 H. W. Cayatineto
H. W. Cayatineto S. T. Hakim
S. T. Hakim- Hakim’s Lab, Department of Biology, School of STEM, Diné College, Tuba City, AZ, United States
Introduction: Flaviviridae comprise a group of enveloped, positive-stranded RNA viruses that are mainly transmitted through either mosquitoes or tick bites and/or contaminated blood, blood products, or other body secretions. These viruses cause diseases ranging from mild to severe and are considered important human pathogens. MicroRNAs (miRNAs) are non-coding molecules involved in growth, development, cell proliferation, protein synthesis, apoptosis, and pathogenesis. These small molecules are even being used as gene suppressors in antiviral therapeutics, inhibiting viral replication. In the current study, we used bioinformatic tools to predict a possible miRNA sequence that could be complementary to the nucleocapsid (NP) and/or capsid (CP) gene of the Flaviviridae family and provide an inhibitory solution.
Methods: Bioinformatics is a field of science that includes tremendous computational analysis, logarithms, and sequence alignments. To predict the right alignments between miRNA and viral mRNA genomes, we used computational databases such as miRBase, NCBI, and Basic Alignment Search Tool–nucleotides (BLAST-n).
Results: Of the 2,600 mature miRNAs, hsa-miR-548d-3p revealed complementary sequences with the flavivirus capsid gene and bovine viral diarrhea virus (BVDV) capsid gene and was selected as a possible candidate to inhibit flaviviruses.
Conclusion: Although more detailed in vitro and in vivo studies are required to test the possible inhibitory effects of hsa-miR-548d-3p against flaviviruses, this computational study may be the first step to study further, developing a novel therapeutic for lethal viruses within the Flaviviridae family using suggested candidate miRNAs.
Introduction
Arboviruses (arthropod-borne viruses) are a group of viruses that are classified into different taxonomic families such as Flaviviridae, Bunyaviridae, Togaviridae, Rhabdoviridae, Reoviridae, and Asfarviridae, with Flaviviridae, Togaviridae, and Bunyaviridae being the families that cause disease in humans (Giménez-Richarte et al., 2022). In this study, we primarily focused on the Flaviviridae family, which includes four species of pestiviruses, namely, bovine viral diarrhea viruses 1 and 2 (BVDV 1 and BVDV2), classical swine fever virus (CSFV), and border disease virus (BDV) (Mari et al., 2016; Maurer et al., 2004; Warrener and Collett, 1995; Schweize and Peterhans, 2001). The Flavivirus genus also includes global human pathogens such as Zika virus (ZIKV), West Nile virus (WNV), Japanese encephalitis virus (JENV), dengue virus (DENV), yellow fever virus (YFV), and tick-borne encephalitis virus (TBEV), which all pose a threat to global public health (Hu et al., 2021; Reed et al., 1998). Hepacivirus, another member of the flavivirus family, or hepatitis C virus or simply HCV, is responsible for non-A and non-B hepatitis among humans (Harada et al., 2000; Merwaiss et al., 2019; Suzich et al., 1993). This genus also includes additional 50 arthropod-borne viruses, which are mainly transmitted via mosquito bites and tick bites (Barrows et al., 2018) (Figure 1). Although the primary vectors are mainly mosquitos and ticks, these viruses have also been detected and isolated from bats and rodents (Junglen et al., 2009).
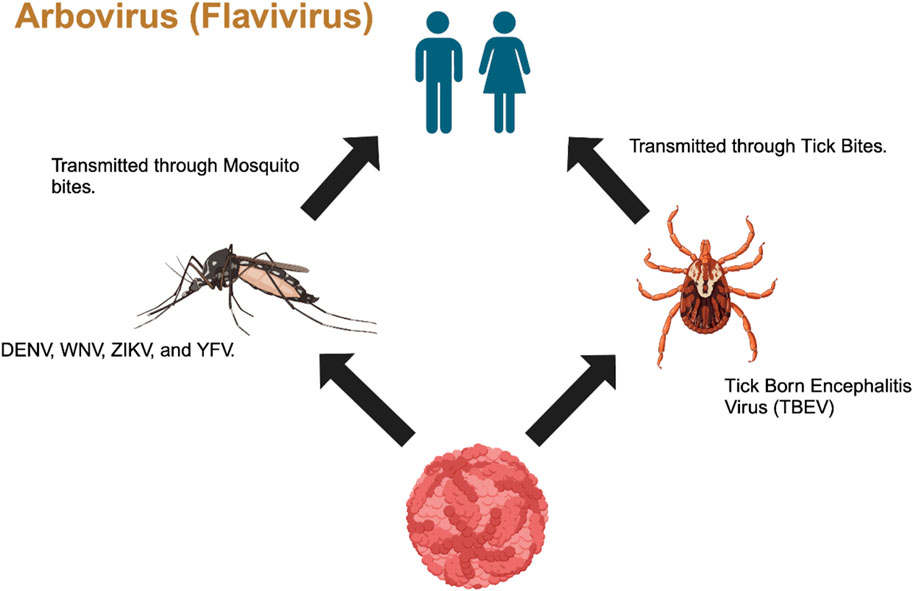
Figure 1. Transmission of Flavivirus from mosquitos and ticks to humans through vector bites (Generated by BioRender).
The incubation period of flavivirus infections in humans can range from 3 to 6 days (Conde et al., 2017), with presentation of acute flavivirus diseases ranging from being mild to severe and can be life-threatening (Pierson and Diamond, 2020). Pierson and Diamond mentioned that the symptoms of mild illness are mostly similar to flu-like symptoms, which includes asymptomatic infection and/or self-limiting febrile episodes, while severe illness includes hemorrhagic fever, shock syndrome, encephalitis, paralysis, congenital defects, hepatitis, and hepatic failure (2020; Benzarti et al., 2019). Although there are vaccines currently available for most of these viruses, which have also been successful, however, due to re-establishment of vectors, globalization, and urbanizations, epidemics continue to occur, which restricts effectiveness of these vaccines (Julander et al., 2009; de Oliveira Figueiredo et al., 2020; van Leur et al., 2021). People infected with DENV can develop more severe manifestations like dengue hemorrhagic fever (DHF) and dengue shock syndrome (DSS), which includes vascular leakage or hypovolemic shock and coagulopathy, followed by bleeding, organ impartment, and death (Simmons et al., 2012; Conde et al., 2017).
Of these flaviviruses, WNV and JENV are known as neurotropic viruses and cause acute encephalopathy, causing severe neuroinflammation of the central nervous system (CNS) and the blood–brain barrier (Li et al., 2015). In WNV, symptoms include flaccid paralysis, convulsions, cranial neuropathies, optic neuritis, ataxia, stiffness, rigidity spasms, and tremors that might cause long-term neurological changes (World Health Organization, 2019). JENV shows symptoms similar to that of WNV but is rare and has a much higher fatality rate of 30% (World Health Organization, 2019). TBEV, is also a member of the encephalitis virus family like WNV and JENV, but on the contrary, it is not transmitted by mosquitos like the other members of the arbovirus family, rather it is transmitted by infected tick (Ixodes ricinus) bites that can spread from animals to humans (Turtle et al., 2012).
HCV, another member of flaviviruses, attacks the hepatocytes (liver cells) in humans (Song et al., 2001), causing inflammation of the liver. HCV is a blood-borne virus and is transmitted primarily through infected blood and/or blood products or contaminated body fluids. One example of possible HCV transmission is the sharing of needles among drug abusers who use needle injections. In the mid 2000s, HCV transmission has also occurred among men who have sexual encounters with other men, also known as Men sex with other men (MSM) (Nijmeijer et al., 2019) (Figure 2). Despite treatments currently available, there is no vaccine for HCV (Duncan et al., 2020). When left untreated, HCV can lead to liver cirrhosis and chronic hepatitis C infection, leading to liver carcinoma (Isken et al., 2007). Progression of HCV is rather slow and can remain unnoticed (asymptomatic) for decades until the patient develops liver disease, which results in delay in diagnosis and treatment (Babiker et al., 2017).
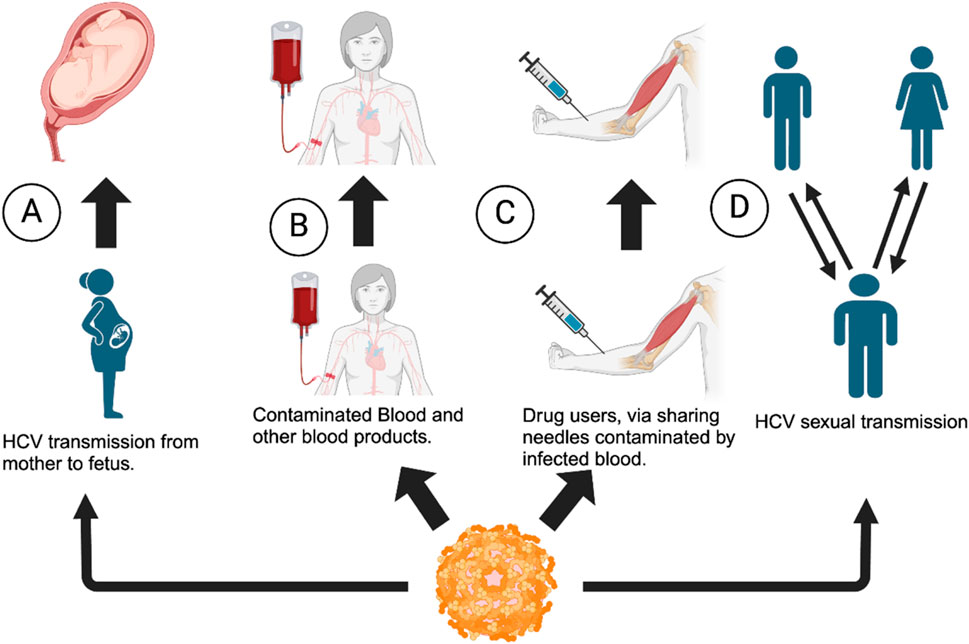
Figure 2. Transmission of HCV virus among individuals. (A) From the mother to fetus (B) via contaminated blood products, (C) via needle sharing among drug abusers and needle injury, and (D) via sexual contact: man to man, man to woman, or vice versa (Flowchart generated using BioRender).
