 Valeria Novelli
Valeria Novelli Patrick Gambelli
Patrick Gambelli Mirella Memmi
Mirella Memmi Carlo Napolitano
Carlo Napolitano
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
OPINION article
Front. Cardiovasc. Med., 12 September 2016
Sec. Cardiovascular Genetics and Systems Medicine
Volume 3 - 2016 | https://doi.org/10.3389/fcvm.2016.00029
This article is part of the Research TopicCurrent Challenges in Cardiovascular Molecular DiagnosticsView all 16 articles
Inherited arrhythmogenic diseases (IADs – also called cardiac channelopathies) are defined as a group of genetic diseases characterized by electrically unstable substrate in a structurally normal heart (1). Genetic testing in cardiac channelopathies has completed its transition from a research-based activity to that of a clinical genetic service. In parallel, the advancements of the sequencing technologies are providing ways to sequence several genes at a relatively low cost. This is progressively changing the approach to the genetic diagnosis of IADs. Indeed, while Sanger-based genetic testing was traditionally limited to the well characterized, most prevalent genes, next-generation sequencing (NGS) allows screening even the “minor” disease genes with very short turnaround time. This approach to genetic testing is highly efficient, but it is also generating remarkable interpretative problems mostly related to the high prevalence of variants of unknown significance (VUS), i.e., not clearly related to disease pathophysiology. This issue is relevant to several genetic diseases of the heart but is particularly evident in IADs, where the familial co-segregation analysis is hampered by the incomplete penetrance and the variable expressivity. How to design the most appropriate screening approach is challenging and it requires in-depth knowledge of the specific diseases of interest. In this Opinion article, we will review the available NGS approaches and try to outline the available strategies to optimize the performance of this genetic testing methodology.
Long QT syndrome (LQTS), Brugada syndrome (BrS), and catecholaminergic polymorphic ventricular tachycardia (CPVT) are the main channelopathies that can cause for sudden cardiac death (SCD) in children or young adults. In the last few years, the number of genes and genetic variants associated with these diseases has increased. For example, there are 15 known LQTS1 genes and at least 16 BrS genes.2 Importantly, however, in each disease, there are few major genes and a larger number of genes accounting for few cases each. The “minor” genes are usually poorly characterized in terms of function and pathophysiological role. As a consequence, the identification of mutations in these genes often leads to results that are difficult to interpret. Therefore, the HRS expert consensus statement on the diagnosis and management of patients with inherited arrhythmias syndromes (2) has outlined the indications for genetic testing on the basis of the epidemiological relevance of the genes and the clinical implications of genetic testing for each disease (i.e., how much the identification of the mutation can impact the clinical management).
The NGS, a massive parallel sequencing technology that revolutionized the genetic diagnostics, allows large-scale and rapid assessment of the entire human genome (3). In principle, there are three approaches that can be used: (1) whole genome sequencing (WGS), applied to sequence the entire genome, coding, and non-coding regions; (2) whole exome sequencing (WES) used to analyze only the “exome,” which represents 1% of the whole genome; (3) target resequencing panel (TRS) of genes, adopted to sequence selected gene sets/panels (4).
The first two approaches, WGS and WES, are mainly applied for research purposes, for discovery of new disease genes, while TRS is commonly used for the diagnosis in the clinical setting (5).
Recently, Pua et al. reported a comparison study applying different approaches of sequencing, such as TRS, WES, and WGS (6). Analyzing a custom panel, including 174 genes involved in inherited cardiac disease, they investigated the performances of the approaches across this set of genes. Results showed that TRS approach achieved a higher coverage (>99.8% at ≥20× read depth) compared with the other approaches (88.1 and 99.3%; WES and WGS, respectively, at ≥20× read depth). Furthermore, this approach has been reported to be faster and more affordable.
In the pre-NGS era, the analysis of yield of genetic testing provided a clear evidence of the tight link between the severity of the clinical phenotype and rate of identified mutations. Bai et al. (7) showed a high yield of screening (64, 51, and 13% for LQTS, CPVT, and BrS, respectively) in patients with a conclusive diagnosis compared with the borderline cases (14, 13 and 2%) (7). A similar concept also applies in the NGS era. Clinicians are tempted to use the fast and efficient NGS technology as a diagnostic tool when clinical examinations are inconclusive. This can lead to the identification of a high rate of VUS, especially on minor genes. Thus, the selection of genes to be included in TRS is crucial. In general, there are three NGS strategies available:
(1) Comprehensive cardio panel: 60–180 genes covering all known genes (channelopathies and/or cardiomyopathies).
