxidase/NOX2 organizer: structure, phosphorylation and implication in diseases. Exp Mol Med (2009) 41(4):217–25. doi:10.3858/emm.2009.41.4.058
64. Prosser BL, Khairallah RJ, Ziman AP, Ward CW, Lederer WJ. X-ROS signaling in the heart and skeletal muscle: stretch-dependent local ROS regulates [Ca2+]i. J Mol Cell Cardiol (2013) 58:172–81. doi:10.1016/j.yjmcc.2012.11.011
65. Bendall JK, Cave AC, Heymes C, Gall N, Shah AM. Pivotal role of a gp91phox-containing NADPH oxidase in angiotensin II-induced cardiac hypertrophy in mice. Circulation (2002) 105(3):293–6. doi:10.1161/hc0302.103712
66. Wojnowski L, Kulle B, Schirmer M, Schluter G, Schmidt A, Rosenberger A, et al. NAD(P)H oxidase and multidrug resistance protein genetic polymorphisms are associated with doxorubicin-induced cardiotoxicity. Circulation (2005) 112(24):3754–62. doi:10.1161/CIRCULATIONAHA.105.576850
67. Matsushima S, Tsutsui H, Sadoshima J. Physiological and pathological functions of NADPH oxidases during myocardial ischemia-reperfusion. Trends Cardiovasc Med (2014) 24(5):202–5. doi:10.1016/j.tcm.2014.03.003
68. Prosser BL, Ward CW, Lederer WJ. X-ROS signaling: rapid mechano-chemo transduction in heart. Science (2011) 333(6048):1440–5. doi:10.1126/science.1202768
69. Heymes C, Bendall JK, Ratajczak P, Cave AC, Samuel JL, Hasenfuss G, et al. Increased myocardial NADPH oxidase activity in human heart failure. J Am Coll Cardiol (2003) 41(12):2164–71. doi:10.1016/S0735-1097(03)00471-6
70. Hingtgen SD, Tian X, Yang J, Dunlay SM, Peek AS, Wu Y, et al. Nox2-containing NADPH oxidase and Akt activation play a key role in angiotensin II-induced cardiomyocyte hypertrophy. Physiol Genomics (2006) 26(3):180–91. doi:10.1152/physiolgenomics.00029.2005
71. Shanmugam P, Valente AJ, Prabhu SD, Venkatesan B, Yoshida T, Delafontaine P, et al. Angiotensin-II type 1 receptor and NOX2 mediate TCF/LEF and CREB dependent WISP1 induction and cardiomyocyte hypertrophy. J Mol Cell Cardiol (2011) 50(6):928–38. doi:10.1016/j.yjmcc.2011.02.012
72. Byrne JA, Grieve DJ, Bendall JK, Li JM, Gove C, Lambeth JD, et al. Contrasting roles of NADPH oxidase isoforms in pressure-overload versus angiotensin II-induced cardiac hypertrophy. Circ Res (2003) 93(9):802–5. doi:10.1161/01.RES.0000099504.30207.F5
73. Maytin M, Siwik DA, Ito M, Xiao L, Sawyer DB, Liao R, et al. Pressure overload-induced myocardial hypertrophy in mice does not require gp91phox. Circulation (2004) 109(9):1168–71. doi:10.1161/01.CIR.0000117229.60628.2F
74. Heymans S, Gonzalez A, Pizard A, Papageorgiou AP, Lopez-Andres N, Jaisser F, et al. Searching for new mechanisms of myocardial fibrosis with diagnostic and/or therapeutic potential. Eur J Heart Fail (2015) 17(8):764–71. doi:10.1002/ejhf.312
75. Miguel-Carrasco JL, Baltanas A, Cebrian C, Moreno MU, Lopez B, Hermida N, et al. Blockade of TGF-β1 signalling inhibits cardiac NADPH oxidase overactivity in hypertensive rats. Oxid Med Cell Longev (2012) 2012:726940. doi:10.1155/2012/726940
76. Johar S, Cave AC, Narayanapanicker A, Grieve DJ, Shah AM. Aldosterone mediates angiotensin II-induced interstitial cardiac fibrosis via a Nox2-containing NADPH oxidase. FASEB J (2006) 20(9):1546–8. doi:10.1096/fj.05-4642fje
77. Satoh M, Ogita H, Takeshita K, Mukai Y, Kwiatkowski DJ, Liao JK. Requirement of Rac1 in the development of cardiac hypertrophy. Proc Natl Acad Sci U S A (2006) 103(19):7432–7. doi:10.1073/pnas.0510444103
78. Whaley-Connell A, Govindarajan G, Habibi J, Hayden MR, Cooper SA, Wei Y, et al. Angiotensin II-mediated oxidative stress promotes myocardial tissue remodeling in the transgenic (mRen2) 27 Ren2 rat. Am J Physiol Endocrinol Metab (2007) 293(1):E355–63. doi:10.1152/ajpendo.00632.2006
79. Li J, Stouffs M, Serrander L, Banfi B, Bettiol E, Charnay Y, et al. The NADPH oxidase NOX4 drives cardiac differentiation: role in regulating cardiac transcription factors and MAP kinase activation. Mol Biol Cell (2006) 17(9):3978–88. doi:10.1091/mbc.E05-06-0532
80. Kuroda J, Ago T, Matsushima S, Zhai P, Schneider MD, Sadoshima J. NADPH oxidase 4 (Nox4) is a major source of oxidative stress in the failing heart. Proc Natl Acad Sci U S A (2010) 107(35):15565–70. doi:10.1073/pnas.1002178107
