 Niloofar Shirvanizadeh
Niloofar Shirvanizadeh Mauno Vihinen
Mauno Vihinen1 Introduction
Genetic variation data is nowadays easy to generate. Variation interpretation means the description of the significance of variations, often in relation to disease. This is substantially more difficult a problem than sequence generation. Experimental methods provide verified interpretations; however, due to huge amounts of variations in every individual, computational approaches are widely used. The length of human genome is over 3 billion base pairs (Nurk et al., 2022). Due to individual genetic heterogeneity, 4.1–5.0 million sites differ from the reference genome (Auton et al., 2015). Various types of prediction methods are widely used to interpret the variations, see (Niroula and Vihinen, 2016). Benchmark studies have indicated large differences in the performance of methods developed for the same type of variation prediction tasks, see e.g., (Thusberg et al., 2011; Niroula and Vihinen, 2019; Zhang et al., 2019; Marabotti et al., 2021; Anderson and Lassmann, 2022). Both predictor development and performance assessment are largely dependent on high-quality data. One might think that there is a large number of verified variations as the genetic diagnosis is widely applied; however, that is not the case, especially when considering specific types of variations or mechanisms.
The development and testing of computational methods are dependent on experimental data. Accurate prediction methods can be developed only with reliable experimentally verified cases with a systematic approach and using relevant measures (Vihinen, 2012; Vihinen, 2013). Method performance has to be assessed in comparison to existing knowledge. For that purpose, benchmark data sets with known and verified outcomes are needed. Such data sets can be time-consuming and costly to collect and require many manual steps. Therefore, it is important that the produced data are distributed and reused.
In the variation interpretation field, two databases deliver such data sets. VariBench (Nair et al., 2013; Sarkar et al., 2020) and VariSNP (Schaafsma et al., 2015) contain variation benchmark data. VariSNP is a version of the dbSNP database (Sherry et al., 2001) for short variations from where known disease-causing variants have been filtered away. VariBench is a generic database that contains all types of variations with all kinds of effects. These resources have been widely used for prediction method training and testing.
What requirements and criteria should benchmark data sets fulfill in relation to variation interpretation and in general? We have defined five criteria, discussed in (Nair et al., 2013). They include relevance, representativeness, non-redundancy, inclusion of both positive and negative cases and reusability. VariBench subscribes to the criteria and collects data sets and distributes them freely. VariBench data sets are frequently used to train and test method performance. These sets facilitate also post-publication comparison of methods to published benchmarks (Sarkar et al., 2020).
The bottleneck in sequencing projects has shifted from sequencing to interpretation of obtained results. Experimental studies of variant effects are the gold standard approaches. They are not feasible in many instances and therefore, various computational approaches have been developed. We divide the prediction methods into five categories in VariBench.
First, pathogenicity, also called tolerance, predictions aim to identify disease-related alterations of various types (for details see Table 1).These methods aim just to detect harmful or disease-related variants. Second, effect-specific methods are for the prediction of various effects at DNA, RNA and protein levels. Third, there are also predictors specific for certain molecules or families of molecules, typically for proteins. Fourth, some methods are dedicated to certain diseases. Fifth, some tools predict the phenotype, typically the severity of the variant effect.
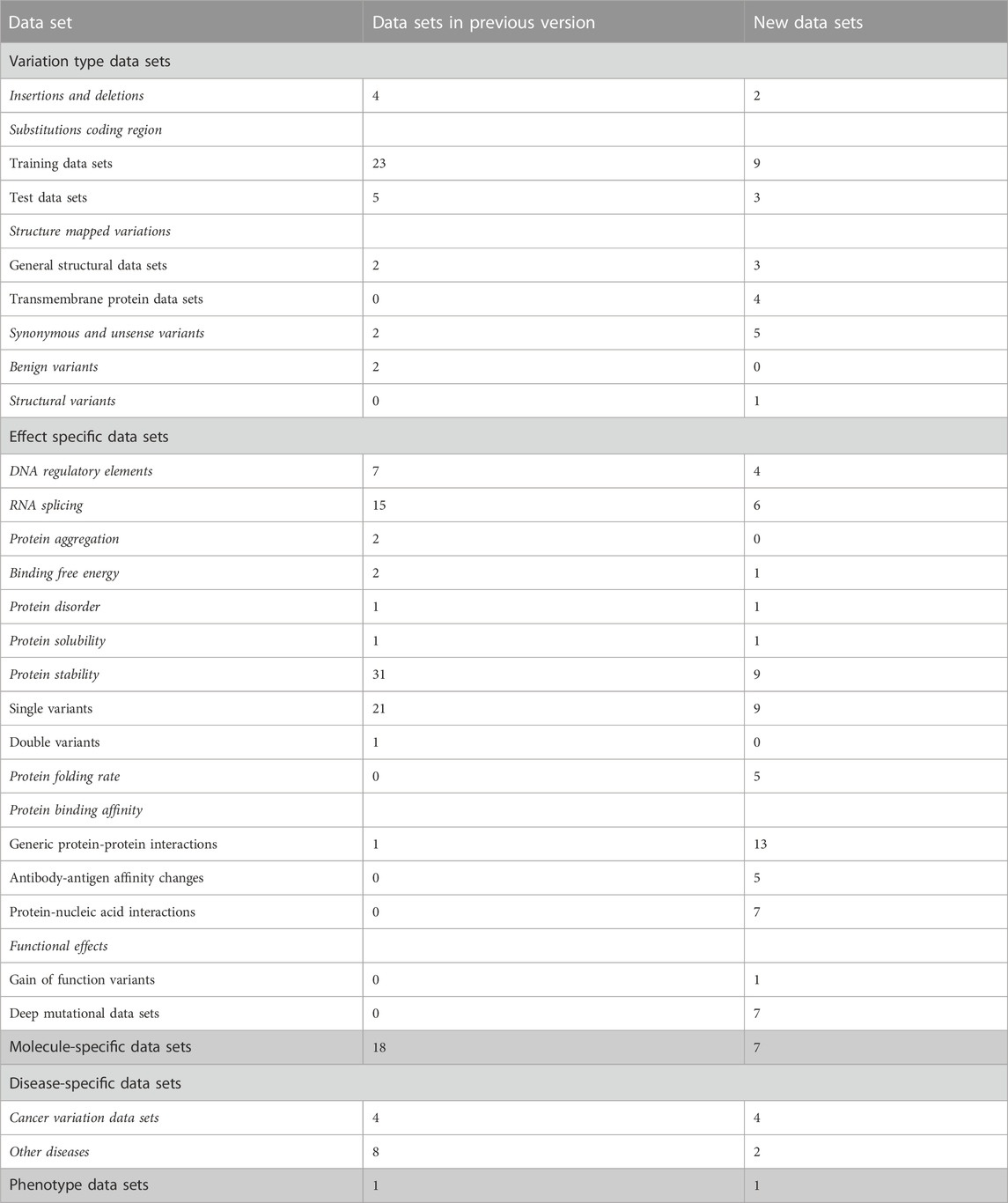
TABLE 1. Types of data sets in VariBench.
