 Shubham Choudhury
Shubham Choudhury Nisha Bajiya
Nisha Bajiya Sumeet Patiyal
Sumeet Patiyal Gajendra P. S. Raghava
Gajendra P. S. Raghava
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Bioinform. , 06 February 2024
Sec. Integrative Bioinformatics
Volume 4 - 2024 | https://doi.org/10.3389/fbinf.2024.1341479
In the past, several methods have been developed for predicting the single-label subcellular localization of messenger RNA (mRNA). However, only limited methods are designed to predict the multi-label subcellular localization of mRNA. Furthermore, the existing methods are slow and cannot be implemented at a transcriptome scale. In this study, a fast and reliable method has been developed for predicting the multi-label subcellular localization of mRNA that can be implemented at a genome scale. Machine learning-based methods have been developed using mRNA sequence composition, where the XGBoost-based classifier achieved an average area under the receiver operator characteristic (AUROC) of 0.709 (0.668–0.732). In addition to alignment-free methods, we developed alignment-based methods using motif search techniques. Finally, a hybrid technique that combines the XGBoost model and the motif-based approach has been developed, achieving an average AUROC of 0.742 (0.708–0.816). Our method—MRSLpred—outperforms the existing state-of-the-art classifier in terms of performance and computation efficiency. A publicly accessible webserver and a standalone tool have been developed to facilitate researchers (webserver: https://webs.iiitd.edu.in/raghava/mrslpred/).
Messenger RNA (mRNA) is a single-stranded RNA, a transcription product that leads to protein synthesis via translation. It carries the cell’s genetic information from the nucleus to the cytoplasm. In the cytoplasm, mRNA is localized to different parts of the cell, resulting in an asymmetric distribution of proteins within the cell (Wang et al., 2021). It plays an important role in several developmental processes, such as neuronal maturation, embryonic patterning, cell migration, cell fate determination, cell adaptation to stress, and the development of body axes in Drosophila melanogaster (Ephrussi et al., 1991; Katz et al., 2012; Medioni et al., 2012; Liu et al., 2019; Tian et al., 2020). Identifying the cellular location of mRNA provides valuable information about the amount of protein synthesis and the location, which correlates with its function (Holt and Bullock, 2009; Tang et al., 2021). Transporting mRNA over a protein has significant advantages, such as transportation cost reduction by the expression of mRNA to generate different types of localized proteins, rapid response to external stimuli, segregation of transcripts to specific organelles or compartments, and prevention of ectopic action of proteins during localization (Kloc et al., 2002; Di Liegro et al., 2014; Tang et al., 2021; Wang et al., 2021). Therefore, knowing the subcellular localization of mRNA is important to understand various biological processes (Tang et al., 2021).
In the past, numerous experimental techniques have been established to identify the location of mRNA in a cell. In vitro visualization can be performed using classical in situ hybridization, MS2-system-based techniques, and RNA (Fagerberg et al., 2014; Wang et al., 2021; Zhang et al., 2021). These experimental techniques are highly sensitive and accurate, capable of detecting subcellular localization with high precision. Despite their accuracy, these techniques cannot be implemented routinely at the genome level due to their cost. Experimental techniques are laborious, have limited applications to specific tissues, are expensive processes, and often require sophisticated instrumentation (Savulescu et al., 2021). Advanced sequencing techniques generate a large amount of information about transcripts. To overcome these challenges, researchers have developed a wide range of in silico methods for predicting the location of mRNA in a cell (Savulescu et al., 2021). Most of these in silico methods are knowledge-based, deriving rules or models from experimental data to predict the location of an mRNA sequence. One can obtain experimental data from major repositories such as RNALocate (Zhang et al., 2017; Cui et al., 2022), lncATLAS (Mas-Ponte et al., 2017), and lncSLdb (Wen et al., 2018).
Over the years, a multitude of methods have been developed for predicting subcellular localization, with the majority relying on machine learning techniques. Examples include RNATracker (Yan et al., 2019), iLoc-mRNA (Zhang et al., 2021), DM3Loc (Wang et al., 2021), mRNALocater (Tang et al., 2021), and mRNALoc (Garg et al., 2020). In most of these methods, datasets have been derived from the popular RNALocate database. One major limitation of these tools is their tendency to predict or assign a single label for a given mRNA. However, in reality, most mRNAs traverse throughout the cell and can be found at multiple locations within it. Lin et al. developed a method named DM3Loc (Wang et al., 2021), specifically designed to predict multiple labels or locations for a given mRNA. This method, referred to as a multi-label subcellular localization prediction method, is based on a deep learning framework. DM3Loc generates features by converting mRNA sequences into one-hot encoded vectors. These vectors serve as input to a CNN classifier, and a multi-head self-attention mechanism is used to enhance its ability to identify sequence regions relevant to localization. Currently, DM3Loc stands as a state-of-the-art multi-label classifier for predicting the subcellular localization of mRNA sequences. Despite its advanced capabilities, DM3Loc has limitations of its own, such as heavy requirements for computational resources and time.
In this study, a novel multi-label classifier is proposed for predicting the subcellular localization of mRNA. This method, named MRSLpred, is based on machine learning and uses the composition features of mRNA sequences for prediction. Importantly, MRSLpred can be implemented at a genome scale as its computational resource requirements are minimal. This proposed method aims to complement existing approaches and address some of the limitations associated with current methods.
The pre-processed dataset was obtained from DM3Loc (Wang et al., 2021), where they have used both experimentally validated and database-curated mRNA sequences of Homo sapiens from RNALocate database v2 (Cui et al., 2022). RNALocate is a database dedicated to providing high-confidence RNA subcellular localization information sourced from the literature, other databases, and RNA-seq datasets. A majority of the mRNAs were localized in more than one subcellular compartment, which is generally the case with most mRNAs in a real-world scenario. In order to ensure that the dataset is non-redundant, CD-HIT was run at a threshold of 80% similarity. Our dataset contains a total of 17,277 non-exclusive human mRNAs spread across six subcellular compartments: the ribosome, cytosol, endoplasmic reticulum (ER), membrane, nucleus, and exosome. The dataset is graphically represented in Figure 1. The number of mRNAs with localization at different compartments is 11,923 for the nucleus, 17,156 for the exosome, 2,338 for the cytosol, 5,210 for the ribosome, 3,232 for the membrane, and 1976 for the ER. The distribution of location labels is depicted in Figure 2.
