 Rebecca Strawn
Rebecca Strawn Parvathi S. Murthy
Parvathi S. Murthy Rüdiger H. Ettrich
Rüdiger H. Ettrich István Pelczer1
István Pelczer1 Jannette Carey
Jannette Carey
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Biophys. , 23 February 2024
Sec. Protein Structure and Dynamics
Volume 2 - 2024 | https://doi.org/10.3389/frbis.2024.1359979
Symmetry was a key concept underlying the MWC model for allostery advanced in 1965 by Monod, Wyman, and Changeux. The reciprocal interactions of symmetrically-arranged identical subunits were proposed to stabilize multimeric assemblies together with the free energy from bound ligands that progressively favor a monomer-like state. Structural symmetry of subunits was assumed to be maintained in the partially-ligated states, even if ligand placement itself is not symmetric. Partially-ligated states can be populated sufficiently for experimental study only in negatively cooperative systems, which were not considered in the MWC model. The work reported here uses 1H, 13C, 15N, and 19F NMR to evaluate the structural symmetry of the hexameric arginine repressor of E. coli, a negatively cooperative system, with a single bound L-arginine ligand. The analysis indicates that the singly-ligated hexamer maintains structural symmetry as probed by these four NMR nuclei. The results are consistent with earlier molecular dynamics simulations suggesting that the global dynamics of the singly-ligated assembly are harnessed to maintain structural symmetry. The results extend MWC symmetry concepts to this negatively cooperative system, and indicate a role for global dynamics in allostery.
Symmetry was a fundamental concept in the MWC model for allostery (Monod et al., 1965). This model envisions an equilibrium between tense (T) and relaxed (R) states of a multimeric protein assembly. The tense state was postulated to arise from mutual constraints upon the subunits that reduce ligand affinity, and is favored over R in the absence of ligand. Ligand binding shifts the equilibrium in favor of the relaxed, monomer-like state with higher affinity. In the canonical case of hemoglobin this shift is thought to be progressive with each additional oxygen ligand, increasing affinity in proportion to the population of targets in the R state. Importantly, structural symmetry was proposed to be maintained in each partially-ligated assembly, i.e., with no mixed R and T states, although the placement of ligands cannot be symmetrical in all intermediate ligation states. The reciprocal interactions among subunits in symmetric assemblies of homomultimers were proposed to be more stabilizing than non-symmetrical interactions, and to maintain the relaxed assembly together with the free energy of ligand binding.
The MWC model is a conceptual, abstract model that provides no clues about the relaxation process, although the authors couched their discussion in presciently dynamic and thermodynamic terms (Monod et al., 1965). Partially-ligated states can be populated sufficiently for experimental study only in systems with negative cooperativity. The antagonized binding of one ligand by another is not accommodated by the MWC model and was not treated in the early work on allostery (Monod and Jacob, 1961; Monod et al., 1963; Monod et al., 1965). Binding data cannot distinguish between antagonized binding and affinity heterogeneity. Affinity heterogeneity is typical in heteromultimeric systems but unexpected in homomultimers. One way affinity heterogeneity can seem to arise even in homomultimers is if a population of target molecules contains an inactive or less active sub-population. Reduced-activity species can occur for many trivial reasons. The KNF model of Koshland, Némethy, and Filmer does accommodate negative cooperativity (Koshland et al., 1966). An example of possible negative cooperativity may have motivated development of the KNF model. That example, of homotetrameric rabbit muscle glyceraldehyde-3-phosphate dehydrogenase (Cook and Koshland, 1970; Mockrin et al., 1975), was later shown to reflect an impure protein preparation (Gennis, 1976) rather than true negative cooperativity.
Since that time many examples of bona fide negative cooperativity have been documented (for a recent review see Wielgus-Kutrowska et al., 2018). The most extreme type of negative cooperativity is part-of-the-sites reactivity, in which even super-saturating substrate concentrations cannot fill some sites when others are occupied, i.e., essentially infinite negative cooperativity. Part-of-the-sites reactivity typically involves direct effects of substrates at active sites rather than allosteric effects of structurally unrelated compounds acting at distant sites. Less extreme examples of negative cooperativity are now known that do appear to operate allosterically. Modest negative cooperativity can offer a potential biological advantage (Carey, 2022) depending on the relative affinities in the binding system (i.e., the magnitude of antagonism) and the dynamic range of cellular ligand and target concentrations. This advantage can accrue when relative affinities and concentrations permit a higher-affinity site to be occupied fully, or nearly fully, while lower-affinity sites remain largely empty. In such a case the system gains a third action level of response to ligand in addition to the empty and saturated states.
The arginine repressor of E. coli, ArgR, is a candidate to make use of such an advantage. ArgR is the feedback regulator of both biosynthesis and catabolism of L-arginine, L-arg (Maas, 1994). L-arg levels in E. coli vary between 0.14 and 1.5 mM in various stages and conditions of cell growth (Caldara et al., 2008). ArgR is additionally a multifunctional protein with a role in plasmid recombination that also depends on L-arg (Stirling et al., 1988). The protein is a ∼100,000 Da hexamer of six identical subunits that binds up to six L-arg ligands. Curiously, DNA operators in the Arg regulon typically present no more than four repeats of a recognition half-site, and footprinting data (Tian et al., 1992) imply that the hexameric protein contacts only four half-sites. It was this symmetry mismatch that initially prompted our curiosity about this system. Prior to the availability of structural information, proteolytic dissection was used to define the domain organization of ArgR (Grandori et al., 1995). The results indicated a two-domain structure for each polypeptide chain, with a central interdomain linker region as the major site of cleavage. The isolated N-terminal domain half of ArgR, ArgRN, is able to bind to operator DNA as a monomer in an L-arg-independent manner (Grandori et al., 1995). The NMR structure of monomeric ArgRN showed it to be a member of the winged-helix-turn-helix family, and four winged-helix domains could account for the footprint of intact ArgR on a natural operator bearing four half-sites (Sunnerhagen et al., 1997). Nevertheless, stoichiometry measurements confirmed that all six subunits are fully active for binding with DNA oligomers bearing partial operator sites (Szwajkajzer et al., 2001).
