 Emma M. Cagwin
Emma M. Cagwin Caitlin M. Padgett
Caitlin M. Padgett Yvonne Lin
Yvonne Lin Wen Zhu
Wen Zhu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Biophys. , 04 April 2025
Sec. Protein Structure and Dynamics
Volume 3 - 2025 | https://doi.org/10.3389/frbis.2025.1560824
TBCK is an essential protein in neurodevelopment. Mutations in the TBCK gene are associated with TBCK Syndrome, a genetic neurological disorder characterized by global developmental delay. TBCK is an enigmatic multidomain protein that contains a pseudokinase domain, a TBC (Tre2-Bub2-Cdc16) domain, and a rhodanese-like domain. Emerging evidence increasingly links TBCK to multiple cellular processes, including mTOR signaling, autophagy, lysosomal function, and mitochondrial maintenance. This review consolidates recent advances in our understanding of TBCK, emphasizing comparative sequence analysis, structural modeling, and its cellular functions. Our analysis shows that both the protein kinase domain and the rhodanese-like domain likely lack catalytic activity and instead primarily function as scaffolds or regulatory domains. The TBC domain contains all the conserved residues, suggesting it may act as a GTPase-activating protein (GAP). These functional hypotheses provide a foundation for further investigations into TBCK’s physiological and pathological roles.
Identifying the genetic root of neurological disorders caused by inborn errors is often challenging because these disorders sometimes present with overlapping features, such as intellectual disability and global developmental delay, despite arising from distinct genetic mutations in hundreds of different genes (Chelly et al., 2006; Rexach et al., 2019). The growing application of next-generation sequencing in clinical settings significantly advanced the molecular etiology of neurological disorders, which enabled the identification and classification of numerous new neurological disorders in recent years (Rexach et al., 2019). Whole genome sequencing not only offers crucial opportunities for early diagnosis, but it also sheds light on the dark proteome that could be valuable for developing targeted treatments (Savatt and Myers, 2021). TBCK Syndrome, named after the human protein TBCK, is a compelling example of such progress (Durham et al., 2023).
TBCK Syndrome, TBCK encephaloneuronopathy or infantile hypotonia with psychomotor retardation and characteristic facies type 3, was first classified as an inborn genetic error in 2015 (Alazami et al., 2015). TBCK was identified as a novel candidate recessive gene associated with TBCK Syndrome through a combination of autozygosity mapping and whole-exome sequencing of samples from a single family that had a history of neurological disorders. This family had a mutation in the TBCK gene (NM 033115:c.1708+1G>A), located on chromosome 4 (Alazami et al., 2015). Following this breakthrough, thirteen individuals from nine unrelated families of Saudi, Syrian, Pakistani, and mixed European descent with intellectual disabilities were identified as carrying biallelic TBCK mutations (Bhoj et al., 2016). These individuals exhibited global developmental delays, affecting motor, speech, and cognitive abilities (Bhoj et al., 2016). Many cases exhibited hypotonia, seizures, unusual facial features, and macrocephaly (Bhoj et al., 2016). Neuroimaging also found distinct white matter changes and abnormalities in the periventricular area in the brain. How and why TBCK mutations lead to these phenotypes remain enigmatic.
TBCK is a protein of unknown function composed of three distinct domains: a protein kinase domain, a TBC (Tre2-Bub2-Cdc16) domain, and a rhodanese-like domain (Figure 1A). When modeled using AlphaFold3, an artificial intelligence (AI)-driven structural prediction tool, these three well-folded domains are predicted to be connected by linker regions with medium to very low confidence scores (pLDDT < 90) (Figure 1B) (Jumper et al., 2021; Varadi et al., 2022). AlphaMissense, another AI-driven large language model that integrates structural predictions with sequence context to assess the pathogenicity of single nucleotide substitutions leading to missense mutations, suggests that pathogenic mutations can occur throughout TBCK, potentially causing structural impairments that disrupt protein function (Cheng et al., 2023). Notably, the distribution of the likely pathogenic variants in TBCK is not uniform but mostly clusters within the TBC domain (Figure 1A). Although these computational tools provide intriguing predictions of the structure and function relationship of TBCK, their accuracy requires experimental validation. While much of the existing literature focuses on the clinical manifestations of TBCK syndrome, this article explores recent advances in sequence and structural homology predictions, molecular and cellular functional analysis, and the disease relevance of the TBCK protein. These insights provide a foundation for future investigations into TBCK’s physiological and pathological roles. A deeper understanding of its biochemical and biophysical properties, as well as its cellular functions of TBCK will also be crucial for evaluating its druggability and therapeutic potential in neurodevelopment disorders.
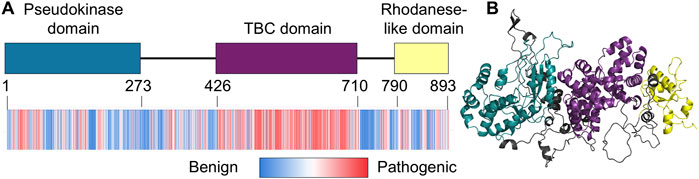
Figure 1. TBCK domain architecture. (A) Domain architecture of human TBCK (UniProt: Q8TEA7), showing the pseudokinase domain (teal), TBC domain (purple), and rhodanese-like domain (yellow). The heat map below highlights benign and pathogenic residues within the TBCK sequence, as predicted by AlphaMissense. (B) Predicted tertiary structure of TBCK (AF-Q8TEA7-F1-v4), with low complexity region (black) and color-coded domains: pseudokinase domain (teal), TBC domain (purple), and rhodanese-like domain (yellow).
TBCK homologs have been identified across various eukaryotic species (Figure 2A). Among the three protein domains, the TBC domain exhibits the highest level of conservation across eukaryotes (Figure 2B). Notably, some organisms, such as Allomyces macrogynus and Stylophora pistillata, possess truncated TBCK homologs lacking rhodanese and the TBC domain, respectively. Since the functional contribution of each domain remains unclear, these shortened homologs may serve distinct roles in their native organisms.
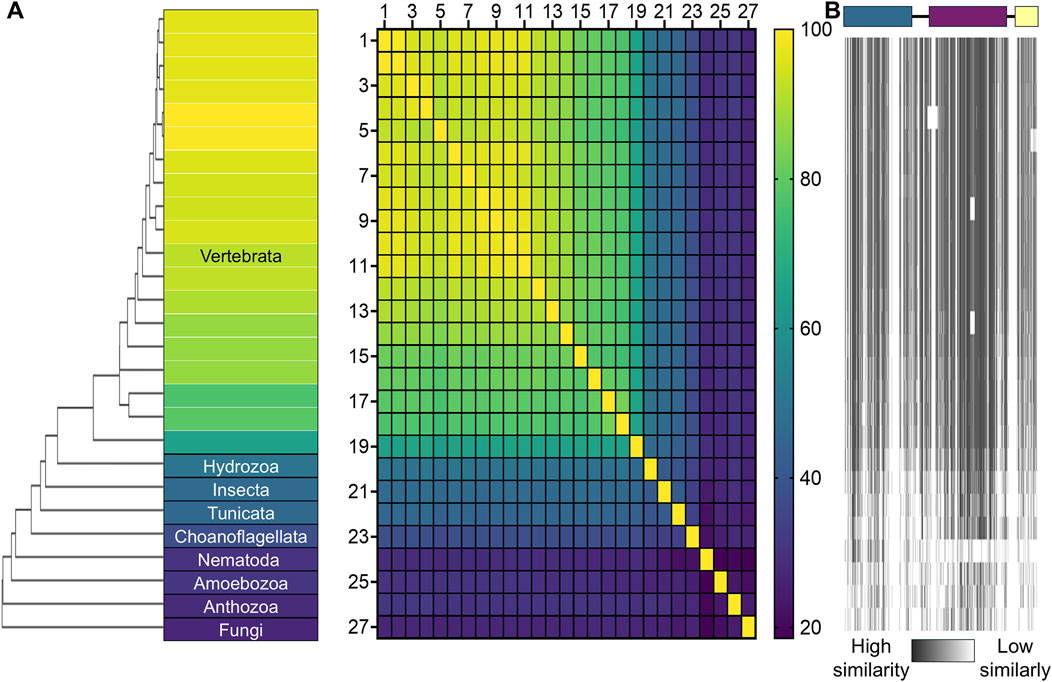
Figure 2. Sequence similarity of TBCK across different organisms. (A) A phylogenetic tree is illustrated using a gradient heat map, where yellow represents 100% sequence identity and dark purple represents 20% identity. The heat map displays the percentage identity matrix for TBCK sequences across 27 species. The species and UniProt IDs are: 1. Homo sapiens (Q8TEA7), 2. Pan troglodytes (K7BBM9), 3. Mus musculus (Q8BM85), 4. Rattus norvegicus (D3ZA89), 5. Trichechus manatus latirostris (A0A2Y9R7R3), 6. Myotis lucifugus (G1PKH5), 7. Equus caballus (F7D9M8), 8. Felis catus (M3WKS8), 9. Canis familiaris (A0A8I3QGU0), 10. Sus scrofa (A0A4X1UU08), 11. Bos taurus (F1MSR5), 12. Phascolarctos cinereus (A0A6P5JH21), 13. Ornithorhynchus anatinus (F7B506), 14. Gallus (Q5F361), 15. Xenopus laevis (Q6DCI5), 16. Callorhinchus milii (A0A4W3I855), 17. Salmo salar (A0A1S3SLA7), 18. Danio rerio (E7F1V3), 19. Petromyzon marinus (A0AAJ7WV02), 20. Hydra vulgaris (T2M976), 21. Drosophila melanogaster (Q9W4F8), 22. Phallusia mammillata (A0A6F9DTU8), 23. Salpingoeca rosetta (F2UF72), 24. Stylophora pistillata (A0A2B4R4K9), 25. Caenorhabditis elegans (Q18386), 26. Social amoeba (Q55G97), and 27. Allomyces macrogynus (A0A0L0SIE5). (B) Sequence similarity of each TBCK domain across these species is visualized, with darker gray indicating higher similarity to the human TBCK sequence. The human TBCK domain architecture is depicted at the top for reference. Color coding follows the same scheme used in Figure 1. Sequence alignments and the visual representation were performed with Clustal Omega v1.2.4 and GraphPad Prism.
The full-length human TBCK protein (UniProt Q8TEA7) consists of 893 amino acids and has a molecular mass of 100.7 kDa. Due to alternative splicing, more than nine distinct transcripts of TBCK can be classified into two major isoform types: long isoforms, which contain all three domains (UniProt: Q8TEA7-2 and Q8TEA7-3), and short isoforms with truncated domains (UniProt: D6RDY5, H0YA45, D6RDG2, D6RC61, D6R950, H0Y8U7, H0Y959, and Q5HYF5) (Wu and Lu, 2020). In the literature, the full-length protein is typically referred to as the canonical sequence. Although the exact mechanism remains poorly understood, each isoform may have a distinct functional role in cellular processes depending on the cell type (Wu and Lu, 2020). To date, no biochemical studies have explored the structure-function relationship of different TBCK isoforms. In this section, we examine the sequence homology of each TBCK domain in the canonical sequence and compare them with related proteins to propose testable strategies for their biochemical functions.
The first 273 amino acids at the N-terminus of TBCK form a serine/threonine-like protein kinase domain (Figure 3A). Serine/threonine kinases catalyze phosphate transfer from ATP to protein substrates in the presence of divalent cations, such as magnesium and manganese (Figure 3B). Catalytically competent kinases typically contain four conserved motifs: a glycine-rich loop (G-loop) with a GXGX2G motif, a VAIK motif, an HRDX4N motif, and a DFG motif (Johnson et al., 1996; Taylor and Kornev, 2011; Murphy et al., 2014; Zhang et al., 2015). The G-loop assists in positioning ATP and binds the γ-phosphate, while the VAIK motif forms a salt bridge with the conserved glutamate residue on the αC helix to facilitate binding of the ⍺- and β-phosphates of ATP. The DFG motif coordinates divalent cations for ATP binding and activation. The aspartate residue of the HRD motif acts as a base in the hydrolysis of ATP (Figure 3C). If one or more of these catalytic motifs are missing or altered, kinases may lose catalytic activity and be classified as pseudokinases. Pseudokinases are thought to have evolved from functional kinases through gene duplication and subsequent mutations that eliminate one or more of the catalytic motifs (Sheetz and Lemmon, 2020). However, there are notable exceptions where missing motifs do not impact the kinase activity. In some cases, nearby residues can compensate for the loss, restoring catalytic activity (Shrestha et al., 2020). A well-documented example is the WNK (“With No K”) kinase, in which the conserved lysine residue in the VAIK motif is absent from the β3 strand but functionally replaced by a lysine in the β2 strand (Xu et al., 2000; Min et al., 2004). Such compensatory mechanisms challenge computational predictions of kinase activity and often yield unexpected experimental results. In particular, many “dark” kinases that were annotated as pseudokinases based solely on sequence comparisons require experimental characterization to determine their true biochemical behavior. This highlights the importance of experimental validation when distinguishing between functional kinases and pseudokinases.