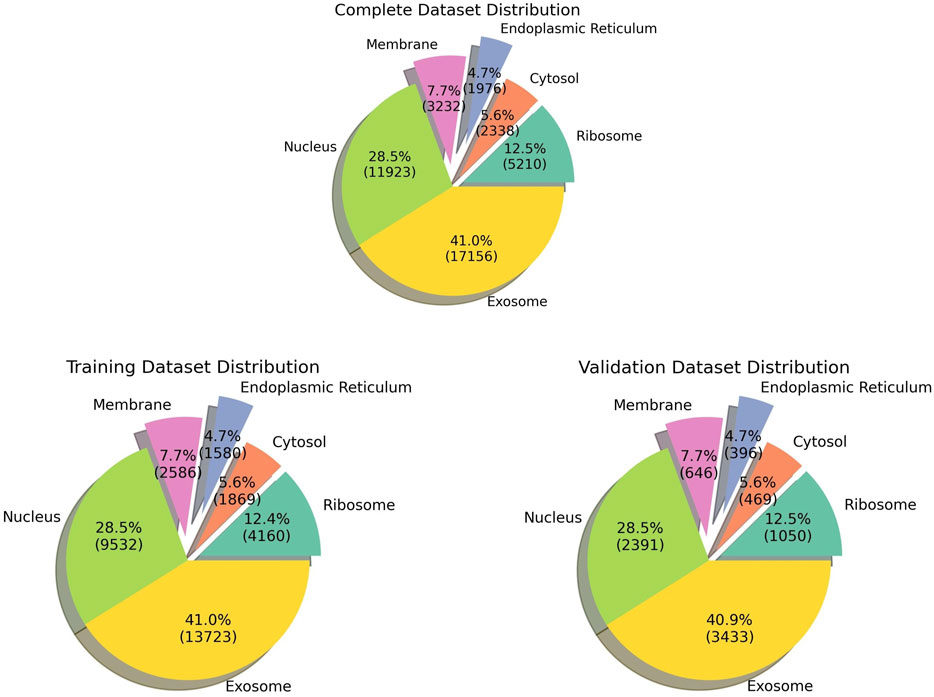
FIGURE 1. Pie chart indicating the data distribution in all the datasets.

FIGURE 2. Upset plot depicting all the possible combinations of subcellular locations.
In order to train our model, we are required to generate features or descriptors corresponding to each mRNA. For the aforementioned purpose, we used the tool ‘Nfeature’ (Mathur et al., 2021) [https://doi.org/10.1101/2021.12.14.472723], which can generate hundreds of features for a single mRNA sequence. These are the two feature classes which were used for training the models:
1. Composition of DNA/RNA for k-mer (CDK): k-mers of length 3 were generated using Nfeature, and the frequency of each k-mer was used as a feature for training the ML model. It was calculated using the following formula:
where
2. Reverse complement of DNA for k-mers (RDK): k-mers of length 4 were generated using Nfeature, and the frequency of the reverse complement of this k-mer will be used as a feature. The formula used for calculating the RDK is similar to the one used for the CDK. However, in this case, instead of counting the occurrence of the k-mer in the input sequence directly, first, the reverse complement of the sequence is generated, and then, the 4-mer frequency in that sequence is calculated.
Both these features were combined together to obtain a vector of 200 features for each mRNA.
Motifs are known to play a significant role in the localization of mRNA within the cell. We used the MERCI tool (Vens et al., 2011) to identify the presence of conserved motifs in the training dataset. For each location, the dataset was split into a positive and a negative dataset based on the localization in that location. Both the positive and negative datasets are then provided as inputs to MERCI (Vens et al., 2011), and then, the tool identifies motifs that can discriminate between positive and negative samples for that location. We acquired six sets of motifs specific to each location, and this information was used to modify the prediction probabilities for each location. For instance, in case a motif specific to ribosome sequences is found within a query sequence, the probability (provided by the ML model) that the query sequence belongs to the ribosome is updated to 1. So, the presence of motifs will practically override the prediction made by the ML model, and those sequences that do not contain any motifs remain unaffected.
The location label of each mRNA was generated using one-hot encoding by converting locations into 0 or 1 s; i.e., if an mRNA is only present in the ribosome and the nucleus, it will have a label like [1,0,0,0,1,0]. Initially, a CNN model was trained using one-hot encoded mRNA sequences. One-hot encoding was performed by converting every mRNA sequence into a
Composition-based features defined above were used to train various machine learning models. Model training was conducted on Python using standard machine learning libraries such as scikit-learn and XGBoost (Chen and Guestrin, 2016). A number of machine learning approaches were used to construct prediction models such as logistic regression, decision tree, random forest classifier, MLP classifier, AdaBoost classifier, Gaussian Naïve Bayes, quadratic discriminant analysis, gradient boosting classifier, and eXtreme Gradient Boosting (XGBoost) classifier. The scikit-learn Python library was used to implement these classification approaches.
Training of the models was conducted using ML classifiers combined with a multioutput classifier. The specialty of this model is that it takes CDK and RDK as features and predicts all the possible locations of the mRNA in one single step.
To ensure the proper model fitting, we used five-fold cross-validation to train and validate the models. The entire dataset was split in an 80:20 ratio, where 80% of the data were used for training and 20% data were used for validation. The training data were further split into five parts, and five-fold cross-validation was performed on the same. In each iteration, a different fold was used for validation, and the remaining four folds were used for validation. The training performance is calculated by taking the average over five iterations. The splitting of data was performed in a stratified manner, ensuring that all the locations were equally distributed within each of the folds. Once the training was conducted, the model was validated using the 20% validation dataset.
