 Nathaly Anto Michel
Nathaly Anto Michel Andreas Zirlik
Andreas Zirlik Dennis Wolf
Dennis Wolf- Faculty of Medicine, Department of Cardiology and Angiology I, Heart Center Freiburg, University of Freiburg, Freiburg, Germany
CD40L (CD154), a member of the tumor necrosis factor superfamily, is a co-stimulatory molecule that was first discovered on activated T cells. Beyond its fundamental role in adaptive immunity—ligation of CD40L to its receptor CD40 is a prerequisite for B cell activation and antibody production—evidence from more than two decades has expanded our understanding of CD40L as a powerful modulator of inflammatory pathways. Although inhibition of CD40L with neutralizing antibodies has induced life-threatening side effects in clinical trials, the discovery of cell-specific effects and novel receptors with distinct functional consequences has opened a new path for therapies that specifically target detrimental properties of CD40L. Here, we carefully evaluate the signaling network of CD40L by gene enrichment analysis and its cell-specific expression, and thoroughly discuss its role in cardiovascular pathologies with a specific emphasis on atherosclerotic and thrombotic disease.
Cardiovascular disease is the major cause of mortality worldwide and is predominantly caused by atherosclerosis, a chronic narrowing of middle sized and large arteries by the buildup of atherosclerotic plaques (1). Subclinical atherosclerosis precedes its potentially life-threatening complications, including acute arterial thrombosis, myocardial infarction, and stroke (2). In the past few decades, it has been well established that inflammatory cues critically fuel the initiation, progression, and complication of atherosclerosis by promoting accumulation of inflammatory leukocytes in the plaque and by driving inflammatory gene expression both systemically and in the atherosclerotic lesion (3). In addition, inflammation exacerbates cardiovascular disease risk factors such as obesity, hypertension, dyslipidemia, and insulin resistance. Therefore, the modulation of inflammatory pathways has been proposed to be a powerful therapeutic strategy against cardiovascular disease (4). Because inflammation is involved in a variety of physiological processes, including host defense, wound healing, hemostasis, and regeneration, the search for pathways and effector molecules that specifically enhance abnormal and dysregulated inflammation has become a major goal. The tumor necrosis factor (TNF) receptor superfamily (TNFRSF) comprises a class of 29 receptors with structural similarities and overlapping functions that can selectively bind one or more of the 19 members of the TNF (ligand) superfamily (TNFSF). These receptor/ligand pairs regulate survival and activation of immune cells and drive the expression of genes that modulate inflammation, immunity, and autoimmunity (5). The ability to modulate inflammation by targeting members of the TNF superfamily is best illustrated by the clinical inhibition of TNF-α, which is now considered to be a hallmark of immunotherapy and anti-inflammatory therapy in a variety of chronic inflammatory pathologies (6).
CD40L—More than a Co-Stimulator of B Cells
CD40L (also known as CD154, gp39, TRAP, TBAM) is a member of the TNF superfamily that was first identified on activated T cells, where it interacts with CD40 receptor on B cells to induce B cell activation, proliferation, and IgG-class switching during co-stimulation (7–10). Mutations of the CD40L gene (Cd40lg) were identified as the cause of the human X-linked immunodeficiency hyper IgM-syndrome (XHIM), a condition characterized by a loss of T cell-dependent humoral immunity and specific IgG antibodies (11). Apart from the 33 kDa full-length version of CD40L that forms trimeric complexes on the cell surface, a truncated 18-kDa version that lacks the cytoplasmic tail, the transmembrane domain, and parts of the extracellular domain is generated by shedding membrane-anchored CD40L by matrix metalloproteinases (MMPs) and certain disintegrin metalloproteinases (ADAMs) (12–14) (Figure 1). This soluble fraction of CD40L (sCD40L) can be detected in the blood circulation, but it also forms multimeric complexes with the membrane-anchored full-length version of CD40L on the cell surface (15). The biological activity of CD40L increases with a higher multimeric organization (16, 17), and it has previously been described that sCD40L is biologically less active than membrane-bound CD40L (16–21). This may in part be caused by an incomplete formation of sCD40L trimers, as sCD40L lacks the transmembrane domain and parts of the extracellular domain that can support trimerization (16, 17). However, the definitive structural organization of sCD40L is still under debate, as trimers of CD40L have been observed to form through interactions independent of the trimerization domain. Accordingly, the spontaneous formation of monomers, dimers, and trimers of sCD40L are detected in the blood circulation (18, 22, 23).
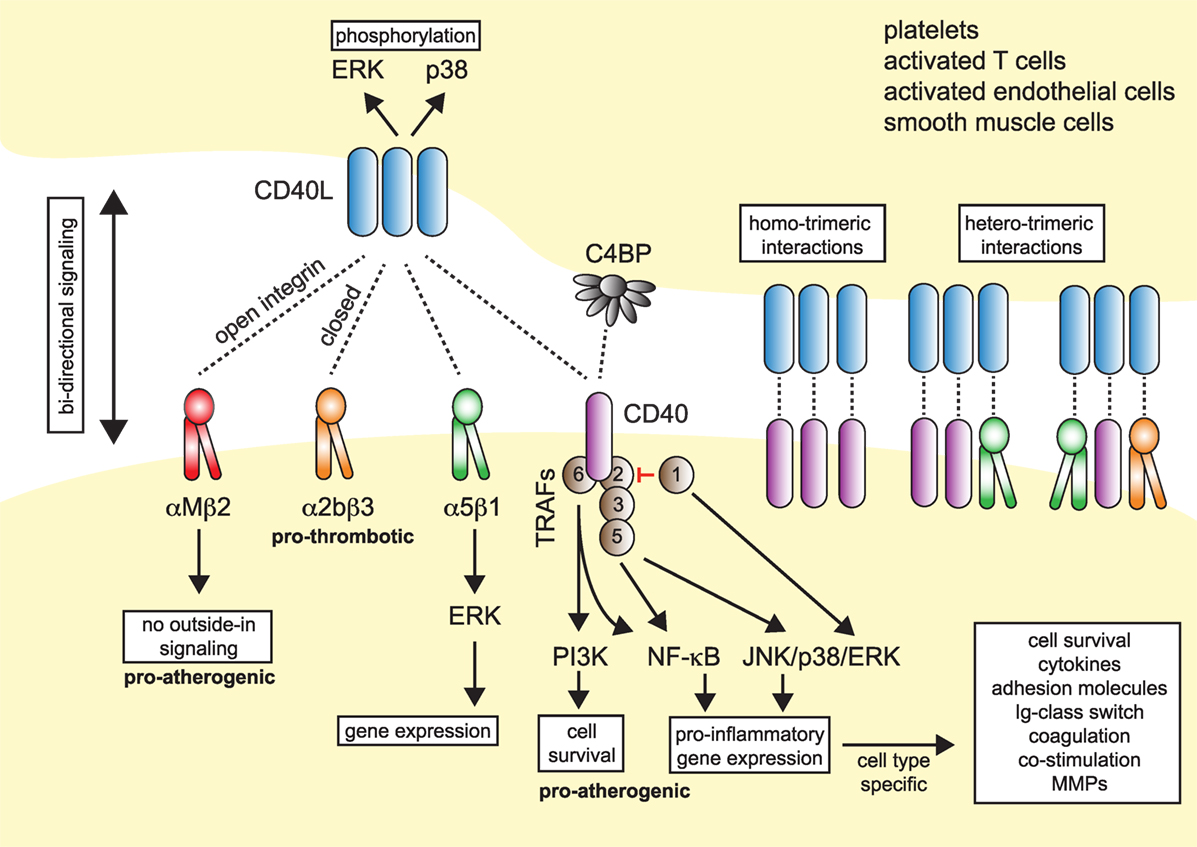
Figure 1. Model of CD40L–receptor interactions. Membrane-bound CD40L forms biologically active trimers that can interact with one of its known receptors, the integrins αIIbβ3, α5β1, αMβ2 (Mac-1), or its “classical” receptor CD40. While binding to CD40 occurs statically, binding to integrins can be enhanced by activation-induced conformational changes of both subunits that expose the ligand-binding site (inside-out signaling). Binding to Mac-1 occurs predominantly through the activated integrin, binding to α5β1 requires no previous cell activation and switching into the open, high-affinity conformation. Binding to αIIbβ3 usually occurs through the open conformation, but exact binding properties to CD40L have not been investigated in detail. Besides CD40L, CD40 can also interact with the complement-factor C4b-binding protein (C4BP). Binding of CD40L to one of its receptors induces downstream signaling events, except for Mac-1, for which CD40L serves as biased agonist without induction of outside-in signaling. Binding to α5β1 causes an activation of MAP-kinase signaling pathways, ligation to CD40 causes activation of mitogen-activated protein kinase (MAPK)-, phosphoinositide 3-kinase (PI3K)-, and nuclear factor-κB (NF-κB)-signaling events and subsequent pro-inflammatory gene expression. The consequences of CD40 signaling depend on the target cell type. On the contrary, ligation of CD40L to a receptor induces bidirectional signaling events in CD40L-bearing cells, e.g., T cells, B cells, or platelets, possibly by induction of MAPK signaling cascades. Binding of CD40L to its ligands can occur in a homotrimeric fashion, where a trimer of CD40L binds three monomers of CD40, or in a heterotrimeric fashion, where each monomer (of a trimer of CD40L) can bind to different receptors, which was demonstrated for CD40, αIIbβ3, and α5β1.
Levels of sCD40L have been proposed as biomarkers of atherothrombosis (24, 25). While it was originally perceived that the expression of CD40L is restricted to T cells, numerous studies have shown that a broad range of hematopoietic cells and vascular/stromal cells can express CD40L in an inducible fashion. Cell types expressing CD40L include T cells, B cells, basophils, eosinophils, monocytes, macrophages, Kupffer cells, natural killer (NK) cells, platelets, mast cells, and dendritic cells (DCs), as well as endothelial cells (ECs), smooth muscle cells (SMCs), and epithelial cells [reviewed in Ref. (7)]. Among hematopoietic cells, gene expression of CD40L is highest in T cells and megakaryocytes, the progenitors of platelets in the bone marrow (Figure 2). The detection of CD40L on cells residing in the atherosclerotic plaque—including ECs, macrophages, foam cells, and SMCs—proposed that CD40L contributes to lesion development and inflammation in atherosclerosis (26, 27). Indeed, work for over two decades has identified CD40L as both a pro-inflammatory surface molecule and soluble cytokine (sCD40L), driving expression of other cytokines, chemokines, adhesion molecules, extracellular matrix-degrading enzymes, and mediators of cell survival. Furthermore, CD40L functions as an adhesion receptor that promotes cell recruitment as well as physical interactions between platelets. A functional involvement of CD40L has been shown in several inflammatory and autoimmune pathologies, including arthritis, nephritis, organ rejection, autoimmune diabetes, inflammatory bowel disease, and systemic lupus erythematosus (SLE) (28–32). Traditionally, CD40L was believed to interact solely with CD40. Recent evidence, however, demonstrates the existence and functional participation of alternative receptors. Here, we will review the functional role of CD40L and its receptors in cardiovascular pathologies.
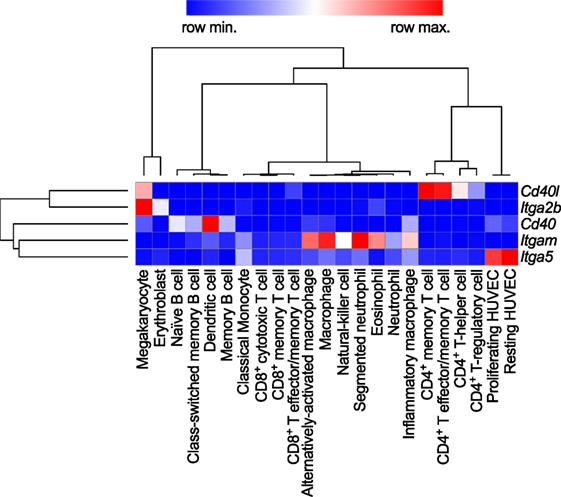
Figure 2. Gene expression patterns of CD40L and its receptors in human immune cell types. Baseline gene expression of different human immune cell types quantified by RNAseq was extracted from the Protein Expression Atlas of the European Bioinformatics Institute (EMBL-EBI) (33). Expression values were retrieved as FPKMs, underwent hierarchical clustering and normalization as row scores by Morpheus (Broad Institute). Gene names are encoding for the proteins as follows: Cd40lg: CD40L; Itgb2: integrin subunit α2b (CD41); Cd40: CD40 receptor; Itgam: integrin subunit αM (CD11b); Itga5: integrin subunit α5 (CD49e).
Receptors for CD40L
Up until now, four different receptors have been identified for CD40L: the three integrins αMβ2 (Mac-1), αIIbβ3, and α5β1, and the “classical” receptor CD40. These are distinguishable by their cellular expression patterns, signaling events, and functional consequences (summarized in Figure 1).
CD40
CD40, member of the TNF receptor superfamily, is a 48-kDa type I transmembrane protein. In addition to B cells, CD40 is expressed constitutively and in an inducible fashion by T cells, DCs, monocytes, platelets, macrophages, SMCs, ECs, and fibroblasts (34–36). Human CD40 gene expression is highest in DCs and intermediate in B cells and megakaryocytes, i.e., platelets (Figure 2). Its expression is regulated by inflammatory cues, including TNF-α, IL-1, IFN-γ, CD40L, and others (7, 21). Of all receptors that ligate CD40L, CD40 has been reported to exhibit the highest affinity for monomeric CD40L, around ~0.5–7nM (37, 38), but higher affinities are suspected for the interaction with trimeric CD40L (39) (Table 1). Whether monomeric sCD40L can act as a competitive antagonist for membrane-bound CD40L is not known, but the biological activity for multimeric, membrane-bound CD40L is higher than that of monomeric CD40L (16–21, 40). Upon binding by CD40L, CD40 monomers on the cell surface are clustered in trimers, a process that is thought to occur more frequently in lipid rafts and by ligation with CD40L itself (41, 42). Consequently, tumor necrosis factor receptor-associated factors (TRAFs), a group of intracellular adapter proteins that also associate with TNF receptors, toll-like receptors (TLRs), and IL-1 receptors, are recruited to the cytoplasmic domain of CD40 (43), where they activate canonical and non-canonical pathways that can result in an engagement of nuclear factor-κB (NF-κB), MAPKs, and phosphoinositide 3-kinase (PI3K), as well as phospholipase Cγ (44). While TRAF-2/3/5/6 activate downstream signaling, TRAF-1 inactivates TRAF-2 by direct binding and is considered an inhibitory TRAF (45). Biological effects of CD40 ligation include cell survival, proliferation, and inflammatory cytokine and chemokine expression (29, 46, 47). CD40 can have distinct effects on different cell types: ligation of CD40 on B cells induces proliferation, activation, and an IgG-class switch (10), while ECs and fibroblasts increase expression of the adhesion factors VCAM-1, ICAM-1, and E-selectin (7). SMCs and ECs release MPP-1, -2, -3, and -9, which are molecules involved in the destabilization of atherosclerotic plaques (7, 48). In contrast, CD40 seems to have an inhibitory role on T cells, as it prevents cytokine release and T cell activation (49).
