 Pietro Enea Lazzerini
Pietro Enea Lazzerini Pier Leopoldo Capecchi
Pier Leopoldo Capecchi Franco Laghi-Pasini
Franco Laghi-Pasini
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med. , 27 May 2015
Sec. Cardiac Rhythmology
Volume 2 - 2015 | https://doi.org/10.3389/fcvm.2015.00026
The long QT syndrome (LQTS), classified as congenital or acquired, is a multi-factorial disorder of myocardial repolarization predisposing to life-threatening ventricular arrhythmias, particularly torsades de pointes. In the latest years, inflammation and immunity have been increasingly recognized as novel factors crucially involved in modulating ventricular repolarization. In the present paper, we critically review the available information on this topic, also analyzing putative mechanisms and potential interplays with the other etiologic factors, either acquired or inherited. Accumulating data indicate inflammatory activation as a potential cause of acquired LQTS. The putative underlying mechanisms are complex but essentially cytokine-mediated, including both direct actions on cardiomyocyte ion channels expression and function, and indirect effects resulting from an increased central nervous system sympathetic drive on the heart. Autoimmunity represents another recently arising cause of acquired LQTS. Indeed, increasing evidence demonstrates that autoantibodies may affect myocardial electric properties by directly cross-reacting with the cardiomyocyte and interfering with specific ion currents as a result of molecular mimicry mechanisms. Intriguingly, recent data suggest that inflammation and immunity may be also involved in modulating the clinical expression of congenital forms of LQTS, possibly triggering or enhancing electrical instability in patients who already are genetically predisposed to arrhythmias. In this view, targeting immuno-inflammatory pathways may in the future represent an attractive therapeutic approach in a number of LQTS patients, thus opening new exciting avenues in antiarrhythmic therapy.
The QT interval indicates the duration of action potential (AP) in ventricles, which represents the sum of ventricular depolarization and repolarization. AP is caused by transmembrane flow of ions, including inward depolarizing currents mainly through sodium and calcium channels, and outward repolarizing currents mainly through potassium channels. More in details, six sequentially activated currents are fundamentally involved: the sodium current (INa), the transient outward current (Ito), the L(long-lasting)-type calcium current (ICaL), the rapid component of the delayed rectifier potassium current (IKr), the slow component of the delayed rectifier potassium current (IKs), and the inward rectifier potassium current (IK1) (Figure 1).
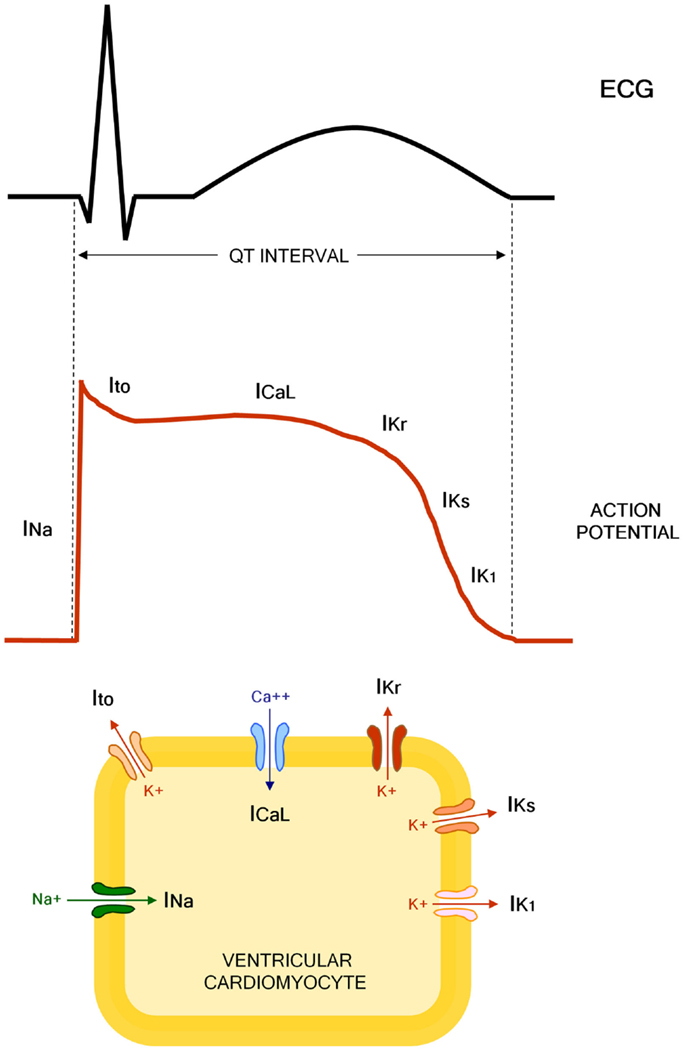
Figure 1. Molecular and electrophysiological basis of QT interval. INa, sodium current; Ito, transient outward current; ICaL, L(long-lasting)-type calcium current; IKr, rapid component of the delayed rectifier potassium current; IKs, slow component of the delayed rectifier potassium current; IK1, inward rectifier potassium current.
The Long QT Syndrome (LQTS) is a multi-factorial disorder of myocardial repolarization characterized by a prolonged corrected QT interval (QTc) on the electrocardiogram (ECG), and predisposing to life-threatening ventricular arrhythmias, particularly torsades de pointes (TdP) (1). The LQTS is traditionally classified as congenital or acquired (1, 2), even though it has becoming clear how in many cases the clinical phenotype is the result of a complex interaction of multiple etiologic factors operating concomitantly in the single patient (3).
Congenital LQTS, which can often be a lethal disorder (2), is caused by genetically determined abnormalities affecting directly or indirectly the function of specific ionic channels involved in ventricular AP, i.e., potassium (loss of function), sodium or calcium channels (gain of function) (4). To date, about 1000 mutations in 13 LQTS-susceptibility genes have been identified; however, only three of these genes, namely KCNQ1 (encoding KvLQT1 channel α-subunit, conducting IKs), KCNH2 (encoding hERG channel α-subunit, conducting IKr), and SCN5A (encoding Nav1.5 channel α-subunit, conducting INa), account by themselves for ~75% of all cases (5). The incidence of the congenital LQTS is not well known, although a recent clinical-genetic analysis on ~45,000 neonates suggests that it may be close to 1:2000 live births (6).
Acquired LQTS is much more prevalent than the congenital form, although its precise incidence and mortality impact in the general population are difficult to be estimated. Nevertheless, recent studies demonstrated that QTc prolongation is highly prevalent (up to 25–30% of hospitalized patients), also preliminarly suggesting that acquired LQTS may be as risky as congenital LQTS (7–10). More frequently, acquired LQTS represents an adverse effect of drugs or the result of electrolyte disturbances interfering with cardiomyocyte electrophysiology (1). In particular, the molecular basis of drug-induced LQTS almost exclusively involves the reduction of IKr through hERG-potassium channel blockade (11). Other currently recognized causes of acquired LQTS include structural heart diseases, bradyarrhythmias, endocrine disorders, liver diseases, nervous system injuries, HIV infection, starvation, hypothermia, and toxins (1, 12–14).
In the latest years, mounting evidence from basic and clinical studies strongly suggests that inflammation and immunity represent further important determinants of acquired LQTS. In the present paper, we review the available data on this topic, also analyzing putative mechanisms and potential interplays with the other etiologic factors, either acquired and inherited.
A list of the causes of acquired LQTS, also including inflammatory- and immune-mediated forms, is proposed in Table 1.

Table 1. Causes of acquired long QT syndrome.
Several lines of evidence support the hypothesis that inflammation, either cardiac or systemic, significantly impacts on QT interval duration and related risk of life-threatening arrhythmias (Table 2).
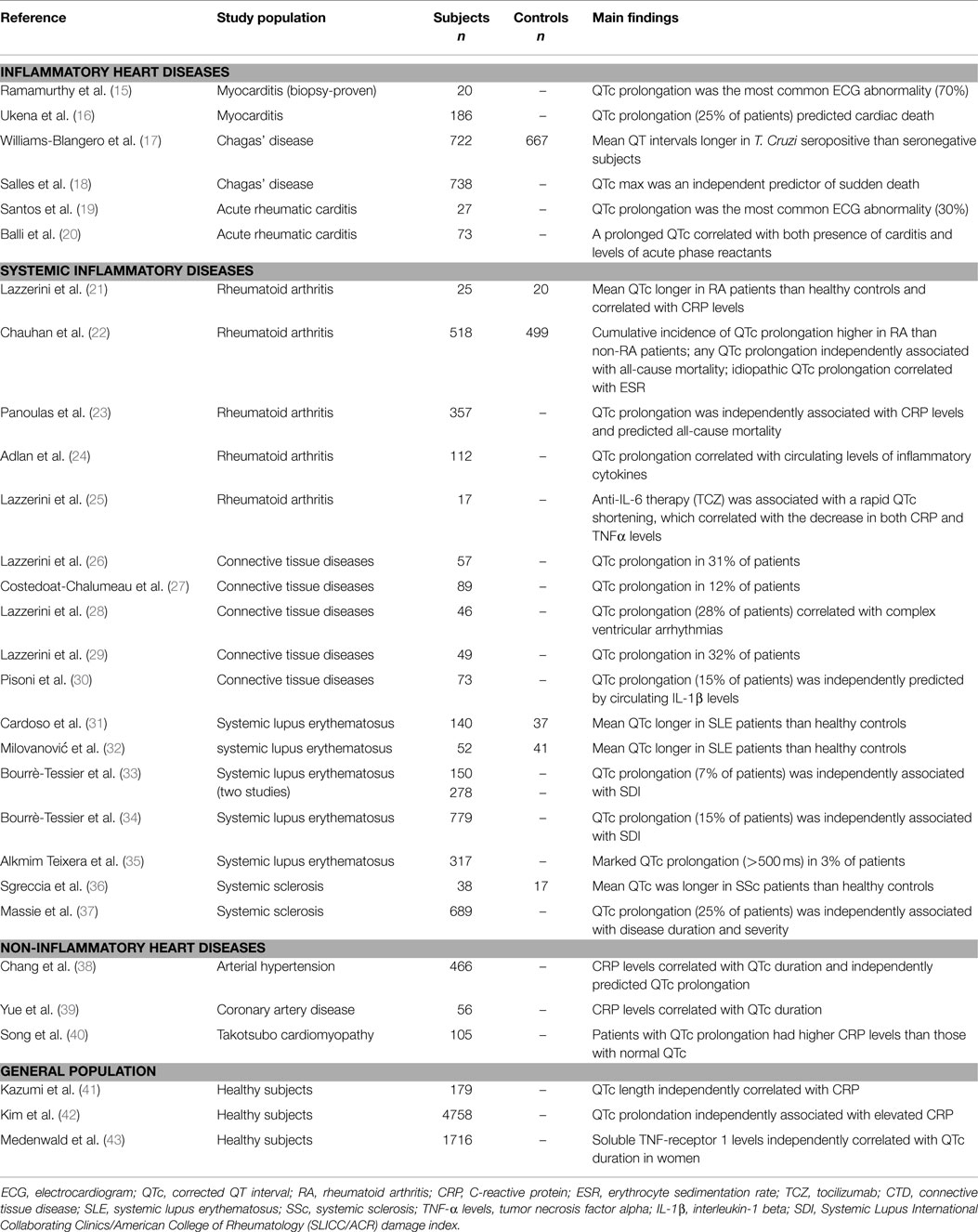
Table 2. Inflammation and QTc prolongation: clinical studies.
First of all, inflammatory heart diseases, particularly myocarditis, are frequently associated with QTc prolongation, in some cases of severe degree. Indeed, in myocarditis patients, a prolonged QTc was found to be the most common electrocardiographic abnormality observed (15), also associating with the occurrence of complex ventricular arrhythmias (44). Accordingly, in a large cohort of 186 patients with myocarditis, Ukena et al. (16) demonstrated that a QTc prolongation ≥440 ms was frequent (~25% of cases) and predicted a poor clinical outcome, including cardiac death. Moreover, several cases of marked QTc prolongation complicated with TdP have been reported in patients with acute infective carditis (myo/endocarditis), independently from the specific etiologic agent involved (45–57). A peculiar form of diffuse myocarditis is Chagas’s disease, triggered by the protozoan Trypanosoma cruzi, and then progressing for (auto)immune-mediated mechanisms (58). In this form, QTc prolongation represents a common and prognostically relevant feature too (17, 18). Noteworthy, in murine models of the disease, a significant correlation between QTc duration and the degree of cardiac inflammation at the histological examination has been demonstrated (59, 60). Finally, evidence also exists that QTc is often prolonged in a purely immune-mediated inflammatory heart disease such as acute rheumatic carditis (61), with some cases even complicating with TdP (62, 63). In particular, an increased QTc has been found to be the most frequent ECG alteration in children with subclinical carditis (~30%) (19). Moreover, in patients with acute rheumatic fever, a prolonged QTc correlated with both the presence of carditis and the level of acute-phase reactants (20).
Not only cardiac, but also systemic inflammation is associated with QT prolongation, as indicated by accumulating data obtained in patients with autoimmune chronic inflammatory diseases, as well as in patients affected with non-inflammatory heart diseases or apparently healthy subjects from general population.
