 Marta Gigli1,2†
Marta Gigli1,2† Rene L. Begay1†
Rene L. Begay1† Gaetano Morea1,2Sharon L. Graw1Gianfranco Sinagra2Matthew R. G. Taylor1Henk Granzier3
Gaetano Morea1,2Sharon L. Graw1Gianfranco Sinagra2Matthew R. G. Taylor1Henk Granzier3 Luisa Mestroni1*
Luisa Mestroni1*
- 1Adult Medical Genetics Program, Cardiovascular Institute, University of Colorado Denver, Aurora, CO, USA
- 2Department of Cardiology, Hospital and University of Trieste, Trieste, Italy
- 3Molecular Cardiovascular Research Program, University of Arizona, Tucson, AZ, USA
Titin (TTN) is known as the largest sarcomeric protein that resides within the heart muscle. Due to alternative splicing of TTN, the heart expresses two major isoforms (N2B and N2BA) that incorporate four distinct regions termed the Z-line, I-band, A-band, and M-line. Next-generation sequencing allows a large number of genes to be sequenced simultaneously and provides the opportunity to easily analyze giant genes such as TTN. Mutations in the TTN gene can cause cardiomyopathies, in particular dilated cardiomyopathy (DCM). DCM is the most common form of cardiomyopathy, and it is characterized by systolic dysfunction and dilation of the left ventricle. TTN truncating variants have been described as the most common cause of DCM, while the real impact of TTN missense variants in the pathogenesis of DCM is still unclear. In a recent population screening study, rare missense variants potentially pathogenic based on bioinformatic filtering represented only 12.6% of the several hundred rare TTN missense variants found, suggesting that missense variants are very common in TTN and are frequently benign. The aim of this review is to understand the clinical role of TTN mutations in DCM and in other cardiomyopathies. Whereas TTN truncations are common in DCM, there is evidence that TTN truncations are rare in the hypertrophic cardiomyopathy (HCM) phenotype. Furthermore, TTN mutations can also cause arrhythmogenic right ventricular cardiomyopathy (ARVC) with distinct clinical features and outcomes. Finally, the identification of a rare TTN missense variant cosegregating with the restrictive cardiomyopathy (RCM) phenotype suggests that TTN is a novel disease-causing gene in this disease. Clinical diagnostic testing is currently able to analyze over 100 cardiomyopathy genes, including TTN; however, the size and presence of extensive genetic variation in TTN presents clinical challenges in determining significant disease-causing mutations. This review discusses the current knowledge of TTN genetic variations in cardiomyopathies and the impact of the diagnosis of TTN pathogenic mutations in the clinical setting.
Introduction
Dilated cardiomyopathy (DCM) is defined by the presence of left ventricular (LV) or biventricular dilatation and systolic dysfunction in the absence of hypertension, valvular disease, or coronary artery disease sufficient to cause global systolic impairment (1). The prevalence of the disease is about 1:2,500, and DCM explains about half of the heart failure cases in the United States. About 35–40% of DCM cases are classified as “idiopathic” or “familial/genetic” cardiomyopathy (2). Other causes of the DCM phenotype are ischemic, congenital, valvular, inflammatory, or cardiotoxic heart disease. Finally, other rare cardiomyopathies, such as hypertrophic cardiomyopathy (HCM), arrhythmogenic right ventricular cardiomyopathy (ARVC), and restrictive cardiomyopathy (RCM), have genetic causes.
In this setting, genetics can justify a significant proportion of DCM cases (up to 25%), so the disease can be classified into genetic and non-genetic forms (3). In DCM, the most common form of cardiomyopathy, more than 50 genes have been associated with the phenotype, usually with incomplete penetrance and variable expressivity, and frequently with familial transmission (2–4). Evidence suggests that familial DCM is inherited in an autosomal dominant pattern in about 90% of cases, but few cases follow an autosomal recessive, x-linked, or mitochondrial pattern of inheritance (5–7). Genes most frequently involved in the disease are encoding structural proteins of the sarcomere (titin and myosin heavy chain), cytoskeleton (desmin), nuclear membrane (lamin A/C), membrane proteins and ion channels (phospholamban and presenilin), protein of the dystrophin-glycoprotein complex (dystrophin and sarcoglycan), desmosomes (desmoplakin and desmoglein), mitochondrial proteins (frataxin), and extracellular matrix proteins (alpha-laminin) (8).
Titin (TTN) encodes the largest human protein, whose name stems from the word Titans, giants of Greek mythology. Among the genes involved in cardiomyopathies, TTN plays a central role because of its frequency and the key structural, mechanical, and regulatory role within the sarcomere in the striated muscle (9). The TTN gene consists of 364 exons, located on chromosome 2q31, that produces maximally a 4,200-kDa protein which is composed of ~38,000 amino acid residues. The size and complex structure of the TTN protein provides architectural support, maintaining the sarcomeric organization during contraction, and developing passive tension during muscle stretching. It also has a sensory and signaling role through the multiple TTN-binding proteins that are organized in signaling hot spots (10–12). The protein is organized in four structural and functional regions: the N-terminal Z-line (anchor to the sarcomeric Z-disk), the I-band (responsible for elastic properties), A-band regions (with a stabilizer role of the thick filament), and the C-terminal M-line extremity (overlap in antiparallel orientation with another C-terminal TTN molecule; modulation of TTN expression and turnover with the tyrosine kinase domain) (10).
Truncation mutations of TTN are the most frequent in DCM where 25% of cases are familial forms and 18% are sporadic forms of DCM (13). However, it remains to be confirmed that TTN truncating mutations are always pathogenic (3, 14). Interestingly, truncations in the A-band region of TTN accounts for up to 25% of DCM cases (15). Furthermore, TTN is involved in the pathogenesis of other cardiomyopathies such as HCM and ARVC that is considered to be a genetic disease (30–50% of cases are familial), and RCM.
After the introduction of next-generation sequencing (NGS), the study of TTN gene mutations, previously difficult to analyze due to its size and complexity, has now allowed the identification of more than 60,000 TTN missense variants (reported in the 1000 Genomes Project) (16, 17). The aim of this review is to discuss the challenges in diagnosing the correlation between TTN mutations and the different types of cardiomyopathy in the clinical setting.