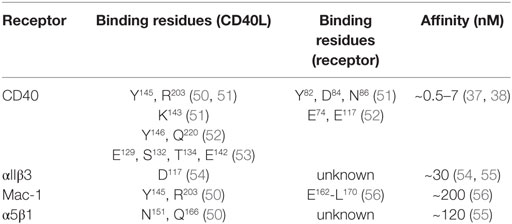
Table 1. Binding properties of CD40L’s receptors.
Apart from CD40L, CD40 has also been reported to bind the complement-associated C4b-binding protein (C4BP), an interaction that occurs on a separate site from the CD40L binding epitope (57), although C4BP binding may partially be mediated by cross-binding to CD40L (58). B cells stimulated with C4BP show enhanced proliferation, adhesion receptor expression, and an IL-4-dependent IgE-class switch.
αMβ2 (Mac-1)
Mac-1 (CD11b/CD18) is a member of the β2-integrin family and a heterodimer of the αM (CD11b) and β2 (CD18) integrin subunit. Mac-1 is predominantly expressed on myeloid cells, such as monocytes, macrophages, and neutrophils, but is also present on NK cells, and, to a smaller extent, on some B cell subsets (B1); however, the highest mRNA abundance of the CD11b αM subunit (Itgam) is confined to neutrophils and macrophages (Figure 2). Mac-1 is required for the firm adhesion and slow rolling of leukocytes on ECs (59, 60). Mac-1 interacts with a broad repertoire of different ligands, including C3bi (61), ICAM-1 (59), fibrinogen (62), fibronectin (63), vitronectin (63), heparin (64), GPIbα (65), RAGE (66), endothelial protein C receptor (EPCR) (67), and others (68). We have recently shown that Mac-1 interacts with CD40L by utilizing a binding site on the major ligand-binding I-domain within the αM subunit (E162-L170), which is distinct from other ligand-binding sites and, thus, is highly selective for CD40L (56, 69). Binding of CD40L by Mac-1 is enhanced by the open, high-affinity conformation of the integrin (50), suggesting that Mac-1/CD40L binding can be regulated by integrin inside-out signaling (70). Peptide inhibition studies have revealed that the CD40L/Mac-1 interaction primarily serves as an adhesive receptor–ligand pair: CD40L expressed on activated ECs binds to Mac-1 on rolling leukocytes to allow their firm adhesion (26, 56, 71). Whether Mac-1 binds to a specific binding site on CD40L is controversial; while Mac-1 binding does not compete with binding of CD40L to CD40 in competitive binding assays (56), a mutant version of CD40L, which lacked the binding site for CD40, also abolished Mac-1 binding (50). These findings suggest that common residues in CD40L involved in the binding of Mac-1 and CD40 exist (Table 1). The main endothelial receptors for Mac-1 include ICAM-1, CD40L, RAGE, and EPCR. In tissue-resident macrophages, where Mac-1 is highly expressed (71, 72), ligation of the integrin promotes cell activation, inflammatory gene expression, and participates in phagocytosis (71–73). While most Mac-1 ligands induce cell activation upon binding—a process referred to as outside-in signaling—CD40L serves as biased agonist that can bind to the integrin without inducing cellular activation (68). Mac-1 is required for many physiological and pathogenic processes involved in inflammation, host defense, and wound healing (72). Inhibition of Mac-1 reduces atherosclerosis (69), neointima formation (74, 75), and thrombotic glomerulonephritis (76) in mice. In humans, a mutation of the β2-subunit, which affects the functionality of Mac-1, LFA-1, and CD11c, is responsible for an immune deficiency known as leukocyte adhesion deficiency (77).
α5β1
The integrin α5β1 (VLA-5) is a heterodimer of the integrin subunits α5 (CD49e) and β1 (CD29). It serves as a primary receptor for fibronectin and vitronectin through an RGD sequence in both molecules (78, 79). It is expressed in monocytes, macrophages, and ECs. The gene transcript of the α5 subunit (Itga5) is most abundant in ECs (Figure 2). In addition, stromal cells such as fibroblasts, chondrocytes, and synovial cells (80) have been reported to express VLA-5. Ligation of the integrin induces MAPK signaling via ERK and stimulates cell survival, proliferation, and inflammatory gene expression (81). ECs require functional VLA-5 to attach, spread, and proliferate within the extracellular matrix (82). VLA-5 also interacts with VEGFR-1 (82), angiopoietin-2 (83), and endostatin (84), which may explain its requirement for angiogenesis (85). Leveille et al. identified CD40L as a ligand for α5β1 that unexpectedly—and in contrast to most integrin ligands—binds to the inactivated conformation of the integrin. Binding of CD40L to VLA-5 induced ERK-signaling pathways and IL-8 expression in a human monocytic cell line (80), similarly to the response caused by fibronectin binding to the integrin. Besides a cross talk of cells with the extracellular matrix, the CD40L/VLA-5 interaction was also recently shown to mediate cytokine production and the adhesion of CD40L+ T cells with fibroblasts (86) and to inhibit apoptosis in T cells (87). Binding sites of CD40 and α5β1 were mapped to different regions within CD40L, effectively allowing both receptors to simultaneously bind to CD40L trimers (55, 80). Specific inhibitors of the CD40L/VLA-5 interaction and their specific impact on cardiovascular pathologies have not been reported yet.
αIIbβ3
Expression of the integrin αIIbβ3 (CD49b/CD61, GPIIb3a) and the transcript Itga2b, which encodes for the αIIb (CD61) subunit of the integrin, are restricted to the megakaryocyte lineage (Figure 2). In addition, the β3 subunit (Itgab3) can form the integrin αVβ3 on platelets and myeloid cells. Platelets express αIIbβ3 on the cell surface, but additional integrin heterodimers are stored in platelet α- and dense granules. These are translocated to the cell surface during platelet activation and degranulation (88). Binding of soluble fibrinogen and von Willebrand factor by αIIbβ3 supports platelet adhesion and aggregation (89). Ligand binding to αIIbβ3 occurs through a site within the α- and β-subunit (90). Clinically, αIIbβ3 is targeted by several platelet aggregation and thrombus blocking drugs, including abciximab, tirofiban, and eptifibatide during cardiac catheterization (89). The main binding site within fibrinogen comprises an Arg–Gly–Asp (RGD) motif that is mimicked by tirofiban and eptifibatide and thus occupies the binding site on αIIbβ3 (91). Ligand binding is regulated by activation-induced conformational changes between the α- and β-subunit and is facilitated by an open, extended confirmation of the integrin, which allows for high-affinity binding (92, 93). Andre et al. established CD40L as a ligand for αIIbβ3 through a KGD-binding motif in CD40L (54). Binding affinity ranges between that of CD40 and Mac-1, VLA-5 (~30nM) (54, 55). sCD40L binding to αIIbβ3 induces signaling events in platelets through p38 and ERK1/2 (50, 54, 94).
Integrated Model of CD40L–Receptor Interactions
Traditionally, CD40 was believed to be the only receptor for CD40L. Newer evidence, however, has identified at least three alternative receptors of the integrin family: Mac-1, αIIbβ3, and α5β1 (Figure 1). These observations, along with the predominantly cell-specific expression of each of these receptors—αIIbβ3 on platelets, α5β1 on ECs, and Mac-1 on the myeloid cell lineage (Figure 1)—have sparked the notion that CD40L has distinct effects on different cellular subsets: mediating thrombotic and hemostatic properties through its interaction with αIIbβ3, cellular adhesion through Mac-1, immune functions (including B cell activation) through CD40, and activation of ECs through interaction with α5β1. Blocking specific binding sites on one of the participating molecules may enable to therapeutically abrogate distinct effector functions of CD40L. This concept is best illustrated by the M7 peptide that specifically blocks the interaction site for Mac-1 on CD40L without interfering with the CD40L–CD40 interaction (56). CD40L/Mac-1 primarily functions in leukocyte adhesion to the endothelium, but its inhibition by M7 does not affect CD40L’s effect on immunity or thrombosis. Such a concept may also enable inflammation-specific therapy, because (1) CD40L protein levels are upregulated rapidly, either transcriptionally or by mobilization of granule stores, mostly in inflammatory microenvironments and (2) integrins, such as Mac-1 and αIIbβ3, can exist in several conformations (95, 96): Mac-1 and αIIbβ3 primarily bind ligands in the activated conformation, which could be targeted by activation-specific blocking strategies (50, 97). This model is further complicated because CD40L binding to a receptor is not exclusive but instead occurs in a heteromeric manner, where a trimer of membrane-bound CD40L binds to two or more of its ligands simultaneously (Figure 1). For instance, platelets express CD40, αIIbβ3, and α5β1. Recent observations indicate that binding sites on CD40L for at least some of its receptors are distinct and suggest that each monomer (of a trimer) may bind to one of its receptors (21, 50, 55). However, the effect of different affinities between CD40L and its receptors—with the highest affinity recorded for CD40 and the lowest for Mac-1 (Table 1)—on heteromeric binding patterns and subsequent signaling is yet to be determined.
CD40L in Atherosclerosis
Clinical Association between Atherosclerosis and CD40L/CD40 Signaling
Atherosclerosis is a chronic inflammatory disease of medium to large arteries that is characterized by the buildup of fibro-fatty plaques in the intimal layer of the vessel, which limits blood flow into the distant vascular bed or results in total thrombotic occlusion during myocardial infarction or stroke (1). Atherosclerotic plaque development is critically driven by the accumulation of lipids, mostly low-density lipoprotein, and the subsequent accumulation of immune and inflammatory cells, such as lymphocytes and cells of the myeloid lineage (98). Activation of infiltrating leukocytes by tissue-resident cells or other leukocytes, secretion of pro-inflammatory mediators such as cytokines, chemokines, or matrix-degrading enzymes, and the recruitment of new blood cells through cell adhesion cascades are the functional hallmarks of atherogenesis (3). The expression of CD40L and its classical ligand CD40 on many of the participating cells, including various leukocyte subpopulations, ECs, and SMCs (26), has led to the hypothesis that the CD40L/CD40 dyad may contribute to atherosclerosis. CD40L and CD40 were identified in human atherosclerotic lesions at every developmental stage (27, 34), and CD40 expression in lesional macrophages and SMCs correlates with the stage of atherosclerosis (34). Relatively, CD40L expression is highest in ruptured atherosclerotic lesions (99), likely due to the deposition of CD40L-expressing platelets at the site of the rupture, or enhanced CD40L gene expression in rupture-prone plaques. The latter is supported by gene set enrichment analysis (GSEA) of the core CD40-signaling gene signature found in transcriptomes of both stable and ruptured atherosclerotic plaques from laser-microdissected macrophage-rich regions of human carotid endarterectomy specimens (100). In ruptured plaques, the expression of signaling molecules downstream of CD40, including NF-κB, TRAFs, and MAP kinases, significantly increased compared to stable human plaques (Figure 3), suggesting enhanced CD40 signaling in unstable plaques.
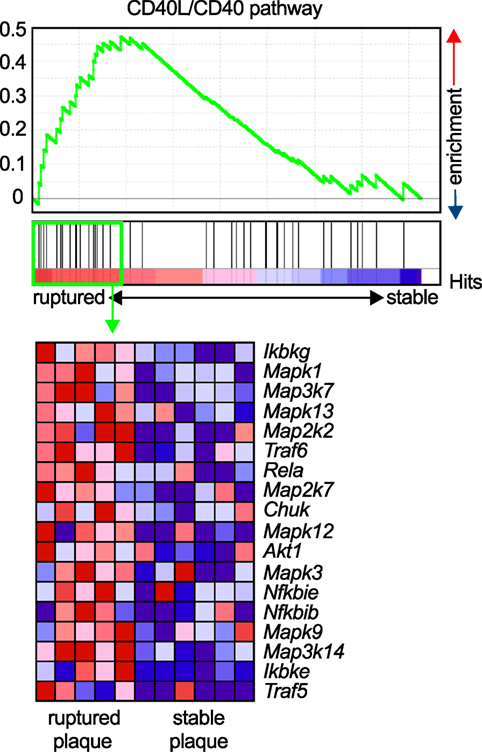
Figure 3. Enrichment of the CD40L/CD40 pathway in unstable human plaques. Gene set enrichment of the core CD40-signaling gene signature (Table 3) was tested on published transcriptomes of human stable and ruptured atherosclerotic plaques from laser-microdissected macrophage-rich regions of carotid endarterectomy specimen. Enrichment was tested between ruptured and stable plaques. The expression of the enrichment-driving genes (lower panel) was blotted on a heatmap and colored by a row-score (blue = lowest, red = highest gene expression per row).
Moreover, plasma levels of soluble CD40L have been proposed as a biomarker for cardiovascular risk; in particular, several studies have established a positive correlation of sCD40L with cardio-metabolic risk factors, such as dyslipidemia, diabetes, and obesity (112–114). The association with cardiovascular disease and outcome, however, is less clear: Lemos et al. found only a minimal correlation of sCD40L plasma levels with hyperlipidemia and no correlation with subclinical atherosclerosis as assessed by coronary calcification and aortic plaque size (114); however, sCD40L levels predicted future cardiovascular events in a case control study in women (115). Other nested case–control studies found a positive correlation with a worsened outcome after myocardial infarction (116), but these findings could not be confirmed in larger cohorts from the same clinical study population (117). It was speculated that the observed clinical associations are caused not by atherosclerosis itself but instead by enhanced platelet release of sCD40L in the setting of pro-thrombotic disease (114). These observations could render sCD40L as a biomarker of thrombotic events and risk, particularly in the setting of myocardial infarction (118–120) and hypercholesterolemia (24).
Data from genome-wide association studies have suggested that certain polymorphisms in the CD40 gene (rs1535045, rs3765459, rs4810485, rs1883832) may be associated with traits of cardiovascular disease, although studies have yielded inconsistent results. rs1535045 and rs3765459 correlated with the calcification score of coronary arteries from diabetic (121) and CVD patients (122). rs4810485, but not rs1535045, correlated with the number of coronary lesions in patients with Kawasaki syndrome (123). The SNP rs1535045 correlates with carotid intima-media thickness, a surrogate marker for atherosclerosis, in patients with rheumatoid arthritis (124). Other studies have also identified a correlation between CVD and rs1883832 (125) and rs4239702 (122). All together, these clinical findings have established a strong association between CD40L/CD40 signaling and cardiovascular disease, particularly in the setting of acute cardiovascular events and unstable atherosclerotic plaques.
