
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cardiovasc. Med. , 02 March 2016
Sec. Cardiac Rhythmology
Volume 3 - 2016 | https://doi.org/10.3389/fcvm.2016.00005
This article is part of the Research Topic Strategies to Improve Management of Ventricular Tachycardias View all 5 articles
 Mohamed Boulaksil1,2,3
Mohamed Boulaksil1,2,3 Marti F. A. Bierhuizen2Markus A. Engelen2,4Mèra Stein2,5Bart J. M. Kok2Shirley C. M. van Amersfoorth6
Marti F. A. Bierhuizen2Markus A. Engelen2,4Mèra Stein2,5Bart J. M. Kok2Shirley C. M. van Amersfoorth6 Marc A. Vos2
Marc A. Vos2 Harold V. M. van Rijen2
Harold V. M. van Rijen2 Jacques M. T. de Bakker1,2,6†
Jacques M. T. de Bakker1,2,6† Toon A. B. van Veen2*†
Toon A. B. van Veen2*†
Background: Ventricular remodeling increases the propensity of ventricular tachyarrhythmias and sudden death in patients. We studied the mechanism underlying these fatal arrhythmias, electrical and structural cardiac remodeling, as well as arrhythmogeneity during early, compensated hypertrophy in a rat model of chronic pressure overload.
Methods: Twenty-six Wistar rats were subjected to transverse aortic constriction (TAC) (n = 13) or sham operation (n = 13). Four weeks postoperative, echo- and electrocardiography was performed. Epicardial (208 or 455 sites) and transmural (30 sites) ventricular activation mapping was performed on Langendorff perfused hearts. Subsequently, hearts were processed for (immuno)histological and molecular analyses.
Results: TAC rats showed significant hypertrophy with preserved left ventricular (LV) function. Epicardial conduction velocity (CV) was similar, but more dispersed in TAC. Transmural CV was slowed in TAC (37.6 ± 2.9 cm s−1) compared to sham (58.5 ± 3.9 cm s−1; P < 0.01). Sustained polymorphic ventricular tachycardias were induced from LV in 8/13 TAC and in 0/13 sham rats. During VT, electrical activation patterns showed variable sites of earliest epicardial activation and altering sites of functional conduction block. Wandering epicardial reentrant activation was sporadically observed. Collagen deposition was significantly higher in TAC compared to sham, but not different between arrhythmogenic and non-arrhythmogenic TAC animals. Connexin43 (Cx43) expression was heterogeneous with a higher prevalence of non-phosphorylated Cx43 in arrhythmogenic TAC animals.
Conclusion: In TAC rats with compensated cardiac hypertrophy, dispersion of conduction correlated to arrhythmogenesis, an increased heterogeneity of Cx43, and a partial substitution with non-phosphorylated Cx43. These alterations may result in the increased vulnerability to polymorphic VTs.
During cardiac pathophysiology, both the compensated and decompensated heart is characterized by structural and electrical remodeling, which is accompanied by a high vulnerability to ventricular arrhythmias and sudden cardiac death (1–4). Cardiac remodeling may be associated with conduction slowing, which increases the propensity for reentrant arrhythmias (5). Not necessarily related to conduction slowing, in addition, local heterogeneous differences in impulse conduction due to spatial differences in remodeling may lead to increased dispersion of impulse conduction, which is proarrhythmogenic as well (6, 7).
Impulse conduction in ventricular myocardium is primarily determined by 1) sodium current (INa), mainly directed by the Nav1.5 sodium ion channel protein, 2) tissue architecture, primarily determined by collagen deposition and cell size, and 3) cell-to-cell coupling, which is mainly mediated in ventricular myocardium by channels that is composed of the gap junction protein connexin43 (Cx43) (5).
Currently, it is unknown to what extent abnormal conduction characteristics contribute to arrhythmogenesis in early, compensated stages of cardiac remodeling. Several studies have shown that abnormal distribution of Cx43 is a key feature of the hypertrophied heart (8, 9). In that respect, heterogeneous distribution of Cx43 resulted in conduction defects and ventricular arrhythmias in a chimeric mouse model (10). In more advanced stages of cardiac disease, heterogeneous distribution of Cx43 has strongly been associated with the occurrence of arrhythmias in both patients and mice (11, 12). Adding to this aspect of heterogeneous remodeling, Cx43 is a phosphoprotein with several phosphorylation states, which determine its gating properties. Both phosphorylating protein kinases and dephosphorylating protein phosphatases are modulators of Cx43 phosphorylation. Dephosphorylation of Cx43 accounts for reduced intercellular coupling that may slow conduction and enhance arrhythmia susceptibility (13–16). It is not clear, however, whether heterogeneous expression of Cx43 is accompanied by, or even results from, a heterogeneous process of Cx43 dephosphorylation. Therefore, we hypothesized that in compensated stages of cardiac remodeling, inhomogeneous dephosphorylation of Cx43 and heterogeneous spatial differences in Cx43 distribution would primarily lead to more dispersed ventricular impulse conduction and an increased propensity for ventricular arrhythmias. In order to acquire in-depth knowledge in this respect, the current study addressed our hypothesis through determination of characteristics of epicardial and transmural impulse propagation and distribution/status of Cx43 gap junction channels.
Animal experiments were performed according to institutional guidelines and the Dutch Experiments on Animals Act and approved by the local Animal Experiments Committee. The study complies with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (No. 85-23, revised in 1996). Wistar rats were housed at 21°C and 60% humidity with an artificial 12:12 h light–dark cycle and were fed with ad libitum standard chow and water.
Twelve-week-old female Wistar rats were transverse aortic constriction (TAC) (n = 13) or sham (n = 13) operated, as described previously (11).
To study compensated remodeling, 4 weeks after TAC or sham operation, echocardiography was performed to assess 1) the extent of aortic constriction, 2) left ventricular (LV) dimensions and wall thickness, and 3) LV function. After a mild isoflurane anesthesia, rats were placed in supine position to record an ECG.
Four weeks after operation, hearts were extracorporated for Langendorff perfusion, and extracellular electrograms were recorded with a 247 or 208 point multiterminal electrode (19 × 13 and 16 × 13 grid, respectively). Recordings of LV and right ventricle (RV) were made during stimulation from the center of the grid on LV at a basic cycle length (BCL) of 150 ms. To map ventricular arrhythmias in both ventricles simultaneously, in 4 of the 13 TAC and 5 of the 13 sham-operated rat hearts, simultaneous epicardial recordings were made from LV (19 × 13 grid) and RV (16 × 13 grid). In the remaining hearts, LV and RV were mapped sequentially with the 19 × 13 electrode.