High-quality variation data sets are difficult and laborious to generate. VariBench collects, organizes, and integrates additional information and distributes different types of variation data sets. It is a unique database. We have updated the resource with 143 new data sets, which include more than 90 million variants. During the update, some new categories of variations and effects have been included. There are currently variations in 5 main categories, 17 subgroups and 11 groups.
2 Data sets and quality
VariBench collects from literature, databases and predictors data sets, which have been used to train methods or assess their performance. There are no selection criteria for the inclusion of data sets. This is because of several reasons. The data sets can be used as such, or they can be further cleaned and pruned to use in additional tasks, be extended with new cases, etc. A good benchmark data set should fulfill several requirements (Vihinen, 2012; Vihinen, 2013), including good coverage, representativeness and containing both positive and negative cases that are experimentally determined. The representativeness of amino acid substitution data sets was investigated (Schaafsma and Vihinen, 2018) and found not to be optimal.
The quality of data sets in VariBench is variable. We include even known low-quality data sets, since they may be valuable when building new data sets and for other applications. We have performed some quality tests, including consistency; however, it is the duty of the users of the data to evaluate whether the data are suitable for intended use. One of the goals of VariBench is to provide existing data sets, even when problematic, e.g., for comparative purposes.
Systematics is an integral part of data and database quality. It is quite common that due to errors and lack of systematics, all variants in an existing data set cannot be reused as they cannot be mapped to reference sequences.
An example of the importance of data quality is in the field of protein stability predictions. Most of the existing predictors are based on a single database, ProTherm, which was shown to contain numerous problems (Yang et al., 2018). Recently, new and higher-quality databases have emerged in this field (Stourac et al., 2021; Turina et al., 2021).
3 Uses of VariBench data
VariBench data sets have been widely used especially to train and test variation interpretation predictors (pathogenicity/tolerance, protein stability, solubility, melting temperature, gene/protein/disease-specific predictors, and interaction and structural effects on folded and disordered regions and proteins), but also in the benchmarking performance of tools for various types and effects. In addition to human, plant and animal-related predictors and benchmarks have benefitted from VariBench (Yang et al., 2022). The data has also facilitated the interpretation of variants according to the guidelines of American College of Medical Genetics and Genomics, and the Association for Molecular Pathology (ACMG/AMP) (Richards et al., 2015) and benchmarking such annotations.
4 Data sets in VariBench
VariBench contains now 559 files for separate data sets from 295 studies and covers a wide range of variations (Tables 1, 2). The data sets were collected from literature, websites and databases. They have been used for predictive purposes, most often to develop novel predictors for different types or effects of variants. Some data sets have been specifically collected for benchmarking purposes.
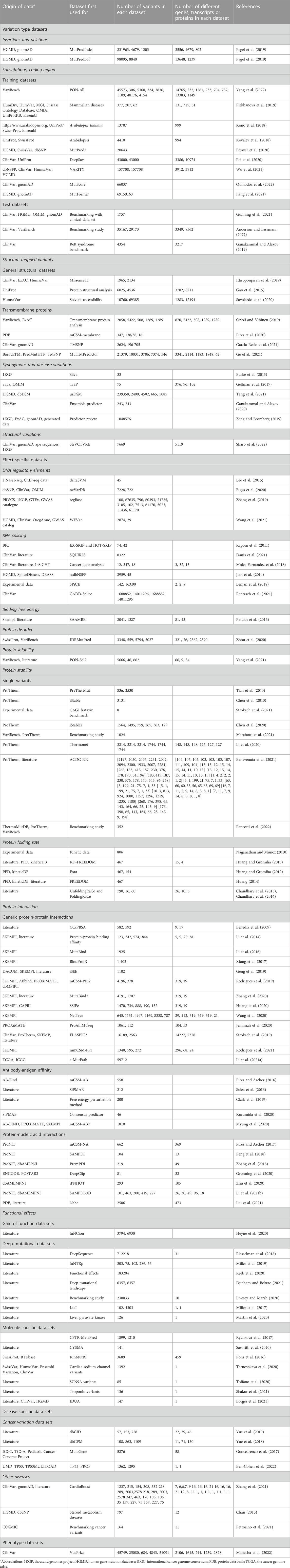
TABLE 2. New data sets in VariBench.