The C-terminal domain, ArgRC, is the locus of L-arg binding as well as subunit assembly. ArgRC was highly resistant to further proteolysis (Grandori et al., 1995), likely because it forms a hexameric assembly, as later confirmed by X-ray diffraction of crystals in which unintended proteolysis of ArgR yielded crystalline ArgRC, apparently with loss of ArgRN (van Duyne et al., 1996). A crystal structure is available for only the C-terminal domain of the E. coli protein, showing ArgRC to be a symmetric hexamer of ∼50,000 Da in presence or absence of six L-arg ligands. ArgRC hexamers comprise two trimers stacked in alignment upon each other with 3-2 symmetry. Each L-arg ligand makes extensive interactions with three of the six subunits, both within and between trimers. The crystal structures of E. coli apo- and holoArgRC have only very minor differences, offering no insight into the mechanism of activation by L-arg. Crystal structures of some intact ArgR homologs (Ni et al., 1999; Dennis et al., 2002; Cherney et al., 2009; Cherney et al., 2010) show hexameric ArgRC domains essentially identical to those of E. coli, but with rotational variance in the alignment of trimers among some homologs, and/or between their apo- and holoArgR states. Six symmetrically-disposed peripheral DNA-binding domains, also essentially identical to those of E. coli, surround ArgRC in the homologs. Major sequence and/or structural differences in the interdomain linker region distinguish the homologs from each other, and from E. coli ArgR (Pandey et al., 2020).
Quantitative study of L-arg binding by intact E. coli ArgR or sub-cloned ArgRC using isothermal titration calorimetry (ITC) led to the conclusion that the six equivalents of L-arg bind to the symmetric hexamer asymmetrically, i.e., with negative cooperativity (Jin et al., 2005). According to model-fitting of extensive ITC data, the first equivalent of L-arg binds with an affinity of approximately 1 µM, and the remaining five ligands bind with an affinity of approximately 100 µM, with identical results for both proteins. Although negative cooperativity can be mistaken for affinity heterogeneity caused by damage to the target sample, exhaustive control experiments ruled out artifactual causes. Among other evidence, both ArgR and ArgRC, which are purified under very different conditions, show the same negatively cooperative binding with L-arg as well as with the L-arg analog L-canavanine (L-can) that binds orders of magnitude more weakly. Furthermore, binding of the first equivalent of L-arg or L-can to ArgR or ArgRC is quantitatively correlated with a unique endothermic component of the reaction heat, followed by exothermic heats associated with filling of the remaining five sites.
In the presence of excess L-arg adequate to fill all six binding sites the E. coli holoArgR hexamer has approximately ten-fold faster cleavage in the linker region compared to apoArgR (Grandori et al., 1995). With the aim to increase confidence in the 1 + 5 binding model derived from the ITC results, the ∼100-fold difference in affinity between the stronger first event and the remaining five binding events was exploited to prepare samples for proteolysis in which mainly the single stronger site is predicted to be occupied. The binding model was used to predict concentrations of ArgR and L-arg that populate the strong site to ∼97% of ligand saturation while limiting occupancy of the weaker sites to ∼6% each on average. The singly-ligated state of ArgR thus prepared showed an approximate tenfold increase in proteolytic sensitivity in the linker region compared to apoArgR (Jin et al., 2005), the same as holo ArgR with all six L-arg binding sites occupied. Thus, the singly-ligated state has a proteolytically detectable conformational alteration in the linker region that is very similar to that of fully-ligated ArgR.
In work published previously (Strawn et al., 2010), molecular dynamics simulations (MD) were used to augment the calorimetric and proteolytic data in the hope of shedding light on the mechanisms of negative cooperativity and L-arg activation. Long timescale, fully atomistic MD (Strawn et al., 2010) shows the two trimers of the E. coli apoArgRC hexamer undergoing an oscillatory rotational motion across the trimer interface reminiscent of the static differences in alignment observed in crystals of some ArgR homologs. This motion was observed to be driven by the repeated formation and release of doubly-hydrogen-bonded ion pairs between Arg and Asp sidechains that face each other in each of the six empty L-arg binding sites (Strawn et al., 2010). When paired with Asp the Arg sidechain occupies the binding site in a manner virtually identical to that of the L-arg ligand, blocking access to the site. As calculated from the MD results, the structures of the six apoArgRC monomers during the simulations, and the center-of-mass of each monomer relative to the common hexamer center-of-mass, indicate that apoArgRC is fully symmetric (Strawn et al., 2010).
Release of an Arg-Asp ion pair opens a binding site that allows occasional entry of an L-arg ligand. The ligand makes all the same interactions with multiple subunits as the Arg sidechain, as well as many novel interactions involving its free alpha-amino and alpha-carboxylate substituents. Binding of the first L-arg ligand resets the affinities of the remaining five empty sites by severely restricting the oscillatory motion of the assembly. The Arg and Asp residues in the empty binding sites continue to reach toward each other, blocking the binding sites and limiting access of L-arg at the remaining five binding sites, which open less frequently once one L-arg is bound (Pandey et al., 2014).
