
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Biomater. Sci., 20 December 2023
Sec. Bio-interactions and Bio-compatibility
Volume 2 - 2023 | https://doi.org/10.3389/fbiom.2023.1272913
 Nicholas Cornell
Nicholas Cornell Donald Griffin*
Donald Griffin*Glycosaminoglycans (GAGs) are linear polysaccharides commonly used to impart bioactivity into synthetic hydrogels through their broad electrostatic-based protein-binding capabilities. In vivo, GAGs are immobilized through a single linkage point and function as semi-rigid ligands that are capable of limited conformation to proteins to enable high affinity interactions, concentration gradients, and co-signaling. Most GAG immobilization strategies in biomaterials target modification of the GAG repeat unit and produce multiple linkage points which effectively turns the GAG into a multifunctional crosslinker. In this study, we utilize real-time monitoring of binding kinetics to investigate the effects of GAG immobilization approach on GAG-protein binding. We show that GAGs immobilized through a single linkage point (GAGSingle) possess enhanced protein binding compared with GAGs immobilized at several points (GAG¬Multi¬). This effect is demonstrated for multiple GAG and protein types, indicating a broad applicability and importance to GAG use in biomaterials.
Glycosaminoglycans (GAGs) have emerged as a versatile tool for adding bioactivity to biomaterials, including hydrogels (Menezes et al., 2022). GAGs exist naturally as linear polysaccharides that influence a diverse array of cellular processes (e.g., proliferation, differentiation, adhesion, immunomodulation) through protein-binding interactions, creating signaling gradients and protecting growth factors from protease degradation (Liang and Kiick, 2014; Sakiyama-Elbert, 2014; Gockel et al., 2022). These interactions are complex and are driven, in part, by electrostatics via alignment of negatively charged chemical moieties (e.g., sulfate groups, carboxylic acids) along the GAG backbone with positively charged amino acid residues of proteins.
When incorporated into hydrogels, the most common approach for GAG incorporation is to covalently immobilize the GAGs within the material network. This strategy provides retention of the GAGs for prolonged bioactivity compared to methods such as charge-based adsorption (Liang and Kiick, 2014). Tethering of several different GAG types into a material network, such as heparin or chondroitin sulfate, has been shown to benefit cell functionality and sustain release of soluble growth factors (Liang and Kiick, 2014; Sakiyama-Elbert, 2014). Most covalent approaches target a modification of the GAG repeat unit that produces multiple attachment points along the GAG backbone and effectively turns the GAG into a multifunctional crosslinker. Importantly, the immobilization of GAGs through multiple attachments differs from their in vivo presentation, wherein they are immobilized via a single end-point attachment.
Sulfated GAGs (e.g., heparan sulfate, chondroitin sulfate, dermatan sulfate) are natively presented as linear chains that are attached to a serine residue of a core protein in the ECM or on the cell surface (e.g., perlecan or aggrecan) via a single tetrasaccharide linker region (Menezes et al., 2022; Gandhi and Mancera, 2008; Huang et al., 2021). Single attachment, combined with some limited structural flexibility (i.e., lacking the restrictive intramolecular bonds found in proteins) is important for their broad protein binding capacity (Figure 1A). (Sasisekharan et al., 2006; Gandhi and Mancera, 2008; Xu and Esko, 2014) Given that the high negative charge density present in GAG structure already limits its flexibility and ability to align its charge with potential protein partners, further confining GAG conformational freedom via multiple attachment points when incorporating into biomaterials may be limiting its ability to form high affinity interactions and to function as bioactive co-signaling molecules (Sasisekharan et al., 2006). Pérez, et al., produced an excellent database, GAG-DB, that serves to model these crucial interactions in silica. Pérez et al. (2020) In this paper, we perform a series of biomaterial-relevant experiments to test the hypothesis that incorporation of GAGs by methods that use multiple covalent attachment points increases steric hinderance and, subsequently, reduces the conformational flexibility of the immobilized chains to interact closely with proteins (i.e., decreases GAG-protein affinity). To validate this hypothesis, we set out to use quantitative Bio-Layer Interferometry measurements to demonstrate that reducing immobilization of GAGs to a single covalent attachment point increases relative GAG-protein association. Previous work has shown that immobilization strategy can be refined to enhance the bioactivity of immobilized GAGs (e.g., GAG clustering (Novoa-Carballal et al., 2018), coupling chemistry (Thakar et al., 2014), choice of modified functional moiety (Osmond et al., 2002)), and our specific hypothesis is supported by studies in vascular biomaterial development focused on heparin, a GAG that is normally presented as a free GAG chain physiologically (i.e., not tethered as a proteoglycan) outside of mast cell presentation (Mulloy et al., 2017), and its binding with anti-thrombin (Gore et al., 2014; Bao et al., 2015; Qiu et al., 2017). These papers compared multi-point attached (MPA) heparin with endpoint attached (EPA) heparin created through a digestion-based generation of terminal aldehyde groups on the end of heparin chains. These functionally focused studies showed materials coated with EPA-heparin had prolonged bioactivity, including enhanced anti-thrombin specific binding, total protein adsorption, and hemocompatibility compared to MPA-heparin coatings. The depth of these studies was somewhat limited by their specific focus (e.g., only heparin and its role as an anti-coagulant) or lack of highly controlled conditions (e.g., differing immobilization chemistries and surface concentrations for MPA and EPA heparin). An additional noteworthy study showed evidence of enhanced bioactivity for EPA immobilization (termed end-on) of chondroitin sulfate and hyaluronan through testing of GAG degradability and aggrecan binding (Altgärde et al., 2013). This work further supports the idea that limiting GAGs conformational flexibility impacts bioactivity through affecting the affinity of GAG-protein interactions.