YFV infections occur in 12% individuals with a 95% confidence interval and in 5 to 26% individuals with manifestations of jaundice, hemorrhage, and organ failure (Waggoneret al., 2018). Mosquitoes are primary carriers in areas of endemicity and are mainly recorded in Africa and South America, and despite successful vaccinations, outbreaks continue and lead to significant high morbidity and mortality rates (Julander et al., 2009). Julander et al. stated that despite vaccinations, there is a great need for more therapies as there is no antiviral agent available for YFV (2009).
Similar to all flaviviruses, ZIKV is another member, which is transmitted to humans by Aedes (Stegomyia subgenus) mosquitoes (Hills et al., 2017), and the disease caused by ZIKV can range from mild to severe, with a 3–12-day incubation period (Basarab et al., 2016; Musso and Gubler, 2016). Basarab et al. stated that ZIKV symptoms can include fever, conjunctivitis, arthralgia, myalgia, and itchy rashes (Musso and Gubler, 2016; Musso and Nhan, 2015; Hamel et al., 2015). However, Basarab et al. claimed that symptoms also include headache, retro-orbital pain, peripheral edema, joint pain, and even gastrointestinal disturbances (2016).
Overall, flaviviruses are small positive-sense, single-stranded RNA viruses that harbor structural proteins such as the capsid (C), which is responsible for protecting the viral genome; the pre-membrane protein (prM); the envelope protein (E); and non-structural (NS) proteins that are categorized as NS2A, NS2B, NS3, NS4A, 2K, NA4B, and NS5 (Mutebi et al., 2004) with a genome of approximately 11 kb (Laureti et al., 2018). During viral entry, replication occurs in the endoplasmic reticulum, where ribosomes are present (Figure 3). Because the genome of these viruses can act as a messenger RNA (mRNA), the genome is readily translated into proteins, making more virus particles (van den Elsen et al., 2021). Viral attachment is accomplished by the E protein attachment to the cognate receptors (Laureti et al., 2018). Laureti et al. conferred that the E protein binds to receptors such as glycosaminoglycans that increase the viral density on the host cell surface, allowing for more effective receptor binding (2018; Perera-Lecoin et al., 2013). On the surface of the E protein, the ectodomain harbors three domains, namely, E-D1, E-2, and E-D3, where E-D3 interacts with attachment factors and receptors and is mainly the target of neutralizing antibodies (Laureti et al., 2018; Pierson Kielian, 2013).
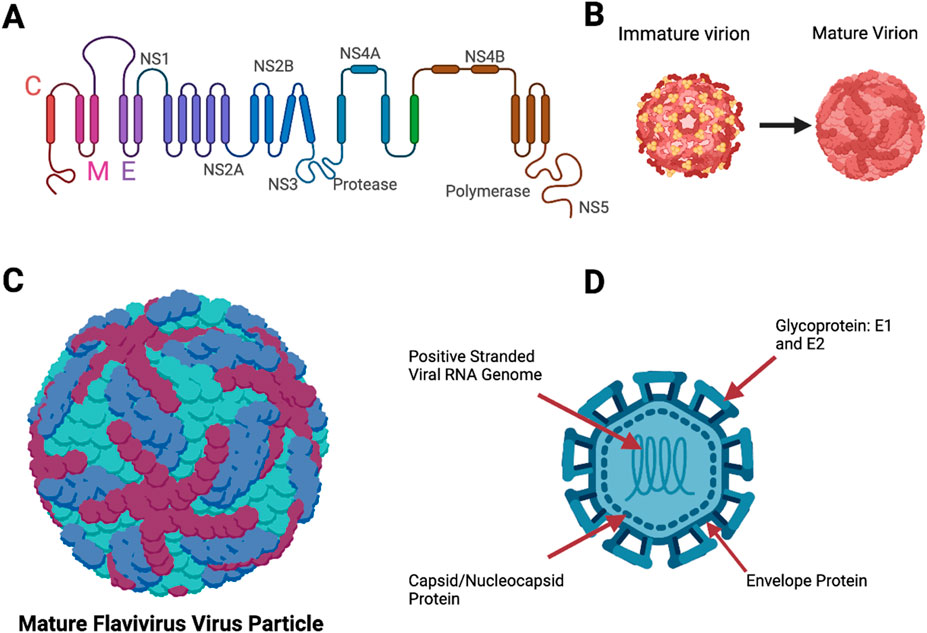
Figure 3. Structures of a flavivirus. (A) Polyprotein that makes up most of the viral structure. (B) Immature flavivirus particle and a mature flavivirus particle during the replication cycle. (C) Mature flavivirus particle with the surface proteins E1 and E2, and envelope structures. E1 is represented in maroon and dark blue colors, and the envelope protein is represented in turquoise color. (D) Complete labeled structure of flavivirus, (our target in this study) with the nucleocapsid/capsid protein (Figure 3 was designed using BioRender).
BVDV is a causative agent of bovine diarrhea and mucosal disease and hemorrhagic syndrome with high mortality among cattle (Jackova et al., 2008). The virion size ranges between 40 and 60 nm (Li et al., 2013), and the genome is approximately 13.3 kb in size (Murrayet al., 2008). The viral proteins of BVDV are organized in the following order: NH2-Npro-C-Erns-E1-E2-p7-NS2- NS3-NS4A-NS4B-NS5A-NS5B-COOH (Tellinghuisen et al., 2006; Neill, 2013; Becheret al., 1998; Chi et al., 2022), which is very similar to that of flavivirus polyprotein.
Transmission of BVDV among cattle includes fomites, such as contaminated feed, water, and equipment, and among other surfaces such as the nose; tongue; milk bottle nipples; needles; palpitations; secretions; and excretion of urine feces, mucus, milk, and other contaminated materials (Niskanen et al., 2000) (Figure 4). When cattle are exposed, they usually recover over time and shed the virus temporarily; however, pregnant cattle are more susceptible, and the outcome depends on the gestational stage of the fetus (Fulton et al. (2000). Although cows are the main host, BVDV infects various cattle, including bisons, and can cause immune dysfunction and result in asymptomatic infections and seroconversion, including fatal mucosal disease (Hause et al., 2021).
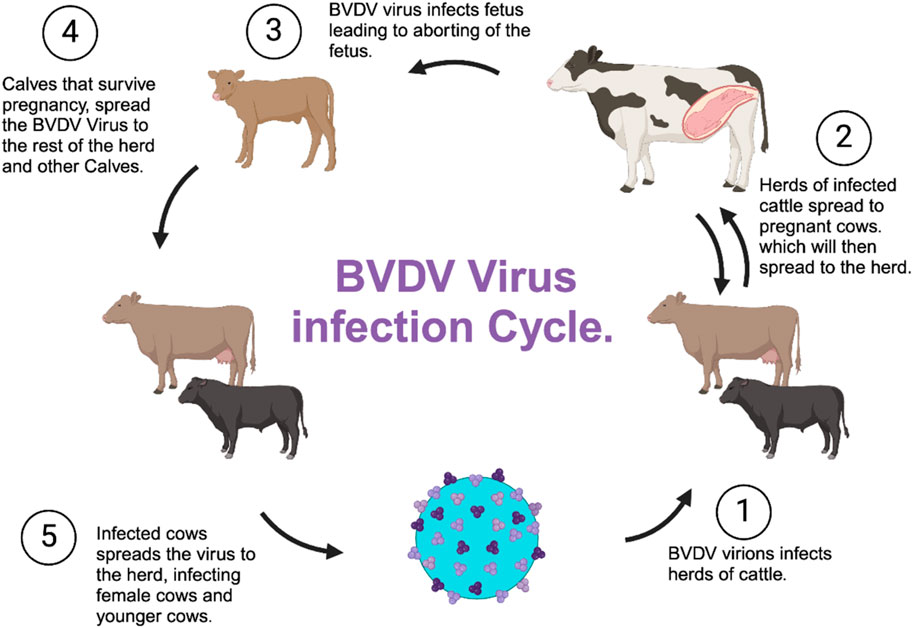
Figure 4. Transmission of the BVDV among a herd of cattle (Generated using BioRender).
Diseases associated with BVDV can range from clinically inappropriate to severe, even with the availability of vaccines (Xue et al., 2009). Acute and persistent BVDV infections among pregnant cows are often accompanied by transmission into the fetuses, resulting in abortions, teratogenic changes, or delivery of persistently infected, immunotolerant calves, depending on the gestation period (Kosinova et al., 2007). In the transmission process, if a cow is pregnant and is infected with the virus, the virus is transmitted to the fetus (Khodekaram-Tafti and Farjanikish, 2017). The virus has the ability to cause transplacental infection, resulting in different outcomes depending on the stage, which includes fetal death, malformation, acute syndromes of the neonate, immune tolerance, and lifelong viral persistence (Peterhans et al., 2003).
In the 90s, two small RNAs were discovered in Caenorhabditis elegans (C. elegans); it was later identified that the longer RNA, about 70 nucleotides, was the precursor of shorter RNAs that were about 22 nucleotides, which were classified as microRNAs (miRNAs) due to their short length (Ardekani and Naeini, 2010). miRNAs are small non-coding segments of RNAs that, unlike mRNAs, which encodes proteins, control various levels of important roles such as animal and human growth regulation, development, gene expression, cell proliferation, apoptosis, and even serves as an initiator for protein synthesis (Ardekani and Naeini, 2010; Ranganathan and Sivasankar, 2014; Finnegan and Pasquinelli, 2013; Fu et al., 2013). Most miRNAs are transcribed from DNA sequences into primary miRNAs (pri-miRNAs), then processed into precursor miRNAs (pre-miRNAs), and then into mature miRNAs (Ha and Kim, 2014; O'Brien et al., 2018).