There are 247 new data files that contain total 90,886,959 variants. Together with previous versions, there are 105,181,219 variants, the increase is more than seven-fold from the original number of 14,294,260 variants. The number of data sets is high because many articles contain more than one data set. Many of the data sets are redundant as they contain data from the same origin. The most common sources of variants are ClinVar (Landrum et al., 2018) database of variants and their disease relationship, ProTherm thermodynamic database (Kumar et al., 2006), and VariBench itself. The number of unique variants is significantly lower than the sum of the variants in the data sets.
The data sets are divided into 5 categories, 17 subgroups and 11 groups (Table 1). The amount of data items varies for independent sets and is dependent on the original data. Data items irrelevant to VariBench (i.e., not describing variants or their effects) were removed when sets were included to the database. In many data sets, variants are described at three molecular levels (DNA, RNA and protein) and sometimes also at protein structural level. One of the aims of VariBench is to facilitate the reuse of existing data sets, therefore the data are provided in as many levels as possible. Further, the data can be used for various purposes, beyond the original application, such as benchmarking, developing different types of predictors, bioinformatics reviews and analyses of variation types, clinical variation interpretation, etc. When doing such an extension, the users must be cautious and aware of the possible limitations of the data sets and to understand how they have been collected.
The main categories of variation type data sets are insertions and deletions, substitutions in coding and non-coding regions, structure-mapped variants, synonymous and unsense variants, benign variants, and DNA structural variants (See Tables 1, 2). Unsense variants are a new category for exonic alterations that may look synonymous, but affect the protein or its expression, typically due to aberrant splicing or miRNA binding alterations (Vihinen, 2022; Vihinen, 2023a; Vihinen, 2023b). Effect-specific data sets include DNA regulatory elements, RNA splicing, and protein property for aggregation, binding free energy, disorder, solubility, stability, folding rate, interactions, and functional effects. Molecule- and disease-specific data sets include information for individual genes, proteins, gene/protein families or diseases. Phenotype data sets are for a disease feature, severity of the phenotype.
Almost all the categories contain new data sets. In addition, we have 6 new variation categories including structural variations in DNA (1 data set), protein folding rate (5 data sets in six publications), antibody-antigen affinity changes (5 articles and sets), protein-nucleic acid interactions (6 articles), gain of function variants (Nurk et al., 2022), and deep mutational data sets (7 studies).
One of the new categories is for functional effects under the effect-specific category. These sets are mainly for massively parallel reporter assays (saturation mutagenesis) experiments. Users of these data have to be careful since the included data sets display a measured effect; however, their relevance to biological effect is not always clear, see (Vihinen, 2021). The functional effect does not necessarily mean biological effect. One would likely say that a reduction of more than 50% of e.g., enzyme activity has a functional effect. There are several diseases where 90% or more of the normal activity has to be lost for an individual to have a disease and show the effect on biological activity (Vihinen, 2021). Examples include hemophilias due to factor II, VII, IX, X or XII variations and severe immunodeficiency caused by adenosine deaminase alterations.
Data availability statement
Publicly available datasets were analyzed in this study. This data can be found here: http://structure.bmc.lu.se/VariBench.
Author contributions
MV conceived the project; NS collected the data sets and developed the web site; NS and MV wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
Financial support from Vetenskapsrådet (2019-01403) and the Swedish Cancer Society (grant number CAN 20 1350) is gratefully acknowledged.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Anderson, D., and Lassmann, T. (2022). An expanded phenotype centric benchmark of variant prioritisation tools. Hum. Mutat. 43, 539–546. doi:10.1002/humu.24362
Auton, A., Brooks, L. D., Durbin, R. M., Garrison, E. P., Kang, H. M., Korbel, J. O., et al. (2015). A global reference for human genetic variation. Nature 526, 68–74. doi:10.1038/nature15393
Ben-Cohen, G., Doffe, F., Devir, M., Leroy, B., Soussi, T., and Rosenberg, S. (2022). TP53_PROF: A machine learning model to predict impact of missense mutations in TP53. Brief. Bioinform 23, bbab524. doi:10.1093/bib/bbab524
Benedix, A., Becker, C. M., de Groot, B. L., Caflisch, A., and Böckmann, R. A. (2009). Predicting free energy changes using structural ensembles. Nat. Methods 6, 3–4. doi:10.1038/nmeth0109-3
Benevenuta, S., Pancotti, C., Fariselli, P., Birolo, G., and Sanavia, T. (2021). An antisymmetric neural network to predict free energy changes in protein variants. J. Phys. D. Appl. Phys. 54, 245403. doi:10.1088/1361-6463/abedfb
Biggs, H., Parthasarathy, P., Gavryushkina, A., and Gardner, P. P. (2020). ncVarDB: a manually curated database for pathogenic non-coding variants and benign controls. Oxford: Database, 2020.