Figure 3. Structural and sequence analysis of the pseudokinase domain in human TBCK. (A) The pseudokinase domain of human TBCK, spanning residues 1–273, is highlighted in teal. (B) Mechanism of kinase activity: the hydroxyl group of a serine/threonine residue in the substrate protein (white sphere) is phosphorylated by a serine protein kinase (gray) in the presence of ATP. (C) Structure of serine/threonine protein kinase human CDK7 (PDB: 1UA2), a catalytically competent protein kinase, highlighting key residues in functional motifs: G-loop (green), VAIK (magenta), ATP binding site, HRD (blue), and DFG (cyan). (D) Sequence alignment of the TBCK pseudokinase domain compared to other pseudokinases and active kinases, showing missing residues in key motifs, including the G-loop, VAIK, HRD, and DFG motifs. The UniPort codes for the sequences aligned are hTBCK (Q8TEA7), hULK4 (Q96C45), hJAK2_JH2 (O60674), hHER3 (P21860), hPRKACA (P17612), and hCDK7 (P50613). (E) Comparison of kinase and pseudokinase structures, including hULK4 (PDB: 6TSZ, cyan), pseudokinase domain of hJAK2-JH2 (PDB: 4FVQ, blue), hHER3 (PDB: 3KEX, green), tyrosine protein kinase hPRKACA (PDB: 2GU8, yellow). (F) AlphaFold3-predicted hTBCK (gray) with its pseudokinase domain (teal) and its pseudo-active site residues. (G) Three phosphorylation sites (orange) and the pseudo-active site (red dashed circle) in the pseudokinase domain of TBCK. Sequence alignments and the visual representation were performed with Clustal Omega v1.2.4, Jalview, and Pymol.
Pseudokinases are classified into four categories based on their ligand interaction features, particularly focusing on the binding capabilities of their pseudo-active sites: Class I no/low nucleotide or cation binding, Class II nucleotide binding only, Class III cation binding only, and Class IV both nucleotide and cation binding without the ability to catalyze phosphoryl transfer (Kung and Jura, 2019). Sequence alignment of TBCK’s kinase domain with representative pseudokinases and kinases reveals sequence identities of 29.3% with human CDK6, 29.2% with TAO1, and 27.4% with MARK2, all of which are serine/threonine kinases. None of the key catalytic motifs found in functionally active kinases are predicted to be present in TBCK (Figure 3D). As a result, TBCK’s kinase domain is classified as a Class I pseudokinase, likely lacking both nucleotide-binding and catalytic function (Vella et al., 2022; Jin and Jianjun, 2024). The structural model predicted by AlphaFold 3 further supports this classification (Figures 3D–F). First, the N-lobe of the pseudokinase domain in TBCK is shorter than that of most serine/threonine-like pseudokinases, missing the β1 and β2 strands. This could impair ATP and bivalent metal recruitment to the nucleotide-binding pocket. Although a GSNG motif near the αC helix may resemble an atypical glycine-rich loop, AlphaFold structural predictions suggest this region exists in a flexible loop with low confidence. Second, the catalytic lysine in the VAIK motif is absent from the TBCK β3 strand. Finally, the HRD and DFG motifs are replaced by HRA and KFG, respectively. Similarly to the Class I pseudokinase TRIB1, TBCK possesses a short αC helix and lacks the typical β1 and β2 strands, which contributes to a shallow nucleotide-binding pocket and makes it unlikely to bind nucleotides. Mass spectrometry-based proteomics screening has identified three phosphorylation sites, S118, Y153, and T169, in the pseudokinase domain of TBCK across various cancer cell lines (Figure 3G) (Hornbeck et al., 2015). Both S118 and Y153 are predicted to reside in the pseudo-active site of the pseudokinase domain. T169 is located in a loop within the AlphaFold-predicted structure, where confidence is low (70 > pLDDT >50). An ubiquitination site at K271 has also been detected in the immortalized T lymphocytes (Udeshi et al., 2013). The biological significance and function of these post-translational modifications remain unclear.
Given these sequence characteristics, TBCK’s pseudokinase domain is most likely to function through protein-protein interactions rather than catalysis. Pseudokinases, typically found as regulatory domains within larger proteins, serve as allosteric regulators, protein scaffolds, and signaling switches (Sheetz and Lemmon, 2022). For instance, Janus kinases (JAKs) use their pseudokinase domain as an autoinhibitory regulator to control phosphorylation of the signal transducer and activator of transcription (STAT) at its kinase domain, thus modulating inflammatory and immune responses (Wilmes et al., 2020; Glassman et al., 2022). Disruption of autoinhibition, as in the JAK2 V617F variant, alters dimerization at the pseudokinase-kinase interface and leads to myeloproliferative neoplasms (Abraham et al., 2024). Similarly, pseudokinases, such as IRAK3 and MLKL, utilize dimerization, triggered by ligand binding or protein-protein interactions, as a regulatory mechanism (Lange et al., 2021; Zhang et al., 2021). While some pseudokinases play critical roles in oncology, neurology, and immunology, many “dark” pseudokinases await experimental characterization with unknown functions awaiting experimental characterization (Zeqiraj and Van Aalten, 2010; Murphy et al., 2014; Mace and Murphy, 2021).
The TBC domain (residues 426–710) is the second domain of TBCK (Figure 4A). TBC domain-containing proteins are widely distributed among eukaryotes (Strom et al., 1993; Du et al., 1998). In humans, more than 40 TBC-containing proteins have been identified, nearly half of which exist as fusion proteins with additional domains that direct their localization and interactions with other cellular components (Fukuda, 2011). Notably, TBCK is the only human TBC protein containing a pseudokinase domain (Fukuda, 2011). Sequence similarity suggests that the TBC domain in TBCK is evolutionarily related to TBC1D23 and TBC1D21 (Figure 4B). TBC1D23 is a TBC protein functioning in endosome-Golgi trafficking, leading to pontocerebellar hypoplasia (Itoh and Fukuda, 2006; Huang et al., 2019). Whereas TBC1D21 is a protein related to the mitochondrial function of sperm and male infertility (Wang et al., 2020; Chen et al., 2022).
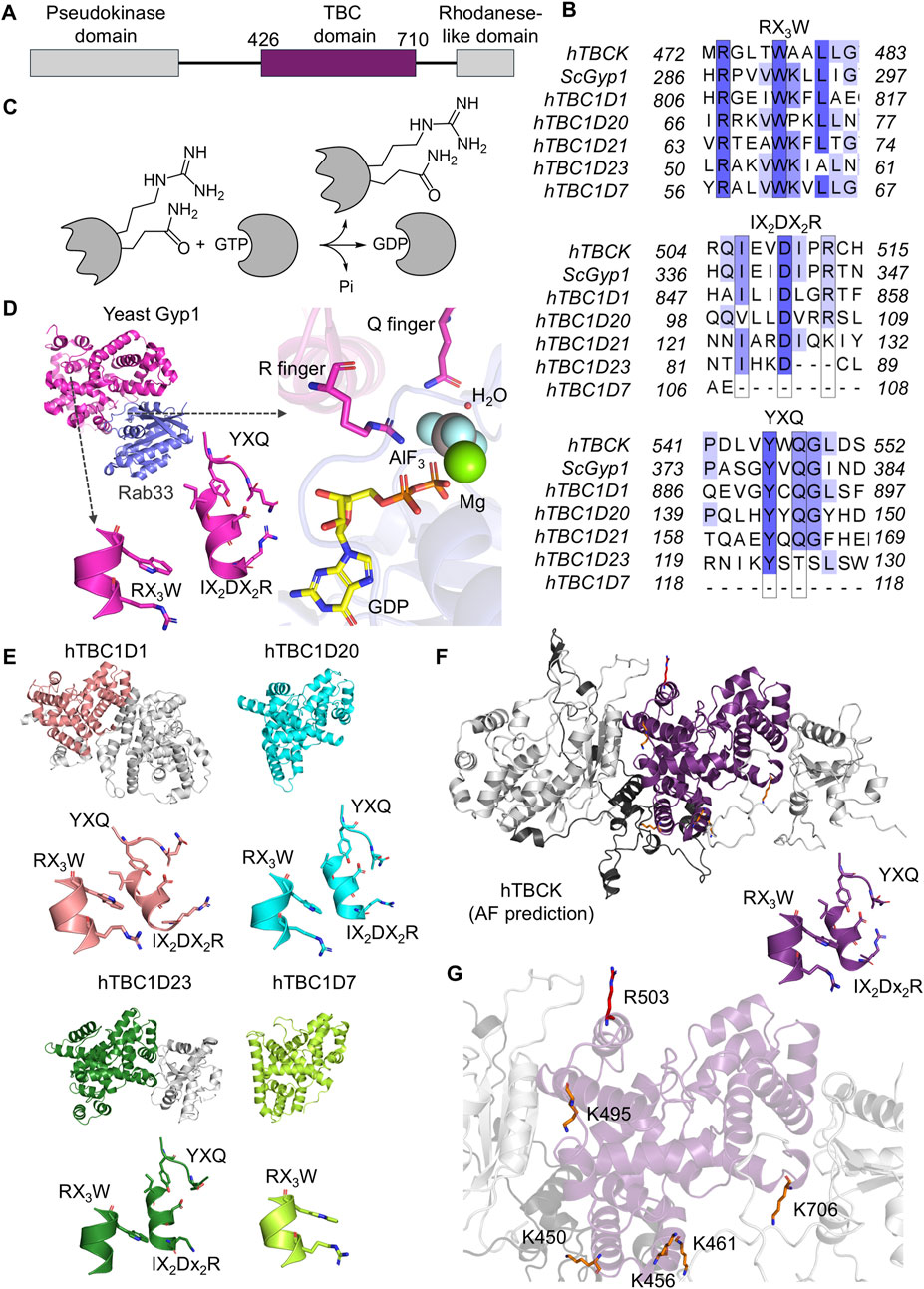
Figure 4. Structural and sequence analysis of the TBC domain in human TBCK. (A) The TBC domain of human TBCK is highlighted in purple, spanning residues 426–710. (B) Sequence alignment of the TBC domain in TBCK compared to other TBC-containing proteins, showing characteristic motifs, including the R finger (IX2DX2R), the Q finger (YXQ), and the conserved RX3W motif. The UniProt codes for the sequences aligned are hTBCK (Q8TEA7), ScGyp1 (Q08484), hTBC1D1 (Q86TI0), hTBC1D20 (Q96BZ9), hTBC1D21 (Q8IYX1), hTBC1D23 (Q9NUY8), and hTBC1D7 (Q9P0N9). (C) GTPase mechanism: the TBC domain facilitates the hydrolysis of GTP in GTPase to produce GDP and inorganic phosphate (Pi). (D) Structure of yeast Gyp1 (PDB: 2G77, magenta) highlighting key residues of the conserved motifs. The interaction between yeast Gyp1 and Rab33 (purple) is also shown. (E) Structural comparison of TBC domains, including hTBC1D1 (PDB: 3QYE, salmon), hTBC1D20 (PDB: 4HL4, cyan), hTBC1D23 (PDB: 6JL7, dark green), and hTBC1D7 (PDB: 3QWL, light green). Key motifs are displayed below each structure. (F) AlphaFold3-predicted human TBCK (gray) with the TBC domain highlighted in purple and the predicted unstructured region with low confidence in black. Key motifs are displayed below the structure. (G) The seven post-translational modifications have been highlighted in the TBC domain of TBCK, including K450, K456, K461, K495, R503 and K706. Lysine and arginine residues are represented in orange and red, respectively. Sequence alignments and the visual representation were performed with Clustal Omega v1.2.4, Jalview, and Pymol.
The primary function of known TBC proteins is to act as GTPase-activating proteins (GAPs) for members of the Rab family. Rab proteins, with over 60 members in humans, are small GTPases that catalyze GTP hydrolysis (Homma et al., 2021). As key players in vesicular transport, Rab proteins are implicated in various diseases, including neuropathy, type 2 diabetes, intellectual disabilities, blindness, immunodeficiencies, and cancer (Stenmark, 2009; Homma et al., 2021). Although many Rabs display intrinsic GTP-hydrolyzing capability, Rab-GAPs, such as TBC-containing proteins, can significantly accelerate GTP hydrolysis in vivo and in vitro. Structural studies of human TBC1D20 and yeast Gyp1 illustrated how TBC domains facilitate GTP hydrolysis (Pan et al., 2006; Gavriljuk et al., 2012). TBC-containing proteins stabilize the intermediate state of Rab proteins in the GTP-bound form, by donating a pair of catalytic residues, arginine and glutamine (R and Q fingers), thereby switching off the Rab signaling pathway (Figure 4C) (Pan et al., 2006). The R finger in the IX2DX2R motif interacts with the β-γ bridging oxygen of GTP. The Q finger in the YXQ motif aligns the water molecule necessary for GTP hydrolysis thereby replacing the role of the conserved glutamine in the DX2GQ motif in the Ras and Rho families (Figure 4D) (Barr and Lambright, 2010; Gavriljuk et al., 2012). TBC proteins have been categorized into five groups based on the presence or absence (designated as X) of the R and G fingers: R-Q, R-R, R-X, X-R, and X-X (Frasa et al., 2012). Group R-Q contains most of the identified TBC-containing proteins and is one of the best-characterized groups to date. Many of them are known to be catalytically competent GAPs, such as TBC1D1, which is a GAP for multiple Rabs involving type 2 diabetes and obesity (Meyre et al., 2008; Peck et al., 2009; Frasa et al., 2012). Lacking one or both residues, however, does not always abolish a TBC-containing protein’s ability to bind Rabs or modulate downstream pathways. For example, TBC1D7, an X-X group member, can still stimulate the GTPase activity of Rab17 via an unknown mechanism and forms a stable complex with other small GTPases to function as an effector (Yoshimura et al., 2007; Qin et al., 2016; Lupi et al., 2024). Another characteristic feature of TBC domains is a conserved RX3W motif near the N-terminus, which presumably helps maintain structural integrity (Pan et al., 2006). Sequence comparisons between TBCK and other characterized TBC-containing proteins suggest that TBCK contains all catalytically important motifs for potential GAP activity (Figures 4B,E,F). Additionally, seven post-translational modifications have been identified in the TBC domain of TBCK, including ubiquitination at K450, K456, K461, K495, and K706, as well as monomethylation at R503 in HEK-293 epithelial cells (Figure 4G) (Udeshi et al., 2013; Hornbeck et al., 2015; Larsen et al., 2016; Akimov et al., 2018). Specifically, R503 is located on the surface of the TBC domain and shares the same α-helix as the R finger. If TBCK functions as a GAP, the modification at R503 could potentially disrupt the interaction between TBCK and Rabs.