There are three distinct types of miRNAs: small interference RNA (siRNA), RNA interferences (RNAi), and miRNAs (Qian et al., 2022). These molecules not only regulate gene expression or growth and development but can also suppress viral replication by targeting specific genes, resulting in inhibiting viral growth in its host. In the process of suppressing viral replication, mature miRNAs bind to complementary sequences on the 3′ end of the target mRNAs (Skalsky ans Cullen, 2010). Perfect complementarity miRNAs generally lead to potential cleavage of the mRNA genome, while imperfect complementarity results in repression and destabilization or degradation (Skalsky and Cullen, 2010; Baek et al., 2008; Selbach et al., 2008). In this study, we utilize advance bioinformatics tools to identify a possible complementary miRNA sequence to the nucleocapsid (NP) and/or capsid (CP) gene sequences of the flavivirus family.
Methods
Collection of Flavivirus genome sequences from NCBI
Complementary alignments were carried out using viral genome sequences that are responsible for the nucleocapsid and capsid protein synthesis of BVDV and all flaviviruses and were obtained from the National Center for Biotechnology Information (NCBI) database (Table 1). Figure 5 shows the flowchart of the computational analysis and multiple sequence alignments using miRBase, NCBI, and Basic Local Alignment Search Tool–nucleotides (BLAST-n).

Table 1. Name of the virus genome, with the accession number, name of genome, and the sequence that were retrieved from the NCBI database.
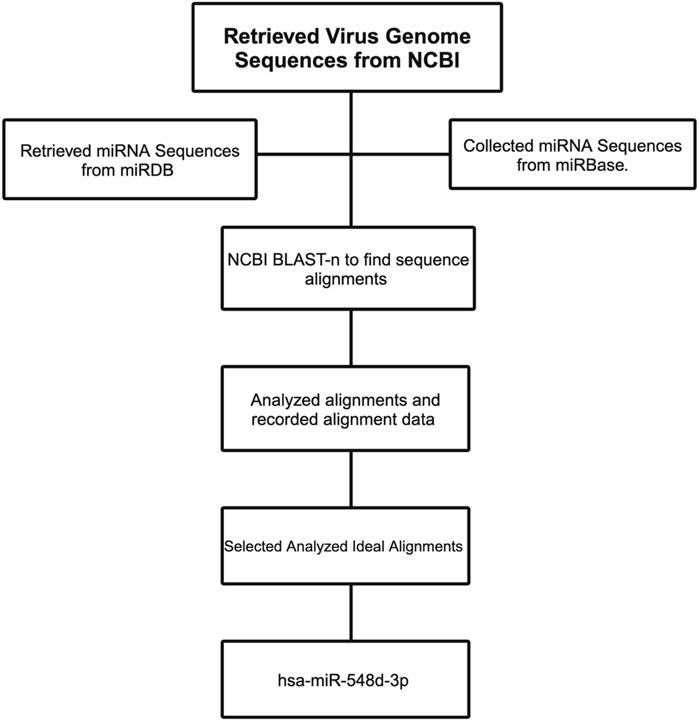
Figure 5. Flowchart demonstrating the workflow to select the right miRNA candidate against the flavivirus genome.
Collection of miRNAs from miRBase and sequence alignments
Figures 8, 6 show the method of predicting the right miRNA sequence using miRBase (https://mirbase.org/) and NCBI BLAST–n) (https://blast.ncbi.nlm.nih.gov/Blast.cgi) (https://blast.ncbi.nlm.nih.gov/Blast.cgi?PROGRAM=blastn&PAGE_TYPE=BlastSearch&LINK_LOC=blasthome). For launching alignments, the miRNA sequences were entered in the Query section of BLAST and the viral mRNA sequence was entered in the Subject section.

Figure 6. (A) Webpage of NCBI BLASTn when finding miRNA–mRNA sequence alignments. (B) Method of using BLASTn. The figure demonstrates that the miRNA sequences were entered into the query box and the viral mRNA genome was entered into the subject box.
Results
After running series of alignments, our results revealed that hsa-miR-548d-3p (MI0003668) showed complementary sequence structures with the viral genome sequences that are responsible for the nucleocapsid gene of the BVDV and flavivirus capsid protein synthesis. Table 2 shows the details about miR-548d-3p, which includes, name, species, accession number, tissues, sequence, and website. According to BLAST-n, hsa-miR-548d-3p exhibited 100% similarities and showed the highest numbers of alignment positions on the YFV capsid gene (7 locations), as compared to the BVDV NP gene and ZIKV CP gene (Figure 7; 5 locations), and 4 alignment positions on the WNV capsid gene. Figures 8A, B shows the number of alignment positions of hsa-miR-548d-3p on our virus' genome. Table 3 shows all alignment data exhibited by hsa-miR-548d-3p on viral genomes of DENV (Figures 9), HCV, and the other flavivirus members, and Figures 10–16 show the number alignment locations of hsa-miR-548d-3p on the flavivirus family's genome.
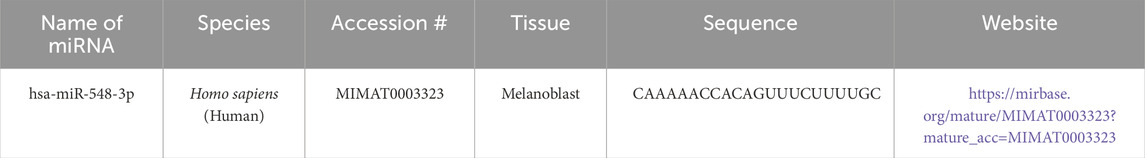
Table 2. Details about the hsa-miR-548-3p sequence.

Figure 7. Complete genome sequence of Zika virus retrieved from NCBI GenBank with accession # KX443145.1 with nucleotides 1 through 10,741. The highlighted area indicates the capsid gene at locations 107–418 nucleotides.
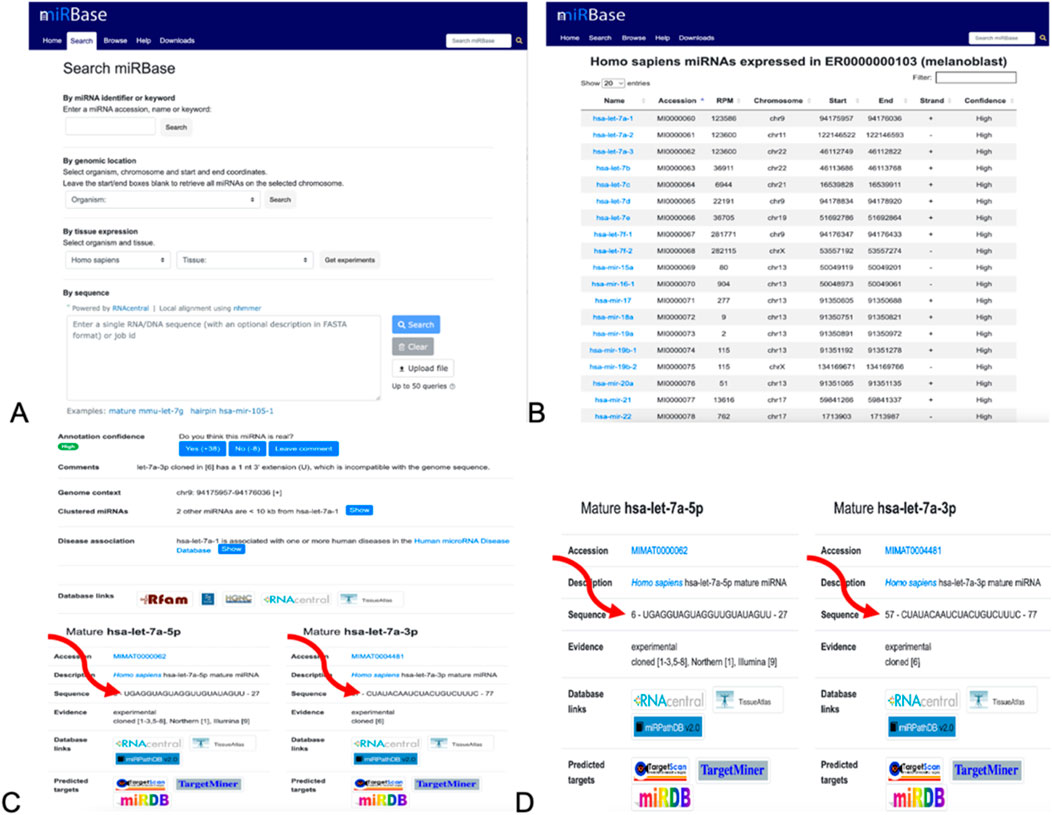
Figure 8. Method of collecting the miRNA sequences from miRBase. (A) Homepage. The organism was switched to Homo sapiens, exploring the tissues and studying the experiment. (B) List of miRNA sequences after performing experiments after selecting the organisms and tissues. (C) Result page after performing experiments, where the sequences of the miRNA are indicated by the arrows. (D) Information about the mature miRNAs and its sequences. It is these sequences that used using BLASTn.