Among systemic autoimmune diseases, the largest evidence regards rheumatoid arthritis (RA) and connective tissue diseases (CTDs). In RA, representing a paradigmatic example of chronic disease with high-grade inflammatory burden, the risk of sudden cardiac death (SCD) is approximately two times higher than in non-RA subjects (64). Recent studies demonstrated that in RA patients, QTc is frequently prolonged, associates with disease severity and inflammatory markers, and predicts mortality (65). In a cohort of 101 patients with chronic inflammatory arthritis, in which a significant positive correlation between C-reactive protein (CRP) values and QTc duration was demonstrated, we found that RA patients had a longer QTc when compared with both spondyloarthritis patients and healthy controls (21). These findings were very recently confirmed in a larger retrospective, population-based cohort study involving 650 RA patients and 650 age- and sex-matched non-RA patients. During follow-up, the cumulative incidence of QTc prolongation at 20 years after RA incidence (or after index date for controls) was higher among RA than non-RA subjects. Notably, in RA patients, erythrocyte sedimentation rate (ESR) at diagnosis was significantly associated with risk of idiopathic QTc prolongation, i.e., excluding prolongations explained by ECG changes, medications, etc. (22). In another prospective study carried out on 357 RA patients, it was found that prolonged QTc is a strong predictor of death as a 50 ms increase in QTc interval associated with a doubling of the hazard for all-cause mortality (22). The evidence that in this population, QTc prolongation was independently associated with CRP levels, and that the significance of the association between QTc and all-cause mortality was lost after the adjustment for CRP, once more and robustly supported the hypothesis that systemic inflammation plays a key mechanistic role in the phenomenon. As a further confirmation of this view, Adlan et al. (24) found that in RA patients circulating levels of inflammatory cytokines (TNFα, IL-1β, IL-6, IL-10) correlated with QTc duration. Moreover, we demonstrated that in RA anti-cytokine therapy with the anti-interleukin 6-receptor antibody, tocilizumab was associated with a rapid and significant QTc shortening, which correlated throughout the study time with the decrease in both CRP, and, more strongly, circulating TNFα levels (25).
Several studies performed in patients with different CTDs reported a high overall prevalence of QTc prolongation (up to ~30%) (26–30), with circulating IL-1β levels independently predicting the presence of a prolonged QTc (30). As regards specific CTD forms, it has been demonstrated that systemic lupus erythematosus (SLE) patients display longer mean QTc than controls (31, 32), and data obtained from large SLE cohorts found a 7–15% incidence of QTc prolongation (33, 34) [marked QTc prolongation, i.e., >500 ms, in ~3% (35)], with a significant association between QTc and overall inflammatory burden, as reflected by SLICC/ACR damage index (SDI) (34, 35). Noteworthy, 10 cases of drug-induced TdP in SLE patients were reported (66–75), and although CRP was specifically assessed only in two cases, nevertheless it was elevated in both (70, 74). Maximum QTc is also increased in systemic sclerosis (SSc) patients when compared to healthy controls (36). Furthermore, a recent study on 689 SSc patients demonstrated that QTc prolongation occurs in 25% of the cases, also independently correlating with disease duration and disease severity (37). Finally, preliminary results suggest that an increased frequency of QTc prolongation may be observed in other chronic inflammatory diseases, particularly inflammatory bowel disease and psoriasis (76, 77).
Systemic inflammation can also be involved in the pathogenesis of QTc prolongation in some non-inflammatory heart diseases. By analyzing 466 hypertensive patients, Chang et al. (38) found that CRP level correlated with QTc and independently predicted QTc prolongation presence. Similarly, in patients with coronary artery disease, a significant association between QTc duration and circulating CRP was observed (39). Moreover, a study involving 105 patients with Takotsubo cardiomyopathy demonstrated that subjects presented with QTc prolongation had higher CRP levels than those with normal QTc (40).
Finally, a significant relationship between the degree of systemic inflammatory activation and QTc duration is also observed in apparently healthy subjects in general population. The first report by Kazumi et al. (41) showed that QTc length was independently correlated with CRP in 179 males aged 18–22 years. More recently, two large population-based studies involving middle-aged or elderly subjects confirmed these findings. In the first one, Kim et al. (42) analyzed 4758 individuals (40–69 years) and concluded that prolonged QTc was significantly associated with elevated CRP (about twofold increase in the odds of QTc being ≥440 ms), independent of confunders. Accordingly, in the CARdiovascular diseases, Living, and Aging in Halle (CARLA) Study involving 1716 subjects aged 45–83 years, parameters of inflammation correlated with QTc duration, particularly soluble TNF-receptor-1 levels (sTNF-R1, a circulating stabile marker of TNFα system activation) in women (43). The concomitant evidence from large prospective community-based studies that inflammatory markers (CRP, IL-6) predict SCD in apparently healthy persons (78, 79) suggests that this association, at least in part, may be explained by a higher propensity to develop long QT-associated malignant arrhythmias.
An increasing body of evidence indicates that inflammatory activation profoundly impacts the electrophysiological properties of cardiomyocytes via multiple effects, ultimately resulting in a prolongation of AP duration (APD), and thereby of the QTc on ECG. In this scenario, the key mediators seem to be inflammatory cytokines (particularly TNFα, IL-6, IL-1β), which may affect myocardium either directly, by modulating specific ion channels critically involved in APD, and indirectly, by increasing central nervous system sympathetic drive on the heart (Figure 2).
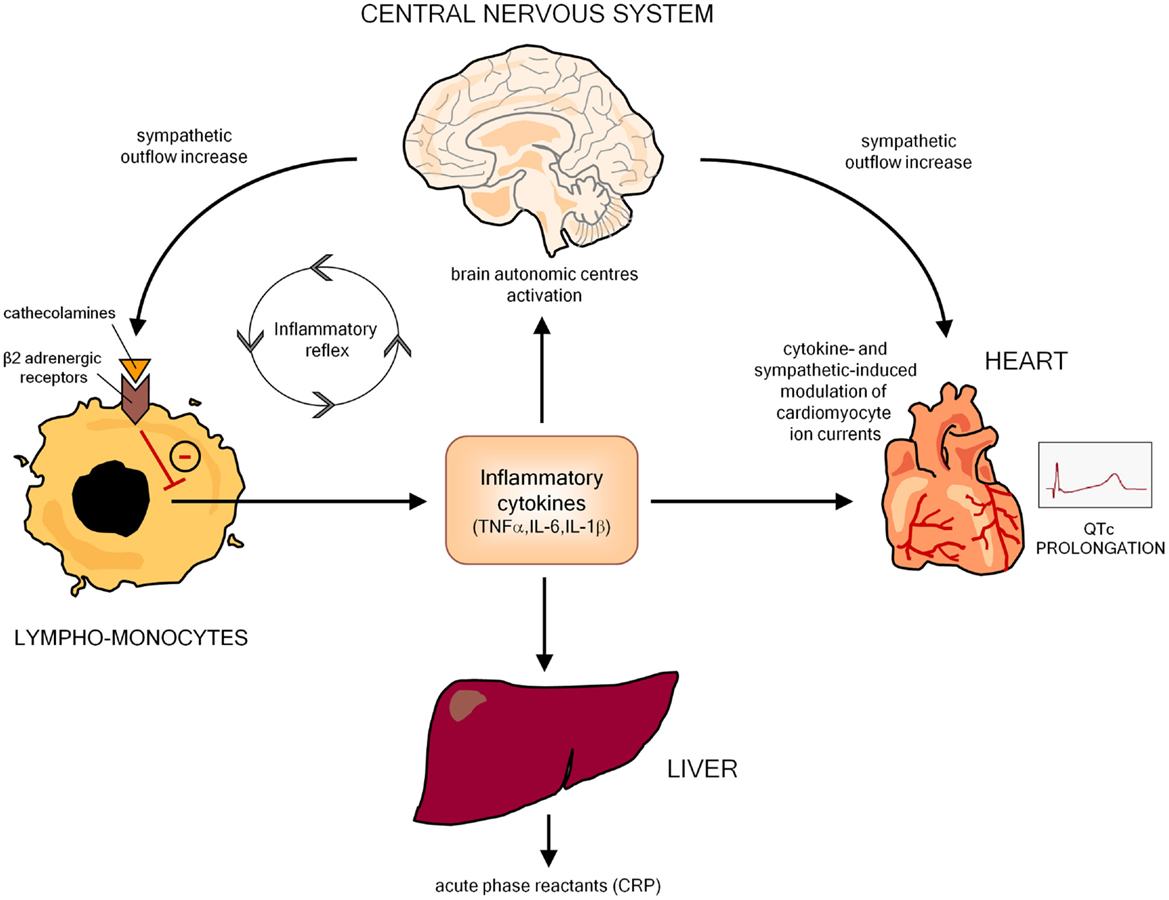
Figure 2. Potential mechanisms responsible for inflammation-mediated QTc prolongation. CRP, C-reactive protein.
A number of basic studies demonstrated significant direct effects of inflammatory cytokines on cardiac electrophysiology, particularly inducing changes in the expression and function of potassium and calcium channels (Table 3). Perfused hearts from transgenic mice overexpressing TNFα exhibited a prolonged APD and re-entrant ventricular arrhythmias (80); left ventricular myocytes isolated from these animals revealed a robust decrease of Ito and a reduced expression of the corresponding potassium channel protein (81). Several authors reported consistent findings when rat ventricular myocytes were cultured with TNFα, also demonstrating the involvement of a molecular cascade including iNOS overexpression, oxidant species generation, NFκB activation, and potassium-channel-interacting protein 2 (KChIP-2) inhibition (82–84). Moreover, Wang and coll (85) showed that TNFα down-regulates in vitro IKr by impairing the function of the hERG potassium channel via the stimulation of reactive oxygen species. Although it is far probable that similar effects on potassium channels are also exerted by the other main pro-inflammatory cytokines IL-6 and IL-1, no specific studies evaluated this topic as yet. Nevertheless, experiments on pig and mouse ventricular cells clearly demonstrated the ability of both these cytokines to prolong APD, possibly by enhancing ICaL (86, 87). Finally, no data exist about possible effects of cytokines on sodium channels. This area needs further evaluation, given that an increase in the INa current may theoretically contribute to cytokine-induced APD prolongation. Although not fully elucidated, the previously reported evidence that circulating inflammatory cytokine levels correlated with QTc duration in patients with RA (24, 25), CTDs (30), as well as in healthy subjects (43) strongly indicate that also in vivo, these pathophysiological mechanisms are of crucial importance.

Table 3. Effects of inflammatory cytokines on cardiomyocyte action potential: electrophysiological and molecular mechanisms.
Animal models of cardiac or systemic inflammation confirm and expand the relevance of these data. In isolated ventricular myocytes from mice with experimental autoimmune myocarditis (EAM), APD was markedly prolonged and Ito density significantly reduced when compared to controls (88). An increased APD (with a higher susceptibility to triggered ventricular arrhythmias) was consistently reported by Park et al. (89) by analyzing EAM rat hearts in which an elevated tissue expression of IL-6 and TNFα was demonstrated. Notably, the authors also proved that both cytokine myocardial accumulation and electrophysiological changes were prevented by prednisone administration. Similar preventive effects, associated with a significant attenuation of Ito inhibition, were also reported by Tang et al. (90) by treating the animals with statins (atorvastatin), whose anti-inflammatory potential in myocarditis has being increasingly recognized (91). Moreover, relevant cardiac electrophysiological alterations have been recently demonstrated in a murine model of myocardial infarction (MI) in which a state of systemic inflammation was induced. In MI mice, intraperitoneal injection with lipopolysaccharide (LPS) was associated with repolarization and APD prolongation, and higher ventricular arrhythmia propensity than non-LPS-injected animals. Notably, LPS-treated mice showed increased inflammatory macrophage activity transmurally in the heart, with a strong relationship between the degree of local myocardial inflammation and electric remodeling. Furthermore, the authors provided indirect evidence of a link between electrophysiological abnormalities and higher IL-1β expression in the myocardium (92). Besides inducing macrophage-derived cytokine production, LPS may also prolong APD by directly downregulating Ito via toll-like receptor 4 activation, as recently demonstrated in isolated rat ventricular myocytes (93).
Inflammation can also produce cardiac electrophysiology changes leading to QTc prolongation in an indirect manner, by inducing autonomic nervous system (ANS) dysfunction. Indeed, many basic and clinical studies demonstrated that, by targeting the autonomic centers of the brain, inflammatory cytokines increase the sympathetic outflow overdrive that in turn inhibits cytokine production and immuno-inflammatory activation by stimulating the β2-adrenergic receptors expressed in circulating lymphocytes and monocytes. Such a self-controlling loop is a crucial component of the so-called inflammatory reflex, and in this context sympathetic activation putatively represents an adaptive response to damping the immuno-inflammatory response (94–97). However, central sympathetic system, when activated, affects not only the immune system, but also all the body districts under its control, including the heart, with relevant electrophysiological consequences on the myocardium (Figure 2). Indeed, cardiomyocyte β-adrenergic receptor activation profoundly and complexly affects calcium (ICaL) and potassium (IKs, IKr) conductance with a net effect of increase in APD (98). Accordingly, cardiac sympathetic denervation shortens APD in rats (99). Moreover, increased catecholamine levels typically prolong QTc in healthy individuals (100–102), and intravenous adrenaline produces increase in QTc length in congenital LQTS patients (103).