CD40L in Experimental Atherosclerosis: A Controversy
Several studies have demonstrated a functional relevance of CD40L, its receptors, and signaling intermediates in experimental atherosclerosis (overview in Table 2). In 1998, Mach et al. first showed that inhibition of CD40L with a neutralizing anti-CD40L antibody reduced the size of atherosclerotic plaques in Lldr−/− mice (101). Atherosclerotic plaques of anti-CD40L-treated mice showed a marked reduction of lipid-positive areas, as well as a reduction of macrophage and T cell markers in immunohistochemistry—features that are linked to a stable, and therefore less likely to rupture, atherosclerotic plaque in humans. The observation that leukocyte recruitment was dampened after CD40L blockade was explained by the authors with a decreased expression of the adhesion molecule VCAM-1, which could hinder the recruitment of these cells to the atherosclerotic lesions. Lutgens et al. later confirmed that Apoe−/− mice treated with an anti-CD40L antibody possessed a stable plaque phenotype—rich in collagen and less populated by macrophages and T-cells. Mechanistically, these effects were explained by enhanced TGF-β signaling (103); however, lesion size was not changed by the anti-CD40L treatment in this study. In another study, CD40L-knockout mice on an Apoe−/− background were not protected from de novo lesion formation but showed a reduction of established atherosclerotic lesions and features of plaque stability (102). Another study in Ldlr−/− mice with established atherosclerotic lesions showed that the treatment with a blocking anti-CD40L antibody protected from further disease progression, although it did not induce plaque regression (104). While these studies have uniformly established that CD40L affects the cellular and extracellular composition of the atherosclerotic plaque, the impact of CD40L on the size of atherosclerotic lesions remains controversial; some studies show that blocking CD40L (by genetic knockouts or antibodies) decreases lesion size (101, 104, 105), while others show that lesion size remains unaffected (102, 103). This disparity in findings could likely be attributed to the different knockouts, genetic backgrounds (Apoe−/− vs. Ldlr−/−), and diet regimens (high-cholesterol diet vs. standard chow diet) tested. In addition, it remains unclear as to whether the blocking antibodies tested would be able to equally target all cell types, as cells within atherosclerotic plaques are less likely to be exposed to the same concentration of antibodies as circulating cells are. However, the common finding that CD40L blockade attenuates lesional accumulation of leukocytes, along with the predominant expression of CD40L on cells of the hematopoietic lineage, suggests that CD40L therapies would block leukocyte migration into the plaque. Yet, in bone marrow transplantation studies where CD40L-deficient bone marrow was transplanted into Ldlr−/− mice, neither lesion size nor the cellular composition in the plaque were changed (105), indicating that stromal cells are more likely to be the cellular source of bioactive CD40L.
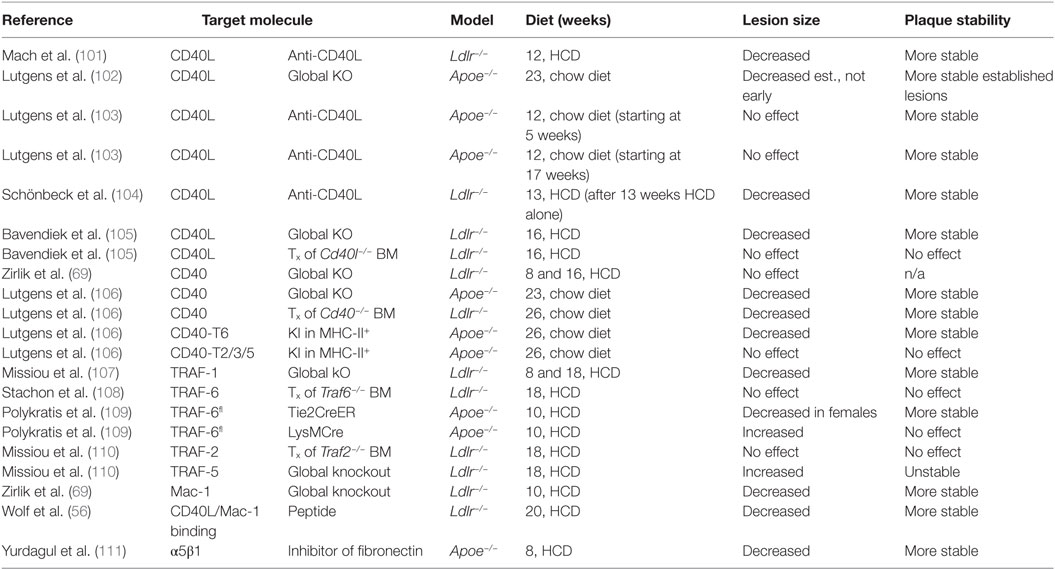
Table 2. CD40L-associated molecules in experimental atherosclerosis.
Is CD40 the Atherogenic Counter Receptor for CD40L?
The observation of enhanced thromboembolic complications after CD40L blockade in human lupus-associated glomerulonephritis (126, 127) has fueled the search for alternative strategies to neutralize CD40L(-signaling). Thus, CD40 receptor was proposed as a potential target. We have recently shown that CD40-knockout mice on an Ldlr−/− background fed with a high fat diet were not protected from de novo atherosclerosis (69); however, another study reported that CD40-deficient Apoe−/− mice on a standard chow diet for 26 weeks developed reduced atherosclerotic lesions with stable features and lowered leukocyte infiltration. In addition, a transplantation of CD40-deficient bone marrow into Ldlr−/− mice in the same study was atheroprotective (106). The finding that CD40 deficiency did not protect from atherosclerosis in at least one of these two studies has raised the possibility that CD40 may not be an exclusive receptor for CD40L. In line with this speculation, we recently demonstrated that CD40L interacts with the leukocyte integrin Mac-1 (69). Treatment with blocking anti-Mac-1 antibodies (69), depletion of Mac-1 expressing cells (128), and treatment with a specific inhibitor of this interaction—the peptide M7 (56)—protected from de novo atherosclerosis in Ldlr−/− mice on a high fat diet and reduced the number of lesional leukocytes, suggesting that this interaction may be a driver of leukocyte mobilization into the plaque. In the same study, we found that leukocytes were incapable of adhering to CD40L-deficient ECs (56). These results suggest that CD40L is a relevant adhesion factor on ECs. These findings confirm previous observations that CD40L is required for leukocyte accumulation in the plaque (101–105). Whether the CD40L–α5β1 interaction modulates atherosclerosis is currently unknown. Recent evidence has suggested that blocking α5β1 by a peptide mimetic protects from atherosclerotic lesion development, although the compound used was specific for the fibronectin binding site on α5β1, and not for the interaction to CD40L (111).
CD40-Dependent Signaling Events in Atherosclerosis
CD40 downstream signaling events are carried out through a group of intracellular signaling molecules known as TRAFs, which activate MAPKs and NF-κB. Signaling through the seven known TRAFs is not exclusive for CD40, because TRAFS are also engaged by TNF receptors, IL-1 receptor, or TLRs. It has been shown that ECs engage TRAF-1, -2, -3, -5, and -6 after stimulation with CD40L, which results in increased expression of inflammatory cytokines (129). In atherosclerotic plaques from Ldlr-deficient mice, TRAF-1/2/3/5/6 are overexpressed (129). Different TRAFs activate distinct downstream signaling events: for instance, CD40 signaling induces PIK3-dependent signaling events through TRAF-6, while TRAF-2/3/5 activate JNK, p38, and ERK signaling. In an elegant study by Lutgens et al., the specific binding site for TRAF-6 or TRAF-2/3/5 within CD40 was deleted in MHCII-expressing cells in knockin mice. Surprisingly, a deletion for the CD40-TRAF-2/3/5 interaction had no effect on atherosclerosis, while a deletion for CD40-TRAF-6 inhibited atherosclerotic plaque development and lowered the numbers of macrophages and T cells in the plaque (106). The role of TRAF-6, however, needs to be carefully evaluated for cell-type specific effects: Polykratis et al. found that a conditional knockout of TRAF-6 in ECs was atheroprotective by inhibiting NF-κB signaling. On the contrary, a specific TRAF-6 knockout in myeloid cells abrogated atheroprotective IL-10 signaling and exacerbated atherosclerosis (109). These findings suggest cell-specific anti- and pro-inflammatory functions of TRAF-6. In line with this, we have recently shown that a global knockout of TRAF-6 had no effect on atherosclerosis, which could be explained by competing pro- and anti-atherosclerotic effects (108). Although inhibition of the CD40-TRAF-2/3/5 axis did not impact atherosclerosis, a knockout of TRAF-5 that was not specific for the CD40-binding site exacerbated atherosclerosis in mice by promoting leukocyte accumulation and foam cell generation in the plaque (110). Likewise, disrupting TRAF-1, a negative inhibitor of TRAF-2, protected from atherosclerosis (107), while the genetic inhibition of TRAF-2 did not change atherosclerotic lesion burden (108). The effects of CD40 signaling are challenging to resolve, because of the complex interplay of distinct upstream receptors for TRAFs and various cell-type specific effects (130). In addition, atherogenesis may also be driven by diet-induced metabolic changes, and the functional role of TRAFs could diverge in metabolism. This is best illustrated by the findings that CD40 is expressed on adipocytes and functions as pro-inflammatory stimulator (131), while T cell CD40 in the setting of dysmetabolism and adipose tissue inflammation is limiting inflammation (49), likely by a TRAF-2/3/5-dependent mechanism (132). In contrast, knocking out the CD40-TRAF-6 binding site protected from dysmetabolism and adipose tissue inflammation (132). These mechanisms may directly and indirectly impact on atherosclerotic lesion burden. A common observation, however, is the modulation of leukocyte infiltration in CD40- or TRAF-deficient animals by regulating adhesion factor expression on ECs and leukocytes (106, 107, 110, 133).
CD40L in Thrombosis and Hemostasis
CD40 Ligation Induces Platelet Activation and Aggregation
Beyond their classical role in hemostasis and thrombosis, platelets contribute to acute and chronic inflammatory pathologies, such as atherosclerosis (134, 135). Key mechanisms to support inflammation include adhesion of platelets to the inflamed endothelium, where they can bind leukocytes and drive their recruitment by supporting rolling and firm adhesion (135–137). Platelets can also support inflammation by releasing inflammatory cytokines and chemokines that are stored in α-granules, including CCL5, CXCL4, and IL-1β. CD40L is an immediate activation marker of platelets that is stored in α-granules and mobilized to the cell surface (138), where it can be detected by flow cytometry within seconds after stimulation in vitro (40, 138). Beside T cells, CD40L gene expression can be detected in megakaryocytes, the progenitors of blood platelets (Figure 2). It is unclear whether CD40L is actively transcribed in platelets. Notably, in one study Cd40lg transcript were detected in platelets (138). However, the more important functional regulation seems to occur by mobilization of granule stored CD40L to the cell surface. Membrane-bound CD40L is subsequently shed by MMPs, likely ADAM10, -17 (12), MMP-2 (13), and -9 (14) within minutes to hours in an integrin αIIβ3-dependent mechanism. This process is also enhanced after ligation of CD40 on platelets (40). CD40L released to the blood circulation is usually referred to as soluble CD40L (sCD40L) (139). 95% of sCD40L in the blood circulation is estimated to originate from platelets (25, 120, 140), but T cells (12) or activated ECs (26) are a possible alternative source.
(s)CD40L has been shown to be platelet agonist itself, promoting activation and aggregation of platelets, thereby driving thrombus formation, growth, and stability (54, 94, 140–142). Clinically, levels of sCD40L correlate with thrombotic events, likely reflecting enhanced platelet activation and a release of CD40L in thrombotic disease (24). Neutralizing CD40L by monoclonal antibodies in clinical trials induced an enhanced rate of thromboembolic complications in humans and primates, suggesting that CD40L is required for stabilizing thrombi (126, 127) (also see Therapeutic Inhibition of CD40L in Clinical Disease). Cd40l−/− mice showed a delayed arterial occlusion time in a thrombosis model—an effect that could be reversed by a transfusion with sCD40L (54). CD40L deficiency resulted in smaller thrombi in vitro (143). In addition, mice with a genetic deficiency of CD40L (144) or after treatment with a blocking anti-CD40L antibody (56, 145) show prolonged bleeding time, although it is not clear whether these effects are platelet dependent or not.
Platelet Receptors for CD40L
The integrins αIIβ3, α5β1 (80), and its “classical” binding partner CD40 (40, 142) have been identified as platelet-expressed receptors for CD40L. It is not entirely clear whether platelet CD40L engages all three identified receptors on platelets simultaneously. Interestingly, formation and stabilization of thrombi by CD40L occurred primarily through binding of αIIβ3 and subsequent tyrosine phosphorylation of the integrin cellular domain (54, 94). These effects were absent when platelets were activated with recombinant CD40L lacking the KGD β3-recognition motif. CD40L stimulation induced αIIβ3 outside-in signaling, Akt-phosphorylation, and glycoprotein VI-induced platelet aggregation in a PI3K-dependent manner but did not require IKKα (146). Interestingly, CD40L promoted thrombus growth in the absence of CD40, suggesting that CD40L induces thrombosis and platelet aggregation via binding to αIIβ3 or α5β1, but not via CD40.
Some of CD40L’s effects may also be caused by binding to CD40, albeit experimental evidence is controversial in this regard. For instance, thrombus formation was not changed in Cd40−/− mice (54, 141) but delayed after treatment with a blocking anti-CD40L or an anti-CD40 antibody (56), as well as in Cd40−/− mice in another study (141). This discrepancy could be caused by tissue- and vessel type-specific expression of CD40 (147). Of note, CD40L induces platelet activation, the release of α- and dense granules (142), as well as aggregation of platelets (141, 148) in a CD40-dependent manner. CD40 ligation on platelets regulated expression of the platelet chemokine CXCL4 (PF4) (148). Recombinant, mutated CD40L lacking the CD40-binding site failed to induce platelet activation (141). Moreover, an inhibiting antibody to CD40 prolonged bleeding time (145). These reports, taken together, suggest that platelet CD40 is required to fully exhibit platelet activation, while αIIβ3 may be dispensable for platelet activation (142), although experimental evidence is contradicting in this regard (149). A potential explanation is that a blockade of αIIβ3 may abrogate CD40L shedding and the release of soluble CD40L (139). Subsequently, decreased levels of sCD40L would limit a possible auto-stimulation of platelets by self-expressed sCD40L (40, 142). While downstream signaling events are mediated primarily via TRAF-6 in atherosclerosis, Donners et al. found that mice with a genetic inhibition of CD40/TRAF-6 signaling did not alter platelet deposition and thrombus formation in an in vitro flow chamber assay (133), suggesting an involvement of TRAF-2/3/5 instead. Indeed, another study found CD40 ligation in platelets activates TRAF-2, the GTPase Rac1, and p38 MAPK (141).
Some newer evidence also suggests the integrin α5β1 in CD40L-mediated platelet activation: blocking antibodies against α5β1 prevented CD40L-induced platelet activation and lowered expression of P-selectin and PAC-1, the activation epitope of αIIβ3 in human platelets (149), raising the possibility that at least α5β1 and CD40 (and maybe partially αIIβ3) cooperate in CD40L-induced platelet activation. In contrast, the fourth receptor for CD40L, αMβ2 (CD11b) (56, 69), is not expressed by platelets. Some observations have raised the possibility that CD40L binding may not be exclusive to certain receptors but instead occurs in a heterotrimeric fashion, by which trimeric (surface-expressed) CD40L simultaneously binds to two or more receptors (50). The involvement of CD40L-associated pathways in thrombosis and hemostasis is summarized in Figure 4.