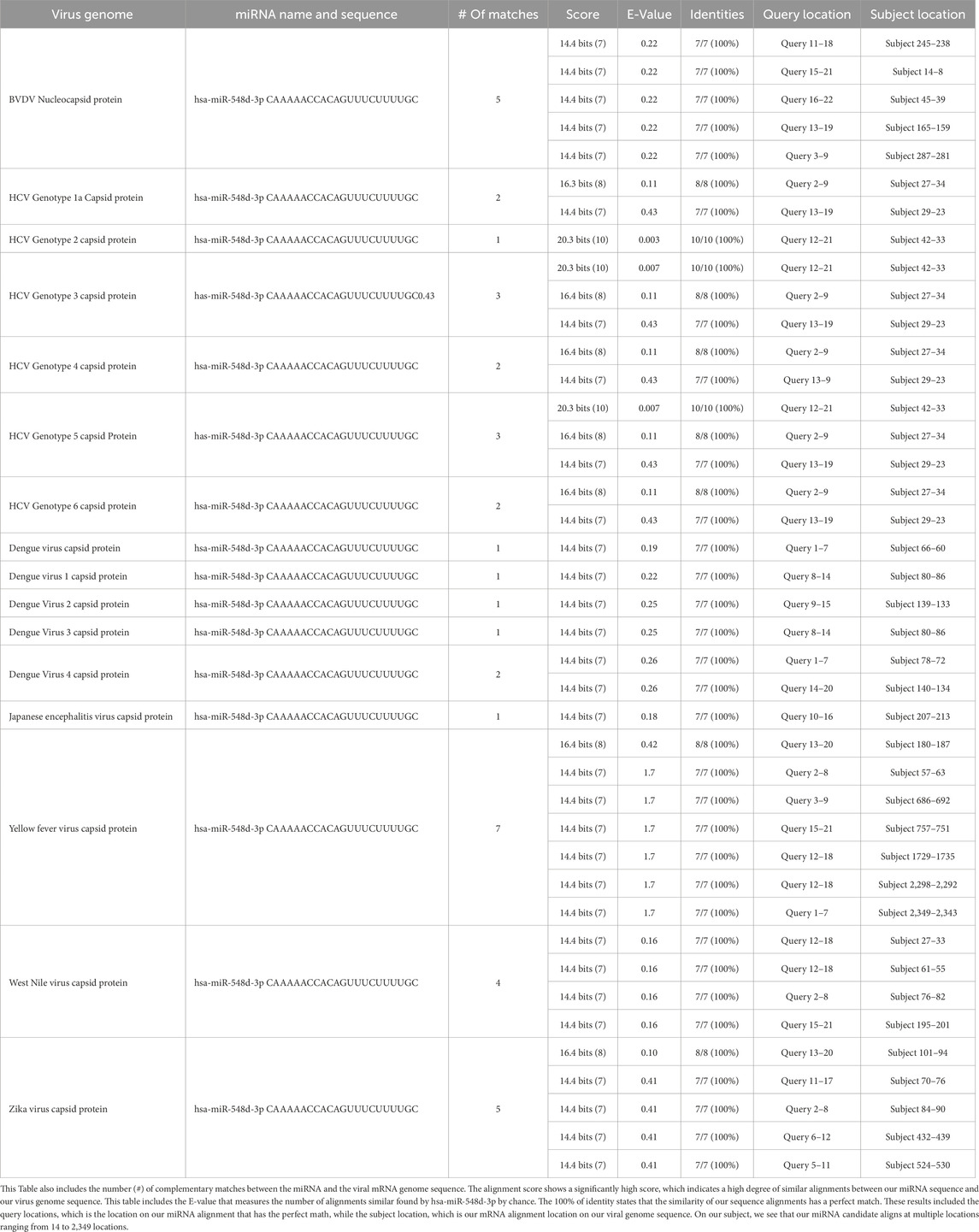
Table 3. All the alignment data on hsa-miR-548-3p to the capsid genome of all members of the flaviviruses retrieved from NCBI BLASTn, including the name of the genome and the name of the miRNA hsa-miR-548d-3p.

Figure 9. Complete genome sequence of dengue virus retrieved from NCBI GenBank with accession KY346993.1 with nucleotides 1 through 10,681. The highlighted area indicates the capsid gene at locations 95–384 nucleotides.
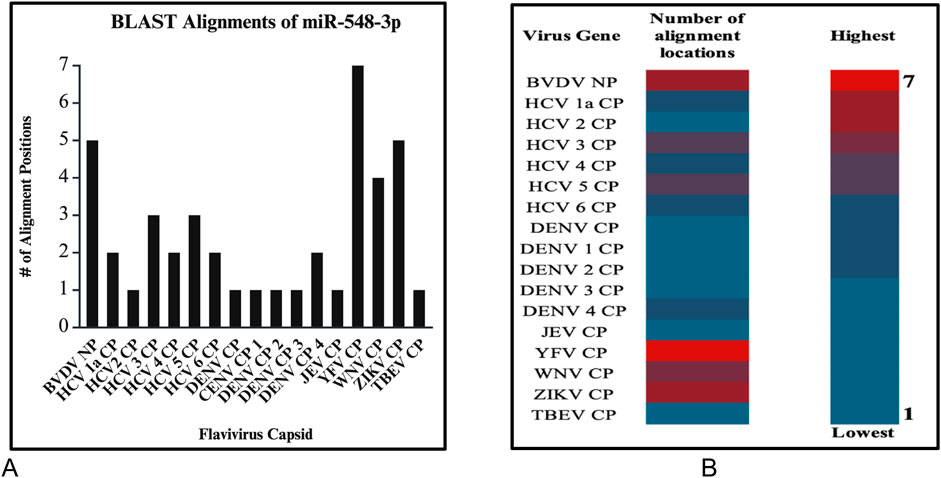
Figure 10. (A) Graphs demonstrate the number of perfect alignment position of hsa-miR-548d-3p on the flavivirus capsid genome. The y-axis is the number of alignment positions, and the x-axis represents the group of viruses in the flavivirus group. (B) Heatmap that shows the highest number of predicted miRNA alignment locations (alignment hits) ranging from one to seven locations along the capsid genome of all members of the flaviviruses.
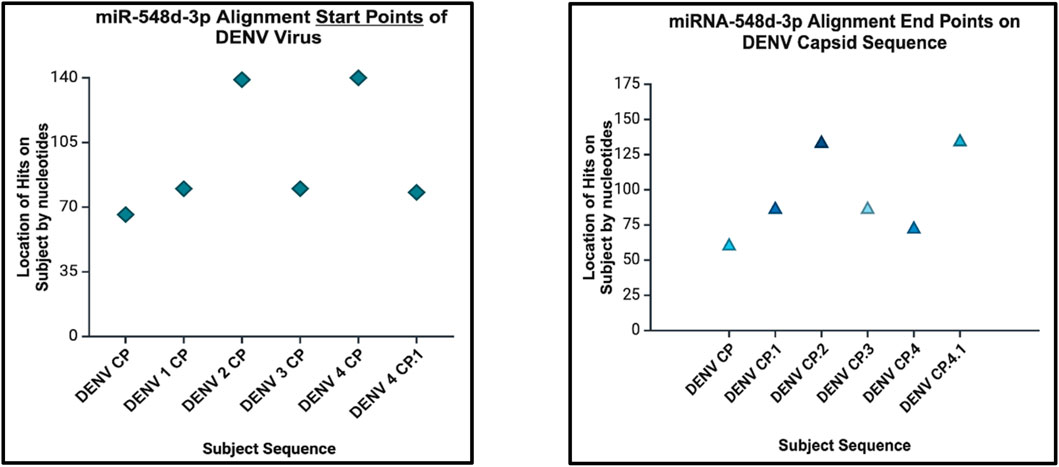
Figure 11. Dot plot that demonstrates the alignment start point (left) and end points (right) of miR-548d-3p on the DENV genome responsible for the capsid protein.
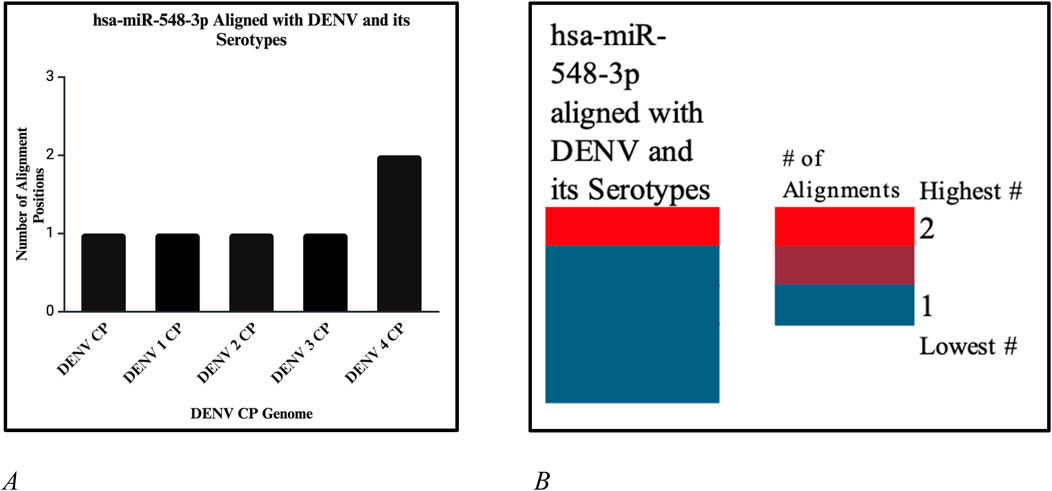
Figure 12. (A) Number of alignment positions that hsa-miR-548d-3p reveals on the capsid genome of DENV and its serotypes. On DENV and DENV 1–3, hsa-miR-548d-3p has only 1 alignment position, as compared to DENV 4, where hsa-miR-548d-3p has two alignment locations on the capsid genome. (B) Heatmap that shows the highest number of alignment positions between hsa-miR-548d-3p and the capsid genome sequence of the DENV virus and serotypes.