A large body of evidence demonstrates a strict relationship between the degree and duration of inflammatory activation and the severity of ANS dysfunction. In particular, many data focused on heart rate variability (HRV), a non-invasive method to detect early cardiovascular autonomic impairment by assessing the effects of the sympatho-vagal balance on the heart (104). Reduced HRV, indicating an increase in sympathetic and a reduction in parasympathetic nervous system activity (104), is a common finding in several systemic inflammatory immuno-mediated diseases, including chronic inflammatory arthritis (21, 65, 105, 106) and CTDs (26, 105), as well as in heart inflammatory diseases, including viral myocarditis (107) and acute rheumatic fever (108). Moreover, HRV parameters inversely correlated with circulating CRP (and/or inflammatory cytokines) in healthy individuals as well as in patients with cardiovascular diseases (109, 110). The amount of data on this topic available in RA is of particular significance. In these patients, cardiac ANS dysfunction is highly prevalent (~60%) with a main pattern indicative of elevated sympathetic activity and reduced parasympathetic activity (65, 106). Autonomic impairment (particularly HRV) associated with disease duration, disease activity, or inflammatory markers (65), and treatment with infliximab (a TNFα-antagonist monoclonal antibody) produced rapid and evident HRV changes, i.e., decrease in the sympathetic tone with a shift toward a relative vagal prevalence (97). Noteworthy, in patients with chronic inflammatory arthritis, systemic inflammation degree, as assessed by CRP, and HRV depression severity significantly correlated one each other and both with QTc duration (21).
In the last years, accumulating evidence indicates that autoimmune mechanisms are involved in the pathogenesis of cardiac arrhythmias (111). Indeed, a number of autoantibodies can deeply interfere with the bioelectric properties of the heart by directly targeting specific receptors, ion channels, or enzymes expressed on the cardiomyocyte surface (112, 113). In particular, increasing data demonstrated that some of these autoantibodies can increase the arrhythmic risk by inducing an acquired LQTS of autoimmune origin. Although most studies relate to anti-Ro/SSA antibodies, some data suggest that other autoantibodies may lead to QTc prolongation and related arrhythmias.
Anti-Ro/SSA antibodies (anti-Ro/SSA) consist of two fundamental subtypes, i.e., anti-Ro/SSA-52kD and anti-Ro/SSA-60kD, whose detection is frequent in CTDs, particularly Sjögren’s syndrome (30–95%) and SLE (30–50%), but also in 0.5–2.7% of the apparently healthy population (114). Large evidence links the trans-placental passage of anti-Ro/SSA from mother to fetus with the risk of developing congenital atrioventricular block (AVB) (115). Although traditionally considered as invulnerable, recent data suggest that also the adult conduction system may be affected by these antibodies (116). Moreover, increasing data indicate that anti-Ro/SSA significantly interfere with ventricular repolarization and promote QTc prolongation (114) (Table 4). In 2000, Cimaz et al. (117) for the first time reported a high prevalence (42%) of prolonged QTc in anti-Ro/SSA-positive infants without congenital-AVB. Later on, the same investigators demonstrated a concomitant disappearance of ECG abnormality and acquired maternal autoantibodies during their first year of life (118). Moreover, Gordon et al. (119) found that the QTc was significantly longer in children of anti-Ro/SSA-positive mothers compared with children of negative mothers, with a further increase in those with siblings with congenital-AVB. Consistent findings were obtained by several following studies performed in adults. In a cohort of adult CTD patients, we found that more than 50% of anti-Ro/SSA-positive subjects displayed a prolonged QTc, with mean QTc values significantly longer in positive vs negative patients (26). Accordingly, a similar prevalence of anti-Ro/SSA-associated QTc prolongation (46%) was demonstrated in a further 24-h ECG monitoring study on 46 CTD patients also showing that this ECG abnormality was associated with the occurrence of complex ventricular arrhythmias (28). More recently, Bourré-Tessier et al. (33) performed two consecutive large studies on 150 and 278 SLE patients, respectively. The authors found a 5.1- to 12.6-times higher risk of QTc prolongation in anti-Ro/SSA-positive group than in negative patients, and each 10 U/ml increase in anti-Ro/SSA titer was associated with a parallel increase in the risk of having prolonged QTc. The existence of a strict relationship between QTc length and antibody levels, as well as subtype specificity, was confirmed in a further study on 49 CTD patients performed in our institution. In this cohort, it was demonstrated a direct correlation between anti-Ro/SSA concentration and QTc duration, but with the anti-Ro/SSA-52kD subtype only when the two subtypes were considered separately (29). Very recently, Pisoni et al. (30) reported that among 73 CTD patients, 20% of anti-Ro/SSA-positive vs 0% of anti-Ro/SSA-negative subjects had QTc prolongation. Notably, in patients with prolonged QTc (all anti-Ro/SSA-positive), IL-1β levels were significantly higher than patients with normal QTc, thus intriguingly suggesting a synergistic interplay between autoantibodies and inflammatory cytokines on QTc duration. Furthermore, Nakamura et al. (120) described the case of a anti-Ro/SSA-positive woman with severe QTc prolongation and TdP in which clear evidence of a direct mechanistic link between circulating antibodies and QTc prolongation was provided (see Mechanisms). In this patient, no genetic or acquired known causes of QT prolongation were detected, although a polymorphism (D85N) in KCNE1 gene was found. Noteworthy, she was totally asymptomatic for autoimmune diseases. Since anti-Ro/SSA is the most frequent autoantibody found in general population, but in most cases (60–70%) totally asymptomatic (114), an intriguing speculation is that by reducing the repolarization reserve anti-Ro/SSA may be silently involved as a predisposing factor in a number of “idiopathic” life-threatening arrhythmias, including drug-induced TdP, and sudden unexpected deaths occurring in apparently healthy people.
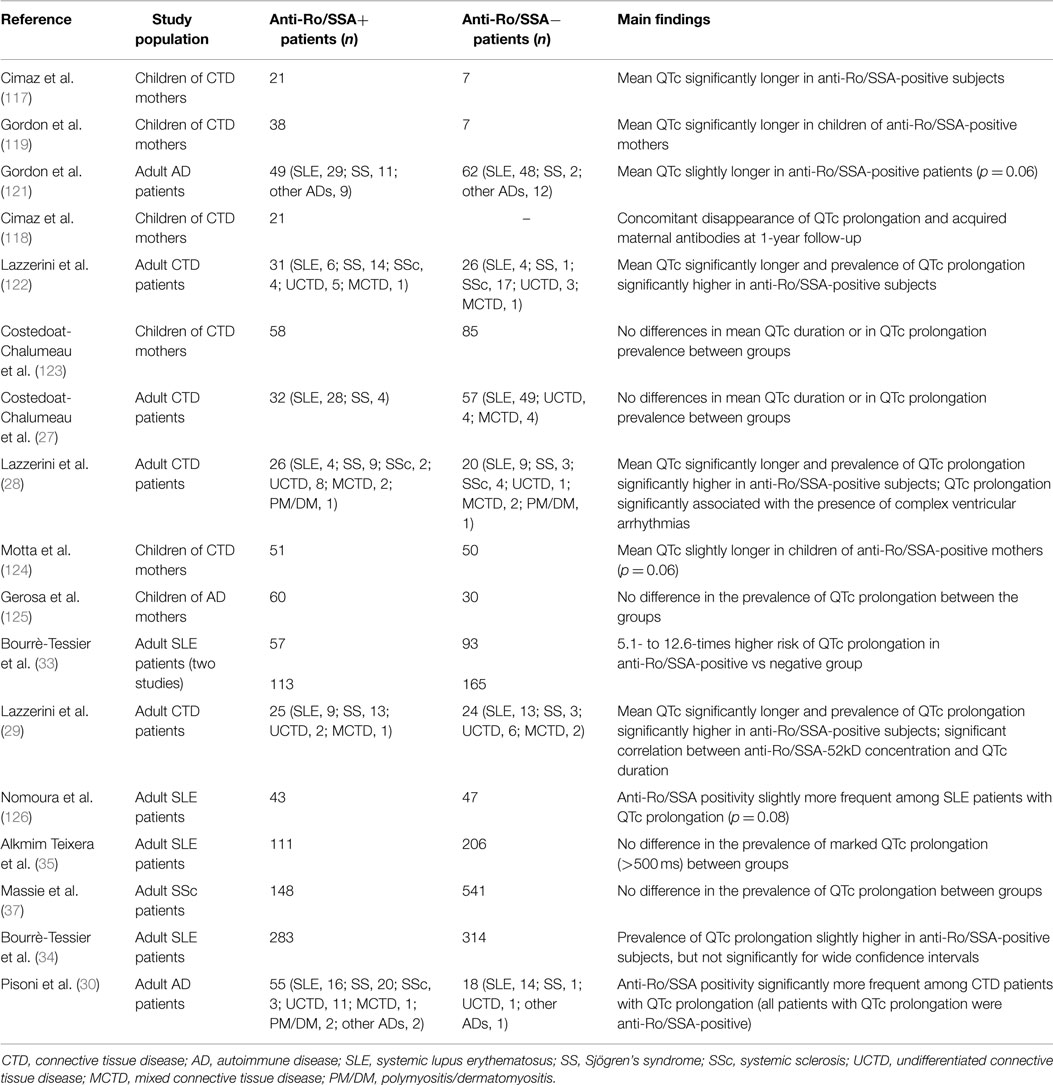
Table 4. Clinical studies on anti-Ro/SSA antibodies and QTc interval.
In addition to the above reported forthrightly supporting data, there are other studies that although not observing significant differences between anti-Ro/SSA-positive and negative patients in terms of mean QTc length and/or QTc prolongation prevalence, nevertheless found differences in these parameters that were very close to statistical significance. This is the case of the pediatric study of Motta et al. (124) (QTc of infants of anti-Ro/SSA-positive mothers slightly prolonged vs control group, p = 0.06), as well as of the adult studies of Gordon et al. (121) (QTc slightly longer in the anti-Ro/SSA-positive CTD group, p = 0.06), Nomura et al. (126) (anti-Ro/SSA positivity slightly more frequent among SLE patients with QTc prolongation, p = 0.08), and Bourrè-Tessier et al. (34) [proportion of SLE patients with prolonged QTc slightly higher in anti-Ro/SSA-52kD-positive group, although not reaching significance for wide confidence intervals].
Although the majority of the data point to an association between anti-Ro/SSA and QTc prolongation, there are conflicting results from other studies, either in children (123, 125) and adults (27, 35, 37). However, it should be noted that one of these studies (37) was performed in SSc patients, who frequently display anti-Ro/SSA but at a low level (127), thus possibly not high enough for the threshold level required for QTc prolongation manifestation (29, 128); in another one (35), involving SLE patients, the authors used a cutoff for QTc prolongation (>500 ms) probably not adequate to detect the phenomenon in this setting, as previous studies consistently demonstrated that in the large majority of the anti-Ro/SSA-positive CTD patients with QTc prolongation values ranged from 440 to 500 ms.
Besides anti-Ro/SSA, some lines of evidence suggest that other autoantibodies, i.e., anti-beta1-adrenergic receptor antibodies (anti-β1) and anti-voltage-gated potassium channel Kv1.4 antibodies (anti-Kv1.4), may be responsible of immuno-mediated forms of acquired LQTS.
Anti-β1 are frequently detected in idiopathic dilated cardiomyopathy (IDC, 30–50%), but also in Chagas’disease and in subjects with primary electrical disturbances (112). IDC is often complicated by ventricular arrhythmias [including TdP (129–134)] and SCD (135), with QT dynamicity representing an independent predictor of major arrhythmic events (136). Since the underlying mechanisms of such electrical instability are not fully clarified, and increasing evidence indicates that autoimmunity plays a relevant role in IDC pathogenesis (137), a possible link between anti-β1 and arrhythmic risk has been investigated. In IDC patients, circulating anti-β1 are associated with increased all-cause and cardiovascular mortality risk (138). Moreover, by studying 104 IDC subjects, Iwata et al. (139) demonstrated that the presence of anti-β1 independently predicted ventricular tachycardia and SCD. The following evidence in an animal model that induction of anti-β1 autoimmunity was concomitantly associated with QT/RR interval prolongation (with a parallel increase in APD at ex vivo electrophysiological examination), sustained ventricular tachycardia, and SCD (140) suggests that these antibodies may increase in vivo the risk of life-threatening arrhythmias at least in part by prolonging QTc.
Finally, an association of anti-Kv1.4 and LQTS has been demonstrated in patients affected with myasthenia gravis (MG), an autoimmune disease primarily affecting the neuromuscular function (141). The Kv1.4 protein is one of the forming α-subunits (Kv) of the voltage-gated potassium channel (VGKC), which plays a crucial role in the acetylcholine presynaptic release, but also in the cardiac repolarization (142). Indeed, Kv1.4 is also expressed in ventricular cardiomyocytes as pore-forming subunit of the channel responsible for the slowly recovering component of Ito, the main current of the phase 1 (early repolarization) of cardiac AP (143). Recent studies indicate that anti-Kv1.4 are relatively frequently detected in MG patients and their presence associates with QTc prolongation (~15–35% of positive cases) (144, 145). Moreover, in a cohort of 650 MG patients, Suzuki et al. (144) reported that among 70 anti-Kv1.4-positive subjects (14%), two died of lethal QT-associated arrhythmias (TdP in one case, SCD in a patient who had QTc prolongation in the other one). Notably, at least two further reports of TdP in MG patients are present in the literature (146, 147).
Although mechanisms underlying autoimmune-mediated LQTS are not fully known, accumulating evidence indicates that autoantibodies may directly affect cardiomyocyte electric properties by interfering on ion channels function (Figure 3).