Evaluation of the model performance was carried out using standard performance metrics. The performance metrics used were sensitivity, specificity, accuracy, Matthews’ correlation coefficient, F1-score, and area under the receiver operator characteristic (AUROC). Out of these metrics, only the AUROC is threshold-independent, whereas all the remaining metrics are dependent on the threshold cut-off. The cut-off for the probabilities was determined by balancing out the sensitivity and specificity as follows:
where TP is true positive, FP is false positive, FN is false negative, and TN is true negative.
In this study, we used a total of 17,277 mRNA sequences with non-exclusive locations for training our model. The primary objective is to develop a model that can accurately predict multiple locations for a single mRNA, mimicking a practical scenario within the cell. So, we developed a multi-label classifier to predict the subcellular localization of mRNA. The outline of the study is shown in Figure 3.
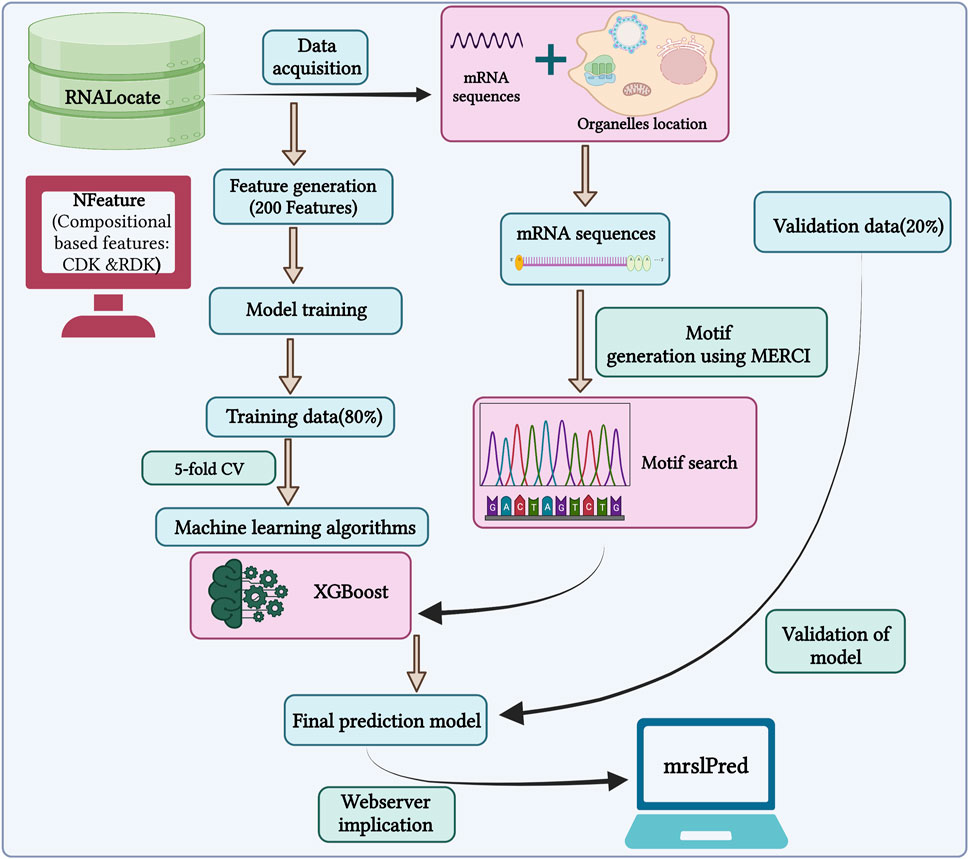
FIGURE 3. Outline of the methodology followed by MRSLpred.
Nucleotide motifs are known to affect mRNA localization, and in this study, we tried to implement this ideology. Discriminatory motifs for each location were identified using the training dataset, and the presence of these motifs was then used to assign location labels to each mRNA in the validation dataset. The motifs that are unique to the positive dataset for each location are searched in the validation dataset, and if any one of these motifs is found within the sequence, then the label for that mRNA in that location is assigned as 1 or 0. The number of hits for motifs in each individual location was as follows—ribosome: 33, cytosol: 25, endoplasmic reticulum: 66, membrane: 29, nucleus: 96, and exosome: 1,655. The top 10 motifs found in each location are provided in Table 1. A large number of motifs were predominantly identified in the mRNA sequences that were assigned to exosomes and nucleus. This may be possibly due to the relatively high number of sequences assigned to the exosome location. This can be seen in Table 2, where the number of unique motifs identified by MERCI in each location is provided, as well as the total number of hits for these motifs found in the training dataset.