Mechanistic Studies of TTN
Titin is the largest human protein. Two TTN filaments with opposite polarity span each sarcomere, namely, the contractile unit in striated muscle cells. TTN is responsible for sarcomere passive stiffness generation (18). TTN is composed of a Z-disk at its N-terminus, whereas the remaining part of the molecule is composed of the elastic I-band region (consisting of tandem Ig segments of serially-linked Ig-like domains), the spring-like PEVK region (is composed of proline (P), glutamate (E), valine (V), and lysine (K)), three unique sequences of Novex1, 2, and 3, cardiac-specific N2B and N2A domains, a thick A-band region, and a M-band region embedding the C-terminus (Figures 1 and 2) (19–21). The extensible I-band region gradually lengthens and develops passive tension when the sarcomere is stretched during diastole (15). The inextensible A-band binds myosin and myosin-binding protein C (MyBP-C), whereas the M-band contains a kinase that affects gene expression and cardiac remodeling (22).
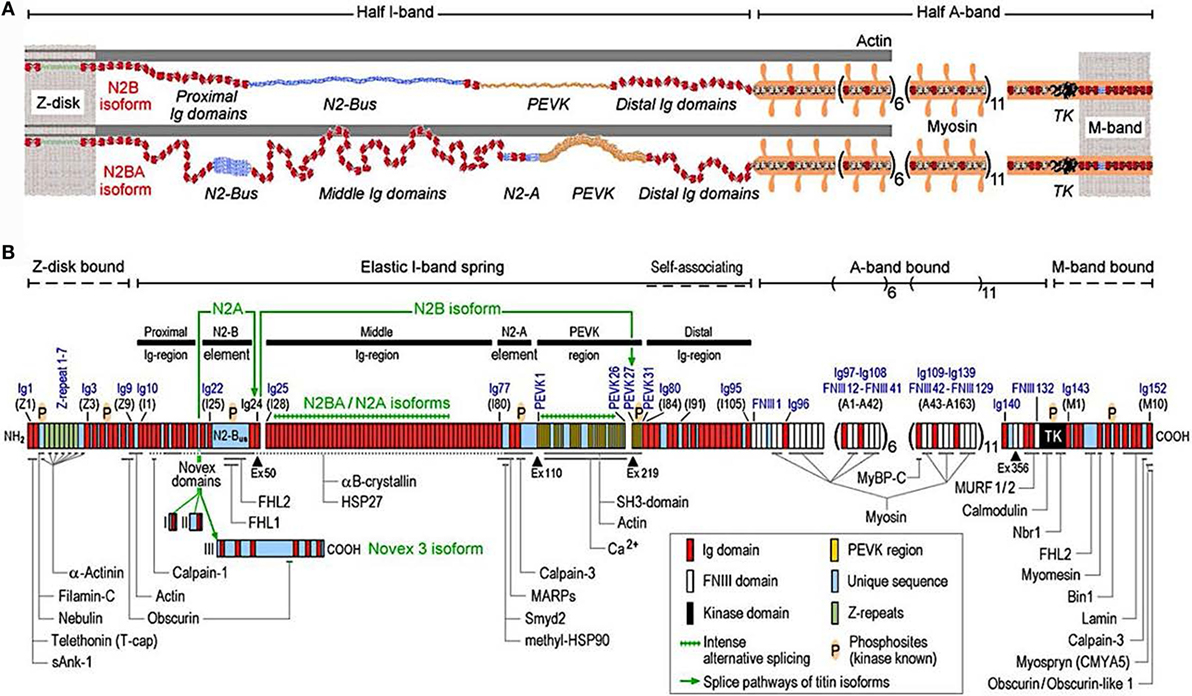
Figure 1. Domain structure of titin isoforms and binding sites of titin ligands. (A) N2B and N2BA titin isoforms represented in the cardiac half-sarcomere, (B) Domain structure of titin sequence, Q8WZ42-1, with ligand binding sites represented (from Linke and Hamdani, with permission) (60).
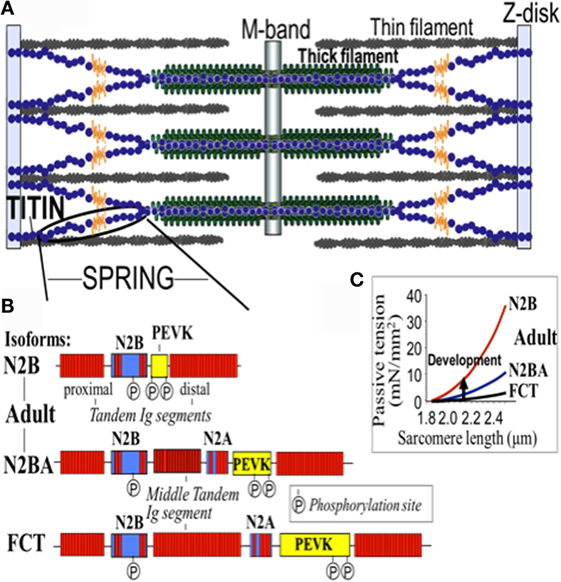
Figure 2. Domain structure of titin isoforms. (A) The spring segment, (B) difference in the domain structure of different isoforms, and (C) the relationship of the passive tension with the sarcomere length in the different isoforms. FTC, fetal cardiac titin.
The 364 exons of TTN undergo extensive alternative splicing to encode different isoforms. In cardiomyocytes, three different isoforms of titin are expressed: adult N2BA, adult N2B, and the fetal cardiac titin (FCT) isoforms. The I-band sequence defines the different proprieties of each isoform, whereas the Z-disk, A-band, and M-line regions are extremely conserved (22). The isoforms, N2BA and N2B are expressed 30–40 and 60–70% respectively, within the TTN protein in healthy adult human heart. The ratio between these two isoforms is a major determinant of the cardiomyocyte stiffness (18). Due to the longer extensible I-band region, the N2BA titin isoform is more compliant than N2B titin (23–25). The compliant N2BA contains additional spring elements in the PEVK and tandem Ig regions and is therefore associated with low cardiomyocyte passive tension (25). The TTN-based passive tension is established by the TTN expression ratio in the human heart. There is a strong relationship between the TTN-based passive tension and the size of the I-band region: the larger the elastic I-band region and the lower the passive tension (22). Variable isoform expression and TTN splicing have become of great importance in different cardiac diseases, including DCM, whereby the compliant N2BA isoform is upregulated and is associated with decreasing passive stiffness and increasing chamber compliance (23, 24, 26, 27).