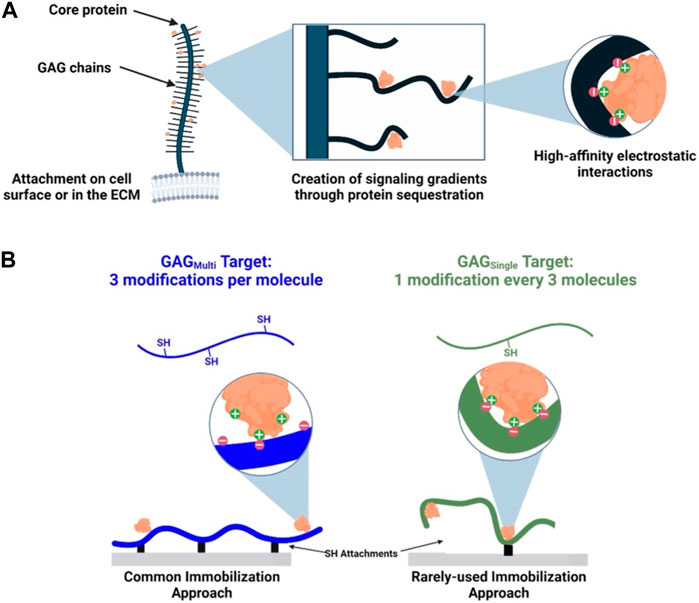
FIGURE 1. Native and synthetic immobilization of GAGs. (A) GAG chains are immobilized at a single point in the ECM or on the cell surface. Some limited conformational flexibility and negative charges along the GAG backbones enable high-affinity electrostatic interactions with proteins. (B) GAGs immobilized via multiple (GAGMulti) or a single attachment (GAGSingle) present respectively as conformationally more restricted or less restricted ligands for protein binding.
In this study, we are investigating beyond a single GAG-protein interaction to make a more general claim on the effects of GAG immobilization approach on subsequent protein binding. Our approach utilizes a single immobilization chemistry to create presentations of GAGs as both multi-point tethered (GAGMulti) and single-point tethered (GAGSingle) ligands (Figure 1B). To our knowledge, our study is the first to investigate the impact of immobilization extent by controlling for immobilization chemistry and then directly probing the interactions of a range of GAGs and proteins (note: the intentional use of a single chemical modification approach for multi- and single-point attachment limited our ability to exactly mimic the most biologically-relevant terminal attachment to a core protein, which we expect would provide the greatest level of conformational freedom). By using multiple GAGs and proteins, we intend to support a more general argument that single-point attachment of GAGs provides enhanced protein interactions relative to the commonly used multiple-point attachment.
We chose to use heparin as our initial GAG for this study given its well-characterized protein interactions and wide-spread use in biomaterials (Schlessinger et al., 2000; Zhang et al., 2001; Liang and Kiick, 2014). Heparin has higher affinity electrostatic interactions with proteins than other GAGs due primarily to its status as the most negatively charged GAG in the body.
To create distinct heparin presentations, we utilized a modified version of a heparin thiolation we have previously described (Pruett et al., 2021). Briefly, heparin was dissolved in ultra-pure water and the carboxylic acids of the heparin were activated via 4-(4,6-Dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride (DMTMM) addition. To control the extent of functionalization, and subsequently the number of immobilization points, the heparin was then reacted with different molar equivalents of 3-(2-pyridyldithiol) propionyl hydrazide (PDPH) compared to overall heparin repeat units (RUs). These reactions were targeted to yield at least 3 modifications per heparin chain (HepMulti, multiple immobilization points) or 1 modification every 3 heparin chains (HepSingle, single immobilization point). Following PDPH attachment, the internal disulfide bond of the PDPH was reduced via TCEP and yielded a thiol group which could participate in Michael-type addition.
Heparin modifications were characterized at multiple points of synthesis. Following PDPH addition, modification was confirmed via 1H-NMR showing addition of the PDPH ring structure to the heparin chains (Figure 2A). Upon reduction of the PDPH internal disulfide bond, number of modifications per chain was determined via a deprotection absorbance assay that quantified the concentration of the leaving PDPH ring structure (Figure 2B). Thiol content of the final product was measured via an Ellman’s assay (Figure 2C). These results confirmed repeatable and tunable thiolation of heparin to enable our desired immobilization presentations.