Conduction velocity (CV) parallel (θL) and perpendicular (θT) to fiber direction was determined from the paced activation maps. Dispersion of epicardial conduction was assessed by calculating the maximal difference at each recording site with neighboring activation times (so-called phase differences). Local phase differences were plotted in a phase map, showing the spatial distribution of inhomogeneity’s in conduction, so-called dispersion in conduction. From each map, a total index of dispersion was calculated (17). Transmural CV (θTM, perpendicular to epicardium) was calculated from local activation times derived from electrograms recorded with three separate needle electrodes.
The susceptibility for arrhythmias was tested by programed stimulation from the center of the grid electrodes. Subsequently, hearts were divided into arrhythmogenic (TAC+) and non-arrhythmogenic (TAC−). Details on recording and analysis of electrograms, and testing of arrhythmia susceptibility were described previously (11).
Following electrical recordings during Langendorff perfusion, hearts were immediately frozen in liquid nitrogen and stored at −80°C. TAC (n = 8) and sham (n = 6) hearts were sectioned serially in 10 μm slices parallel to the epicardial surface. All slices were mounted on AAS (aminopropyltriethoxysilane)-coated glass slides.
The amount of fibrosis was quantified by picro Sirius Red staining. From each stained section, up to 40 digital photomicrographs from different areas were taken, depending upon section size. For fibrosis content, the percentage of picro Sirius Red staining in each photomicrograph was determined. For each section, staining in all photomicrographs was averaged.
Cell size was determined in transversal sections after labeling of sections with anti-dystrophin. Photomicrographs (more than five photomicrographs of different areas from each section) of anti-dystrophin labeled sections at 20× magnification were used to measure transversal cell surface area (TCSA) in square micrometer. Sodium channel expression by anti-Nav1.5 antibody was determined, as described previously (18).
The protein level and distribution of total Cx43 were determined in parallel sections by immunohistochemistry, as described previously (19). The protein level and distribution of non-phosphorylated Cx43 were revealed by a monoclonal mouse anti-Cx43 antibody (Zymed 13-8300). Immunostaining intensity was quantified, as described previously (11). Heterogeneity of Cx43 distribution was quantified in 1280 × 1024 pixel size photomicrographs of the Cx43 labeling. These photomicrographs were transformed into eight-bit black (Cx43) and white (background) pictures. A custom written script in Matlab (The MathWorks Inc., USA) was applied to assess the shortest distance for each Cx43 pixel to its neighboring Cx43 pixel. SD of all shortest distances of all Cx43 pixels was used as a measure of spatial Cx43 heterogeneity.
Total Cx43 protein levels were determined by Western blot analysis, as described previously (19). A Cx43-specific C-terminal antibody was utilized to visualize the unphosphorylated isoform of Cx43 (Cx43-P0) (20).
Data were analyzed using SPSS software (SPSS 17, Chicago, IL, USA). Statistical significance of differences was evaluated by unpaired Student’s t-test or ANOVA followed by post hoc Bonferroni test as appropriate. Arrhythmia incidence was compared with Fisher’s exact test. Two-sided P values <0.05 were considered statistically significant. Data are expressed as mean ± SEM.
After 4 weeks of pressure overload, TAC rats showed hypertrophy with preserved LV function. Diastolic LV posterior wall thickness and heart/body weight ratio increased significantly (Table S1 in Supplementary Material). Fractional shortening improved in this early compensated hypertrophic state. QRS duration was significantly longer in TAC (20.4 ± 0.38 ms) compared to sham-operated rats (17.8 ± 0.44 ms; P < 0.01).
In 8/13 TAC rats (62%), polymorphic ventricular tachyarrhythmias (pVT) were induced by premature or burst stimulation compared to 0/13 in sham rats (P = 0.001). During VT, epicardial electrical activation patterns of LV during VT showed variable sites of earliest epicardial activation of consecutive beats, and altering sites of functional conduction block (Figure 1). In contrast, RV epicardial activation maps showed broad and fast activation fronts. Wandering epicardial reentrant activation was sporadically observed. Figure 2 shows a rare example of a single reentrant pathway on the LV epicardium, which starts 520 ms after induction of VT and completes at t = 580 ms. However, following up on this, the area where reentry occurred was activated almost simultaneously during the next beat (electrograms at electrode sites 2–7 occur almost simultaneously).
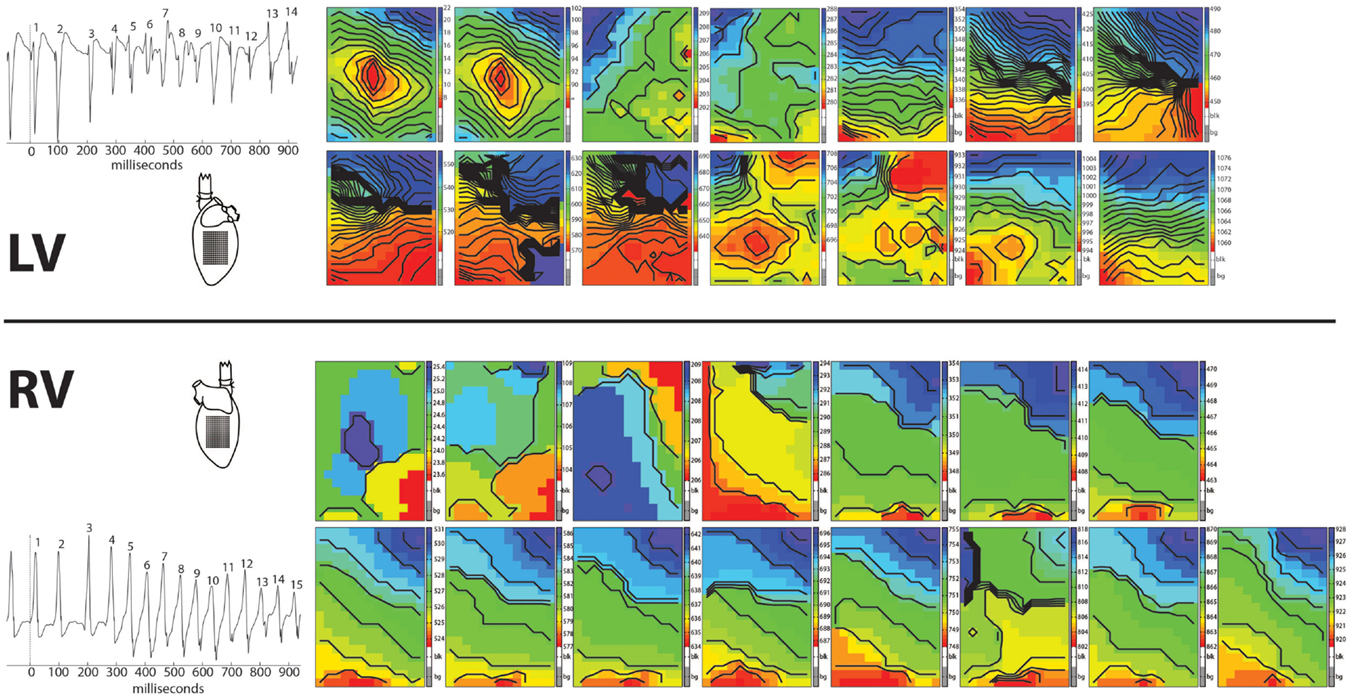
Figure 1. Arrhythmia. Epicardial activation maps of a polymorphic tachycardia induced in the left ventricle of a Langendorff perfused TAC rat heart. Simultaneously recorded activation maps of LV and RV are displayed in chronical order to show the evolution of the arrhythmia. Tachycardia was induced by one premature stimulus applied at the center of the recording electrode on LV. First two activation maps are from the last basic and the premature stimulus. Note the centrifugal spread of activation of these two maps of LV (red earliest activation, blue latest activation). Activation patterns of LV are more complex then of RV, showing multiple and varying areas of conduction block/slowing (areas of crowding isochronal lines). In contrast, activation patterns in RV show broad fronts, sweeping over the epicardium at high speed.