A recent study by Roberts et al. suggested that the clinical significance of TTN truncating variants is largely predicated by the exon usage and variant location (the distance of the truncating variant from the protein N-terminus) (28). Furthermore, the authors compared TTN truncating variants among different isoforms and found TTN truncating variants altering both N2BA and N2B were overrepresented in DCM patients versus controls and more strongly associated with DCM as compared with the TTN truncations involving the N2BA isoform only. Conversely, the TTN truncations of the controls were composed of exons not incorporated into N2BA and N2B transcripts (28).
The TTN gene structure is organized to accommodate extensive splicing events. Roberts et al. defined a percentage spliced in (PSI) score based on RNA sequencing data from end-stage DCM and donor heart in order to find the mean usage of each TTN exon (28). The PSI estimates the proportion of transcripts that incorporate a given exon. A high PSI was given to an exon constitutively expressed and present in all TTN isoforms, while a low PSI was usually present only in one isoform and had a lower expression. Moreover, exon symmetry was related to PSI: only 3 exons among the 175 with PSI < 0.99 were asymmetric versus 27% of those with PSI > 0.99. Interestingly, the authors found that more than 80% of all TTN exons were symmetric and that their exclusion would not alter the translational reading frame. For instance, in the I-band, the region with the lower PSI, 93% of alternately spliced exons were symmetric: a truncating variant in that region will fall in exons spliced out or not expressed in the majority of the transcripts and should not have such a deleterious effect. While the stiffness of TTN is defined primarily by the I-band segment sequence of each isoform, it is well known that the cardiac passive tension can be affected by multiple post-translational modifications of contractile and regulatory proteins (29). Few studies have discovered that protein kinase phosphorylation significantly alters the stiffness of N2B and PEVK spring elements (30, 31). The N2B spring element is phosphorylated by PKA and PKG with a reduction in passive tension (29, 32).
The mechanisms responsible for the changes in TTN isoform expression are still not completely understood; however, it has been shown that RNA-Binding Motif Protein 20 (RBM20), a RNA splicing factor, plays an important role in this process and a reduced expression of RBM20 can alter TTN splicing and isoform expression in human (33) and mice (34), leading to DCM.
Therefore, the TTN-based myocardial stiffness is determined by the TTN isoform composition and the phosphorylation state of TTN’s elastic I-band. Different kinases can modify the TTN elasticity in different ways; indeed, it is known that changes in post-translational modification (in particular hypophosphorylation) plays a role in the pathophysiology of heart disease (13).
Titin in the Pathogenesis of Dilated Cardiomyopathy
Dilated cardiomyopathy is a primary myocardial disease with variable natural history and clinical presentation affecting young individuals with a potential long life expectancy. A genetic etiology is demonstrated in ~30% of cases (35), and the giant muscle TTN protein has been recognized as the major human disease-causing gene for DCM (9). The advances in contemporary DNA sequencing and the introduction of NGS have allowed the screening of TTN in large cohorts of patients with DCM and in the past few years have been prolific in the description of new DCM-related TTN mutations. A comprehensive cohort study by Herman et al. (16) on 312 DCM patients reported TTN truncating mutations to be the cause of DCM in 25 and 18% of familial DCM and sporadic cases, respectively. TTN truncating mutations found in subjects with DCM were overrepresented in the A-band region and were absent from the Z-disk and M-band regions. Interestingly, TTN truncation variants were also present in up to 2% of the control population, but the control subjects were less enriched for the A-band region of TTN including the Z-band variants. A recent study by Pugh et al. (36) confirmed the presence of truncating variants in the general population (1.65%) and demonstrated that truncating variants located in the A-band are more common in patients with DCM compared with controls. The rate of TTN truncating variants found by Pugh et al., in the DCM cohort was ~14%. In addition, a reduced frequency of variants in the I-band was identified in probands compared with controls, whereas no differences were detected in the Z and M bands.
The TTN gene has also been evaluated in the European Atlas study of 639 patients with sporadic or familial DCM by NGS. Mutations in TTN were identified in 19% of familial and 11% of sporadic cases (37). Noteworthy, 44% of patients with a truncating TTN variant also presented an additional known disease-causing variant in at least one other gene involved in the pathogenesis of DCM; thus in these cases, the TTN variant may not be the only contributor leading to the pathogenesis of DCM (37).
A large study recently compared the burden of rare TTN variants across five cohorts of healthy volunteers, participants in the Framingham Heart Study, participants in the Jackson Heart Study, cohort of unselected ambulatory patients with DCM, and end-stage DCM cases. The authors confirmed that TTN truncations were not uniformly distributed within and between study groups, being more common in patients with DCM (22%), but with a rate in the healthy volunteers ranging between 1 and 2.9% (28). The TTN truncation variants in the DCM cohort were located predominantly in the A-band, as already described in previous studies mentioned above (16, 36).
The role of TTN truncation mutations in the pathogenesis of DCM has been largely recognized. However, the high prevalence of missense variants and the potential modifier effects make it difficult to elucidate the effective role of TTN missense variants in DCM. Some of these variants are proposed to be pathogenic, but other variants are of unknown significance (VUS). In order to address this challenge, a recent multicenter study sequenced the TTN gene in a cohort of 147 DCM patients (38). In this cohort, 13 TTN truncating variants had previously been reported (16), and 348 missense variants were filtered by bioinformatic algorithms resulting in 44 out of 348 (involving 37 probands) classified as “severe” or likely pathogenic. Among the nine families with TTN variants classified as “severe,” five were considered false positives due to discordant cosegregation analysis among affected relatives, whereas four families had “severe” TTN variants that cosegregated with the DCM phenotype. The remaining 28 probands harbored “severe” variants that could not be assessed by cosegregation (possibly pathogenic). Furthermore, the outcome of TTN missense variants carriers did not differ significantly from the other DCM patients (Figure 3). Interestingly, the distribution of the likely and possibly TTN severe missense variants across TTN domains was again non-random and was overrepresented in the A-band region of TTN. Specifically, variants were overrepresented in the C-zone of the A-band, which consists of a super repeat of 11 immunoglobulin-like domains and Fn-III domains shown to bind to MyBP-C and subfragment myosin-1, and is essential for the length dependency of force development and calcium sensitivity (39). Therefore, although the real impact of TTN missense variants in the pathogenesis of DCM is still unclear, the clustering of variants in the A-band in DCM may suggest that some A-band missense variants may have a functional detrimental effect on contractility and should be further investigated.