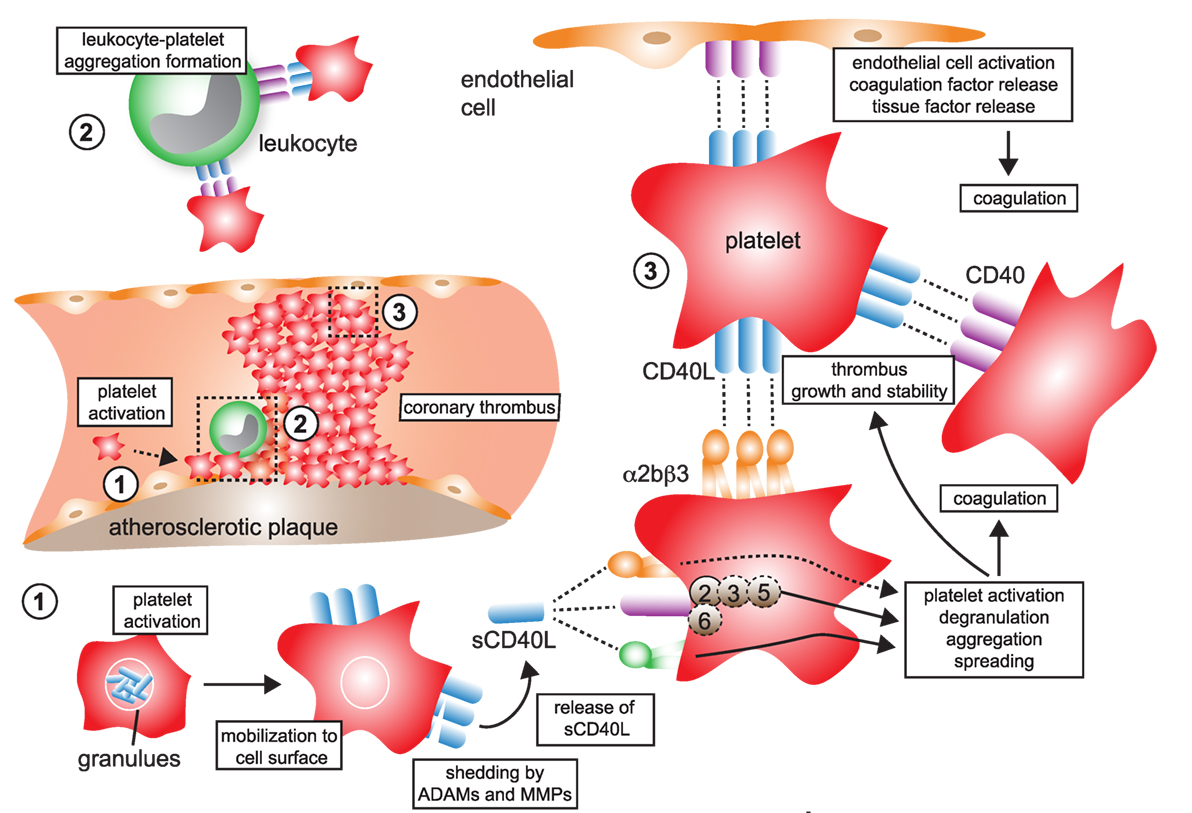
Figure 4. CD40L and its receptors in thrombosis and hemostasis. (1) Monomeric CD40L is stored in α- and dense granules of platelets. Upon platelet activation and degranulation, CD40L is mobilized to the cell surface and forms biologically active trimers. These are shed within minutes to hours by ADAMs and matrix metalloproteinases (MMPs) and released as monomers of soluble CD40L (sCD40L). These have been reported to form dimers and trimers in circulation that a biologically active. (2) Platelet-expressed CD40L binds to its receptors on leukocytes, α5β1 and CD40, but not αMβ2 (Mac-1), to form platelet–leukocyte aggregates. Notably, platelet-expressed CD40 can bind to CD40L-expressing leukocytes, such as activated T cells. (3) Ligation of platelet-expressed receptors, α5β1 and CD40, by CD40L initiates platelet activation cascades that eventually result in the surface expression of platelet receptors, such as P-selectin, degranulation, aggregation, and spreading. CD40 ligation on platelets activates a TRAF-2 dependent, but not TRAF-6 dependent, signaling pathway. It is a controversy whether ligation of α2bβ3 by CD40L participates in platelet activation. However, physical interactions between platelet-expressed α2bβ3 and CD40L, as well as between CD40 and CD40L, help to stabilize platelet aggregates and fuel thrombus growth. Binding of platelet CD40L and of sCD40L to its endothelial receptors CD40 and α5β1 promotes endothelial cell activation, pro-inflammatory gene expression, and secretion of coagulation factors, including tissue factor, which in turn can amplify thrombosis and coagulation.
Inflammatory and Immune Effects of Platelet CD40L/CD40 Signaling
It has been challenging to specifically attribute platelet-expressed CD40 and CD40L to atherosclerosis due to the limited availability of conditional knockout models in the past. However, two recent reports have addressed this question by transfusing platelets deficient for either of those molecules in competent recipients. After transferring Cd40−/−Apoe−/− platelets, Gerdes et al. found a twofold reduction of atherosclerosis in recipient Apoe−/− mice compared with a transfer of WT platelets. This effect was accompanied by a reduction of macrophages and neutrophils in atherosclerotic aortas in animals that received Cd40−/− platelets. The authors speculated whether this decrease of myeloid cell accumulation in the lesion was caused by an inability of CD40-deficient platelets to adhere to the endothelium and to recruit leukocytes and/or by reduced platelet–leukocyte aggregates. Indeed, it was observed in the same study that a lack of CD40 decreased platelet adhesion to the carotid endothelium and lowered formation of leukocyte–platelet aggregates (148). CD40-deficient mice also demonstrated reduced leukocyte–platelet complexes in another study (150). Formation of leukocyte–platelet aggregates—likely by a P-selectin-dependent mechanism—has previously been shown to enhance leukocyte recruitment into the atherosclerotic plaque (135–137). Also, an adoptive transfer of Cd40l−/− platelets protected from atherosclerosis when compared with WT platelets in another study (143). Taken together, both studies emphasize a role for the CD40L–CD40 interaction in the leukocytes-platelet cross talk.
In contrast, we found earlier that the CD40L/Mac-1 interaction does not mediate leukocyte–platelet aggregate formation (56), suggesting not all receptors for CD40L are fully capable to mediate firm binding between leukocytes and platelets. Beyond promoting leukocyte–platelet aggregation, Henn et al. demonstrated that platelet-expressed CD40L activates ECs and stimulated the secretion of pro-inflammatory cytokines, such as TNF-α and IL-1β, as well as to promote the expression of adhesion molecules that will ultimately support leukocyte recruitment (40, 138). Interestingly, some further studies have revealed that many of the pro-inflammatory consequences of platelet–endothelial interactions in inflammation depend on CD40 signaling (151–153).
Atherosclerosis is partially driven by adaptive immunity, which initiates and maintains a T cell response to autoantigens in the plaque and promotes the formation of autoantibodies (98). CD40L is a potent co-stimulatory molecule that is required in the T-cell-dependent activation of B cells. It is therefore fascinating to speculate whether platelet CD40L would participate in this response. Indeed, it was shown that platelet CD40L is sufficient to provide a co-stimulatory signal to drive T and B cell immunity in allograft rejection, viral, and bacterial infection (154–156). However, the specific impact of platelet CD40L in atherosclerosis-associated immunity is largely unknown.
Mechanistic Hallmarks of CD40L
Role of CD40L in Lymphocyte Recruitment to the Plaque
Recruitment of lymphocytes and myeloid cells initiates atherogenesis and maintains plaque inflammation (157). Several findings support that CD40L regulates leukocyte accumulation in the plaque (Figure 5), as evidenced by altered numbers of lesional macrophages, T cells, and other leukocytes after modulation of CD40L, its receptors, or signaling intermediates (Table 2). A smaller number of studies have interrogated how CD40L regulates accumulation, adhesion, and migration of leukocytes functionally. In summary, four different mechanisms can be gathered from these studies:
– CD40L is expressed on activated ECs (26, 151, 152) and functions as adhesion factor itself. We have previously shown an impairment of Cd40l−/− ECs in an in vitro flow chamber system independent of other adhesion factors expressed on ECs (56). Endothelial CD40L interacts with Mac-1 (56, 69) (Figure 5-1a), CD40 (158, 159), and α5β1 (86) (Figure 5-1b).
– Endothelial or circulating CD40L binds to its receptors (CD40, α5β1) on circulating, rolling, or arrested leukocytes, which activates pro-inflammatory signaling cascades in these (Figure 5-2). As a results, leukocytes upregulate adhesion molecules that are required for rolling, adhesion, or transmigration (160): ligation of monocyte CD40 by endothelial CD40L increases pro-inflammatory cytokine expression (152); circulating CD40L promotes the activation of neutrophils and integrin activation in a CD40-dependent fashion (161); and blocking CD40L abolishes leukocyte recruitment (162). CD40L furthermore activates myeloid cells by engagement of α5β1 (80). On the contrary, CD40L is a biased agonist for Mac-1 not activating outside-in signaling.
– Circulating CD40L, leukocyte-, platelet- (40, 138), or microparticle-bound CD40L (163) stimulates adhesion factor expression (44, 164), cytokine- and chemokine expression (152, 165, 166) in ECs in a CD40- (165, 167) and α5β1- (80) dependent mechanism (Figure 5-3). Activation of ECs promotes recruitment of leukocytes. Inflammatory cytokines (44) and modified lipids (168) were shown to increase expression of CD40 on ECs, explaining why CD40L-dependent activation of ECs is enhanced in the pro-inflammatory milieu of atherosclerosis.
– Circulating CD40L activates platelets or leukocytes and favors the formation of platelet–leukocyte aggregates (Figure 4-2).
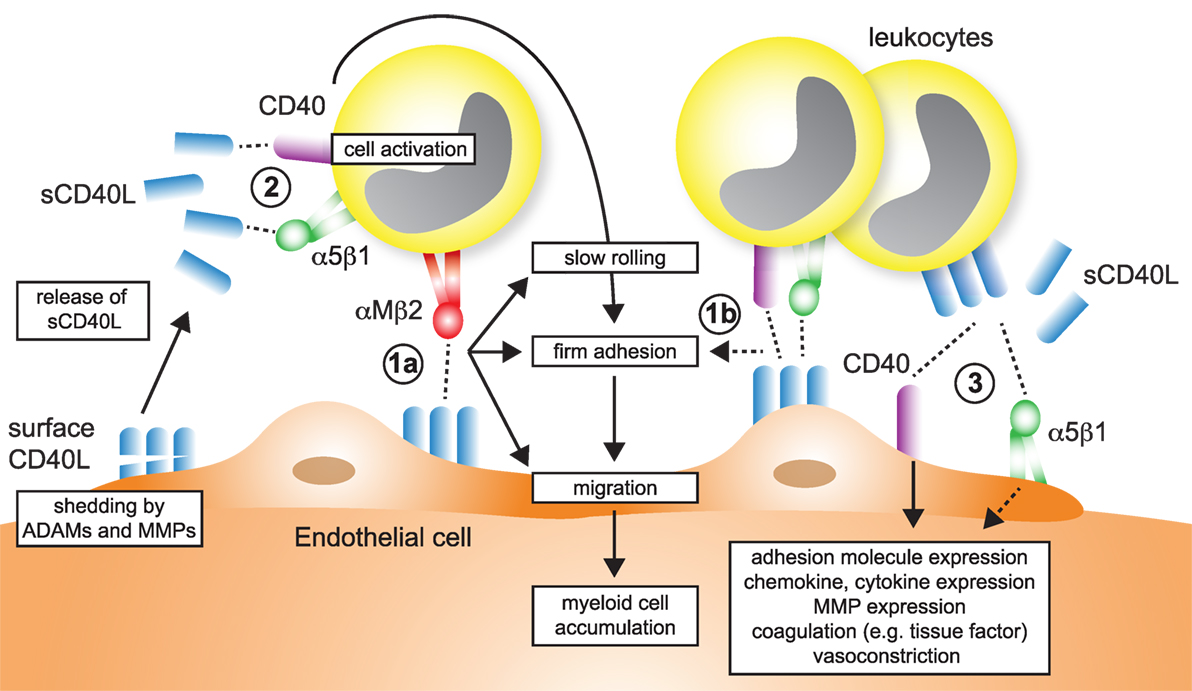
Figure 5. Participation of CD40L pathways in leukocyte recruitment. Several simultaneous mechanisms that contribute to the recruitment and transmigration of inflammatory leukocytes in inflamed tissue have been identified: (1a) endothelial cells (ECs) express membrane-bound CD40L in inflammation. The exact dynamics and stability of endothelial-expressed CD40L is unknown, but shedding by ADAMs and matrix metalloproteinases (MMPs) has been described. The interaction of αMβ2 (Mac-1) with endothelial CD40L mediates slow rolling and firm adhesion, which ultimately favors myeloid cell accumulation in tissues. (1b) It has been proposed that besides αMβ2, also CD40 and α5β1, expressed on leukocytes may participate in rolling and adhesion. (2) Ligation of α5β1 and CD40 on leukocytes, either by sCD40L or membrane-bound CD40L from leukocytes, platelets, or ECs activate pro-inflammatory signaling in leukocytes and increased expression of adhesion receptors that will in turn enhance cell recruitment. (3) α5β1 and CD40 expressed on ECs is ligated by sCD40L or cell-bound CD40L. As a result, ECs upregulate adhesion molecule expression and secretion of chemokines, besides other pro-inflammatory changes.
Mechanisms of CD40L-Mediated Plaque Instability
In human atherosclerosis, a stable plaque is characterized by a thick fibrous cap with intact collagen fibers and a small necrotic core, while an unstable or vulnerable plaque is characterized by thin cap, collagen breakdown, and a large necrotic core. Although mice do not develop spontaneous plaque rupture, an increased accumulation of macrophages and lipids, and lowered numbers of SMCs and collagen resemble a less stable plaque in mouse atherosclerosis (1). The complex interplay of stabilizing factors and destabilizing factors, such as expression of inflammatory cytokines and matrix-disintegrating enzymes is believed to orchestrate the composition and physical stability of the plaque (44, 169). In gene enrichment analysis, CD40-dependent signaling pathways are enriched in macrophage-rich regions of unstable human plaques (Figure 3). CD40L has been shown to modulate several facets of plaque instability: all major cell types that build the scaffold or populate the core of the atherosclerotic plaque, lymphocytes, macrophages, SMCs, and ECs, express CD40L and CD40 (26, 27, 34, 36, 159, 164, 170–172). MMPs are the main destabilizing collagenases in the plaque (169). MMPs were shown to be induced by CD40L and CD40 ligation in SMCs, ECs, and macrophages (44). CD40 in human plaques not only colocalizes with MMPs and SMCs but its ligation also upregulates the expression of MMP-1/2/3/8/9/13 and pro-inflammatory cytokines (173–176). CD40 expression in SMCs increases after stimulation with pro-inflammatory cytokines such as TNF-α, IL-1β, and INF-γ (26). CD40 ligation in SMCs enhances the expression of the adhesion molecules VCAM-1, ICAM-1, E-selectin, and the chemokine receptors CCR1, CCR5, and CXCR4, which in turn increase the recruitment of leukocytes into the plaque (44). Also, IL-1β-converting enzyme and tissue factor are secreted by SMCs in a CD40-dependent manner (177, 178). Interestingly, the interaction of CD40L to Mac-1 is not important for SMC functioning. We have recently tested a genetic knockout for CD40 in the setting of neointima formation after a surgical wire injury in the carotid artery, which induces SMC proliferation. CD40-deficient animal showed a smaller neointima, but not animals treated with the CD40L/Mac-1 blocker, M7, suggesting that CD40-dependent signaling pathways predominate in SMC activation and proliferation (150). Besides modulation of stabilizing collagen and fibrous cap formation, CD40L has also been implicated in necrotic core formation: ligation of CD40 induced proapoptotic pathways in lymphocytes and macrophages (7, 179) along with increased expressed of pro-inflammatory cytokines and collagenases (160). In addition, several studies have also suggested that the thrombotic potential of the plaque is regulated by CD40L, mainly by expression of pro-thrombotic tissue factor in tissue-resident cells, macrophages, SMCs, and ECs (160, 180–182).