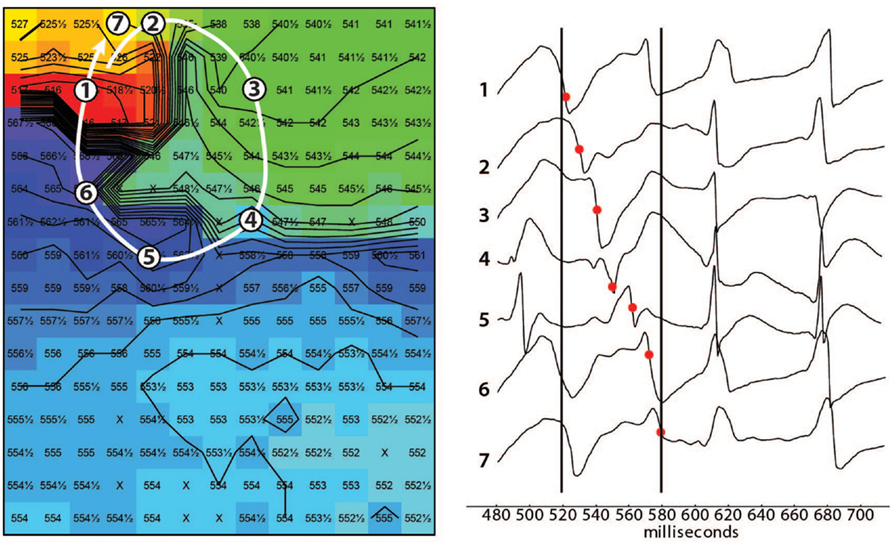
Figure 2. Epicardial activation map showing reentry. Example of reentrant activation (white arrow) at the LV epicardium occurring 520 ms after induction of a tachycardia by a single premature stimulus. Note the sequence of activation indicated by the black dots in the tracings. The following beat activates the area where reentry occurred almost simultaneously (electrograms after the right black line occur almost simultaneously). Black lines are isochronal lines at distances of 2 mm, numbers are activation times with respect to the premature stimulus that induced the tachycardia. Reentry occurred frequently but was never stable.
Epicardial CV longitudinal and transversal to fiber direction was not different between sham- and TAC-operated animals, neither for RV nor LV (Table 1). However, transmural CV was slowed in TAC-operated rats compared to sham-operated animals (37.6 ± 2.9 vs. 58.5 ± 3.9 cm s−1 respectively; P < 0.01). ERP in both RV and LV was longer in TAC compared to sham-operated animals, but the difference did not reach significance (Table 1).
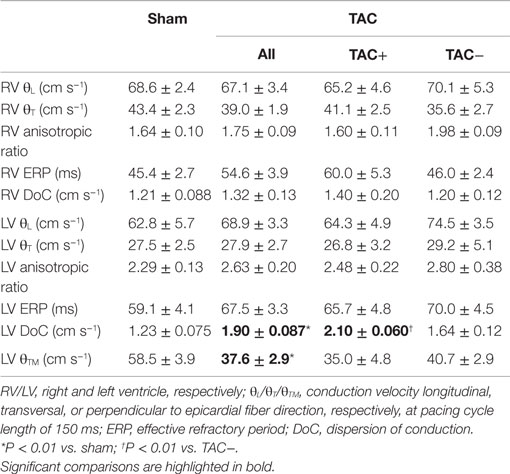
Table 1. Mapping data: data of electrical mapping study during Langendorff perfusion.
To determine parameters associated with arrhythmogeneity, we measured cell size, collagen content, sodium channel (SCN5a), and Cx43 protein levels and distribution. In TAC rats, TCSA was significantly larger in both LV (12%) and RV (11%) compared to sham rats (Table 2). The amount of collagen was significantly more abundant in LV (63%) and RV (156%) of TAC rats compared to sham-operated animals (Figure 3; Table 2). Sodium channel expression as determined by quantification of Nav1.5 immunofluorescence was not different between TAC and sham (Table 2).
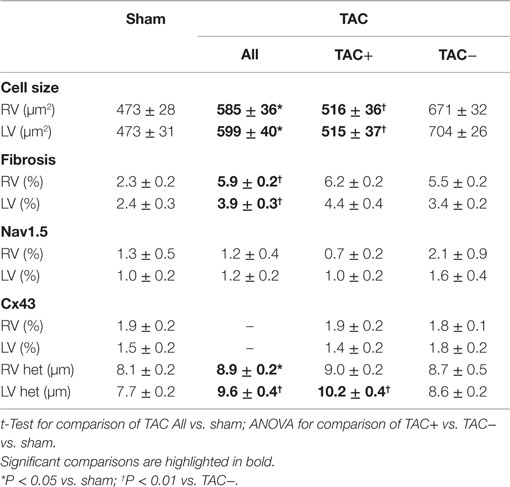
Table 2. Cellular data and fibrosis: data of cell size measured as transversal cell surface area (TCSA), percentage of interstitial fibrosis, Nav1.5 sodium channel, Cx43 gap junction channel, and its heterogeneity (Cx43 het) as derived from immunofluorescent signals.