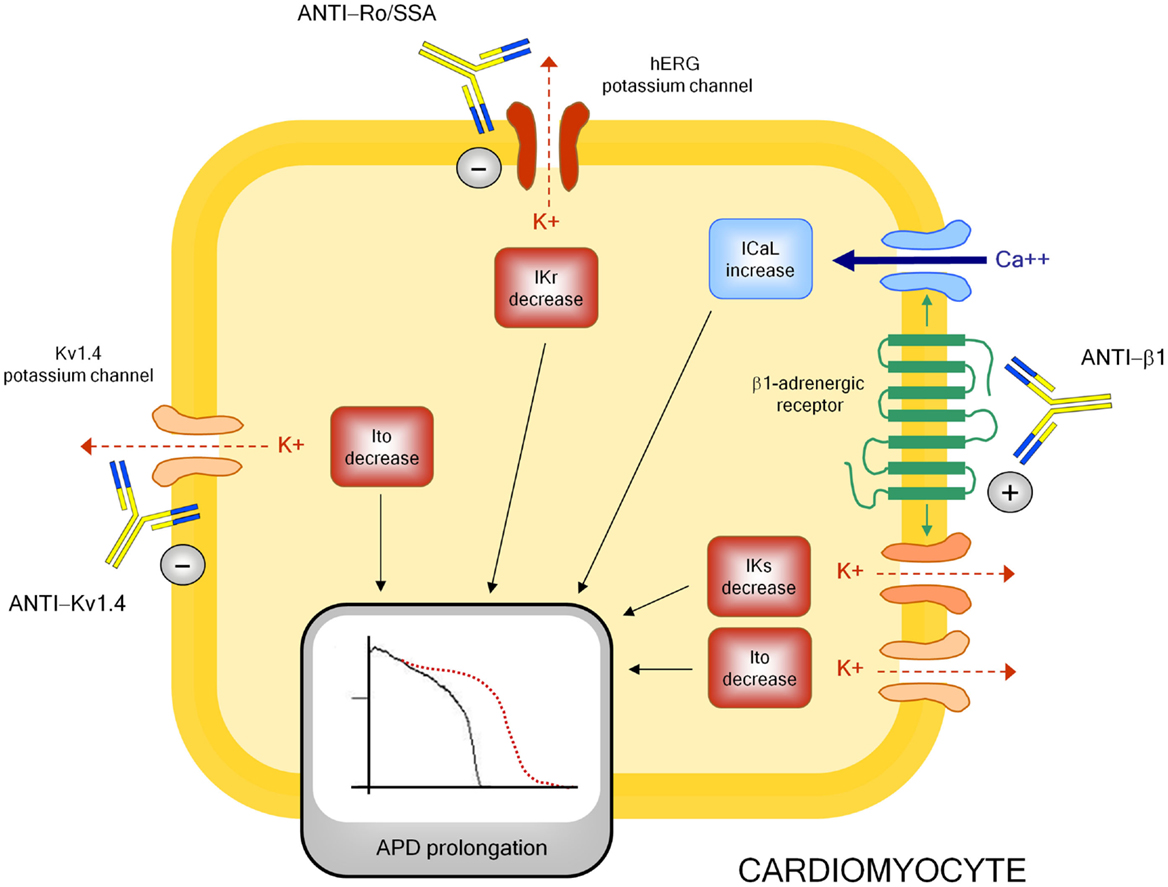
Figure 3. Autoantibody-mediated QTc prolongation: molecular targets and electrophysiological consequences. Anti-β1, anti β1-adrenergic receptor antibodies; Ito, transient outward potassium current; IKr, rapid component of the delayed rectifier current; IKs, slow component of the delayed rectifier current; ICaL, L-type calcium current; APD, action potential duration.
The electrophysiological effects of anti-Ro/SSA are largely recognized, but mostly in the setting of congenital-AVB. Experimental studies clearly demonstrated the ability of anti-Ro/SSA from mothers with children with congenital-AVB in biochemically cross-reacting with l-type and T-type calcium-channels, thus significantly inhibiting the related currents (ICaL, ICaT), which both play a key role in the AP of heart conduction system cells (148). More in detail, it has been proven that anti-Ro/SSA specifically recognize the α1 pore-forming subunit of the calcium channels through a binding site localized on the extracellular loop of domain I S5–S6 (122, 149–153). The hypothesis is that Ro protein (particularly Ro52-kD) shares structural similarities with calcium channels, thus explaining a cross-reactivity of anti-Ro/SSA as a result of molecular mimicry mechanisms. Keeping this in mind, and in consideration of the fact that calcium and potassium channels belong to the same superfamily of voltage-gated ion channels in which, in particular, the structure of the voltage-sensor sequence is highly conserved (154), it is conceivable that a concomitant inhibitory cross-reactivity with potassium channels may be responsible of the effects of anti-Ro/SSA on QTc interval by impairing ventricular repolarization. In accordance with this view, some recent data suggest that hERG-potassium channel, conducting IKr, may represent a further specific target of anti-Ro/SSA. As already cited, Nakamura et al. (120) demonstrated that both serum and purified IgGs from an anti-Ro/SSA-positive woman with extreme QTc prolongation and TdPs specifically reacted with hERG-channel and induced a concentration-dependent and fully reversible inhibition of IKr. In a very recent study performed in the laboratory of Boutjdir in collaboration with our institution, these findings have been confirmed and expanded in a larger number of subjects, by comparing anti-Ro/SSA-positive vs negative CTD patients, as well as in an animal model. In particular, electrophysiological and biochemical evidence is provided that anti-Ro/SSA inhibit IKr and prolong APD by directly binding to the hERG-channel protein, likely at the pore region where homology with Ro-52kD antigen is present. Moreover, Ro-52kD antigen immunized guinea-pigs showed QTc prolongation on ECG after developing high titers of anti-Ro/SSA (155). In accordance with these results, strongly suggestive of a mechanism dependent on a purely electrophysiological interference on the heart, recent preliminary data from single case reports demonstrated the effectiveness of immunosuppressive therapy in reversing anti-Ro/SSA-associated electrocardiographic abnormalities in vivo, at least in adults (116, 156–158).
Despite this evidence, clinical studies analyzing the relationship between anti-Ro/SSA and QTc showed some degrees of discrepancy. Moreover, even among studies demonstrating significant association, markedly different percentages of QTc prolongation in anti-Ro/SSA-positive CTD patients were observed (~10–60%) (29). Although previously reported data (29, 30) suggest that it may be explained, at least in part, by differences among CTD cohorts in terms of autoantibody concentration and specificity [high levels of anti-Ro/SSA-52kD are particularly frequent in SS, much less in SLE and rarely in SSc (159)], or disease-related inflammatory burden (and thus cytokine levels), the above electrophysiological data, by indicating that anti-Ro/SSA inhibit both calcium and hERG-potassium currents, provide a further pathophysiological mechanism possibly contributing to differences observed. Indeed, it is well recognized that calcium and potassium channels have conflicting effects on APD, thus on QTc length. A block of the inward ICaL during the plateau phase shortens, while an inhibition of outward IKr during repolarization prolongs APD (160). Thus, it is conceivable that a concomitant inhibitory effect of anti-Ro/SSA on calcium channels can partially counteract the IKr inihibition-dependent APD prolonging effects in vivo, thus reducing the actual extent of QTc prolongation observed (128). In this view, intrinsic (inherited or acquired) differences in potassium and calcium channel expression on patient’s cardiomyocytes may participate in the QTc variability observed. In conclusion, evidence indicates that anti-Ro/SSA inhibit IKr, but the clinical phenotype may not be the same for each patient as a result of several modifying factors, including the anti-Ro/SSA level, the degree of systemic inflammation, and the peculiar cardiomyocyte ion channels’ profile.
A modulating activity on ion channel function seems to be also critically involved in the mechanism by which anti-β1 prolong APD and QTc, although in this case the effect is indirect via a stimulating interaction with the myocardial β1-adrenergic receptor. Indeed, some basic studies demonstrated that anti-β1 produced a profound electrical remodeling of the cardiomyocyte, mainly involving potassium and calcium conductance. Christ et al. (161) found that purified anti-β1, obtained from IDC patients, increased APD and ICaL in isolated rat and human cardiomyocytes. Later on, by analyzing isolated ventricular myocytes from rabbits immunized with a synthetic peptide corresponding to the second extracellular loop of β1-adrenergic receptors, Fukuda et al. (140) showed a significant decrease (~35–45%) of Ito1 and Iks. Moreover, they demonstrated APD prolongation and early afterdepolarization in the right ventricular papillary muscle, as well as a longer QT/RR interval ratio and a higher prevalence of sustained ventricular tachycardia in immunized vs control rabbits.
Recent data intriguingly suggest that inflammation and immunity may be also involved in modulating the clinical expression of congenital LQTS, possibly triggering or enhancing electrical instability in patients already genetically predisposed to arrhythmias.
Rizzo et al. (162) performed a histopathologic study on stellate ganglia specimens obtained from 12 patients, 8 with different forms of congenital LQTS and 4 with catecholaminergic polymorphic ventricular tachycardia (CPVT), who underwent left cardiac sympathetic denervation for malignant intractable arrhythmias. Indeed, all the patients were severely sympthomatic before the ganglionectomy, with most patients having had multiple shocks from a previously implantable cardioverter defibrillator (including arrhythmic storms), and the procedure resulted in a rhythm stabilization in almost all the cases. Examination of patients’ stellate ganglia revealed low-grade but distinct inflammatory infiltrates composed by activated T lymphocytes and macrophages, indicative of a chronic T-cell-mediated ganglionitis. Moreover, morphometric analysis demonstrated that the number of T cells/mm2 were significantly higher in the ganglia of these patients when compared with those obtained from 10 sex- and age-matched control subjects accidentally died. On the basis of these findings, the authors speculated that a T-cell-mediated cytotoxicity toward ganglion cells may boost adrenergic activity through release of inflammatory mediators in ganglia and in this manner contribute to the electric instability in LQTS/CPVT patients, particularly in those who are heavily symptomatic. In accordance with this view, intracardiac ganglionitis and its pro-arrythmic potential have been previously described in LQTS patients who died suddenly, the first time over 35 years ago (163–165). Moreover, although the origin of inflammatory infiltrates remains unknown, Rizzo et al. (162) put forward the hypothesis of a viral (however not herpes-virus DNA was found in specimens) or autoimmune pathogenesis. As concerns this latter mechanism, Moss et al. (166) underlined how all patients had recurrent syncope and/or many defibrillator shocks, and both transient hypoperfusion and recurrent shocks could cause ganglionic cell injury with protein damage putatively resulting in a secondary autoimmune reaction with manifestations of ganglionitis. In any case, independently whether ganglionitis is a primary event or a phenomenon secondarily occurring after first severe arrhythmic episodes (thus triggering a self-aggravating loop), it is conceivable that it may have played a role in precipitating life-threatening tachyarrhythmias since stellectomy induced rhythm stabilization in almost all patients. These findings, although preliminary, intriguingly suggest that immuno-inflammatory pathways could in the future represent a novel target in the therapeutic approach to congenital LQTS, particularly in patients with intractable arrhythmias despite appropriate standard therapy.
Although these considerations primarily point to the therapeutic potential of interventions lowering the degree of the immuno-inflammatory response, a very recent study from the group of Nattel (167) suggests that a selective stimulation of the immune system may be also theoretically useful in the treatment of congenital LQTS. Starting from previous evidence demonstrating that in a subpopulation of IDC patients, autoantibodies against the KCNQ1-encoded Kv7.1 potassium channel were associated with QTc shortening possibly by increasing IKs conductance (168). Li et al. (167) immunized rabbits with KCNQ1-channel peptide, thus inducing high circulating anti-KCNQ1 antibody titers. As expected, these animals developed significant QTc shortening compared to controls, as well as APD decrease and IKs densities enhancement in left ventricular cardiomyocytes when isolated. Since these findings indicate that KCNQ1 autoimmunity accelerates cardiac repolarization by increasing channel function, the potential consequences of this immunization were tested in a well-recognized rabbit model of human LQTS, induced by infusion of methoxamine and dofetilide. KCNQ1-immunized animals showed much less striking ECG changes with significantly less severe QTc prolongation, compared to sham-immunized animals upon drug challenge (17.5 vs 73.4% increase). Moreover, life-threatening ventricular arrhythmias, particularly TdP, were observed in the sham-group only. On the basis of these results, the authors speculated that by enhancing repolarization reserve KCNQ1 vaccination may be therapeutically useful in patients with congenital LQTS resistant to conventional treatments, thus opening new exciting avenues in antiarrhythmic therapy (167).
In the latest years, inflammation and immunity have been increasingly recognized as novel factors crucially involved in modulating arrhythmic risk, an effect in part resulting from a significant impact on ventricular repolarization.
A number of considerations suggest that these phenomena may have relevant clinical implications, also in terms of therapeutic perspectives.
First, although inflammatory and autoimmune mechanisms are in most cases probably not per se able to induce a QTc prolongation as critical as to induce TdP [but actually this is true for all recognized causes of LQTS, when present alone (130, 169)], nevertheless they can reduce the ventricular repolarization reserve, thereby significantly increasing the risk of life-threatening arrhythmias in the presence of other classical QT-prolonging factors (drugs, electrolyte imbalances, genetic polymorphisms, etc.). While it is well conceivable that these events may take place in patients with autoimmune chronic inflammatory diseases, thus putatively contributing to explain the increased risk of sudden death observed in the course of RA (64) and CTDs (170), nevertheless asymptomatic low-grade chronic inflammation and/or circulating anti-Ro/SSA may be also silently involved, as a predisposing factor, in a number of unexpected life-threatening arrhythmias, including drug-induced TdP, and SCDs occurring in general population.