(2) Comprehensive arrhythmias panel: 20–60 genes restricted to arrhythmogenic conditions (Table 1).
(3) “Key gene” panels: few genes (3–6 genes), with high evidence, related to a specific phenotype.
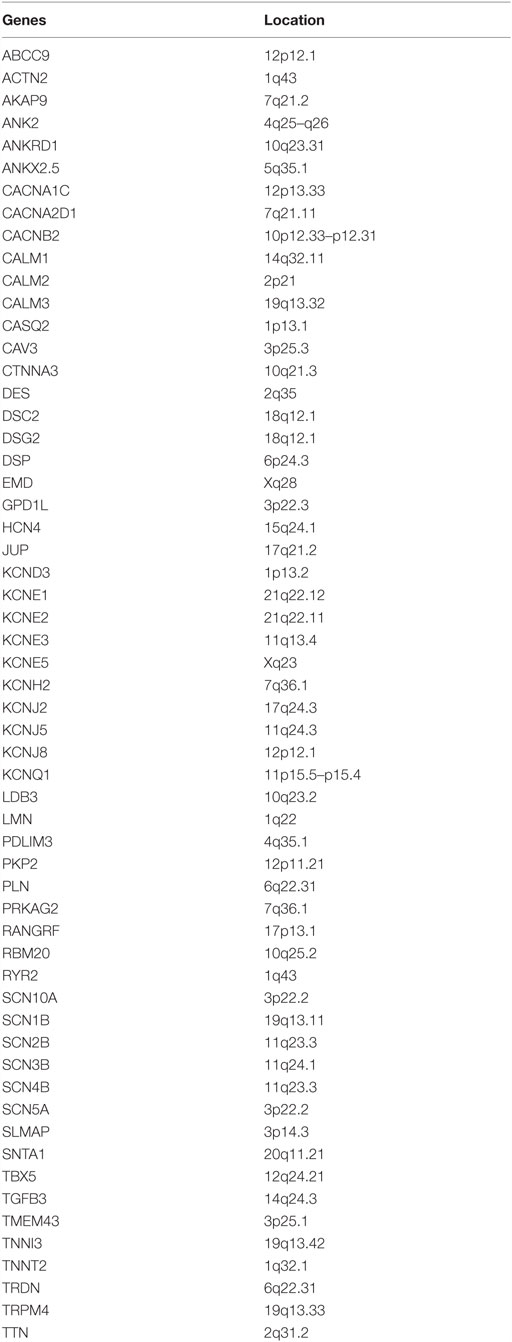
Table 1. Genes included in comprehensive arrhythmias panels.
In patients with conclusive diagnosis, use of TRS panels with a limited set of well-characterized genes should be considered the first step to reduce the number of tests with uncertain findings (first tier) (Figure 1).
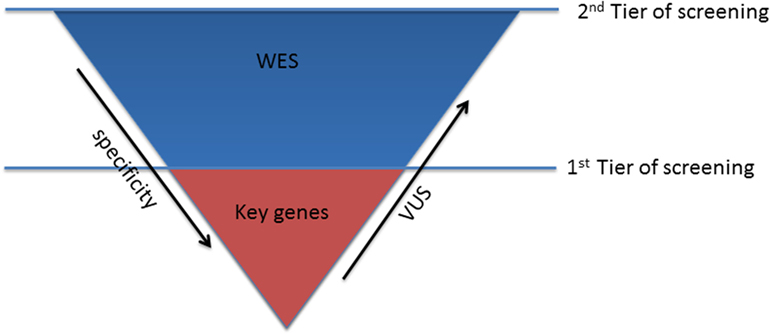
Figure 1. Sequencing strategy for genetic testing.
The optimal strategy in subjects, who turn out negative in the first step, is much less defined. After the exclusion of the keys genes, the second tier of screening, using WES approach, can be considered (Figure 1). This choice might be preferable over the use of comprehensive cardio panels, due to the limited evidence of the minor genes associated with channelopathies, which cannot justify the investment required for the design and production of the second larger disease-specific gene panel. Therefore, WES will guarantee the consideration of all the mutations in the minor genes that have not been unraveled yet and consider also rare genetic variants in novel genes, still unrelated with the phenotype.