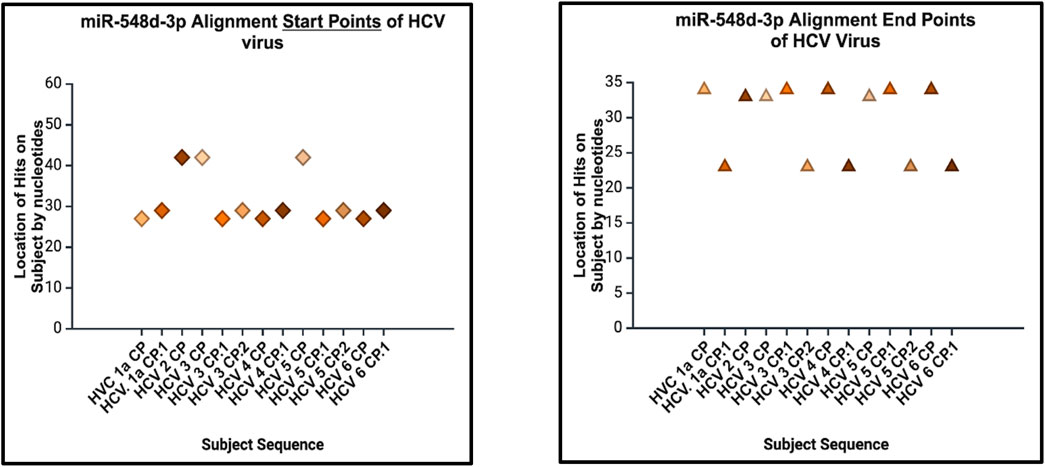
Figure 13. Sequence alignment start points (left) and end points (right) of hsa-miR-548d-3p on the capsid genome of HCV and its genotypes. The location of alignments is indicated on the y-axis, and the viral HCV genome sequences for the capsid protein are indicated on the x-axis. Figure 13 shows that miR-548d-3p has identical matches on identical locations on the genome sequences responsible for the capsid gene on all genotypes of HCV.
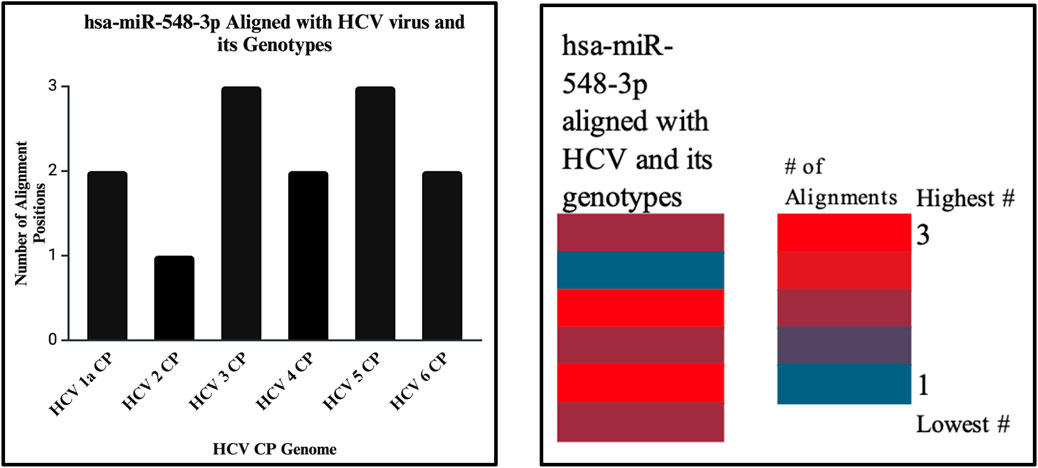
Figure 14. The bar graph (right) shows the highest and lowest number of alignments demonstrated between our miRNA and the genome of the HCV virus. The heatmap (left) shows that the highest number is color coded in red, while the lowest is encoded in blue. In this analysis, both show that the highest number of alignment locations on the HCV virus capsid sequence is 3.
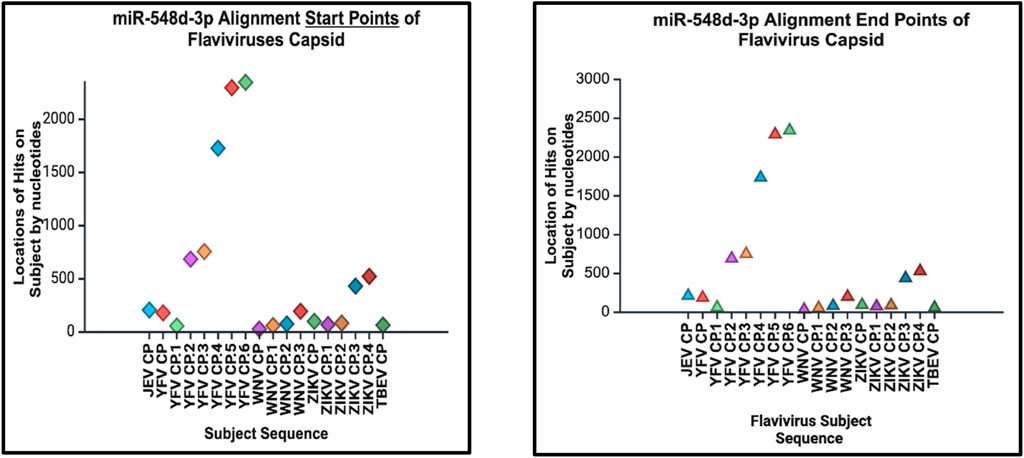
Figure 15. Alignment start points (right) and end points (left) of our candidate miRNA. The location of alignment hit points is indicated on the y–axis, and the viral Flavivirus genome sequences for the capsid protein are indicated on the x–axis. Start points are indicated by diamonds, and the end points are indicated by triangles.
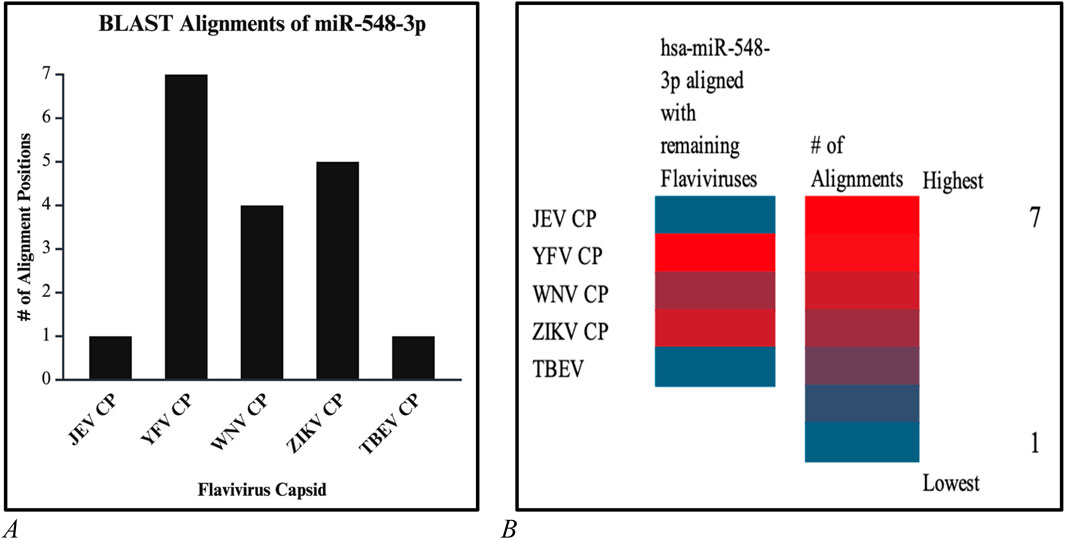
Figure 16. (A) Bar graph and (B) and heatmap chart that depicted the number of alignment positions that the candidate miRNA displays. Both reveal that the highest number of alignments is 7 (red) and the lowest number of alignments is 1 (blue).
hsa-miR-548d-3p aligned with dengue virus
Our results showed that the hsa-miR-548d-3p sequence is identical to the capsid sequence of dengue virus (DENV) and its four serotypes (Figures 9, 10; Table 1). We found that miR-548-3p has a 100% perfect match on DENV virus genome sequences, as displayed in Table 3. Figures 12A, B show that out of the four serotypes of DENV, 1 out of 5 (20%) showed 2 alignment locations and aligns at 2 nucleotides on the DENV 4 genome at subject locations 78–72 and 140–134.
hsa-miR-548d-3p alignment with HCV
The alignment analysis also revealed multiple sequence alignments between hsa-miR-548d-3p and the capsid sequence of the HCV virus and its genotypes. Figure 15 shows that out of the six genotypes of HCV, the highest number of alignment locations is observed between our miRNA and HCV genotype 3 and genotype 5 (33.33%) and the second highest number of locations is observed on genotype 1, genotype 4, and genotype 6 (50%).
hsa-miR-548d-3p aligned with Flavivirus capsid gene
Additional alignments were carried out, and it showcased that miR-548d-3p harbors sequence similarities with the capsid genome of the remaining Flavivirus members [ZIKV (Figure 7), YFV, WNV, and TBEV]. Figure 15 shows that our candidate miRNA shows similarities on various sections on the flavivirus capsid genome, and Figures 16A, B demonstrate that hsa-miR-548d-3p has the highest number of alignments on the YFV capsid genome (7 alignment locations), 4 alignment positions on the WNV capsid genome, and 5 alignment positions on the ZIKV capsid genome, depicted by this sequence alignment analysis.
Discussion
miRNAs are a class of small non-coding RNA segments, ranging up to 22 nt long, and serve as possible inhibiting regulators against viral mRNA expression during virus replication (Hasan et al., 2014). A perfect complementary sequence between miRNA and mRNA regions is believed to be sufficient for successful cleavage or degradation of the mRNA sequence, but imperfect alignments may block viral translation (Casal et al., 2004; Hasan et al., 2014). We studied the sequence homology of our miRNA sequence and the flavivirus genome sequences, and we found that after numerous sequence alignments, this study confirms the significant complementary sequence of our candidate miRNA sequence on the flavivirus capsid genome. After careful analysis, we have analyzed that hsa-miR-548d-3p showed identical alignment locations on the capsid gene of DENV 1, 3, and 4 viruses with some minor differences (Figure 11; Table 3). Figure 13 also confirms that hsa-miR-548d-3p also has identical alignment locations on the HCV virus and its genotypes. Hence, those miRNAs are used as antiviral therapeutics; these findings suggest that hsa-miR-548d-3p may be a possible candidate as a universal antiviral therapeutic agent against infections caused by the flavivirus family.