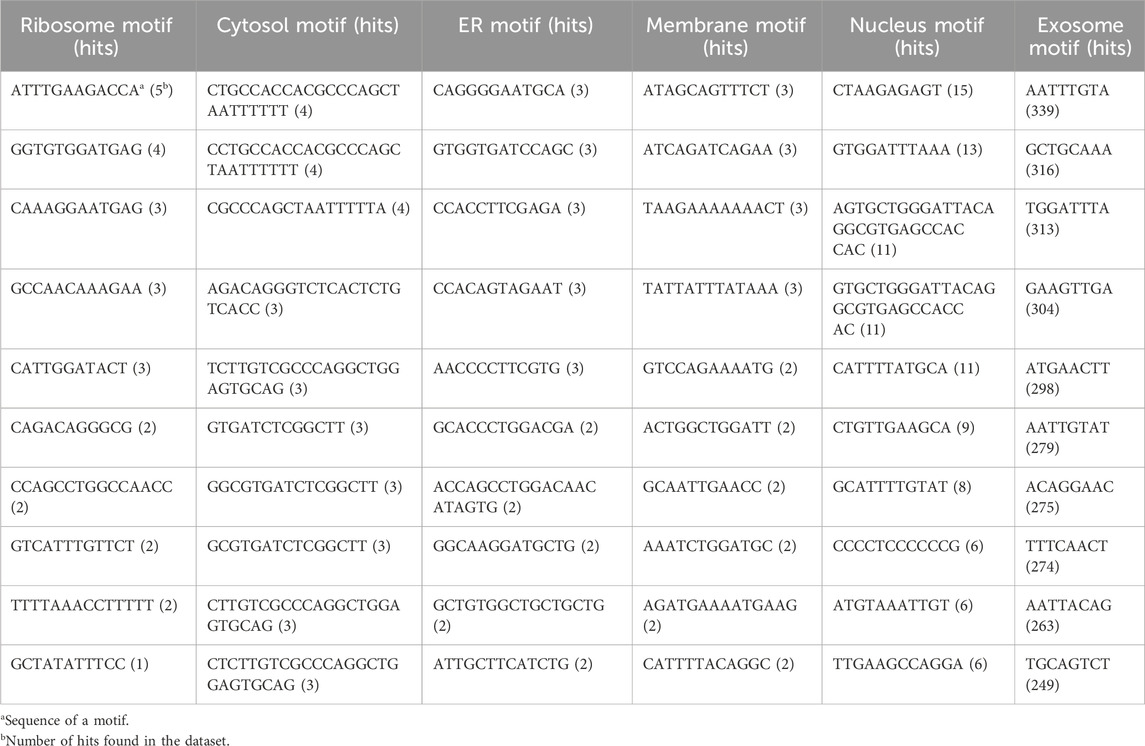
TABLE 1. Sequence of top 10 motifs exclusively found in each subcellular location; all these motifs were discovered in the training dataset using MERCI software.
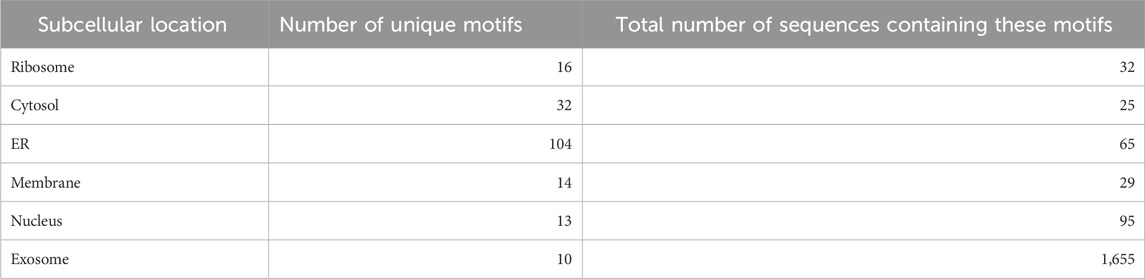
TABLE 2. Number of location-specific unique motifs found in the training dataset and the number of sequences in the training dataset containing these motifs (coverage).
Different composition-based features were used to train the ML model, and we achieved maximum average AUROCs of 0.705 and 0.691 and average MCCs of 0.213 and 0.195, respectively, in the cases of CDK and RDK. Upon the combination of these two features, the model performance further improved, giving an AUROC in the range of 0.709 and an MCC of 0.216.
Training of models was conducted using standard python packages—decision tree, random forest classifier, MLP classifier, AdaBoost classifier, Gaussian Naïve Bayes, quadratic discriminant analysis, gradient boosting classifier, and eXtreme Gradient Boosting classifier. The results for all the models are shown in Supplementary Table S1. Initially, composition-based features were used for training the models, and the XGBoost classifier had the best performance among all the ML models. The hyperparameters for the XGBoost model that reported the best performance were as follows: MultiOutputClassifier [XGBClassifier (n_estimators = 1,000, learning_rate = 0.01, random_state = 1, max_delta_step = 1, and n_jobs = −1)]. The metrics for the best performing model are shown in Table 3.
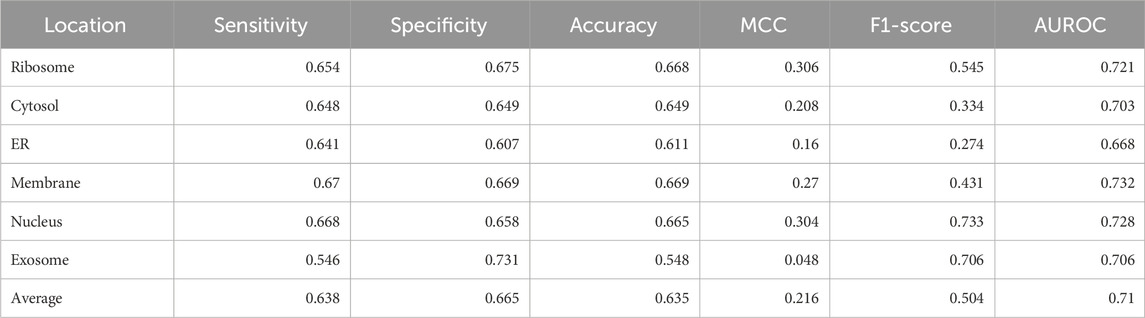
TABLE 3. Performance of the native XGBoost multi-label classifier on CDK3+RDK4 features.
The best performance was obtained by combining the alignment-free XGBoost model with the motif module. Once the ML model makes the prediction, the presence of these motifs was then used to tweak the prediction. If any motif is found in the query sequences, the prediction probability from the ML model is switched to 1. For instance, if a motif unique to the ribosome is found within a query sequence, and the prediction probability for that location by the ML model is 0.4, then in the final model, it will be assigned a probability of 1. Furthermore, if no motifs were found for the same query sequence, then the prediction probability assigned by the ML model remains unchanged.
Significant performance improvement was observed upon implementing this module. The performance metrics for the ML + motif module are provided in Table 4. The area under the receiver operator characteristic curve for each of the individual locations is shown in Figure 4.