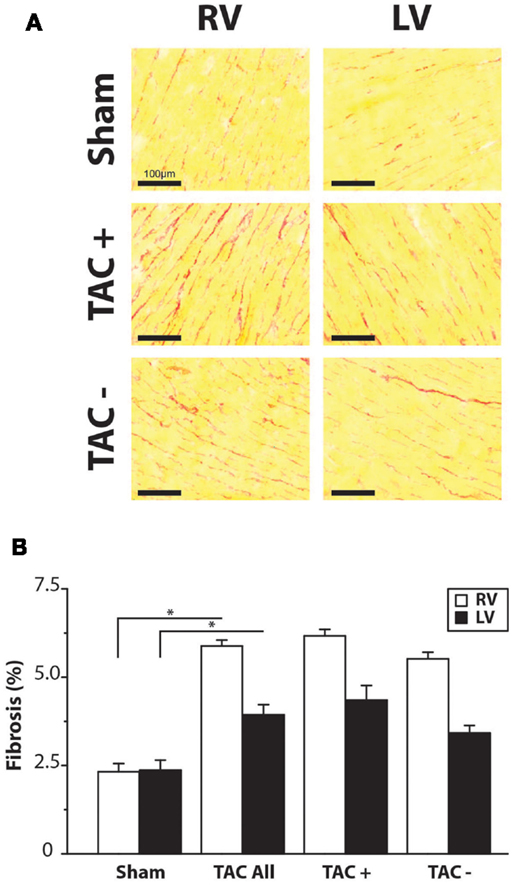
Figure 3. Fibrosis. (A) Representative examples of Sirius Red staining. Right and left ventricle (RV/LV) of sham and TAC rats. (B) Quantifications of collagen content in percentage of total slide area. Collagen content was significantly higher in TAC compared to sham animals. There was no difference between TAC+ and TAC−.
Immunolabeling of Cx43 protein displayed a significantly more heterogeneous spatial distribution in TAC compared to sham-operated animals (Table 2; Figures 4A,B). Some areas exhibited normal homogeneous Cx43 labeling, whereas in other regions, Cx43 labeling was heterogeneous or even nearly absent. In addition, Western blot analysis of total Cx43 protein levels revealed a lower level of Cx43 in TAC rats (Figure 4C, and quantified after normalization to total protein in Figure 4D). However, when total Cx43 protein signals in the different groups were corrected for their respective TCSA values (cell size, Table 2), this reduction could not be confirmed (Figure 4E).
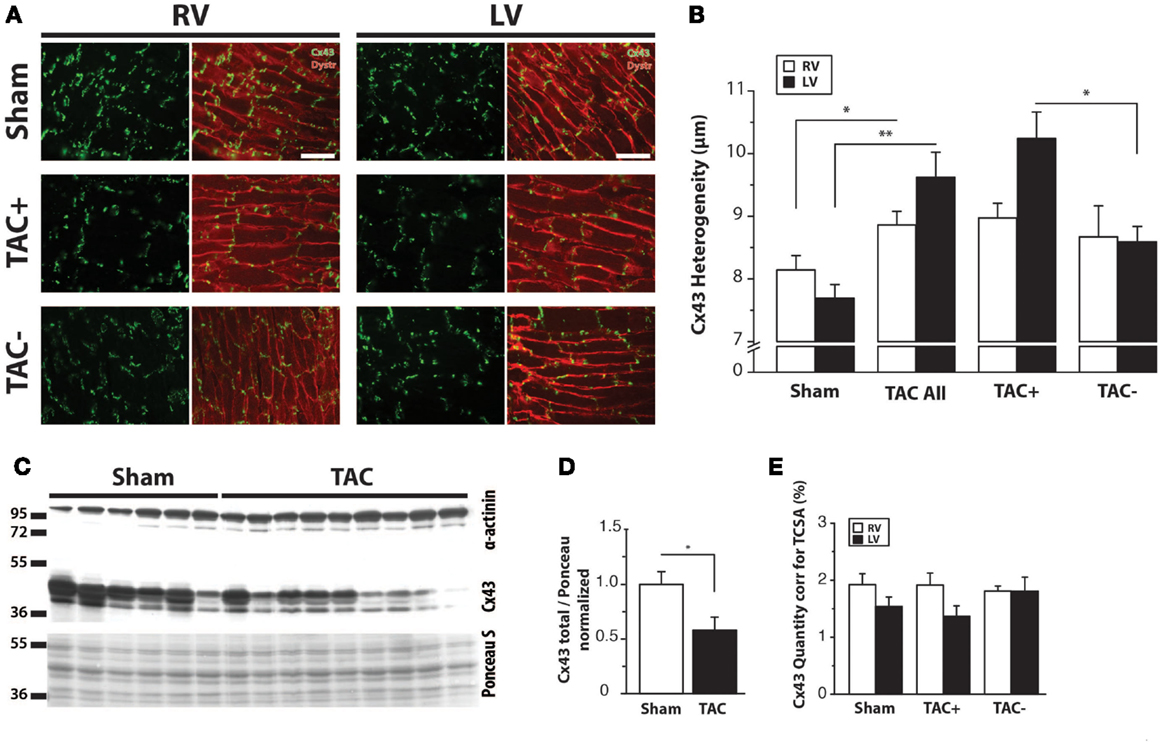
Figure 4. Cx43 distribution. (A) Typical examples of anti-Cx43 labeling along with merged photographs of both anti-Cx43 and anti-dystrophin labeling from right and left ventricle (RV/LV) of sham- and TAC-operated rats with or without arrhythmia susceptibility (TAC+/TAC−). Scale bar = 100 μm. (B) Quantifications of heterogeneity of Cx43 distribution are shown (see also Table 2). *P < 0.05 vs. same ventricle; **P < 0.01 vs. same ventricle. (C) Western blot showing Cx43 including the bands for different phosphorylation states in LV myocardial samples from TAC and sham rats. Ponceau S blot is for validation of equal loading. (D) Quantifications of total Cx43 expression, normalized for sham. (E) Quantifications of total Cx43 expression, normalized for cell size (TCSA, Table 2).
Regarding the heterogeneity of Cx43 remodeling, Figure 5A shows that in areas where total Cx43 signals were reduced or apparently even absent, the non-phosphorylated isoform of Cx43 (Cx43-NP) appeared still present and predominant. This observation was further substantiated via Western blot analysis, which revealed a significantly increased level of Cx43-NP in TAC animals (Figures 5B,C).