These two opposing effects—attempted ion pairing across the five empty sites frustrated by a single bound L-arg that impedes rotation—create a shuddering motion that is propagated through the hexameric ArgRC assembly (Strawn et al., 2010). Intense motion is manifested at the surface of the hexamer as judged by B-factors calculated from the MD results (Strawn et al., 2010). Little motion is detectable near the center of the assembly, where the single L-arg maintains its interactions with three of the six subunits (Strawn et al., 2010). No internal rearrangements are evident within any monomer of the assembly (Strawn et al., 2010). A macroscopic analog of the dynamic ArgRC assembly in the singly-ligated state is a bouquet of balloons in strong wind (Figure 1A). The bouquet represents the C-terminal domains (Figure 1B) tethered by their reciprocal subunit interactions and by the single bound ligand. The periphery of each balloon is mobile enough to create a colored halo that represents the excursions of each ArgRC subunit. As in singly-ligated ArgRC, far less motion is detectable at the point where the bouquet is tethered. As calculated from the MD results, the structures of the ArgRC monomers, as well as the center-of-mass of each monomer relative to the common hexamer center-of-mass, both support the conclusion that the singly-ligated state is completely symmetric (Strawn et al., 2010). Apparently, any asymmetry introduced by the binding of only one ligand is averaged out by the intense shuddering motion of the assembly.
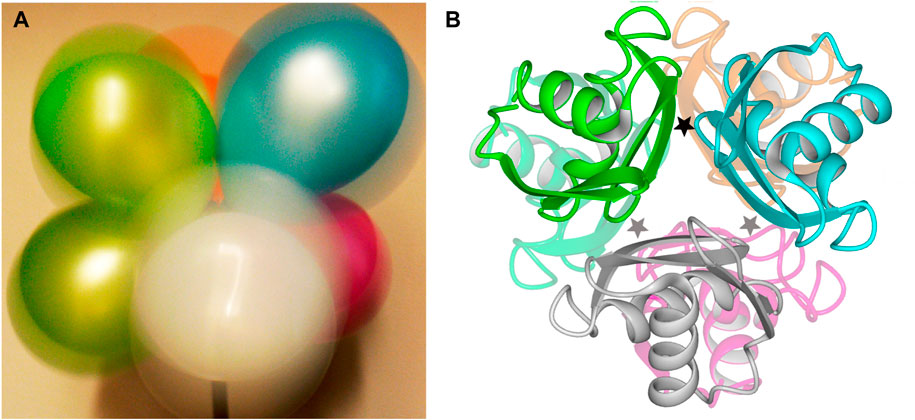
FIGURE 1. Structure of E. coli ArgRC. (A). Macroscopic model for shuddering motion of ArgRC with a single bound L-arg. When photographed with long exposure time, a bouquet of balloons in strong wind tethered at one common central point (bottom) displays intense peripheral motion evident as a colored halo around the top of each balloon. The tethered points experience far less motion, as can be seen at the bottoms of the upper green and blue balloons. (B). Crystal structure of ArgRC. Ribbon model based on PDB 1XXC (Van Duyne et al., 1996). Subunits are colored to match the balloons in the symbolic view of panel A. The locations of the L-arg binding sites are indicated by stars; because the two trimers lie precisely atop each other in this crystal structure, only three stars are shown for the six sites. One star is black to represent the single L-arg ligand.
To further evaluate the MD-predicted symmetry of the singly-ligated state of intact E. coli ArgR and its L-arg-binding domain ArgRC, NMR is used in the work reported here to compare samples with zero vs. one L-arg ligand bound. The apo- and holoproteins are fully symmetric as judged by X-ray diffraction (Van Duyne et al., 1996) and MD (Strawn et al., 2010). As shown here by NMR, apoArgR and apoArgRC are also symmetric in solution. The NMR evidence for this symmetry is that both proteins display the spectral complexity of far smaller proteins, consistent with molecular weights corresponding approximately to each respective subunit (∼17,000 Da or ∼ 8,000 Da), indicating that all six subunits behave equivalently, i.e., the hexamers are structurally symmetric. The premise of the present approach is that if the binding of a single ligand breaks the structural symmetry of the hexameric protein, then the NMR spectra will present additional resonances and/or changes of chemical shifts reflecting the distinct structure of a singly-ligated subunit.
Proteins were produced and purified as described previously (Jin et al., 2005). Labeled proteins were produced in M9 minimal medium with addition of 15NH4Cl instead of NH4Cl, or with addition upon induction of 19F-Tyr DL-m-fluorotyrosine (Sigma). The 19F-Tyr-labeled ArgR used here was produced for another study that required introduction of a mutation, Cys68Ser. The Cys68Ser ArgR protein was expressed and purified as for wild type ArgR. Reverse titrations used guanidino-labeled 15N-L-arg (Sigma).
As in the earlier proteolysis experiments, the affinities recovered from fitting the 1 + 5 binding model to the ITC data were used to predict ligand occupancies in the stronger and weaker sites at specific concentrations of protein and ligand. In contrast to the proteolysis experiments, at protein target concentrations used in NMR ligand-binding theory predicts a more unique population of higher-affinity states and more limited occupancy of weaker-affinity sites. This outcome is possible because when target concentrations are high relative to the ligand dissociation equilibrium constant, Kd, mass action makes the ligand more effective in converting the target to the bound state. This effect makes it possible to approach saturation of the high-affinity site with little excess free ligand; the low concentration of excess free ligand limits the occupancy of the lower-affinity sites. Judicious choice of the concentrations used in most experiments here (given in each figure legend) predicts ∼95% occupancy of the high-affinity single site and ∼5% total occupancy of the five remaining weaker sites (∼1% each). The greatly reduced solubility of the intact ArgR holoprotein at these concentrations limited its NMR analysis. The solubility of ArgRC is greater than that of ArgR, and its affinity for L-arg is equivalent to that of ArgR according to ITC data.
