 Tehrim Motiwala
Tehrim Motiwala Babalwa Nyide
Babalwa Nyide Thandeka Khoza
Thandeka Khoza
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Bioinform., 25 March 2025
Sec. Protein Bioinformatics
Volume 5 - 2025 | https://doi.org/10.3389/fbinf.2025.1498916
This article is part of the Research TopicComputational protein function prediction based on sequence and/or structural dataView all 5 articles
The Enterobacter cloacae complex (ECC) consists of six Enterobacter species (E. cloacae, hormaechei, kobei, ludwigii, nimipressuralis and asburiae) that have emerged as nosocomial pathogens of interest, with E. cloacae and Enterobacter hormachei being the most frequently isolated ECC species in human clinical specimens and intensive care unit (ICU) patients. Many nosocomial outbreaks of E. cloacae have been related to transmission through contaminated surgical equipment and operative cleaning solutions. As this pathogen evades the action of antibiotics, it is important to find alternative targets to limit the devastating effects of these pathogens. ClpV is a Clp ATPase which dissociates and recycles the contracted sheath of the bacterial type VI secretion system (T6SS), thereby regulating bacterial populations and facilitating environmental colonization. Seventy-one Enterobacter strains were mined for Clp ATPase proteins. All the investigated strains contained ClpA, ClpB, ClpX and ClpV while only 20% contained ClpK. All the investigated strains contained more than one ClpV protein, and the ClpV proteins showed significant variations. Three ClpV proteins from E. cloacae strain E3442 were then investigated to determine the structural difference between each protein. Homology modelling showed the proteins to be structurally similar to each other, however the physicochemical characteristics of the proteins vary. Additionally, physicochemical analysis and molecular dynamic simulations showed that the proteins were highly dynamic and not significantly different from each other. Further investigation of the proteins in silico and in vitro in the presence and absence of various ligands and proteins could be performed to determine whether the proteins all interact with their surroundings in the same manner. This would allow one to determine why multiple homologs of the same protein are expressed by pathogens.
Enterobacter spp. are a genus of Gram-negative bacteria belonging to the Enterobacteriaceae family. Microorganisms of this genus form part of the commensal microflora of the gastrointestinal (GI) tract of humans and animals (Davin-Regli et al., 2019; Mezzatesta et al., 2012). In more recent years, the Enterobacter spp. have become subject to increasing clinical scrutiny and are recognized as one of the bacterial pathogens described as ESKAPE pathogens which evade the effects of many different classes of antibiotics (Davin-Regli et al., 2019; De Oliveira D. M. et al., 2020; Pendleton et al., 2013; De Oliveira D. M. P. et al., 2020).
Furthermore, the Enterobacter spp. have been identified as the causative agents of post-surgical peritonitis, hospital-acquired sepsis, nosocomial urinary tract infections, neonatal pneumonia cases, bloodstream infections, endocarditis, septic arthritis, and intra-abdominal infection and therefore emerged as nosocomial pathogens of interest (De Oliveira D. M. et al., 2020; Mezzatesta et al., 2012; Ghazvini and Farsiani, 2020; Davin-Regli et al., 2019). From the six Enterobacter spp. of the Enterobacter cloacae complex (ECC), E. cloacae and Enterobacter hormachei are the most frequently isolated ECC species in human clinical specimens and intensive care unit (ICU) patients. E. cloacae outbreaks have been related to transmission through contaminated surgical equipment and operative cleaning solutions. This may be due to their ability to colonize medical devices and intravenous hospital equipment among other objects. More interestingly, there is evidence to suggest that healthcare workers can act as reservoirs for transmission of E. cloacae given that this bacterium is commonly contracted through the skin (Van Nierop et al., 1998; Mezzatesta et al., 2012).
E. cloacae are resilient, dynamic, and opportunistic pathogens with the ability to upregulate resistance determinants or evolve and adapt to overcome arising challenges in the environment. Their diverse arsenal of strategies includes (but is not limited to) the acquisition of resistance-conferring genes through horizontal gene transfer, and the ability to overproduce AmpC β-lactamases by mutation or depression of a chromosomal gene (Davin-Regli et al., 2019; Huang et al., 2012; Jacoby, 2009; Kaneko et al., 2005). Moreover, through the action of the Bush group 1 inducible natural cephalosporinase, E. cloacae are intrinsically resistance to various antimicrobial compounds including amoxicillin, amoxicillin-clavulanic acid, ampicillin, cefoxitin and cephalothin (Davin-Regli and Pagès, 2015; Mezzatesta et al., 2012). There is a growing body of evidence that links caseinolytic proteins (Clp) and pathogenicity of ESKAPE pathogens and this has unearthed exciting new avenues for anti-microbial drug discovery efforts targeting Clp proteins (D’andrea et al., 2022; Dhiman and Singh, 2018; Motiwala et al., 2022).
Clp proteins belong to the AAA+ (ATPases associated with diverse cellular activities) superfamily that facilitate protein unfolding, refolding, and tolerance to heat stress and oxidative stress (Bouchnak and Van Wijk, 2021; Sen et al., 2020; Voos, 2013). Clp proteins consist of the peptidase (ClpP) and the regulatory (Clp ATPase). Clp ATPases are characterized by their distinct nucleotide-stabilized ring-shaped multimeric structures (Neuwald et al., 1999; Bouchnak and Van Wijk, 2021). Clp ATPases are classified based on the number of nucleotide binding domains (NBDs) they possess; Class I members (Clps A, B, C, D, E, K, L and V) contain two NBDs, whereas Class II members (Clps M, N, X and Y) contain only one (Motiwala et al., 2022; Schlieker et al., 2005). The NBD is characterized by a conserved sequence of between 200–250 amino acids that make up the Walker A and Walker B motifs, which are responsible for ATP binding and hydrolysis (Bouchnak and Van Wijk, 2021; Schlieker et al., 2005; Seraphim and Houry, 2020).
To execute their functions, Clp ATPases can operate via two mechanisms that depend on the presence or absence of the tripeptide consensus sequence (IGF/L) on a helix-loop-helix motif of the NBD (Kim et al., 2001; Schlieker et al., 2005). Clp ATPases of the ClpA sub-family commonly possess this tripeptide, which is required for interaction with the Clp peptidase, ClpP (Kim et al., 2001; Schlieker et al., 2005). These ATPases recruit, refold, and reactivate misfolded proteins. Alternatively, they recruit misfolded proteins and then bind to inactive ClpP, a serine protease comprising two heptameric rings, thereby triggering a conformational change that realigns the residues of the catalytic triad and activates the protease site of ClpP (Brötz-Oesterhelt and Sass, 2014; Capestany et al., 2008). Proteins that are unsuccessfully reactivated are degraded through this proteolytic complex, which comprises of one ClpP tetradecamer sandwiched between two regulatory ATPase hexamers (Frees et al., 2007; Kirstein et al., 2009; Schlieker et al., 2005). By contrast, Clp ATPases of the ClpB/Hsp104 sub-family, which lack this tripeptide, can resolubilize and reactivate misfolded proteins alone or through cooperation with chaperones of the DnaK/DnaJ/GrpE system (Motiwala et al., 2022; Schlieker et al., 2005; Wickner et al., 1994; Zavilgelsky et al., 2004; Zolkiewski, 1999).
ClpV dissociates and recycles the contracted sheath of the bacterial type VI secretion system (T6SS) for new injection cycles (Förster et al., 2014; Douzi et al., 2016). T6SS is an offensive system used by bacteria to compete with environmental rivals and is responsible for the transport and delivery of toxins to target cells, thus regulating bacterial populations (Douzi et al., 2016; Smith-Roberge et al., 2018). Overall, members of the ClpB/Hsp104 and ClpV sub-families share approximately 30%–35% primary sequence identity (Schlieker et al., 2005). Like the ClpB/Hsp104 sub-family, homologs of the ClpV sub-family lack the IGF/L tripeptide sequence, suggesting that ClpV ATPases function independently of ClpP and the substrate proteolysis mechanism (Schlieker et al., 2005). The widespread distribution of Clp proteins across many microbial genera is a testament to their importance. Evidently, the role of maintaining protein homeostasis is crucial for cell survival, making a strong case for the targeting of Clp proteins for antimicrobial drug studies (Brötz-Oesterhelt and Sass, 2014; Frees et al., 2007). Here, we show the distribution of Clp proteins among strains of the Enterobacter genus, and the phylogenetic analysis of ClpV, the dominant Clp ATPase within the clinically significant E. cloacae species.
The National Center for Biotechnology Information (NCBI) Genome database (https://www.ncbi.nlm.nih.gov/home/genomes/) was browsed by organism to collect protein genomes of 71 Enterobacter strains (17 species, complete draft). The 71 Enterobacter strains which were investigated included; 27 Enterobacter hormaechei strains, 1 Enterobacter cancerogenus strain, 6 Enterobacter ludwigii strains, 6 Enterobacter roggenkampii strains, 2 Enterobacter bugandensis strains, 13 E. cloacae strains, 4 Enterobacter asburiae strains, 3 Enterobacter kobei strains, 1 Enterobacter chengduensis strain, 1 Enterobacter sichuanensis strain, 1 Enterobacter oligotrophicus strain, 1 E. N18-03635 strain, 1 E. JBIWA003 strain, 1 E. JBIWA005 strain, 1 E. JBIWA008 strain, 1 E. DNB-S2 strain and 1 E. SGAir0187 strain.