Borges, P., Pasqualim, G., and Matte, U. (2021). Which is the best in silico program for the missense variations in idua gene? A comparison of 33 programs plus a conservation score and evaluation of 586 missense variants. Front. Mol. Biosci. 8, 752797. doi:10.3389/fmolb.2021.752797
Buske, O. J., Manickaraj, A., Mital, S., Ray, P. N., and Brudno, M. (2013). Identification of deleterious synonymous variants in human genomes. Bioinformatics 29, 1843–1850. doi:10.1093/bioinformatics/btt308
Chan, A. O. (2013). Performance of in silico analysis in predicting the effect of non-synonymous variants in inherited steroid metabolic diseases. Steroids 78, 726–730. doi:10.1016/j.steroids.2013.04.002
Chaudhary, P., Naganathan, A. N., and Gromiha, M. M. (2015). Folding RaCe: A robust method for predicting changes in protein folding rates upon point mutations. Bioinformatics 31, 2091–2097. doi:10.1093/bioinformatics/btv091
Chaudhary, P., Naganathan, A. N., and Gromiha, M. M. (2016). Prediction of change in protein unfolding rates upon point mutations in two state proteins. Biochim. Biophys. Acta 1864, 1104–1109. doi:10.1016/j.bbapap.2016.06.001
Chen, C. W., Lin, J., and Chu, Y. W. (2013). iStable: off-the-shelf predictor integration for predicting protein stability changes. BMC Bioinforma. 14, S5. Suppl 2. doi:10.1186/1471-2105-14-s2-s5
Chen, C. W., Lin, M. H., Liao, C. C., Chang, H. P., and Chu, Y. W. (2020). iStable 2.0: predicting protein thermal stability changes by integrating various characteristic modules. Comput. Struct. Biotechnol. J. 18, 622–630. doi:10.1016/j.csbj.2020.02.021
Clark, A. J., Negron, C., Hauser, K., Sun, M., Wang, L., Abel, R., et al. (2019). Relative binding affinity prediction of charge-changing sequence mutations with FEP in protein-protein interfaces. J. Mol. Biol. 431, 1481–1493. doi:10.1016/j.jmb.2019.02.003
Danis, D., Jacobsen, J. O. B., Carmody, L. C., Gargano, M. A., McMurry, J. A., Hegde, A., et al. (2021). Interpretable prioritization of splice variants in diagnostic next-generation sequencing. Am. J. Hum. Genet. 108, 1564–1577. doi:10.1016/j.ajhg.2021.06.014
Dunham, A. S., and Beltrao, P. (2021). Exploring amino acid functions in a deep mutational landscape. Mol. Syst. Biol. 17, e10305. doi:10.15252/msb.202110305
Ganakammal, S. R., and Alexov, E. (2020). An ensemble approach to predict the pathogenicity of synonymous variants. Genes. (Basel), 11. doi:10.3390/genes11091102
Ganakammal, S. R., and Alexov, E. (2019). Evaluation of performance of leading algorithms for variant pathogenicity predictions and designing a combinatory predictor method: application to rett syndrome variants. PeerJ 7, e8106. doi:10.7717/peerj.8106
Gao, M., Zhou, H., and Skolnick, J. (2015). Insights into disease-associated mutations in the human proteome through protein structural analysis. Structure 23, 1362–1369. doi:10.1016/j.str.2015.03.028
Garcia-Recio, A., Gómez-Tamayo, J. C., Reina, I., Campillo, M., Cordomí, A., Olivella, M., et al. (2021). Tmsnp: A web server to predict pathogenesis of missense mutations in the transmembrane region of membrane proteins. Nar. Genom Bioinform 3, lqab008. doi:10.1093/nargab/lqab008
Ge, F., Zhu, Y. H., Xu, J., Muhammad, A., Song, J., and Yu, D. J. (2021). MutTMPredictor: robust and accurate cascade xgboost classifier for prediction of mutations in transmembrane proteins. Comput. Struct. Biotechnol. J. 19, 6400–6416. doi:10.1016/j.csbj.2021.11.024
Gelfman, S., Wang, Q., McSweeney, K. M., Ren, Z., La Carpia, F., Halvorsen, M., et al. (2017). Annotating pathogenic non-coding variants in genic regions. Nat. Commun. 8, 236. doi:10.1038/s41467-017-00141-2
Geng, C., Vangone, A., Folkers, G. E., Xue, L. C., and Bonvin, A. (2019). iSEE: interface structure, evolution, and energy-based machine learning predictor of binding affinity changes upon mutations. Proteins 87, 110–119. doi:10.1002/prot.25630
Goncearenco, A., Rager, S. L., Li, M., Sang, Q. X., Rogozin, I. B., and Panchenko, A. R. (2017). Exploring background mutational processes to decipher cancer genetic heterogeneity. Nucleic Acids Res. 45, W514–w522. doi:10.1093/nar/gkx367
Grønning, A. G. B., Doktor, T. K., Larsen, S. J., Petersen, U. S. S., Holm, L. L., Bruun, G. H., et al. (2020). DeepCLIP: predicting the effect of mutations on protein-rna binding with deep learning. Nucleic Acids Res. 48, 7099–7118. doi:10.1093/nar/gkaa530
Gunning, A. C., Fryer, V., Fasham, J., Crosby, A. H., Ellard, S., Baple, E. L., et al. (2021). Assessing performance of pathogenicity predictors using clinically relevant variant datasets. J. Med. Genet. 58, 547–555. doi:10.1136/jmedgenet-2020-107003
Heyne, H. O., Baez-Nieto, D., Iqbal, S., Palmer, D. S., Brunklaus, A., May, P., et al. (2020). Predicting functional effects of missense variants in voltage-gated sodium and calcium channels. Sci. Transl. Med. 12, eaay6848. doi:10.1126/scitranslmed.aay6848
Huang, L. T. (2014). Finding simple rules for discriminating folding rate change upon single mutation by statistical and learning methods. Protein Pept. Lett. 21, 743–751. doi:10.2174/09298665113209990070
Huang, L. T., and Gromiha, M. M. (2010). First insight into the prediction of protein folding rate change upon point mutation. Bioinformatics 26, 2121–2127. doi:10.1093/bioinformatics/btq350
Huang, L. T., and Gromiha, M. M. (2012). Real value prediction of protein folding rate change upon point mutation. J. Comput. Aided Mol. Des. 26, 339–347. doi:10.1007/s10822-012-9560-3
Huang, X., Zheng, W., Pearce, R., and Zhang, Y. (2020). SSIPe: accurately estimating protein-protein binding affinity change upon mutations using evolutionary profiles in combination with an optimized physical energy function. Bioinformatics 36, 2429–2437. doi:10.1093/bioinformatics/btz926
Ittisoponpisan, S., Islam, S. A., Khanna, T., Alhuzimi, E., David, A., and Sternberg, M. J. E. (2019). Can predicted protein 3D structures provide reliable insights into whether missense variants are disease associated? J. Mol. Biol. 431, 2197–2212. doi:10.1016/j.jmb.2019.04.009
Jemimah, S., Sekijima, M., and Gromiha, M. M. (2020). ProAffiMuSeq: sequence-based method to predict the binding free energy change of protein-protein complexes upon mutation using functional classification. Bioinformatics 36, 1725–1730. doi:10.1093/bioinformatics/btz829
Jian, X., Boerwinkle, E., and Liu, X. (2014). In silico prediction of splice-altering single nucleotide variants in the human genome. Nucleic Acids Res. 42, 13534–13544. doi:10.1093/nar/gku1206
Jiang, T., Fang, L., and Wang, K. (2021). MutFormer: A context-dependent transformer-based model to predict pathogenic missense mutations. Available at: https://arxiv.org/abs/2110.14746.