Moreover, as also preliminarily suggested by a recent histopathology study in stellate ganglia (161), it is also probable that inflammation and immunity may enhance arrhythmic risk in patients with congenital LQTS. Indeed, it should be underlined that an acute inflammatory illness is recently recognized among the possible precipitant factors of malignant arrhythmias and electrical storms in these subjects (171–173). Although it has been demonstrated that fever has per se a role by influencing temperature-sensitive biophysical properties of mutant channels (particularly in LQTS2) (173, 174), it can also be speculated that in patients with congenital LQTS episodes of systemic inflammation may further increase arrhythmias susceptibility due to circulating cytokines directly affecting cardiomyocyte APD, and indirectly increasing sympathetic output from central and peripheral ANS (Figure 4). In this view, it is also conceivable that some acquired LQTS patients are occult (latent) carriers of mutations in LQTS-susceptibility genes that are unmasked under inflammatory/autoimmune conditions, with a potential different impact of immunotherapies on QTc.
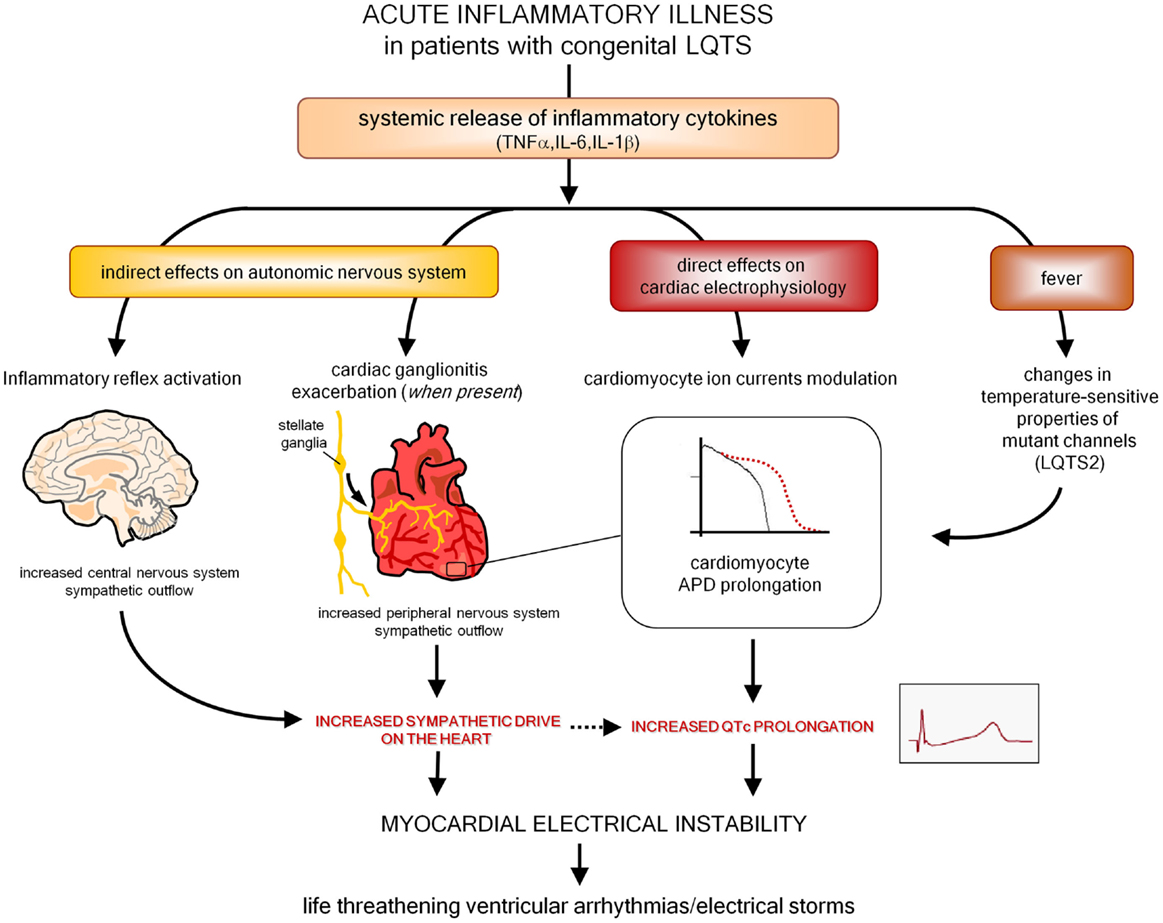
Figure 4. Putative pathways involved in exacerbating myocardial electrical instability in patients with congenital LQTS during an acute inflammatory illness. LQTS, long QT syndrome; APD, action potential duration.
Thus, in terms of therapeutic perspectives, in patients with inflammatory/autoimmune disease-associated QTc prolongation, besides avoiding any further acquired factor potentially prolonging QTc, and carefully balancing pro and contra when introducing any QT-prolonging drug, available data highlight the importance of minimizing systemic immuno-inflammatory burden through a tight control of disease activity, a goal now more feasible after the introduction of potent biologic therapies targeting the immune system.
As concerns QTc prolongation occurring in general population subjects displaying chronic low-grade systemic inflammation and/or specific autoantibodies in the absence of a clinically evident inflammatory/autoimmune disease, as well as malignant intractable arrhythmias in congenital LQTS patients with signs of immuno-inflammatory activation, no data are currently available on the therapeutic role of anti-inflammatory or immunomodulatory interventions. Nevertheless, accumulating evidence reviewed in this paper underlines the need for further specific investigations on this topic.
In conclusion, the potential impact of inflammatory and immunologic mechanisms on ventricular repolarization should be always carefully kept in mind, not only in the presence of a manifest immune-inflammatory disease, but also in subjects with QTc prolongation of unclear origin, or in patients with an already recognized LQTS (inherited or acquired) as a possible trigger for electrical instability. In this view, targeting immuno-inflammatory pathways may represent an attractive and innovative therapeutic approach in a number of LQTS patients.
We do not have any financial support or other benefits from commercial sources for the work reported on in the manuscript, or any other financial interests which could create a potential conflict of interest or the appearance of a conflict of interest with regard to the work.
1. Khan IA. Clinical and therapeutic aspects of congenital and acquired long QT syndrome. Am J Med (2002) 112:58–66. doi:10.1016/S0002-9343(01)01011-7
2. Schwartz PJ, Crotti L, Insolia R. Long-QT syndrome: from genetics to management. Circ Arrhythm Electrophysiol (2012) 5:868–77. doi:10.1161/CIRCEP.111.962019
3. Roden DM, Viswanathan PC. Genetics of acquired long QT syndrome. J Clin Invest (2005) 115:2025–32. doi:10.1172/JCI25539
4. Ruan Y, Liu N, Napolitano C, Priori SG. Therapeutic strategies for long-QT syndrome: does the molecular substrate matter? Circ Arrhythm Electrophysiol (2008) 1:290–7. doi:10.1161/CIRCEP.108.795617
5. Schwartz PJ, Ackerman MJ. The long QT syndrome: a transatlantic clinical approach to diagnosis and therapy. Eur Heart J (2013) 34:3109–16. doi:10.1093/eurheartj/eht089
6. Schwartz PJ, Stramba-Badiale M, Crotti L, Pedrazzini M, Besana A, Bosi G, et al. Prevalence of the congenital long-QT syndrome. Circulation (2009) 120:1761–7. doi:10.1161/CIRCULATIONAHA.109.863209
7. Tisdale JE, Wroblewski HA, Overholser BR, Kingery JR, Trujillo TN, Kovacs RJ. Prevalence of QT interval prolongation in patients admitted to cardiac care units and frequency of subsequent administration of QT interval-prolonging drugs: a prospective, observational study in a large urban academic medical center in the US. Drug Saf (2012) 35:459–70. doi:10.2165/11598160-000000000-00000
8. Pasquier M, Pantet O, Hugli O, Pruvot E, Buclin T, Waeber G, et al. Prevalence and determinants of QT interval prolongation in medical inpatients. Intern Med J (2012) 42:933–40. doi:10.1111/j.1445-5994.2011.02447.x
9. Mönnig G, Köbe J, Löher A, Wasmer K, Milberg P, Zellerhoff S, et al. Role of implantable cardioverter defibrillator therapy in patients with acquired long QT syndrome: a long-term follow-up. Europace (2012) 14:396–401. doi:10.1093/europace/eur316
10. Wong LC, Behr ER. Acquired long QT syndrome: as risky as congenital long QT syndrome? Europace (2012) 14:310–1. doi:10.1093/europace/eur372
11. Kannankeril P, Roden DM, Darbar D. Drug-induced long QT syndrome. Pharmacol Rev (2010) 62:760–81. doi:10.1124/pr.110.003723
12. Viskin S. Long QT syndromes and torsade de pointes. Lancet (1999) 354:1625–33. doi:10.1016/S0140-6736(99)02107-8
13. Møller S, Bernardi M. Interactions of the heart and the liver. Eur Heart J (2013) 34:2804–11. doi:10.1093/eurheartj/eht246
14. Gonzalez CD, de Sereday M, Sinay I, Santoro S. Endocrine therapies and QTc prolongation. Curr Drug Saf (2010) 5:79–84. doi:10.2174/157488610789869157
15. Ramamurthy S, Talwar KK, Goswami KC, Shrivastava S, Chopra P, Broor S, et al. Clinical profile of biopsy proven idiopathic myocarditis. Int J Cardiol (1993) 41:225–32. doi:10.1016/0167-5273(93)90119-2
16. Ukena C, Mahfoud F, Kindermann I, Kandolf R, Kindermann M, Böhm M. Prognostic electrocardiographic parameters in patients with suspected myocarditis. Eur J Heart Fail (2011) 13:398–405. doi:10.1093/eurjhf/hfq229
17. Williams-Blangero S, Magalhaes T, Rainwater E, Blangero J, Corrêa-Oliveira R, Vandeberg JL. Electrocardiographic characteristics in a population with high rates of seropositivity for Trypanosoma cruzi infection. Am J Trop Med Hyg (2007) 77:495–9.
18. Salles G, Xavier S, Sousa A, Hasslocher-Moreno A, Cardoso C. Prognostic value of QT interval parameters for mortality risk stratification in Chagas’ disease: results of a long-term follow-up study. Circulation (2003) 108:305–12. doi:10.1161/01.CIR.0000079174.13444.9C
19. Santos CC, Santos EC, Ribeiro LC, Ribeiro MV. Clinical, eletrocardiographic and echocardiographic aspects of subclinical carditis. Front Pediatr (2015).
20. Balli S, Oflaz MB, Kibar AE, Ece I. Rhythm and conduction analysis of patients with acute rheumatic fever. Pediatr Cardiol (2013) 34:383–9. doi:10.1007/s00246-012-0467-5
21. Lazzerini PE, Acampa M, Capecchi PL, Hammoud M, Maffei S, Bisogno S, et al. Association between high sensitivity C-reactive protein, heart rate variability and corrected QT interval in patients with chronic inflammatory arthritis. Eur J Intern Med (2013) 24:368–74. doi:10.1016/j.ejim.2013.02.009
22. Chauhan K, Ackerman M, Crowson CS, Matteson EL, Gabriel SE. Population-based study of QT interval prolongation in patients with rheumatoid arthritis. Clin Exp Rheumatol (2015) 33:84–9.
23. Panoulas VF, Toms TE, Douglas KM, Sandoo A, Metsios GS, Stavropoulos-Kalinoglou A, et al. Prolonged QTc interval predicts all-cause mortality in patients with rheumatoid arthritis: an association driven by high inflammatory burden. Rheumatology (2014) 53:131–7. doi:10.1093/rheumatology/ket338
24. Adlan AM, Panoulas VF, Smith JP, Fisher JP, Kitas GD. Association between corrected QT interval and inflammatory cytokines in rheumatoid arthritis. J Rheumatol (2015) 42:421–8. doi:10.3899/jrheum.140861
25. Lazzerini PE, Acampa M, Capecchi PL, Fineschi I, Selvi E, Moscadelli V, et al. Antiarrhythmic potential of anti-cytokine therapy in rheumatoid arthritis: tocilizumab reduces QTc interval by controlling systemic inflammation. Arthritis Care Res (2015) 67:332–9. doi:10.1002/acr.22455
26. Lazzerini PE, Acampa M, Guideri F, Capecchi PL, Campanella V, Morozzi G, et al. Prolongation of the corrected QT interval in adult patients with anti-Ro/SSA-positive connective tissue diseases. Arthritis Rheum (2004) 50:1248–52. doi:10.1002/art.20130
27. Costedoat-Chalumeau N, Amoura Z, Hulot JS, Cohen L, Piette JC. Letter in response to “prolongation of the corrected QT interval in adult patients with anti-Ro/SSA-positive connective tissue diseases” by Lazzerini et al. in Arthritis Rheum 2004;50:1248-52. Arthritis Rheum (2005) 52:676–7. doi:10.1002/art.20845; author reply 677-678[letter]
28. Lazzerini PE, Capecchi PL, Guideri F, Bellisai F, Selvi E, Acampa M, et al. Comparison of frequency of complex ventricular arrhythmias in patients with positive versus negative anti-Ro/SSA and connective tissue disease. Am J Cardiol (2007) 100:1029–34. doi:10.1016/j.amjcard.2007.04.048
29. Lazzerini PE, Capecchi PL, Acampa M, Morozzi G, Bellisai F, Bacarelli MR, et al. Anti-Ro/SSA-associated corrected QT interval prolongation in adults: the role of antibody level and specificity. Arthritis Care Res (2011) 63:1463–70. doi:10.1002/acr.20540
30. Pisoni CN, Reina S, Arakaki D, Eimon A, Carrizo C, Borda E. Elevated IL-1beta levels in anti-Ro/SSA-positive connective tissue disease patients with prolonged corrected QT interval. Clin Exp Rheumatol (2015).