Clp ATPases were mined from different Enterobacter strains by first obtaining the protein file for each strain from NCBI. Each file was then converted from.gz to FASTA format using an online server (https://www.convertfiles.com/file_type_description/GZ_Archive_FIle.html). The FASTA files were then individually searched for the presence of Clp ATPases. The Clp ATPase sequences were separated from the main file and used for further analyses.
The Clp ATPase protein sequences were aligned using MAFFT v6.864 embedded on the T-rex web server (http://www.trex.uqam.ca/) (Katoh et al., 2005; Boc et al., 2012). The alignments were deduced by the T-rex server, following this the file for the best tree was downloaded, visualized and colored using the interactive Tree of Life (iTOL) v6 server (https://itol.embl.de/) (Letunic and Bork, 2021). The phylogenetic tree (Neighbor-joining, BLOSUM62) of ClpV in comparison with ClpVI from Pseudomonas aeruginosa (UniProt: Q9I742) and Vibrio cholera ClpV1 (UniProt: A0A395TZY1) was constructed using JalView v 2.11.2.2 (Waterhouse et al., 2009a).
Clustal omega (https://www.ebi.ac.uk/jdispatcher/msa/clustalo) was used in four ways during the investigation. Firstly, the percentage identity between the various Clp ATPase classes was analyzed. Secondly, the percentage identity between the ClpV mined from the E. cloacae strains, P. aeruginosa ClpV1 (UniProt: Q9I742), and V. cholera ClpV1 (UniProt: A0A395TZY1) was compared. Thirdly, the percentage identity between the E. cloacae E3442 ClpV homologs were analyzed. Lastly, the percentage identity between the three E3442 ClpV proteins and V. cholera ClpV1 (UniProt: A0A395TZY1) was investigated (Sievers et al., 2011). The domains were assigned to the ClpV proteins using the annotations provided through InterPro (Blum et al., 2020).
The three ClpV proteins mined from E. cloacae E3342 were modelled using the template 1qvrA through Swiss Model (https://swissmodel.expasy.org/) (Waterhouse et al., 2018). The structures were refined using the Protein REFinement via Molecular Dynamics (PREFMD) web server (https://feig.bch.msu.edu/prefmd) (Heo and Feig, 2017). Structural assessment of the refined structures was then performed using the Swiss-Model assessment tool (https://swissmodel.expasy.org/assess) and MolProbity (http://molprobity.biochem.duke.edu/) (Waterhouse et al., 2018; Williams et al., 2018). The refined structures were superimposed with the modelling template (PDB 1qvrA) using PyMol (Schrödinger, 2020).
The ProtParam tool on the Expasy server (https://web.expasy.org/protparam/) was used to compare the physicochemical properties of the three ClpV proteins using the respective protein sequences (Gasteiger et al., 2003). ClpV protein disorder was predicted using the IUPred3 server (https://iupred3.elte.hu/) (Erdős et al., 2021). ClpV sequences were analyzed using Mafft on the Jalview software and the InterPro server (http://www.ebi.ac.uk/interpro/) was used to investigate the protein families, domains and function of the ClpV sequences (Waterhouse et al., 2009b; Apweiler et al., 2001). Normal Mode Analysis (NMA) from the DynaMut server (https://biosig.lab.uq.edu.au/dynamut/analysis) was performed to compare the conformation of the ClpV proteins (Rodrigues et al., 2018). The ClpV sequences were annotated as per the InterPro server and Lee et al. (2003). The 3D structures of the ClpV proteins were then compared using the Dali server (http://ekhidna2.biocenter.helsinki.fi/dali/) (Holm, 2022). Each ClpV protein sequence was analyzed using the STRING v11.5 server (https://string-db.org/) for protein-protein association analysis (Szklarczyk et al., 2019). The minimum required interaction score was set to medium (0.400). DogSite3 was used through the ProteinsPlus server (https://proteins.plus/) to predict binding sites on the ClpV proteins (Graef et al., 2023; Schöning-Stierand et al., 2022; Schöning-Stierand et al., 2020; Fährrolfes et al., 2017).
Molecular dynamic (MD) simulations were performed using Maestro v12.2 through the implemented GPU-enabled Desmond molecular dynamics simulation engine (Schrodinger, 2006). The modelled ClpV structures were saved as Protein Data Bank (PDB) files and submitted to the Linux (Ubuntu) desktop server for molecular simulations studies. The system builder module was used to incorporate the TIP3P solvent model with the OPLS forcefield. The proteins were placed in an orthorhombic box (distance from the box face to the outermost protein atom was set to 10 Å, the box angle was α = β = γ = 90°). The volume box containing the proteins was minimized, and counter ions were added to neutralize the system. In addition, 0.15 M NaCl was added into the solvent box for physiological conditioning. The system was then submitted for MD simulations for 250 ns.
Post dynamic analyses of the trajectories derived from the MD simulation studies were performed using Schrodinger Maestro v12.2. Firstly, Simulation Quality Analysis was used to analyze the quality of simulations through the average energy, pressure, temperature, and volume analysis. Secondly, Simulation Interaction Diagram algorithm was used to analyze the RMSD of the alpha carbon atoms (Cα), the root mean square fluctuation (RMSF) of the residues, and secondary structure element analysis. Lastly, the Simulation Events Analysis algorithm (implemented in Maestro v12.2) was used to calculate the radius of gyration (Rg).
Advancement into sequencing technologies enables researchers to study the presence and distribution of genes in microorganisms to shed light into relationships amongst species (Pareek et al., 2011). Clp ATPases are essential for bacterial survival and pathogenicity and are emerging as potential drug targets (Culp and Wright, 2017). However, it was observed that the presence and distribution of Clp ATPases in the Enterobacter species has not been fully investigated hence, genome wide mining was performed to gain an insight into the diversity of Clp ATPases in the Enterobacter species.
The data mined and tallied for the 71 Enterobacter species shows that all the studied species have both Class I (ClpA and ClpB) and Class II (ClpX and ClpV) Clp ATPase members (Figure 1). It was also observed that fewer Enterobacter species (20%) contained ClpK, a class II member of Clp ATPases that has been recently linked to thermotolerance properties of another ESKAPE pathogen member, Klebsiella pneumonia (Bojer et al., 2010; Motiwala et al., 2021). It was very interesting to observe from genome data mining that all investigated strains contained one gene copy for ClpA, ClpB and ClpX. Contrastingly, certain Enterobacter strains contained more than one copy of the ClpV gene.
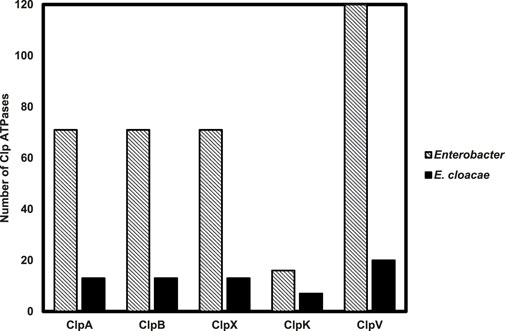
Figure 1. Distribution of Clp ATPases across 71 Enterobacter strains with a focus on the Clp ATPases identified in 13 Enterobacter cloacae strains. The number of Clp ATPases mined from the 71 Enterobacter strains and 13 Enterobacter cloacae strains through the NCBI genome database were tallied.
It then became necessary to ascertain the evolutionary relationship of the Clp ATPase species in Enterobacter species. Subsequently, we constructed a phylogenetic tree of the mined Clp ATPases. Figure 2 showed that the ClpA, ClpB, ClpK and ClpV proteins diverge from one point. This is expected as all these proteins contain two nucleotide domains and belong to Clp ATPase Class I. Therefore, one would assume that they would be more closely related to each other than to ClpX which belongs to Clp ATPase Class II (Figure 2A). Analysis of the phylogenetic tree shows that there is not a great amount of genetic variation among the ClpA, ClpB, ClpX and ClpK proteins, however, there appears to be significant genetic variation among the ClpV proteins (Figure 2A). This correlates with Clustal omega analysis of each protein group (Figure 2B).