For analysis of structural symmetry in the singly-ligated state, four kinds of NMR data were collected for proteins with zero or with one L-arg ligand bound: one-dimensional 1H spectra of ArgRC (Figure 2); 13C spectra at natural abundance with unlabeled ArgRC (Figure 3); 19F spectra of Cys68Ser intact ArgR labeled with 19F-Tyr (Figure 4); and HSQC spectra of 15N-labeled ArgRC (Figure 5). These NMR experiments aimed to evaluate symmetry of the hexamers on the wide range of timescales of NMR relaxation inherent to each observed NMR-active nucleus, ranging from ∼40 MHz for 15N to ∼400 MHz for 1H resonances. Figures 2–5 compare paired spectra of unligated and singly-ligated protein, and display a difference spectrum (one L-arg minus zero L-arg) between each pair wherever feasible. In all cases the spectra of unligated and singly-ligated protein are the same in every detail, with no sign of additional resonances or chemical shift changes that would indicate a breaking of symmetry in the hexamer upon binding of a single L-arg. In addition, the difference spectra give no indication of structural changes. These results indicate that binding of one equivalent of ligand does not break the symmetry of the hexamer, consistent with the results from MD simulations. Limited NMR analysis of ArgRC with higher partial occupancies of L-arg binding sites, or with all 6 L-arg sites occupied, also showed no evidence of the breaking of symmetry (data not shown).
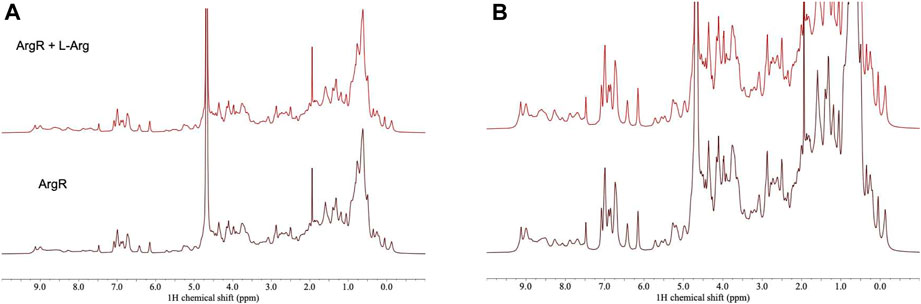
FIGURE 2. 1H NMR spectra of ArgRC with zero or one L-arg bound. ArgRC concentration 13 mM monomer, L-arg concentration zero or 2.17 mM, yielding approximately 99% occupancy of the single strong site and negligible occupancy of the five weaker sites. Bottom, zero L-arg; top, one L-arg. (A) Spectral intensity is truncated at the height of the water resonance at ∼4.6 ppm equal to the maximum height of the methylene envelope centered around ∼0.75 ppm. At this scale, fine details are difficult to compare in the two spectra. (B) Spectra are truncated at an arbitrary height in the methylene envelope centered at ∼1.5 ppm. At this scale, fine details can be better compared than in panel A. Spectra were acquired in 20 mM sodium phosphate buffer, pH 7.5, ∼5% D2O using a 500-MHz Bruker AVANCE-III spectrometer with a TCI (triple resonance H/C/N) cryoprobe.
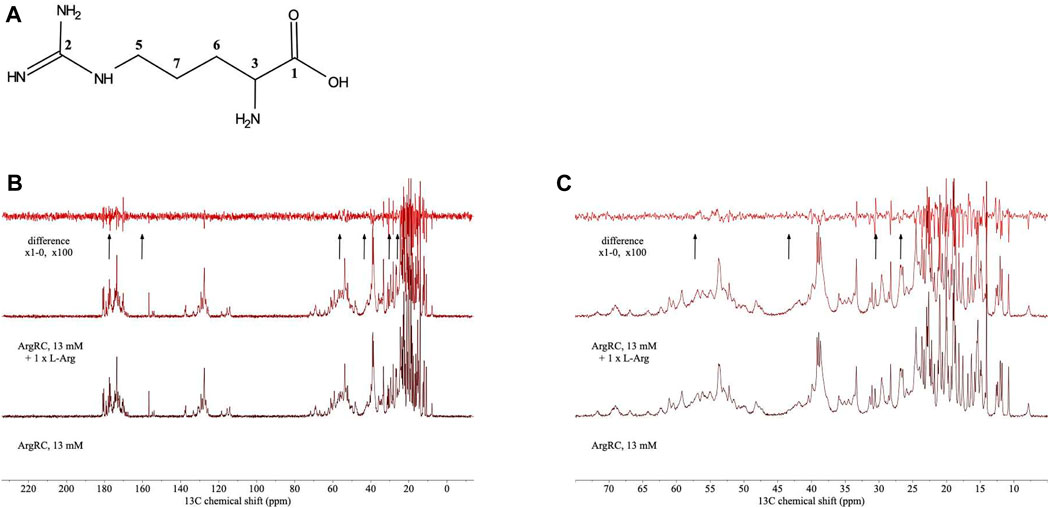
FIGURE 3. 13C NMR spectra of unlabeled ArgRC with zero or one L-arg bound. (A) Numbering of 13C carbon atoms of free L-arg. (B) 13C NMR spectra. ArgRC concentration 13 mM monomer, L-arg concentration zero or 2.17 mM, yielding approximately 99% occupancy of the single strong site and negligible occupancy of the five weaker sites as calculated from the 1+5 binding model. Bottom, zero L-arg; middle, one L-arg; upper, difference spectrum (one L-arg minus zero L-arg). Arrows below the difference spectrum mark (left to right) the expected resonance positions of 13C carbon atoms of free L-arg numbered 1 to 7 in panel A at ∼ 177, 160, 57, 43, 30, and 26 ppm (Human Metabolome Database; Wishart et al., 2022). (C) Zoomed-in view of the upfield region from panel B. Spectra were acquired at natural abundance in 20 mM sodium phosphate buffer, pH 7.5, ∼100% D2O using a 500-MHz Bruker AVANCE-III spectrometer equipped with 13C-optimized DCH cryoprobe.