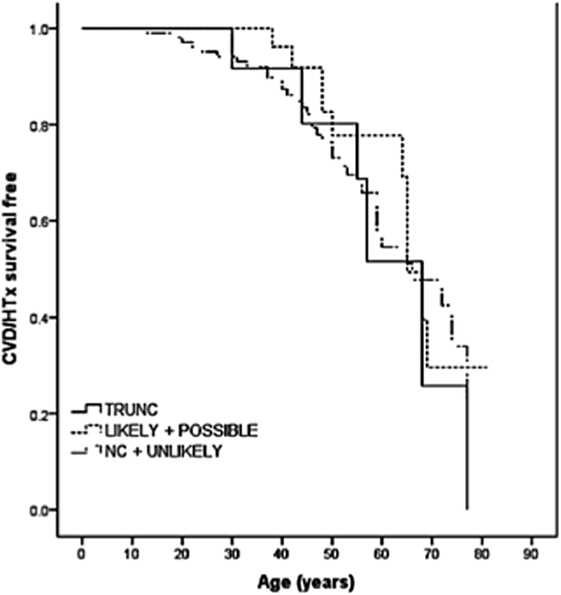
Figure 3. Long-term survival curves in TTN variant carriers. Kaplan–Meier event-free survival for cardiovascular death (CVD) or heart transplantation (HTx) based on TTN variant categories: truncations (TRUNC); “likely” and “possibly” missense variants; non-carriers (NC), and “unlikely” with lack of cosegregation. TTN indicates titin gene (from Begay et al., with permission) (38).
Titin in Other Forms of Cardiomyopathy
Hypertrophic Cardiomyopathy
Hypertrophic cardiomyopathy is a common and inherited cardiomyopathy with a prevalence of 1 in 500 (40). HCM presents as an unexplained LV hypertrophy, myocardial disarray, and fibrosis that translate in increased risk of life-threatening ventricular arrhythmias, sudden cardiac death, and an increased life-long risk of heart failure (41, 42). In the majority of cases, HCM has an autosomal dominant trait and mutations in at least 11 different genes. These genes encode for sarcomeric proteins that are responsible for 50–65% of familial cases (9). While TTN truncation mutations are common in DCM, there is evidence that TTN truncations are rare in the HCM phenotype, with a frequency similar to control populations (16). Using high-throughout sequencing in 142 HCM probands, Lopes et al. found 219 TTN rare variants with 209 being novel missense variants (43). However, this cohort of individuals potentially had a sarcomeric gene mutation that likely caused HCM, and the actual pathogenic role of these TTN variants in unknown.
Restrictive Cardiomyopathy
Restrictive cardiomyopathy is a very rare form of cardiomyopathy, characterized by preserved biventricular systolic function and a restrictive physiology determining an impaired LV filling despite normal cavity size and frequently normal wall thickness. RCM can be secondary to idiopathic or system disease. It is believed that a significant proportion of RCM cases are genetically determined (42). The pattern of inheritance can be autosomal dominant, autosomal recessive, or x-linked (44). The overall prognosis of RCM is poor, usually resulting in progressive biventricular heart failure with a high mortality rate in the absence of heart transplantation. Interestingly, RCM overlaps in clinical features with HCM (42). Recently, a study using linkage analysis that reported a TTN missense variant (TTN: c.22862A>G) cosegregating with RCM in six affected individuals of a family. The most common genes were excluded due to lack of complete cosegregation. Interestingly, some healthy individuals also harbored the TTN missense variant resulting in an incomplete penetrance (44). The identification of a rare missense variant in TTN cosegregating with the RCM disease phenotype suggests that TTN is a novel disease-causing gene for RCM.
Arrhythmogenic Right Ventricular Cardiomyopathy
Arrhythmogenic right ventricular cardiomyopathy is considered to be a genetic disease (30–50% of cases) mainly with autosomal dominant pattern of inheritance (45). ARVC is characterized by fibrofatty replacement of the myocardium, predominantly of the right ventricle, although the left ventricle can also be involved. Typical symptoms include palpitations, cardiac syncope, and cardiac arrest due to ventricular arrhythmias. Heart failure may develop later in life as a result of this disease (46). One study by Taylor et al. in which the investigators analyzed by direct sequencing of 312 exons of TTN (311 expressing TTN protein) found TTN mutations to be associated with the ARVC phenotype (47). Among seven different probands with an ARVC phenotype, eight TTN rare variants (two TTN variants present in one proband) were identified (47). In addition to this study, another investigation by Brun et al. compared the clinical outcomes of ARVC patients with TTN mutations, desmosomal mutations, and patients with no identifiable mutation (non-carriers) (45). In this study, 13% of TTN rare variants were accounted for in their population of subjects. Among the 67 ARVC affected patients (39 ARVC families), 11 harbored rare TTN variants and 8 desmosomal genes variants. The TTN carriers had increased supraventricular arrhythmias, and conduction disease compared with non-carriers (45), while desmosomal gene variant carriers had the worse prognosis. In conclusion, these studies suggest that TTN mutations can cause ARVC and TTN mutation carriers have distinct clinical features and outcomes.
Titin as a Gene Modifier
TTN variants are very frequent; of them, pathogenic mutations are relatively rare and most variants are probably benign. However, a portion of these variants could have a modifier gene effect. For instance, TTN has been proposed as a modifier gene in combination with the Lamin A/C (LMNA) gene (48, 49). A modifier gene is not the causal gene, but it may affect the phenotypic expression (50). In a study by Roncarati et al., the authors reported a TTN missense mutation modifying the DCM phenotype primarily caused by a LMNA mutation. The authors analyzed 41 Italian patients using whole exome sequencing (WES). Fourteen individuals harbored LMNA: c.656A>C mutation, and of those five also carried a novel TTN missense mutation (TTN: c.14563C>T) as well (48). LMNA gene mutations are known to be causative of a specific phenotype expression of DCM (51). According to Taylor et al., patients carrying a LMNA mutation show a poor prognosis and experience high event-rates compared with non-carriers of a LMNA mutation (52). Upholding the structure of the nucleus, chromatin arrangement, and gene expression is encoded by the LMNA gene for the A-type lamins (53). In a study by Roncarati et al., the presence of the TTN variant and the LMNA mutation carriers modified DCM patients’ clinical course and disease severity, with double heterozygotes requiring earlier heart transplantation (four individuals) compared with those only harboring the LMNA mutation alone. Furthermore, histological studies showed more evidence that double heterozygote individuals had worse outcomes on a cellular level (48). In conclusion, this study suggests a modifier role of TTN variants that contribute to the complexity of the DCM phenotype.