81. Zhang M, Brewer AC, Schroder K, Santos CX, Grieve DJ, Wang M, et al. NADPH oxidase-4 mediates protection against chronic load-induced stress in mouse hearts by enhancing angiogenesis. Proc Natl Acad Sci U S A (2010) 107(42):18121–6. doi:10.1073/pnas.1009700107
82. Langbein H, Brunssen C, Hofmann A, Cimalla P, Brux M, Bornstein SR, et al. NADPH oxidase 4 protects against development of endothelial dysfunction and atherosclerosis in LDL receptor deficient mice. Eur Heart J (2016) 37(22):1753–61. doi:10.1093/eurheartj/ehv564
83. Schroder K, Zhang M, Benkhoff S, Mieth A, Pliquett R, Kosowski J, et al. Nox4 is a protective reactive oxygen species generating vascular NADPH oxidase. Circ Res (2012) 110(9):1217–25. doi:10.1161/CIRCRESAHA.112.267054
84. Matsuno K, Iwata K, Matsumoto M, Katsuyama M, Cui W, Murata A, et al. NOX1/NADPH oxidase is involved in endotoxin-induced cardiomyocyte apoptosis. Free Radic Biol Med (2012) 53(9):1718–28. doi:10.1016/j.freeradbiomed.2012.08.590
85. Brechard S, Tschirhart EJ. Regulation of superoxide production in neutrophils: role of calcium influx. J Leukoc Biol (2008) 84(5):1223–37. doi:10.1189/jlb.0807553
86. Foyouzi-Youssefi R, Petersson F, Lew DP, Krause KH, Nusse O. Chemoattractant-induced respiratory burst: increases in cytosolic Ca2+ concentrations are essential and synergize with a kinetically distinct second signal. Biochem J (1997) 322(Pt 3):709–18. doi:10.1042/bj3220709
87. Gallois A, Bueb JL, Tschirhart E. Effect of SK&F 96365 on extracellular Ca2+-dependent O2- production in neutrophil-like HL-60 cells. Eur J Pharmacol (1998) 361(2–3):293–8. doi:10.1016/S0014-2999(98)00728-6
88. Brechard S, Melchior C, Plancon S, Schenten V, Tschirhart EJ. Store-operated Ca2+ channels formed by TRPC1, TRPC6 and Orai1 and non-store-operated channels formed by TRPC3 are involved in the regulation of NADPH oxidase in HL-60 granulocytes. Cell Calcium (2008) 44(5):492–506. doi:10.1016/j.ceca.2008.03.002
89. Noubade R, Wong K, Ota N, Rutz S, Eidenschenk C, Valdez PA, et al. NRROS negatively regulates reactive oxygen species during host defence and autoimmunity. Nature (2014) 509(7499):235–9. doi:10.1038/nature13152
90. Kim EY, Anderson M, Dryer SE. Insulin increases surface expression of TRPC6 channels in podocytes: role of NADPH oxidases and reactive oxygen species. Am J Physiol Renal Physiol (2012) 302(3):F298–307. doi:10.1152/ajprenal.00423.2011
91. Kim EY, Anderson M, Dryer SE. Sustained activation of N-methyl-d-aspartate receptors in podoctyes leads to oxidative stress, mobilization of transient receptor potential canonical 6 channels, nuclear factor of activated T cells activation, and apoptotic cell death. Mol Pharmacol (2012) 82(4):728–37. doi:10.1124/mol.112.079376
92. Wang Z, Wei X, Zhang Y, Ma X, Li B, Zhang S, et al. NADPH oxidase-derived ROS contributes to upregulation of TRPC6 expression in puromycin aminonucleoside-induced podocyte injury. Cell Physiol Biochem (2009) 24(5–6):619–26. doi:10.1159/000257517
93. Levy D, Kenchaiah S, Larson MG, Benjamin EJ, Kupka MJ, Ho KK, et al. Long-term trends in the incidence of and survival with heart failure. N Engl J Med (2002) 347(18):1397–402. doi:10.1056/NEJMoa020265
94. Liu C, Cao F, Tang QZ, Yan L, Dong YG, Zhu LH, et al. Allicin protects against cardiac hypertrophy and fibrosis via attenuating reactive oxygen species-dependent signaling pathways. J Nutr Biochem (2010) 21(12):1238–50. doi:10.1016/j.jnutbio.2009.11.001
95. Liu X, Gai Y, Liu F, Gao W, Zhang Y, Xu M, et al. Trimetazidine inhibits pressure overload-induced cardiac fibrosis through NADPH oxidase-ROS-CTGF pathway. Cardiovasc Res (2010) 88(1):150–8. doi:10.1093/cvr/cvq181
Keywords: Ca2+, canonical transient receptor potential, reactive oxygen species, NADPH oxidase, cardiac remodeling, cardiac fibrosis
Citation: Numaga-Tomita T, Oda S, Shimauchi T, Nishimura A, Mangmool S and Nishida M (2017) TRPC3 Channels in Cardiac Fibrosis. Front. Cardiovasc. Med. 4:56. doi: 10.3389/fcvm.2017.00056
Received: 02 May 2017; Accepted: 21 August 2017;
Published: 07 September 2017
Edited by:
Andrew James Webb, King’s College London, United KingdomReviewed by:
Suowen Xu, University of Rochester, United StatesTom Van Agtmael, University of Glasgow, United Kingdom
Copyright: © 2017 Numaga-Tomita, Oda, Shimauchi, Nishimura, Mangmool and Nishida. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Motohiro Nishida, bmlzaGlkYUBuaXBzLmFjLmpw