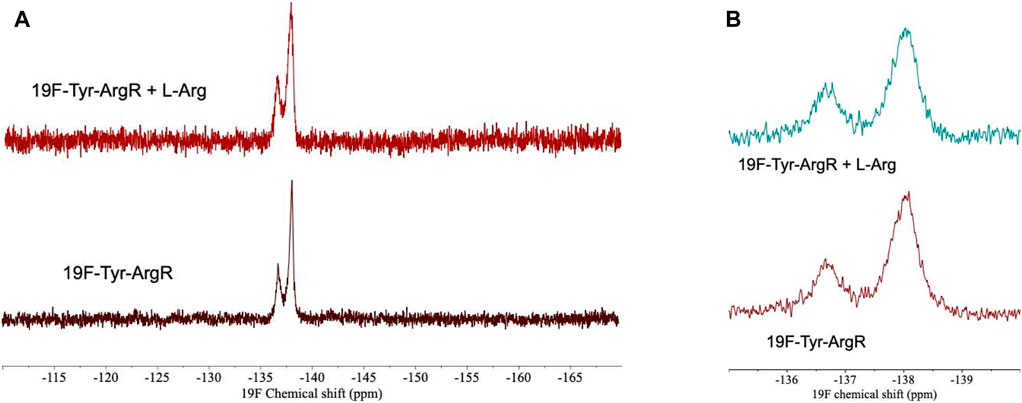
FIGURE 4. 19F-Tyr NMR spectra of intact Cys68Ser ArgR with zero or one L-arg bound. ArgR concentration 480 µM monomer, L-arg concentration 80 µM, yielding approximately 94% occupancy of the stronger single site and negligible occupancy of the weaker five sites. (A) Full spectral window. Upper, one L-arg; peak widths are 420 and 355 Hz (full width at half maximum, left to right). Lower, zero L-arg; peak widths are 300 and 200 Hz. (B) Zoom in on 135–140 ppm region. Spectra were acquired in 0.1 M tris buffer, pH 7.5, ∼10% D2O using a 600 MHz Varian INOVA spectrometer. NMR spectra at pH 8.5 were used to assign the downfield peak to Tyr67 on the N-terminal side of the interdomain linker by comparison of wild type and Cys68Ser mutant 19F-Tyr-labeled ArgR. Tyr91 and Tyr 145 in the upfield peak pack together in the folded C-terminal domain on the side opposite the L-arg binding site.
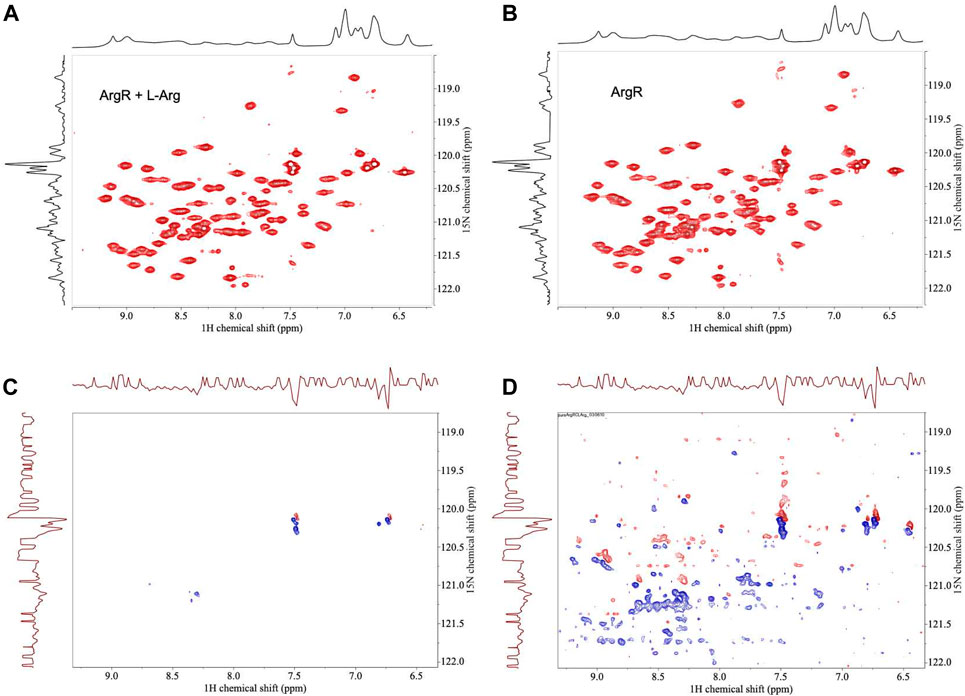
FIGURE 5. HSQC NMR spectra of 15N-labeled ArgRC with zero or one L-arg bound. 15N-labeled ArgRC concentration 80 µM monomer, L-arg (unlabled) concentration zero or 20 μM, yielding approximately 90% occupancy of the single stronger site and ∼5% occupancy of the weaker five sites (∼1% each). (A) L-arg added. (B) No L-arg added. (C) Difference spectrum (one L-arg minus zero L-arg) using the baselines shown in the upper spectra. (D) Difference spectrum (one L-arg minus zero L-arg) by cutting the first contour level close to the noise floor. In the difference spectra negative crosspeaks (blue) indicate excess intensities present in the zero L-arg spectrum; positive crosspeaks (red) indicate resonances present in the one L-arg spectrum. The x-axes display 1H chemical shift; the y-axes are 15N chemical shift. The corresponding one-dimensional spectrum is presented along each respective axis: the 1H spectrum shown was collected independently; the 15N spectrum shown is the projection of the 15N crosspeaks onto the axis. Spectra were acquired in 20 mM sodium phosphate buffer, pH 7.5, ∼5% D2O using a 500-MHz Bruker AVANCE-III spectrometer equipped with a QNP cryoprobe.