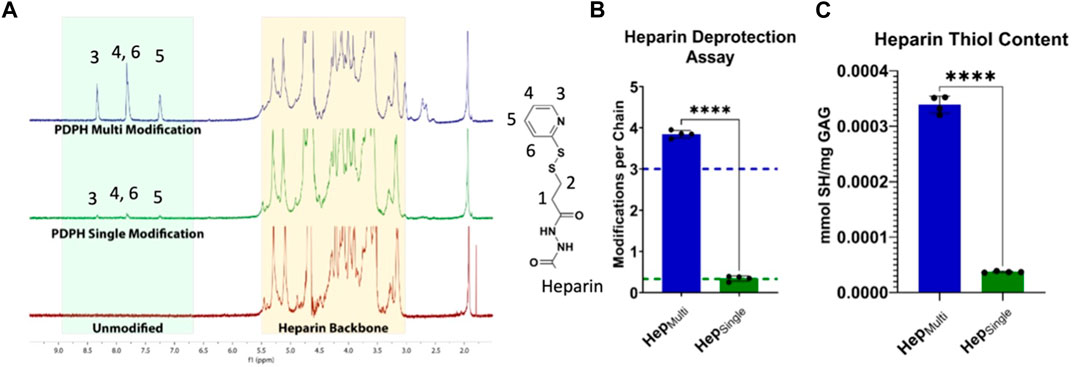
FIGURE 2. Characterization of heparin modifications. (A) 1H-NMR spectroscopy showed differential levels of PDPH modification for HepMulti and HepSingle. (B) PDPH deprotection absorbance assay confirmed desired targets (shown as dotted lines) of more than 3 modifications per HepMulti chain and approximately 0.333 modifications per HepSingle chain. (C) Ellman’s assay quantification of thiol groups following deprotection confirmed a ∼10X increase in active thiol groups for HepMulti compared to HepSingle. Statistics: Unpaired t-tests, N = 4, ****p < 0.0001.
To investigate the impact of heparin immobilization approach on protein interaction we utilized Bio-Layer Interferometry (BLI), which is a label-free technology to monitor real-time interaction between an immobilized biomolecule and a soluble binding partner (Shah and Duncan, 2014). BLI probes utilize optical interference to quantify the spectral shift between an internal reference layer and the surface of the probe (the biolayer). As more biomolecules bind to the surface, this spectral shift grows, enabling quantification of binding kinetics. HepMulti and HepSingle were biotinylated with a maleimide-functionalized biotin through Michael-type addition. This enabled subsequent immobilization of the heparin forms onto streptavidin coated BLI biosensors (Figure 3A). Probes were equilibrated in solution before introducing different heparin forms. Heparin concentrations were optimized to achieve sufficient binding without saturating the BLI probes (Supplementary Figure S3). Following heparin loading, probes were equilibrated again in buffer to remove unbound heparin. We chose basic fibroblast growth factor (FGF-2) as an initial binding partner for heparin given its published high-affinity and well-characterized interaction with heparin during cellular uptake (Schlessinger et al., 2000; Koledova et al., 2019). FGF-2 was incubated with the heparin-labeled BLI probes at several protein concentrations before transferring the probes back to buffer for dissociation. To decouple the effects of chemical modification from immobilized presentation, we created a third form of heparin (termed HepCap) in which we capped >90% of thiol groups on HepMulti with 6-maleimidohexanoic acid prior to biotinylation (Figure 3B). As a result, HepCap was chemically modified by PDPH to the same extent as HepMulti but was physically immobilized at a single point similar to HepSingle. BLI experiments were performed to characterize FGF-2 binding with HepMulti, HepSingle, and HepCap. Resulting binding curves were fit globally using non-linear regression to determine binding kinetics for association and dissociation (Supplementary Table S4). (Shah and Duncan, 2014; Chen et al., 2022). The equilibrium dissociation constant (KD) of FGF-2 binding with HepMulti (∼1.6 nM) matched well with previous literature values (Zhang et al., 2019) while that of HepSingle was found to be significantly lower (∼0.97 nM), indicating higher affinity binding between the HepSingle and FGF-2 (Figure 3C). Crucially, the KD of HepCap (∼0.83 nM) was not only significantly lower than HepMulti but statistically equivalent to that of HepSingle. This finding confirmed that preserving conformational freedom enhances heparin-protein affinity. Therefore, the differences observed in FGF-2 affinity between HepMulti and HepSingle were due to the immobilization approach.