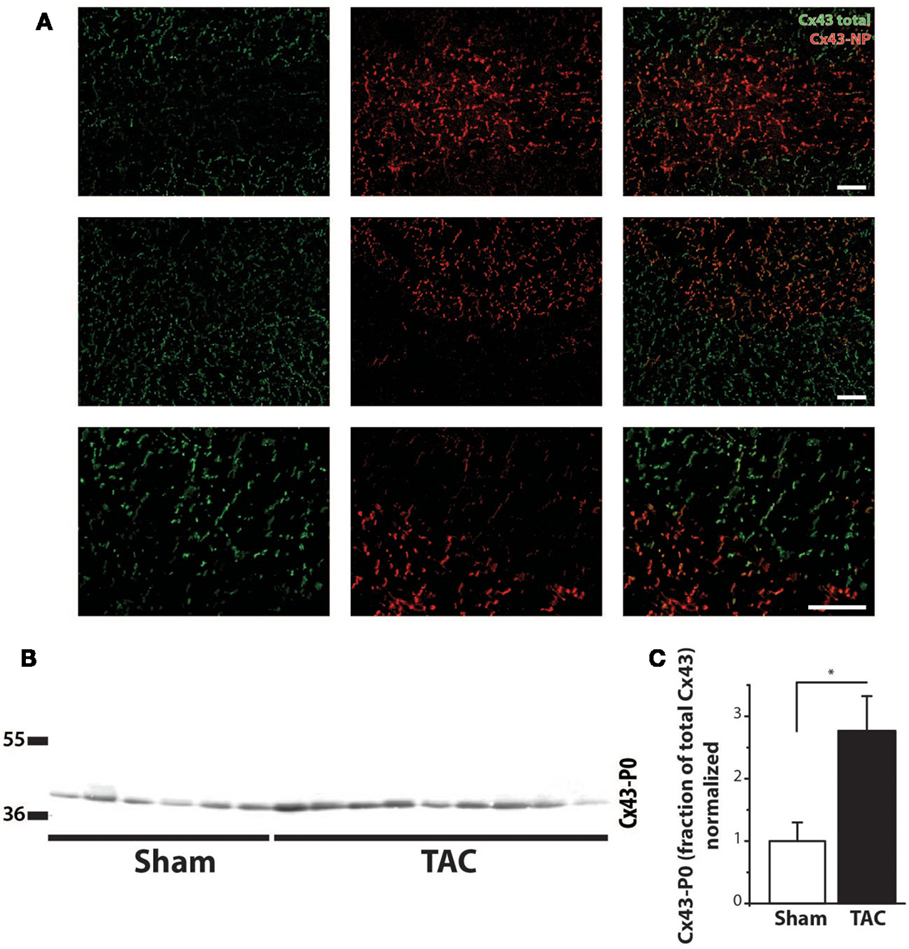
Figure 5. Cx43 dephosphorylation. (A) Typical examples of immunolabeling with anti-Cx43 and anti-non-phosphorylated Cx43 (Cx43-NP) along with merged photographs of both labelings from LV of two different TAC+ rats (upper two rows). Lowest row shows a higher magnification of a border zone between normal Cx43 expression and diminished Cx43 with substitution by Cx43-NP. (B) Western blot of non-phosphorylated Cx43 expression (Cx43-P0) in the same LV myocardial samples as in Figure 4C. (C) Quantifications of Cx43-NP expression, normalized to sham. Scale bar = 100 μm. *P < 0.05 vs. sham.
To determine the relevance of these changes for arrhythmogenesis, we compared TAC rats with (TAC+) and without (TAC−) arrhythmias. QRS duration was not different between TAC+ and TAC− rats (Table S1 in Supplementary Material). Furthermore, TAC+ rats and TAC− rats did not show any difference in ventricular function, although calculated LV mass was significantly lower in TAC+ compared to TAC− (340.7 ± 32.1 vs. 443.9 ± 27.4 μg; P = 0.028; Table S1 in Supplementary Material). In line with that finding, TCSA showed that cell size in LV and RV was significantly smaller in TAC+ as compared to TAC− animals (Table 2).
On the other hand, no significant difference existed in the collagen content of either LV or RV between TAC+ and TAC− animals (Table 2). The same accounted for the protein levels of total Cx43 and the sodium channel (Figure 4E; Table 2).
Although transmural CV was lower in TAC+ compared to TAC− animals (35.0 ± 4.8 vs. 40.7 ± 2.9 cm s−1, Table 1), this difference did not reach significance. On the other hand, dispersion of conduction (DoC) in LV was significantly higher in TAC hearts and did differ between TAC+ and TAC− animals, with a higher DoC in TAC+ (2.10 ± 0.060 cm s−1) compared to TAC− rats (1.64 ± 0.12 cm s−1; P < 0.01, Table 1). Fitting to these data on DoC, spatial Cx43 expression was most heterogeneous in TAC+, as compared to TAC− rats (P < 0.05; Table 2; Figure 4B). Additionally, areas with an apparently reduced Cx43 protein level showed an increased fraction of non-phosphorylated Cx43 (Figure 5A), but this shift was not significantly different between TAC+ and TAC−. Finally, in LV but not RV, dispersion in conduction was linearly correlated to the level of Cx43 heterogeneity (R2 = 0.769; P < 0.001; Figure 6).
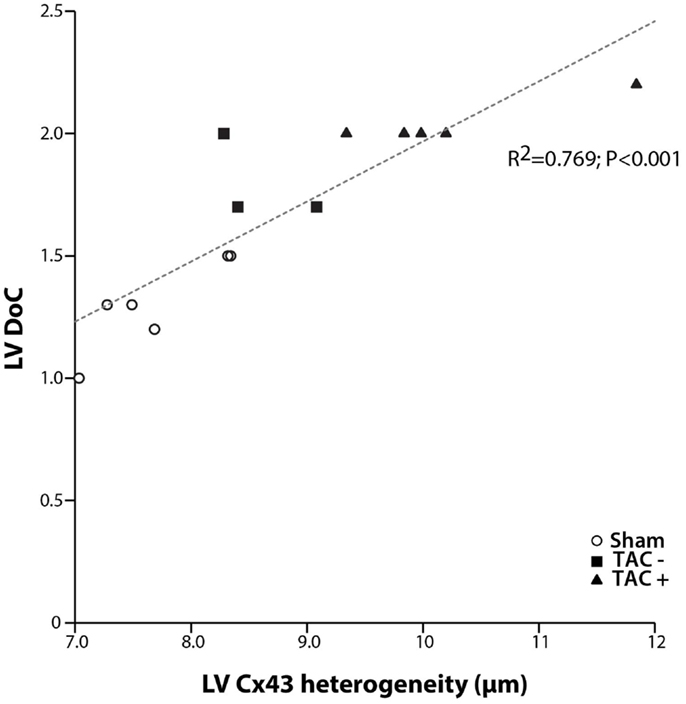
Figure 6. Dispersion of conduction and Cx43 heterogeneity. Graph showing the relation between dispersion of conduction (DoC) and Cx43 heterogeneity. A clear linear relation is shown with a high correlation coefficient (R2 = 0.769; P < 0.001). With increasing spatial heterogeneity of Cx43 distribution, dispersion of conduction becomes higher.