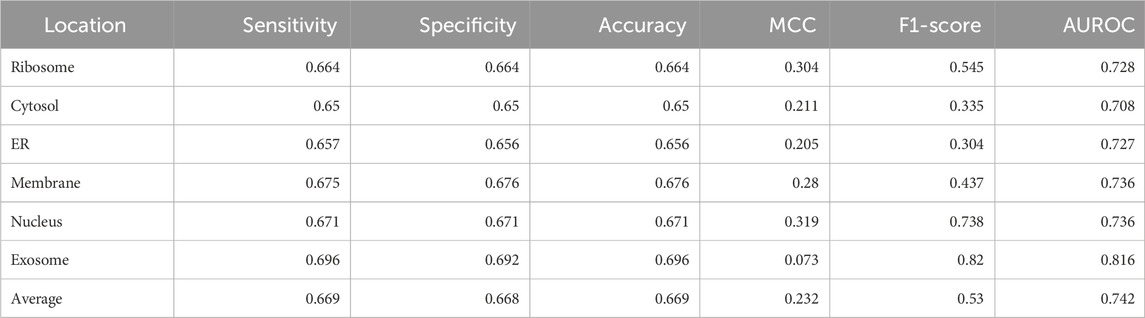
TABLE 4. Performance of the XGBoost multi-label classifier combined with the motif module (final model).
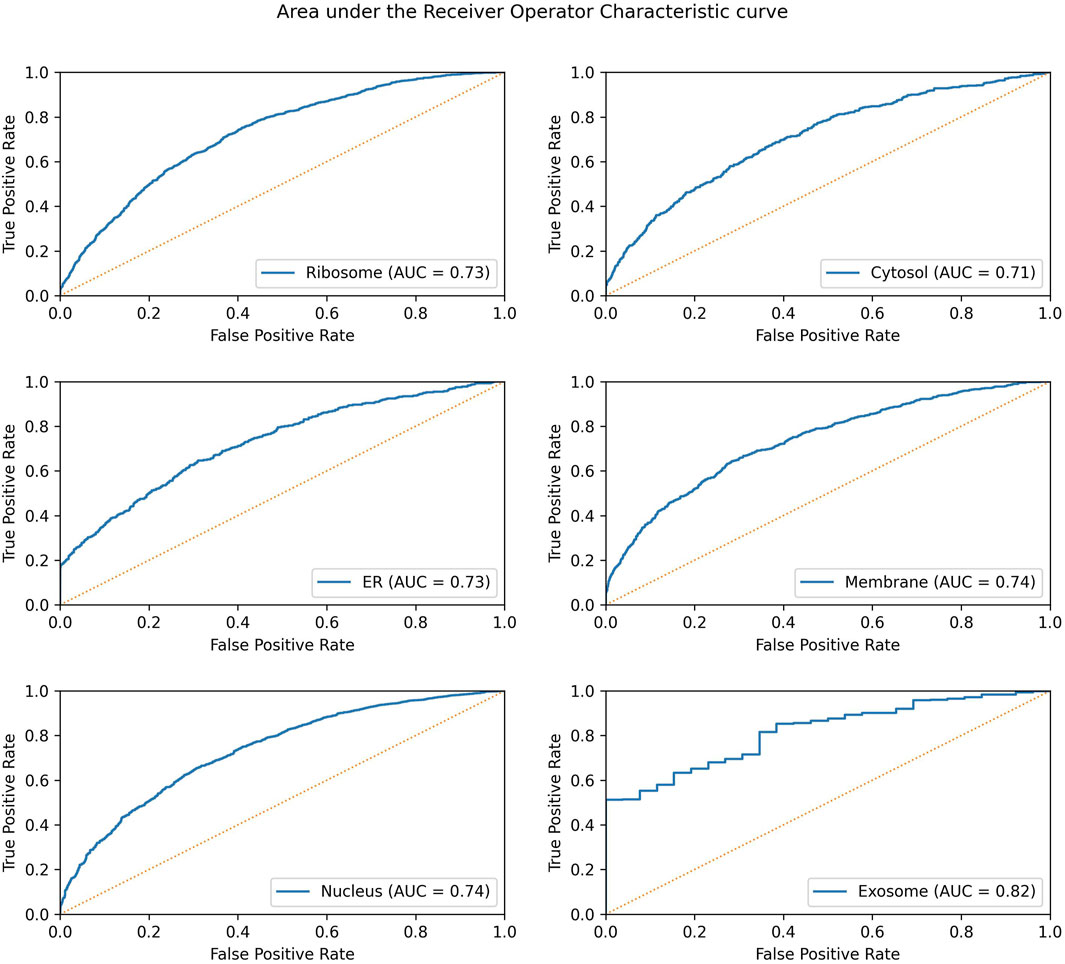
FIGURE 4. AUROC for the final model in each individual location. The AUROC is calculated on the validation dataset, and the yellow dotted line represents the AUROC for a random prediction (AUROC = 0.5).
Currently, DM3Loc is the only tool that performs multi-label classification out of the box and is developed on the latest version of RNALocate (version 2). We also used the same non-redundant dataset that is used in DM3Loc. However, the dataset splitting is performed differently, maintaining a similar proportion of subcellular locations in each split. The performance of both DM3Loc and MRSLpred was evaluated on the validation dataset, and it was observed that DM3Loc performs better than MRSLpred in terms of AUROC and MCC. However, the better performance of DM3Loc can be attributed to the bias in favor of DM3Loc as some of the sequences in the validation dataset are present within the training dataset used in DM3Loc. This leads to an overreported result when evaluating DM3Loc on our validation result.
Both MRSLpred and DM3Loc have been designed as multi-label classifiers where more than one location can be assigned to a single sequence. On the other hand, other existing methods perform multiclass classification, assigning only one location to a single sequence, and comparing MRSLpred/DM3Loc with these tools would be unfair to them. However, for the sake of comparison, we evaluated the performance of all these tools on the validation dataset used in MRSLpred. The detailed comparison of MRSLpred with DM3Loc and all other tools which do not support multi-label classification is provided in Table 5.