Nevertheless, it is clear that there may be hurdles also in the interpretations of WES data. Independently from the screening approach, it should be considered that for diagnostic purposes the presence of (1) a clear pathophysiological link between the genetic variant and the phenotype and (2) the co-segregation within families, still remain crucial for the interpretation (8).
An example of the first tier strategy was recently reported by Millat et al. (9). Analyzing a cohort of 15 LQTS with a key panel, including only the main five genes associated with LQTS (KCNQ1, KCNH2, SCN5A, KCNE1, and KCNE2), they compared TRS and Sanger sequencing. The results showed that Sanger efficiently sequenced all the 69 exons compared with the TRS that sequenced 55/69 exons (86% of the targeted regions). NGS–TRS showed cost and turnaround time advantages over Sanger method. The study by Millat et al. highlights a very relevant problem, which is common to all NGS platforms: lack of coverage of specific regions of genes. In some cases, the problem can be particularly relevant. For example, several exons of KCNH2, a highly prevalent LQTS gene (~35% of patients), are not completely sequenced due to their high CG rich sequence. Thus, integration with Sanger sequencing of uncovered regions is often required with a consequence impact on costs and turnaround time.
Another interesting study on the evaluation of the first tier approach has been reported by Steffensen et al. in a cohort of 39 patients analyzed for the main genes associated with LQTS (KCNQ1, KCNH2, SCN5A, and KCNE1) (10). Results showed a high percentage of patients (17 patients; 44%) carrying variants classified as pathogenic compared with patients carrying VUS or VUS likely pathogenic (11 patients; 28%) and no alterations (13 patients; 34%).
Enlarging the screening to other seven minor genes (ANK2, KCNJ2, CACNA1C, CAV3, SCN4B, AKAP9, and SNTA1) associated with LQTS, the authors identified only three more variants, two classified as VUS and one as likely benign, demonstrating a very limited contribution, when including minor genes in the screening but a significant increase in the cost of the genetic tests.
The use of large panels, inclusive of all the minor genes may have additional limitations, as reported by Alfares et al. in patients with hypertrophic cardiomyopathy (HCM) (11). They tested over 9 years, 2,912 probands referred for clinical HCM genetic testing with different approaches: 11-gene panel, 18-gene panel, and a 50-gene pan cardiomyopathy panel. Results showed that the majority of positive tests were due to pathogenic or likely pathogenic variants in the MYBPC3 and MYH7 genes (83%), the two well-characterized genes routinely screened even with Sanger sequencing. Furthermore, analyzing a subset of 202 HCM patients with 18-gene panel and the pan cardiomyopathy panel, none of the probands had a causative variant outside the 18 “classic” HCM genes, suggested that use of the extended cardiomyopathy gene panel is useless for patients with HCM and should be reserved for patients with atypical clinical phenotypes (12).
Next-generation sequencing technology has improved significantly over the past few years. However, there are still some limitations that need to be considered in terms of sensitivity of the uncovered regions (lower than Sanger sequencing). Moreover, the high-throughput capability is revealing itself as a double edge sword: on the one hand, it allows amazingly short turnaround time and reduced costs, but on the other hand, it reveals an increased rate of VUS and tests that are not conclusive (and therefore clinically irrelevant). A way to overcome this problem can be the implementation of a shared knowledge based on VUS. International collaborative efforts for the annotation of genetic variants are currently being explored as a mean to improve the interpretation capabilities for NGS results (13). The ClinVar database3 is a publicly available tool for deposition and retrieval of variant data and annotations (14). This effort is expected to support the decision on the pathogenicity of identified variants and, most importantly, to resolve the classification of VUS. Meanwhile, the most appropriate use of NGS is that of a phenotype-driven approach with sequencing panels with a limited number of well-known genes and used in patients with clear clinical indications for genetic testing. Therefore, if causative mutations are not identified on the “key” disease IAD genes, the analysis should take a “research track” with the use of WES. However, patients should be counseled accordingly.