Kono, T. J. Y., Lei, L., Shih, C. H., Hoffman, P. J., Morrell, P. L., and Fay, J. C. (2018). Comparative genomics approaches accurately predict deleterious variants in plants. G3 (Bethesda) 8, 3321–3329. doi:10.1534/g3.118.200563
Kovalev, M. S., Igolkina, A. A., Samsonova, M. G., and Nuzhdin, S. V. (2018). A pipeline for classifying deleterious coding mutations in agricultural plants. Front. Plant Sci. 9, 1734. doi:10.3389/fpls.2018.01734
Kumar, M. D., Bava, K. A., Gromiha, M. M., Prabakaran, P., Kitajima, K., Uedaira, H., et al. (2006). ProTherm and ProNIT: thermodynamic databases for proteins and protein-nucleic acid interactions. Nucleic Acids Res. 34, D204–D206. doi:10.1093/nar/gkj103
Kurumida, Y., Saito, Y., and Kameda, T. (2020). Predicting antibody affinity changes upon mutations by combining multiple predictors. Sci. Rep. 10, 19533. doi:10.1038/s41598-020-76369-8
Landrum, M. J., Lee, J. M., Benson, M., Brown, G. R., Chao, C., Chitipiralla, S., et al. (2018). ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 46, D1062–d1067. doi:10.1093/nar/gkx1153
Lee, D., Gorkin, D. U., Baker, M., Strober, B. J., Asoni, A. L., McCallion, A. S., et al. (2015). A method to predict the impact of regulatory variants from DNA sequence. Nat. Genet. 47, 955–961. doi:10.1038/ng.3331
Leman, R., Gaildrat, P., Le Gac, G., Ka, C., Fichou, Y., Audrezet, M. P., et al. (2018). Novel diagnostic tool for prediction of variant spliceogenicity derived from a set of 395 combined in silico/in vitro studies: an international collaborative effort. Nucleic Acids Res. 46, 7913–7923. doi:10.1093/nar/gky372
Li, B., Yang, Y. T., Capra, J. A., and Gerstein, M. B. (2020). Predicting changes in protein thermodynamic stability upon point mutation with deep 3D convolutional neural networks. PLoS Comput. Biol. 16, e1008291. doi:10.1371/journal.pcbi.1008291
Li, G., Panday, S. K., Peng, Y., and Alexov, E. (2021b). SAMPDI-3D: predicting the effects of protein and dna mutations on protein-dna interactions. Bioinformatics 37, 3760–3765. doi:10.1093/bioinformatics/btab567
Li, M., Petukh, M., Alexov, E., and Panchenko, A. R. (2014). Predicting the impact of missense mutations on protein-protein binding affinity. J. Chem. Theory Comput. 10, 1770–1780. doi:10.1021/ct401022c
Li, M., Simonetti, F. L., Goncearenco, A., and Panchenko, A. R. (2016). MutaBind estimates and interprets the effects of sequence variants on protein-protein interactions. Nucleic Acids Res. 44, W494–W501. doi:10.1093/nar/gkw374
Li, Y., Burgman, B., Khatri, I. S., Pentaparthi, S. R., Su, Z., McGrail, D. J., et al. (2021a). e-MutPath: computational modeling reveals the functional landscape of genetic mutations rewiring interactome networks. Nucleic Acids Res. 49, e2. doi:10.1093/nar/gkaa1015
Liu, J., Liu, S., Liu, C., Zhang, Y., Pan, Y., Wang, Z., et al. (2021). Nabe: An energetic database of amino acid mutations in protein-nucleic acid binding interfaces. Oxford: Database, 2021.