The main and novel results from this study are 1) polymorphic tachycardias can be induced by programed stimulation from the LV in 62% of rat hearts in which compensated hypertrophy was induced by TAC. 2) Activation patterns during polymorphic tachycardias were more complex in LV than RV showing areas of conduction slowing and conduction block, unstable reentrant circuits, and multiple sites of epicardial breakthrough in LV. Wave fronts in RV were broad and propagated fast suggesting a prominent role for the LV in the detected arrhythmias. 3) Transmural conduction slowing with a generally preserved epicardial CV. 4) Heterogeneous Cx43 expression is associated with spatially dispersed impulse conduction, which enhances vulnerability to polymorphic ventricular tachycardias. 5) The heterogeneous Cx43 expression is correlated with a shift toward predominance of the non-phosphorylated isoform in regions with reduced Cx43 expression.
The mapping data suggest that initiation of ventricular tachycardias by premature stimulation in this rat model of compensated hypertrophy is based on a focal mechanism rather than on reentry. However, conduction delay and epicardial reentrant activation were observed in later phases of the tachycardias, which indicate that the reentry might play a role in perpetuation of the arrhythmia. No support for a reentrant mechanism in the initiation of the tachycardia was found, since there was no evidence for unidirectional conduction block near the site of stimulation during initiation of the tachycardia. Earliest epicardial activation of the initial ectopic beats of the VTs always occurred distant from the site of stimulation.
A focal initiation of VT may result from triggered activity arising from either early- or delayed-afterdepolarizations (EADs or DADs). Action potential duration was increased in the hypertrophied hearts compared to hearts of sham-operated animals, which enhances the possibility for triggered activity (21). In patients with end-stage dilated cardiomyopathy, a focal mechanism could be associated with a polymorphic ventricular tachycardia (22). In our study, the TAC hearts displayed an increased collagen deposition, a heterogeneously decreased Cx43 expression, and a reduced phosphorylation status of Cx43, which most likely results in a reduced electrical coupling. Reduced electrical coupling not only increases the possibility for reentrant arrhythmias but also favors the occurrence of focal activity (23), and, as explained, both mechanisms might have been active in this model of hypertrophy.
The occurrence of focal activity and reduced phosphorylation status of Cx43 are compatible with the observation made by Jin et al., who found afterdepolarizations as trigger for arrhythmias using optical mapping in a similar model (24).
Furthermore, we observed that following ectopic beats had longer activation times (latest − earliest activation of a beat within the recording area), which increased from 5 to 40 ms within a few beats (Figure 1). Isochrones became more crowded, indicative for a rundown of the CV. In addition, zones of conduction block arose. These changes set up the possibility for reentry, which we indeed observed in LV, as illustrated in Figure 2.
The high susceptibility to polymorphic VTs in rats with compensated hypertrophy was linked to increased dispersion of LV CV and heterogeneous Cx43 expression.
The relationship between the occurrence of VT and dispersion in Cx43 expression has been shown in patients (11, 12), dogs (25, 26), rabbits (6), and mice (10, 11). Heterogeneous electrical uncoupling in our model is associated with increased electrical heterogeneities in conduction and repolarization, which may create a substrate for reentry. This implies that the polymorphic character of VTs in our TAC rats is most likely due to inhomogeneous conduction, which by itself is caused by dispersed expression of Cx43. Indeed, the increased DoC in LV highly paralleled to increased Cx43 heterogeneity (R2 = 0.769, Figure 6).
It is unclear to what extent the downregulation of Cx43 or the increased levels of fibrosis potentiate the effects of heterogeneous Cx43 expression on dispersed conduction and polymorphic VTs.
Several studies in transgenic mice with reduced Cx43 have shown that a moderate and homogeneous decrease of 50% in Cx43 expression by itself does not necessarily lead to conduction impairment (27, 28). In our TAC rats, Cx43 expression was down regulated by 42%, which did not lead to significant impairment of epicardial CV. The observed increase in QRS duration is regarded to be caused by the increase in ventricular mass (Table S1 in Supplementary Material) in a similar fashion as we showed before (29). LV transmural conduction slowing was associated with increased LV fibrosis, which is in agreement to a mild reduction in transversal conduction as observed in other studies (30). In addition, the heart may become more sensitive to spatial differences in intercellular coupling in the background of other impairments in conduction parameters. It is known that the heart has “conduction reserve,” as result of which the heart is able to maintain normal conduction velocities even under conditions of moderate changes in Cx43, SCN5a, or collagen levels (18, 31, 32). Moderate changes in Cx43 or collagen, however, may reduce this conduction reserve and sensitize the heart to local differences in electrical coupling and thus to local differences in CV.
This heterogeneous conduction substrate resulted in the occurrence of pVT. Polymorphic ventricular tachycardia in the healthy, isolated rabbit heart has been shown to result from either a single or paired (“figure-of-eight”) non-stationary scroll wave (33). Polymorphic VT in patients after myocardial infarction (MI) has been shown to be triggered and possibly maintained by activity originating from the distal Purkinje system localized in the MI border zone and could successfully be abolished by radiofrequency ablation (34). In TdP, triggered activity and wandering reentrant activation have been proposed to explain its polymorphic character (35).
As mentioned before, we observed that areas with normal Cx43 expression neighbored zones with apparently almost no Cx43 immunofluorescent signal. It appeared, however, that in these areas with poor levels of total Cx43, a shift occurred toward non-phosphorylated Cx43. Phosphorylation of Cx43 is a key mechanism in its functional gating properties. In general, dephosphorylation of Cx43 leads to impaired coupling (15), which results in slowed conduction and the genesis of unidirectional block by which the susceptibility to reentrant arrhythmias is increased (16, 36). However, as described before, reduced electrical coupling may also unmask ectopic foci, which may serve as focal drivers of arrhythmias (23).
Increased collagen amount also gives rise to reduced cell-to-cell coupling, impaired conduction, and propensity for triggered activation. Conduction slowing by (interstitial) fibrosis mainly occurs in the transversal direction, while the effect on conduction in longitudinal direction is less prominent. This is explained by the fact that interstitial fibrosis is most often oriented parallel to the fiber direction, thereby separating cardiomyocytes on the long side. Myocardial fibers are oriented parallel to the epicardium and their direction rotates over an angle of about 120° from the epicardial plane to the endocardial plane (37). Because of this fiber rotation, measurements of transversal and longitudinal epicardial conduction velocities not only reflect epicardial conduction velocities but also those of deeper layers in different fiber angles (37). However, during transmural conduction, the activation front is nearly perpendicular to fiber orientation, and most sensitive to increased levels of interstitial fibrosis. Indeed, in our model, transmural CV was significantly reduced. However, although collagen expression was significantly increased in TAC rats, no differences were observed between TAC+ and TAC− rats. As such, fibrosis was not identified as a discriminating factor explaining the observed arrhythmogeneity. As mentioned before, increased levels of fibrosis itself may contribute to a compromised conduction reserve and increased propensity for polymorphic VTs.