Following a segment comprised of polar residues, which AlphaFold predicted to be intrinsically disordered (residues 711–789), the C-terminal region (residues 790–891) of TBCK adopts a rhodanese-like fold (Figure 5A). Rhodaneses catalyze the reaction between cyanide and thiosulfate to yield thiocyanate and sulfite. They are vital in sulfur trafficking, redox regulation, thiol modifications, and cofactor biosynthesis (Alsohaibani et al., 2023). Its enzymatic activity relies on a CX4-5 R/T motif, wherein the cysteine side chain enables sulfur transfer (Figure 5B) (Bordo and Bork, 2002). Another conserved feature is the DXR motif, which helps to position the catalytic residues and stabilize the overall fold. Beyond sulfur transfer, rhodanese-like proteins can sometimes act as phosphatases, transferring phosphate or arsenate (Zhang et al., 2022; Burroughs and Aravind, 2023). In such cases, the arginine residue within the CX4-5 R/T motif helps stabilize the transition state. Rhodanese homologs can exhibit higher functional diversity across different kingdoms. In prokaryotes, a rhodanese fold can be found in selenouridine synthase to assist with tRNA modification (Wolfe et al., 2004). Some yeasts and plants also contain rhodanese-like arsenate reductases for detoxification. Human Cdc25B and Cdc25C phosphatases are rhodanese-like proteins that can adventitiously reduce arsenate but do not primarily function in arsenate detoxification (Bhattacharjee et al., 2010; Zhang et al., 2022). Importantly, all of these functional rhodanese-like proteins contain catalytic cysteines (Alsohaibani et al., 2023; Burroughs and Aravind, 2023).
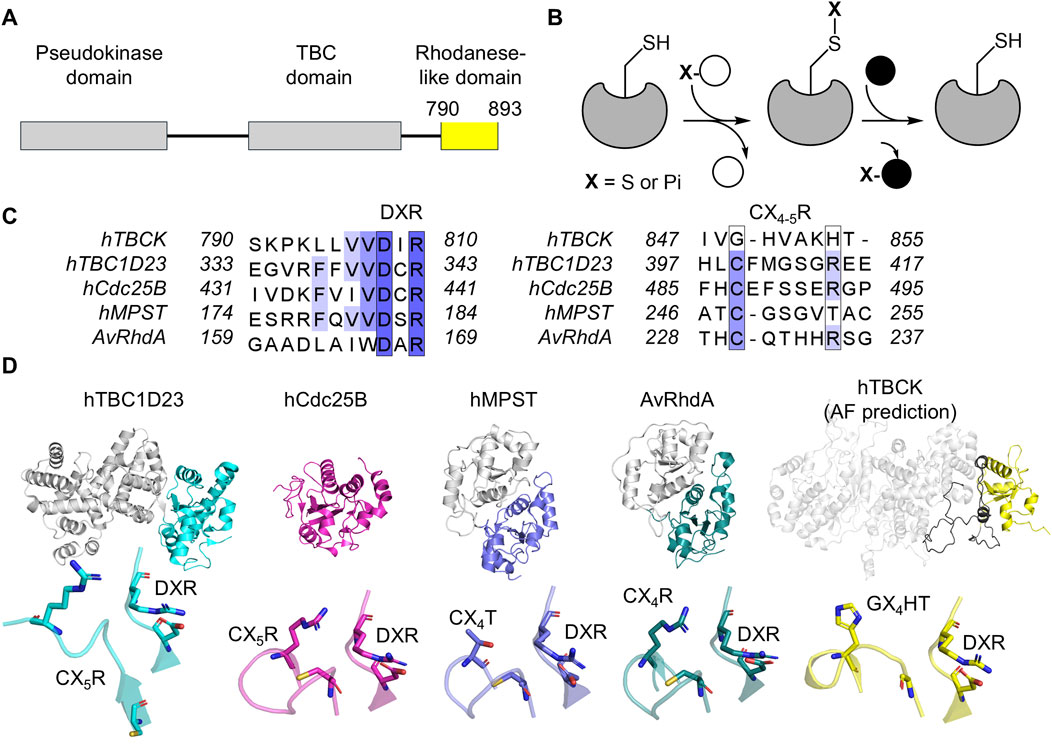
Figure 5. Structural and sequence analysis of the rhodanese-like domain in human TBCK. (A) The rhodanese-like domain of human TBCK is highlighted in yellow, spanning residues 790–893. (B) Mechanism of rhodanese activity: the enzyme features a conserved cysteine residue in its active site (left complex). This residue reacts with a sulfur donor (white sphere), forming a persulfide intermediate (middle complex) and releasing the first product. The sulfur atom from the persulfide intermediate is then transferred to an acceptor molecule (right complex), producing the final product. (C) Sequence alignment of the rhodanese-like domain in TBCK compared to other rhodanese domains, highlighting characteristic motifs. The UniProt codes for the sequences aligned are hTBCK (Q8TEA7), hTBC1D23 (Q9NUY8), hCdc25B (P30305), human mercaptopyruvate sulfurtransferase (MPST) (P25325), and Azotobacter vinelandii RhdA (P52197). (D) Structural comparison of rhodanese domains, including human TBC1D23 (PDB: 6JL7, cyan), human Cdc25D (PDB: 1YMK, magenta), hMPST (PDB: 3OLH, blue), AvRhdA (PDB: 1H4K, teal), and AlphaFold3-predicted human TBCK (gray). The rhodanese-like domain in TBCK is highlighted in yellow. The predicted unstructured region with low confidence is indicated in black. Key motifs identified in the sequence are displayed below each structure. Sequence alignments and visual representation were performed with Clustal Omega v1.2.4, Jalview, and Pymol.
In our sequence alignment, the rhodanese-like domain appears to display the lowest overall conservation across various organisms of TBCK’s three domains. The TBCK rhodanese-like domain likely lacks canonical sulfurtransferase activity because the critical CX4-5 R/T motif is replaced by GX4H. Further, the residue substitution is not consistently conserved across species (Figures 5C,D). Nevertheless, the DXR motif remains intact, suggesting it may still maintain a stable rhodanese fold for mediating protein-protein interactions. Only one ubiquitination site, K881, has been identified in the rhodanese-like domain of TBCK in the Jurkat T lymphocyte cell line (Udeshi et al., 2013). Additionally, S890, in the C-terminal tail, can be phosphorylated across a variety of model cell lines for breast cancer, lung cancer, ovarian cancer, and melanoma (Klammer et al., 2012; Mertins et al., 2014; Stuart et al., 2015; NCI CPTAC et al., 2016). TBC1D23 is another human TBC-containing protein with a rhodanese-like domain, but it lacks a pseudokinase domain (Figure 5C) (Liu et al., 2020). TBC1D23 is known for its involvement in endosome-derived vesicle transfer and is linked to neurodevelopmental disorders (Marin-Valencia et al., 2017; Liu et al., 2020). Although TBC1D23 retains a CX4-5 R/T motif and a DXR motif, it has no detectable sulfurtransferase or phosphatase activity. Instead, its rhodanese domain, packs against its TBC domain, forming a binding cleft for golgin-97 and golgin-245 at the Golgi apparatus (Liu et al., 2020). Through this interaction, TBC1D23 bridges golgin-97/245 and the WASH/FAM21 complex, facilitating endosome-to-Golgi trafficking (Shin et al., 2017; Navarro Negredo et al., 2018; Huang et al., 2019; Tu et al., 2024). This provides insight into the potential non-catalytic function of the rhodanese-like domain in TBCK. Whether the rhodanese-like domain of TBCK functions strictly as a protein-protein interface or has retained any catalytic functions remains an open question.
The three domains in human TBCK are connected by structurally flexible and partially disordered linker regions, which span residues 274–425 and 710–790 (Figure 1A). Several short α-helices are present in these regions. These segments exhibit low structural confidence in the AlphaFold3-predicted structure (pLDDT <70). Sequence alignment reveals that the residues in the linker regions are conserved among mammalian TBCKs, but their conservation drops significantly when comparing human TBCK across other eukaryotic organisms. Post-translational modifications have been discovered in the linker regions of TBCK. A total of eleven sites have been identified or predicted to be modified by phosphorylation and ubiquitination. Among them, three ubiquitination sites (K285, K343, K349) have been found in the linker between the pseudokinase and TBC domains. K343 shows the highest frequency of ubiquitination among these three residues in three separate studies (Mertins et al., 2013; Udeshi et al., 2013; Akimov et al., 2018). Residues K310 and T349 are predicted ubiquitination and phosphorylation sites, respectively, since their cognate positions in mouse TBCK have been reported to be modified (Wang et al., 2011; Hornbeck et al., 2015). In the linker region between the TBC and rhodanese-like domains, Y732 can be phosphorylated in T cell and myeloma (St-Germain et al., 2009; Arneja et al., 2014). Studies on the mouse TBCK ortholog further suggest that T775, T782, and S784 in human TBCK are potential phosphorylation sites (Hornbeck et al., 2015). Many multidomain proteins contain low-complexity regions, which are typically enriched in repeating sequences containing glycine, serine, glutamine, proline, or lysine (Fondon and Garner, 2004; Gemayel et al., 2015; Pelassa et al., 2019; Chavali et al., 2020). While glutamine-rich repeats are often related to neurodegenerative diseases, they have been shown to modulate protein activity in a length-dependent manner (Gemayel et al., 2015). Increasing evidence shows that these regions play functional roles in protein-protein interactions and liquid-liquid phase separation (Vaglietti et al., 2023). However, TBCK’s linker regions do not contain homopolymeric amino acid repeats. Whether these linkers contribute to TBCK’s biological function beyond providing structural flexibility remains to be explored through further investigation.
Recent breakthroughs in the molecular characterization of TBCK have revealed its unexpected role as part of the Five-subunit Early endosomal Rab5 and RNA/ribosome intermediarY (FERRY) complex, which regulates early endosomal mRNA trafficking in neurons through a small GTPase-guided pathway (Figure 6A) (Quentin et al., 2023; Schuhmacher et al., 2023). The FERRY complex is comprised of TBCK (Fy-1), Ppp1r21 (Fy-2), C12orf4 (Fy-3), Cryzl1 (Fy-4), and Gatd1 (Fy-5) (Schuhmacher et al., 2023). The FERRY complex was initially identified through pull-down experiments aimed at finding Rab5 effectors. Rab5 is a small GTPase central to vesicular trafficking, and the FERRY complex as a whole shows strong binding to the Rab5:GTPγS complex in pull-down assays (Horiuchi et al., 1997; Quentin et al., 2023; Schuhmacher et al., 2023). While the clamp-shaped core of the FERRY complex, formed by Ppp1r21, Cryzl1, and Gatd1, was well-resolved structurally in the cryogenic electron microscopy (cryo-EM) study, the details of the interactions between TBCK, C12orf4, and Rab5 were not (Quentin et al., 2023). Further cross-linking mass spectrometry experiments on FERRY-associated cellular components reveal that the FERRY complex primarily interacts with ribosomal proteins and mRNAs, potentially influencing the cellular transcriptome (Schuhmacher et al., 2023).