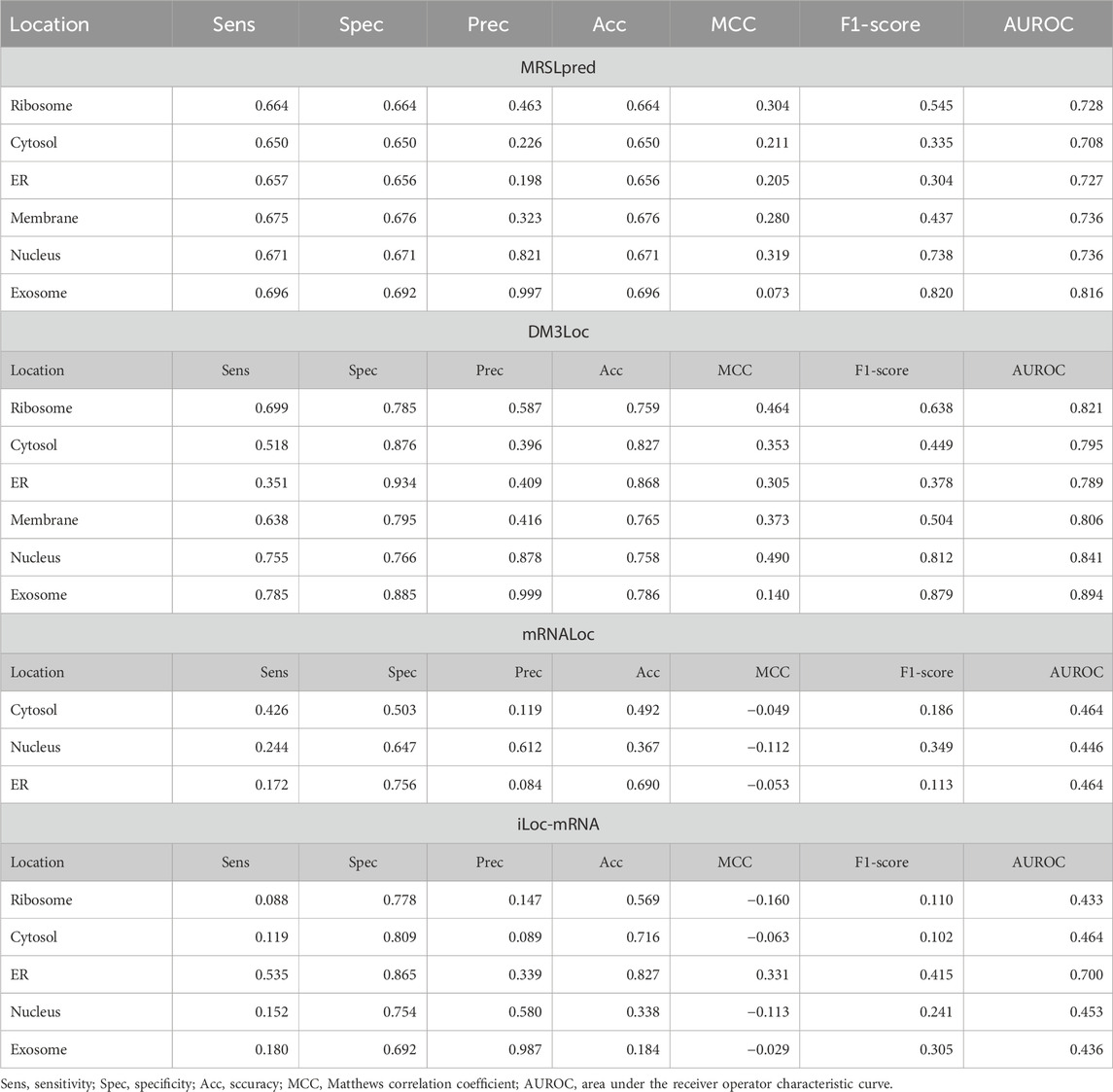
TABLE 5. Benchmarking of MRSLpred with existing prediction tools on the validation dataset used in MRSLpred.
Another significant advantage of MRSLpred is that it is computationally very efficient and consumes very less time. A time comparison between both the methods was performed on an iMac (21.5-inch, Late 2015 model, 2.8 GHz Quad-Core Intel Core i5, 8 GB DDR3 RAM, and Intel Iris Pro Graphics 6,200–536 MB). The detailed comparison of MRSLpred and DM3Loc based on the time taken to generate predictions is provided in Table 6.
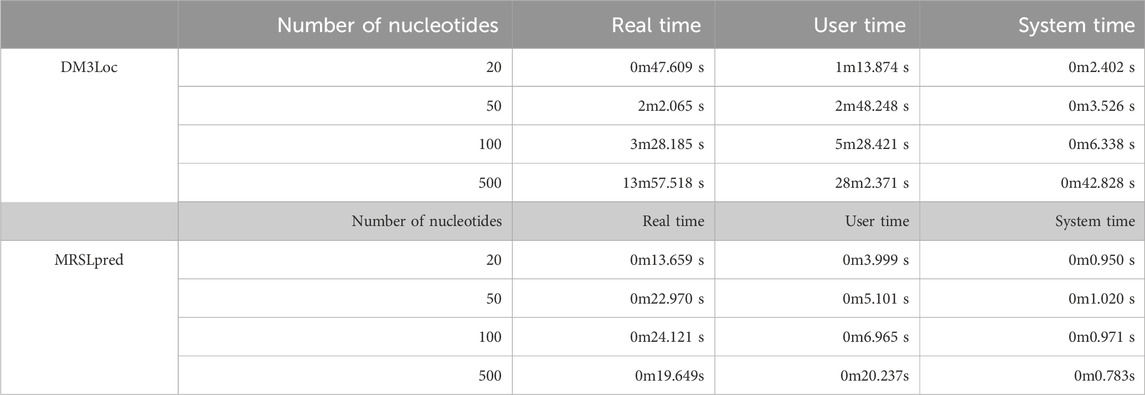
TABLE 6. Comparison of MRSLpred with DM3Loc based on the time taken for similar datasets.
mRNA localization is a relevant biological process that controls the concentration of mRNA at different locations. This, in turn, exerts control on the level of protein translation at various locations within the cell (Kindler et al., 2005). Understanding mRNA subcellular localization will provide a better perspective on how protein synthesis is regulated at the mRNA level. Spatial localization of mRNA is also gathering massive interest in the field of development biology. The majority of developmental processes rely on cellular polarity generated by mRNA localization to undergo differentiation (Lécuyer et al., 2007; Claußen et al., 2015). A lot of research work is focused on understanding these spatial processes, which, when perturbed, can lead to major developmental disorders.