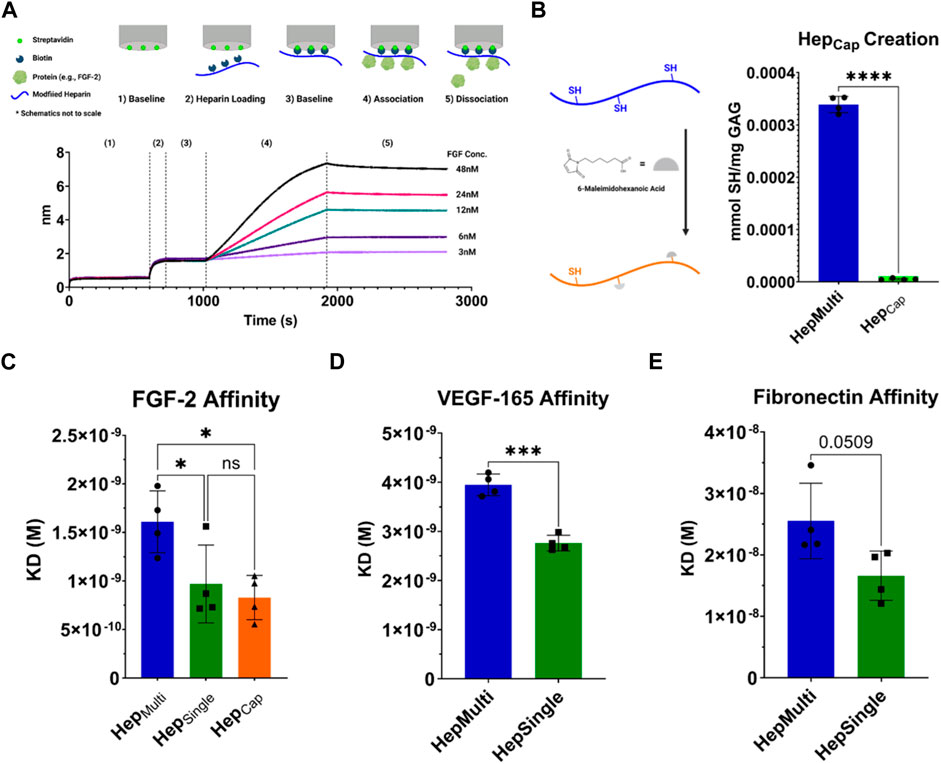
FIGURE 3. Bio-Layer Interferometry (BLI) for heparin-protein binding affinity. (A) Schematic of BLI experiment and representative trace. Experimental data shown is from a HepMulti/FGF-2 experiment. (B) Creation of HepCap to decouple modification from immobilization. HepMulti is modified with 6-maleimidohexanoic acid to cap >90% of thiol groups. Reduction of thiols was confirmed with Ellman’s assay. (C) Equilibrium dissociation constants (KD) for FGF-2 binding with HepMulti, HepSingle, and HepCap measured via BLI. (D) KD for VEGF-165 binding to HepMulti and HepSingle measured via BLI. (E) KD for full-length fibronectin binding to HepMulti and HepSingle measured via BLI, p = 0.0509. Statistics: ANOVA for FGF-2 results, multiple comparisons post-hoc tests (Tukey HSD), N = 4, *p < 0.05. Unpaired t-test for VEGF-165 and fibronectin, N = 4, ***p < 0.001.
To validate our assumption that the impact of immobilization approach extended beyond heparin binding to FGF-2, we first investigated the effects of HepMulti and HepSingle with other proteins known to bind heparin. VEGF-165 is a crucial angiogenic factor and demonstrated similar results to FGF-2, with HepSingle having a significantly lower KD (∼2.76 nM) than HepMulti (∼3.95 nM) (Figure 3D). A similar effect was observed with the full-length ECM protein fibronectin (FN); however, the difference was not statistically significant (HepSingle: ∼16.6nM, HepMulti: ∼25.5nM, p = 0.0509) (Figure 3E). We hypothesize that heterogeneity in chain length, sulfation pattern, and monosaccharide units (iduronic acid versus glucuronic acid) of our chosen unfractionated heparin source likely influenced the signal to noise ratio of these affinity results and that future studies would benefit from using highly fractionated (less heterogeneous) sources. Despite this difficulty, these results confirm that the benefits of maintaining heparin’s limited conformational flexibility when immobilizing applies beyond the canonical heparin-FGF-2 relationship and is seen in other heparin binding partners.
Broadening our investigation of GAG immobilization beyond heparin, we hypothesized that the enhanced bioactivity of singular attachment for HepSingle could be replicated with other sulfated GAGs, such as chondroitin sulfate (CS) that also interacts with proteins through electrostatic binding. Notably, chondroitin sulfate is composed of different monosaccharides than heparin and, thus, presents a different sulfation pattern. We utilized the same thiolation chemical approach (i.e., thiolation via DMTMM mediated amide coupling of PDPH to free carboxylic acid) and characterization used for our heparin studies to create two immobilized forms of chondroitin sulfate A, termed CS-AMulti and CS-ASingle (Figure 4A). We again performed BLI experiments to generate the KD of FGF-2 with our different presentations of CS-A. CS-ASingle displayed a significantly lower KD (∼2.51 nM) than CS-AMulti (∼4.11 nM), demonstrating that the enhanced bioactivity seen through GAGSingle immobilization is replicable across different sulfated GAGs (Figure 4B). The KD of both CS-A forms were larger than that of HepMulti and HepSingle, which we expected given that heparin is more negatively charged and, thus has greater electrostatic binding potential with FGF-2.
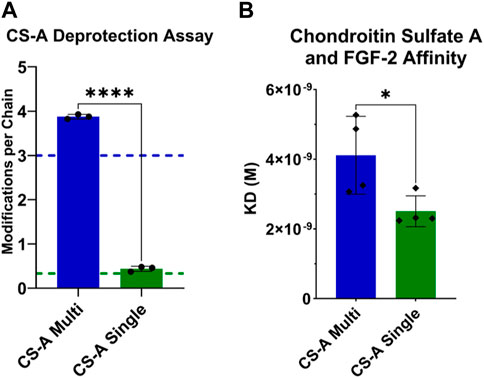
FIGURE 4. Characterizing the modification and FGF-2 binding kinetics of CS-AMulti and CS-ASingle. (A) PDPH deprotection absorbance assay confirmed desired targets (shown as dotted lines) of at least 3 modifications per CS-AMulti chain and approximately 0.333 modifications per CS-ASingle chain. (B) KD for FGF-2 binding to CS-AMulti and CS-ASingle measured via BLI. Statistics: Unpaired t-test, N = 4, *p < 0.05.