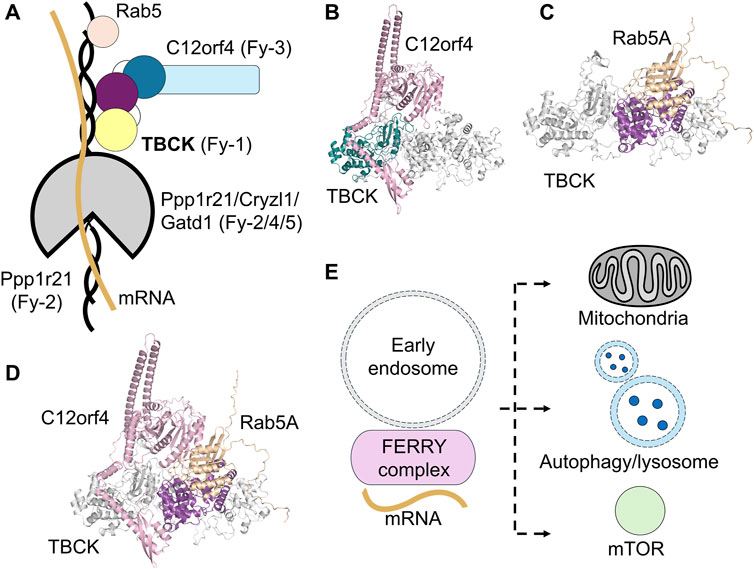
Figure 6. Proposed roles of TBCK in cellular processes. (A) TBCK is associated with the FERRY complex, potentially playing a role in endosomal trafficking and mRNA transport. The FERRY complex comprised of TBCK (Fy-1, yellow/purple/dark blue circles), Ppp1r21 (Fy-2, black/gray), C12orf4 (Fy-3, sky blue), Cryzl1 (Fy-4, gray), and Gatd1 (Fy-5, gray) interacts with Rab5 (wheat circle), and mRNA (yellow line). (B) The AlphaFold3-predicted structural model of human TBCK (gray and teal) and C12orf4 (pink) complex model. C12orf4 could potentially interact with the pseudokinase domain of TBCK. (C) AlphaFold3-predicted structural model of human TBCK (gray) with its TBC domain (purple) bound to Rab5A (wheat). (D) AlphaFold3-predicted structural model of human TBCK (gray) with its TBC domain (purple) bound to Rab5A (wheat) and C12orf4 (pink). (E) TBCK and the FERRY complex are implicated in mitochondrial maintenance, autophagy, lysosomal function, and the mTOR signaling pathway.
Among the five FERRY subunits, TBCK appears to be the most evolutionarily ancient. The other components exhibit lower conservation in organisms, such as fungi and flatworms, suggesting they emerged more recently (Schuhmacher et al., 2023). Despite TBCK’s evolutionary prominence, its functional role within the FERRY complex remains poorly understood. Notably, TBCK does not appear to be essential for the complex’s structural integrity or mRNA binding (Quentin et al., 2023). Given TBCK’s non-essential role in mRNA binding, TBCK could potentially contribute to the subunit interactions. Interestingly, AlphaFold predicts that C12orf4, a FERRY subunit, may interact closely with TBCK’s pseudokinase domain (Figure 6B). This is consistent with the role of many pseudokinases to act as scaffolds that facilitate protein-protein interactions.
As demonstrated in the previous section, it is likely that TBCK’s function is carried out by its TBC domain being a GAP of Rab. This is supported by evidence that substituting the R finger with histidine in TBCK leads to TBCK syndrome (Fukuda, 2011; Chong et al., 2016). This hypothesis is further supported by AlphaFold models of TBCK:Rab5 complex (Figure 6C). Additionally, the Rab5 docking site does not overlap with the predicated C12orf4 binding site (Figure 6D). The AlphaFold model predicts that the R and Q fingers point toward the Rab5 active site in a manner that closely resembles other known Rab-GAP complexes. Experimental findings regarding TBCK’s interaction with Rab5 are inconsistent. One study using the in vivo proximity labeling technique, MitoID, and in vitro pull-down assays did suggest that TBCK directly interacts with Rab5A:GTP (Gillingham et al., 2019), while a later pull-down study did not detect any interaction (Schuhmacher et al., 2023). A yeast two-hybrid interaction study also showed that TBCK does not interact with any Rab nor exhibit GAP activity (Itoh et al., 2006). Further studies confirmed that TBCK does not interact with Rab27A, a Rab involved in melanosome aggregation (Itoh and Fukuda, 2006). The challenges of identifying a specific Rab-TBC pair are derived from the weak correlation between different experimental methods, such as pull-down assays, yeast two-hybrid experiments, and RNAi knockdown-based validations under different conditions (Gabernet-Castello et al., 2013). These discrepancies may also reflect the transient nature of Rab-GAP interaction. Moreover, demonstrating physical binding does not conclusively prove catalytic activity. Indeed, GAP-deficient mutants of TBC proteins often bind Rab:GTP more tightly than their wild-type counterparts (Frasa et al., 2012). Further biochemical analyses are needed to clarify the exact relationship between TBCK’s TBC domain and Rab5.
In recent years, there has been a growing number of reports on the cellular function of TBCK, although much of its role remains unknown. TBCK is classified as a cytosolic protein. A report published in 2013 on TBCK’s potential function in cancer cell lines found that TBCK is broadly expressed in various human cell line models, including HEK293 and HeLa, and is localized near the nucleus and centrosomes (Liu et al., 2013). This aligns with the observation that TBCK is localized on the surface of myofibers and colocalized with nuclei in the myotubes of muscle cells in healthy individuals but is absent in samples collected from individuals lacking TBCK (Saredi et al., 2020). Further investigations demonstrated that TBCK is a cytoplasmic protein that accumulates at the mitotic apparatus during mitosis and meiosis. This supports the idea that TBCK may play a role in regulating cell proliferation (Wu et al., 2014).
The discovery of somatic frameshift TBCK mutations in tumor cells initially led researchers to suspect that TBCK might play a role in oncology (Greenman et al., 2007; Gao et al., 2021). This suspicion was further supported by the identification of miR-1208, a tumor suppressor microRNA that binds to TBCK’s 3′-untranslated region and negatively regulates TBCK expression in renal cancer cell lines (Kim et al., 2019). Notably, lowering TBCK levels enhances the sensitivity of cancer cells to chemotherapy, e.g., factor-related apoptosis-inducing ligand (TRAIL), cisplatin, and doxorubicin, suggesting therapeutic potential in modulating TBCK levels in oncology (Kim et al., 2019; Park et al., 2023). The drug sensitivity, however, does not necessarily mean that TBCK is an oncogene actively promoting cell proliferation in tumors (Wu et al., 2014). Further studies revealed that TRAIL sensitivity is likely due to the reduced TBCK level, which directly or indirectly inhibits PDK1, ERK, and AKT signaling pathways by suppressing the phosphorylation of these enzymes (Park et al., 2023). Additionally, TBCK expression levels are correlated with the expression of genes related to lipoproteins, such as apolipoprotein B in porcine hepatic tissue, suggesting TBCK’s potential role in lipid metabolism (Ballester et al., 2017).
A potential link was drawn between TBCK and mTOR signaling pathways because when TBCK is depleted, the phosphorylation of mTOR Complex1 (mTORC1) downstream targets decreases (Liu et al., 2013). The regulation of mTOR by TBCK is believed to be at the transcription level (Liu et al., 2013). If the mRNA of one of the mTOR proteins is a FERRY complex cargo, the mRNA binding capability of FERRY might explain the earlier observations that the transcription level of mTOR components was reduced in TBCK knockouts (Liu et al., 2013; Tan et al., 2022). The mTOR pathway is known to play a role in many neurological disorders, such as autism, epilepsy, intellectual disability, and macrocephaly, which are comorbid with TBCK Syndrome (Liu et al., 2013), (Lipton and Sahin, 2014). Studies have shown that rapamycin-mediated mTOR inhibition can prevent seizures in neurodevelopmental disorders caused by STRADα mutations, another pseudokinase that regulates neuronal pathfinding and migration (Parker et al., 2013). TBCK knockouts led to HEK293 cells becoming smaller, likely stemming from disrupted actin organization (Liu et al., 2013). The correlation between mTOR signaling and the function of TBCK in cell size and proliferation is logical because the mTOR pathway regulates gene transcription and protein synthesis to control cell growth (Saxton and Sabatini, 2017). Due to the link between TBCK and mTORC1, it was suspected that activating mTOR through leucine supplementation could be used to treat TBCK Syndrome, like many other mTOR-related disorders (Bhoj et al., 2016). Studies have shown that leucine activates mTOR pathway by binding to Sestrin2, an upstream regulator of mTORC1 (Wolfson et al., 2016). Leucine’s metabolite, acetyl-coenzyme A, also positively regulates mTORC1 activity by acetylating the mTORC1 regulator, Raptor (Son et al., 2019). Indeed, a 2016 study found the mTOR pathway was rescued when leucine was supplied to immortalized lymphoblastoid cells derived from a patient carrying the homozygous TBCK frameshift variant (Bhoj et al., 2016). However, a recent study suggests that mTORC1 inhibition observed in TBCK patient-derived cells and HeLa cells varies across different patients and cell types, indicating that the link between TBCK and mTOR may be cell type-specific (Angireddy et al., 2024).
TBCK deficiency leads to autophagy dysregulation, which is characterized by accumulation of autophagosomes (Tintos-Hernández et al., 2021). Since mTORC1 negatively regulates autophagy, dysfunction of TBCK could induce a pro-autophagic state and lead to increased autophagic flux (Mushtaq et al., 2023). Biochemical and morphological analyses of patient samples with the TBCK c.304C>T (p.Gln102*) mutation reveal vacuolated B-cells and prominent lipofuscin-like storage deposits in neurons and other organs (Beck-Wödl et al., 2018). Fibroblasts from patients harboring the TBCK mutations also display a significant accumulation of autophagosomes and heightened autophagic flux (Ortiz-González et al., 2018). Further investigations, using TBCK patient-derived induced pluripotent stem cells differentiated into neuroprogenitor cells, revealed severe impairments in endoplasmic reticulum-to-Golgi vesicular transport and autophagosome biogenesis coupled with slowed cell cycle progression and migration (Moreira et al., 2022). Consistently, TBCK knockdown in neuroprogenitor cell lines hinders their differentiation into neurons and astrocytes (Angireddy et al., 2024). Additional evidence linking TBCK to autophagy is the upregulation of TBCK expression during enterovirus 71 and coxsackievirus A16 infections, the viruses responsible for hand, foot, and mouth disease (Song et al., 2020). Moreover, disrupted oligosaccharide degradation aligns with aberrant autophagic and lysosomal activity (Ortiz-González et al., 2018). Lysosomal dysfunction contributes to the accumulation of mitophagosomes, which may explain the blockage in the degradation of autophagosomes and mitophagosomes (Tintos-Hernández et al., 2021). TBCK dysfunction can be classified as a lysosomal storage disorder due to the increased autophagosome formation and impaired autophagosome-lysosome fusion.
Mitochondrial function and mitophagosomes can also be impaired by TBCK dysfunction (Tintos-Hernández et al., 2021). Fibroblasts from TBCK patients also show reduced mitochondrial DNA and respiratory function with elevated mitophagy, which cannot be rescued by mTOR activation, suggesting that the disease mechanism could be multi-faceted (Tintos-Hernández et al., 2021). Reduced mitochondrial DNA copy number impairs the synthesis of essential respiratory chain components, leading to decreased respiratory capacity and compromised energy production (Tintos-Hernández et al., 2021). Elevated mitophagy is observed, possibly as a compensatory mechanism for dysfunctional mitochondria, evidenced by extensive colocalization of mitochondrial and lysosomal markers (Tintos-Hernández et al., 2021). Mitochondrial co-localization and membrane biogenesis during mitophagy are known to be influenced by other proteins in the same superfamily with TBCK, such as TBC1D15 (Yamano et al., 2014). Furthermore, the mRNA-loaded FERRY complex colocalizes with mitochondria in neurons. This suggests that TBCK can play a role in delivering nuclear-encoded transcripts of mitochondrial proteins (Quentin et al., 2023; Schuhmacher et al., 2023). This is supported by findings that, in TBCK knockdown neural progenitor cells, neurons, and astrocytes, altered mitochondrial membrane potential and dynamics can be rescued by a mitochondrial fission inhibitor (Angireddy et al., 2024).
Collectively, these studies establish a close link between TBCK, autophagy, lysosomes, and mitochondrial function. This aligns with the neurodegenerative manifestations of TBCK Syndrome because lysosomal impairment-induced mitochondrial dysfunction is a common feature of many neurodegenerative diseases (Figure 6E). These findings provide mechanistic support for both the neurodevelopmental and neurodegenerative phenotypes observed in patients.
More than 60 clinical cases of TBCK Syndrome have been reported worldwide since the first classification of TBCK Syndrome, spanning numerous countries and ethnic backgrounds (Table 1) (Chong et al., 2016; Guerreiro et al., 2016; Mandel et al., 2017; Reuter et al., 2017; Beck-Wödl et al., 2018, p. 1; Hartley et al., 2018; Ortiz-González et al., 2018; Sumathipala et al., 2019; Zapata-Aldana et al., 2019; Nass et al., 2020; Saredi et al., 2020; Tsang et al., 2020; Bertoli-Avella et al., 2021; Liu et al., 2021; Miyamoto et al., 2021; Murdock et al., 2021; Dai et al., 2022; Elliott et al., 2022; Maha and Shaza, 2022; Tan et al., 2022; De Luca-Ramirez, Rosado Fernández and Torres, 2023; Durham et al., 2023; Nair et al., 2023; Prem et al., 2023; Sabanathan et al., 2023; Jacob et al., 2024). Additionally, over 800 TBCK mutations have been identified through genome sequencing and added to clinical genetic databases, such as ClinVar (Landrum et al., 2024). More than 100 of these mutations are classified as pathogenic or likely pathogenic based on their expected impact on the integrity of the protein (Table 2 and 3). However, other variants classified as likely benign or of uncertain significance may still impair cellular function through different mechanisms. For instance, the compound heterozygous TBCK variants, NM_001163435.2:c.1201T>C p.(Tyr401His) and c.1588C>T p.(Arg530Cys), have been observed in individuals with corpus callosum anomalies, which overlaps with the phenotype of TBCK Syndrome. However, both variants were annotated as having uncertain significance in ClinVar (Miyamoto et al., 2021). The number of cases of TBCK Syndrome is likely underreported due to limited access to advanced diagnostic tools and neuroimaging (Durham et al., 2023). For example, two retrospective studies on tissue samples collected from individuals who died decades ago as children showcase the powerful genetic screening methods that were previously unavailable (Sumathipala et al., 2019; Jacob et al., 2024). Limited awareness of the disorder may also contribute to the poor understanding of TBCK Syndrome and the potential underreporting of cases (Durham et al., 2023).