Clinical Assessment of Titin Variants
Titin has been known to be cause a DCM phenotype for many years; however, the systematic analysis and the complete meaning of its contribution to DCM have been precluded by its giant size and sequencing technical limitations (54). As discussed earlier, using NGS, Herman et al. found that heterozygous mutations truncating the full-length TTN are the most common causes of DCM; occurring in ~25% of familial cases of DCM and 18% of sporadic cases. However, TTN truncating variants were also found in ~2% of healthy controls (16, 55), raising concern about the correct clinical interpretations of TTN variants. The finding of a TTN truncating variant in a patient before the onset of clinical manifestation of disease thus requires further in-depth analysis to support pathogenicity (9). Additional factors, such as band location and PSI score, might help to differentiate pathogenic truncation mutations from benign variants (28, 36). This is of particular importance considering that most DCM patients present late in the course of the disease (advanced disease presenting with heart failure or sudden cardiac death), while the early detection of asymptomatic DCM might be critical to enable early intervention that may prevent the progression to advanced disease (56). Moreover, TTN truncation variants may be found in association with other disease-related genes, increasing the concerns about the actual role of some TTN mutations (37).
Analysis of a large number of genes has led to the identification of sequence VUS. These VUS are one of the main challenges of NGS, because cardiologists and clinical geneticists are faced with uncertainty of the clinical meaning of VUS findings (57).
To date indeed, a large number of identified TTN truncating variants are still classified as VUS, and the high prevalence of missense variants in TTN, and their potential modifier roles make interpretation difficult in both research and the clinical settings. The location of TTN truncating variants can contribute to a better definition of genetic findings, because as already mentioned, TTN truncating variants associated with DCM are located predominantly in the A-band (16, 38). The availability of multiple family members to test for cosegregation with disease, the absence in population databases (ClinVar, ExAC, 1000 Genomes Project, and NHLBI Exome Sequencing Project), prediction software (PolyPhen, SIFT, GERP), and functional data also add in the understanding of classifying the pathogenicity of TTN variants (9). Most importantly, NGS has to be considered a diagnostic test in development and testing results need to be interpreted cautiously in close collaboration between bioformaticians, cardiologists, molecular biologists, and clinical geneticists preferably in expert centers. Many novel variants identified by NGS and classified as VUS present as an inconclusive test result, pending further evidence (57).
Once the pathogenic effect has been defined, another concern is the variability in phenotype expression based on the presence and type of TTN mutation variants. Roberts et al. found more severe impaired LV function, lower stroke volume, and thinner LV walls in TTN truncating than in TTN truncating negative DCM patients (Figure 4) (28). In this cohort, the TTN genotype independently predicted phenotype severity. Furthermore, TTN truncating positive patients more frequently suffer from sustained ventricular tachycardia (28). In the future, if larger prospective studies confirm these findings, TTN mutations might influence the decision-making process for the selection of candidates to an implantable cardioverter defibrillator (ICD) implantation, such as in other cardiomyopathies at high risk of life-threatening arrhythmias (58).
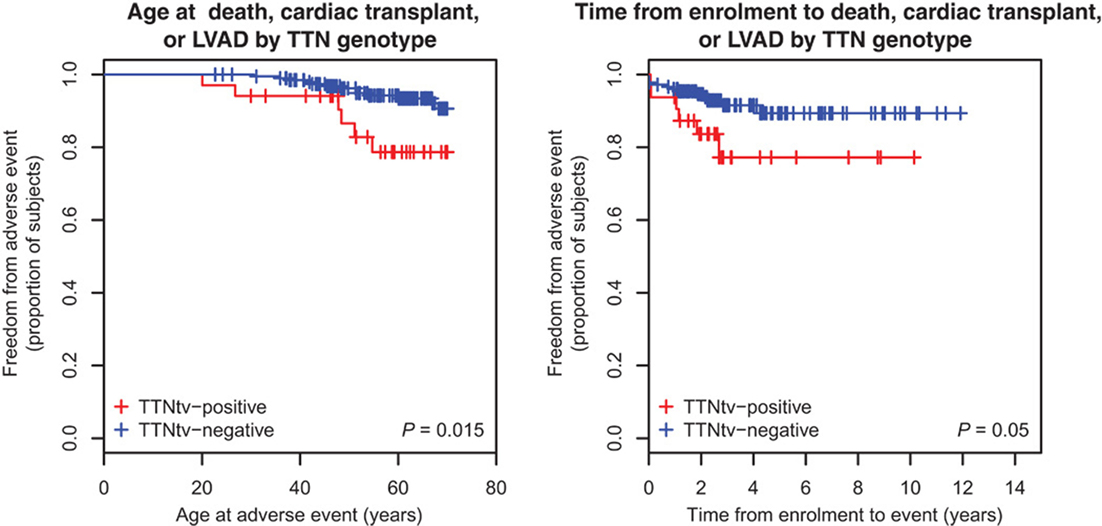
Figure 4. Survival of TTN truncation carriers. Patients carriers of a TTN truncation variant (TTNtv) had a worse clinical outcome when considering the age of adverse event (death, cardiac transplant or left ventricular assisted device) (P = 0.015). They also had a worse clinical outcome when considering the time of event from enrollment (from Roberts et al., with permission) (28).
Mutations in TTN and other proteins affecting TTN splicing are associated with the development of DCM, but these mechanisms are still not completely understood (59). Variable isoform expression and TTN splicing have become of great importance in DCM, and are associated with decreasing passive stiffness and increasing chamber compliance (26). Both mechanisms might be important in the process of DCM in connection to TTN mutations. By genetic approaches or by splicing or posttranslational modifications TTN appears to be a target for future therapeutic interventions (9).
Regarding the universe of TTN missense variants, the situation is even more challenging because TTN missense variants are very common and their real meaning is still unknown. A recent study demonstrated that missense variants did not correlate with the clinical measures of disease severity or progression and indicated that the DCM phenotype caused by TTN missense variants are not distinguishable from other types of DCM (Figure 3). According to the authors, TTN rare missense mutations should not be currently interpreted as disease-causing in most situations (38). Nevertheless, there is some interesting evidence that TTN missense mutations may have a modifier role leading to a greater severity of cardiomyopathy (17, 48). In the future, a better understanding of the TTN missense variants in DCM will be elucidated with large-scale TTN sequencing and functional investigations on TTN variant domains.