A separate, but important, observation when we performed our CS studies was that when performing the same chemical modifications and FGF-2 BLI experiments with a commercial CS mixture (i.e., commercial supplier defined the CS as a mixture of CS-A, CS-C, CS-E, and unsulfated chondroitin), we did not observe enhanced protein affinity for CSSingle compared to CSMulti (Supplementary Figure S8). This CS mixture contained disaccharide RUs with zero, one, or two sulfate groups according to the manufacturer (Sigma-Aldrich). We hypothesize that the heterogenous sulfation pattern may bias chemical modification toward specific CS isomers, which would make the effects of immobilization and sulfation difficult to separate (i.e., CSmulti presented all forms of CS and CSsingle did not). This idea is supported by the more consistent effects of CS-A (uniformly single sulfated RUs) (Djerbal et al., 2017) and heparin (triple sulfated in >70% of RUs) (Meneghetti et al., 2015) however, further studies are needed to confirm this hypothesis.
GAGs are an amazing resource for adding bioactivity into a biomaterial for both in vitro and in vivo applications. GAG natural presentation as semi-rigid polymers with single point attachment provides them with a limited amount of conformational flexibility, which is greatly affected by their structural heterogeneity (i.e., disaccharide composition, sulfation pattern, chain length) and local microenvironment (e.g., pH, salts, etc.) (Samantray et al., 2021). For example, chain flexibility varies greatly between heparin and heparin sulfate due to the increased prevalence of GlcA-GlcNAc sequences. In this manuscript, our intent is not to argue in favor of one GAG or one specific GAG immobilization chemistry. Rather, we argue more broadly that further limitation of molecular freedom by increased covalent tethering (i.e., greater than one) can negatively impact GAG affinity for proteins of interest. We believe our results are a valuable addition to the field of biomaterials, which will allow researchers to intentionally select from the available toolbox of immobilization strategies when designing bioactive material platforms through in increased understanding of the impact of tethering extent. Specifically, we present a highly controlled and tunable approach to control GAG immobilization and presentation to demonstrate that GAGs immobilized via a single tether can enhance protein affinity across different proteins and GAG types. The broad applicability of this knowledge is promising for GAG-based bioactive addition, which is often incorporated with an intent to use GAG physiological function to match the native microenvironment of the target tissue (e.g., CS in cartilage engineering) (Kilmer et al., 2022). We believe the application of this knowledge (i.e., intentional immobilization) within biomaterial approaches is simple and will require minimal changes to established immobilization methods. However, we feel it is important to note that while the trend and impact of immobilization extent (i.e., single versus multi-point attachment) will prove consistent across GAGs, the chosen immobilization chemistry will have a significant impact on absolute affinity values. In future studies, we plan to investigate the functional effects of these GAG immobilization forms on both the cell (in vitro) and tissue (in vivo) level response.
Heparin (MW = 15,000 Da, porcine intestinal mucosa) was purchased from Millipore Sigma. Chondroitin Sulfate (CS, bovine cartilage) and CS-A (bovine trachea) were purchased from Sigma-Aldrich. GAG modification and characterization to create GAGMulti and GAGSingle were based on a protocol previously reported by the author’s group (Pruett et al., 2021). Briefly, GAGs were dissolved in ultrapure water and modified with different amounts of 3-(2-pyridyldithiol) propionyl hydrazide (PDPH, CovaChem) in the presence of 4-(4,6-Dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride (DMTMM, Oakwood Chemical). PDPH was added at a 1:4 and 1:40 molar ratio of heparin repeat units (RU, estimated MW of heparin RU = 619.49 Da (Pomin, 2014)) to create HepMulti and HepSingle respectively. To create CSMulti, CS-AMulti, CSSingle, and CS-ASingle, PDPH was added at a 1:5 molar ratio of RU for the GAGMulti and 1:40 molar ratio of RU for the GAGSingle (estimate MW of CS RU = 513.86 Da (Pomin, 2014), estimated MW of CS-A RU = 459.00Da (Djerbal et al., 2017)). AlexaFluor hydrazide 488 or 555 (Invitrogen) were added to all reactions to fluorescently label heparin. Reactions were dialyzed according to the previous protocol and the product was lyophilized. PDPH-modified GAGs were deprotected to expose thiol groups by mixing with 25 mM tris(2-carboxyethyl)phosphine hydrochloride (TCEP, Alfa Aesar) and modification level was quantified via absorbance at 343 nm using a ThermoFisher Nanodrop 2000c spectrophotometer. Deprotected product was dialyzed, lyophilized, and stored at −20°C until use. Thiol content was measured via Ellman’s assay according to manufacturer protocol (ThermoFisher). Differential modification was further confirmed on the modified GAGS by 1H-NMR prior to deprotection through looking at relative modification signal (e.g., comparing multi-to single-modification via ratio of PDPH integration, δ = ∼8.35–7.25, to the GAG peaks integration, δ = 5.5–3.1). This data was acquired by dissolving GAG forms in deuterium oxide and acquiring 1H-NMR spectra on a Varian Inova 500 NMR spectrometer located in the UVA Biomolecular Magnetic Resonance Facility.