Alignment data of NCBI BLAST-n
To understand the idea of a good alignment between two sequences, it is necessary to understand the score, E-value (expected value), percentage of identity, and gaps. The bits score indicates how significant the alignment is; the higher the score on the alignment, the better. Observing the expected or, simply, the E-value indicates the significance of an alignment; the lower the E-value signifies, the better the alignment between two sequences. According to Tom Madden, if an alignment has an E-value of 0.05, then the similarities have a 5 in 100 possibility of occurring by chance (Madden, 2013). The percentage (%) of identity signifies how perfect the alignment is between two sequences. Table 3 shows that all the alignments demonstrated are 7/7 or 8/8, which indicates two sequences are 100% similar. According to Fassler and Coopet (2008), the percentage of identity of 100% means that the nucleotides of the subject sequence are identical to the reference sequence or the query at every position of the alignment.
Using miRBase for the prediction of miRNA targets
Many bioinformatics tools were developed for biogenesis and to help biologists investigate miRNA biology. Among these tools, miRBase (https://mirbase.org/) is the most widely used software program (Chen, L., et al., 2019), which was developed in the year 2002 (Chen, L., et al., 2019). Later, the name changed from “the microRNA Registry” provided molecular researchers with stable and unique gene names for their novel miRNA discoveries and storages of miRNA sequences (Kozomara and Griffiths-Jones, 2010). miRBase is a primary repository database for retrieving data on miRNA and has three main functions: 1) provides confidential services for independent assignment of miRNA genes, 2) sequences provide miRNA data, annotation, references, and links to other published miRNAs, and 3) provides miRNA target pipelines for the prediction of the target (Griffiths-Jones et al., 2007; Griffiths-Jones, 2006). miRBase also allows searching published pre-miRNA and mature miRNA sequences, in addition to readily available annotation and sequence data that are available for download (Luna Buitrago et al., 2023). Overall, miRBase provides scientists a variety of data on miRNAs when obtaining sequences that include the accession number, symbols, description, and gene family.
Using miRBase and BLASTn
One of the important function of miRbase is to provide a microRNA target pipeline for the prediction of targets for all published animal miRNAs (Griffiths-Jones et al., 2007; Griffiths-Jones, 2006), and all miRNA sequences from this database revealed interactions against 3′-untranslated regions, which are predicted from all available species (Griffiths-Jones, 2006).
We used BLAST to perform alignments for its potential to detect similar regions within parts of long sequences. It is a fast, sensitive, and accurate tool for analyzing sequences for alignments (Altschul, et al., 1990; Lobo, 2008). The reason that miRBase does not run alignment analysis was BLAST was used when searching for the right miRNA sequences over 2,600 miRNAs. Additionally, BLAST was used because it is the most widely used software package in bioinformatics research due to its main function of comparing sequence(s) of interest (Stover and Cavalcanti, 2017).
Roles of hsa-miR-548d-3p in humans
hsa-miR-548d-3p is a mature miRNA that is found in primates and comprises over 69 identified miRNAs that are presented in all human chromosomes, but also as a more poorly conserved miRNA (Ramos-Sanchez et al., 2022), it demonstrates to enhance cell proliferation and inhibit apoptosis in breast cancer (Souza et al., 2016; Souza et al., 2021). Functions can include many biological processes, such as signaling pathways like MAPK, phosphatidylinositol (P13K), p53, B-cell receptor, T-cell receptor, TGF-beta, PPAR, calcium, and insulin signaling pathways, and in human tumorigenesis, such as colorectal cancer, glioma, and non-small cell lung cancer (Ramox-Sanchez, 2022; Liang et al., 2012). hsa-miR-548d-3p is proven to be involved in homeostasis of stress damage, and metabolic and survival pathways for cell proliferation (Cannataro and Cione, 2019; Maiorino et al., 2015). In an experiment done by Rooda L. et al., their results indicated that hsa-miR-548d-3p and its family may play additional roles in humans, such as in ovarian follicle activation, development, granulosa cell differentiation, and proliferation (Rooda et al., 2021).
Bovine viral diarrhea virus as a model for flaviviruses
Prestiviruses are more closely related to HCV than the classical flaviviruses and have been used as surrogate models for HCV (Tellinghuisen et al., 2006; Lackner et al., 2004) to test in vitro infectivity (Durantel et al., 2004). According to Chen et. al. (2022), as one of the most characterized members of the Flaviviridae family, BVDV serves as a good model system to study flaviviruses and has primarily been used as a surrogate model for HCV in identification and characterization of antiviral agents (Finkielsztein et al., 2010). This approach leverages the similarities between BVDV and HCV to develop and test potential treatments for HCV more effectively. Lai et al. (2000) stated that both viruses BVDV and HCV utilize the IRES within the 5′ Untranslated Region (UTR) necessary for translation of viral polyprotein, while NS3 proteases of both viruses require NS4A as a cofactor for polyprotein processing.
Limitations
This was a pure computer-based study using bioinformatic tools to showcase possible miRNA–mRNA sequence similarities. Due to pestiviruses like the BVDV, which is used as a surrogate model for studying HCV virus, we hypothesized that if we can utilize the BVDV genome sequence as our test subject, then we could find a possible universal miRNA-based antiviral therapeutic for the family of flaviviruses. Based on our results and Table 3 and Figures 10–16, we found hsa-miR-548d-3p as a possible candidate due to its perfect match with the genome of all our viruses rather than just one. Again, this is a full bioinformatic-based analytical study, where in vivo lab equipment was not used. Hence, the results are not considered final until proved using in vivo experimentation.
Conclusion
After performing a series of sequence alignments, we predicted hsa-miR-548d-3p, a mature miRNA sequence, as a potential candidate to target flaviviruses showing perfect alignments with BVDV; HCV genotype 1a, 2, 3, 4, 5, and 6; DENV serotype 1, 2, 3, and 4; JENV; WNV; ZIKA; and TBEV. Although more detailed in vitro and in vivo studies are required to utilize hsa-miR-548d-3p as an antiviral therapeutic, this study may be considered a first step to develop a new type of miRNA treatment against a range of viruses within the Flaviviridae family. This study also recognizes that the BVDV may not be the surrogate model for only HCV virus but can also prove to be a good model system for antiviral therapeutic studies against other members of the Flaviviridae family.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/supplementary material.
Author contributions
HC: writing–original draft and writing–review and editing. SH: writing–original draft and writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. The authors gratefully acknowledge the moral support from School of STEM, Diné College, and financial support from MS Biology Grant # NSF-TCUP-ICE-TI-2202023 (PI: Donald K. Robinson).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215 (3), 403–410. doi:10.1006/jmbi.1990.9999
Ardekani, A. M., and Naeini, M. M. (2010). The role of MicroRNAs in human diseases. Avicenna J. Med. Biotechnol. 2 (4), 161–179.
Babiker, A., Jeudy, J., Kligerman, S., Khambaty, M., Shah, A., and Bagchi, S. (2017). Risk of cardiovascular disease due to chronic hepatitis C infection: a review. J. Clin. Transl. hepatology 5 (4), 1–20. doi:10.14218/jcth.2017.00021
Baek, D., Villén, J., Shin, C., Camargo, F. D., Gygi, S. P., and Bartel, D. P. (2008). The impact of microRNAs on protein output. Nature 455 (7209), 64–71. doi:10.1038/nature07242
Barrows, N. J., Campos, R. K., Liao, K. C., Prasanth, K. R., Soto-Acosta, R., Yeh, S. C., et al. (2018). Biochemistry and molecular biology of flaviviruses. Chem. Rev. 118 (8), 4448–4482. doi:10.1021/acs.chemrev.7b00719
Basarab, M., Bowman, C., Aarons, E. J., and Cropley, I. (2016). Zika virus. Bmj 352, i1049. doi:10.1136/bmj.i1049
Becher, P., Orlich, M., and Thiel, H. J. (1998). Complete genomic sequence of border disease virus, a pestivirus from sheep. J. virology 72 (6), 5165–5173. doi:10.1128/jvi.72.6.5165-5173.1998
Benzarti, E., Linden, A., Desmecht, D., and Garigliany, M. (2019). Mosquito-borne epornitic flaviviruses: an update and review. J. General Virology 100 (2), 119–132. doi:10.1099/jgv.0.001203
Cannataro, R., and Cione, E. (2019). Antioxidant and microRNAs: an applied overview. J. Microbiol. Biotechnol. Rep.