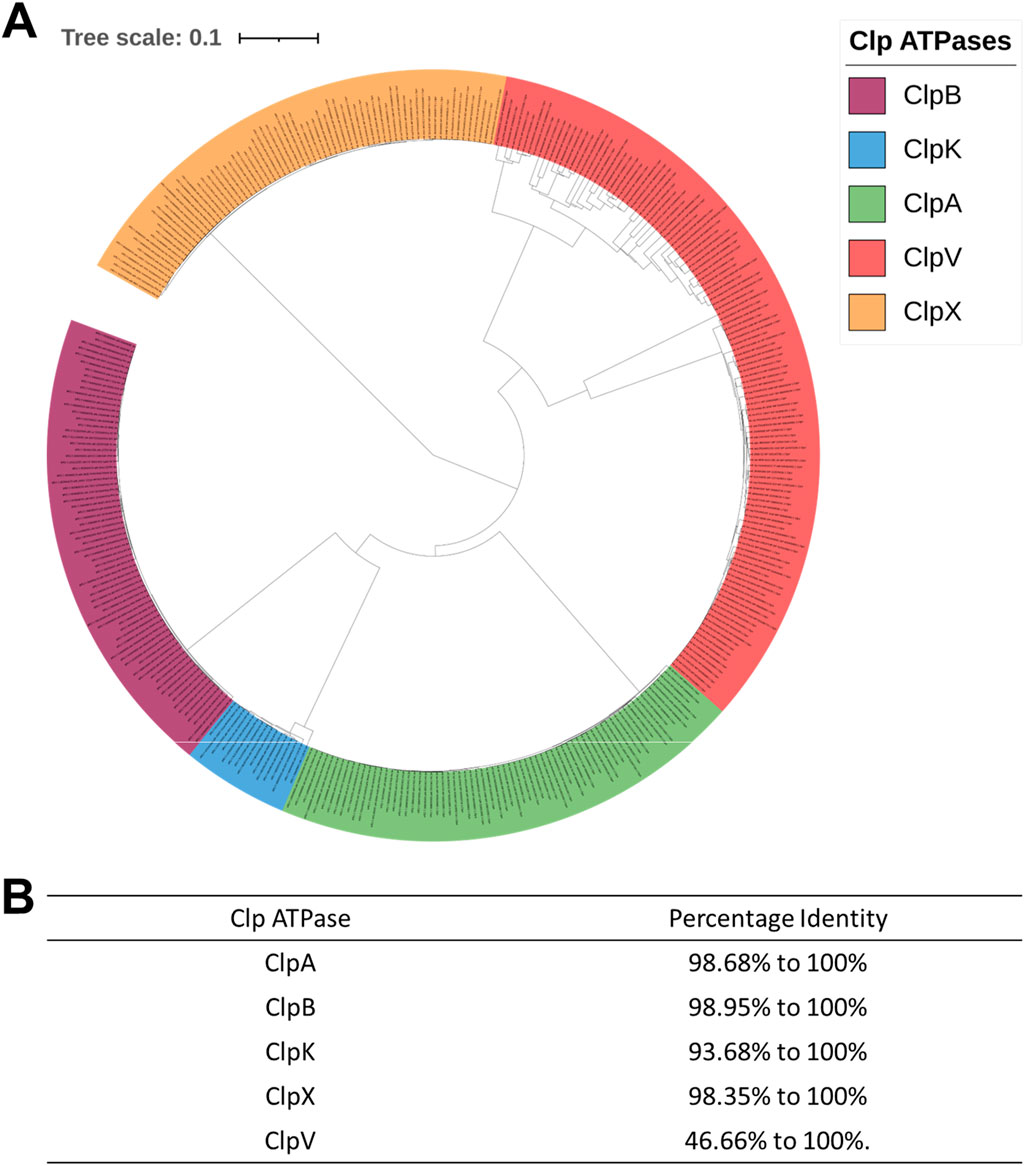
Figure 2. Phylogenetic tree and Clustal analysis of Clp ATPases mined from 71 Enterobacter strains. (A) Phylogenetic tree of the mined Clp ATPases. The alignment for the tree was done using MAFFT embedded on the Trex servers. The tree was visualized and colored using iTOL. The various Clp ATPases are represented in assorted colors. Tree distance scale: 0.1. (B) Clustal analysis of the Clp ATPases mined from the Enterobacter strains.
The role of ClpV proteins has been widely studied in Gram-negative opportunistic pathogens; V. cholera and P. aeruginosa (Smith-Roberge et al., 2018; Sun et al., 2014; Lesic et al., 2009; Li et al., 2020). The ClpV proteins mined from various E. cloacae strains were compared to ClpV, from V. cholera and P. aeruginosa through Clustal omega and phylogenetic analysis. This was done to elucidate whether the ClpV proteins from the E. cloacae strains may have a similar function to those of ClpV1 from V. cholera and P. aeruginosa. Clustal omega showed that ClpV from the various E. cloacae strains had between 43.56% and 100% similarity with ClpV1 from P. aeruginosa, and between 43.31% and 100% similarity with ClpV1 from V. cholera. ClpV from E. cloacae had a percentage identity of greater than 30% in comparison to ClpV1 from both V. cholera and P. aeruginosa, therefore indicating that the sequences are homologous (Pearson, 2013). Additionally, analysis of the phylogenetic tree comparing the evolution of ClpV from E. cloacae and ClpV1 from both V. cholera and P. aeruginosa showed that these proteins all shared a common ancestor, indicated by the number 1 (Figure 3). Figure 3 shows that a group of ClpV from E. cloacae (orange box) are more closely related to ClpVI from V. cholera and P. aeruginosa compared to the other group of ClpV from E. cloacae (blue box). Clustal omega analysis showed that three mined ClpV proteins from the E3442 strain had between 46.82% and 100% similarity, this correlated with the trend observed in Figure 3 which shows that these proteins lie on different branches of the phylogenetic tree.

Figure 3. Neighbor Joining tree of ClpV mined from Enterobacter cloacae in comparison to ClpVI from Pseudomonas aeruginosa and Vibrio cholera. The tree was constructed using the Neighbor Joining method (BLOSUM62) on Jalview v2.11.2.2 (Waterhouse et al., 2009a). The protein codes for each mined ClpV protein are displayed. The orange box outlines one group of ClpV proteins while the blue box outlines the second group. The common point of origin of the ClpV proteins is indicated by the number 1.
To further investigate the uniqueness of the ClpV proteins identified within E. cloacae strain E3442, the three ClpV proteins were compared through homology modelling. The ClpV proteins were modelled using the 1qvrA template and the model quality was assessed (Figure 4; Table 1). The alignment of ClpV1, ClpV2, and ClpV3 with the template (1QVR-A) revealed a root mean square deviation (RMSD) score of 0.270, 0.283 and 0.347, respectively. These scores are close to 0 and indicate that there is a good alignment between the coordinates of the model and template. Therefore, the proteins are structurally similar to the template (Carugo and Pongor, 2001). Secondary and tertiary analysis of the proteins show that they are mainly α-helical (Figure 4).
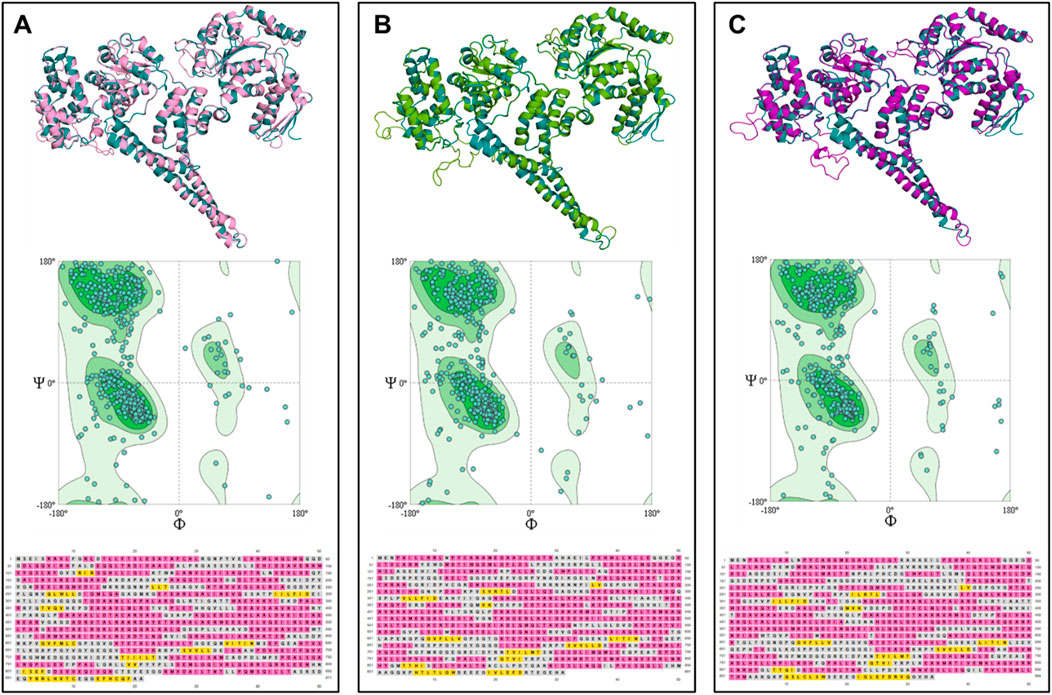
Figure 4. Tertiary structure, Ramachandran plot and secondary structure analysis of three ClpV proteins from E. cloacae strain E3442. (A) ClpV1 (lilac) and 1qvrA template (blue). (B) ClpV2 (green) and 1qvrA template (blue). (C) ClpV3 (purple) and 1qvrA template (blue). The α-helical residues are colored pink and the β-sheets and yellow.
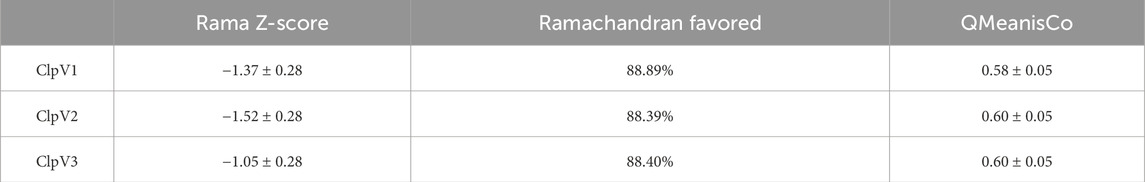
Table 1. Assessment of the model quality of the modelled ClpV proteins from Enterobacter cloacae strain E3442.
The Rama Z-score provides an overall insight into the quality of a modelled protein by comparing the model to a reference set of high-resolution structures (Sobolev et al., 2020). The Rama Z-scores of the modelled proteins were less than two and therefore the modelled structures are confirmed to be of adequate quality (Table 1). The Ramachandran favored scores of the three modelled proteins are greater than 80%, this indicates that the generated models can be trusted and used for further analysis (Maxwell and Popelier, 2017). The QMeanisCo uses statistical potentials of mean force to quantify model quality and modelling errors (Studer et al., 2019). The QMeanisCo values for the three modelled proteins are close to 1, thus once again confirming that the models are of adequate quality (Table 1).