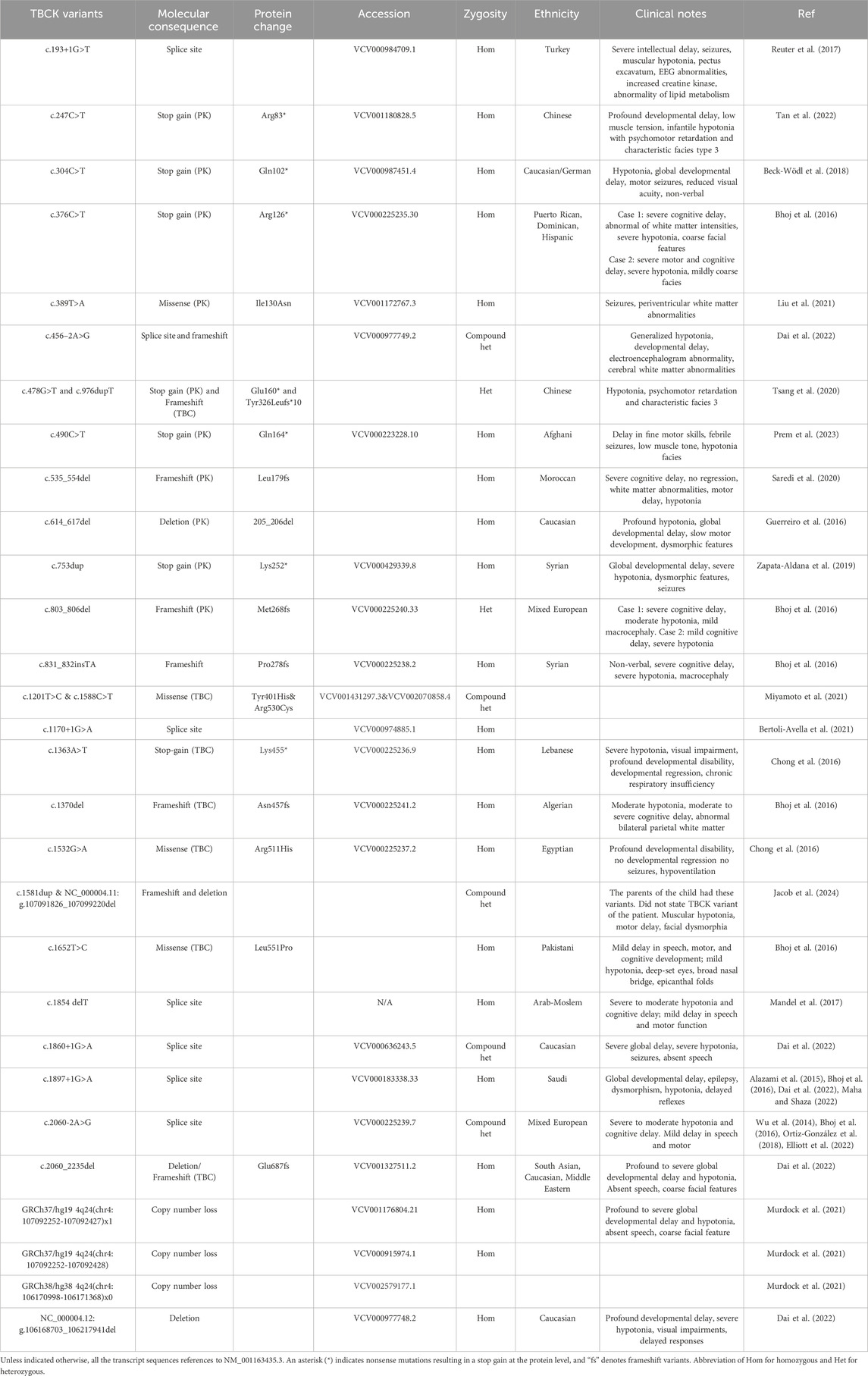
Table 1. Clinical cases of TBCK variants reported in literature.
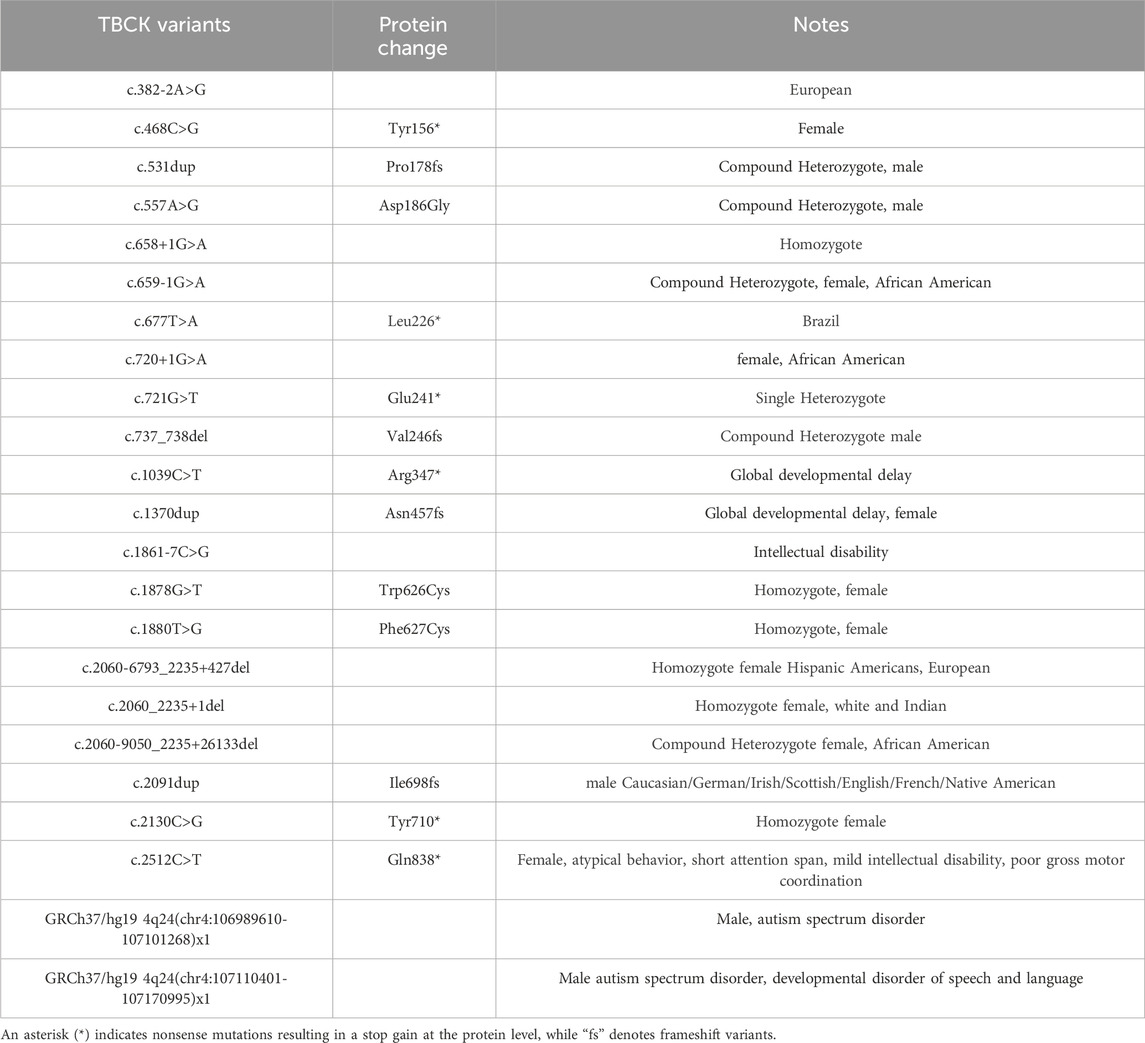
Table 2. TBCK variants reported in ClinVar, including details of the mutations and associated clinical notes describing the affected individuals.

Table 3. Pathogenic or likely pathogenic variants identified in ClinVar but not reported in literature or have a clear clinical description of the affected individuals.
Among the 29 documented disease-associated mutations in TBCK, 7 (21.9%) result in frameshift mutations and 7 (21.9%) introduce premature stop codons in the open reading frame (Table 1; Figure 7A). These mutations could severely compromise protein integrity, potentially leading to biological consequences in two plausible ways: (1) the truncated protein misfolds and is rapidly degraded, preventing it from performing its biological function, and (2) the truncated variants may form toxic aggregates, further disrupting cellular processes, similar to what is observed with other neurodegenerative diseases, such as SOD1-induced amyotrophic lateral sclerosis (Brasil et al., 2019). When Western blotting was performed on immortalized lymphoblastoid cell lines, a distinct TBCK protein band was detected in the lymphoblastoid of the healthy control group, while no detectable protein was observed in lymphoblastoid from diagnosed individuals with frameshift mutations in TBCK (Bhoj et al., 2016). The absence of detectable protein suggests that the mutations likely result in a loss of correctly folded full-length protein, supporting the first proposed scenario in which the misfolded protein is rapidly degraded. The second scenario currently lacks experimental support. The clinical manifestations of TBCK Syndrome, such as macroglossia and coarse facies, resemble other lysosomal storage diseases (Ortiz-González et al., 2018). TBCK Syndrome also resembles diseases related to mitochondrial dysfunction because there are several overlapping clinical presentations with genetic mutations that disrupt mitochondrial protein function (Chong et al., 2016). These clinical presentations are consistent with the cellular function identified in the aforementioned in vitro assays.
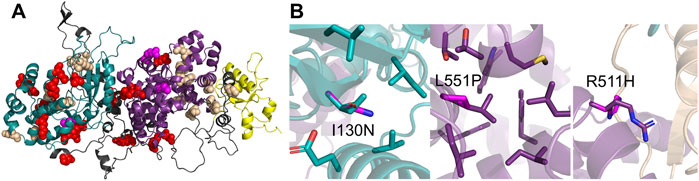
Figure 7. Human TBCK mutations are linked to TBCK Syndrome. (A) Clinical mutations mapped onto the predicted tertiary structure of TBCK, with mutations represented as spheres. The structure is color-coded to match the domains: pseudokinase domain (teal), TBC domain (purple), and rhodanese-like domain (yellow). Red spheres indicate TBCK variants associated with TBCK syndrome, as reported in the literature. Wheat-colored spheres represent disease-associated TBCK variants listed in databases. Magenta spheres indicate the three missense variants I130N, R511H, and L551P. (B) AlphaFold3-predicted structural model of human TBCK and disease-associated variants zooming into missense mutations, I130N, R511H, and L551P (magenta), and nearby interactions in the pseudokinase domain (teal), TBC domain (purple), and Rab5A (wheat). Visual representations were performed with Pymol.
The most frequently reported TBCK mutation is the “Boricua mutation” (R126*), which causes the truncation of the protein at arginine 126 in the pseudokinase domain and accounts for nearly 20% of cases described in the literature and is predominantly found in individuals of Puerto Rican descent. To date, three missense TBCK variants (I130N, R511H, and L551P) have been associated with TBCK Syndrome (Figure 7B) (Bhoj et al., 2016; Chong et al., 2016; Liu et al., 2021). However, the correlation between specific mutation sites and clinical severity is currently considered weak. Individuals with I130N and R511H show symptoms comparable to truncation mutations, such as profound developmental delays and seizures (Chong et al., 2016; Liu et al., 2021). The individual with I130N, who exhibited seizures and white matter abnormalities, eventually died of other complications at 9 months (Liu et al., 2021). In contrast, individuals with the L551P variant generally exhibit milder symptoms, including normal reflexes, less severe developmental delays, and no seizures (Bhoj et al., 2016). The I130N variant resides in the pseudokinase domain, where a hydrophobic residue is replaced by a polar, non-charged residue. I130 is positioned within a hydrophobic pocket surrounded by isoleucine and leucine residues, and its substitution with asparagine may destabilize this local protein environment. The pathogenic substitution, L551P, replaces a branched hydrophobic residue with a cyclic residue, potentially disrupting protein folding or stability. The most notable variant is R511H, where the arginine corresponds to the R finger critical for GAP activity in the TBC domain is replaced by histidine. This clinically identified variant further supports the importance of the R511 residue and the essential function of the TBC domain.