Livesey, B. J., and Marsh, J. A. (2020). Using deep mutational scanning to benchmark variant effect predictors and identify disease mutations. Mol. Syst. Biol. 16, e9380. doi:10.15252/msb.20199380
Mahecha, D., Nuñez, H., Lattig, M. C., and Duitama, J. (2022). Machine learning models for accurate prioritization of variants of uncertain significance. Hum. Mutat. 43, 449–460. doi:10.1002/humu.24339
Marabotti, A., Del Prete, E., Scafuri, B., and Facchiano, A. (2021). Performance of Web tools for predicting changes in protein stability caused by mutations. BMC Bioinforma. 22, 345. doi:10.1186/s12859-021-04238-w
Martin, T. A., Wu, T., Tang, Q., Dougherty, L. L., Parente, D. J., Swint-Kruse, L., et al. (2020). Identification of biochemically neutral positions in liver pyruvate kinase. Proteins 88, 1340–1350. doi:10.1002/prot.25953
Miller, M., Bromberg, Y., and Swint-Kruse, L. (2017). Computational predictors fail to identify amino acid substitution effects at rheostat positions. Sci. Rep. 7, 41329. doi:10.1038/srep41329
Miller, M., Vitale, D., Kahn, P. C., Rost, B., Bromberg, Y., and funtrp, (2019). funtrp: identifying protein positions for variation driven functional tuning. Nucleic Acids Res. 47, e142. doi:10.1093/nar/gkz818
Moles-Fernández, A., Duran-Lozano, L., Montalban, G., Bonache, S., López-Perolio, I., Menéndez, M., et al. (2018). Computational tools for splicing defect prediction in breast/ovarian cancer genes: how efficient are they at predicting rna alterations? Front. Genet. 9, 366. doi:10.3389/fgene.2018.00366
Myung, Y., Rodrigues, C. H. M., Ascher, D. B., Pires, D. E. V., and mCSM-Ab2, (2020). mCSM-AB2: guiding rational antibody design using graph-based signatures. Bioinformatics 36, 1453–1459. doi:10.1093/bioinformatics/btz779
Naganathan, A. N., and Muñoz, V. (2010). Insights into protein folding mechanisms from large scale analysis of mutational effects. Proc. Natl. Acad. Sci. U. S. A. 107, 8611–8616. doi:10.1073/pnas.1000988107
Nair, P. S., Vihinen, M., and VariBench, (2013). VariBench: A benchmark database for variations. Hum. Mutat. 34, 42–49. doi:10.1002/humu.22204
Niroula, A., and Vihinen, M. (2019). How good are pathogenicity predictors in detecting benign variants? PLoS Comput. Biol. 15, e1006481. doi:10.1371/journal.pcbi.1006481
Niroula, A., and Vihinen, M. (2016). Variation interpretation predictors: principles, types, performance, and choice. Hum. Mutat. 37, 579–597. doi:10.1002/humu.22987
Nurk, S., Koren, S., Rhie, A., Rautiainen, M., Bzikadze, A. V., Mikheenko, A., et al. (2022). The complete sequence of a human genome. Science 376, 44–53. doi:10.1126/science.abj6987
Orioli, T., and Vihinen, M. (2019). Benchmarking membrane proteins: subcellular localization and variant tolerance predictors. BMC Genomics 20, 547. doi:10.1186/s12864-019-5865-0
Pagel, K. A., Antaki, D., Lian, A., Mort, M., Cooper, D. N., Sebat, J., et al. (2019). Pathogenicity and functional impact of non-frameshifting insertion/deletion variation in the human genome. PLoS Comput. Biol. 15, e1007112. doi:10.1371/journal.pcbi.1007112
Pancotti, C., Benevenuta, S., Birolo, G., Alberini, V., Repetto, V., Sanavia, T., et al. (2022). Predicting protein stability changes upon single-point mutation: A thorough comparison of the available tools on a new dataset. Brief. Bioinform 23, bbab555. doi:10.1093/bib/bbab555
Pei, J., Kinch, L. N., Otwinowski, Z., and Grishin, N. V. (2020). Mutation severity spectrum of rare alleles in the human genome is predictive of disease type. PLoS Comput. Biol. 16, e1007775. doi:10.1371/journal.pcbi.1007775
Pejaver, V., Urresti, J., Lugo-Martinez, J., Pagel, K. A., Lin, G. N., Nam, H. J., et al. (2020). Inferring the molecular and phenotypic impact of amino acid variants with MutPred2. Nat. Commun. 11, 5918. doi:10.1038/s41467-020-19669-x
Peng, Y., Sun, L., Jia, Z., Li, L., and Alexov, E. (2018). Predicting protein-DNA binding free energy change upon missense mutations using modified MM/PBSA approach: SAMPDI webserver. Bioinformatics 34, 779–786. doi:10.1093/bioinformatics/btx698
Petrosino, M., Novak, L., Pasquo, A., Chiaraluce, R., Turina, P., Capriotti, E., et al. (2021). Analysis and interpretation of the impact of missense variants in cancer. Int. J. Mol. Sci., 22.