Table 2 shows the physicochemical properties of the three ClpV proteins. The instability index of all three proteins is greater than 40, the proteins are therefore considered to be unstable. However, it is essential to remember that the stability of the protein is not only determined by the intrinsic nature of the protein but also by the protein environment (Gamage et al., 2019). The high aliphatic indexes indicate that these proteins are thermally stable. Additionally, the negative Grand average of Hydropathicity (GRAVY) score indicates that the proteins are all hydrophilic (Panda and Chandra, 2012). Figure 5 shows the protein disorder prediction, the disorder score for the residues of all three proteins lie below one and the three proteins are not considered to be disordered. Therefore, these proteins can be expressed and purified (Deng et al., 2012).
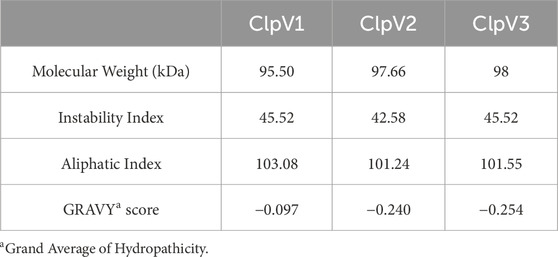
Table 2. Comparison of the physicochemical properties of the three ClpV proteins.
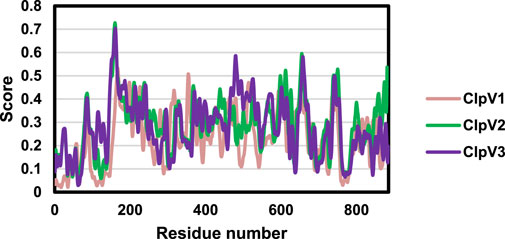
Figure 5. Disorder prediction of the three ClpV proteins. The disorder of ClpV1 (pink), ClpV2 (green), and ClpV3 (purple) proteins was predicted using the IUPred3 server.
Following analysis of the physicochemical characteristics and disorder prediction of the proteins, the difference in the protein sequence and 3D structure were further investigated. Figure 6A shows significant sequence similarity and conservation, especially between ClpV2 and ClpV3. The difference observed between the protein sequences indicate that these proteins may have variable enzyme activity and binding affinities. The 3D structure analysis corresponds with the primary sequence analysis in that ClpV2 and ClpV3 fall on the same branch of the phylogenetic tree (Figures 6B,C). Additionally, the Dali Z-score of 44.3 is greater than 20 and confirms that the three ClpV structures are homologous to each other (Holm et al., 2008).
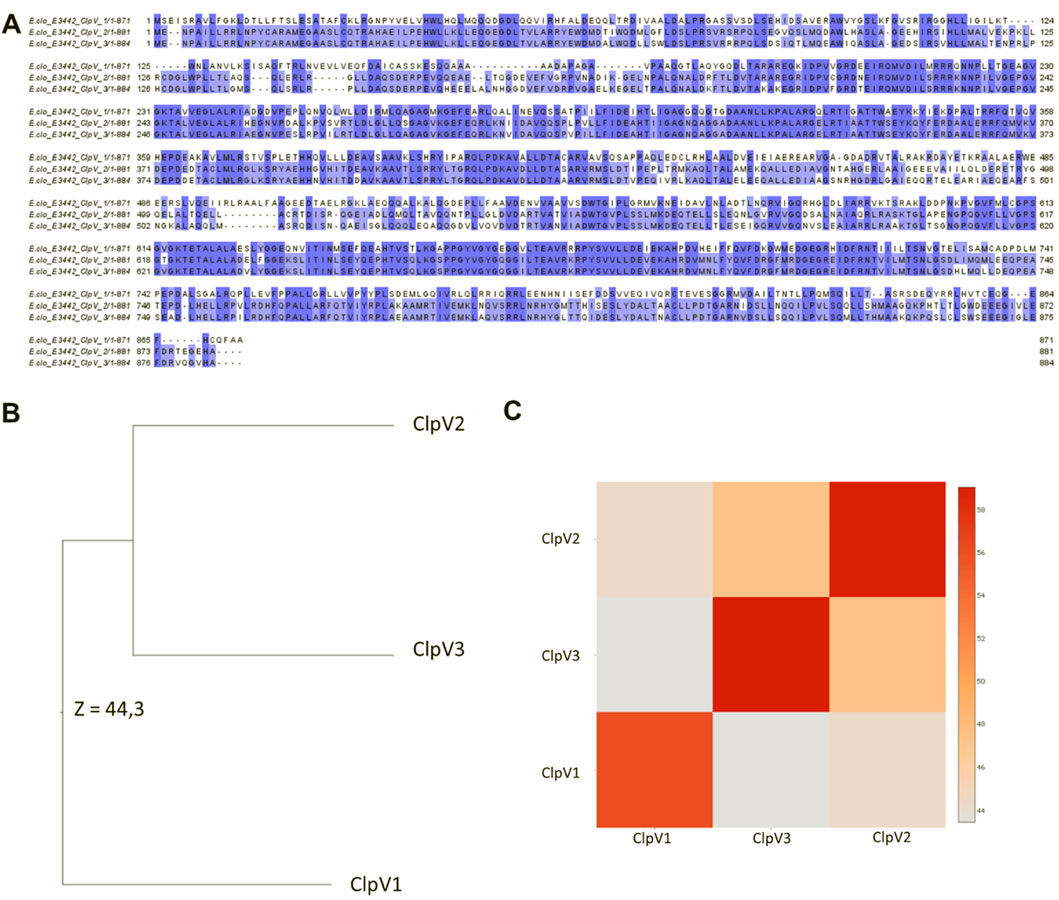
Figure 6. Comparison of the sequence and 3D structures of the three ClpV proteins using the Jalview software and Dali server, respectively. (A) Multisequence alignment performed using Mafft on the Jalview software. Alignment colored according to percentage identity. (B) Average linkage clustering of the Dali Z-scores is used to construct a Dendrogram. (C) Dali Z-scores are used to construct a heatmap.
The InterPro server analysis identified protein families, domains, and functional sites in ClpV proteins (Apweiler et al., 2001). ClpV1 and ClpV2 contain the repeat domain, which plays a key role in substrate specificity, a characteristic feature of the Clp A/B family (Lo et al., 2001). The nucleotide binding domains (NDB I and II) which are crucial for ATP binding and are well conserved among Clp A/B family members (Gottesman et al., 1990). They exhibit high sequence identity, with only minor variations across the three ClpV proteins. A highly conserved site (highlighted in orange) is situated in NBDI and remains identical across the three ClpV proteins (Figure 7). Similar to ClpB and ClpK, ClpV proteins contain a middle linker region that separates the NBDs and is essential for chaperone activity (Lee et al., 2003; Motiwala et al., 2021). Additionally, the AAA + lid domain, located in the middle domain of ClpV proteins, acts as a regulatory “gatekeeper” for substrate access through the central pore (Miller and Enemark, 2016).

Figure 7. Alignment of the three ClpV proteins. The annotations were done according to the InterPro server and Lee et al. (2003). Asterisks (*), colons (:) and full stops (.) represent the identical residues, sequence homologies, and weak similarities, respectively. The residues are highlighted blue, green, pink, red, orange, purple, olive and yellow. These represent the repeat domain, Nucleotide Binding Domain (NBD) 1, Walker A motif, Walker B motif, conserved site 1, AAA lid domain, NBD II, and conserved regions of the Clp A/B family. (A) First half of the alignment (residues 1 to 496). (B) Second half of alignment.
Further conserved regions include the Walker A (highlighted in pink) and Walker B (highlighted in red) motifs, located within NBD I and NBD II. The Walker A motif, which is responsible for ATP binding, is largely conserved, though a single amino acid variation suggests potential differences in ATP interactions among the three proteins. The Walker B motif, which facilitates metal ion binding, remains identical and homologous across ClpV proteins, indicating a shared metal ion interaction mechanism (Lee et al., 2003; Motiwala et al., 2021). Conserved residues (highlighted in yellow) further support the structural and functional similarity of the ClpV proteins (Figure 7).
Potential binding pockets on the three ClpV proteins were analyzed using the DogSite3 module on the ProteinsPlus server. This was done following confirmation on the difference in the primary and 3D structure of the proteins. Interestingly, the best binding pocket was observed to be around the same area for all three proteins with ClpV1 having the biggest binding pocket (Figure 8). The difference in the size of the binding pockets indicates that ClpV1 may bind bigger proteins or ligands, while ClpV2 and ClpV3 bind to ligands or proteins of similar size. The difference in size between the binding pockets could also be attributed to the presence of the repeat domain (Figure 7).
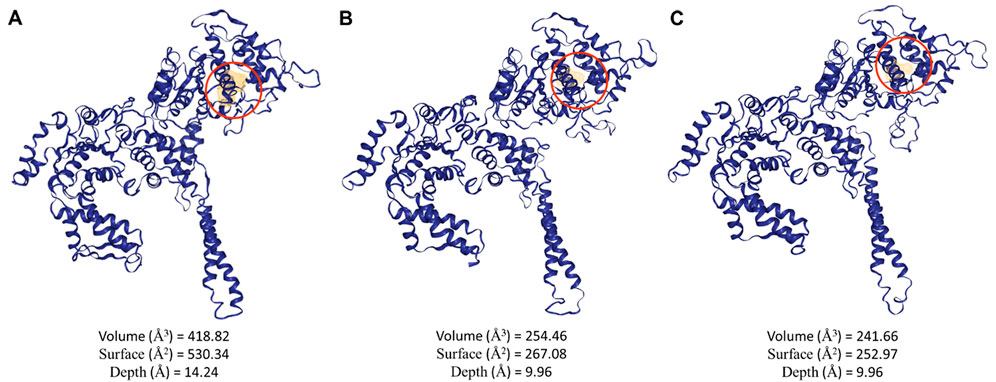
Figure 8. Potential binding pockets of the three ClpV proteins identified using the DogSite3 module on the ProteinsPlus server. (A) volume, surface and depth of the best potential binding site of ClpV1, (B) volume, surface and depth of the best potential binding site of ClpV2, and (C) volume, surface and depth of the best potential binding site of ClpV3.