Casal, M. L., Dambach, D. M., Meister, T., Jezyk, P. F., Patterson, D. F., and Henthorn, P. S. (2004). Familial glomerulonephropathy in the bullmastiff. Veterinary pathol. 41 (4), 319–325. doi:10.1354/vp.41-4-319
Chen, L., Heikkinen, L., Wang, C., Yang, Y., Sun, H., and Wong, G. (2019). Trends in the development of miRNA bioinformatics tools. Briefings Bioinforma. 20 (5), 1836–1852. doi:10.1093/bib/bby054
Chen, N., Liu, Y., Bai, T., Chen, J., Zhao, Z., Li, J., et al. (2022). Quercetin inhibits Hsp70 blocking of bovine viral diarrhea virus infection and replication in the early stage of virus infection. Viruses 14 (11), 2365. doi:10.3390/v14112365
Chi, S., Chen, S., Jia, W., He, Y., Ren, L., and Wang, X. (2022). Non-structural proteins of bovine viral diarrhea virus. Virus genes 58 (6), 491–500. doi:10.1007/s11262-022-01914-8
Conde, J. N., Silva, E. M., Barbosa, A. S., and Mohana-Borges, R. (2017). The complement system in flavivirus infections. Front. Microbiol. 8, 213. doi:10.3389/fmicb.2017.00213
de Oliveira Figueiredo, P., Stoffella-Dutra, A. G., Barbosa Costa, G., Silva de Oliveira, J., Dourado Amaral, C., Duarte Santos, J., et al. (2020). Re-emergence of yellow fever in Brazil during 2016-2019: challenges, lessons learned, and perspectives. Viruses 12 (11), 1233. doi:10.3390/v12111233
Duncan, J. D., Urbanowicz, R. A., Tarr, A. W., and Ball, J. K. (2020). Hepatitis C virus vaccine: challenges and prospects. Vaccines 8 (1), 90. doi:10.3390/vaccines8010090
Durantel, D., Carrouée-Durantel, S., Branza-Nichita, N., Dwek, R. A., and Zitzmann, N. (2004). Effects of interferon, ribavirin, and iminosugar derivatives on cells persistently infected with noncytopathic bovine viral diarrhea virus. Antimicrob. agents Chemother. 48 (2), 497–504. doi:10.1128/aac.48.2.497-504.2004
Fassler, J., and Cooper, P. (2008). “BLAST glossary. 2011 jul 14,” in BLAST® help (Bethesda (MD): National Center for Biotechnology Information US). Available at: https://www.ncbi.nlm.nih.gov/books/NBK62051/.
Finkielsztein, L. M., Moltrasio, G., Caputto, M., Castro, E., Cavallaro, L., and Moglioni, A. (2010). What is known about the antiviral agents active against bovine viral diarrhea virus (BVDV)? Curr. Med. Chem. 17 (26), 2933–2955. doi:10.2174/092986710792065036
Finnegan, E. F., and Pasquinelli, A. E. (2013). MicroRNA biogenesis: regulating the regulators. Crit. Rev. Biochem. Mol. Biol. 48 (1), 51–68. doi:10.3109/10409238.2012.738643
Fu, G., Brkić, J., Hayder, H., and Peng, C. (2013). MicroRNAs in human placental development and pregnancy complications. Int. J. Mol. Sci. 14 (3), 5519–5544. doi:10.3390/ijms14035519
Fulton, R. W., Purdy, C. W., Confer, A. W., Saliki, J. T., Loan, R. W., Briggs, R. E., et al. (2000). Bovine viral diarrhea viral infections in feeder calves with respiratory disease: interactions with Pasteurella spp., parainfluenza-3 virus, and bovine respiratory syncytial virus. Can. J. veterinary Res. = Revue Can. de recherche veterinaire 64 (3), 151–159.
Giménez-Richarte, Á., Ortiz de Salazar, M. I., Giménez-Richarte, M. P., Collado, M., Fernández, P. L., Clavijo, C., et al. (2022). Transfusion-transmitted arboviruses: update and systematic review. PLOS Neglected Trop. Dis. 16 (10), e0010843. doi:10.1371/journal.pntd.0010843
Griffiths-Jones, S., et al. (2006). miRBase: microRNA sequences, targets and gene nomenclature. Nucleic acids Res. 34 (Database issue), D140–D144. doi:10.1093/nar/gkj112
Griffiths-Jones, S., Saini, H. K., van Dongen, S., and Enright, A. J. (2007). miRBase: tools for microRNA genomics. Nucleic acids Res. 36 (Suppl. l_1), D154–D158. doi:10.1093/nar/gkm952
Ha, M., and Kim, V. N. (2014). Regulation of microRNA biogenesis. Nat. Rev. Mol. cell Biol. 15 (8), 509–524. doi:10.1038/nrm3838
Hamel, R., Dejarnac, O., Wichit, S., Ekchariyawat, P., Neyret, A., Luplertlop, N., et al. (2015). Biology of Zika virus infection in human skin cells. J. virology 89 (17), 8880–8896. doi:10.1128/jvi.00354-15
Harada, T., Tautz, N., and Thiel, H. J. (2000). E2-p7 region of the bovine viral diarrhea virus polyprotein: processing and functional studies. J. virology 74 (20), 9498–9506. doi:10.1128/jvi.74.20.9498-9506.2000
Hasan, M. M., Akter, R., Ullah, M. S., Abedin, M. J., Ullah, G. M. A., and Hossain, M. Z. (2014). A computational approach for predicting role of human microRNAs in MERS-CoV genome. Adv. Bioinforma. 2014 (1), 1–8. doi:10.1155/2014/967946
Hills, S. L., Fischer, M., and Petersen, L. R. (2017). Epidemiology of Zika virus infection. J. Infect. Dis. 216 (Suppl. l_10), S868–S874. doi:10.1093/infdis/jix434
Hu, T., Wu, Z., Wu, S., Chen, S., and Cheng, A. (2021). The key amino acids of E protein involved in early flavivirus infection: viral entry. Virology J. 18 (1), 136. doi:10.1186/s12985-021-01611-2
Hause, B. M, Pillatzki, A., Clement, T., Bragg, T., Ridpath, J., and Chase, C. C. L. (2021). Persistent infection of American bison (Bison bison) with bovine viral diarrhea virus and bosavirus. Vet. Microbiol. 252, 108949. doi:10.1016/j.vetmic.2020.108949
Isken, O., Baroth, M., Grassmann, C. W., Weinlich, S., Ostareck, D. H., Ostareck-Lederer, A., et al. (2007). Nuclear factors are involved in hepatitis C virus RNA replication. Rna 13 (10), 1675–1692. doi:10.1261/rna.594207
Jackova, A., Novackova, M., Pelletier, C., Audeval, C., Gueneau, E., Haffar, A., et al. (2008). The extended genetic diversity of BVDV-1: typing of BVDV isolates from France. Veterinary Res. Commun. 32 (1), 7–11. doi:10.1007/s11259-007-9012-z
Julander, J. G., Shafer, K., Smee, D. F., Morrey, J. D., and Furuta, Y. (2009). Activity of T-705 in a hamster model of yellow fever virus infection in comparison with that of a chemically related compound, T-1106. Antimicrob. agents Chemother. 53 (1), 202–209. doi:10.1128/aac.01074-08
Junglen, S., Kopp, A., Kurth, A., Pauli, G., Ellerbrok, H., and Leendertz, F. H. (2009). A new flavivirus and a new vector: characterization of a novel flavivirus isolated from uranotaenia mosquitoes from a tropical rain forest. J. virology 83 (9), 4462–4468. doi:10.1128/jvi.00014-09
Khodakaram-Tafti, A., and Farjanikish, G. H. (2017). Persistent bovine viral diarrhea virus (BVDV) infection in cattle herds. Iran. J. veterinary Res. 18 (3), 154–163.
Kosinova, E., Psikal, I., Robesova, B., and Kovarcik, K. (2007). Real-time PCR for quantitation of bovine viral diarrhea virus RNA using SYBR Green I fluorimetry. VETERINARNI MEDICINA-PRAHA- 52 (6), 253–261. doi:10.17221/1882-vetmed
Kozomara, A., and Griffiths-Jones, S. (2010). miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic acids Res. 39 (Suppl. l_1), D152–D157. doi:10.1093/nar/gkq1027
Lackner, T., Müller, A., Pankraz, A., Becher, P., Thiel, H. J., Gorbalenya, A. E., et al. (2004). Temporal modulation of an auto protease is crucial for replication and pathogenicity of an RNA virus. J. virology 78 (19), 10765–10775. doi:10.1128/jvi.78.19.10765-10775.2004
Lai, V. C., Zhong, W., Skelton, A., Ingravallo, P., Vassilev, V., Donis, R. O., et al. (2000). Generation and characterization of a hepatitis C virus NS3 protease-dependent bovine viral diarrhea virus. J. virology 74 (14), 6339–6347. doi:10.1128/jvi.74.14.6339-6347.2000
Laureti, M., Narayanan, D., Rodriguez-Andres, J., Fazakerley, J. K., and Kedzierski, L. (2018). Flavivirus receptors: diversity, identity, and cell entry. Front. Immunol. 9, 2180. doi:10.3389/fimmu.2018.02180
Li, F., Wang, Y., Yu, L., Cao, S., Wang, K., Yuan, J., et al. (2015). Viral infection of the central nervous system and neuroinflammation precede blood-brain barrier disruption during Japanese encephalitis virus infection. J. virology 89 (10), 5602–5614. doi:10.1128/jvi.00143-15
Li, Y., Wang, J., Kanai, R., and Modis, Y. (2013). Crystal structure of glycoprotein E2 from bovine viral diarrhea virus. Proc. Natl. Acad. Sci. 110 (17), 6805–6810. doi:10.1073/pnas.1300524110
Liang, T., Guo, L., and Liu, C. (2012). Genome-wide analysis of mir-548 gene family reveals evolutionary and functional implications. BioMed Res. Int. 2012 (1), 1–8. doi:10.1155/2012/679563
Luna Buitrago, D., Lovering, R. C., and Caporali, A. (2023). Insights into online microRNA bioinformatics tools. Non-coding RNA 9 (2), 18. doi:10.3390/ncrna9020018