Petukh, M., Dai, L., and Alexov, E. (2016). Saambe: webserver to predict the charge of binding free energy caused by amino acids mutations. Int. J. Mol. Sci. 17, 547. doi:10.3390/ijms17040547
Pires, D. E., and Ascher, D. B. (2016). mCSM-AB: a web server for predicting antibody-antigen affinity changes upon mutation with graph-based signatures. Nucleic Acids Res. 44, W469–W473. doi:10.1093/nar/gkw458
Pires, D. E. V., and Ascher, D. B. (2017). mCSM-NA: predicting the effects of mutations on protein-nucleic acids interactions. Nucleic Acids Res. 45, W241–w246. doi:10.1093/nar/gkx236
Pires, D. E. V., Rodrigues, C. H. M., and Ascher, D. B. (2020). mCSM-membrane: predicting the effects of mutations on transmembrane proteins. Nucleic Acids Res. 48, W147–w153. doi:10.1093/nar/gkaa416
Plekhanova, E., Nuzhdin, S. V., Utkin, L. V., and Samsonova, M. G. (2019). Prediction of deleterious mutations in coding regions of mammals with transfer learning. Evol. Appl. 12, 18–28. doi:10.1111/eva.12607
Pons, T., Vazquez, M., Matey-Hernandez, M. L., Brunak, S., Valencia, A., and Izarzugaza, J. M. (2016). KinMutRF: A random forest classifier of sequence variants in the human protein kinase superfamily. BMC Genomics 17, 396. Suppl 2. doi:10.1186/s12864-016-2723-1
Quinodoz, M., Peter, V. G., Cisarova, K., Royer-Bertrand, B., Stenson, P. D., Cooper, D. N., et al. (2022). Analysis of missense variants in the human genome reveals widespread gene-specific clustering and improves prediction of pathogenicity. Am. J. Hum. Genet. 109, 457–470. doi:10.1016/j.ajhg.2022.01.006
Raponi, M., Kralovicova, J., Copson, E., Divina, P., Eccles, D., Johnson, P., et al. (2011). Prediction of single-nucleotide substitutions that result in exon skipping: identification of a splicing silencer in brca1 exon 6. Hum. Mutat. 32, 436–444. doi:10.1002/humu.21458
Reeb, J., Wirth, T., and Rost, B. (2020). Variant effect predictions capture some aspects of deep mutational scanning experiments. BMC Bioinforma. 21, 107. doi:10.1186/s12859-020-3439-4
Rentzsch, P., Schubach, M., Shendure, J., and Kircher, M. (2021). CADD-Splice-improving genome-wide variant effect prediction using deep learning-derived splice scores. Genome Med. 13, 31. doi:10.1186/s13073-021-00835-9
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of medical genetics and genomics and the association for molecular Pathology. Genet. Med. 17, 405–424. doi:10.1038/gim.2015.30
Riesselman, A. J., Ingraham, J. B., and Marks, D. S. (2018). Deep generative models of genetic variation capture the effects of mutations. Nat. Methods 15, 816–822. doi:10.1038/s41592-018-0138-4
Rodrigues, C. H. M., Myung, Y., Pires, D. E. V., Ascher, D. B., and mCSM-Ppi2, (2019). mCSM-PPI2: predicting the effects of mutations on protein–protein interactions. Nucleic Acids Res. 47, W338–w344. doi:10.1093/nar/gkz383
Rodrigues, C. H. M., Pires, D. E. V., Ascher, D. B., and mmCSM-Ppi, (2021). mmCSM-PPI: predicting the effects of multiple point mutations on protein–protein interactions. Nucleic Acids Res. 49, W417–w424. doi:10.1093/nar/gkab273
Rychkova, A., Buu, M., Scharfe, C., Lefterova, M., Odegaard, J., Schrijver, I., et al. (2017). Developing gene-specific meta-predictor of variant pathogenicity.
Sarkar, A., Yang, Y., and Vihinen, M. (2020). Variation benchmark datasets: update, criteria, quality and applications. Database 2020, baz117. doi:10.1093/database/baz117
Sasorith, S., Baux, D., Bergougnoux, A., Paulet, D., Lahure, A., Bareil, C., et al. (2020). The CYSMA web server: an example of integrative tool for in silico analysis of missense variants identified in mendelian disorders. Hum. Mutat. 41, 375–386. doi:10.1002/humu.23941
Savojardo, C., Manfredi, M., Martelli, P. L., and Casadio, R. (2020). Solvent accessibility of residues undergoing pathogenic variations in humans: from protein structures to protein sequences. Front. Mol. Biosci. 7, 626363. doi:10.3389/fmolb.2020.626363
Schaafsma, G. C., and Vihinen, M. (2018). Representativeness of variation benchmark datasets. BMC Bioinforma. 19 (1), 461. doi:10.1186/s12859-018-2478-6
Schaafsma, G. C., Vihinen, M., and VariSNP, (2015). VariSNP, A benchmark database for variations from dbSNP. Hum. Mutat. 36, 161–166. doi:10.1002/humu.22727
Shakur, R., Ochoa, J. P., Robinson, A. J., Niroula, A., Chandran, A., Rahman, T., et al. (2021). Prognostic implications of troponin T variations in inherited cardiomyopathies using systems biology. NPJ Genom Med. 6, 47. doi:10.1038/s41525-021-00204-w
Sharo, A. G., Hu, Z., Sunyaev, S. R., and Brenner, S. E. (2022). StrVCTVRE: A supervised learning method to predict the pathogenicity of human genome structural variants. Am. J. Hum. Genet. 109, 195–209. doi:10.1016/j.ajhg.2021.12.007
Sherry, S. T., Ward, M. H., Kholodov, M., Baker, J., Phan, L., Smigielski, E. M., et al. (2001). dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 29, 308–311. doi:10.1093/nar/29.1.308
Stourac, J., Dubrava, J., Musil, M., Horackova, J., Damborsky, J., Mazurenko, S., et al. (2021). FireProtDB: database of manually curated protein stability data. Nucleic Acids Res. 49, D319–d324. doi:10.1093/nar/gkaa981
Strokach, A., Corbi-Verge, C., and Kim, P. M. (2019). Predicting changes in protein stability caused by mutation using sequence-and structure-based methods in a CAGI5 blind challenge. Hum. Mutat. 40, 1414–1423. doi:10.1002/humu.23852