Heparin modification—1H NMR (500 MHz, D2O): δ = 8.3 ppm (s, 1H), 7.85 ppm (s, 2H), 7.25 ppm (s, 1H), 5.5–3.1 (heparin, m).
Chondroitin sulfate modification—1H NMR (500 MHz, D2O): δ = 8.3 ppm (s, 1H), 7.85 ppm (s, 2H), 7.25 ppm (s, 1H), 4.75–3.15 (chondroitin sulfate, m).
To create HepMulti-Capped (HepCap), HepMulti and 6-Maleimidohexanoic acid (Sigma-Aldrich) were dissolved separately in pH 7.4 1X PBS at 10 mg/mL (4.35 mM immobilized thiol groups) and 0.735 mg/mL (3.48 mM maleimide groups) respectively to yield a thiol to maleimide molar ratio of 1.0:0.8. Solutions were mixed 1:1 and incubated for 15min at 37°C under agitation. Reaction product was then filtered to remove unbound 6-maleimidohexanoic acid using ZEBA desalting columns (7 kDa MWCO, Thermo) and the manufacturer’s protocol. Thiol content was measured via Ellman’s assay.
GAGs were dissolved at 1 mg/mL in pH 7.4 1X PBS. Biotin-maleimide (Bio-MAL, TCI) was diluted in pH 7.4 1X PBS to molar concentrations matching the thiol content of each GAG form based on previous PDPH deprotection absorbance results (HepMulti and HepCap–0.435mM, HepSingle–0.0284mM, CSMulti - 0.2257mM, CSSingle–0.0296mM, CS-AMulti–0.2586mM, CS-ASingle–0.0292 mM). GAG and Bio-MAL solutions were mixed 1:1 and reacted at room temperature for 15min under agitation. Biotinylated GAGs were isolated from unbound Bio-MAL by filtering through ZEBA desalting columns using the manufacturer’s protocol. HepCap was isolated by dialyzing into pH 7.4 1X PBS overnight using a Slide-A-Lyzer® Dialysis Cassette (2 kDa MWCO, Thermo Scientific). Biotinylated GAG forms were stored at −20°C until use.
BLI experiments were conducted on an Octet RED96 system (ForteBio) located in the UVA Biomolecular Analysis Facility. An assay buffer consisting of 10 mM HEPES pH 7.4, 100 mM NaCl, 0.001% BSA, and 0.01% Tween 20 was used for all experiments. Kinetics experiments followed the same format with small differences in protein concentration and timing based on the protein being tested (Table 1). All experiments were conducted at 30°C at an agitation speed of 1000rpm as recommended in the Octet user manual. Streptavidin Biosensors (Sartorius) were initially equilibrated in assay buffer then loaded with biotinylated GAG at optimized concentrations for each GAG form (HepSingle, CSSingle, and CS-ASingle–10ug/mL, HepMulti, HepCap, CSMulti and CS-AMulti–1 ug/mL). Following loading, probes were equilibrated in assay buffer, associated with various protein conditions (Table 1), and then again transferred into assay buffer for protein dissociation. VEGF165 equilbration steps were abbreviated due to the low solution stability of the protein. Each experiment used a reference probe exposed solely to assay buffer throughout the experiment to account for instrument drift. Heparin-FGF-2 experiments were run in triplicate with probes being regenerated to heparin-only with pH 2.0 10 mM glycine dissolved in assay buffer (technical repeats). Each experimental setup for a GAG-protein pair was repeated 4 times with new sets of probes (experimental repeats). FGF-2 was purchased from R&D Systems, human fibronectin (FN) and VEGF165 were purchased from Gibco.
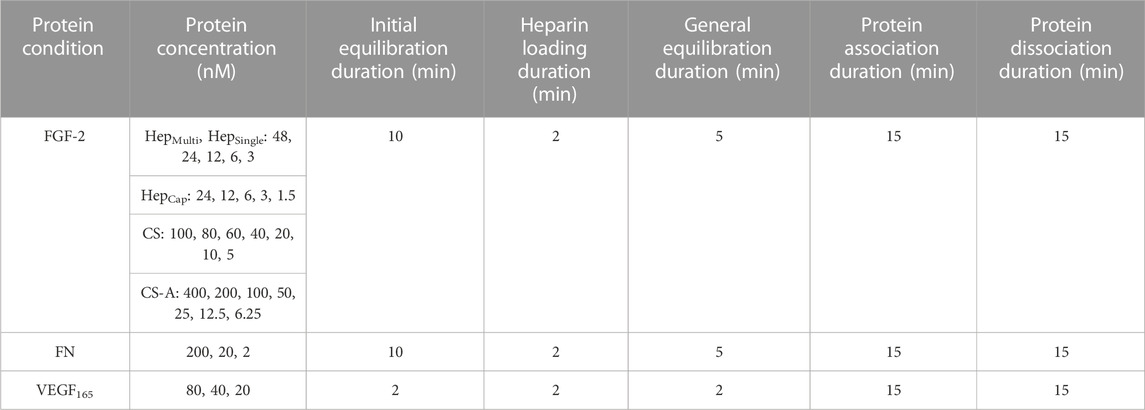
TABLE 1. Experimental parameters for BLI studies.