Normal Mode Analysis (NMA) was performed to investigate the conformational changes between the ClpV proteins. The color transformations from blue to red in Figure 9 represents the conformational changes and flexibility of the three protein structures (Chen et al., 2025). The most flexible region of the protein appears to be the middle domain with ClpV1 having the most flexible middle region compared to ClpV2 and ClpV3.
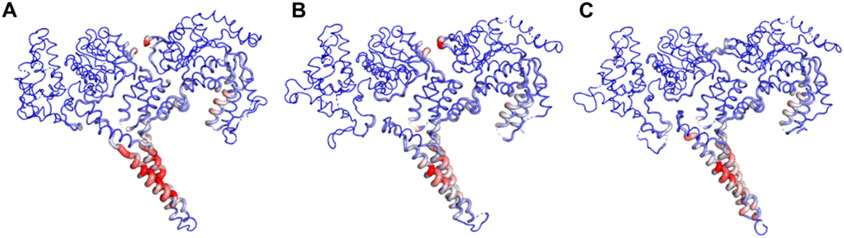
Figure 9. Normal Mode Analysis of ClpV proteins using the DynaMut server. (A) ClpV1, (B) ClpV2, and (C) ClpV3.
Finally, the three ClpV proteins were analyzed using the STRING server to examine their protein-protein interactions (Figure 9). This analysis aimed to explore the similarities and differences in the interacting partners of each ClpV protein. Interestingly, Figure 9 reveals that all three ClpV proteins interact with ClpP, which is unexpected given that ClpV lacks the tripeptide sequence required for ClpP interaction (Schlieker et al., 2005). The mechanism of interaction requires further investigation through in vitro studies or additional in silico protein-protein binding analyses. However, the observed interaction may result from indirect binding or alternative interaction motifs (Förster et al., 2014).
Our findings indicate that the three ClpV proteins are not structurally identical. Among them, ClpV1 and ClpV2 interact with ClpX, suggesting that the presence of a ClpX-binding site is another distinguishing feature of these proteins. As noted earlier, each strain contains a single copy of ClpX. However, since cells often produce multiple copies of the same protein, ClpX could potentially interact with two different types of ClpV proteins (Alberts et al., 2002). ClpX along with ClpA and ClpB are assumed to play a role in the functioning of the T6SS of Klebsiella pneumoniae, however the function of these Clp ATPases in the functioning of this system is yet to be elucidated (Barbosa and Lery, 2019).
MD simulations were performed for the three modelled ClpV proteins. Potential energy, radius of gyration, root mean square deviation (RMSD) and root mean square fluctuation (RMSF) were analyzed to explore the dynamic nature and stability of the proteins (Figure 11). The potential energy profiles of the three proteins remained stable throughout the simulation time of 250 ns (Figure 11A). The radius of gyration (rg) gives an indication of the compactness of the protein, the rg values of all three proteins did not exceed 1 Å therefore indicating that the proteins are compact and stable (Figure 11B). The RMSF shows the residues of the proteins which are the most flexible during the period of simulation (Mohan et al., 2022). The flexibility of all three proteins is significant (>1Å), thus indicating that the proteins are highly dynamic (Figure 11C). The RMSD further confirms the stability of the three proteins, ClpV2 and ClpV3 appear to reach equilibration around 10 ns, while ClpV1 seems to stabilize around 10 ns and continues fluctuating until 100 ns (Figure 11D). The MD run was extended to 400 ns and the dynamic nature of the protein remained consistent with the 250 ns run (data not shown). This along with the RMSF, Rg, and RMSD analysis at 250 ns confirmed that the proteins are stable, and compact. Radial distribution function (RDF) analysis also showed that the proteins had reached full conformational sampling as the systems equilibrated over time (data not shown).
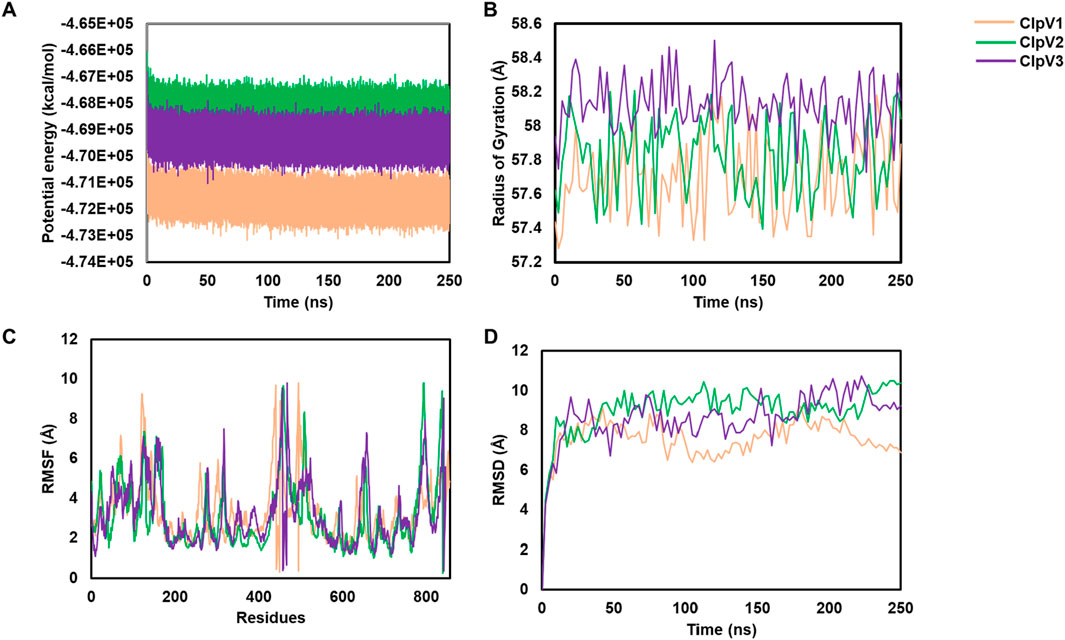
Figure 11. Using potential energy, radius of gyration, RMSF and RMSD to analyze molecular simulations of three ClpV proteins. ClpV1 (pink), ClpV2 (green) and ClpV3 (purple). (A) potential energy of alpha carbons observed over 250 ns. (B) radius of gyration of alpha carbons observed over 250 ns. (C) RMSF for each protein residue. (D) RMSD of alpha carbons observed over 250 ns.
The Enterobacter species are a pathogenic group of organisms which evade the action of antibiotics. This makes these pathogens a tremendous burden on the healthcare system and therefore it is important to find alternative targets for drug development to combat their spread. Clp ATPases are a group of proteins which play a role in protein homeostasis, and therefore drugs can essentially be designed to target these proteins and disrupt protein homeostasis. Before drug design, it is important to identify and characterize proteins. In this study, the distribution of Clp ATPases amongst the Enterobacter species was investigated to gain an insight into the prevalence of these proteins in the pathogens. Out of the 71 investigated strains it was observed that all contain ClpA, ClpB, ClpX and ClpV, while only 20% contained ClpK (Figure 1). ClpK not being found in all the strains could indicate that a number of the Enterobacter strains may not be subjected to heat stress and therefore have not taken up this protein which is easily obtainable through horizontal gene transfer. Additionally, it is possible that there are low numbers of ClpK present due to the presence of ClpB which also plays a role in the survival of bacteria under heat stress (Alam et al., 2021). It was interesting to observe that the pattern of divergence observed between the Enterobacter ClpA, ClpB, ClpK and ClpX proteins correlates with the pattern of divergence observed between the Klebsiella ClpA, ClpB, ClpK and ClpX proteins (Figure 2) (Motiwala et al., 2022). The ClpV proteins were observed to be genetically variable in comparison to the other Clp ATPase proteins (Figure 2).
The genetic variation observed amongst the investigated ClpV proteins led to further investigation of the proteins. ClpV resets the Type VI Secretion System (T6SS/TssH) by dissembling a sheath which contracts to provide energy for the entry of effector molecules in the cell (Kapitein et al., 2013). The T6SS has been identified to be present in Gram-negative bacteria and plays a role in inducing diarrhea and therefore contributes to the replication of V. cholera in the intestine (Kapitein et al., 2013; Förster et al., 2014). Additionally, ClpV has been found to contribute to the pathogenicity and biofilm formation of P. aeruginosa in vivo (Förster et al., 2014; Li et al., 2020). The ClpV proteins from Enterobacter shared more than 40% similarity with the ClpV proteins from P. aeruginosa and V. cholera and the proteins lie close to each other on the phylogenetic tree (Figure 3). This suggests that Enterobacter ClpV may play a similar role to ClpV from P. aeruginosa and V. cholera, the ClpV proteins would need to be further investigated through in vitro characterization.