Initially, TBCK mutations were believed to cause disease exclusively in an autosomal recessive manner (Guerreiro et al., 2016). However, emerging evidence suggests that TBCK heterozygotes, containing one functional wild-type TBCK in one allele, may also present mild neurological symptoms. Inspired by findings from TBCK+/− mouse models, which demonstrated reduced exploratory and investigatory behaviors, further investigations revealed sex-based differences in haploinsufficiency in mice (Nair et al., 2023). Consistent with this observation, a large-scale analysis of 10,900 unrelated individuals allows researchers to identify 19 TBCK+/− heterozygotes encompassing 16 unique loss-of-function variants (Nair et al., 2023). These individuals exhibited a significantly higher incidence of acquired toe deformities (Nair et al., 2023). TBCK variants are also connected to heart development, as evidenced by their association with congenital heart disease in genome analyses (Ma et al., 2023). Studies using the zebrafish model have revealed TBCK’s role in the formation of the atrioventricular canal (Ma et al., 2023). Collectively, these studies underscore TBCK’s essential role in axonal neuropathy and organ development.
Benefiting from genomic sequencing, previous studies have uncovered the genetic cause of TBCK Syndrome. Most reported cases carry mutations that yield truncated TBCK. However, diagnosis and classification are only the first steps in the long journey to improve the quality of life and life expectancy of individuals affected by this disorder. Much about TBCK’s biological function needs to be uncovered in order to develop effective therapeutic strategies to alleviate the burden on patients and their families. In-depth biological and biochemical characterization of TBCK, both structurally and functionally, is a critical next step. Based on sequence similarity, we propose that the protein kinase domain and the rhodanese-like domain likely function as structural scaffolds by interacting with other protein-binding partners. The TBC domain may function as a GAP for an as-yet-unidentified Rab protein. A comprehensive understanding of TBCK’s interactions, catalytic capabilities, and regulatory mechanisms, particularly within the FERRY complex, the mTOR pathway, and Rab-mediated early endosome trafficking, is needed to deepen our fundamental knowledge of this enigmatic protein. Such insights may ultimately pave the way for novel therapeutic approaches for TBCK-related disorders.
EC: Writing–original draft, Writing–review and editing. CP: Visualization, Writing–review and editing. YL: Data curation, Investigation, Writing–review and editing. WZ: Supervision, Visualization, Writing–original draft, Writing–review and editing.
The author(s) declare that financial support was received for the research and/or publication of this article.
We acknowledge support from Florida State University start-up funding provided to the WZ Group and the Florida State University Institute for Pediatric Rare Diseases Award. We also thank Miss Chloe T. Allen (Department of Chemistry, Florida A&M University, Tallahassee) for her assistance with the literature search..
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abraham, B. G., Haikarainen, T., Vuorio, J., Girych, M., Virtanen, A. T., Kurttila, A., et al. (2024). Molecular basis of JAK2 activation in erythropoietin receptor and pathogenic JAK2 signaling. Sci. Adv. 10 (10), eadl2097. doi:10.1126/sciadv.adl2097
Akimov, V., Barrio-Hernandez, I., Hansen, S. V. F., Hallenborg, P., Pedersen, A. K., Bekker-Jensen, D. B., et al. (2018). UbiSite approach for comprehensive mapping of lysine and N-terminal ubiquitination sites. Nat. Struct. and Mol. Biol. 25 (7), 631–640. doi:10.1038/s41594-018-0084-y
Alazami, A. M., Patel, N., Shamseldin, H., Anazi, S., Al-Dosari, M., Alzahrani, F., et al. (2015). Accelerating novel candidate gene discovery in neurogenetic disorders via whole-exome sequencing of prescreened multiplex consanguineous families. Cell Rep. 10 (2), 148–161. doi:10.1016/j.celrep.2014.12.015
Alsohaibani, R., Claudel, A. L., Perchat-Varlet, R., Boutserin, S., Talfournier, F., Boschi-Muller, S., et al. (2023). Rhodanese-Fold containing proteins in humans: not just key players in sulfur trafficking. Antioxidants 12 (4), 843. doi:10.3390/antiox12040843
Angireddy, R., Karisetty, B. C., Katsura, K. A., Díaz, A., Murali, S., Smith, S., et al. (2024). A novel human TBCK- neuronal cell model results in severe neurodegeneration and partial rescue with mitochondrial fission inhibition. bioRxiv, 2024. doi:10.1101/2024.10.30.621078
Arneja, A., Johnson, H., Gabrovsek, L., Lauffenburger, D. A., and White, F. M. (2014). Qualitatively different T cell phenotypic responses to IL-2 versus IL-15 are unified by identical dependences on receptor signal strength and duration. J. Immunol. 192 (1), 123–135. doi:10.4049/jimmunol.1302291
Ballester, M., Ramayo-Caldas, Y., Revilla, M., Corominas, J., Castelló, A., Estellé, J., et al. (2017). Integration of liver gene co-expression networks and eGWAs analyses highlighted candidate regulators implicated in lipid metabolism in pigs. Sci. Rep. 7 (1), 46539. doi:10.1038/srep46539
Barr, F., and Lambright, D. G. (2010). Rab GEFs and GAPs. Curr. Opin. Cell Biol. 22 (4), 461–470. doi:10.1016/j.ceb.2010.04.007
Beck-Wödl, S., Harzer, K., Sturm, M., Buchert, R., Rieß, O., Mennel, H. D., et al. (2018). Homozygous TBC1 domain-containing kinase (TBCK) mutation causes a novel lysosomal storage disease – a new type of neuronal ceroid lipofuscinosis (CLN15)? Acta Neuropathol. Commun. 6 (1), 145. doi:10.1186/s40478-018-0646-6
Bertoli-Avella, A. M., Beetz, C., Ameziane, N., Rocha, M. E., Guatibonza, P., Pereira, C., et al. (2021). Successful application of genome sequencing in a diagnostic setting: 1007 index cases from a clinically heterogeneous cohort. Eur. J. Hum. Genet. 29 (1), 141–153. doi:10.1038/s41431-020-00713-9
Bhattacharjee, H., Sheng, J., Ajees, A. A., Mukhopadhyay, R., and Rosen, B. P. (2010). Adventitious arsenate reductase activity of the catalytic domain of the human Cdc25B and Cdc25C phosphatases. Biochemistry 49 (4), 802–809. doi:10.1021/bi9019127
Bhoj, E. J., Li, D., Harr, M., Edvardson, S., Elpeleg, O., Chisholm, E., et al. (2016). Mutations in TBCK, encoding TBC1-domain-containing kinase, lead to a recognizable syndrome of intellectual disability and hypotonia. Am. J. Hum. Genet. 98 (4), 782–788. doi:10.1016/j.ajhg.2016.03.016
Bordo, D., and Bork, P. (2002). The rhodanese/Cdc25 phosphatase superfamily: sequence–structure–function relations. EMBO Rep. 3 (8), 741–746. doi:10.1093/embo-reports/kvf150
Brasil, A. D. A., de Carvalho, M. D. C., Gerhardt, E., Queiroz, D. D., Pereira, M. D., Outeiro, T. F., et al. (2019). Characterization of the activity, aggregation, and toxicity of heterodimers of WT and ALS-associated mutant Sod1. Proc. Natl. Acad. Sci. 116 (51), 25991–26000. doi:10.1073/pnas.1902483116
Burroughs, A. M., and Aravind, L. (2023). New biochemistry in the Rhodanese-phosphatase superfamily: emerging roles in diverse metabolic processes, nucleic acid modifications, and biological conflicts. NAR Genomics Bioinforma. 5 (1), lqad029. doi:10.1093/nargab/lqad029
Chavali, S., Singh, A. K., Santhanam, B., and Babu, M. M. (2020). Amino acid homorepeats in proteins. Nat. Rev. Chem. 4 (8), 420–434. doi:10.1038/s41570-020-0204-1
Chelly, J., Khelfaoui, M., Francis, F., Chérif, B., and Bienvenu, T. (2006). Genetics and pathophysiology of mental retardation. Eur. J. Hum. Genet. 14 (6), 701–713. doi:10.1038/sj.ejhg.5201595
Chen, Y., Chen, X., Zhang, H., Sha, Y., Meng, R., Shao, T., et al. (2022). TBC1D21 is an essential factor for sperm mitochondrial sheath assembly and male fertility. Biol. Reproduction 107 (2), 619–634. doi:10.1093/biolre/ioac069
Cheng, J., Novati, G., Pan, J., Bycroft, C., Žemgulytė, A., Applebaum, T., et al. (2023). Accurate proteome-wide missense variant effect prediction with AlphaMissense. Science 381 (6664), eadg7492. doi:10.1126/science.adg7492
Chong, J. X., Caputo, V., Phelps, I., Stella, L., Worgan, L., Dempsey, J., et al. (2016). Recessive inactivating mutations in TBCK, encoding a Rab GTPase-activating protein, cause severe infantile syndromic encephalopathy. Am. J. Hum. Genet. 98 (4), 772–781. doi:10.1016/j.ajhg.2016.01.016
Dai, H., Zhu, W., Yuan, B., Walley, N., Schoch, K., Jiang, Y., et al. (2022). A recurrent single-exon deletion in TBCK might be under-recognized in patients with infantile hypotonia and psychomotor delay. Hum. Mutat. 43 (12), 1816–1823. doi:10.1002/humu.24497
De Luca-Ramirez, J., Rosado Fernández, S., and Torres, O. A. (2023). Raising awareness of TBC1 domain-containing kinase (TBCK) epileptic encephalopathy among Puerto Rican children. Ann. Child Neurology Soc. 1 (2), 168–171. doi:10.1002/cns3.20023
Du, L.-L., Collins, R. N., and Novick, P. J. (1998). Identification of a Sec4p GTPase-activating protein (GAP) as a novel member of a Rab GAP family. J. Biol. Chem. 273 (6), 3253–3256. doi:10.1074/jbc.273.6.3253
Durham, E. L., Angireddy, R., Black, A., Melendez-Perez, A., Smith, S., Gonzalez, E. M., et al. (2023). TBCK syndrome: a rare multi-organ neurodegenerative disease. Trends Mol. Med. 29 (10), 783–785. doi:10.1016/j.molmed.2023.06.009
Elliott, A. M., Adam, S., du Souich, C., Lehman, A., Nelson, T. N., van Karnebeek, C., et al. (2022). Genome-wide sequencing and the clinical diagnosis of genetic disease: the CAUSES study. Hum. Genet. Genomics Adv. 3 (3), 100108. doi:10.1016/j.xhgg.2022.100108
Fondon, J. W., and Garner, H. R. (2004). Molecular origins of rapid and continuous morphological evolution. Proc. Natl. Acad. Sci. 101 (52), 18058–18063. doi:10.1073/pnas.0408118101
Frasa, M. A. M., Koessmeier, K. T., Ahmadian, M. R., and Braga, V. M. M. (2012). Illuminating the functional and structural repertoire of human TBC/RABGAPs. Nat. Rev. Mol. Cell Biol. 13 (2), 67–73. doi:10.1038/nrm3267
Fukuda, M. (2011). TBC proteins: GAPs for mammalian small GTPase Rab? Biosci. Rep. 31 (3), 159–168. doi:10.1042/BSR20100112
Gabernet-Castello, C., O’Reilly, A. J., Dacks, J. B., and Field, M. C. (2013). Evolution of tre-2/bub2/cdc16 (TBC) Rab GTPase-activating proteins Mol. Biol. Cell 24, 1574–1583. doi:10.1091/mbc.e12-07-0557
Gao, J., Xi, L., Yu, R., Xu, H., Wu, M., and Huang, H. (2021). Differential mutation detection capability through capture-based targeted sequencing in plasma samples in hepatocellular carcinoma. Front. Oncol. 11, 596789. doi:10.3389/fonc.2021.596789
Gavriljuk, K., Gazdag, E. M., Itzen, A., Kötting, C., Goody, R. S., and Gerwert, K. (2012). Catalytic mechanism of a mammalian Rab·RabGAP complex in atomic detail. Proc. Natl. Acad. Sci. 109 (52), 21348–21353. doi:10.1073/pnas.1214431110
Gemayel, R., Chavali, S., Pougach, K., Legendre, M., Zhu, B., Boeynaems, S., et al. (2015). Variable glutamine-rich repeats modulate transcription factor activity. Mol. Cell 59 (4), 615–627. doi:10.1016/j.molcel.2015.07.003
Gillingham, A. K., Bertram, J., Begum, F., and Munro, S. (2019). In vivo identification of GTPase interactors by mitochondrial relocalization and proximity biotinylation. eLife 8, e45916. doi:10.7554/eLife.45916