Finally, despite the recent advances in genetic studies and in the understanding of the different effects of specific gene mutations in the pathogenesis of DCM, the clinical approach to diagnosing cardiomyopathy affected families remains largely based on the general recommendations for heart failure management, familiar screening programs, and systematic follow-up. The continuous improvement in technologies, such as the increasing evidence concerning the clinical expression of different gene variants might lead in the future to an individualized clinical approach to identifying carriers of different mutations.
Conclusion
Titin is the largest protein in striated muscle. TTN variants have been shown to cause the following cardiac diseases: DCM, RCM, HCM, and ARVC. The advancement of NGS has allowed researchers to analyze the whole TTN gene, which has revealed the leading role of this gene in DCM. Challenges are the high genetic variability of the gene, the large number of missense and truncation variants found in control populations, and the criteria for clinical diagnosis of many variants demand individualized clinical diagnosis platforms for TTN carriers. Future studies will clarify whether the early identification of TTN-related cardiomyopathies might positively influence the natural history of disease by the early initiation of therapeutic management.
Author Contributions
All authors have contributed significantly, read, and approved the manuscript. In particular, MG, RB, and GM: drafting of the manuscript; GS, MT, HG, and LM: revising critically the manuscript for important intellectual content.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This study was supported by the EU FP7-PEOPLE-2011-IRSES 291834 SarcoSI, NIH grants UL1 RR025780, UL1 TR001082, R01 HL69071, R01 116906 to LM; CCTSI K23, JL067915, and R01HL109209 to MT; HL062881 to HG. This work was supported in part by a Trans-Atlantic Network of Excellence grant from the Leducq Foundation (14-CVD 03).
References
1. Elliott P, Andersson B, Arbustini E, Bilinska Z, Cecchi F, Charron P, et al. Classification of the cardiomyopathies: a position statement from the European Society Of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J (2008) 29(2):270–6. doi:10.1093/eurheartj/ehm342
2. Hershberger RE, Morales A, Siegfried JD. Clinical and genetic issues in dilated cardiomyopathy: a review for genetics professionals. Genet Med (2010) 12(11):655–67. doi:10.1097/GIM.0b013e3181f2481f
3. Pinto YM, Elliott PM, Arbustini E, Adler Y, Anastasakis A, Böhm M, et al. Proposal for a revised definition of dilated cardiomyopathy, hypokinetic non-dilated cardiomyopathy, and its implications for clinical practice: a position statement of the ESC working group on myocardial and pericardial diseases. Eur Heart J (2016) 37(23):1850–8. doi:10.1093/eurheartj/ehv727
4. Hershberger RE, Hedges DJ, Morales A. Dilated cardiomyopathy: the complexity of a diverse genetic architecture. Nat Rev Cardiol (2013) 10(9):531–47. doi:10.1038/nrcardio.2013.105
5. Burkett EL, Hershberger RE. Clinical and genetic issues in familial dilated cardiomyopathy. J Am Coll Cardiol (2005) 45(7):969–81. doi:10.1016/j.jacc.2004.11.066
6. Hershberger RE, Cowan J, Morales A, Siegfried JD. Progress with genetic cardiomyopathies: screening, counseling, and testing in dilated, hypertrophic, and arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circ Heart Fail (2009) 2(3):253–61. doi:10.1161/CIRCHEARTFAILURE.108.817346
7. Hershberger RE, Lindenfeld J, Mestroni L, Seidman CE, Taylor MR, Towbin JA. Genetic evaluation of cardiomyopathy – a Heart Failure Society of America practice guideline. J Card Fail (2009) 15(2):83–97. doi:10.1016/j.cardfail.2009.01.006
8. Sinagra G, Mestroni L, Camerini F. Genetic Cardiomyopathies. A Clinical Approach. London: Springer (2012).
9. Gerull B. The rapidly evolving role of titin in cardiac physiology and cardiomyopathy. Can J Cardiol (2015) 31(11):1351–9. doi:10.1016/j.cjca.2015.08.016
10. Chauveau C, Rowell J, Ferreiro A. A rising titan: TTN review and mutation update. Hum Mutat (2014) 35(9):1046–59. doi:10.1002/humu.22611
11. Witt CC, Ono Y, Puschmann E, McNabb M, Wu Y, Gotthardt M, et al. Induction and myofibrillar targeting of CARP, and suppression of the Nkx2.5 pathway in the MDM mouse with impaired titin-based signaling. J Mol Biol (2004) 336(1):145–54. doi:10.1016/j.jmb.2003.12.021
12. Granzier H, Wu Y, Siegfried L, LeWinter M. Titin: physiological function and role in cardiomyopathy and failure. Heart Fail Rev (2005) 10(3):211–23. doi:10.1007/s10741-005-5251-7
13. LeWinter MM, Granzier HL. Titin is a major human disease gene. Circulation (2013) 127(8):938–44. doi:10.1161/CIRCULATIONAHA.112.139717
14. Akinrinade O, Ollila L, Vattulainen S, Tallila J, Gentile M, Salmenperä P, et al. Genetics and genotype-phenotype correlations in Finnish patients with dilated cardiomyopathy. Eur Heart J (2015) 36(34):2327–37. doi:10.1093/eurheartj/ehv253
15. Granzier HL, Irving TC. Passive tension in cardiac muscle: contribution of collagen, titin, microtubules, and intermediate filaments. Biophys J (1995) 68(3):1027–44. doi:10.1016/S0006-3495(95)80278-X
16. Herman DS, Lam L, Taylor MR, Wang L, Teekakirikul P, Christodoulou D, et al. Truncations of titin causing dilated cardiomyopathy. N Engl J Med (2012) 366(7):619–28. doi:10.1056/NEJMoa1110186