L-arg itself is not detected in any of the spectra. This result is not unexpected because the calculations based on ITC affinity indicate little or no free L-arg is predicted at the concentrations used for the singly-ligated spectra, and the resonances of free L-arg lie under protein resonances in any case. Therefore, to provide evidence that the ligand is indeed bound under the conditions of the NMR experiments, two kinds of control experiment were conducted. A reverse titration of ArgRC protein into L-arg was carried out (Figure 6). This series of spectra shows that the proton signals from the ligand are detectably broadened upon binding to the protein and their intensities are reduced progressively. The reverse titration results are quantitatively consistent with the affinities derived from the 1 + 5 binding model (not shown), further supporting that model. The second control experiment was designed to take advantage of the ability of bound L-arg to prevent the exchange of trimers between intact ArgR and domain fragment ArgRC to form mixed hexamers of intermediate mass. The exchange reaction can be visualized on native polyacrylamide gels (van Duyne et al., 1996; Szwajkajzer et al., 2001). Exchange was found to be blocked in samples from a titration series like that in Figure 6 (not shown), indicating the presence of L-arg in the bound state.
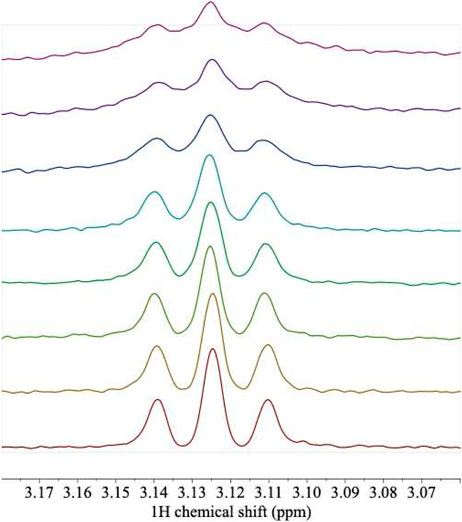
FIGURE 6. 1H NMR spectra of L-arg titrated with unlabeled ArgRC. A sample of L-arg at an initial concentration of 100 μM was titrated with aliquots from a 1.04 mM monomer stock of unlabeled ArgRC, yielding the following predicted fraction of total L-arg bound (%), calculated from the 1 + 5 binding model (bottom to top): 0, 5.7, 10.8, 16.7, 19.4, 33.4, 41.2, and 49.2%. Solution conditions were 20 mM sodium phosphate, pH 7.5, with 5% D2O. Protein proton peaks that overlap L-arg were corrected by local baseline correction. The triplet shown corresponds to the protons on L-arg C-δ. The experiment used an L-Arg sample labeled with 15N in the two terminal positions; labeling had no detectable effect on the covalently bound protons in remote locations, including C-δ. Data were collected on a 500-MHz Bruker AVANCE-III spectrometer equipped with a TCI cryoprobe.
In all cases the solution NMR experiments described in this report are fully consistent with the prediction based on previously published MD results (Strawn et al., 2010) that the singly-ligated hexamer is structurally symmetric. The intense shuddering observed in the MD simulations apparently averages out any structural differences caused by the bound ligand. The extensive interactions of the single L-arg ligand with multiple subunits, together with the symmetric interactions among subunits, may enable the assembly to survive the intense shuddering that symmetrizes its structure. It is equally important to note that the free energy lowering by the bound ligand is a distributed property of the whole system, and does not reside at the binding site itself.
It is unclear whether the symmetry of partially-ligated states, or the maintenance of symmetry by motions that can average out ligand-induced asymmetries, may be more general. Few other negatively cooperative systems have been studied with the range of approaches applied to ArgR. An intriguing case with persistent knowledge gaps despite extensive study is that of the cyclic AMP receptor protein, CRP, where asymmetry of ligand binding has long been thought to play a role in transcriptional activation of the protein by cAMP (Takahashi et al., 1980; Heyduk and Lee, 1989). ITC data for CRP-cAMP binding (e.g., Gorshkova et al., 1995; Rodgers et al., 2013) present unexplained anomalies reminiscent of those found with ArgR that complicate evaluation of affinities and assignment of positive or negative cooperativity. Further analysis of CRP ITC behavior and its relationship to CRP dynamics may expand our understanding of allosteric mechanisms and of CRP itself. It is not clear if activation by cAMP is allosteric, because two of its four cAMP ligands per dimer make contact directly with DNA; the roles of its distinct ligand-binding sites in transcription activation have not been uniquely resolved. One extant possibility is that the cAMP ligands relevant for activation are those bound directly at the DNA interface, and not those bound in the distant canonical cAMP-binding domains. In that case CRP may be similar to the E. coli tryptophan repressor dimer, TrpR, which binds its two L-tryptophan ligands at the DNA-binding surface where they make direct contacts to the operator (Otwinowski et al., 1988; Lawson and Carey, 1993), i.e., their action is not allosteric.
The symmetry of the singly-ligated state of ArgR documented here by NMR and visualized previously by MD simulation (Strawn et al., 2010) is fully consistent with the monomer-like relaxed state proposed in the MWC model, despite the fact that ArgR displays antagonized, rather than facilitated, binding at the remaining empty sites. This inference extends dramatically the extraordinary prescience of the MWC model, which contemplated proteins as dynamic and thermodynamic entities at a time when the usual view was of rigid crystalline solids at best, and at worst as being possibly possessed by vitalism, a doubt that in some corners may have been dispelled only later (Nicholson and Gawne, 2015), e.g., by the total chemical synthesis of ribonuclease A (Gutte and Merrifield, 1969). As anticipated by the MWC model, symmetric subunit interactions and multidentate interactions of the ligand with several subunits appear to offer stability to the assembly for maintaining the relaxed, monomer-like state of the partially-ligated system, which in this case must survive the intense motion that averages out any asymmetry. Thus the conceptual symmetry principles of the MWC model appear to encompass the case of ArgR even though the model did not consider negative cooperativity and does not accommodate it mathematically. The results open the possibility that the distinction between R and T states reflects dynamic rather than static structural differences, consistent with the entities contemplated in the MWC model.