31. Cardoso CR, Sales MA, Papi JA, Salles GF. QT-interval parameters are increased in systemic lupus erythematosus patients. Lupus (2005) 14:846–52. doi:10.1191/0961203305lu2225oa
32. Milovanović B, Stojanović L, Milićevik N, Vasić K, Bjelaković B, Krotin M. Cardiac autonomic dysfunction in patients with systemic lupus, rheumatoid arthritis and sudden death risk. Srp Arh Celok Lek (2010) 138:26–32. doi:10.2298/SARH1002026M
33. Bourré-Tessier J, Clarke AE, Huynh T, Bernatsky S, Joseph L, Belisle P, et al. Prolonged corrected QT interval in anti-Ro/SSA-positive adults with systemic lupus erythematosus. Arthritis Care Res (2011) 63:1031–7. doi:10.1002/acr.20470
34. Bourré-Tessier J, Urowitz MB, Clarke AE, Bernatsky S, Krantz MJ, Huynh T, et al. Electrocardiographic findings in systemic lupus erythematosus: data from an international inception cohort. Arthritis Care Res (2015) 67:128–35. doi:10.1002/acr.22370
35. Teixeira RA, Borba EF, Pedrosa A, Nishioka S, Viana VS, Ramires JA, et al. Evidence for cardiac safety and antiarrhythmic potential of chloroquine in systemic lupus erythematosus. Europace (2014) 16:887–92. doi:10.1093/europace/eut290
36. Sgreccia A, Morelli S, Ferrante L, Perrone C, De Marzio P, De Vincentiis G, et al. QT interval and QT dispersion in systemic sclerosis (scleroderma). J Intern Med (1998) 243:127–32.
37. Massie C, Hudson M, Tatibouet S, Steele R, Huynh T, Fritzler MJ, et al. Absence of an association between anti-Ro antibodies and prolonged QTc interval in systemic sclerosis: a multicenter study of 689 patients. Semin Arthritis Rheum (2014) 44:338–44. doi:10.1016/j.semarthrit.2014.07.001
38. Chang KT, Shu HS, Chu CY, Lee WH, Hsu PC, Su HM, et al. Association between C-reactive protein, corrected QT interval and presence of QT prolongation in hypertensive patients. Kaohsiung J Med Sci (2014) 30:310–5. doi:10.1016/j.kjms.2014.02.012
39. Yue W, Schneider A, Rückerl R, Koenig W, Marder V, Wang S, et al. Relationship between electrocardiographic and biochemical variables in coronary artery disease. Int J Cardiol (2007) 119:185–91. doi:10.1016/j.ijcard.2006.07.129
40. Song BG, Chung SM, Kim SH, Kim HJ, Kang GH, Park YH, et al. The QT prolongation and clinical features in patients with takotsubo cardiomyopathy: experiences of two tertiary cardiovascular centers. Anadolu Kardiyol Derg (2014) 14:162–9. doi:10.5152/akd.2013.4745
41. Kazumi T, Kawaguchi A, Hirano T, Yoshino G. C-reactive protein in young, apparently healthy men: associations with serum leptin, QTc interval, and high-density lipoprotein-cholesterol. Metabolism (2003) 52:1113–6. doi:10.1016/S0026-0495(03)00184-7
42. Kim E, Joo S, Kim J, Ahn J, Kim J, Kimm K, et al. Association between C-reactive protein and QTc interval in middle-aged men and women. Eur J Epidemiol (2006) 21:653–9. doi:10.1007/s10654-006-9034-9
43. Medenwald D, Kors JA, Loppnow H, Thiery J, Kluttig A, Nuding S, et al. Inflammation and prolonged QT time: results from the cardiovascular disease, living and ageing in halle (CARLA) study. PLoS One (2014) 9:e95994. doi:10.1371/journal.pone.0095994
44. Karjalainen J, Viitasalo M, Kala R, Heikkilä J. 24-Hour electrocardiographic recordings in mild acute infectious myocarditis. Ann Clin Res (1984) 16:34–9.
45. Finley JP, Radford DJ, Freedom RM. Torsade de pointes ventricular tachycardia in a newborn infant. Br Heart J (1978) 40:421–4. doi:10.1136/hrt.40.4.421
46. Mitamura E, Mifune J, Kanamori K, Hifumi N, Shimizu O, Yamamura I, et al. A case of torsade de pointes tachycardia complicating diphtheria. Kokyu To Junkan (1985) 33:223–8.
47. Sareli P, Schamroth CL, Passias J, Schamroth L. Torsade de pointes due to coxsackie B3 myocarditis. Clin Cardiol (1987) 10:361–2. doi:10.1002/clc.4960100514
48. Devriendt J, Staroukine M, Schils E, Sivaciyan B, Van Beers D. Legionellosis and “torsades de pointes”. Acta Cardiol (1990) 45:329–33.
49. Badorff C, Zeiher AM, Hohnloser SH. Torsade de pointes tachycardia as a rare manifestation of acute enteroviral myocarditis. Heart (2001) 86:489–90. doi:10.1136/heart.86.5.489
50. Izawa A, Yazaki Y, Hayashi S, Imamura H, Kusama Y, Isobe M. Transient left ventricular aneurysm and hypertrophy accompanied by polymorphic ventricular tachycardia in a patient suspected of acute myocarditis. Jpn Heart J (2000) 41:97–102. doi:10.1536/jhj.41.97
51. Gowani SA, Kumar A, Arora S, Lahiri B. Legionella pneumonia complicated by myocarditis and torsades de pointes: a case report and review of literature. Conn Med (2013) 77:331–4.
52. Jensen TB, Dalsgaard D, Johansen JB. Cardiac arrest due to torsades de pointes ventricular tachycardia in a patient with Lyme carditis. Ugeskr Laeger (2014) 176(35).
53. Khazan M, Mathis AS. Probable case of torsades de pointes induced by fluconazole. Pharmacotherapy (2002) 22:1632–7. doi:10.1592/phco.22.17.1632.34129
54. Sayar N, Terzi S, Yilmaz HY, Atmaca H, Kocak F, Dayi SU, et al. A case of prosthetic mitral valve Brucella endocarditis complicated with torsades de pointes. Heart Vessels (2006) 21:331–3. doi:10.1007/s00380-006-0907-3
55. Irie T, Kaneko Y, Nakajima T, Saito A, Kurabayashi M. QT interval prolongation and torsade de pointes induced by propofol and hypoalbuminemia. Int Heart J (2010) 51:365–6. doi:10.1536/ihj.51.365
56. Aypar E, Kendirli T, Tutar E, Ciftçi E, Ince E, Ileri T, et al. Voriconazole-induced QT interval prolongation and torsades de pointes. Pediatr Int (2011) 53:761–3. doi:10.1111/j.1442-200X.2010.03321.x
57. Tounsi A, Abid L, Akrout M, Hentati M, Kammoun S. QT prolongation complicated with torsades de pointes in prosthetic mitral valve endocarditis: a case report. Case Rep Med (2012) 2012:574923. doi:10.1155/2012/574923
58. Cunha-Neto E, Chevillard C. Chagas disease cardiomyopathy: immunopathology and genetics. Mediators Inflamm (2014) 2014:683230. doi:10.1155/2014/683230
59. Garcia S, Ramos CO, Senra JF, Vilas-Boas F, Rodrigues MM, Campos-de-Carvalho AC, et al. Treatment with benznidazole during the chronic phase of experimental Chagas’ disease decreases cardiac alterations. Antimicrob Agents Chemother (2005) 49:1521–8. doi:10.1128/AAC.49.4.1521-1528.2005
60. Eickhoff CS, Lawrence CT, Sagartz JE, Bryant LA, Labovitz AJ, Gala SS, et al. ECG detection of murine chagasic cardiomyopathy. J Parasitol (2010) 96:758–64. doi:10.1645/GE-2396.1
61. Saraiva LR, Santos CL, de Aguiar IR. The prolongation of the QT interval in the acute rheumatic carditis: an enigma. Arq Bras Cardiol (2006) 87:e254–6. doi:10.1590/S0066-782X2006001900025
62. Kaul UA, Gambhir DS, Khalilullah M. Torsade de pointes: manifestation of acute rheumatic carditis. Indian Heart J (1983) 35:117–9.
63. Liberman L, Hordof AJ, Alfayyadh M, Salafia CM, Pass RH. Torsade de pointes in a child with acute rheumatic fever. J Pediatr (2001) 138:280–2. doi:10.1067/mpd.2001.110302
64. Maradit-Kremers H, Crowson CS, Nicola PJ, Ballman KV, Ballman KV, Roger VL, et al. Increased unrecognized coronary heart disease and sudden death in rheumatoid arthritis. A population-based cohort study. Arthritis Rheum (2005) 52:402–11. doi:10.1002/art.20878
65. Lazzerini PE, Capecchi PL, Acampa M, Galeazzi M, Laghi-Pasini F. Arrhythmic risk in rheumatoid arthritis: the driving role of systemic inflammation. Autoimmun Rev (2014) 13:936–44. doi:10.1016/j.autrev.2014.05.007
66. Hodak SP, Moubarak JB, Rodriguez I, Gelfand MC, Alijani MR, Tracy CM. QT prolongation and near fatal cardiac arrhythmia after intravenous tacrolimus administration: a case report. Transplantation (1998) 66:535–7. doi:10.1097/00007890-199808270-00021
67. Amankwa K, Krishnan SC, Tisdale JE. Torsades de pointes associated with fluoroquinolones: importance of concomitant risk factors. Clin Pharmacol Ther (2004) 75(3):242–7. doi:10.1016/j.clpt.2003.11.376
68. Letsas KP, Alexanian IP, Pappas LK, Kounas SP, Efremidis M, Sideris A, et al. QT interval prolongation and torsade de pointes associated with indapamide. Int J Cardiol (2006) 112:373–4. doi:10.1016/j.ijcard.2005.07.055
69. Chen CY, Wang FL, Lin CC. Chronic hydroxychloroquine use associated with QT prolongation and refractory ventricular arrhythmia. Clin Toxicol (2006) 44:173–5. doi:10.1080/15563650500514558
70. Pham CP, de Feiter PW, van der Kuy PH, van Mook WN. Long QTc interval and torsade de pointes caused by fluconazole. Ann Pharmacother (2006) 40:1456–61. doi:10.1345/aph.1G741
71. Heinrich TW, Biblo LA, Schneider J. Torsades de pointes associated with ziprasidone. Psychosomatics (2006) 47:264–8. doi:10.1176/appi.psy.47.3.264
72. Ahmed N, Rai R, Riaz K, Khalid MA, Wase A. Levofloxacin-induced torsades de pointes. Resid Staff Physician (2007) 53:51–4.