17. Merlo M, Sinagra G, Carniel E, Slavov D, Zhu X, Barbati G, et al. Poor prognosis of rare sarcomeric gene variants in patients with dilated cardiomyopathy. Clin Transl Sci (2013) 6(6):424–8. doi:10.1111/cts.12116
18. Hidalgo C, Granzier H. Tuning the molecular giant titin through phosphorylation: role in health and disease. Trends Cardiovasc Med (2013) 23(5):165–71. doi:10.1016/j.tcm.2012.10.005
19. Bang ML, Centner T, Fornoff F, Geach AJ, Gotthardt M, McNabb M, et al. The complete gene sequence of titin, expression of an unusual approximately 700-kDa titin isoform, and its interaction with obscurin identify a novel Z-line to I-band linking system. Circ Res (2001) 89(11):1065–72. doi:10.1161/hh2301.100981
20. Labeit S, Barlow DP, Gautel M, Gibson T, Holt J, Hsieh CL, et al. A regular pattern of two types of 100-residue motif in the sequence of titin. Nature (1990) 345(6272):273–6. doi:10.1038/345273a0
21. Lange S, Xiang F, Yakovenko A, Vihola A, Hackman P, Rostkova E, et al. The kinase domain of titin controls muscle gene expression and protein turnover. Science (2005) 308(5728):1599–603. doi:10.1126/science.1110463
22. LeWinter MM, Granzier H. Cardiac titin: a multifunctional giant. Circulation (2010) 121(19):2137–45. doi:10.1161/CIRCULATIONAHA.109.860171
23. Neagoe C, Opitz CA, Makarenko I, Linke WA. Gigantic variety: expression patterns of titin isoforms in striated muscles and consequences for myofibrillar passive stiffness. J Muscle Res Cell Motil (2003) 24(2–3):175–89. doi:10.1023/A:1026053530766
24. Cazorla O, Freiburg A, Helmes M, Centner T, McNabb M, Wu Y, et al. Differential expression of cardiac titin isoforms and modulation of cellular stiffness. Circ Res (2000) 86(1):59–67. doi:10.1161/01.RES.86.1.59
25. Lahmers S, Wu Y, Call DR, Labeit S, Granzier H. Developmental control of titin isoform expression and passive stiffness in fetal and neonatal myocardium. Circ Res (2004) 94(4):505–13. doi:10.1161/01.RES.0000115522.52554.86
26. Nagueh SF, Shah G, Wu Y, Torre-Amione G, King NM, Lahmers S, et al. Altered titin expression, myocardial stiffness, and left ventricular function in patients with dilated cardiomyopathy. Circulation (2004) 110(2):155–62. doi:10.1161/01.CIR.0000135591.37759.AF
27. Opitz CA, Leake MC, Makarenko I, Benes V, Linke WA. Developmentally regulated switching of titin size alters myofibrillar stiffness in the perinatal heart. Circ Res (2004) 94(7):967–75. doi:10.1161/01.RES.0000124301.48193.E1
28. Roberts AM, Ware JS, Herman DS, Schafer S, Baksi J, Bick AG, et al. Integrated allelic, transcriptional, and phenomic dissection of the cardiac effects of titin truncations in health and disease. Sci Transl Med (2015) 7(270):270ra276. doi:10.1126/scitranslmed.3010134
29. Yamasaki R, Wu Y, McNabb M, Greaser M, Labeit S, Granzier H. Protein kinase A phosphorylates titin’s cardiac-specific N2B domain and reduces passive tension in rat cardiac myocytes. Circ Res (2002) 90(11):1181–8. doi:10.1161/01.RES.0000021115.24712.99
30. Belin RJ, Sumandea MP, Allen EJ, Schoenfelt K, Wang H, Solaro RJ, et al. Augmented protein kinase C-alpha-induced myofilament protein phosphorylation contributes to myofilament dysfunction in experimental congestive heart failure. Circ Res (2007) 101(2):195–204. doi:10.1161/CIRCRESAHA.107.148288
31. Hidalgo C, Hudson B, Bogomolovas J, Zhu Y, Anderson B, Greaser M, et al. PKC phosphorylation of titin’s PEVK element: a novel and conserved pathway for modulating myocardial stiffness. Circ Res (2009) 105(7):631–638, 617 p following 638. doi:10.1161/CIRCRESAHA.109.198465
32. Krüger M, Kötter S, Grützner A, Lang P, Andresen C, Redfield MM, et al. Protein kinase G modulates human myocardial passive stiffness by phosphorylation of the titin springs. Circ Res (2009) 104(1):87–94. doi:10.1161/CIRCRESAHA.108.184408
33. Guo W, Schafer S, Greaser ML, Radke MH, Liss M, Govindarajan T, et al. RBM20, a gene for hereditary cardiomyopathy, regulates titin splicing. Nat Med (2012) 18(5):766–73. doi:10.1038/nm.2693
34. Methawasin M, Hutchinson KR, Lee EJ, Smith JE III, Saripalli C, Hidalgo CG, et al. Experimentally increasing titin compliance in a novel mouse model attenuates the Frank-Starling mechanism but has a beneficial effect on diastole. Circulation (2014) 129(19):1924–36. doi:10.1161/CIRCULATIONAHA.113.005610
35. Morita H, Seidman J, Seidman CE. Genetic causes of human heart failure. J Clin Invest (2005) 115(3):518–26. doi:10.1172/JCI24351
36. Pugh TJ, Kelly MA, Gowrisankar S, Hynes E, Seidman MA, Baxter SM, et al. The landscape of genetic variation in dilated cardiomyopathy as surveyed by clinical DNA sequencing. Genet Med (2014) 16(8):601–8. doi:10.1038/gim.2013.204
37. Haas J, Frese KS, Peil B, Kloos W, Keller A, Nietsch R, et al. Atlas of the clinical genetics of human dilated cardiomyopathy. Eur Heart J (2015) 36(18):1123a–35a. doi:10.1093/eurheartj/ehu301
38. Begay RL, Graw S, Sinagra G, Merlo M, Slavov D, Gowan K, et al. Role of titin missense variants in dilated cardiomyopathy. J Am Heart Assoc (2015) 4(11):e002645. doi:10.1161/JAHA.115.002645