The raw data supporting the conclusion of this article will be made available by the authors, without undue reservation.
RS: Formal Analysis, Methodology, Validation, Writing–review and editing, Data curation, Investigation, Visualization. PM: Data curation, Formal Analysis, Investigation, Methodology, Validation, Writing–review and editing. RE: Formal Analysis, Writing–review and editing, Resources, Visualization. IP: Formal Analysis, Resources, Writing–review and editing, Visualization, Data curation, Investigation, Methodology, Software, Validation. JC: Formal Analysis, Methodology, Resources, Validation, Writing–review and editing, Conceptualization, Funding acquisition, Project administration, Supervision, Writing–original draft.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
PM thanks Georgian Court University for an award that enabled her sabbatical visit at Princeton University. RE acknowledges the Institute of Systems Biology and Ecology, Czech Academy of Sciences, Zamek 136, Nové Hrady, Czech Republic. JC acknowledges with gratitude NSF awards 0853423 and 1358737 that supported this work. We thank our colleagues Angela Creager and Fred Hughson for their comments on the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer RG declared a shared parent affiliation with the authors RS, PM, RE, IP, and JC to the handling editor at the time of review.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Caldara, M., Dupont, G., Leroy, F., Goldbeter, A., De Vuyst, L., and Cunin, R. (2008). Arginine biosynthesis in Escherichia coli: experimental perturbation and mathematical modeling. J. Biol. Chem. 283 (10), 6347–6358. doi:10.1074/jbc.M705884200
Carey, J. (2022). Affinity, specificity, and cooperativity of DNA binding by bacterial gene regulatory proteins. Int. J. Mol. Sci. 23 (1), 562. doi:10.3390/ijms23010562
Cherney, L. T., Cherney, M. M., Garen, C. R., and James, M. N. (2009). The structure of the arginine repressor from Mycobacterium tuberculosis bound with its DNA operator and co-repressor, L-arginine. J. Mol. Biol. 388 (1), 85–97. doi:10.1016/j.jmb.2009.02.053
Cherney, L. T., Cherney, M. M., Garen, C. R., and James, M. N. (2010). Crystal structure of the intermediate complex of the arginine repressor from Mycobacterium tuberculosis bound with its DNA operator reveals detailed mechanism of arginine repression. J. Mol. Biol. 399 (2), 240–254. doi:10.1016/j.jmb.2010.03.065
Cook, R. A., and Koshland, D. E. (1970). Positive and negative cooperativity in yeast glyceraldehyde 3-phosphate dehydrogenase. Biochemistry 9 (17), 3337–3342. doi:10.1021/bi00819a007
Dennis, C. A., Glykos, N. M., Parsons, M. R., and Phillips, S. E. (2002). The structure of AhrC, the arginine repressor/activator protein from Bacillus subtilis. Acta Crystallogr. Sect. D. Biol. Crystallogr. 58 (Pt 3), 421–430. doi:10.1107/s0907444901021692
Gennis, L. S. (1976). Negative homotropic cooperativity and affinity heterogeneity: preparation of yeast glyceraldehyde-3-phosphate dehydrogenase with maximal affinity homogeneity. Proc. Natl. Acad. Sci. U. S. A. 73 (11), 3928–3932. doi:10.1073/pnas.73.11.3928
Gorshkova, I., Moore, J. L., McKenney, K. H., and Schwarz, F. P. (1995). Thermodynamics of cyclic nucleotide binding to the cAMP receptor protein and its T127L mutant. J. Biol. Chem. 270 (37), 21679–21683. doi:10.1074/jbc.270.37.21679
Grandori, R., Lavoie, T. A., Pflumm, M., Tian, G., Niersbach, H., Maas, W. K., et al. (1995). The DNA-binding domain of the hexameric arginine repressor. J. Mol. Biol. 254 (2), 150–162. doi:10.1006/jmbi.1995.0607
Gutte, B., and Merrifield, R. B. (1969). Total synthesis of an enzyme with ribonuclease A activity. J. Am. Chem. Soc. 91 (2), 501–502. doi:10.1021/ja01030a050
Heyduk, T., and Lee, J. C. (1989). Escherichia coli cAMP receptor protein: evidence for three protein conformational states with different promoter binding affinities. Biochemistry 28 (17), 6914–6924. doi:10.1021/bi00443a021
Jin, L., Xue, W. F., Fukayama, J. W., Yetter, J., Pickering, M., and Carey, J. (2005). Asymmetric allosteric activation of the symmetric ArgR hexamer. J. Mol. Biol. 346 (1), 43–56. Epub 2004 Dec 21. Erratum in: J Mol Biol. 2005 Mar 25;347(2):479. doi:10.1016/j.jmb.2004.11.031
Koshland, D. E., Némethy, G., and Filmer, D. (1966). Comparison of experimental binding data and theoretical models in proteins containing subunits. Biochemistry 5 (1), 365–385. doi:10.1021/bi00865a047
Lawson, C. L., and Carey, J. (1993). Tandem binding in crystals of a trp represser/operator half-site complex. Nature 366 (6451), 178–182. doi:10.1038/366178a0
Maas, W. K. (1994). The arginine repressor of Escherichia coli. Microbiol. Rev. 58 (4), 631–640. doi:10.1128/mr.58.4.631-640.1994