Kinetic binding curves from BLI experiments were analyzed in GraphPad Prism software (version 9.3.0). The protein association and dissociation steps were isolated from raw traces of spectral shift and reference probes were subtracted to eliminate assay drift. Curves were fit using an “association then dissociation” non-linear regression model with global fitting for Kon and Koff. For all experiments, Kon was constrained to be positive and “ambiguous” fits were included. For heparin-FGF-2 experiments, KDs were averaged first by technical repeats then by experimental repeats. All KDs represent the average and standard deviation of N = 4 experimental repeats.
The raw data supporting the conclusion of this article will be made available by the authors, without undue reservation.
NC: Conceptualization, Data curation, Formal Analysis, Methodology, Visualization, Writing–original draft. DG: Conceptualization, Funding acquisition, Resources, Supervision, Writing–review and editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The authors would like to thank the University of Virginia Biomedical Engineering Department for providing lab start-up funds and the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK; R01 10297936) for their financial support of the described work.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fbiom.2023.1272913/full#supplementary-material
Altgärde, N., Nilebäck, E., de Battice, L., Pashkuleva, I., Reis, R. L., Becher, J., et al. (2013). Probing the biofunctionality of biotinylated hyaluronan and chondroitin sulfate by hyaluronidase degradation and aggrecan interaction. Acta Biomater. 9 (9), 8158–8166. doi:10.1016/j.actbio.2013.05.031
Bao, J., Wu, Q., Sun, J., Zhou, Y., Wang, Y., Jiang, X., et al. (2015). Hemocompatibility improvement of perfusion-decellularized clinical-scale liver scaffold through heparin immobilization. Sci. Rep. 5 (1), 10756. doi:10.1038/srep10756
Chen, Q., Li, F., Wang, H., Bu, C., Shi, F., Jin, L., et al. (2022). Evaluating the immunogenicity of heparin and heparin derivatives by measuring their binding to platelet factor 4 using biolayer interferometry. Front. Mol. Biosci. 9, 966754. doi:10.3389/fmolb.2022.966754
Djerbal, L., Lortat-Jacob, H., and Kwok, J. (2017). Chondroitin sulfates and their binding molecules in the central nervous system. Glycoconj. J. 34 (3), 363–376. doi:10.1007/s10719-017-9761-z
Gandhi, N. S., and Mancera, R. L. (2008). The structure of glycosaminoglycans and their interactions with proteins. Chem. Biol. Drug Des. 72 (6), 455–482. doi:10.1111/j.1747-0285.2008.00741.x
Gockel, L. M., Nekipelov, K., Ferro, V., Bendas, G., and Schlesinger, M. (2022). Tumour cell-activated platelets modulate the immunological activity of CD4+, CD8+, and NK cells, which is efficiently antagonized by heparin. Cancer Immunol. Immunother. 71 (10), 2523–2533. doi:10.1007/s00262-022-03186-5
Gore, S., Andersson, J., Biran, R., Underwood, C., and Riesenfeld, J. (2014). Heparin surfaces: impact of immobilization chemistry on hemocompatibility and protein adsorption. J. Biomed. Mat. Res. B Appl. Biomater. 102 (8), 1817–1824. doi:10.1002/jbm.b.33154
Huang, Y.-F., Mizumoto, S., and Fujita, M. (2021). Novel insight into glycosaminoglycan biosynthesis based on gene expression profiles. Front. Cell Dev. Biol. 9, 709018. doi:10.3389/fcell.2021.709018
Kilmer, C. E., Walimbe, T., Panitch, A., and Liu, J. C. (2022). Incorporation of a collagen-binding chondroitin sulfate molecule to a collagen type I and II blend hydrogel for cartilage tissue engineering. ACS Biomater. Sci. Eng. 8 (3), 1247–1257. doi:10.1021/acsbiomaterials.1c01248
Koledova, Z., Sumbal, J., Rabata, A., de La Bourdonnaye, G., Chaloupkova, R., Hrdlickova, B., et al. (2019). Fibroblast growth factor 2 protein stability provides decreased dependence on heparin for induction of FGFR signaling and alters ERK signaling dynamics. Front. Cell Dev. Biol. 7, 331. doi:10.3389/fcell.2019.00331
Liang, Y., and Kiick, K. L. (2014). Heparin-functionalized polymeric biomaterials in tissue engineering and drug delivery applications. Acta Biomater. 10 (4), 1588–1600. doi:10.1016/j.actbio.2013.07.031
Meneghetti, M. C. Z., Hughes, A. J., Rudd, T. R., Nader, H. B., Powell, A. K., Yates, E. A., et al. (2015). Heparan sulfate and heparin interactions with proteins. J. R. Soc. Interface 12 (110), 20150589. doi:10.1098/rsif.2015.0589