Upon observing the variation of ClpV in the Enterobacter species it was important to gain an insight into the structural difference of the protein isomers identified in the Enterobacter strains. Additionally, it is interesting to note that the one ClpV protein identified from the same strain does not lie next to the other, or on the same branch as the other on the phylogenetic tree (Figure 3). This indicates that despite belonging to the same strain, the ClpV proteins are different to each other (Figure 3). This could be due the presence of distinct T6SS classes in Gram-negative bacteria, each with structural alterations unique to the ClpV protein which binds to them (Förster et al., 2014).
Despite being different to each other in terms of sequence, the three ClpV proteins are similar in terms of structure, disorder prediction and physiochemical properties (Figures 4, 5; Table 2). A considerably low percentage of ClpV residues were predicted to be disordered (Figure 5), therefore these proteins can be expressed, purified and used for subsequent biophysical characterisation. Additionally, it was observed that ClpV2 and ClpV3 are more alike in terms of structure, conserved residues, flexibility regions and potential binding sites (Figures 6–9). However, their differences are once again highlighted when analyzing the potential proteins that these ClpV proteins interact with (Figure 10). Furthermore, it was interesting to observe that the most flexible regions were the middle domain of the three proteins, which has been identified as being critical for the disaggregase activity of ClpB and mediates interactions with the Hsp70 chaperone system (Desantis and Shorter, 2012).
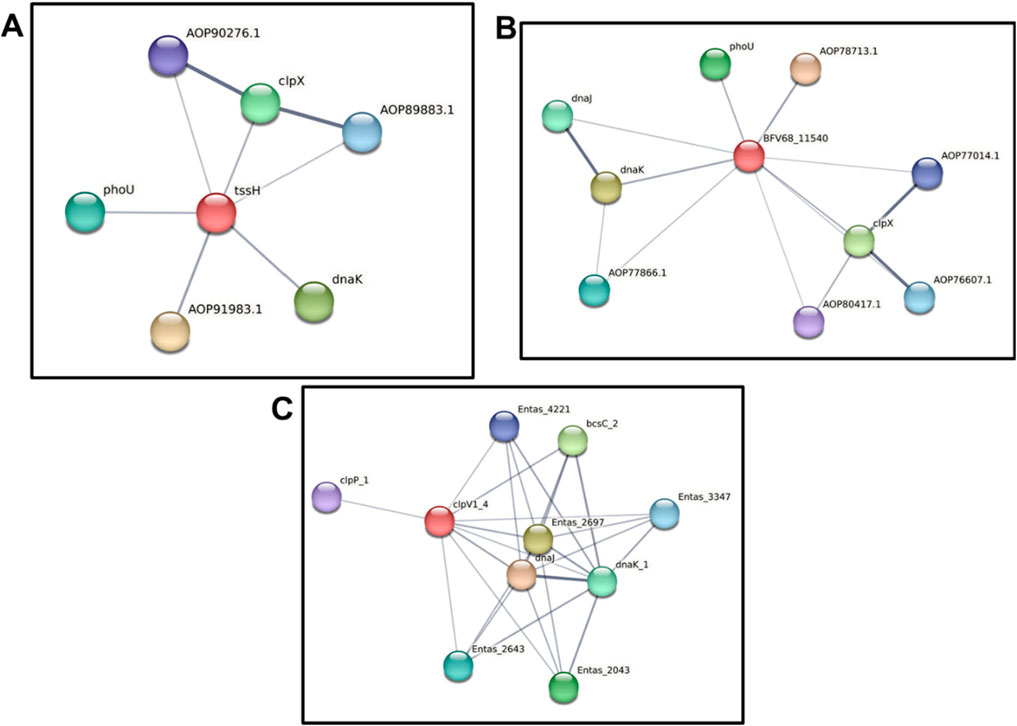
Figure 10. Protein-protein interaction analysis of three ClpV proteins using the STRING server. (A) Protein-protein interactions of ClpV1 (red circle), AOP91983.1 is a NAD-dependent epimerase, AOP90276.1 and AOP89883.1 are ATP-dependent Clp protease proteolytic subunits. (B) Protein-protein interactions of ClpV2 (red circle), AOP78713.1 is a NAD-dependent epimerase, AOP77866.1 is an antitermination protein, AOP80417.1 is peptidase S14, AOP76607.1 and AOP77014.1 are ATP-dependent Clp protease proteolytic subunits. (C) Protein-protein interactions of ClpV3 (red circle), the Entas proteins play a role in the KEGG pathway. The grey lines indicate the interaction confidence, with a thicker line indicating a higher confidence.
The potential energy profile of the three modelled models were investigated and indicated a slight, insignificant shift (Figure 11A), indicating that the models were relatively stable as there was no extreme force experienced by any atom due to the positioning other atoms (Alagu Lakshmi et al., 2020; Hollingsworth and Dror, 2018). Two analyses were used to confirm the dynamic nature of the modelled ClpV proteins. Firstly, the increase in the RMSD values observed over 250 ns indicated confirmational changes (Tiwari and Mohanty, 2013; Buchner, 2019) (Figure 11D). Secondly, the features of the radius of gyration profiles of the ClpV proteins indicated structural transformation, suggesting the constant transformation of the proteins during simulation (Figure 11). This is expected as these proteins are characterised as being chaperones and are thus expected to be highly dynamic in nature (Sučec et al., 2021).
Following the in the preliminary in silico investigation of these proteins, future studies could focus on investigating ligands and proteins that interact with these Clp ATPases. Additionally, it would be interesting to investigate whether these proteins all interact with the same Type VI Secretion System (T6SS/TssH). This investigation could allow for the development of potential drugs to target the secretion system-ClpV complex or drugs to target each ClpV homolog could be developed.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
TM: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Project administration, Visualization, Writing–original draft, Writing–review and editing. BN: Writing–review and editing. TK: Conceptualization, Funding acquisition, Project administration, Supervision, Validation, Writing–review and editing.
The author(s) declare that financial support was received for the research and/or publication of this article. Tehrim Motiwala would like to thank Department of Science and Technology-National Research Foundation (DST-NRF), South Africa for the Doctoral Scholarship grant number MND210602605517. Babalwa Nyide would like to thank the Poliomyelitis Research Foundation (PRF) for the Doctoral scholarship funding grant number 24/64. Thandeka Khoza also thanks the National Research Foundation grant number 121275, South African Medical Research Council-SIR grant and University of KwaZulu-Natal for research grants.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Alagu Lakshmi, S., Shafreen, R., Priya, A., and Shunmugiah, K. (2020). Ethnomedicines of Indian origin for combating COVID-19 infection by hampering the viral replication: using structure-based drug discovery approach. J. Biomol. Struct. Dyn. 39, 4594–4609. doi:10.1080/07391102.2020.1778537
Alam, A., Bröms, J. E., Kumar, R., and Sjöstedt, A. (2001). The role of ClpB in bacterial stress responses and virulence. Front. Mol. Biosci. 8, 668910.
Alberts, B., Johnson, A., Lewis, J., Raff, M., Roberts, K., and Walter, P. (2002). “From DNA to RNA,” in Molecular biology of the cell. 4th Edn (New York, NY: Garland Science). Available online at: https://www.ncbi.nlm.nih.gov/books/NBK26887/.