All authors (VN, MM, PG, and CN) contributed extensively to the work presented in this opinion article.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Bezzina CR, Lahrouchi N, Priori SG. Genetics of sudden cardiac death. Circ Res (2015) 116(12):1919–36. doi: 10.1161/CIRCRESAHA.116.304030
2. Priori SG, Wilde AA, Horie M, Cho Y, Behr ER, Berul C, et al. HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes: document endorsed by HRS, EHRA, and APHRS in May 2013 and by ACCF, AHA, PACES, and AEPC in June 2013. Heart Rhythm (2013) 10(12):1932–63. doi:10.1016/j.hrthm.2013.05.014
3. Lubitz SA, Ellinor PT. Next-generation sequencing for the diagnosis of cardiac arrhythmia syndromes. Heart Rhythm (2015) 12(5):1062–70. doi:10.1016/j.hrthm.2015.01.011
4. de Koning TJ, Jongbloed JD, Sikkema-Raddatz B, Sinke RJ. Targeted next-generation sequencing panels for monogenetic disorders in clinical diagnostics: the opportunities and challenges. Expert Rev Mol Diagn (2015) 15(1):61–70. doi:10.1586/14737159.2015.976555
5. Rehm HL. Disease-targeted sequencing: a cornerstone in the clinic. Nat Rev Genet (2013) 14(4):295–300. doi:10.1038/nrg3463
6. Pua CJ, Pua CJ, Bhalshankar J, Miao K, Walsh R, John S, et al. Development of a comprehensive sequencing assay for inherited cardiac condition genes. J Cardiovasc Transl Res (2016) 9(1):3–11. doi:10.1007/s12265-016-9673-5
7. Bai R, Napolitano C, Bloise R, Monteforte N, Priori SG. Yield of genetic screening in inherited cardiac channelopathies: how to prioritize access to genetic testing. Circ Arrhythm Electrophysiol (2009) 2(1):6–15. doi:10.1161/CIRCEP.108.782888
8. Rabbani B, Tekin M, Mahdieh N. The promise of whole-exome sequencing in medical genetics. J Hum Genet (2014) 59(1):5–15. doi:10.1038/jhg.2013.114
9. Millat G, Chanavat V, Rousson R. Evaluation of a new high-throughput next-generation sequencing method based on a custom AmpliSeq library and ion torrent PGM sequencing for the rapid detection of genetic variations in long QT syndrome. Mol Diagn Ther (2014) 18(5):533–9. doi:10.1007/s40291-014-0099-y
10. Steffensen AB, Refaat MM, David JP, Mujezinovic A, Calloe K, Wojciak J, et al. High incidence of functional ion-channel abnormalities in a consecutive Long QT cohort with novel missense genetic variants of unknown significance. Sci Rep (2015) 5:10009. doi:10.1038/srep10009
11. Alfares AA, Kelly MA, McDermott G, Funke BH, Lebo MS, Baxter SB, et al. Results of clinical genetic testing of 2,912 probands with hypertrophic cardiomyopathy: expanded panels offer limited additional sensitivity. Genet Med (2015) 17(11):880–8. doi:10.1038/gim.2014.205
12. Ackerman MJ. Genetic purgatory and the cardiac channelopathies: exposing the variants of uncertain/unknown significance issue. Heart Rhythm (2015) 12(11):2325–31. doi:10.1016/j.hrthm.2015.07.002
13. Rehm HL, Berg JS, Brooks LD, Bustamante CD, Evans JP, Landrum MJ, et al. ClinGen – the clinical genome resource. N Engl J Med (2015) 372(23):2235–42. doi:10.1056/NEJMsr1406261
Keywords: genetic variants, next-generation sequencing, channelopathy genetic testing, genetic panel
Citation: Novelli V, Gambelli P, Memmi M and Napolitano C (2016) Challenges in Molecular Diagnostics of Channelopathies in the Next-Generation Sequencing Era: Less Is More? Front. Cardiovasc. Med. 3:29. doi: 10.3389/fcvm.2016.00029
Received: 14 March 2016; Accepted: 26 August 2016;
Published: 12 September 2016
Edited by:
Edward J. Lesnefsky, Virginia Commonwealth University and McGuire Veterans Affairs Medical Center, USAReviewed by:
Fadi G. Akar, Icahn School of Medicine at Mount Sinai, USACopyright: © 2016 Novelli, Gambelli, Memmi and Napolitano. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carlo Napolitano, Y2FybG8ubmFwb2xpdGFub0Bmc20uaXQ=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.