Therapeutic Inhibition of CD40L in Clinical Disease
The participation of CD40L in pro-inflammatory and autoimmune disease, such as arthritis, nephritis, organ rejection, autoimmune diabetes, inflammatory bowel disease, and SLE (28–32) has led to the clinical evaluation of antibody-mediated neutralization of CD40L in human disease. For instance, BG9588, a humanized antihuman CD40L antibody, was tested in a phase II study in 28 patients with lupus glomerulonephritis. Although treatment with BG9588 and other antihuman CD40L antibodies showed a significant reduction in disease-specific parameters (127, 183), including levels of autoantibodies and renal function, its clinical evaluation was stopped because of an increased rate of thromboembolic events in one study, where two cases of myocardial infarction were recorded (127). In addition, CD40L blockade with the anti-CD40L clone (ATTC 5C8.33) caused multiple thrombotic events in monkeys (126). On the contrary, reports on different antihuman CD40L clones and preparations (e.g., IDEC-131) have demonstrated clinical safety (184), suggesting that antibody clones and/or preparations may contribute to its side effects. Thus, the mechanism by which CD40L raised thromboembolism is controversial: many experimental studies in the mouse have supported the idea that CD40L acts as a cross-linker of platelets in spontaneous formed thrombi (54) and that its inhibition destabilizes thrombi and favors thromboembolism. However, it is unknown whether this mechanism is the cause for thromboembolic events in humans. Finally, a study employing the anti-CD40L antibody CDP7657 (a PEGylated Fab fragment of the antibody) failed to induce thromboembolism in Cynomolgus monkeys, while dose-dependently inhibiting CD40L-dependent immune function (185). Likewise, a version of the antihuman CD40L antibody clone hu5c8 that does not bind to the Fc receptor was clinically effective in the same study without affecting platelet function in vitro (185). Both results suggest that clinical side effects of hu5c8 may be caused by binding to the Fc receptor on platelets through FcγRIIa and, therefore, in part independent of CD40L itself (186).
Concluding Remarks
A considerable number of studies over the last two decades have demonstrated that CD40L is a powerful mediator of inflammation and thrombosis. Its functional repertoire is fine tuned by the differential expression of CD40L itself and of its receptors in various cell types. Clinically, CD40L was proposed as biomarker of atherothrombosis, while its receptor CD40 associates with cardiovascular disease in GWAS. The receptor-unspecific therapeutic inhibition of CD40L, however, is unfavorable, because CD40L is required for many physiological processes in immunity and hemostasis. Its broad functional repertoire may, however, enable to develop cell- and function specific inhibition strategies. This was best illustrated by M7, a specific inhibitor of the CD40L/Mac-1 interaction that mediates leukocyte recruitment, while not interfering with CD40 binding. Yet, little is known about the cell-type specific role of CD40L and its receptor, but the development of conditional and tissue-specific knockout models will help to clarify these in future. The emerging concept of cell- and function-specific blockade could even be expanded to specific cell signaling pathways engaged by CD40L/CD40 signaling. An interesting study by van den Berg et al. lately demonstrated the potency of a selective blocker of the CD40-TRAF-6 interaction that protected from diet-induced obesity (187). Such therapeutic strategies, along with others, have revived the search for selective tools to block CD40L in cardiovascular pathologies and may ultimately point to novel therapeutic strategies against human cardiovascular disease.
Methods
Differential Gene Expression
Baseline gene expression of different human immune cell types quantified by RNAseq was extracted from the Protein Expression Atlas of the European Bioinformatics Institute (EMBL-EBI) (33). Expression values were retrieved as FPKMs, underwent hierarchical clustering, and normalization as row scores by Morpheus (Broad Institute).
Gene Set Enrichment Analysis
GenePattern 2.0 (188) was used to process RNA array data for GSEA (189), which was run with the default settings (100 iterations, weighted). Gene enrichment was tested for the core CD40-signaling gene signature (Table 3) on published transcriptomes of human stable and ruptured atherosclerotic plaques from laser microdissected macrophage-rich regions of carotid endarterectomy specimen [accession number Array Express E-GEOD-41571 (100)]. Enrichment was tested between ruptured and stable plaques. A P-value of 0.2 was considered significant. The expression of the enrichment-driving genes was blotted on a heatmap and colored by a row score.

Table 3. CD40-signaling core gene signature.
Author Contributions
DW, NM, and AZ analyzed data and designed and wrote the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Ross R. Atherosclerosis – an inflammatory disease. N Engl J Med (1999) 340(2):115–26. doi:10.1056/NEJM199901143400207
2. Lozano R, Naghavi M, Foreman K, Lim S, Shibuya K, Aboyans V, et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet (2012) 380(9859):2095–128. doi:10.1016/S0140-6736(12)61728-0
3. Libby P. Inflammation in atherosclerosis. Nature (2002) 420(6917):868–74. doi:10.1038/nature01323
4. Charo IF, Taub R. Anti-inflammatory therapeutics for the treatment of atherosclerosis. Nat Rev Drug Discov (2011) 10(5):365–76. doi:10.1038/nrd3444
5. Croft M. The role of TNF superfamily members in T-cell function and diseases. Nat Rev Immunol (2009) 9(4):271–85. doi:10.1038/nri2526
6. Kalliolias GD, Ivashkiv LB. TNF biology, pathogenic mechanisms and emerging therapeutic strategies. Nat Rev Rheumatol (2016) 12(1):49–62. doi:10.1038/nrrheum.2015.169
7. Schönbeck U, Libby P. The CD40/CD154 receptor/ligand dyad. Cell Mol Life Sci (2001) 58(1):4–43. doi:10.1007/PL00000776
8. Foy TM, Shepherd DM, Durie FH, Aruffo A, Ledbetter JA, Noelle RJ. In vivo CD40-gp39 interactions are essential for thymus-dependent humoral immunity. II. Prolonged suppression of the humoral immune response by an antibody to the ligand for CD40, gp39. J Exp Med (1993) 178(5):1567–75. doi:10.1084/jem.178.5.1567
9. Lesley R, Kelly LM, Xu Y, Cyster JG. Naive CD4 T cells constitutively express CD40L and augment autoreactive B cell survival. Proc Natl Acad Sci U S A (2006) 103(28):10717–22. doi:10.1073/pnas.0601539103
10. Murphy K, Travers P, Walport M, Janeway C. Janeway’s Immunobiology. New York: Garland Science (2008).
11. Notarangelo LD, Duse M, Ugazio AG. Immunodeficiency with hyper-IgM (HIM). Immunodefic Rev (1992) 3(2):101–21.
12. Yacoub D, Benslimane N, Al-Zoobi L, Hassan G, Nadiri A, Mourad W. CD154 is released from T-cells by a disintegrin and metalloproteinase domain-containing protein 10 (ADAM10) and ADAM17 in a CD40 protein-dependent manner. J Biol Chem (2013) 288(50):36083–93. doi:10.1074/jbc.M113.506220
13. Choi WS, Jeon OH, Kim DS. CD40 ligand shedding is regulated by interaction between matrix metalloproteinase-2 and platelet integrin alpha(IIb)beta(3). J Thromb Haemost (2010) 8(6):1364–71. doi:10.1111/j.1538-7836.2010.03837.x
14. Rahman M, Zhang S, Chew M, Syk I, Jeppsson B, Thorlacius H. Platelet shedding of CD40L is regulated by matrix metalloproteinase-9 in abdominal sepsis. J Thromb Haemost (2013) 11(7):1385–98. doi:10.1111/jth.12273
15. Hsu YM, Lucci J, Su L, Ehrenfels B, Garber E, Thomas D. Heteromultimeric complexes of CD40 ligand are present on the cell surface of human T lymphocytes. J Biol Chem (1997) 272(2):911–5. doi:10.1074/jbc.272.2.911
16. Fanslow WC, Srinivasan S, Paxton R, Gibson MG, Spriggs MK, Armitage RJ. Structural characteristics of CD40 ligand that determine biological function. Semin Immunol (1994) 6(5):267–78. doi:10.1006/smim.1994.1035
17. Morris AE, Remmele RL Jr, Klinke R, Macduff BM, Fanslow WC, Armitage RJ. Incorporation of an isoleucine zipper motif enhances the biological activity of soluble CD40L (CD154). J Biol Chem (1999) 274(1):418–23. doi:10.1074/jbc.274.1.418
18. Miller EA, Gopal R, Valdes V, Berger JS, Bhardwaj N, O’Brien MP. Soluble CD40 ligand contributes to dendritic cell-mediated T-cell dysfunction in HIV-1 infection. AIDS (2015) 29(11):1287–96. doi:10.1097/QAD.0000000000000698
19. Naito M, Hainz U, Burkhardt UE, Fu B, Ahove D, Stevenson KE, et al. CD40L-Tri, a novel formulation of recombinant human CD40L that effectively activates B cells. Cancer Immunol Immunother (2013) 62(2):347–57. doi:10.1007/s00262-012-1331-4
20. Haswell LE, Glennie MJ, Al-Shamkhani A. Analysis of the oligomeric requirement for signaling by CD40 using soluble multimeric forms of its ligand, CD154. Eur J Immunol (2001) 31(10):3094–100. doi:10.1002/1521-4141(2001010)31:10<3094::AID-IMMU3094>3.0.CO;2-F
21. Hassan GS, Merhi Y, Mourad WM. CD154 and its receptors in inflammatory vascular pathologies. Trends Immunol (2009) 30(4):165–72. doi:10.1016/j.it.2009.01.004
22. Mazzei GJ, Edgerton MD, Losberger C, Lecoanet-Henchoz S, Graber P, Durandy A, et al. Recombinant soluble trimeric CD40 ligand is biologically active. J Biol Chem (1995) 270(13):7025–8. doi:10.1074/jbc.270.13.7025
23. Pietravalle F, Lecoanet-Henchoz S, Blasey H, Aubry JP, Elson G, Edgerton MD, et al. Human native soluble CD40L is a biologically active trimer, processed inside microsomes. J Biol Chem (1996) 271(11):5965–7. doi:10.1074/jbc.271.11.5965
24. Cipollone F, Mezzetti A, Porreca E, Di Febbo C, Nutini M, Fazia M, et al. Association between enhanced soluble CD40L and prothrombotic state in hypercholesterolemia: effects of statin therapy. Circulation (2002) 106(4):399–402. doi:10.1161/01.CIR.0000025419.95769.F0
25. Heeschen C, Dimmeler S, Hamm CW, van den Brand MJ, Boersma E, Zeiher AM, et al. Soluble CD40 ligand in acute coronary syndromes. N Engl J Med (2003) 348(12):1104–11. doi:10.1056/NEJMoa022600
26. Mach F, Schonbeck U, Sukhova GK, Bourcier T, Bonnefoy JY, Pober JS, et al. Functional CD40 ligand is expressed on human vascular endothelial cells, smooth muscle cells, and macrophages: implications for CD40-CD40 ligand signaling in atherosclerosis. Proc Natl Acad Sci U S A (1997) 94(5):1931–6. doi:10.1073/pnas.94.5.1931
27. Hakkinen T, Karkola K, Yla-Herttuala S. Macrophages, smooth muscle cells, endothelial cells, and T-cells express CD40 and CD40L in fatty streaks and more advanced human atherosclerotic lesions. Colocalization with epitopes of oxidized low-density lipoprotein, scavenger receptor, and CD16 (Fc gammaRIII). Virchows Arch (2000) 437(4):396–405. doi:10.1007/s004280000239
28. Quezada SA, Jarvinen LZ, Lind EF, Noelle RJ. CD40/CD154 interactions at the interface of tolerance and immunity. Annu Rev Immunol (2004) 22:307–28. doi:10.1146/annurev.immunol.22.012703.104533
29. Danese S, Sans M, Fiocchi C. The CD40/CD40L costimulatory pathway in inflammatory bowel disease. Gut (2004) 53(7):1035–43. doi:10.1136/gut.2003.026278
30. Durie FH, Fava RA, Foy TM, Aruffo A, Ledbetter JA, Noelle RJ. Prevention of collagen-induced arthritis with an antibody to gp39, the ligand for CD40. Science (1993) 261(5126):1328–30. doi:10.1126/science.7689748
31. Mohan C, Shi Y, Laman JD, Datta SK. Interaction between CD40 and its ligand gp39 in the development of murine lupus nephritis. J Immunol (1995) 154(3):1470–80.