Apweiler, R., Attwood, T. K., Bairoch, A., Bateman, A., Birney, E., Biswas, M., et al. (2001). The InterPro database, an integrated documentation resource for protein families, domains and functional sites. Nucleic Acids Res. 29, 37–40. doi:10.1093/nar/29.1.37
Barbosa, V. a. A., and Lery, L. M. S. (2019). Insights into Klebsiella pneumoniae type VI secretion system transcriptional regulation. BMC Genomics 20, 506. doi:10.1186/s12864-019-5885-9
Blum, M., Chang, H.-Y., Chuguransky, S., Grego, T., Kandasaamy, S., Mitchell, A., et al. (2020). The InterPro protein families and domains database: 20 years on. Nucleic Acids Res. 49, D344–D354. doi:10.1093/nar/gkaa977
Boc, A., Diallo, A. B., and Makarenkov, V. (2012). T-REX: a web server for inferring, validating and visualizing phylogenetic trees and networks. Nucleic Acids Res. 40, W573–W579. doi:10.1093/nar/gks485
Bojer, M. S., Struve, C., Ingmer, H., Hansen, D. S., and Krogfelt, K. A. (2010). Heat resistance mediated by a new plasmid encoded Clp ATPase, ClpK, as a possible novel mechanism for nosocomial persistence of Klebsiella pneumoniae. PLoS One 5, e15467. doi:10.1371/journal.pone.0015467
Bouchnak, I., and Van Wijk, K. J. (2021). Structure, function, and substrates of Clp AAA+ protease systems in cyanobacteria, plastids, and apicoplasts: a comparative analysis. J. Biol. Chem. 296, 100338. doi:10.1016/j.jbc.2021.100338
Brötz-Oesterhelt, H., and Sass, P. (2014). Bacterial caseinolytic proteases as novel targets for antibacterial treatment. Int. J. Med. Microbiol. 304, 23–30. doi:10.1016/j.ijmm.2013.09.001
Buchner, J. (2019). Molecular chaperones and protein quality control: an introduction to the JBC Reviews thematic series. J. Biol. Chem. 294, 2074–2075. doi:10.1074/jbc.rev118.006739
Capestany, C. A., Tribble, G. D., Maeda, K., Demuth, D. R., and Lamont, R. J. (2008). Role of the Clp system in stress tolerance, biofilm formation, and intracellular invasion in Porphyromonas gingivalis. J. Bacteriol. 190, 1436–1446. doi:10.1128/jb.01632-07
Carugo, O., and Pongor, S. (2001). A normalized root-mean-spuare distance for comparing protein three-dimensional structures. Protein Sci. 10, 1470–1473. doi:10.1110/ps.690101
Chen, J., Wang, J., Yang, W., Zhao, L., and Xu, X. (2025). Identifying inhibitor-SARS-CoV2-3CLpro binding mechanism through molecular docking, GaMD simulations, correlation network analysis and MM-GBSA calculations. Molecules 30, 805. doi:10.3390/molecules30040805
Culp, E., and Wright, G. D. (2017). Bacterial proteases, untapped antimicrobial drug targets. J. Antibiotics 70, 366–377. doi:10.1038/ja.2016.138
D’andrea, F. B., Poulton, N. C., Froom, R., Tam, K., Campbell, E. A., and Rock, J. M. (2022). The essential M. tuberculosis Clp protease is functionally asymmetric in vivo. Sci. Adv. 8, eabn7943. doi:10.1126/sciadv.abn7943
Davin-Regli, A., Lavigne, J.-P., and Pagès, J.-M. (2019). Enterobacter spp.: update on taxonomy, clinical aspects, and emerging antimicrobial resistance. Clin. Microbiol. Rev. 32, e00002–e00019. doi:10.1128/cmr.00002-19
Davin-Regli, A., and Pagès, J.-M. (2015). Enterobacter aerogenes and Enterobacter cloacae; versatile bacterial pathogens confronting antibiotic treatment. Front. Microbiol. 6, 392. doi:10.3389/fmicb.2015.00392
Deng, X., Eickholt, J., and Cheng, J. (2012). A comprehensive overview of computational protein disorder prediction methods. Mol. Biosyst. 8, 114–121. doi:10.1039/c1mb05207a
De Oliveira, D. M., Forde, B. M., Kidd, T. J., Harris, P. N., Schembri, M. A., Beatson, S. A., et al. (2020a). Antimicrobial resistance in ESKAPE pathogens. Clin. Microbiol. Rev. 33, 001811-19–e219. doi:10.1128/cmr.00181-19
De Oliveira, D. M. P., Forde, B. M., Kidd, T. J., Harris, P. N. A., Schembri, M. A., Beatson, S. A., et al. (2020b). Antimicrobial resistance in ESKAPE pathogens. Clin. Microbiol. Rev. 33, e00181. doi:10.1128/cmr.00181-19
Desantis, M. E., and Shorter, J. (2012). The elusive middle domain of Hsp104 and ClpB: location and function. Biochim. Biophys. Acta 1823, 29–39. doi:10.1016/j.bbamcr.2011.07.014
Dhiman, R., and Singh, R. (2018). Recent advances for identification of new scaffolds and drug targets for Mycobacterium tuberculosis. IUBMB Life 70, 905–916. doi:10.1002/iub.1863
Douzi, B., Brunet, Y. R., Spinelli, S., Lensi, V., Legrand, P., Blangy, S., et al. (2016). Structure and specificity of the Type VI secretion system ClpV-TssC interaction in enteroaggregative Escherichia coli. Sci. Rep. 6, 34405. doi:10.1038/srep34405
Erdős, G., Pajkos, M., and Dosztányi, Z. (2021). IUPred3: prediction of protein disorder enhanced with unambiguous experimental annotation and visualization of evolutionary conservation. Nucleic Acids Res. 49, W297–W303. doi:10.1093/nar/gkab408
Fährrolfes, R., Bietz, S., Flachsenberg, F., Meyder, A., Nittinger, E., Otto, T., et al. (2017). ProteinsPlus: a web portal for structure analysis of macromolecules. Nucleic Acids Res. 45, W337–W343. doi:10.1093/nar/gkx333
Förster, A., Planamente, S., Manoli, E., Lossi, N. S., Freemont, P. S., and Filloux, A. (2014). Coevolution of the ATPase ClpV, the sheath proteins TssB and TssC, and the accessory protein TagJ/HsiE1 distinguishes type VI secretion classes. J. Biol. Chem. 289, 33032–33043. doi:10.1074/jbc.m114.600510
Frees, D., Savijoki, K., Varmanen, P., and Ingmer, H. (2007). Clp ATPases and ClpP proteolytic complexes regulate vital biological processes in low GC, Gram-positive bacteria. Mol. Microbiol. 63, 1285–1295. doi:10.1111/j.1365-2958.2007.05598.x
Gamage, D. G., Gunaratne, A., Periyannan, G. R., and Russell, T. G. (2019). Applicability of instability index for in vitro protein stability prediction. Protein Pept. Lett. 26, 339–347. doi:10.2174/0929866526666190228144219
Gasteiger, E., Gattiker, A., Hoogland, C., Ivanyi, I., Appel, R. D., and Bairoch, A. (2003). ExPASy: the proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 31, 3784–3788. doi:10.1093/nar/gkg563
Gottesman, S., Squires, C., Pichersky, E., Carrington, M., Hobbs, M., Mattick, J. S., et al. (1990). Conservation of the regulatory subunit for the Clp ATP-dependent protease in prokaryotes and eukaryotes. Proc. Natl. Acad. Sci. U. S. A. 87, 3513–3517. doi:10.1073/pnas.87.9.3513
Graef, J., Ehrt, C., and Rarey, M. (2023). Binding site detection remastered: enabling fast, robust, and reliable binding site detection and descriptor calculation with DoGSite3. J. Chem. Inf. Model. 63, 3128–3137. doi:10.1021/acs.jcim.3c00336
Heo, L., and Feig, M. (2017). PREFMD: a web server for protein structure refinement via molecular dynamics simulations. Bioinformatics 34, 1063–1065. doi:10.1093/bioinformatics/btx726
Hollingsworth, S. A., and Dror, R. O. (2018). Molecular dynamics simulation for all. Neuron 99, 1129–1143. doi:10.1016/j.neuron.2018.08.011
Holm, L. (2022). Dali server: structural unification of protein families. Nucleic Acids Res. 50, W210–W215. doi:10.1093/nar/gkac387
Holm, L., Kääriäinen, S., Rosenström, P., and Schenkel, A. (2008). Searching protein structure databases with DaliLite v.3. Bioinformatics 24, 2780–2781. doi:10.1093/bioinformatics/btn507
Huang, S., Dai, W., Sun, S., Zhang, X., and Zhang, L. (2012). Prevalence of plasmid-mediated quinolone resistance and aminoglycoside resistance determinants among carbapeneme non-susceptible Enterobacter cloacae. PloS one 7, e47636. doi:10.1371/journal.pone.0047636
Jacoby, G. A. (2009). AmpC beta-lactamases. Clin. Microbiol. Rev. 22, 161–182. doi:10.1128/cmr.00036-08
Kaneko, K., Okamoto, R., Nakano, R., Kawakami, S., and Inoue, M. (2005). Gene mutations responsible for overexpression of AmpC beta-lactamase in some clinical isolates of Enterobacter cloacae. J. Clin. Microbiol. 43, 2955–2958. doi:10.1128/jcm.43.6.2955-2958.2005
Kapitein, N., Bönemann, G., Pietrosiuk, A., Seyffer, F., Hausser, I., Locker, J. K., et al. (2013). ClpV recycles VipA/VipB tubules and prevents non-productive tubule formation to ensure efficient type VI protein secretion. Mol. Microbiol. 87, 1013–1028. doi:10.1111/mmi.12147
Katoh, K., Kuma, K.-I., Toh, H., and Miyata, T. (2005). MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic Acids Res. 33, 511–518. doi:10.1093/nar/gki198
Kim, Y.-I., Levchenko, I., Fraczkowska, K., Woodruff, R. V., Sauer, R. T., and Baker, T. A. (2001). Molecular determinants of complex formation between Clp/Hsp100 ATPases and the ClpP peptidase. Nat. Struct. Biol. 8, 230–233. doi:10.1038/84967
Kirstein, J., Molière, N., Dougan, D. A., and Turgay, K. (2009). Adapting the machine: adaptor proteins for Hsp100/Clp and AAA+ proteases. Nat. Rev. Microbiol. 7, 589–599. doi:10.1038/nrmicro2185
Lee, S., Sowa, M. E., Watanabe, Y.-H., Sigler, P. B., Chiu, W., Yoshida, M., et al. (2003). The structure of ClpB: a molecular chaperone that rescues proteins from an aggregated state. Cell 115, 229–240. doi:10.1016/s0092-8674(03)00807-9