73. Stas P, Faes D, Noyens P. Conduction disorder and QT prolongation secondary to long-term treatment with chloroquine. Int J Cardiol (2008) 127:e80–2. doi:10.1016/j.ijcard.2007.04.055
74. Newton-Cheh C, Lin AE, Baggish AL, Wang H. Case records of the Massachusetts General Hospital. Case 11-2011. A 47-year-old man with systemic lupus erythematosus and heart failure. N Engl J Med (2011) 364:1450–60. doi:10.1056/NEJMcpc1011319
75. Kandan SR, Saha M. Severe primary hypothyroidism presenting with torsades de pointes. BMJ Case Rep (2012) 2012. doi:10.1136/bcr.12.2011.5306
76. Curione M, Aratari A, Amato S, Colotto M, Barbato M, Leone S, et al. A study on QT interval in patients affected with inflammatory bowel disease without cardiac involvement. Intern Emerg Med (2010) 5:307–10. doi:10.1007/s11739-010-0382-9
77. Simsek H, Sahin M, Akyol A, Akdag S, Ozkol HU, Gumrukcuoglu HA, et al. Increased risk of atrial and ventricular arrhythmia in long-lasting psoriasis patients. ScientificWorldJournal (2013) 2013:901215. doi:10.1155/2013/901215
78. Albert CM, Ma J, Rifai N, Stampfer MJ, Ridker PM. Prospective study of C-reactive protein, homocysteine, and plasma lipid levels as predictors of sudden cardiac death. Circulation (2002) 105:2595–9. doi:10.1161/01.CIR.0000017493.03108.1C
79. Hussein AA, Gottdiener JS, Bartz TM, Sotoodehnia N, DeFilippi C, See V, et al. Inflammation and sudden cardiac death in a community-based population of older adults: the cardiovascular health study. Heart Rhythm (2013) 10:1425–32. doi:10.1016/j.hrthm.2013.07.004
80. London B, Baker LC, Lee JS, Shusterman V, Choi BR, Kubota T, et al. Calcium-dependent arrhythmias in transgenic mice with heart failure. Am J Physiol Heart Circ Physiol (2003) 284:H431–41. doi:10.1152/ajpheart.00431.2002
81. Petkova-Kirova PS, Gursoy E, Mehdi H, McTiernan CF, London B, Salama G. Electrical remodeling of cardiac myocytes from mice with heart failure due to the overexpression of tumor necrosis factor-alpha. Am J Physiol Heart Circ Physiol (2006) 290:H2098–107. doi:10.1152/ajpheart.00097.2005
82. Kawada H, Niwano S, Niwano H, Yumoto Y, Wakisaka Y, Yuge M, et al. Tumor necrosis factor-alpha downregulates the voltage gated outward K+ current in cultured neonatal rat cardiomyocytes: a possible cause of electrical remodeling in diseased hearts. Circ J (2006) 70:605–9. doi:10.1253/circj.70.605
83. Fernández-Velasco M, Ruiz-Hurtado G, Hurtado O, Moro MA, Delgado C. TNF-alpha downregulates transient outward potassium current in rat ventricular myocytes through iNOS overexpression and oxidant species generation. Am J Physiol Heart Circ Physiol (2007) 293:H238–45. doi:10.1152/ajpheart.01122.2006
84. Panama BK, Latour-Villamil D, Farman GP, Zhao D, Bolz SS, Kirshenbaum LA, et al. Nuclear factor kappaB downregulates the transient outward potassium current I(to, f) through control of KChIP2 expression. Circ Res (2011) 108:537–43. doi:10.1161/CIRCRESAHA.110.229112
85. Wang J, Wang H, Zhang Y, Gao H, Nattel S, Wang Z. Impairment of HERG K(+) channel function by tumor necrosis factor-alpha: role of reactive oxygen species as a mediator. J Biol Chem (2004) 279:13289–92. doi:10.1074/jbc.C400025200
86. Li YH, Rozanski GJ. Effects of human recombinant interleukin-1 on electrical properties of guinea pig ventricular cells. Cardiovasc Res (1993) 27:525–30. doi:10.1093/cvr/27.3.525
87. Hagiwara Y, Miyoshi S, Fukuda K, Nishiyama N, Ikegami Y, Tanimoto K, et al. SHP2-mediated signaling cascade through gp130 is essential for LIF-dependent I CaL, [Ca2+]i transient, and APD increase in cardiomyocytes. J Mol Cell Cardiol (2007) 43:710–6. doi:10.1016/j.yjmcc.2007.09.004
88. Less H, Shilkrut M, Rubinstein I, Berke G, Binah O. Cardiac dysfunction in murine autoimmune myocarditis. J Autoimmun (1999) 12:209–20. doi:10.1006/jaut.1998.0273
89. Park H, Park H, Lee D, Oh S, Lim J, Hwang HJ, et al. Increased phosphorylation of Ca(2+) handling proteins as a proarrhythmic mechanism in myocarditis. Circ J (2014) 78:2292–301. doi:10.1253/circj.CJ-14-0277
90. Tang Q, Huang J, Qian H, Chen L, Wang T, Wang H, et al. Antiarrhythmic effect of atorvastatin on autoimmune myocarditis is mediated by improving myocardial repolarization. Life Sci (2007) 80:601–8. doi:10.1016/j.lfs.2006.11.045
91. Lazzerini PE, Capecchi PL, Laghi-Pasini F. Statins as a new therapeutic perspective in myocarditis and postmyocarditis dilated cardiomyopathy. Cardiovasc Drugs Ther (2013) 27:365–9. doi:10.1007/s10557-013-6475-8
92. De Jesus NM, Wang L, Herren AW, Wang J, Shenasa F, Bers DM, et al. Atherosclerosis exacerbates arrhythmia after myocardial infarction: role of myocardial inflammation. Heart Rhythm (2014) 12:169–78. doi:10.1016/j.hrthm.2014.10.007
93. Monnerat-Cahli G, Alonso H, Gallego M, Alarcón ML, Bassani RA, Casis O, et al. Toll-like receptor 4 activation promotes cardiac arrhythmias by decreasing the transient outward potassium current (Ito) through an IRF3-dependent and MyD88-independent pathway. J Mol Cell Cardiol (2014) 76:116–25. doi:10.1016/j.yjmcc.2014.08.012
94. Elenkov IJ, Wilder RL, Chrousos GP, Vizi S. The sympathetic nerve – an integrative interface between two supersystems: the brain and the immune system. Pharmacol Rev (2000) 52:595–638.
96. Martelli D, Yao ST, McKinley MJ, McAllen RM. Reflex control of inflammation by sympathetic nerves, not the vagus. J Physiol (2014) 592:1677–86. doi:10.1113/jphysiol.2013.268573
97. Lazzerini PE, Acampa M, Hammoud M, Maffei S, Capecchi PL, Selvi E, et al. Arrhythmic risk during acute infusion of infliximab: a prospective, single-blind, placebo-controlled, crossover study in patients with chronic arthritis. J Rheumatol (2008) 35:1958–65.
98. Wu CT, Nattel S. Triggering of cardiac arrhythmic events in long QT syndrome: lessons from funny bunnies. J Physiol (2012) 590:1311–2. doi:10.1113/jphysiol.2012.229344
99. Xie X, Visweswaran R, Guzman PA, Smith RM, Osborn JW, Tolkacheva EG. The effect of cardiac sympathetic denervation through bilateral stellate ganglionectomy on electrical properties of the heart. Am J Physiol Heart Circ Physiol (2011) 301:H192–9. doi:10.1152/ajpheart.01149.2010
100. Baumert M, Lambert GW, Dawood T, Lambert EA, Esler MD, McGrane M, et al. QT interval variability and cardiac norepinephrine spillover in patients with depression and panic disorder. Am J Physiol Heart Circ Physiol (2008) 295:H962–8. doi:10.1152/ajpheart.00301.2008
101. Kaufman ES, Gorodeski EZ, Dettmer MM, Dikshteyn M. Use of autonomic maneuvers to probe phenotype/genotype discordance in congenital long QT syndrome. Am J Cardiol (2005) 96:1425–30. doi:10.1016/j.amjcard.2005.07.046
102. Vyas H, Hejlik J, Ackerman MJ. Epinephrine QT stress testing in the evaluation of congenital long-QT syndrome: diagnostic accuracy of the paradoxical QT response. Circulation (2006) 113:1385–92. doi:10.1161/CIRCULATIONAHA.105.600445
103. Noda T, Takaki H, Kurita T, Suyama K, Nagaya N, Taguchi A, et al. Gene-specific response of dynamic ventricular repolarization to sympathetic stimulation in LQT1, LQT2 and LQT3 forms of congenital long QT syndrome. Eur Heart J (2002) 23:975–83. doi:10.1053/euhj.2001.3079
104. Stein PK, Bosner MS, Kleiger RE, Conger BM. Heart rate variability: a measure of cardiac autonomic tone. Am Heart J (1994) 127:1376–81. doi:10.1016/0002-8703(94)90059-0
105. Stojanovich L. Autonomic dysfunction in autoimmune rheumatic disease. Autoimmun Rev (2009) 8:569–72. doi:10.1016/j.autrev.2009.01.018
106. Adlan AM, Lip GY, Paton JF, Kitas GD, Fisher JP. Autonomic function and rheumatoid arthritis-A systematic review. Semin Arthritis Rheum (2014) 44:283–304. doi:10.1016/j.semarthrit.2014.06.003
107. Gao X, Peng L, Zeng Q, Wu ZK. Autonomic nervous function and arrhythmias in patients with acute viral myocarditis during a 6-month follow-up period. Cardiology (2009) 113:66–71. doi:10.1159/000167794
108. Karacan M, Ceviz N, Olgun H. Heart rate variability in children with acute rheumatic fever. Cardiol Young (2012) 22:285–92. doi:10.1017/S1047951111001429
109. Haensel A, Mills PJ, Nelesen RA, Ziegler MG, Dimsdale JE. The relationship between heart rate variability and inflammatory markers in cardiovascular diseases. Psychoneuroendocrinology (2008) 33:1305–12. doi:10.1016/j.psyneuen.2008.08.007
110. Malave HA, Taylor AA, Nattama J, Deswal A, Mann DL. Circulating levels of tumor necrosis factor correlate with indexes of depressed heart rate variability: a study in patients with mild-to-moderate heart failure. Chest (2003) 123:716–24. doi:10.1378/chest.123.3.716
111. Lazzerini PE, Capecchi PL, Guideri F, Acampa M, Galeazzi M, Laghi Pasini F. Connective tissue diseases and cardiac rhythm disorders: an overview. Autoimmun Rev (2006) 5:306–13. doi:10.1016/j.autrev.2005.11.002
112. Lazzerini PE, Capecchi PL, Guideri F, Acampa M, Selvi E, Bisogno S, et al. Autoantibody-mediated cardiac arrhythmias: mechanisms and clinical implications. Basic Res Cardiol (2008) 103:1–11. doi:10.1007/s00395-007-0686-8
113. Lee HC, Huang KT, Wang XL, Shen WK. Autoantibodies and cardiac arrhythmias. Heart Rhythm (2011) 8:1788–95. doi:10.1016/j.hrthm.2011.06.032
114. Lazzerini PE, Capecchi PL, Laghi Pasini F. Anti-Ro/SSA antibodies and cardiac arrhythmias in the adult: facts and hypotheses. Scand J Immunol (2010) 72:213–22. doi:10.1111/j.1365-3083.2010.02428.x
115. Ambrosi A, Sonesson SE, Wahren-Herlenius M. Molecular mechanisms of congenital heart block. Exp Cell Res (2014) 325:2–9. doi:10.1016/j.yexcr.2014.01.003
116. Lazzerini PE, Capecchi PL, Laghi-Pasini F. Isolated heart block of unknown origin in adults and anti-Ro/SSA antibodies: clinical evidence, putative mechanisms and therapeutic implications. Heart Rhythm (2015) 12:449–54. doi:10.1016/j.hrthm.2014.10.031
117. Cimaz R, Stramba-Badiale M, Brucato A, Catelli L, Panzeri P, Meroni PL. QT interval prolongation in asymptomatic anti-SSA/Ro–positive infants without congenital heart block. Arthritis Rheum (2000) 43:1049–53. doi:10.1002/1529-0131(200005)43:5<1049::AID-ANR13>3.0.CO;2-X
118. Cimaz R, Meroni PL, Brucato A, Fesstova V, Panzeri P, Goulene K, et al. Concomitant disappearance of electrocardiographic abnormalities and of acquired maternal autoantibodies during the first year of life in infants who had QT interval prolongation and anti-SSA/Ro positivity without congenital heart block at birth. Arthritis Rheum (2003) 48:266–8. doi:10.1002/art.10700
119. Gordon PA, Khamashta MA, Hughes GR, Rosenthal E. Increase in the heart rate-corrected QT interval in children of anti-Ro-positive mothers, with a further increase in those with siblings with congenital heart block: comment on the article by Cimaz et al. Arthritis Rheum (2001) 44:242–3. doi:10.1002/1529-0131(200101)44:1<242::AID-ANR34>3.0.CO;2-S
120. Nakamura K, Katayama Y, Kusano KF, Haraoka K, Tani Y, Nagase S, et al. Anti-KCNH2 antibody-induced long QT syndrome: novel acquired form of long QT syndrome. J Am Coll Cardiol (2007) 50:1808–9. doi:10.1016/j.jacc.2007.07.037
121. Gordon PA, Rosenthal E, Khamashta MA, Hughes GR. Absence of conduction defects in the electrocardiograms [correction of echocardiograms] of mothers with children with congenital complete heart block. J Rheumatol (2001) 28:366–9.
122. Qu Y, Xiao GQ, Chen L, Boutjdir M. Autoantibodies from mothers of children with congenital heart block downregulate cardiac L-type Ca channels. J Mol Cell Cardiol (2001) 33:1153–63. doi:10.1006/jmcc.2001.1379
123. Costedoat-Chalumeau N, Amoura Z, Lupoglazoff JM, Huong DL, Denjoy I, Vauthier D, et al. Outcome of pregnancies in patients with anti-SSA/Ro antibodies: a study of 165 pregnancies, with special focus on electrocardiographic variations in the children and comparison with a control group. Arthritis Rheum (2004) 50:3187–94. doi:10.1002/art.20554
124. Motta M, Rodriguez-Perez C, Tincani A, Lojacono A, Chirico G. Outcome of infants from mothers with anti-SSA/Ro antibodies. J Perinatol (2007) 27:278–83. doi:10.1038/sj.jp.7211688
125. Gerosa M, Cimaz R, Stramba-Badiale M, Goulene K, Meregalli E, Trespidi L, et al. Electrocardiographic abnormalities in infants born from mothers with autoimmune diseases-a multicentre prospective study. Rheumatology (2007) 46:1285–9. doi:10.1093/rheumatology/kem073
126. Nomura A, Kishimoto M, Takahashi O, Deshpande GA, Yamaguchi K, Okada M. Prolongation of heart rate-corrected QT interval is a predictor of cardiac autonomic dysfunction in patients with systemic lupus erythematosus. Rheumatol Int (2014) 34:643–7. doi:10.1007/s00296-013-2718-z
127. Parker JC, Burlingame RW, Bunn CC. Prevalence of antibodies to Ro-52 in a serologically defined population of patients with systemic sclerosis. J Autoimmune Dis (2009) 6:2. doi:10.1186/1740-2557-6-2
128. Lazzerini PE, Capecchi PL, Boutjdir M, Laghi-Pasini F. Comment on “absence of an association between anti-Ro antibodies and prolonged QTc interval in systemic sclerosis: a multicenter study of 689 patients”. Semin Arthritis Rheum (2015) 44:e16–7. doi:10.1016/j.semarthrit.2014.10.002
129. Akerström F, Arias MA, Casares-Medrano J, Pachón M, Puchol A, Rodríguez-Picón B, et al. Biventricular pacing-induced torsade de pointes. J Am Coll Cardiol (2012) 60:e23. doi:10.1016/j.jacc.2012.02.086
130. Digby GC, Pérez Riera AR, Barbosa Barros R, Simpson CS, Redfearn DP, Methot M, et al. Acquired long QT interval: a case series of multifactorial QT prolongation. Clin Cardiol (2011) 34:577–82. doi:10.1002/clc.20945
131. Silvetti MS, Drago F, Bevilacqua M, Ragonese P. Amiodarone-induced torsade de pointes in a child with dilated cardiomyopathy. Ital Heart J (2001) 2:231–6.