However, in vitro investigation of mRNA localization is a costly business and labor-intensive at the same time. Computational techniques can provide a viable solution to this problem, as they are swift, inexpensive, and reliable. Modern machine learning and deep learning techniques manage to perform well on biological data and are pretty accurate.
Many tools have already been developed for mRNA subcellular localization prediction. DM3Loc (Wang et al., 2021) is a popular tool that deploys a multi-head self-attention mechanism with a CNN model. DM3Loc uses a one-hot encoding vector as a feature for the CNN model and achieves an AUROC in the range of 0.698–0.773 (average: 0.742) and an MCC in the range of 0.074–0.386 (average: 0.270) on their own validation dataset. However, the performance of DM3Loc on our validation dataset painted a different picture. It only achieved an AUROC in the range of 0.557–0.776 (average: 0.704) and an MCC in the range of 0–0.353 (average: 0.081). As of now, only DM3Loc performs multi-label classification, whereas all the other tools are multiclass classifiers. Due to its complex architecture, DM3Loc requires very intensive computational power that includes a dedicated graphics card. For many sequences, one-hot encoding mRNA sequences is a herculean task, as most of the time, the RAM crashes due to the increasing size of the vector.
MRSLpred achieves an AUROC between 0.708 and 0.816 (average: 0.742) and an MCC between 0.073 and 0.319 (average: 0.232) on the validation dataset. MRSLpred uses a much simpler approach, combining an XGBoost model with a motif-based module and using compositional features for the model. Due to this, the method is extremely fast and computationally inexpensive while achieving comparable performance with the more complex method—DM3Loc. The time taken by MRSLpred for predicting the location of 500 mRNA sequences is less than 1 min, whereas for the same dataset, DM3Loc takes approximately 32 min.
Sequence-based classification has its drawbacks as it tends to lose structural information, which is also known to play an essential role in subcellular location. However, in order to use structure-based features for prediction, more computational power may be required. Furthermore, the dataset used in this study had an inherent class imbalance, which makes the model biased to classes represented in larger numbers (the nucleus and exosome). In the future version of our tool, we expect to obtain a more balanced dataset where all locations have equal representation.
It is worth mentioning that we could have achieved better performance with deep learning methods. We tried to implement a CNN classifier based on one-hot encoded sequences, but the maximum average AUROC achieved for all the locations was 0.584 on the validation dataset. The CNN classifier performed very well on the training dataset (average AUROC = 0.837) but failed miserably on the validation dataset.
In this tool, we managed to develop a multi-label subcellular localization prediction tool that can accurately identify all the possible subcellular locations that an mRNA could move to. Our final model was based on an XGBoost multi-label classifier that also deploys a motif module to improve our prediction. We managed to achieve an AUROC of 0.708–0.816 (average: 0.742) and an MCC of 0.073–0.319 (average: 0.232). Due to the simpler architecture of our tool, it is extremely fast, and the standalone can be run on very minimal computational power. We believe that this will prove to be a valuable tool for biologists who work with mRNA localization. MRSLpred is available online at https://webs.iiitd.edu.in/raghava/mrslpred/, and its standalone can be downloaded from https://webs.iiitd.edu.in/raghava/mrslpred/standalone.php. The standalone is also available on GitHub and is accessible at https://github.com/raghavagps/mrslpred.
Publicly available datasets were analyzed in this study. These data can be found online at: https://webs.iiitd.edu.in/raghava/mrslpred/downloads.php.
SC: conceptualization, data curation, formal analysis, investigation, methodology, software, validation, visualization, writing–original draft, and writing–review and editing. NB: formal analysis, visualization, writing–original draft, and writing–review and editing. SP: formal analysis, software, and writing–review and editing. GR: conceptualization, formal analysis, investigation, methodology, supervision, writing–original draft, and writing–review and editing.
The authors declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by the BIC grant from the Department of Biotechnology, India [BT/PR40158/BTIS/137/24/2021].