Glassman, C. R., Tsutsumi, N., Saxton, R. A., Lupardus, P. J., Jude, K. M., and Garcia, K. C. (2022). Structure of a Janus kinase cytokine receptor complex reveals the basis for dimeric activation. Science 376 (6589), 163–169. doi:10.1126/science.abn8933
Greenman, C., Stephens, P., Smith, R., Dalgliesh, G. L., Hunter, C., Bignell, G., et al. (2007). Patterns of somatic mutation in human cancer genomes. Nature 446 (7132), 153–158. doi:10.1038/nature05610
Guerreiro, R. J., Brown, R., Dian, D., de Goede, C., Bras, J., and Mole, S. E. (2016). Mutation of TBCK causes a rare recessive developmental disorder. Neurol. Genet. 2 (3), e76. doi:10.1212/NXG.0000000000000076
Hartley, T., Wagner, J., Warman-Chardon, J., Tétreault, M., Brady, L., Baker, S., et al. (2018). Whole-exome sequencing is a valuable diagnostic tool for inherited peripheral neuropathies: outcomes from a cohort of 50 families. Clin. Genet. 93 (2), 301–309. doi:10.1111/cge.13101
Homma, Y., Hiragi, S., and Fukuda, M. (2021). Rab family of small GTPases: an updated view on their regulation and functions. FEBS J. 288 (1), 36–55. doi:10.1111/febs.15453
Horiuchi, H., Lippé, R., McBride, H. M., Rubino, M., Woodman, P., Stenmark, H., et al. (1997). A novel Rab5 GDP/GTP exchange factor complexed to rabaptin-5 links nucleotide exchange to effector recruitment and function. Cell 90 (6), 1149–1159. doi:10.1016/S0092-8674(00)80380-3
Hornbeck, P. V., Zhang, B., Murray, B., Kornhauser, J. M., Latham, V., and Skrzypek, E. (2015). PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res. 43 (D1), D512–D520. doi:10.1093/nar/gku1267
Huang, W., Liu, Z., Yang, F., Zhou, H., Yong, X., Yang, X., et al. (2019). Structural and functional studies of TBC1D23 C-terminal domain provide a link between endosomal trafficking and PCH. Proc. Natl. Acad. Sci. 116 (45), 22598–22608. doi:10.1073/pnas.1909316116
Itoh, T., and Fukuda, M. (2006). Identification of EPI64 as a GTPase-activating protein specific for Rab27A. J. Biol. Chem. 281 (42), 31823–31831. doi:10.1016/S0021-9258(19)84097-8
Itoh, T., Satoh, M., Kanno, E., and Fukuda, M. (2006). Screening for target Rabs of TBC (Tre-2/Bub2/Cdc16) domain-containing proteins based on their Rab-binding activity. Genes Cells 11 (9), 1023–1037. doi:10.1111/j.1365-2443.2006.00997.x
Jacob, M., Brugger, M., Andres, S., Wagner, M., Graf, E., Berutti, R., et al. (2024). Genome sequencing for cases unsolved by exome sequencing: identifying a single-exon deletion in TBCK in a case from 30 Years ago. Neuropediatrics 55 (04), 260–264. doi:10.1055/s-0044-1782680
Jin, W., and Jianjun, Z. (2024). Revisit TBCK-A pseudo kinase or a true kinase. IgMin Res. 2 (8), 720–725. doi:10.61927/igmin238
Johnson, L. N., Noble, M. E. M., and Owen, D. J. (1996). Active and inactive protein kinases: structural basis for regulation. Cell 85 (2), 149–158. doi:10.1016/S0092-8674(00)81092-2
Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., et al. (2021). Highly accurate protein structure prediction with AlphaFold. Nature 596 (7873), 583–589. doi:10.1038/s41586-021-03819-2
Kim, E.-A., Jang, J. H., Sung, E. G., Song, I. H., Kim, J. Y., and Lee, T. J. (2019). MiR-1208 increases the sensitivity to cisplatin by targeting TBCK in renal cancer cells. Int. J. Mol. Sci. 20 (14), 3540. doi:10.3390/ijms20143540
Klammer, M., Kaminski, M., Zedler, A., Oppermann, F., Blencke, S., Marx, S., et al. (2012). Phosphosignature predicts dasatinib response in non-small cell lung cancer. Mol. and Cell. Proteomics 11 (9), 651–668. doi:10.1074/mcp.M111.016410
Kung, J. E., and Jura, N. (2019). Prospects for pharmacological targeting of pseudokinases. Nat. Rev. Drug Discov. 18, 501–526. doi:10.1038/s41573-019-0018-3
Landrum, M. J., Chitipiralla, S., Kaur, K., Brown, G., Chen, C., Hart, J., et al. (2024). ClinVar: updates to support classifications of both germline and somatic variants. Nucleic Acids Res. 53, D1313–D1321. doi:10.1093/nar/gkae1090
Lange, S. M., Nelen, M. I., Cohen, P., and Kulathu, Y. (2021). Dimeric structure of the pseudokinase IRAK3 suggests an allosteric mechanism for negative regulation. Structure 29 (3), 238–251.e4. doi:10.1016/j.str.2020.11.004
Larsen, S. C., Sylvestersen, K. B., Mund, A., Lyon, D., Mullari, M., Madsen, M. V., et al. (2016). Proteome-wide analysis of arginine monomethylation reveals widespread occurrence in human cells. Sci. Signal. 9 (443), rs9. doi:10.1126/scisignal.aaf7329
Lipton, J. O., and Sahin, M. (2014). The Neurology of mTOR. Neuron 84 (2), 275–291. doi:10.1016/j.neuron.2014.09.034
Liu, D., Yang, F., Liu, Z., Wang, J., Huang, W., Meng, W., et al. (2020). Structure of TBC1D23 N-terminus reveals a novel role for rhodanese domain PLoS Biol. 18, e3000746. doi:10.1371/journal.pbio.3000746
Liu, J., Zheng, Y., Huang, J., Zhu, D., Zang, P., Luo, Z., et al. (2021). Expanding the genotypes and phenotypes for 19 rare diseases by exome sequencing performed in pediatric intensive care unit. Hum. Mutat. 42 (11), 1443–1460. doi:10.1002/humu.24266
Liu, Y., Yan, X., and Zhou, T. (2013). TBCK influences cell proliferation, cell size and mTOR signaling pathway PLoS One 8, e71349. doi:10.1371/journal.pone.0071349
Lupi, M., Avanzato, D., Confalonieri, S., Martino, F., Pennisi, R., Pupo, E., et al. (2024). TBC1 domain-containing proteins are frequently involved in triple-negative breast cancers in connection with the induction of a glycolytic phenotype. Cell Death and Dis. 15 (9), 647. doi:10.1038/s41419-024-07037-2
Ma, J., Gu, Y., Liu, J., Song, J., Zhou, T., Jiang, M., et al. (2023). Functional screening of congenital heart disease risk loci identifies 5 genes essential for heart development in zebrafish. Cell. Mol. Life Sci. 80 (1), 19. doi:10.1007/s00018-022-04669-5
Mace, P. D., and Murphy, J. M. (2021). ‘There’s more to death than life: noncatalytic functions in kinase and pseudokinase signaling. J. Biol. Chem. 296, 100705. doi:10.1016/j.jbc.2021.100705
Maha, A., and Shaza, M. A. (2022). Variants in TBCK cause global developmental delay, dysmorphism, hypotonia. Int. J. Pediatr. Res. 8 (1). doi:10.23937/2469-5769/1510095
Mandel, H., Khayat, M., Chervinsky, E., Elpeleg, O., and Shalev, S. (2017). TBCK-related intellectual disability syndrome: case study of two patients. Am. J. Med. Genet. Part A 173 (2), 491–494. doi:10.1002/ajmg.a.38019
Marin-Valencia, I., Gerondopoulos, A., Zaki, M. S., Ben-Omran, T., Almureikhi, M., Demir, E., et al. (2017). Homozygous mutations in TBC1D23 lead to a non-degenerative form of pontocerebellar hypoplasia. Am. J. Hum. Genet. 101 (3), 441–450. doi:10.1016/j.ajhg.2017.07.015
Mertins, P., Qiao, J. W., Patel, J., Udeshi, N. D., Clauser, K. R., Mani, D. R., et al. (2013). Integrated proteomic analysis of post-translational modifications by serial enrichment. Nat. Methods 10 (7), 634–637. doi:10.1038/nmeth.2518
Mertins, P., Yang, F., Liu, T., Mani, D., Petyuk, V. A., Gillette, M. A., et al. (2014). Ischemia in tumors induces early and sustained phosphorylation changes in stress kinase pathways but does not affect global protein levels. Mol. and Cell. Proteomics 13 (7), 1690–1704. doi:10.1074/mcp.M113.036392
Meyre, D., Farge, M., Lecoeur, C., Proenca, C., Durand, E., Allegaert, F., et al. (2008). R125W coding variant in TBC1D1 confers risk for familial obesity and contributes to linkage on chromosome 4p14 in the French population. Hum. Mol. Genet. 17 (12), 1798–1802. doi:10.1093/hmg/ddn070
Min, X., Lee, B. H., Cobb, M. H., and Goldsmith, E. J. (2004). Crystal structure of the kinase domain of WNK1, a kinase that causes a hereditary form of hypertension. Structure 12 (7), 1303–1311. doi:10.1016/j.str.2004.04.014
Miyamoto, S., Kato, M., Hiraide, T., Shiohama, T., Goto, T., Hojo, A., et al. (2021). Comprehensive genetic analysis confers high diagnostic yield in 16 Japanese patients with corpus callosum anomalies. J. Hum. Genet. 66 (11), 1061–1068. doi:10.1038/s10038-021-00932-y
Moreira, D. D. P., Suzuki, A. M., Silva, A. L., Varella-Branco, E., Meneghetti, M. C. Z., Kobayashi, G. S., et al. (2022). Neuroprogenitor cells from patients with TBCK encephalopathy suggest deregulation of early secretory vesicle transport. Front. Cell. Neurosci. 15, 803302. doi:10.3389/fncel.2021.803302
Murdock, D. R., Dai, H., Burrage, L. C., Rosenfeld, J. A., Ketkar, S., Müller, M. F., et al. (2021). Transcriptome-directed analysis for Mendelian disease diagnosis overcomes limitations of conventional genomic testing. J. Clin. Investigation 131 (1), e141500. doi:10.1172/JCI141500
Murphy, J. M., Zhang, Q., Young, S., Reese, M., Bailey, F., Eyers, P., et al. (2014). A robust methodology to subclassify pseudokinases based on their nucleotide-binding properties. Biochem. J. 457 (2), 323–334. doi:10.1042/BJ20131174
Mushtaq, A., Ashraf, N. U., and Altaf, M. (2023). The mTORC1–G9a–H3K9me2 axis negatively regulates autophagy in fatty acid–induced hepatocellular lipotoxicity. J. Biol. Chem. 299 (3), 102937. doi:10.1016/j.jbc.2023.102937
Nair, D., Diaz-Rosado, A., Varella-Branco, E., Ramos, I., Black, A., Angireddy, R., et al. (2023). Heterozygous variants in TBCK cause a mild neurologic syndrome in humans and mice. Am. J. Med. Genet. Part A 191 (10), 2508–2517. doi:10.1002/ajmg.a.63320
Nass, K., Gorel, A., Abdullah, M. M., V. Martin, A., Kloos, M., Marinelli, A., et al. (2020). Structural dynamics in proteins induced by and probed with X-ray free-electron laser pulses. Nat. Commun. 11 (1), 1814. doi:10.1038/s41467-020-15610-4
Navarro Negredo, P., Edgar, J. R., Manna, P. T., Antrobus, R., and Robinson, M. S. (2018). The WDR11 complex facilitates the tethering of AP-1-derived vesicles. Nat. Commun. 9 (1), 596. doi:10.1038/s41467-018-02919-4
NCI CPTA, C Mertins, P., Mani, D. R., Ruggles, K. V., Gillette, M. A., Clauser, K. R., Wang, P., et al. (2016). Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 534 (7605), 55–62. doi:10.1038/nature18003
Ortiz-González, X. R., Tintos-Hernández, J. A., Keller, K., Li, X., Foley, A. R., Bharucha-Goebel, D. X., et al. (2018). Homozygous boricua TBCK mutation causes neurodegeneration and aberrant autophagy: TBCK-encephaloneuronopathy Disrupts Autophagy. Ann. Neurology 83 (1), 153–165. doi:10.1002/ana.25130
Pan, X., Eathiraj, S., Munson, M., and Lambright, D. G. (2006). TBC-domain GAPs for Rab GTPases accelerate GTP hydrolysis by a dual-finger mechanism. Nature 442 (7100), 303–306. doi:10.1038/nature04847
Park, C.-Y., Jang, J. H., Song, I. H., Kim, J. Y., Doh, K. O., and Lee, T. J. (2023). Suppression of TBCK enhances TRAIL-mediated apoptosis by causing the inactivation of the akt signaling pathway in human renal carcinoma Caki-1 cells. Genes and Genomics 45 (11), 1357–1365. doi:10.1007/s13258-023-01453-y
Parker, W. E., Orlova, K. A., Parker, W. H., Birnbaum, J. F., Krymskaya, V. P., Goncharov, D. A., et al. (2013). Rapamycin prevents seizures after depletion of STRADA in a rare neurodevelopmental disorder. Sci. Transl. Med. 5 (182), 182ra53. doi:10.1126/scitranslmed.3005271
Peck, G. R., Chavez, J. A., Roach, W. G., Budnik, B. A., Lane, W. S., Karlsson, H. K., et al. (2009). Insulin-stimulated phosphorylation of the Rab GTPase-activating protein TBC1D1 regulates GLUT4 translocation. J. Biol. Chem. 284 (44), 30016–30023. doi:10.1074/jbc.M109.035568