Mockrin, S. C., Byers, L. D., and Koshland, D. E. (1975). Subunit interactions in yeast glyceraldehyde-3-phosphate dehydrogenase. Biochemistry 14 (25), 5428–5437. doi:10.1021/bi00696a008
Monod, J., Changeux, J. P., and Jacob, F. (1963). Allosteric proteins and cellular control systems. J. Mol. Biol. 6, 306–329. doi:10.1016/s0022-2836(63)80091-1
Monod, J., and Jacob, F. (1961). General conclusions: teleonomic mechanisms in cellular metabolism, growth, and differentiation. Cold Spring Harb. Symposia Quantitative Biol. 26, 389–401. doi:10.1101/sqb.1961.026.01.048
Monod, J., Wyman, J., and Changeux, J. P. (1965). On the nature of allosteric transitions: a plausible model. J. Mol. Biol. 12, 88–118. doi:10.1016/s0022-2836(65)80285-6
Ni, J., Sakanyan, V., Charlier, D., Glansdorff, N., and Van Duyne, G. D. (1999). Structure of the arginine repressor from Bacillus stearothermophilus. Nat. Struct. Biol. 6 (5), 427–432. doi:10.1038/8229
Nicholson, D. J., and Gawne, R. (2015). Neither logical empiricism nor vitalism, but organicism: what the philosophy of biology was. Hist. Philosophy Life Sci. 37 (4), 345–381. doi:10.1007/s40656-015-0085-7
Otwinowski, Z., Schevitz, R. W., Zhang, R. G., Lawson, C. L., Joachimiak, A., Marmorstein, R. Q., et al. (1988). Crystal structure of trp represser/operator complex at atomic resolution. Nature 335 (6188), 321–329. Erratum in: Nature 1988 Oct 27;335(6193):837. PMID: 3419502. doi:10.1038/335321a0
Pandey, S. K., Melichercik, M., Řeha, D., Ettrich, R. H., and Carey, J. (2020). Conserved dynamic mechanism of allosteric response to L-arg in divergent bacterial arginine repressors. Molecules 25 (9), 2247. doi:10.3390/molecules25092247
Pandey, S. K., Řeha, D., Zayats, V., Melichercik, M., Carey, J., and Ettrich, R. (2014). Binding-competent states for L-arginine in E. coli arginine repressor apoprotein. J. Mol. Model 20 (7), 2330. doi:10.1007/s00894-014-2330-5
Rodgers, T. L., Townsend, P. D., Burnell, D., Jones, M. L., Richards, S. A., McLeish, T. C., et al. (2013). Modulation of global low-frequency motions underlies allosteric regulation: demonstration in CRP/FNR family transcription factors. PLoS Biol. 11 (9), e1001651. doi:10.1371/journal.pbio.1001651
Stirling, C. J., Stewart, G., and Sherratt, D. J. (1988). Multicopy plasmid stability in Escherichia coli requires host-encoded functions that lead to plasmid site-specific recombination. Mol. Gen. Genet. 214 (1), 80–84. doi:10.1007/BF00340183
Strawn, R., Melichercik, M., Green, M., Stockner, T., Carey, J., and Ettrich, R. (2010). Symmetric allosteric mechanism of hexameric Escherichia coli arginine repressor exploits competition between L-arginine ligands and resident arginine residues. PLoS Comput. Biol. 6 (6), e1000801. doi:10.1371/journal.pcbi.1000801
Sunnerhagen, M., Nilges, M., Otting, G., and Carey, J. (1997). Solution structure of the DNA-binding domain and model for the complex of multifunctiona hexameric arginine represser with DNA. Nat. Struct. Biol. 4 (10), 819–826. doi:10.1038/nsb1097-819
Szwajkajzer, D., Dai, L., Fukayama, J. W., Abramczyk, B., Fairman, R., and Carey, J. (2001). Quantitative analysis of DNA binding by the Escherichia coli arginine repressor11Edited by D. E. Draper. J. Mol. Biol. 312 (5), 949–962. doi:10.1006/jmbi.2001.4941
Tian, G., Lim, D., Carey, J., and Maas, W. K. (1992). Binding of the arginine repressor of Escherichia coli K12 to its operator sites. J. Mol. Biol. 226 (2), 387–397. doi:10.1016/0022-2836(92)90954-i
Van Duyne, G. D., Ghosh, G., Maas, W. K., and Sigler, P. B. (1996). Structure of the oligomerization andL-arginine binding domain of the arginine repressor of. J. Mol. Biol. 256 (2), 377–391. doi:10.1006/jmbi.1996.0093
Wielgus-Kutrowska, B., Grycuk, T., and Bzowska, A. (2018). Part-of-the-sites binding and reactivity in the homooligomeric enzymes - facts and artifacts. Archives Biochem. Biophysics 642, 31–45. doi:10.1016/j.abb.2018.01.011
Keywords: ligand-binding theory, affinity heterogeneity, equilibrium dissociation constant, isothermal titration calorimetry ITC, cyclic AMP receptor protein CRP
Citation: Strawn R, Murthy PS, Ettrich RH, Pelczer I and Carey J (2024) Symmetry of a partially-ligated state maintained by dynamics in a negatively cooperative system. Front. Biophys. 2:1359979. doi: 10.3389/frbis.2024.1359979
Received: 22 December 2023; Accepted: 07 February 2024;
Published: 23 February 2024.
Edited by:
Rita Grandori, University of Milano-Bicocca, ItalyReviewed by:
Hiroshi Fujisaki, Nippon Medical School, JapanCopyright © 2024 Strawn, Murthy, Ettrich, Pelczer and Carey. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jannette Carey, amNhcmV5QHByaW5jZXRvbi5lZHU=
†Present address: Rebecca Strawn, Johnson and Johnson Innovative Medicine, Malvern, PA, United States
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.