The authors are thankful to the Department of Biotechnology (DBT), the Department of Science and Technology (DST-INSPIRE), and the Council of Scientific and Industrial Research (CSIR) for fellowships and financial support. They are also thankful to the Department of Computational Biology, IIITD, New Delhi, for infrastructure and facilities.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors declare that they were editorial board members of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fbinf.2024.1341479/full#supplementary-material
Chen, T., and Guestrin, C. (2016). “XGBoost,” in Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining. KDD ’16: The 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, New York, NY, USA (ACM). doi:10.1145/2939672.2939785
Claußen, M., Lingner, T., Pommerenke, C., Opitz, L., Salinas, G., and Pieler, T. (2015). Global analysis of asymmetric RNA enrichment in oocytes reveals low conservation between closely related Xenopus species, Mol. Biol. Cell. 26 (21), 3777–3787. doi:10.1091/mbc.E15-02-0115
Cui, T., Dou, Y., Tan, P., Ni, Z., Liu, T., Wang, D., et al. (2022). RNALocate v2.0: an updated resource for RNA subcellular localization with increased coverage and annotation. Nucleic acids Res. 50 (D1), D333–D339. doi:10.1093/nar/gkab825
Di Liegro, C. M., Schiera, G., and Di Liegro, I. (2014). Regulation of mRNA transport, localization and translation in the nervous system of mammals (Review). Int. J. Mol. Med. 33 (4), 747–762. doi:10.3892/ijmm.2014.1629
Ephrussi, A., Dickinson, L. K., and Lehmann, R. (1991). Oskar organizes the germ plasm and directs localization of the posterior determinant nanos, Cell. 66 (1), 37–50. doi:10.1016/0092-8674(91)90137-n
Fagerberg, L., Hallström, B. M., Oksvold, P., Kampf, C., Djureinovic, D., Odeberg, J., et al. (2014). Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell. proteomics MCP 13 (2), 397–406. doi:10.1074/mcp.M113.035600
Garg, A., Singhal, N., Kumar, R., and Kumar, M. (2020). mRNALoc: a novel machine-learning based in-silico tool to predict mRNA subcellular localization. Nucleic acids Res. 48 (W1), W239–W243. doi:10.1093/nar/gkaa385
Holt, C. E., and Bullock, S. L. (2009). Subcellular mRNA localization in animal cells and why it matters. Sci. (New York, N.Y.) 326 (5957), 1212–1216. doi:10.1126/science.1176488
Katz, Z. B., Wells, A. L., Park, H. Y., Wu, B., Shenoy, S. M., and Singer, R. H. (2012). β-Actin mRNA compartmentalization enhances focal adhesion stability and directs cell migration, Genes and Dev. 26 (17), 1885–1890. doi:10.1101/gad.190413.112
Kindler, S., Wang, H., Richter, D., and Tiedge, H. (2005). RNA transport and local control of translation, Annu. Rev. Cell. Dev. Biol. 21 (1), 223–245. doi:10.1146/annurev.cellbio.21.122303.120653
Kloc, M., Zearfoss, N. R., and Etkin, L. D. (2002). Mechanisms of subcellular mRNA localization, Cell. 108 (4), 533–544. doi:10.1016/s0092-8674(02)00651-7
Lécuyer, E., Yoshida, H., Parthasarathy, N., Alm, C., Babak, T., Cerovina, T., et al. (2007). Global analysis of mRNA localization reveals a prominent role in organizing cellular architecture and function. Cell. 131 (1), 174–187. doi:10.1016/j.cell.2007.08.003
Liu, D., Li, G., and Zuo, Y. (2019). Function determinants of TET proteins: the arrangements of sequence motifs with specific codes, Briefings Bioinforma. 20 (5), 1826–1835. doi:10.1093/bib/bby053
Mas-Ponte, D., Carlevaro-Fita, J., Palumbo, E., Hermoso Pulido, T., Guigo, R., and Johnson, R. (2017). LncATLAS database for subcellular localization of long noncoding RNAs. RNA (New York, N.Y.) 23 (7), 1080–1087. doi:10.1261/rna.060814.117
Mathur, M., Patiyal, S., Dhall, A., Jain, S., Tomer, R., Arora, A., et al. (2021). Nfeature: a platform for computing features of nucleotide sequences. bioRxiv. doi:10.1101/2021.12.14.472723
Medioni, C., Mowry, K., and Besse, F. (2012). Principles and roles of mRNA localization in animal development. Development 139 (18), 3263–3276. doi:10.1242/dev.078626
Savulescu, A. F., Bouilhol, E., Beaume, N., and Nikolski, M. (2021). Prediction of RNA subcellular localization: learning from heterogeneous data sources, iScience 24 (11), 103298. doi:10.1016/j.isci.2021.103298
Tang, Q., Nie, F., Kang, J., and Chen, W. (2021). mRNALocater: enhance the prediction accuracy of eukaryotic mRNA subcellular localization by using model fusion strategy. Mol. Ther. J. Am. Soc. Gene Ther. 29 (8), 2617–2623. doi:10.1016/j.ymthe.2021.04.004
Tian, L., Chou, H. L., Fukuda, M., Kumamaru, T., and Okita, T. W. (2020). MRNA localization in plant cells, Plant physiol. 182 (1), 97–109. doi:10.1104/pp.19.00972
Vens, C., Rosso, M.-N., and Danchin, E. G. J. (2011). Identifying discriminative classification-based motifs in biological sequences. Bioinformatics 27 (9), 1231–1238. doi:10.1093/bioinformatics/btr110
Wang, D., Zhang, Z., Jiang, Y., Mao, Z., Lin, H., and Xu, D. (2021). DM3Loc: multi-label mRNA subcellular localization prediction and analysis based on multi-head self-attention mechanism. Nucleic acids Res. 49 (8), e46. doi:10.1093/nar/gkab016
Wen, X., Gao, L., Guo, X., Li, X., Huang, X., Wang, Y., et al. (2018). lncSLdb: a resource for long non-coding RNA subcellular localization. Database (Oxford) 2018, 1–6. doi:10.1093/database/bay085
Yan, Z., Lécuyer, E., and Blanchette, M. (2019). Prediction of mRNA subcellular localization using deep recurrent neural networks. Bioinformatics 35 (14), i333–i342. doi:10.1093/bioinformatics/btz337
Zhang, T., Tan, P., Wang, L., Jin, N., Li, Y., Zhang, L., et al. (2017). RNALocate: a resource for RNA subcellular localizations. Nucleic acids Res. 45 (D1), D135–D138. doi:10.1093/nar/gkw728
Keywords: subcellular localization, multi-label, motif search, messenger RNA, machine learning
Citation: Choudhury S, Bajiya N, Patiyal S and Raghava GPS (2024) MRSLpred—a hybrid approach for predicting multi-label subcellular localization of mRNA at the genome scale. Front. Bioinform. 4:1341479. doi: 10.3389/fbinf.2024.1341479
Received: 20 November 2023; Accepted: 15 January 2024;
Published: 06 February 2024.
Edited by:
Sophia Tsoka, King’s College London, United KingdomReviewed by:
Yuexu Jiang, University of Missouri, United StatesCopyright © 2024 Choudhury, Bajiya, Patiyal and Raghava. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gajendra P. S. Raghava, cmFnaGF2YUBpaWl0ZC5hYy5pbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.