Lesic, B., Starkey, M., He, J., Hazan, R., and Rahme, L. G. (2009). Quorum sensing differentially regulates Pseudomonas aeruginosa type VI secretion locus I and homologous loci II and III, which are required for pathogenesis. Microbiol. Read. 155, 2845–2855. doi:10.1099/mic.0.029082-0
Letunic, I., and Bork, P. (2021). Interactive Tree of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–W296. doi:10.1093/nar/gkab301
Li, Y., Chen, L., Zhang, P., Bhagirath, A. Y., and Duan, K. (2020). ClpV3 of the H3-type VI secretion system (H3-T6SS) affects multiple virulence factors in Pseudomonas aeruginosa. Front. Microbiol. 11, 1096. doi:10.3389/fmicb.2020.01096
Lo, J. H., Baker, T. A., and Sauer, R. T. (2001). Characterization of the N-terminal repeat domain of Escherichia coli ClpA-A class I Clp/HSP100 ATPase. Protein Sci. 10, 551–559. doi:10.1110/ps.41401
Maxwell, P., and Popelier, P. (2017). Unfavorable regions in the ramachandran plot: is it really steric hindrance? The interacting quantum atoms perspective. J. Comput. Chem. 38, 2459–2474. doi:10.1002/jcc.24904
Mezzatesta, M. L., Gona, F., and Stefani, S. (2012). Enterobacter cloacae complex: clinical impact and emerging antibiotic resistance. Future Microbiol. 7, 887–902. doi:10.2217/fmb.12.61
Miller, J. M., and Enemark, E. J. (2016). Fundamental characteristics of AAA+ protein family structure and function. Archaea 2016, 1–12. doi:10.1155/2016/9294307
Mohan, A., Shaji, S., Padmanabhan, S., Naisam, S., and Sreekumar, N. (2022). The potentials of Calotropis procera against filarial elephantiasis: an in-silico approach. J. Parasit. Dis. 46, 384–394. doi:10.1007/s12639-021-01456-0
Motiwala, T., Akumadu, B. O., Zuma, S., Mfeka, M. S., Chen, W., Achilonu, I., et al. (2021). Caseinolytic proteins (Clp) in the genus Klebsiella: special focus on ClpK. Molecules 27, 200. doi:10.3390/molecules27010200
Motiwala, T., Akumadu, B. O., Zuma, S., Mfeka, M. S., Chen, W., Achilonu, I., et al. (2022). Caseinolytic proteins (Clp) in the genus Klebsiella: special focus on ClpK. Molecules 27, 200. doi:10.3390/molecules27010200
Neuwald, A. F., Aravind, L., Spouge, J. L., and Koonin, E. V. (1999). AAA+: a class of chaperone-like ATPases associated with the assembly, operation, and disassembly of protein complexes. Genome Res. 9, 27–43. doi:10.1101/gr.9.1.27
Panda, S., and Chandra, G. (2012). Physicochemical characterization and functional analysis of some snake venom toxin proteins and related non-toxin proteins of other chordates. Bioinformation 8, 891–896. doi:10.6026/97320630008891
Pareek, C. S., Smoczynski, R., and Tretyn, A. (2011). Sequencing technologies and genome sequencing. J. Appl. Genet. 52, 413–435. doi:10.1007/s13353-011-0057-x
Pearson, W. R. (2013). An introduction to sequence similarity (“homology”) searching. Curr. Protoc. Bioinforma. 42, 3.1.1–3.1.8. doi:10.1002/0471250953.bi0301s42
Pendleton, J. N., Gorman, S. P., and Gilmore, B. F. (2013). Clinical relevance of the ESKAPE pathogens. Expert Rev. anti-infective Ther. 11, 297–308. doi:10.1586/eri.13.12
Rizi, K. S., Ghazvini, K., and Farsiani, H. (2020). Clinical and pathogenesis overview of Enterobacter infections. Rev. Clin. Med. 6, 146–154. doi:10.22038/rcm.2020.44468.1296
Rodrigues, C. H., Pires, D. E., and Ascher, D. B. (2018). DynaMut: predicting the impact of mutations on protein conformation, flexibility and stability. Nucleic Acids Res. 46, W350–W355. doi:10.1093/nar/gky300
Schlieker, C., Zentgraf, H., Dersch, P., and Mogk, A. (2005). ClpV, a unique Hsp100/Clp member of pathogenic proteobacteria. Biol. Chem. 386, 1115–1127. doi:10.1515/bc.2005.128
Schöning-Stierand, K., Diedrich, K., Ehrt, C., Flachsenberg, F., Graef, J., Sieg, J., et al. (2022). ProteinsPlus: a comprehensive collection of web-based molecular modeling tools. Nucleic Acids Res. 50, W611–W615. doi:10.1093/nar/gkac305
Schöning-Stierand, K., Diedrich, K., Fährrolfes, R., Flachsenberg, F., Meyder, A., Nittinger, E., et al. (2020). ProteinsPlus: interactive analysis of protein–ligand binding interfaces. Nucleic Acids Res. 48, W48–W53. doi:10.1093/nar/gkaa235
Schrodinger (2006). Proceedings of the 2006 ACM/IEEE conference on supercomputing. Tampa, Florida: Association for Computing Machinery.
Sen, A., Zhou, Y., and Imlay, J. A. (2020). During oxidative stress the Clp proteins of Escherichia coli ensure that iron pools remain sufficient to reactivate oxidized metalloenzymes. J. Bacteriol. 202, e00235–20. doi:10.1128/jb.00235-20
Seraphim, T. V., and Houry, W. A. (2020). AAA+ proteins. Curr. Biol. 30, R251–R257. doi:10.1016/j.cub.2020.01.044
Sievers, F., Wilm, A., Dineen, D., Gibson, T. J., Karplus, K., Li, W., et al. (2011). Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539. doi:10.1038/msb.2011.75
Smith-Roberge, J., Stietz, M. S., Dong, T. G., and Ingalls, B. (2018). ATPase binding and consequent disassembly of the type VI secretion system in Vibrio cholerae: a dynamic model. IFAC-PapersOnLine 51, 82–83. doi:10.1016/j.ifacol.2018.09.035
Sobolev, O. V., Afonine, P. V., Moriarty, N. W., Hekkelman, M. L., Joosten, R. P., Perrakis, A., et al. (2020). A global ramachandran score identifies protein structures with unlikely stereochemistry. Structure 28, 1249–1258.e2. doi:10.1016/j.str.2020.08.005
Studer, G., Rempfer, C., Waterhouse, A. M., Gumienny, R., Haas, J., and Schwede, T. (2019). QMEANDisCo—distance constraints applied on model quality estimation. Bioinformatics 36, 2647–1771. doi:10.1093/bioinformatics/btaa058
Sučec, I., Bersch, B., and Schanda, P. (2021). How do chaperones bind (partly) unfolded client proteins? Front. Mol. Biosci. 8, 762005. doi:10.3389/fmolb.2021.762005
Sun, K., Bröms, J., Lavander, M., Gurram Bharat, K., Enquist, P.-A., Andersson, C. D., et al. (2014). Screening for inhibition of Vibrio cholerae VipA-VipB interaction identifies small-molecule compounds active against type VI secretion. Antimicrob. Agents Chemother. 58, 4123–4130. doi:10.1128/aac.02819-13
Szklarczyk, D., Gable, A. L., Lyon, D., Junge, A., Wyder, S., Huerta-Cepas, J., et al. (2019). STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 47, D607–d613. doi:10.1093/nar/gky1131
Tiwari, G., and Mohanty, D. (2013). An in silico analysis of the binding modes and binding affinities of small molecule modulators of PDZ-peptide interactions. PLOS ONE 8, e71340. doi:10.1371/journal.pone.0071340
Van Nierop, W., Duse, A., Stewart, R., Bilgeri, Y., and Koornhof, H. (1998). Molecular epidemiology of an outbreak of Enterobacter cloacae in the neonatal intensive care unit of a provincial hospital in Gauteng, South Africa. J. Clin. Microbiol. 36, 3085–3087. doi:10.1128/jcm.36.10.3085-3087.1998
Voos, W. (2013). Chaperone–protease networks in mitochondrial protein homeostasis. Biochimica Biophysica Acta (BBA) - Mol. Cell Res. 1833, 388–399. doi:10.1016/j.bbamcr.2012.06.005
Waterhouse, A., Bertoni, M., Bienert, S., Studer, G., Tauriello, G., Gumienny, R., et al. (2018). SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 46, W296–W303. doi:10.1093/nar/gky427
Waterhouse, A. M., Procter, J. B., Martin, D. M., Clamp, M., and Barton, G. J. (2009a). Jalview Version 2--a multiple sequence alignment editor and analysis workbench. Bioinformatics 25, 1189–1191. doi:10.1093/bioinformatics/btp033
Waterhouse, A. M., Procter, J. B., Martin, D. M. A., Clamp, M., and Barton, G. J. (2009b). Jalview Version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics 25, 1189–1191. doi:10.1093/bioinformatics/btp033
Wickner, S., Gottesman, S., Skowyra, D., Hoskins, J., Mckenney, K., and Maurizi, M. R. (1994). “A molecular chaperone, ClpA, functions like DnaK and DnaJ,” Proc. Natl. Acad. Sci. U. S. A. 91. 12218–12222. doi:10.1073/pnas.91.25.12218
Williams, C. J., Headd, J. J., Moriarty, N. W., Prisant, M. G., Videau, L. L., Deis, L. N., et al. (2018). MolProbity: more and better reference data for improved all-atom structure validation. Protein Sci. 27, 293–315. doi:10.1002/pro.3330
Zavilgelsky, G., Kotova, V. Y., Mazhul, M., and Manukhov, I. (2004). The effect of Clp proteins on DnaK-dependent refolding of bacterial luciferases. Mol. Biol. 38, 427–433. doi:10.1023/b:mbil.0000032215.53875.f8
Keywords: Enterobacter, Enterobacter cloacae, Clp ATPases, ClpV, antibiotic resistance, alternative therapy
Citation: Motiwala T, Nyide B and Khoza T (2025) Molecular dynamic simulations to assess the structural variability of ClpV from Enterobacter cloacae. Front. Bioinform. 5:1498916. doi: 10.3389/fbinf.2025.1498916
Received: 19 September 2024; Accepted: 17 March 2025;
Published: 25 March 2025.
Edited by:
Yaan J. Jang, University of Oxford, United KingdomReviewed by:
Jianzhong Chen, Shandong Jiaotong University, ChinaCopyright © 2025 Motiwala, Nyide and Khoza. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Thandeka Khoza, a2hvemF0MUB1a3puLmFjLnph
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.