We have shown a significant correlation between heterogeneous Cx43 expression, on one hand, and dispersed impulse conduction and enhanced vulnerability to polymorphic ventricular tachycardias, on the other hand. The heterogeneous Cx43 expression was furthermore associated with a shift toward the non-phosphorylated isoform of Cx43 in the regions with reduced Cx43 expression.
Conducting the experiments: MB, ME, MS, BK, SA, HR, and TV; design of the study: MV, HR, JB, and TV; data analysis: MB, MFAB, MV, HR, JB, and TV; writing and critical revision of the manuscript: MB, MFAB, MV, HR, JB, and TV.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by the Netherlands Heart Foundation (Grant numbers M96.001 and 2003B128). We also acknowledge the support from the Netherlands CardioVascular Research Initiative: the Dutch Heart Foundation, Dutch Federation of University Medical Centres, the Netherlands Organisation for Health Research and Development and the Royal Netherlands Academy of Sciences (CVON-PREDICT).
The Supplementary Material for this article can be found online at http://journal.frontiersin.org/article/10.3389/fcvm.2016.00005
1. Levy D, Garrison RJ, Savage DD, Kannel WB, Castelli WP. Prognostic implications of echocardiographically determined left ventricular mass in the Framingham heart study. N Engl J Med (1990) 322:1561–6. doi: 10.1056/NEJM199005313222203
2. Ghali JK, Kadakia S, Cooper RS, Liao YL. Impact of left ventricular hypertrophy on ventricular arrhythmias in the absence of coronary artery disease. J Am Coll Cardiol (1991) 17:1277–82. doi:10.1016/S0735-1097(10)80135-4
3. Zipes DP, Wellens HJ. Sudden cardiac death. Circulation (1998) 98:2334–51. doi:10.1161/01.CIR.98.21.2334
4. van Opstal JM, Verduyn SC, Leunissen HD, de Groot SH, Wellens HJ, Vos MA. Electrophysiological parameters indicative of sudden cardiac death in the dog with chronic complete AV-block. Cardiovasc Res (2001) 50:354–61. doi:10.1016/S0008-6363(01)00226-7
5. Kleber AG, Rudy Y. Basic mechanisms of cardiac impulse propagation and associated arrhythmias. Physiol Rev (2004) 84:431–88. doi:10.1152/physrev.00025.2003
6. Wiegerinck RF, van Veen TA, Belterman CN, Schumacher CA, Noorman M, de Bakker JM, et al. Transmural dispersion of refractoriness and conduction velocity is associated with heterogeneously reduced connexin43 in a rabbit model of heart failure. Heart Rhythm (2008) 5:1178–85. doi:10.1016/j.hrthm.2008.04.026
7. Jansen JA, Noorman M, Musa H, Stein M, de Jong S, van der Nagel R, et al. Reduced heterogeneous expression of cx43 results in decreased nav1.5 expression and reduced sodium current that accounts for arrhythmia vulnerability in conditional cx43 knockout mice. Heart Rhythm (2012) 9:600–7. doi:10.1016/j.hrthm.2011.11.025
8. Uzzaman M, Honjo H, Takagishi Y, Emdad L, Magee AI, Severs NJ, et al. Remodeling of gap junctional coupling in hypertrophied right ventricles of rats with monocrotaline-induced pulmonary hypertension. Circ Res (2000) 86:871–8. doi:10.1161/01.RES.86.8.871
9. Qu J, Volpicelli FM, Garcia LI, Sandeep N, Zhang J, Marquez-Rosado L, et al. Gap junction remodeling and spironolactone-dependent reverse remodeling in the hypertrophied heart. Circ Res (2009) 104:365–71. doi:10.1161/CIRCRESAHA.108.184044
10. Gutstein DE, Morley GE, Vaidya D, Liu F, Chen FL, Stuhlmann H, et al. Heterogeneous expression of gap junction channels in the heart leads to conduction defects and ventricular dysfunction. Circulation (2001) 104:1194–9. doi:10.1161/hc3601.093990
11. Boulaksil M, Winckels SK, Engelen MA, Stein M, van Veen TA, Jansen JA, et al. Heterogeneous connexin43 distribution in heart failure is associated with dispersed conduction and enhanced susceptibility to ventricular arrhythmias. Eur J Heart Fail (2010) 12:913–21. doi:10.1093/eurjhf/hfq092
12. Kitamura H, Ohnishi Y, Yoshida A, Okajima K, Azumi H, Ishida A, et al. Heterogeneous loss of connexin43 protein in nonischemic dilated cardiomyopathy with ventricular tachycardia. J Cardiovasc Electrophysiol (2002) 13:865–70. doi:10.1046/j.1540-8167.2002.00865.x
13. Burstein B, Comtois P, Michael G, Nishida K, Villeneuve L, Yeh YH, et al. Changes in connexin expression and the atrial fibrillation substrate in congestive heart failure. Circ Res (2009) 105:1213–22. doi:10.1161/CIRCRESAHA.108.183400
14. Ai X, Pogwizd SM. Connexin 43 downregulation and dephosphorylation in nonischemic heart failure is associated with enhanced colocalized protein phosphatase type 2A. Circ Res (2005) 96:54–63. doi:10.1161/01.RES.0000152325.07495.5a
15. Beardslee MA, Lerner DL, Tadros PN, Laing JG, Beyer EC, Yamada KA, et al. Dephosphorylation and intracellular redistribution of ventricular connexin43 during electrical uncoupling induced by ischemia. Circ Res (2000) 87:656–62. doi:10.1161/01.RES.87.8.656
16. Remo BF, Qu J, Volpicelli FM, Giovannone S, Shin D, Lader J, et al. Phosphatase-resistant gap junctions inhibit pathological remodeling and prevent arrhythmias. Circ Res (2011) 108:1459–66. doi:10.1161/CIRCRESAHA.111.244046
17. Lammers WJ, Schalij MJ, Kirchhof CJ, Allessie MA. Quantification of spatial inhomogeneity in conduction and initiation of reentrant atrial arrhythmias. Am J Physiol (1990) 259:H1254–63.