132. Yamada S, Kuga K, Yamaguchi I. Torsade de pointes induced by intravenous and long-term oral amiodarone therapy in a patient with dilated cardiomyopathy. Jpn Circ J (2001) 65:236–8. doi:10.1253/jcj.65.236
133. Vecchia L, Ometto R, Finocchi G, Vincenzi M. Torsade de pointes ventricular tachycardia during low dose intermittent dobutamine treatment in a patient with dilated cardiomyopathy and congestive heart failure. Pacing Clin Electrophysiol (1999) 22:397–9. doi:10.1111/j.1540-8159.1999.tb00461.x
134. Faber TS, Zehender M, Van de Loo A, Hohnloser S, Just H. Torsade de pointes complicating drug treatment of low-malignant forms of arrhythmia: four cases reports. Clin Cardiol (1994) 17:197–202. doi:10.1002/clc.4960170410
135. European Heart Rhythm Association, Heart Rhythm Society, Zipes DP, Camm AJ, Borggrefe M, Buxton AE, et al. ACC/AHA/ESC 2006 guidelines for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: a report of the American College of Cardiology/American Heart Association Task Force and the European Society of Cardiology Committee for practice guidelines (writing committee to develop guidelines for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death). J Am Coll Cardiol (2006) 48:e247–346.
136. Iacoviello M, Forleo C, Guida P, Romito R, Sorgente A, Sorrentino S, et al. Ventricular repolarization dynamicity provides independent prognostic information toward major arrhythmic events in patients with idiopathic dilated cardiomyopathy. J Am Coll Cardiol (2007) 50:225–31. doi:10.1016/j.jacc.2007.02.071
137. Lappé JM, Pelfrey CM, Tang WH. Recent insights into the role of autoimmunity in idiopathic dilated cardiomyopathy. J Card Fail (2008) 14:521–30. doi:10.1016/j.cardfail.2008.02.016
138. Störk S, Boivin V, Horf R, Hein L, Lohse MJ, Angermann CE, et al. Stimulating autoantibodies directed against the cardiac beta1-adrenergic receptor predict increased mortality in idiopathic cardiomyopathy. Am Heart J (2006) 152:697–704. doi:10.1016/j.ahj.2006.05.004
139. Iwata M, Yoshikawa T, Baba A, Anzai T, Mitamura H, Ogawa S. Autoantibodies against the second extracellular loop of beta1-adrenergic receptors predict ventricular tachycardia and sudden death in patients with idiopathic dilated cardiomyopathy. J Am Coll Cardiol (2001) 37:418–24. doi:10.1016/S0735-1097(00)01109-8
140. Fukuda Y, Miyoshi S, Tanimoto K, Oota K, Fujikura K, Iwata M, et al. Autoimmunity against the second extracellular loop of beta(1)-adrenergic receptors induces early afterdepolarization and decreases in K-channel density in rabbits. J Am Coll Cardiol (2004) 43:1090–100. doi:10.1016/j.jacc.2003.09.057
141. Suzuki S, Utsugisawa K, Yoshikawa H, Motomura M, Matsubara S, Yokoyama K, et al. Autoimmune targets of heart and skeletal muscles in myasthenia gravis. Arch Neurol (2009) 66:1334–8. doi:10.1001/archneurol.2009.229
142. Yellen G. The voltage-gated potassium channels and their relatives. Nature (2002) 419:35–42. doi:10.1038/nature00978
143. Niwa N, Nerbonne JM. Molecular determinants of cardiac transient outward potassium current (I(to)) expression and regulation. J Mol Cell Cardiol (2010) 48:12–25. doi:10.1016/j.yjmcc.2009.07.013
144. Suzuki S, Satoh T, Yasuoka H, Hamaguchi Y, Tanaka K, Kawakami Y, et al. Novel autoantibodies to a voltage-gated potassium channel Kv1.4 in a severe form of myasthenia gravis. J Neuroimmunol (2005) 170:141–9. doi:10.1016/j.jneuroim.2005.08.017
145. Suzuki S, Baba A, Kaida K, Utsugisawa K, Kita Y, Tsugawa J, et al. Cardiac involvements in myasthenia gravis associated with anti-Kv1.4 antibodies. Eur J Neurol (2014) 21:223–30. doi:10.1111/ene.12234
146. Hirose K, Yamaguchi H, Oshima Y, Choraku M, Hirono A, Takamori N, et al. Severe respiratory failure and torsades de pointes induced by disopyramide in a patient with myasthenia gravis. Intern Med (2008) 47:1703–8. doi:10.2169/internalmedicine.47.0976
147. Valbusa A, Ingrassia S, Rosa GM, Infante MT, Schenone A, Montecucco F, et al. Takotsubo cardiomyopathy and torsade de pointes in myasthenic crisis: be aware of QT prolongation. Am J Emerg Med (2013) 31:1717–8. doi:10.1016/j.ajem.2013.09.030
148. Karnabi E, Boutjdir M. Role of calcium channels in congenital heart block. Scand J Immunol (2010) 72:226–34. doi:10.1111/j.1365-3083.2010.02439.x
149. Qu Y, Baroudi G, Yue Y, Boutjdir M. Novel molecular mechanism involving α1D (Cav 1.3) L-Type calcium channel in autoimmune-associated sinus bradycardia. Circulation (2005) 111:3034–41. doi:10.1161/CIRCULATIONAHA.104.517326
150. Karnabi E, Qu Y, Wadgaonkar R, Mancarella S, Yue Y, Chahine M, et al. Congenital heart block: identification of autoantibody binding site on the extracellular loop (domain I, S5-S6) of alpha(1D) L-type Ca channel. J Autoimmun (2010) 34:80–6. doi:10.1016/j.jaut.2009.06.005
151. Strandberg LS, Cui X, Rath A, Liu J, Silverman ED, Liu X, et al. Congenital heart block maternal sera autoantibodies target an extracellular epitope on the α1G T-type calcium channel in human fetal hearts. PLoS One (2013) 8:e72668. doi:10.1371/journal.pone.0072668
152. Boutjdir M, Chen L, Zhang ZH, Tseng CE, DiDonato F, Rashbaum W, et al. Arrhythogenicity of IgG and anti-52-kD SSA/Ro affinity-purified antibodies from mothers of children with congenital heart block. Circ Res (1997) 80:354–62. doi:10.1161/01.RES.80.3.354
153. Xiao GQ, Qu Y, Hu K, Boutjdir M. Down-regulation of L-type calcium channel in pups born to 52 kDa SSA/Ro immunized rabbits. FASEB J (2001) 15:1539–45. doi:10.1096/fj.01-0052com
154. Pohorille A, Schweighofer K, Wilson MA. The origin and early evolution of membrane channels. Astrobiology (2005) 5:1–17. doi:10.1089/ast.2005.5.1
155. Yue Y, Castrichini M, Srivastava U, Fabris F, Shah K, Li Z, et al. Pathogenesis of the novel autoimmune-associated long QT syndrome. Circulation (2015). doi:10.1161/CIRCULATIONAHA.115.009800
156. Saribayev M, Tufan F, Oz F, Erer B, Ozpolat T, Ozturk GB, et al. Corticosteroid treatment normalizes QTc prolongation and improves heart block in an elderly patient with anti-Ro-positive systemic lupus erythematosus. Aging Clin Exp Res (2014) 26:337–9. doi:10.1007/s40520-013-0168-9
157. Santos-Pardo I, Martínez-Morillo M, Villuendas R, Bayes-Genis A. Anti-Ro antibodies and reversible atrioventricular block. N Engl J Med (2013) 368:2335–7. doi:10.1056/NEJMc1300484
158. Santos-Pardo I, Villuendas R, Salvador I, Martinez-Morillo M, Olivè A, Bayes-Genis A. Anti-Ro/SSA antibodies and cardiac rhythm disturbances: present and future perspectives. Int J Cardiol (2015) 184:244–50. doi:10.1016/j.ijcard.2014.11.002
159. Dugar M, Cox S, Limaye V, Gordon TP, Roberts-Thomson PJ. Diagnostic utility of anti-Ro52 detection in systemic autoimmunity. Postgrad Med J (2010) 86:79–82. doi:10.1136/pgmj.2009.089656
160. Patel C, Antzelevitch C. Pharmacological approach to the treatment of long and short QT syndromes. Pharmacol Ther (2008) 118:138–51. doi:10.1016/j.pharmthera.2008.02.001
161. Christ T, Wettwer E, Dobrev D, Adolph E, Knaut M, Wallukat G, et al. Autoantibodies against the beta1 adrenoceptor from patients with dilated cardiomyopathy prolong action potential duration and enhance contractility in isolated cardiomyocytes. J Mol Cell Cardiol (2001) 33:1515–25. doi:10.1006/jmcc.2001.1414
162. Rizzo S, Basso C, Troost D, Aronica E, Frigo AC, Driessen AH, et al. T-cell-mediated inflammatory activity in the stellate ganglia of patients with ion-channel disease and severe ventricular arrhythmias. Circ Arrhythm Electrophysiol (2014) 7:224–9. doi:10.1161/CIRCEP.113.001184
163. James TN, Zipes DP, Finegan RE, Eisele JW, Carter JE. Cardiac ganglionitis associated with sudden unexpected death. Ann Intern Med (1979) 91:727–30. doi:10.7326/0003-4819-91-5-727
164. James TN, Froggatt P, Atkinson WJ Jr, Lurie PR, McNamara DG, Miller WW, et al. De subitaneis mortibus. XXX. Observations on the pathophysiology of the long QT syndromes with special reference to the neuropathology of the heart. Circulation (1978) 57:1221–31. doi:10.1161/01.CIR.57.6.1221
165. Pfeiffer D, Fiehring H, Henkel HG, Rostock KJ, Rathgen K. Long QT syndrome associated with inflammatory degeneration of the stellate ganglia. Clin Cardiol (1989) 12:222–4. doi:10.1002/clc.4960120408
166. Moss AJ, Ryan DH, Yeaney GA. Ganglionitis and genetic cardiac arrhythmias: more questions than answers. Circ Arrhythm Electrophysiol (2014) 7:190–1. doi:10.1161/CIRCEP.114.001589
167. Li J, Maguy A, Duverger JE, Vigneault P, Comtois P, Shi Y, et al. Induced KCNQ1 autoimmunity accelerates cardiac repolarization in rabbits: potential significance in arrhythmogenesis and antiarrhythmic therapy. Heart Rhythm (2014) 11:2092–100. doi:10.1016/j.hrthm.2014.07.040
168. Li J, Seyler C, Wiedmann F, Schmidt C, Schweizer PA, Becker R, et al. Anti-KCNQ1 K+ channel autoantibodies increase IKs current and are associated with QT interval shortening in dilated cardiomyopathy. Cardiovasc Res (2013) 98:496–503. doi:10.1093/cvr/cvt046
169. Drew BJ, Ackerman MJ, Funk M, Gibler WB, Kligfield P, Menon V, et al. Prevention of torsade de pointes in hospital settings: a scientific statement from the American Heart Association and the American College of Cardiology Foundation. J Am Coll Cardiol (2010) 55:934–47. doi:10.1016/j.jacc.2010.01.001
170. Abu-Shakra M, Urowitz MB, Gladman DD, Gough J. Mortality studies in systemic lupus erythematosus. Results from a single center. I. Causes of death. J Rheumatol (1995) 22(7):1259–64.
171. Silva Marques J, Veiga A, Nóbrega J, Correia MJ, de Sousa J. Electrical storm induced by H1N1 A influenza infection. Europace (2010) 12:294–5. doi:10.1093/europace/eup430
172. Lim SM, Pak HN, Lee MH, Kim SS, Joung B. Fever-induced QTc prolongation and ventricular fibrillation in a healthy young man. Yonsei Med J (2011) 52:1025–7. doi:10.3349/ymj.2011.52.6.1025
173. Amin AS, Klemens CA, Verkerk AO, Meregalli PG, Asghari-Roodsari A, de Bakker JM, et al. Fever-triggered ventricular arrhythmias in Brugada syndrome and type 2 long-QT syndrome. Neth Heart J (2010) 18:165–9. doi:10.1007/BF03091755
Keywords: long QT syndrome, inflammation, cytokines, immunity, autoantibodies, anti-Ro/SSA
Citation: Lazzerini PE, Capecchi PL and Laghi-Pasini F (2015) Long QT syndrome: an emerging role for inflammation and immunity. Front. Cardiovasc. Med. 2:26. doi: 10.3389/fcvm.2015.00026
Received: 16 February 2015; Accepted: 08 May 2015;
Published: 27 May 2015
Edited by:
Theofilos M. Kolettis, University of Ioannina, GreeceReviewed by:
Luigi Venetucci, University of Manchester, UKCopyright: © 2015 Lazzerini, Capecchi and Laghi-Pasini. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pietro Enea Lazzerini, Department of Medical Sciences, Surgery and Neurosciences, University of Siena, Policlinico “Le Scotte”, Viale Bracci, Siena, 53100, Italy,bGF6emVyaW5pN0B1bmlzaS5pdA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.