Menezes, R., Vincent, R., Osorno, L., Hu, P., and Arinzeh, T. L. (2022). Biomaterials and tissue engineering approaches using glycosaminoglycans for tissue repair: lessons learned from the native extracellular matrix. Acta Biomater. 163, 210–227. doi:10.1016/j.actbio.2022.09.064
Mulloy, B., Lever, R., and Page, C. P. (2017). Mast cell glycosaminoglycans. Glycoconj. J. 34 (3), 351–361. doi:10.1007/s10719-016-9749-0
Novoa-Carballal, R., Carretero, A., Pacheco, R., Reis, R. L., and Pashkuleva, I. (2018). Star-like glycosaminoglycans with superior bioactivity assemble with proteins into microfibers. Chem. – Eur. J. 24 (54), 14341–14345. doi:10.1002/chem.201802243
Osmond, R. I. W., Kett, W. C., Skett, S. E., and Coombe, D. R. (2002). Protein-heparin interactions measured by BIAcore 2000 are affected by the method of heparin immobilization. Anal. Biochem. 310 (2), 199–207. doi:10.1016/s0003-2697(02)00396-2
Pérez, S., Bonnardel, F., Lisacek, F., Imberty, A., Ricard Blum, S., and Makshakova, O. (2020). GAG-DB, the new interface of the three-dimensional landscape of glycosaminoglycans. Biomolecules 10 (12), 1660. doi:10.3390/biom10121660
Pomin, V. H. (2014). NMR chemical shifts in structural biology of glycosaminoglycans. Anal. Chem. 86 (1), 65–94. doi:10.1021/ac401791h
Pruett, L. J., Jenkins, C. H., Singh, N. S., Catallo, K. J., and Griffin, D. R. (2021). Heparin microislands in microporous annealed particle scaffolds for accelerated diabetic wound healing. Adv. Funct. Mat. 31, 2104337. doi:10.1002/adfm.202104337
Qiu, X., Lee, B. L.-P., Ning, X., Murthy, N., Dong, N., and Li, S. (2017). End-point immobilization of heparin on plasma-treated surface of electrospun polycarbonate-urethane vascular graft. Acta Biomater. 51, 138–147. doi:10.1016/j.actbio.2017.01.012
Sakiyama-Elbert, S. E. (2014). Incorporation of heparin into biomaterials. Acta Biomater. 10 (4), 1581–1587. doi:10.1016/j.actbio.2013.08.045
Samantray, S., Olubiyi, O. O., and Strodel, B. (2021). The influences of sulphation, salt type, and salt concentration on the structural heterogeneity of glycosaminoglycans. Int. J. Mol. Sci. 22 (21), 11529. doi:10.3390/ijms222111529
Sasisekharan, R., Raman, R., and Prabhakar, V. (2006). Glycomics approach to structure-function relationships of glycosaminoglycans. Annu. Rev. Biomed. Eng. 8 (1), 181–231. doi:10.1146/annurev.bioeng.8.061505.095745
Schlessinger, J., Plotnikov, A. N., Ibrahimi, O. A., Eliseenkova, A. V., Yeh, B. K., Yayon, A., et al. (2000). Crystal structure of a ternary FGF-FGFR-heparin complex reveals a dual role for heparin in FGFR binding and dimerization. Mol. Cell 6 (3), 743–750. doi:10.1016/S1097-2765(00)00073-3
Shah, N. B., and Duncan, T. M. (2014). Bio-layer interferometry for measuring kinetics of protein-protein interactions and allosteric ligand effects. J. Vis. Exp. 84, e51383. doi:10.3791/51383
Thakar, D., Migliorini, E., Coche-Guerente, L., Sadir, R., Lortat-Jacob, H., Boturyn, D., et al. (2014). A quartz crystal microbalance method to study the terminal functionalization of glycosaminoglycans. Chem. Commun. 50 (96), 15148–15151. doi:10.1039/C4CC06905F
Xu, D., and Esko, J. D. (2014). Demystifying heparan sulfate–protein interactions. Annu. Rev. Biochem. 83 (1), 129–157. doi:10.1146/annurev-biochem-060713-035314
Zhang, F., Zheng, L., Cheng, S., Peng, Y., Fu, L., Zhang, X., et al. (2019). Comparison of the interactions of different growth factors and glycosaminoglycans. Mol. Basel Switz. 24 (18), 3360. doi:10.3390/molecules24183360
Keywords: heparin, chondroitin sulfate, thiolation, Glycosaminoclycans, immobilization, methods
Citation: Cornell N and Griffin D (2023) Investigating the role between glycosaminoglycan immobilization approach and protein affinity. Front. Front. Biomater. Sci. 2:1272913. doi: 10.3389/fbiom.2023.1272913
Received: 04 August 2023; Accepted: 01 December 2023;
Published: 20 December 2023.
Edited by:
Maria Grazia Raucci, National Research Council (IPCB-CNR), ItalyReviewed by:
Deirdre R. Coombe, Curtin University, AustraliaCopyright © 2023 Cornell and Griffin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Donald Griffin, ZHJncmlmZmluQHZpcmdpbmlhLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.