32. Gerritse K, Laman JD, Noelle RJ, Aruffo A, Ledbetter JA, Boersma WJ, et al. CD40-CD40 ligand interactions in experimental allergic encephalomyelitis and multiple sclerosis. Proc Natl Acad Sci U S A (1996) 93(6):2499–504. doi:10.1073/pnas.93.6.2499
33. Petryszak R, Keays M, Tang YA, Fonseca NA, Barrera E, Burdett T, et al. Expression Atlas update – an integrated database of gene and protein expression in humans, animals and plants. Nucleic Acids Res (2016) 44(D1):D746–52. doi:10.1093/nar/gkv1045
34. Bruemmer D, Riggers U, Holzmeister J, Grill M, Lippek F, Settmacher U, et al. Expression of CD40 in vascular smooth muscle cells and macrophages is associated with early development of human atherosclerotic lesions. Am J Cardiol (2001) 87(1):21–7. doi:10.1016/S0002-9149(00)01266-2
35. Banchereau J, Dubois B, Fayette J, Burdin N, Briere F, Miossec P, et al. Functional CD40 antigen on B cells, dendritic cells and fibroblasts. Adv Exp Med Biol (1995) 378:79–83. doi:10.1007/978-1-4615-1971-3_16
36. Alderson MR, Armitage RJ, Tough TW, Strockbine L, Fanslow WC, Spriggs MK. CD40 expression by human monocytes: regulation by cytokines and activation of monocytes by the ligand for CD40. J Exp Med (1993) 178(2):669–74. doi:10.1084/jem.178.2.669
37. Lang I, Fullsack S, Wyzgol A, Fick A, Trebing J, Arana JA, et al. Binding studies of TNF receptor superfamily (TNFRSF) receptors on intact cells. J Biol Chem (2016) 291(10):5022–37. doi:10.1074/jbc.M115.683946
38. Armitage RJ, Sato TA, Macduff BM, Clifford KN, Alpert AR, Smith CA, et al. Identification of a source of biologically active CD40 ligand. Eur J Immunol (1992) 22(8):2071–6. doi:10.1002/eji.1830220817
39. Wyzgol A, Muller N, Fick A, Munkel S, Grigoleit GU, Pfizenmaier K, et al. Trimer stabilization, oligomerization, and antibody-mediated cell surface immobilization improve the activity of soluble trimers of CD27L, CD40L, 41BBL, and glucocorticoid-induced TNF receptor ligand. J Immunol (2009) 183(3):1851–61. doi:10.4049/jimmunol.0802597
40. Henn V, Steinbach S, Buchner K, Presek P, Kroczek RA. The inflammatory action of CD40 ligand (CD154) expressed on activated human platelets is temporally limited by coexpressed CD40. Blood (2001) 98(4):1047–54. doi:10.1182/blood.V98.4.1047
41. Reyes-Moreno C, Girouard J, Lapointe R, Darveau A, Mourad W. CD40/CD40 homodimers are required for CD40-induced phosphatidylinositol 3-kinase-dependent expression of B7.2 by human B lymphocytes. J Biol Chem (2004) 279(9):7799–806. doi:10.1074/jbc.M313168200
42. Reyes-Moreno C, Sharif-Askari E, Girouard J, Leveille C, Jundi M, Akoum A, et al. Requirement of oxidation-dependent CD40 homodimers for CD154/CD40 bidirectional signaling. J Biol Chem (2007) 282(27):19473–80. doi:10.1074/jbc.M701076200
43. Xie P. TRAF molecules in cell signaling and in human diseases. J Mol Signal (2013) 8(1):7. doi:10.1186/1750-2187-8-7
44. Schonbeck U, Libby P. CD40 signaling and plaque instability. Circ Res (2001) 89(12):1092–103. doi:10.1161/hh2401.101272
45. Tsitsikov EN, Laouini D, Dunn IF, Sannikova TY, Davidson L, Alt FW, et al. TRAF1 is a negative regulator of TNF signaling. Enhanced TNF signaling in TRAF1-deficient mice. Immunity (2001) 15(4):647–57. doi:10.1016/S1074-7613(01)00207-2
46. Elgueta R, Benson MJ, de Vries VC, Wasiuk A, Guo Y, Noelle RJ. Molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunol Rev (2009) 229(1):152–72. doi:10.1111/j.1600-065X.2009.00782.x
47. Neron S, Suck G, Ma XZ, Sakac D, Roy A, Katsman Y, et al. B cell proliferation following CD40 stimulation results in the expression and activation of Src protein tyrosine kinase. Int Immunol (2006) 18(2):375–87. doi:10.1093/intimm/dxh377
48. Schönbeck U, Mach F, Sukhova GK, Atkinson E, Levesque E, Herman M, et al. Expression of stromelysin-3 in atherosclerotic lesions: regulation via CD40-CD40 ligand signaling in vitro and in vivo. J Exp Med (1999) 189(5):843. doi:10.1084/jem.189.5.843
49. Wolf D, Jehle F, Anto Michel N, Bukosza EN, Rivera J, Chen YC, et al. Co-inhibitory suppression of T cell activation by CD40 protects from obesity and adipose tissue inflammation in mice. Circulation (2014) 129(23):2414–25. doi:10.1161/CIRCULATIONAHA.113.008055
50. Alturaihi H, Hassan GS, Al-Zoobi L, Salti S, Darif Y, Yacoub D, et al. Interaction of CD154 with different receptors and its role in bidirectional signals. Eur J Immunol (2015) 45(2):592–602. doi:10.1002/eji.201444941
51. Bajorath J, Chalupny NJ, Marken JS, Siadak AW, Skonier J, Gordon M, et al. Identification of residues on CD40 and its ligand which are critical for the receptor-ligand interaction. Biochemistry (1995) 34(6):1833–44. doi:10.1021/bi00006a003
52. Bajorath J, Marken JS, Chalupny NJ, Spoon TL, Siadak AW, Gordon M, et al. Analysis of gp39/CD40 interactions using molecular models and site-directed mutagenesis. Biochemistry (1995) 34(31):9884–92. doi:10.1021/bi00031a009
53. An HJ, Kim YJ, Song DH, Park BS, Kim HM, Lee JD, et al. Crystallographic and mutational analysis of the CD40-CD154 complex and its implications for receptor activation. J Biol Chem (2011) 286(13):11226–35. doi:10.1074/jbc.M110.208215
54. Andre P, Prasad KS, Denis CV, He M, Papalia JM, Hynes RO, et al. CD40L stabilizes arterial thrombi by a beta3 integrin – dependent mechanism. Nat Med (2002) 8(3):247–52. doi:10.1038/nm0302-247
55. El Fakhry Y, Alturaihi H, Yacoub D, Liu L, Guo W, Leveille C, et al. Functional interaction of CD154 protein with alpha5beta1 integrin is totally independent from its binding to alphaIIbbeta3 integrin and CD40 molecules. J Biol Chem (2012) 287(22):18055–66. doi:10.1074/jbc.M111.333989
56. Wolf D, Hohmann JD, Wiedemann A, Bledzka K, Blankenbach H, Marchini T, et al. Binding of CD40L to Mac-1’s I-domain involves the EQLKKSKTL motif and mediates leukocyte recruitment and atherosclerosis – but does not affect immunity and thrombosis in mice. Circ Res (2011) 109(11):1269–79. doi:10.1161/CIRCRESAHA.111.247684
57. Brodeur SR, Angelini F, Bacharier LB, Blom AM, Mizoguchi E, Fujiwara H, et al. C4b-binding protein (C4BP) activates B cells through the CD40 receptor. Immunity (2003) 18(6):837–48. doi:10.1016/S1074-7613(03)00149-3
58. Williams KT, Young SP, Negus A, Young LS, Adams DH, Afford SC. C4b binding protein binds to CD154 preventing CD40 mediated cholangiocyte apoptosis: a novel link between complement and epithelial cell survival. PLoS One (2007) 2(1):e159. doi:10.1371/journal.pone.0000159
59. Dunne JL, Ballantyne CM, Beaudet AL, Ley K. Control of leukocyte rolling velocity in TNF-alpha-induced inflammation by LFA-1 and Mac-1. Blood (2002) 99(1):336–41. doi:10.1182/blood.V99.1.336
60. Dunne JL, Collins RG, Beaudet AL, Ballantyne CM, Ley K. Mac-1, but not LFA-1, uses intercellular adhesion molecule-1 to mediate slow leukocyte rolling in TNF-alpha-induced inflammation. J Immunol (2003) 171(11):6105–11. doi:10.4049/jimmunol.171.11.6105
61. Ross GD, Lambris JD. Identification of a C3bi-specific membrane complement receptor that is expressed on lymphocytes, monocytes, neutrophils, and erythrocytes. J Exp Med (1982) 155(1):96–110. doi:10.1084/jem.155.1.96
62. Altieri DC, Agbanyo FR, Plescia J, Ginsberg MH, Edgington TS, Plow EF. A unique recognition site mediates the interaction of fibrinogen with the leukocyte integrin Mac-1 (CD11b/CD18). J Biol Chem (1990) 265(21):12119–22.
63. Kanse SM, Matz RL, Preissner KT, Peter K. Promotion of leukocyte adhesion by a novel interaction between vitronectin and the beta2 integrin Mac-1 (alphaMbeta2, CD11b/CD18). Arterioscler Thromb Vasc Biol (2004) 24(12):2251–6. doi:10.1161/01.ATV.0000146529.68729.8b
64. Peter K, Schwarz M, Conradt C, Nordt T, Moser M, Kubler W, et al. Heparin inhibits ligand binding to the leukocyte integrin Mac-1 (CD11b/CD18). Circulation (1999) 100(14):1533–9. doi:10.1161/01.CIR.100.14.1533
65. Ehlers R, Ustinov V, Chen Z, Zhang X, Rao R, Luscinskas FW, et al. Targeting platelet-leukocyte interactions: identification of the integrin Mac-1 binding site for the platelet counter receptor glycoprotein Ibalpha. J Exp Med (2003) 198(7):1077–88. doi:10.1084/jem.20022181
66. Chavakis T, Bierhaus A, Al-Fakhri N, Schneider D, Witte S, Linn T, et al. The pattern recognition receptor (RAGE) is a counterreceptor for leukocyte integrins: a novel pathway for inflammatory cell recruitment. J Exp Med (2003) 198(10):1507–15. doi:10.1084/jem.20030800
67. Fink K, Busch HJ, Bourgeois N, Schwarz M, Wolf D, Zirlik A, et al. Muhlen: Mac-1 directly binds to the endothelial protein C-receptor: a link between the protein C anticoagulant pathway and inflammation? PLoS One (2013) 8(2):e53103. doi:10.1371/journal.pone.0053103
68. Simon DI. Opening the field of integrin biology to “biased agonism”. Circ Res (2011) 109(11):1199–201. doi:10.1161/CIRCRESAHA.111.257980
69. Zirlik A, Maier C, Gerdes N, MacFarlane L, Soosairajah J, Bavendiek U, et al. CD40 ligand mediates inflammation independently of CD40 by interaction with Mac-1. Circulation (2007) 115(12):1571–80. doi:10.1161/CIRCULATIONAHA.106.683201
70. Fan Z, Ley K. Leukocyte arrest: biomechanics and molecular mechanisms of beta2 integrin activation. Biorheology (2015) 52(5–6):353–77. doi:10.3233/BIR-15085
71. Wolf D, Bukosza N, Engel D, Poggi M, Jehle F, Anto Michel N, et al. Inflammation, but not recruitment, of adipose tissue macrophages requires signalling through Mac-1 (CD11b/CD18) in diet-induced obesity (DIO). Thromb Haemost (2016) 117(2):325–38. doi:10.1160/TH16-07-0553
72. Rosetti F, Mayadas TN. The many faces of Mac-1 in autoimmune disease. Immunol Rev (2016) 269(1):175–93. doi:10.1111/imr.12373
73. Ding A, Wright SD, Nathan C. Activation of mouse peritoneal macrophages by monoclonal antibodies to Mac-1 (complement receptor type 3). J Exp Med (1987) 165(3):733–49. doi:10.1084/jem.165.3.733
74. Rogers C, Edelman ER, Simon DI. A mAb to the beta2-leukocyte integrin Mac-1 (CD11b/CD18) reduces intimal thickening after angioplasty or stent implantation in rabbits. Proc Natl Acad Sci U S A (1998) 95(17):10134–9. doi:10.1073/pnas.95.17.10134
75. Simon DI, Dhen Z, Seifert P, Edelman ER, Ballantyne CM, Rogers C. Decreased neointimal formation in Mac-1(−/−) mice reveals a role for inflammation in vascular repair after angioplasty. J Clin Invest (2000) 105(3):293–300. doi:10.1172/JCI7811
76. Hirahashi J, Hishikawa K, Kaname S, Tsuboi N, Wang Y, Simon DI, et al. Mac-1 (CD11b/CD18) links inflammation and thrombosis after glomerular injury. Circulation (2009) 120(13):1255–65. doi:10.1161/CIRCULATIONAHA.109.873695
77. Kishimoto TK, Hollander N, Roberts TM, Anderson DC, Springer TA. Heterogeneous mutations in the beta subunit common to the LFA-1, Mac-1, and p150,95 glycoproteins cause leukocyte adhesion deficiency. Cell (1987) 50(2):193–202. doi:10.1016/0092-8674(87)90215-7
78. Obara M, Kang MS, Yamada KM. Site-directed mutagenesis of the cell-binding domain of human fibronectin: separable, synergistic sites mediate adhesive function. Cell (1988) 53(4):649–57. doi:10.1016/0092-8674(88)90580-6
79. Mould AP, Askari JA, Humphries MJ. Molecular basis of ligand recognition by integrin alpha 5beta 1. I. Specificity of ligand binding is determined by amino acid sequences in the second and third NH2-terminal repeats of the alpha subunit. J Biol Chem (2000) 275(27):20324–36. doi:10.1074/jbc.M000572200
80. Leveille C, Bouillon M, Guo W, Bolduc J, Sharif-Askari E, El-Fakhry Y, et al. CD40 ligand binds to alpha5beta1 integrin and triggers cell signaling. J Biol Chem (2007) 282(8):5143–51. doi:10.1074/jbc.M608342200
81. Hassan GS, Merhi Y, Mourad W. CD40 ligand: a neo-inflammatory molecule in vascular diseases. Immunobiology (2012) 217(5):521–32. doi:10.1016/j.imbio.2011.03.010
82. Orecchia A, Lacal PM, Schietroma C, Morea V, Zambruno G, Failla CM. Vascular endothelial growth factor receptor-1 is deposited in the extracellular matrix by endothelial cells and is a ligand for the alpha 5 beta 1 integrin. J Cell Sci (2003) 116(Pt 17):3479–89. doi:10.1242/jcs.00673
83. Felcht M, Luck R, Schering A, Seidel P, Srivastava K, Hu J, et al. Angiopoietin-2 differentially regulates angiogenesis through TIE2 and integrin signaling. J Clin Invest (2012) 122(6):1991–2005. doi:10.1172/jci58832
84. Sudhakar A, Sugimoto H, Yang C, Lively J, Zeisberg M, Kalluri R. Human tumstatin and human endostatin exhibit distinct antiangiogenic activities mediated by alpha v beta 3 and alpha 5 beta 1 integrins. Proc Natl Acad Sci U S A (2003) 100(8):4766–71. doi:10.1073/pnas.0730882100
85. Mittal A, Pulina M, Hou S-Y, Astrof S. Fibronectin and integrin alpha 5 play requisite roles in cardiac morphogenesis. Dev Biol (2013) 381(1):73–82. doi:10.1016/j.ydbio.2013.06.010