Pelassa, I., Cibelli, M., Villeri, V., Lilliu, E., Vaglietti, S., Olocco, F., et al. (2019). Compound dynamics and combinatorial patterns of amino acid repeats encode a system of evolutionary and developmental markers Genome Biol. Evol. 11, 3159–3178. doi:10.1093/gbe/evz216
Prem, C., Sulaiman, A., and Kirmani, S. (2023). Mutation of TBC1 Domain containing kinase (TBCK) with associated intellectual disability and hypotonia. J. Pak. Med. Assoc. 73 (10), 2083–2085. doi:10.47391/JPMA.6733
Qin, J., Wang, Z., Hoogeveen-Westerveld, M., Shen, G., Gong, W., Nellist, M., et al. (2016). Structural basis of the interaction between tuberous sclerosis complex 1 (TSC1) and tre2-bub2-cdc16 domain family member 7 (TBC1D7). J. Biol. Chem. 291 (16), 8591–8601. doi:10.1074/jbc.M115.701870
Quentin, D., Schuhmacher, J. S., Klink, B. U., Lauer, J., Shaikh, T. R., Huis in ’t Veld, P. J., et al. (2023). Structural basis of mRNA binding by the human FERRY Rab5 effector complex. Mol. Cell 83 (11), 1856–1871.e9. doi:10.1016/j.molcel.2023.05.009
Reuter, M. S., Tawamie, H., Buchert, R., Hosny Gebril, O., Froukh, T., Thiel, C., et al. (2017). Diagnostic yield and novel candidate genes by exome sequencing in 152 consanguineous families with neurodevelopmental disorders. JAMA Psychiatry 74 (3), 293. doi:10.1001/jamapsychiatry.2016.3798
Rexach, J., Lee, H., Martinez-Agosto, J. A., Németh, A. H., and Fogel, B. L. (2019). Clinical application of next-generation sequencing to the practice of neurology. Lancet Neurology 18 (5), 492–503. doi:10.1016/S1474-4422(19)30033-X
Sabanathan, S., Gulhane, D., Mankad, K., Davison, J., Ong, M. T., Phadke, R., et al. (2023). Expanding the phenotype of children presenting with hypoventilation with biallelic TBCK pathogenic variants and literature review. Neuromuscul. Disord. 33 (1), 50–57. doi:10.1016/j.nmd.2022.10.004
Saredi, S., Cauley, E. S., Ruggieri, A., Spivey, T. M., Ardissone, A., Mora, M., et al. (2020). Myopathic changes associated with psychomotor delay and seizures caused by a novel homozygous mutation in TBCK. Muscle and Nerve 62 (2), 266–271. doi:10.1002/mus.26907
Savatt, J. M., and Myers, S. M. (2021). Genetic testing in neurodevelopmental disorders. Front. Pediatr. 9, 526779. doi:10.3389/fped.2021.526779
Saxton, R. A., and Sabatini, D. M. (2017). mTOR signaling in growth, metabolism, and disease. Cell 168 (6), 960–976. doi:10.1016/j.cell.2017.02.004
Schuhmacher, J. S., tom Dieck, S., Christoforidis, S., Landerer, C., Davila Gallesio, J., Hersemann, L., et al. (2023). The Rab5 effector FERRY links early endosomes with mRNA localization. Mol. Cell 83 (11), 1839–1855.e13. doi:10.1016/j.molcel.2023.05.012
Sheetz, J. B., and Lemmon, M. A. (2022). Looking lively: emerging principles of pseudokinase signaling. Trends Biochem. Sci. 47 (10), 875–891. doi:10.1016/j.tibs.2022.04.011
Shin, J. J. H., Gillingham, A. K., Begum, F., Chadwick, J., and Munro, S. (2017). TBC1D23 is a bridging factor for endosomal vesicle capture by golgins at the trans-Golgi. Nat. Cell Biol. 19 (12), 1424–1432. doi:10.1038/ncb3627
Shrestha, S., Byrne, D. P., Harris, J. A., Kannan, N., and Eyers, P. A. (2020). Cataloguing the dead: breathing new life into pseudokinase research. FEBS J. 287 (19), 4150–4169. doi:10.1111/febs.15246
Son, S. M., Park, S. J., Lee, H., Siddiqi, F., Lee, J. E., Menzies, F. M., et al. (2019). Leucine signals to mTORC1 via its metabolite acetyl-coenzyme A. Cell Metab. 29 (1), 192–201.e7. doi:10.1016/j.cmet.2018.08.013
Song, J., Hu, Y., Li, W., Li, H., Zheng, H., Chen, Y., et al. (2020). Transcriptome analysis following enterovirus 71 and coxsackievirus A16 infection in respiratory epithelial cells. Archives Virol. 165 (12), 2817–2828. doi:10.1007/s00705-020-04821-1
Stenmark, H. (2009). Rab GTPases as coordinators of vesicle traffic. Nat. Rev. Mol. Cell Biol. 10 (8), 513–525. doi:10.1038/nrm2728
St-Germain, J. R., Taylor, P., Tong, J., Jin, L. L., Nikolic, A., Stewart, I. I., et al. (2009). Multiple myeloma phosphotyrosine proteomic profile associated with FGFR3 expression, ligand activation, and drug inhibition. Proc. Natl. Acad. Sci. 106 (47), 20127–20132. doi:10.1073/pnas.0910957106
Strom, M., Vollmer, P., Tan, T. J., and Gallwitz, D. (1993). A yeast GTPase-activating protein that interacts specifically with a member of the Ypt/Rab family. Nature 361 (6414), 736–739. doi:10.1038/361736a0
Stuart, S. A., Houel, S., Lee, T., Wang, N., Old, W. M., and Ahn, N. G. (2015). A phosphoproteomic comparison of B-RAFV600E and MKK1/2 inhibitors in melanoma cells. Mol. and Cell. Proteomics 14 (6), 1599–1615. doi:10.1074/mcp.M114.047233
Sumathipala, D., Strømme, P., Gilissen, C., Corominas, J., Frengen, E., and Misceo, D. (2019). TBCK encephaloneuropathy with abnormal lysosomal storage: use of a structural variant bioinformatics pipeline on whole-genome sequencing Data unravels a 20-year-old clinical mystery. Pediatr. Neurol. 96, 74–75. doi:10.1016/j.pediatrneurol.2019.02.001
Tan, H.-Y., Wang, B., and Song, Y.-Z. (2022). Identification of a novel pathogenic TBCK variant in a Chinese patient with infantile hypotonia with psychomotor retardation and characteristic facies type 3 (IHPRF3): a case report. BMC Pediatr. 22 (1), 612. doi:10.1186/s12887-022-03672-w
Taylor, S. S., and Kornev, A. P. (2011). Protein kinases: evolution of dynamic regulatory proteins. Trends Biochem. Sci. 36 (2), 65–77. doi:10.1016/j.tibs.2010.09.006
Tintos-Hernández, J. A., Santana, A., Keller, K. N., and Ortiz-González, X. R. (2021). Lysosomal dysfunction impairs mitochondrial quality control and is associated with neurodegeneration in TBCK encephaloneuronopathy. Brain Commun. 3 (4), fcab215. doi:10.1093/braincomms/fcab215
Tsang, M. H. Y., Kwong, A. K., Chan, K. L., Fung, J. L., Yu, M. H., Mak, C. C., et al. (2020). Delineation of molecular findings by whole-exome sequencing for suspected cases of paediatric-onset mitochondrial diseases in the Southern Chinese population. Hum. Genomics 14 (1), 28. doi:10.1186/s40246-020-00278-0
Tu, Y., Yang, Q., Tang, M., Gao, L., Wang, Y., Wang, J., et al. (2024). TBC1D23 mediates Golgi-specific LKB1 signaling. Nat. Commun. 15 (1), 1785. doi:10.1038/s41467-024-46166-2
Udeshi, N. D., Svinkina, T., Mertins, P., Kuhn, E., Mani, D., Qiao, J. W., et al. (2013). Refined preparation and use of anti-diglycine remnant (K-ε-GG) antibody enables routine quantification of 10,000s of ubiquitination sites in single proteomics experiments. Mol. and Cell. Proteomics 12 (3), 825–831. doi:10.1074/mcp.O112.027094
Vaglietti, S., Villeri, V., Dell’Oca, M., Marchetti, C., Cesano, F., Rizzo, F., et al. (2023). PolyQ length-based molecular encoding of vocalization frequency in FOXP2. iScience 26 (10), 108036. doi:10.1016/j.isci.2023.108036
Varadi, M., Anyango, S., Deshpande, M., Nair, S., Natassia, C., Yordanova, G., et al. (2022). AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 50 (D1), D439–D444. doi:10.1093/nar/gkab1061
Vella, V., Giamas, G., and Ditsiou, A. (2022). Diving into the dark kinome: lessons learned from LMTK3. Cancer Gene Ther. 29 (8–9), 1077–1079. doi:10.1038/s41417-021-00408-3
Wang, Y., Yang, F., Fu, Y., Huang, X., Wang, W., Jiang, X., et al. (2011). Spatial phosphoprotein profiling reveals a compartmentalized extracellular signal-regulated kinase switch governing neurite growth and retraction. J. Biol. Chem. 286 (20), 18190–18201. doi:10.1074/jbc.M111.236133
Wang, Y.-Y., Ke, C. C., Chen, Y. L., Lin, Y. H., Yu, I. S., Ku, W. C., et al. (2020). Deficiency of the Tbc1d21 gene causes male infertility with morphological abnormalities of the sperm mitochondria and flagellum in mice PLOS Genet. 16, e1009020. doi:10.1371/journal.pgen.1009020
Wilmes, S., Hafer, M., Vuorio, J., Tucker, J. A., Winkelmann, H., Löchte, S., et al. (2020). Mechanism of homodimeric cytokine receptor activation and dysregulation by oncogenic mutations. Science 367 (6478), 643–652. doi:10.1126/science.aaw3242
Wolfe, M. D., Ahmed, F., Lacourciere, G. M., Lauhon, C. T., Stadtman, T. C., and Larson, T. J. (2004). Functional diversity of the rhodanese homology domain. J. Biol. Chem. 279 (3), 1801–1809. doi:10.1074/jbc.M310442200
Wolfson, R. L., Chantranupong, L., Saxton, R. A., Shen, K., Scaria, S. M., Cantor, J. R., et al. (2016). Sestrin2 is a leucine sensor for the mTORC1 pathway. Science 351 (6268), 43–48. doi:10.1126/science.aab2674
Wu, J., Li, Q., Li, Y., Lin, J., Yang, D., Zhu, G., et al. (2014). A long type of TBCK is a novel cytoplasmic and mitotic apparatus-associated protein likely suppressing cell proliferation. J. Genet. Genomics 41 (2), 69–72. doi:10.1016/j.jgg.2013.12.006
Wu, J., and Lu, G. (2020). Multiple functions of TBCK protein in neurodevelopment disorders and tumors (Review). Oncol. Lett. 21 (1), 1. doi:10.3892/ol.2020.12278
Xu, B., English, J. M., Wilsbacher, J. L., Stippec, S., Goldsmith, E. J., and Cobb, M. H. (2000). WNK1, a novel mammalian serine/threonine protein kinase lacking the catalytic lysine in subdomain II. J. Biol. Chem. 275 (22), 16795–16801. doi:10.1074/jbc.275.22.16795
Yamano, K., Fogel, A. I., Wang, C., van der Bliek, A. M., and Youle, R. J. (2014). Mitochondrial Rab GAPs govern autophagosome biogenesis during mitophagy. eLife 3, e01612. doi:10.7554/eLife.01612
Yoshimura, S., Egerer, J., Fuchs, E., Haas, A. K., and Barr, F. A. (2007). Functional dissection of Rab GTPases involved in primary cilium formation. J. Cell Biol. 178 (3), 363–369. doi:10.1083/jcb.200703047
Zapata-Aldana, E., Kim, D. D., Remtulla, S., Prasad, C., Nguyen, C. T., and Campbell, C. (2019). Further delineation of TBCK - infantile hypotonia with psychomotor retardation and characteristic facies type 3. Eur. J. Med. Genet. 62 (4), 273–277. doi:10.1016/j.ejmg.2018.08.004
Zeqiraj, E., and Van Aalten, D. M. (2010). Pseudokinases-remnants of evolution or key allosteric regulators? Curr. Opin. Struct. Biol. 20 (6), 772–781. doi:10.1016/j.sbi.2010.10.001
Zhang, J., Liu, J., Zheng, F., Yu, M., Shabala, S., and Song, W. Y. (2022). Comparative analysis of arsenic transport and tolerance mechanisms: evolution from prokaryote to higher plants. Cells 11 (17), 2741. doi:10.3390/cells11172741
Zhang, L., Wang, J. C., Hou, L., Cao, P. R., Wu, L., Zhang, Q. S., et al. (2015). Functional role of histidine in the conserved his-x-asp motif in the catalytic core of protein kinases. Sci. Rep. 5 (1), 10115. doi:10.1038/srep10115
Keywords: TBCK, protein kinase, TBC domain, rhodanese domain, genetic disorder
Citation: Cagwin EM, Padgett CM, Lin Y and Zhu W (2025) Decoding TBCK: from bioinformatic insights of domain architecture to disease implications. Front. Biophys. 3:1560824. doi: 10.3389/frbis.2025.1560824
Received: 15 January 2025; Accepted: 14 March 2025;
Published: 04 April 2025.
Edited by:
Jannette Carey, Princeton University, United StatesCopyright © 2025 Cagwin, Padgett, Lin and Zhu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wen Zhu, d3podUBjaGVtLmZzdS5lZHU=
†Present addresses: Emma M. Cagwin, School of Law, University of Georgia, Athens, GA, United States
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.