Maiorino, M., Bosello-Travain, V., Cozza, G., Miotto, G., Roveri, A., Toppo, S., et al. (2015). Understanding mammalian glutathione peroxidase 7 in the light of its homologs. Free Radic. Biol. Med. 83, 352–360. doi:10.1016/j.freeradbiomed.2015.02.017
Mari, V. L., Losurdo, M., Lucente, M. S., Lorusso, E., Elia, G., Martella, V., et al. (2016). Multiplex real-time RT-PCR assay for bovine viral diarrhea virus type 1, type 2 and HoBi-like pestivirus. J. virological methods 229, 1–7. doi:10.1016/j.jviromet.2015.12.003
Maurer, K., Krey, T., Moennig, V., Thiel, H. J., and Rümenapf, T. (2004). CD46 is a cellular receptor for bovine viral diarrhea virus. J. virology 78 (4), 1792–1799. doi:10.1128/jvi.78.4.1792-1799.2004
Merwaiss, F., Czibener, C., and Alvarez, D. E. (2019). Cell-to-cell transmission is the main mechanism supporting bovine viral diarrhea virus spread in cell culture. J. Virology 93 (3), 017766-18–e11128. doi:10.1128/jvi.01776-18
Murray, C. L., Marcotrigiano, J., and Rice, C. M. (2008). Bovine viral diarrhea virus core is an intrinsically disordered protein that binds RNA. J. virology 82 (3), 1294–1304. doi:10.1128/jvi.01815-07
Musso, D., and Gubler, D. J. (2016). Zika virus. Clin. Microbiol. Rev. 29 (3), 487–524. doi:10.1128/cmr.00072-15
Musso, D., and Nhan, T. X. (2015). Emergence of Zika virus. Clin. Microbiol. 4, 222. doi:10.4172/2327-5073.1000222
Mutebi, J. P., Rijnbrand, R. C. A., Wang, H., Ryman, K. D., Wang, E., Fulop, L. D., et al. (2004). Genetic relationships and evolution of genotypes of yellow fever virus and other members of the yellow fever virus group within the Flavivirus genus based on the 3′ noncoding region. J. virology 78 (18), 9652–9665. doi:10.1128/jvi.78.18.9652-9665.2004
Neill, J. D. (2013). Molecular biology of bovine viral diarrhea virus. Biologicals 41 (1), 2–7. doi:10.1016/j.biologicals.2012.07.002
Nijmeijer, B. M., Koopsen, J., Schinkel, J., Prins, M., and Geijtenbeek, T. B. (2019). Sexually transmitted hepatitis C virus infections: current trends, and recent advances in understanding the spread in men who have sex with men. J. Int. AIDS Soc. 22 (l), e25348. doi:10.1002/jia2.25348
Niskanen, R., Lindberg, A., Larsson, B., and Alenius, S. (2000). Lack of virus transmission from bovine viral diarrhoea virus infected calves to susceptible peers. Acta Veterinaria Scand. 41 (1), 93–99. doi:10.1186/bf03549659
O'Brien, J., Hayder, H., Zayed, Y., and Peng, C. (2018). Overview of microRNA biogenesis, mechanisms of actions, and circulation. Front. Endocrinol. 9, 402. doi:10.3389/fendo.2018.00402
Perera-Lecoin, M., Meertens, L., Carnec, X., and Amara, A. (2013). Flavivirus entry receptors: an update. Viruses 6 (1), 69–88. doi:10.3390/v6010069
Peterhans, E., Jungi, T. W., and Schweizer, M. (2003). BVDV and innate immunity. Biologicals 31 (2), 107–112. doi:10.1016/s1045-1056(03)00024-1
Pierson, T. C., and Diamond, M. S. (2020). The continued threat of emerging flaviviruses. Nat. Microbiol. 5 (6), 796–812. doi:10.1038/s41564-020-0714-0
Pierson, T. C., and Kielian, M. (2013). Flaviviruses: braking the entering. Curr. Opin. virology 3 (1), 3–12. doi:10.1016/j.coviro.2012.12.001
Qian, Q., Xu, R., and Wang, Y., (2022). “The NS4A protein of classical swine fever virus suppresses RNA silencing in mammalian cells.” J. Virology, 96(15), 018744-e1921. doi:10.1128/jvi.01874-21
Ramos-Sanchez, E. M., Reis, L. C., Souza, M. A., Muxel, S. M., Santos, K. R., Lagos, D., et al. (2022). miR-548d-3p is up-regulated in human visceral leishmaniasis and suppresses parasite growth in macrophages. Front. Cell. Infect. Microbiol. 12, 826039. doi:10.3389/fcimb.2022.826039
Ranganathan, K., and Sivasankar, V. (2014). MicroRNAs-Biology and clinical applications. J. Oral Maxillofac. Pathology 18 (2), 229–234. doi:10.4103/0973-029x.140762
Reed, K. E., Gorbalenya, A. E., and Rice, C. M. (1998). The NS5A/NS5 proteins of viruses from three genera of the family flaviviridae are phosphorylated by associated serine/threonine kinases. J. virology 72 (7), 6199–6206. doi:10.1128/jvi.72.7.6199-6206.1998
Rooda, I., Kaselt, B., Liivrand, M., Smolander, O. P., Salumets, A., and Velthut-Meikas, A. (2021). Hsa-mir-548 family expression in human reproductive tissues. BMC Genomic Data 22, 40–13. doi:10.1186/s12863-021-00997-w
Schweizer, M., and Peterhans, E. (2001). Noncytopathic bovine viral diarrhea virus inhibits double-stranded RNA-induced apoptosis and interferon synthesis. J. Virology 75 (10), 4692–4698. doi:10.1128/jvi.75.10.4692-4698.2001
Selbach, M., Schwanhäusser, B., Thierfelder, N., Fang, Z., Khanin, R., and Rajewsky, N. (2008). Widespread changes in protein synthesis induced by microRNAs. nature 455 (7209), 58–63. doi:10.1038/nature07228
Simmons, C. P., Farrar, J. J., van Vinh Chau, N., and Wills, B. (2012). Dengue. N. Engl. J. Med. 366 (15), 1423–1432. doi:10.1056/nejmra1110265
Skalsky, R. L., and Cullen, B. R. (2010). Viruses, microRNAs, and host interactions. Annu. Rev. Microbiol. 64, 123–141. doi:10.1146/annurev.micro.112408.134243
Song, Z. Q., Hao, F., Min, F., Ma, Q. Y., and Liu, G. D. (2001). Hepatitis C virus infection of human hepatoma cell line 7721 in vitro. World J. Gastroenterology 7 (5), 685. doi:10.3748/wjg.7.685
Souza, M. A., Almeida, T. M., Castro, M. C. A., Oliveira-Mendes, A. P., Almeida, A. F., Oliveira, B. C., et al. (2016). American tegumentary leishmaniasis: mRNA expression for Th1 and Treg mediators are predominant in patients with recent active disease. Immunobiology 221 (2), 253–259. doi:10.1016/j.imbio.2015.08.009
Souza, M. D. A., Ramos-Sanchez, E. M., Muxel, S. M., Lagos, D., Reis, L. C., Pereira, V. R. A., et al. (2021). miR-548d-3p alters parasite growth and inflammation in leishmania (Viannia) braziliensis infection. Front. Cell. Infect. Microbiol. 11, 687647. doi:10.3389/fcimb.2021.687647
Stover, N. A., and Cavalcanti, A. R. (2017). Using NCBI BLAST. Curr. Protoc. Essent. Lab. Tech. 14 (1), 11–1. doi:10.1002/cpet.8
Suzich, J. A., Tamura, J. K., Palmer-Hill, F., Warrener, P., Grakoui, A., Rice, C. M., et al. (1993). Hepatitis C virus NS3 protein polynucleotide-stimulated nucleoside triphosphatase and comparison with the related pestivirus and flavivirus enzymes. J. virology 67 (10), 6152–6158. doi:10.1128/jvi.67.10.6152-6158.1993
Tellinghuisen, T. L., Paulson, M. S., and Rice, C. M. (2006). The NS5A protein of bovine viral diarrhea virus contains an essential zinc-binding site similar to that of the hepatitis C virus NS5A protein. J. virology 80 (15), 7450–7458. doi:10.1128/jvi.00358-06
Turtle, L., Griffiths, M. J., and Solomon, T. (2012). Encephalitis caused by flaviviruses. QJM Mon. J. Assoc. Physicians 105 (3), 219–223. doi:10.1093/qjmed/hcs013
van den Elsen, K., Quek, J. P., and Luo, D. (2021). Molecular insights into the flavivirus replication complex. Viruses 13 (6), 956. doi:10.3390/v13060956
van Leur, S. W., Heunis, T., Munnur, D., and Sanyal, S. (2021). Pathogenesis and virulence of flavivirus infections. Virulence 12 (1), 2814–2838. doi:10.1080/21505594.2021.1996059
Waggoner, J. J., Rojas, A., and Pinsky, B. A. (2018). Yellow fever virus: diagnostics for a persistent arboviral threat. J. Clin. Microbiol. 56 (10), e00827-18–e01128. doi:10.1128/jcm.00827-18
Warrener, P., and Collett, M. S. (1995). Pestivirus NS3 (p80) protein possesses RNA helicase activity. J. virology 69 (3), 1720–1726. doi:10.1128/jvi.69.3.1720-1726.1995
World Health Organization (2019). Japanese encephalitis. World Health Organization. Available at: https://www.who.int/news-room/fact-sheets/detail/japanese-encephalitis.
Keywords: Flavivirus, miRNA, BLAST, NCBI, alignments, antiviral
Citation: Cayatineto HW and Hakim ST (2025) hsa-miR-548d-3p: a potential microRNA to target nucleocapsid and/or capsid genes in multiple members of the Flaviviridae family . Front. Bioinform. 4:1487292. doi: 10.3389/fbinf.2024.1487292
Received: 27 August 2024; Accepted: 11 December 2024;
Published: 14 January 2025.
Edited by:
Deepak Agarwal, Tamil Nadu Fisheries University, IndiaReviewed by:
Nan Zheng, Nanjing University, ChinaMuhammad Aleem Ashraf, Emerson University Multan, Pakistan
Carolina Polonio, Brigham and Women’s Hospital and Harvard Medical School, United States
Copyright © 2025 Cayatineto and Hakim. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: S. T. Hakim, c3RhYmFzc3VtQGRpbmVjb2xsZWdlLmVkdQ==