86. Loubaki L, Semlali A, Boisvert M, Jacques E, Plante S, Aoudjit F, et al. Crosstalk between T cells and bronchial fibroblasts obtained from asthmatic subjects involves CD40L/alpha 5 beta 1 interaction. Mol Immunol (2010) 47(11–12):2112–8. doi:10.1016/j.molimm.2010.03.011
87. Bachsais M, Naddaf N, Yacoub D, Salti S, Alaaeddine N, Aoudjit F, et al. The interaction of CD154 with the alpha5beta1 integrin inhibits Fas-induced T cell death. PLoS One (2016) 11(7):e0158987. doi:10.1371/journal.pone.0158987
88. Duperray A, Berthier R, Chagnon E, Ryckewaert JJ, Ginsberg M, Plow E, et al. Biosynthesis and processing of platelet GPIIb-IIIa in human megakaryocytes. J Cell Biol (1987) 104(6):1665–73. doi:10.1083/jcb.104.6.1665
89. Lefkovits J, Plow EF, Topol EJ. Platelet glycoprotein IIb/IIIa receptors in cardiovascular medicine. N Engl J Med (1995) 332(23):1553–9. doi:10.1056/NEJM199506083322306
90. Bennett JS. Structure and function of the platelet integrin alphaIIbbeta3. J Clin Invest (2005) 115(12):3363–9. doi:10.1172/JCI26989
91. Topol EJ, Byzova TV, Plow EF. Platelet GPIIb-IIIa blockers. Lancet (1999) 353(9148):227–31. doi:10.1016/S0140-6736(98)11086-3
92. Shattil SJ, Kim C, Ginsberg MH. The final steps of integrin activation: the end game. Nat Rev Mol Cell Biol (2010) 11(4):288–300. doi:10.1038/nrm2871
93. Lau TL, Kim C, Ginsberg MH, Ulmer TS. The structure of the integrin alphaIIbbeta3 transmembrane complex explains integrin transmembrane signalling. EMBO J (2009) 28(9):1351–61. doi:10.1038/emboj.2009.63
94. Prasad KS, Andre P, He M, Bao M, Manganello J, Phillips DR. Soluble CD40 ligand induces beta3 integrin tyrosine phosphorylation and triggers platelet activation by outside-in signaling. Proc Natl Acad Sci U S A (2003) 100(21):12367–71. doi:10.1073/pnas.2032886100
95. Takagi J, Springer TA. Integrin activation and structural rearrangement. Immunol Rev (2002) 186:141–63. doi:10.1034/j.1600-065X.2002.18613.x
96. Fan Z, McArdle S, Marki A, Mikulski Z, Gutierrez E, Engelhardt B, et al. Neutrophil recruitment limited by high-affinity bent beta2 integrin binding ligand in cis. Nat Commun (2016) 7:12658. doi:10.1038/ncomms12658
97. Schwarz M, Meade G, Stoll P, Ylanne J, Bassler N, Chen YC, et al. Conformation-specific blockade of the integrin GPIIb/IIIa: a novel antiplatelet strategy that selectively targets activated platelets. Circ Res (2006) 99(1):25–33. doi:10.1161/01.RES.0000232317.84122.0c
98. Wolf D, Zirlik A, Ley K. Beyond vascular inflammation – recent advances in understanding atherosclerosis. Cell Mol Life Sci (2015) 72(20):3853–69. doi:10.1007/s00018-015-1971-6
99. Lutgens E, Daemen MJ. CD40-CD40L interactions in atherosclerosis. Trends Cardiovasc Med (2002) 12(1):27–32. doi:10.1016/S1050-1738(01)00142-6
100. Lee K, Santibanez-Koref M, Polvikoski T, Birchall D, Mendelow AD, Keavney B. Increased expression of fatty acid binding protein 4 and leptin in resident macrophages characterises atherosclerotic plaque rupture. Atherosclerosis (2013) 226(1):74–81. doi:10.1016/j.atherosclerosis.2012.09.037
101. Mach F, Schonbeck U, Sukhova GK, Atkinson E, Libby P. Reduction of atherosclerosis in mice by inhibition of CD40 signalling. Nature (1998) 394(6689):200–3. doi:10.1038/28204
102. Lutgens E, Gorelik L, Daemen MJ, de Muinck ED, Grewal IS, Koteliansky VE, et al. Requirement for CD154 in the progression of atherosclerosis. Nat Med (1999) 5(11):1313–6. doi:10.1038/15271
103. Lutgens E, Cleutjens KB, Heeneman S, Koteliansky VE, Burkly LC, Daemen MJ. Both early and delayed anti-CD40L antibody treatment induces a stable plaque phenotype. Proc Natl Acad Sci U S A (2000) 97(13):7464–9. doi:10.1073/pnas.97.13.7464
104. Schonbeck U, Sukhova GK, Shimizu K, Mach F, Libby P. Inhibition of CD40 signaling limits evolution of established atherosclerosis in mice. Proc Natl Acad Sci U S A (2000) 97(13):7458–63. doi:10.1073/pnas.97.13.7458
105. Bavendiek U, Zirlik A, LaClair S, MacFarlane L, Libby P, Schonbeck U. Atherogenesis in mice does not require CD40 ligand from bone marrow-derived cells. Arterioscler Thromb Vasc Biol (2005) 25(6):1244–9. doi:10.1161/01.ATV.0000161420.55482.ef
106. Lutgens E, Lievens D, Beckers L, Wijnands E, Soehnlein O, Zernecke A, et al. Deficient CD40-TRAF6 signaling in leukocytes prevents atherosclerosis by skewing the immune response toward an antiinflammatory profile. J Exp Med (2010) 207(2):391–404. doi:10.1084/jem.20091293
107. Missiou A, Kostlin N, Varo N, Rudolf P, Aichele P, Ernst S, et al. Tumor necrosis factor receptor-associated factor 1 (TRAF1) deficiency attenuates atherosclerosis in mice by impairing monocyte recruitment to the vessel wall. Circulation (2010) 121(18):2033–44. doi:10.1161/CIRCULATIONAHA.109.895037
108. Stachon P, Missiou A, Walter C, Varo N, Colberg C, Wolf D, et al. Tumor necrosis factor receptor associated factor 6 is not required for atherogenesis in mice and does not associate with atherosclerosis in humans. PLoS One (2010) 5(7):e11589. doi:10.1371/journal.pone.0011589
109. Polykratis A, van Loo G, Xanthoulea S, Hellmich M, Pasparakis M. Conditional targeting of tumor necrosis factor receptor-associated factor 6 reveals opposing functions of toll-like receptor signaling in endothelial and myeloid cells in a mouse model of atherosclerosis. Circulation (2012) 126(14):1739–51. doi:10.1161/CIRCULATIONAHA.112.100339
110. Missiou A, Rudolf P, Stachon P, Wolf D, Varo N, Aichele P, et al. TRAF5 deficiency accelerates atherogenesis in mice by increasing inflammatory cell recruitment and foam cell formation. Circ Res (2010) 107(6):757–66. doi:10.1161/CIRCRESAHA.110.219295
111. Yurdagul A Jr, Green J, Albert P, McInnis MC, Mazar AP, Orr AW. α5β1 integrin signaling mediates oxidized low-density lipoprotein-induced inflammation and early atherosclerosis. Arterioscler Thromb Vasc Biol (2014) 34(7):1362–73. doi:10.1161/ATVBAHA.114.303863
112. Unek IT, Bayraktar F, Solmaz D, Ellidokuz H, Sisman AR, Yuksel F, et al. The levels of soluble CD40 ligand and C-reactive protein in normal weight, overweight and obese people. Clin Med Res (2010) 8(2):89–95. doi:10.3121/cmr.2010.889
113. Varo N, Vicent D, Libby P, Nuzzo R, Calle-Pascual AL, Bernal MR, et al. Elevated plasma levels of the atherogenic mediator soluble CD40 ligand in diabetic patients: a novel target of thiazolidinediones. Circulation (2003) 107(21):2664–9. doi:10.1161/01.CIR.0000074043.46437.44
114. de Lemos JA, Zirlik A, Schonbeck U, Varo N, Murphy SA, Khera A, et al. Associations between soluble CD40 ligand, atherosclerosis risk factors, and subclinical atherosclerosis: results from the Dallas Heart Study. Arterioscler Thromb Vasc Biol (2005) 25(10):2192–6. doi:10.1161/01.ATV.0000182904.08513.60
115. Schonbeck U, Varo N, Libby P, Buring J, Ridker PM. Soluble CD40L and cardiovascular risk in women. Circulation (2001) 104(19):2266–8. doi:10.1161/hc4401.099447
116. Varo N, de Lemos JA, Libby P, Morrow DA, Murphy SA, Nuzzo R, et al. Soluble CD40L: risk prediction after acute coronary syndromes. Circulation (2003) 108(9):1049–52. doi:10.1161/01.CIR.0000088521.04017.13
117. Olenchock BA, Wiviott SD, Murphy SA, Cannon CP, Rifai N, Braunwald E, et al. Lack of association between soluble CD40L and risk in a large cohort of patients with acute coronary syndrome in OPUS TIMI-16. J Thromb Thrombolysis (2008) 26(2):79–84. doi:10.1007/s11239-007-0156-z
118. Garlichs C, Eskafi S, Raaz D, Schmidt A, Ludwig J, Herrmann M, et al. Patients with acute coronary syndromes express enhanced CD40 ligand/CD154 on platelets. Heart (2001) 86(6):649–55. doi:10.1136/heart.86.6.649
119. Viallard JF, Solanilla A, Gauthier B, Contin C, Dechanet J, Grosset C, et al. Increased soluble and platelet-associated CD40 ligand in essential thrombocythemia and reactive thrombocytosis. Blood (2002) 99(7):2612–4. doi:10.1182/blood.V99.7.2612
120. Ahn ER, Lander G, Jy W, Bidot CJ, Jimenez JJ, Horstman LL, et al. Differences of soluble CD40L in sera and plasma: implications on CD40L assay as a marker of thrombotic risk. Thromb Res (2004) 114(2):143–8. doi:10.1016/j.thromres.2004.06.005
121. Burdon KP, Langefeld CD, Beck SR, Wagenknecht LE, Carr JJ, Rich SS, et al. Variants of the CD40 gene but not of the CD40L gene are associated with coronary artery calcification in the Diabetes Heart Study (DHS). Am Heart J (2006) 151(3):706–11. doi:10.1016/j.ahj.2005.05.004
122. Zhou L, Xie L, Zheng D, Li N, Zhu J, Wang S, et al. Genetic variants of CD40 gene are associated with coronary artery disease and blood lipid levels. Biomed Res Int (2016) 2016:1693619. doi:10.1155/2016/1693619
123. Kuo HC, Chao MC, Hsu YW, Lin YC, Huang YH, Yu HR, et al. CD40 gene polymorphisms associated with susceptibility and coronary artery lesions of Kawasaki disease in the Taiwanese population. ScientificWorldJournal (2012) 2012:520865. doi:10.1100/2012/520865
124. Garcia-Bermudez M, Gonzalez-Juanatey C, Lopez-Mejias R, Teruel M, Corrales A, Miranda-Filloy JA, et al. Study of association of CD40-CD154 gene polymorphisms with disease susceptibility and cardiovascular risk in Spanish rheumatoid arthritis patients. PLoS One (2012) 7(11):e49214. doi:10.1371/journal.pone.0049214
125. Yun Y, Ma C, Ma X. The SNP rs1883832 in CD40 gene and risk of atherosclerosis in Chinese population: a meta-analysis. PLoS One (2014) 9(5):e97289. doi:10.1371/journal.pone.0097289
126. Kawai T, Andrews D, Colvin RB, Sachs DH, Cosimi AB. Thromboembolic complications after treatment with monoclonal antibody against CD40 ligand. Nat Med (2000) 6(2):114. doi:10.1038/72162
127. Boumpas DT, Furie R, Manzi S, Illei GG, Wallace DJ, Balow JE, et al. A short course of BG9588 (anti-CD40 ligand antibody) improves serologic activity and decreases hematuria in patients with proliferative lupus glomerulonephritis. Arthritis Rheum (2003) 48(3):719–27. doi:10.1002/art.10856
128. Stoneman V, Braganza D, Figg N, Mercer J, Lang R, Goddard M, et al. Monocyte/macrophage suppression in CD11b diphtheria toxin receptor transgenic mice differentially affects atherogenesis and established plaques. Circ Res (2007) 100(6):884–93. doi:10.1161/01.RES.0000260802.75766.00
129. Zirlik A, Bavendiek U, Libby P, MacFarlane L, Gerdes N, Jagielska J, et al. TRAF-1, -2, -3, -5, and -6 are induced in atherosclerotic plaques and differentially mediate proinflammatory functions of CD40L in endothelial cells. Arterioscler Thromb Vasc Biol (2007) 27(5):1101–7. doi:10.1161/ATVBAHA.107.140566
130. Weinberg EO, Genco CA. Directing TRAF-ic: cell-specific TRAF6 signaling in chronic inflammation and atherosclerosis. Circulation (2012) 126(14):1678–80. doi:10.1161/CIRCULATIONAHA.112.134379
131. Missiou A, Wolf D, Platzer I, Ernst S, Walter C, Rudolf P, et al. CD40L induces inflammation and adipogenesis in adipose cells – a potential link between metabolic and cardiovascular disease. Thromb Haemost (2010) 103(4):788–96. doi:10.1160/TH09-07-0463
132. Chatzigeorgiou A, Seijkens T, Zarzycka B, Engel D, Poggi M, van den Berg S, et al. Blocking CD40-TRAF6 signaling is a therapeutic target in obesity-associated insulin resistance. Proc Natl Acad Sci U S A (2014) 111(7):2686–91. doi:10.1073/pnas.1400419111
133. Donners MM, Beckers L, Lievens D, Munnix I, Heemskerk J, Janssen BJ, et al. The CD40-TRAF6 axis is the key regulator of the CD40/CD40L system in neointima formation and arterial remodeling. Blood (2008) 111(9):4596–604. doi:10.1182/blood-2007-05-088906
134. Gawaz M, Favaloro EJ. Platelets, inflammation and cardiovascular diseases. New concepts and therapeutic implications. Semin Thromb Hemost (2010) 36(2):129–30. doi:10.1055/s-0030-1251495
135. Huo Y, Schober A, Forlow SB, Smith DF, Hyman MC, Jung S, et al. Circulating activated platelets exacerbate atherosclerosis in mice deficient in apolipoprotein E. Nat Med (2003) 9(1):61–7. doi:10.1038/nm810
136. Gawaz M, Langer H, May AE. Platelets in inflammation and atherogenesis. J Clin Invest (2005) 115(12):3378–84. doi:10.1172/JCI27196
137. von Hundelshausen P, Weber C. Platelets as immune cells: bridging inflammation and cardiovascular disease. Circ Res (2007) 100(1):27–40. doi:10.1161/01.RES.0000252802.25497.b7
138. Henn V, Slupsky JR, Grafe M, Anagnostopoulos I, Forster R, Muller-Berghaus G, et al. CD40 ligand on activated platelets triggers an inflammatory reaction of endothelial cells. Nature (1998) 391(6667):591–4. doi:10.1038/35393
139. Furman MI, Krueger LA, Linden MD, Barnard MR, Frelinger AL III, Michelson AD. Release of soluble CD40L from platelets is regulated by glycoprotein IIb/IIIa and actin polymerization. J Am Coll Cardiol (2004) 43(12):2319–25. doi:10.1016/j.jacc.2003.12.055