18. Stein M, van Veen TA, Remme CA, Boulaksil M, Noorman M, van Stuijvenberg L, et al. Combined reduction of intercellular coupling and membrane excitability differentially affects transverse and longitudinal cardiac conduction. Cardiovasc Res (2009) 83:52–60. doi:10.1093/cvr/cvp124
19. van Veen TA, van Rijen HV, Wiegerinck RF, Opthof T, Colbert MC, Clement S, et al. Remodeling of gap junctions in mouse hearts hypertrophied by forced retinoic acid signaling. J Mol Cell Cardiol (2002) 34:1411–23. doi:10.1006/jmcc.2002.2102
20. Sosinsky GE, Solan JL, Gaietta GM, Ngan L, Lee GJ, Mackey MR, et al. The c-terminus of connexin43 adopts different conformations in the Golgi and gap junction as detected with structure-specific antibodies. Biochem J (2007) 408:375–85. doi:10.1042/BJ20070550
21. Jin H, Lyon AR, Akar FG. Arrhythmia mechanisms in the failing heart. Pacing Clin Electrophysiol (2008) 31:1048–56. doi:10.1111/j.1540-8159.2008.01134.x
22. Pogwizd SM, McKenzie JP, Cain ME. Mechanisms underlying spontaneous and induced ventricular arrhythmias in patients with idiopathic dilated cardiomyopathy. Circulation (1998) 98:2404–14. doi:10.1161/01.CIR.98.22.2404
23. Wilders R, Wagner MB, Golod DA, Kumar R, Wang YG, Goolsby WN, et al. Effects of anisotropy on the development of cardiac arrhythmias associated with focal activity. Pflugers Arch (2000) 441:301–12. doi:10.1007/s004240000413
24. Jin H, Chemaly ER, Lee A, Kho C, Hadri L, Hajjar RJ, et al. Mechanoelectrical remodeling and arrhythmias during progression of hypertrophy. FASEB J (2010) 24:451–63. doi:10.1096/fj.09-136622
25. Poelzing S, Akar FG, Baron E, Rosenbaum DS. Heterogeneous connexin43 expression produces electrophysiological heterogeneities across ventricular wall. Am J Physiol Heart Circ Physiol (2004) 286:H2001–9. doi:10.1152/ajpheart.00987.2003
26. Akar FG, Spragg DD, Tunin RS, Kass DA, Tomaselli GF. Mechanisms underlying conduction slowing and arrhythmogenesis in nonischemic dilated cardiomyopathy. Circ Res (2004) 95:717–25. doi:10.1161/01.RES.0000144125.61927.1c
27. van Rijen HV, Eckardt D, Degen J, Theis M, Ott T, Willecke K, et al. Slow conduction and enhanced anisotropy increase the propensity for ventricular tachyarrhythmias in adult mice with induced deletion of connexin43. Circulation (2004) 109:1048–55. doi:10.1161/01.CIR.0000117402.70689.75
28. Morley GE, Vaidya D, Samie FH, Lo C, Delmar M, Jalife J. Characterization of conduction in the ventricles of normal and heterozygous cx43 knockout mice using optical mapping. J Cardiovasc Electrophysiol (1999) 10:1361–75. doi:10.1111/j.1540-8167.1999.tb00192.x
29. Wiegerinck RF, Verkerk AO, Belterman CN, van Veen TA, Baartscheer A, Opthof T, et al. Larger cell size in rabbits with heart failure increases myocardial conduction velocity and QRS duration. Circulation (2006) 113:806–13. doi:10.1161/CIRCULATIONAHA.105.565804
30. van Veen TA, Stein M, Royer A, Le Quang K, Charpentier F, Colledge WH, et al. Impaired impulse propagation in SCN5a-knockout mice: combined contribution of excitability, connexin expression, and tissue architecture in relation to aging. Circulation (2005) 112:1927–35. doi:10.1161/CIRCULATIONAHA.105.539072
31. Stein M, Boulaksil M, Engelen MA, Van Veen TAB, Hauer RN, de Bakker JM, et al. Conduction reserve and arrhythmias. Neth Heart J (2006) 14:113–6.
32. van Rijen HV, de Bakker JM, van Veen TA. Hypoxia, electrical uncoupling, and conduction slowing: role of conduction reserve. Cardiovasc Res (2005) 66:9–11. doi:10.1016/j.cardiores.2005.02.003
33. Gray RA, Jalife J, Panfilov A, Baxter WT, Cabo C, Davidenko JM, et al. Nonstationary vortexlike reentrant activity as a mechanism of polymorphic ventricular tachycardia in the isolated rabbit heart. Circulation (1995) 91:2454–69. doi:10.1161/01.CIR.91.9.2454
34. Szumowski L, Sanders P, Walczak F, Hocini M, Jais P, Kepski R, et al. Mapping and ablation of polymorphic ventricular tachycardia after myocardial infarction. J Am Coll Cardiol (2004) 44:1700–6. doi:10.1016/j.jacc.2004.08.034
35. Boulaksil M, Jungschleger JG, Antoons G, Houtman MJ, de Boer TP, Wilders R, et al. Drug-induced torsade de pointes arrhythmias in the chronic AV block dog are perpetuated by focal activity. Circ Arrhythm Electrophysiol (2011) 4:566–76. doi:10.1161/CIRCEP.110.958991
36. Quan W, Rudy Y. Unidirectional block and reentry of cardiac excitation: a model study. Circ Res (1990) 66:367–82. doi:10.1161/01.RES.66.2.367
Keywords: arrhythmia, sudden death, electrophysiology, hypertrophy
Citation: Boulaksil M, Bierhuizen MFA, Engelen MA, Stein M, Kok BJM, van Amersfoorth SCM, Vos MA, van Rijen HVM, de Bakker JMT and van Veen TAB (2016) Spatial Heterogeneity of Cx43 is an Arrhythmogenic Substrate of Polymorphic Ventricular Tachycardias during Compensated Cardiac Hypertrophy in Rats. Front. Cardiovasc. Med. 3:5. doi: 10.3389/fcvm.2016.00005
Received: 08 December 2015; Accepted: 18 February 2016;
Published: 02 March 2016
Edited by:
Shimon Rosenheck, Meir Medical Center, IsraelReviewed by:
Fadi G. Akar, Icahn School of Medicine at Mount Sinai, USACopyright: © 2016 Boulaksil, Bierhuizen, Engelen, Stein, Kok, van Amersfoorth, Vos, van Rijen, de Bakker and van Veen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Toon A. B. van Veen, YS5hLmIudmFudmVlbkB1bWN1dHJlY2h0Lm5s
†Jacques M. T. de Bakker and Toon A. B. van Veen contributed equally.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.