39. Muhle-Goll C, Habeck M, Cazorla O, Nilges M, Labeit S, Granzier H. Structural and functional studies of titin’s fn3 modules reveal conserved surface patterns and binding to myosin S1 – a possible role in the Frank-Starling mechanism of the heart. J Mol Biol (2001) 313(2):431–47. doi:10.1006/jmbi.2001.5017
40. Maron BJ, Gardin JM, Flack JM, Gidding SS, Kurosaki TT, Bild DE. Prevalence of hypertrophic cardiomyopathy in a general population of young adults. Echocardiographic analysis of 4111 subjects in the CARDIA Study. Coronary Artery Risk Development in (Young) Adults. Circulation (1995) 92(4):785–9. doi:10.1161/01.CIR.92.4.785
41. Lopes LR, Rahman MS, Elliott PM. A systematic review and meta-analysis of genotype-phenotype associations in patients with hypertrophic cardiomyopathy caused by sarcomeric protein mutations. Heart (2013) 99(24):1800–11. doi:10.1136/heartjnl-2013-303939
42. Cahill TJ, Ashrafian H, Watkins H. Genetic cardiomyopathies causing heart failure. Circ Res (2013) 113(6):660–75. doi:10.1161/CIRCRESAHA.113.300282
43. Lopes LR, Zekavati A, Syrris P, Hubank M, Giambartolomei C, Dalageorgou C, et al. Genetic complexity in hypertrophic cardiomyopathy revealed by high-throughput sequencing. J Med Genet (2013) 50(4):228–39. doi:10.1136/jmedgenet-2012-101270
44. Peled Y, Gramlich M, Yoskovitz G, Feinberg MS, Afek A, Polak-Charcon S, et al. Titin mutation in familial restrictive cardiomyopathy. Int J Cardiol (2014) 171(1):24–30. doi:10.1016/j.ijcard.2013.11.037
45. Brun F, Barnes CV, Sinagra G, Slavov D, Barbati G, Zhu X, et al. Titin and desmosomal genes in the natural history of arrhythmogenic right ventricular cardiomyopathy. J Med Genet (2014) 51(10):669–76. doi:10.1136/jmedgenet-2014-102591
46. Basso C, Corrado D, Marcus FI, Nava A, Thiene G. Arrhythmogenic right ventricular cardiomyopathy. Lancet (2009) 373(9671):1289–300. doi:10.1016/S0140-6736(09)60256-7
47. Taylor M, Graw S, Sinagra G, Barnes C, Slavov D, Brun F, et al. Genetic variation in titin in arrhythmogenic right ventricular cardiomyopathy-overlap syndromes. Circulation (2011) 124(8):876–85. doi:10.1161/CIRCULATIONAHA.110.005405
48. Roncarati R, Viviani Anselmi C, Krawitz P, Lattanzi G, von Kodolitsch Y, Perrot A, et al. Doubly heterozygous LMNA and TTN mutations revealed by exome sequencing in a severe form of dilated cardiomyopathy. Eur J Hum Genet (2013) 21(10):1105–11. doi:10.1038/ejhg.2013.16
49. Lin F, Worman HJ. Structural organization of the human gene encoding nuclear lamin A and nuclear lamin C. J Biol Chem (1993) 268(22):16321–6.
50. Marian AJ. Modifier genes for hypertrophic cardiomyopathy. Curr Opin Cardiol (2002) 17(3):242–52. doi:10.1097/00001573-200205000-00006
51. Tesson F, Saj M, Uvaize MM, Nicolas H, Ploski R, Bilinska Z. Lamin A/C mutations in dilated cardiomyopathy. Cardiol J (2014) 21(4):331–42. doi:10.5603/CJ.a2014.0037
52. Taylor MR, Fain PR, Sinagra G, Robinson ML, Robertson AD, Carniel E, et al. Natural history of dilated cardiomyopathy due to lamin A/C gene mutations. J Am Coll Cardiol (2003) 41(5):771–80. doi:10.1016/S0735-1097(02)02954-6
53. Broers JL, Ramaekers FC, Bonne G, Yaou RB, Hutchison CJ. Nuclear lamins: laminopathies and their role in premature ageing. Physiol Rev (2006) 86(3):967–1008. doi:10.1152/physrev.00047.2005
54. Ho CY, Charron P, Richard P, Girolami F, Van Spaendonck-Zwarts KY, Pinto Y. Genetic advances in sarcomeric cardiomyopathies: state of the art. Cardiovasc Res (2015) 105(4):397–408. doi:10.1093/cvr/cvv025
55. Golbus JR, Puckelwartz MJ, Fahrenbach JP, Dellefave-Castillo LM, Wolfgeher D, McNally EM. Population-based variation in cardiomyopathy genes. Circ Cardiovasc Genet (2012) 5(4):391–9. doi:10.1161/CIRCGENETICS.112.962928
56. Hershberger RE. Cardiovascular genetic medicine: evolving concepts, rationale, and implementation. J Cardiovasc Transl Res (2008) 1(2):137–43. doi:10.1007/s12265-008-9031-3
57. Mogensen J, van Tintelen JP, Fokstuen S, Elliott P, van Langen IM, Meder B, et al. The current role of next-generation DNA sequencing in routine care of patients with hereditary cardiovascular conditions: a viewpoint paper of the European Society of Cardiology working group on myocardial and pericardial diseases and members of the European Society of Human Genetics. Eur Heart J (2015) 36(22):1367–70. doi:10.1093/eurheartj/ehv122
58. Pasotti M, Klersy C, Pilotto A, Marziliano N, Rapezzi C, Serio A, et al. Long-term outcome and risk stratification in dilated cardiolaminopathies. J Am Coll Cardiol (2008) 52(15):1250–60. doi:10.1016/j.jacc.2008.06.044
59. LeWinter MM, Granzier HL. Cardiac titin and heart disease. J Cardiovasc Pharmacol (2014) 63(3):207–12. doi:10.1097/FJC.0000000000000007
Keywords: titin, TTN, familial cardiomyopathy, cardiovascular genetics, clinical genetics, heart failure, clinical diagnosis
Citation: Gigli M, Begay RL, Morea G, Graw SL, Sinagra G, Taylor MRG, Granzier H and Mestroni L (2016) A Review of the Giant Protein Titin in Clinical Molecular Diagnostics of Cardiomyopathies. Front. Cardiovasc. Med. 3:21. doi: 10.3389/fcvm.2016.00021
Received: 24 March 2016; Accepted: 27 June 2016;
Published: 21 July 2016
Edited by:
Georges Nemer, American University of Beirut, LebanonCopyright: © 2016 Gigli, Begay, Morea, Graw, Sinagra, Taylor, Granzier and Mestroni. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Luisa Mestroni, bHVpc2EubWVzdHJvbmlAdWNkZW52ZXIuZWR1
†Marta Gigli and Rene L. Begay contributed equally.