140. Andre P, Nannizzi-Alaimo L, Prasad SK, Phillips DR. Platelet-derived CD40L: the switch-hitting player of cardiovascular disease. Circulation (2002) 106(8):896–9. doi:10.1161/01.CIR.0000028962.04520.01
141. Yacoub D, Hachem A, Theoret JF, Gillis MA, Mourad W, Merhi Y. Enhanced levels of soluble CD40 ligand exacerbate platelet aggregation and thrombus formation through a CD40-dependent tumor necrosis factor receptor-associated factor-2/Rac1/p38 mitogen-activated protein kinase signaling pathway. Arterioscler Thromb Vasc Biol (2010) 30(12):2424–33. doi:10.1161/ATVBAHA.110.216143
142. Inwald DP, McDowall A, Peters MJ, Callard RE, Klein NJ. CD40 is constitutively expressed on platelets and provides a novel mechanism for platelet activation. Circ Res (2003) 92(9):1041–8. doi:10.1161/01.RES.0000070111.98158.6C
143. Lievens D, Zernecke A, Seijkens T, Soehnlein O, Beckers L, Munnix IC, et al. Platelet CD40L mediates thrombotic and inflammatory processes in atherosclerosis. Blood (2010) 116(20):4317–27. doi:10.1182/blood-2010-01-261206
144. Crow AR, Leytin V, Starkey AF, Rand ML, Lazarus AH. CD154 (CD40 ligand)-deficient mice exhibit prolonged bleeding time and decreased shear-induced platelet aggregates. J Thromb Haemost (2003) 1(4):850–2. doi:10.1046/j.1538-7836.2003.t01-1-00115.x
145. Langer F, Ingersoll SB, Amirkhosravi A, Meyer T, Siddiqui FA, Ahmad S, et al. The role of CD40 in CD40L- and antibody-mediated platelet activation. Thromb Haemost (2005) 93(6):1137–46. doi:10.1160/TH04-12-0774
146. Kuijpers MJ, Mattheij NJ, Cipolla L, van Geffen JP, Lawrence T, Donners MM, et al. Platelet CD40L modulates thrombus growth via phosphatidylinositol 3-kinase beta, and not via CD40 and IkappaB kinase alpha. Arterioscler Thromb Vasc Biol (2015) 35(6):1374–81. doi:10.1161/ATVBAHA.114.305127
147. Gavins FN, Li G, Russell J, Perretti M, Granger DN. Microvascular thrombosis and CD40/CD40L signaling. J Thromb Haemost (2011) 9(3):574–81. doi:10.1111/j.1538-7836.2010.04176.x
148. Gerdes N, Seijkens T, Lievens D, Kuijpers MJ, Winkels H, Projahn D, et al. Platelet CD40 exacerbates atherosclerosis by transcellular activation of endothelial cells and leukocytes. Arterioscler Thromb Vasc Biol (2016) 36(3):482–90. doi:10.1161/ATVBAHA.115.307074
149. Simic D, Bogdan N, Teng F, Otieno M. Blocking alpha5beta1 integrin attenuates sCD40L-mediated platelet activation. Clin Appl Thromb Hemost (2015) 1–8. doi:10.1177/1076029615624549
150. Willecke F, Tiwari S, Rupprecht B, Wolf D, Hergeth S, Hoppe N, et al. Interruption of classic CD40L-CD40 signalling but not of the novel CD40L-Mac-1 interaction limits arterial neointima formation in mice. Thromb Haemost (2014) 112(2):379–89. doi:10.1160/TH13-08-0653
151. Danese S, Scaldaferri F, Papa A, Pola R, Gasbarrini A, Sgambato A, et al. CD40L-positive platelets induce CD40L expression de novo in endothelial cells: adding a loop to microvascular inflammation. Arterioscler Thromb Vasc Biol (2004) 24(9):e162. doi:10.1161/01.ATV.0000138073.91195.70
152. Wagner AH, Guldenzoph B, Lienenluke B, Hecker M. CD154/CD40-mediated expression of CD154 in endothelial cells: consequences for endothelial cell-monocyte interaction. Arterioscler Thromb Vasc Biol (2004) 24(4):715–20. doi:10.1161/01.ATV.0000122853.99978.b1
153. Danese S, de la Motte C, Sturm A, Vogel JD, West GA, Strong SA, et al. Platelets trigger a CD40-dependent inflammatory response in the microvasculature of inflammatory bowel disease patients. Gastroenterology (2003) 124(5):1249–64. doi:10.1016/S0016-5085(03)00289-0
154. Xu H, Zhang X, Mannon RB, Kirk AD. Platelet-derived or soluble CD154 induces vascularized allograft rejection independent of cell-bound CD154. J Clin Invest (2006) 116(3):769–74. doi:10.1172/JCI27155
155. Elzey BD, Grant JF, Sinn HW, Nieswandt B, Waldschmidt TJ, Ratliff TL. Cooperation between platelet-derived CD154 and CD4+ T cells for enhanced germinal center formation. J Leukoc Biol (2005) 78(1):80–4. doi:10.1189/jlb.1104669
156. Elzey BD, Schmidt NW, Crist SA, Kresowik TP, Harty JT, Nieswandt B, et al. Platelet-derived CD154 enables T-cell priming and protection against Listeria monocytogenes challenge. Blood (2008) 111(7):3684–91. doi:10.1182/blood-2007-05-091728
157. Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol (2007) 7(9):678–89. doi:10.1038/nri2156
158. Pluvinet R, Petriz J, Torras J, Herrero-Fresneda I, Cruzado JM, Grinyo JM, et al. RNAi-mediated silencing of CD40 prevents leukocyte adhesion on CD154-activated endothelial cells. Blood (2004) 104(12):3642–6. doi:10.1182/blood-2004-03-0817
159. Yellin MJ, Brett J, Baum D, Matsushima A, Szabolcs M, Stern D, et al. Functional interactions of T cells with endothelial cells: the role of CD40L-CD40-mediated signals. J Exp Med (1995) 182(6):1857–64. doi:10.1084/jem.182.6.1857
160. Mach F, Schonbeck U, Bonnefoy JY, Pober JS, Libby P. Activation of monocyte/macrophage functions related to acute atheroma complication by ligation of CD40: induction of collagenase, stromelysin, and tissue factor. Circulation (1997) 96(2):396–9. doi:10.1161/01.CIR.96.2.396
161. Jin R, Yu S, Song Z, Zhu X, Wang C, Yan J, et al. Soluble CD40 ligand stimulates CD40-dependent activation of the beta2 integrin Mac-1 and protein kinase C zeta (PKCzeta) in neutrophils: implications for neutrophil-platelet interactions and neutrophil oxidative burst. PLoS One (2013) 8(6):e64631. doi:10.1371/journal.pone.0064631
162. Li G, Sanders JM, Bevard MH, Sun Z, Chumley JW, Galkina EV, et al. CD40 ligand promotes Mac-1 expression, leukocyte recruitment, and neointima formation after vascular injury. Am J Pathol (2008) 172(4):1141–52. doi:10.2353/ajpath.2008.070633
163. Leroyer AS, Rautou PE, Silvestre JS, Castier Y, Leseche G, Devue C, et al. CD40 ligand+ microparticles from human atherosclerotic plaques stimulate endothelial proliferation and angiogenesis a potential mechanism for intraplaque neovascularization. J Am Coll Cardiol (2008) 52(16):1302–11. doi:10.1016/j.jacc.2008.07.032
164. Hollenbaugh D, Mischel-Petty N, Edwards CP, Simon JC, Denfeld RW, Kiener PA, et al. Expression of functional CD40 by vascular endothelial cells. J Exp Med (1995) 182(1):33–40. doi:10.1084/jem.182.1.33
165. Greene JA, Portillo JA, Lopez Corcino Y, Subauste CS. CD40-TRAF signaling upregulates CX3CL1 and TNF-alpha in human aortic endothelial cells but not in retinal endothelial cells. PLoS One (2015) 10(12):e0144133. doi:10.1371/journal.pone.0144133
166. Omari KM, Chui R, Dorovini-Zis K. Induction of beta-chemokine secretion by human brain microvessel endothelial cells via CD40/CD40L interactions. J Neuroimmunol (2004) 146(1–2):203–8. doi:10.1016/j.jneuroim.2003.10.032
167. Pluvinet R, Olivar R, Krupinski J, Herrero-Fresneda I, Luque A, Torras J, et al. CD40: an upstream master switch for endothelial cell activation uncovered by RNAi-coupled transcriptional profiling. Blood (2008) 112(9):3624–37. doi:10.1182/blood-2008-03-143305
168. Li D, Liu L, Chen H, Sawamura T, Mehta JL. LOX-1, an oxidized LDL endothelial receptor, induces CD40/CD40L signaling in human coronary artery endothelial cells. Arterioscler Thromb Vasc Biol (2003) 23(5):816–21. doi:10.1161/01.ATV.0000066685.13434.FA
169. Libby P, Geng YJ, Sukhova GK, Simon DI, Lee RT. Molecular determinants of atherosclerotic plaque vulnerability. Ann N Y Acad Sci (1997) 811:134–42; discussion 142–5. doi:10.1111/j.1749-6632.1997.tb51996.x
170. Cocks BG, de Waal Malefyt R, Galizzi JP, de Vries JE, Aversa G. IL-13 induces proliferation and differentiation of human B cells activated by the CD40 ligand. Int Immunol (1993) 5(6):657–63. doi:10.1093/intimm/5.6.657
171. Karmann K, Hughes CC, Schechner J, Fanslow WC, Pober JS. CD40 on human endothelial cells: inducibility by cytokines and functional regulation of adhesion molecule expression. Proc Natl Acad Sci U S A (1995) 92(10):4342–6. doi:10.1073/pnas.92.10.4342
172. Krzesz R, Wagner AH, Cattaruzza M, Hecker M. Cytokine-inducible CD40 gene expression in vascular smooth muscle cells is mediated by nuclear factor kappaB and signal transducer and activation of transcription-1. FEBS Lett (1999) 453(1–2):191–6. doi:10.1016/S0014-5793(99)00683-3
173. Mukundan L, Milhorn DM, Matta B, Suttles J. CD40-mediated activation of vascular smooth muscle cell chemokine production through a Src-initiated, MAPK-dependent pathway. Cell Signal (2004) 16(3):375–84. doi:10.1016/j.cellsig.2003.08.008
174. Schonbeck U, Mach F, Sukhova GK, Murphy C, Bonnefoy JY, Fabunmi RP, et al. Regulation of matrix metalloproteinase expression in human vascular smooth muscle cells by T lymphocytes: a role for CD40 signaling in plaque rupture? Circ Res (1997) 81(3):448–54. doi:10.1161/01.RES.81.3.448
175. Sukhova GK, Schonbeck U, Rabkin E, Schoen FJ, Poole AR, Billinghurst RC, et al. Evidence for increased collagenolysis by interstitial collagenases-1 and -3 in vulnerable human atheromatous plaques. Circulation (1999) 99(19):2503–9. doi:10.1161/01.CIR.99.19.2503
176. Herman MP, Sukhova GK, Libby P, Gerdes N, Tang N, Horton DB, et al. Expression of neutrophil collagenase (matrix metalloproteinase-8) in human atheroma: a novel collagenolytic pathway suggested by transcriptional profiling. Circulation (2001) 104(16):1899–904. doi:10.1161/hc4101.097419
177. Schonbeck U, Mach F, Bonnefoy JY, Loppnow H, Flad HD, Libby P. Ligation of CD40 activates interleukin 1beta-converting enzyme (caspase-1) activity in vascular smooth muscle and endothelial cells and promotes elaboration of active interleukin 1beta. J Biol Chem (1997) 272(31):19569–74. doi:10.1074/jbc.272.31.19569
178. Schonbeck U, Mach F, Sukhova GK, Herman M, Graber P, Kehry MR, et al. CD40 ligation induces tissue factor expression in human vascular smooth muscle cells. Am J Pathol (2000) 156(1):7–14. doi:10.1016/S0002-9440(10)64699-8
179. Armitage RJ, Fanslow WC, Strockbine L, Sato TA, Clifford KN, Macduff BM, et al. Molecular and biological characterization of a murine ligand for CD40. Nature (1992) 357(6373):80–2. doi:10.1038/357080a0
180. Zhou L, Stordeur P, de Lavareille A, Thielemans K, Capel P, Goldman M, et al. CD40 engagement on endothelial cells promotes tissue factor-dependent procoagulant activity. Thromb Haemost (1998) 79(5):1025–8.
181. Miller DL, Yaron R, Yellin MJ. CD40L-CD40 interactions regulate endothelial cell surface tissue factor and thrombomodulin expression. J Leukoc Biol (1998) 63(3):373–9.
182. Slupsky JR, Kalbas M, Willuweit A, Henn V, Kroczek RA, Muller-Berghaus G. Activated platelets induce tissue factor expression on human umbilical vein endothelial cells by ligation of CD40. Thromb Haemost (1998) 80(6):1008–14.
183. Huang W, Sinha J, Newman J, Reddy B, Budhai L, Furie R, et al. The effect of anti-CD40 ligand antibody on B cells in human systemic lupus erythematosus. Arthritis Rheum (2002) 46(6):1554–62. doi:10.1002/art.10273
184. Davis JC Jr, Totoritis MC, Rosenberg J, Sklenar TA, Wofsy D. Phase I clinical trial of a monoclonal antibody against CD40-ligand (IDEC-131) in patients with systemic lupus erythematosus. J Rheumatol (2001) 28(1):95–101.
185. Shock A, Burkly L, Wakefield I, Peters C, Garber E, Ferrant J, et al. CDP7657, an anti-CD40L antibody lacking an Fc domain, inhibits CD40L-dependent immune responses without thrombotic complications: an in vivo study. Arthritis Res Ther (2015) 17:234. doi:10.1186/s13075-015-0757-4
186. Robles-Carrillo L, Meyer T, Hatfield M, Desai H, Davila M, Langer F, et al. Anti-CD40L immune complexes potently activate platelets in vitro and cause thrombosis in FCGR2A transgenic mice. J Immunol (2010) 185(3):1577–83. doi:10.4049/jimmunol.0903888
187. van den Berg SM, Seijkens TT, Kusters PJ, Zarzycka B, Beckers L, den Toom M, et al. Blocking CD40-TRAF6 interactions by small-molecule inhibitor 6860766 ameliorates the complications of diet-induced obesity in mice. Int J Obes (Lond) (2015) 39(5):782–90. doi:10.1038/ijo.2014.198
188. Reich M, Liefeld T, Gould J, Lerner J, Tamayo P, Mesirov JP. GenePattern 2.0. Nat Genet (2006) 38(5):500–1. doi:10.1038/ng0506-500
Keywords: CD40L, CD40 signaling, Mac-1, cardiovascular diseases, atherosclerosis, thrombosis, inflammation
Citation: Michel NA, Zirlik A and Wolf D (2017) CD40L and Its Receptors in Atherothrombosis—An Update. Front. Cardiovasc. Med. 4:40. doi: 10.3389/fcvm.2017.00040
Received: 22 March 2017; Accepted: 29 May 2017;
Published: 20 June 2017
Edited by:
Philipp Von Hundelshausen, Ludwig-Maximilians-Universität München, GermanyReviewed by:
Harald Wajant, University Hospital Würzburg, GermanyHector A. Cabrera-Fuentes, Justus Liebig Universität Gießen, Germany
Copyright: © 2017 Michel, Zirlik and Wolf. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dennis Wolf, ZGVubmlzLndvbGZAdW5pdmVyc2l0YWV0cy1oZXJ6emVudHJ1bS5kZQ==