Strokach, A., Lu, T. Y., and Kim, P. M. (2021). ELASPIC2 (EL2): combining contextualized language models and graph neural networks to predict effects of mutations. J. Mol. Biol. 433, 166810. doi:10.1016/j.jmb.2021.166810
Sulea, T., Vivcharuk, V., Corbeil, C. R., Deprez, C., and Purisima, E. O. (2016). Assessment of solvated interaction energy function for ranking antibody-antigen binding affinities. J. Chem. Inf. Model. 56, 1292–1303. doi:10.1021/acs.jcim.6b00043
Tang, X., Zhang, T., Cheng, N., Wang, H., Zheng, C. H., Xia, J., et al. (2021). usDSM: a novel method for deleterious synonymous mutation prediction using undersampling scheme. Brief. Bioinform 22, bbab123. doi:10.1093/bib/bbab123
Tarnovskaya, S. I., Korkosh, V. S., Zhorov, B. S., and Frishman, D. (2020). Predicting novel disease mutations in the cardiac sodium channel. Biochem. Biophys. Res. Commun. 521, 603–611. doi:10.1016/j.bbrc.2019.10.142
Thusberg, J., Olatubosun, A., and Vihinen, M. (2011). Performance of mutation pathogenicity prediction methods on missense variants. Hum. Mutat. 32, 358–368. doi:10.1002/humu.21445
Tian, J., Wu, N., Chu, X., and Fan, Y. (2010). Predicting changes in protein thermostability brought about by single- or multi-site mutations. BMC Bioinforma. 11, 370. doi:10.1186/1471-2105-11-370
Toffano, A. A., Chiarot, G., Zamuner, S., Marchi, M., Salvi, E., Waxman, S. G., et al. (2020). Computational pipeline to probe NaV1.7 gain-of-function variants in neuropathic painful syndromes. Sci. Rep. 10, 17930. doi:10.1038/s41598-020-74591-y
Turina, P., Fariselli, P., and Capriotti, E. (2021). ThermoScan: semi-automatic identification of protein stability data from Pubmed. Front. Mol. Biosci. 8, 620475. doi:10.3389/fmolb.2021.620475
Vihinen, M. (2021). Functional effects of protein variants. Biochimie 180, 104–120. doi:10.1016/j.biochi.2020.10.009
Vihinen, M. (2013). Guidelines for reporting and using prediction tools for genetic variation analysis. Hum. Mutat. 34, 275–282. doi:10.1002/humu.22253
Vihinen, M. (2012). How to evaluate performance of prediction methods? Measures and their interpretation in variation effect analysis. BMC Genomics 13, S2. Suppl 4. doi:10.1186/1471-2164-13-s4-s2
Vihinen, M. (2023b). Nonsynonymous synonymous variants demand for a paradigm shift in genetics. Curr. Genet. 24, 18–23. doi:10.2174/1389202924666230417101020
Vihinen, M. (2023a). Systematic errors in annotations of truncations, loss-of-function and synonymous variants. Front. Genet. 14, 1015017. doi:10.3389/fgene.2023.1015017
Wang, M., Cang, Z., and Wei, G. W. (2020). A topology-based network tree for the prediction of protein-protein binding affinity changes following mutation. Nat. Mach. Intell. 2, 116–123. doi:10.1038/s42256-020-0149-6
Wang, Y., Jiang, Y., Yao, B., Huang, K., Liu, Y., Wang, Y., et al. (2021). WEVar: A novel statistical learning framework for predicting noncoding regulatory variants. Brief. Bioinform 22, bbab189. doi:10.1093/bib/bbab189
Wu, Y., Li, R., Sun, S., Weile, J., and Roth, F. P. (2021). Improved pathogenicity prediction for rare human missense variants. Am. J. Hum. Genet. 108, 1891–1906. doi:10.1016/j.ajhg.2021.08.012
Xiong, P., Zhang, C., Zheng, W., and Zhang, Y. (2017). BindProfX: assessing mutation-induced binding affinity change by protein interface profiles with pseudo-counts. J. Mol. Biol. 429, 426–434. doi:10.1016/j.jmb.2016.11.022
Yang, Y., Shao, A., and Vihinen, M. (2022). PON-All, amino acid substitution tolerance predictor for all organisms. Front. Mol. Biosci. 9, 867572. doi:10.3389/fmolb.2022.867572
Yang, Y., Urolagin, S., Niroula, A., Ding, X., Shen, B., and Vihinen, M. (2018). PON-Tstab: protein variant stability predictor importance of training data quality. Int. J. Mol. Sci. 19, 1009. doi:10.3390/ijms19041009
Yang, Y., Zeng, L., Vihinen, M., and Pon-Sol2, (2021). Prediction of effects of variants on protein solubility. Int. J. Mol. Sci., 22.
Yue, Z., Zhao, L., Cheng, N., Yan, H., and Xia, J. (2019). dbCID: a manually curated resource for exploring the driver indels in human cancer. Brief. Bioinform 20, 1925–1933. doi:10.1093/bib/bby059
Yue, Z., Zhao, L., and Xia, J. (2018). dbCPM: a manually curated database for exploring the cancer passenger mutations. Brief. Bioinform 21, 309–317. doi:10.1093/bib/bby105
Zeng, Z., and Bromberg, Y. (2019). Predicting functional effects of synonymous variants: A systematic review and perspectives. Front. Genet. 10, 914. doi:10.3389/fgene.2019.00914
Zhang, N., Chen, Y., Lu, H., Zhao, F., Alvarez, R. V., Goncearenco, A., et al. (2020). MutaBind2: predicting the impacts of single and multiple mutations on protein-protein interactions. iScience 23, 100939. doi:10.1016/j.isci.2020.100939
Zhang, N., Chen, Y., Zhao, F., Yang, Q., Simonetti, F. L., and Li, M. (2018). PremPDI estimates and interprets the effects of missense mutations on protein-DNA interactions. PLoS Comput. Biol. 14, e1006615. doi:10.1371/journal.pcbi.1006615
Zhang, S., He, Y., Liu, H., Zhai, H., Huang, D., Yi, X., et al. (2019). regBase: whole genome base-wise aggregation and functional prediction for human non-coding regulatory variants. Nucleic Acids Res. 47, e134. doi:10.1093/nar/gkz774
Zhang, X., Walsh, R., Whiffin, N., Buchan, R., Midwinter, W., Wilk, A., et al. (2021). Disease-specific variant pathogenicity prediction significantly improves variant interpretation in inherited cardiac conditions. Genet. Med. 23, 69–79. doi:10.1038/s41436-020-00972-3
Zhou, J. B., Xiong, Y., An, K., Ye, Z. Q., and Wu, Y. D. (2020). IDRMutPred: predicting disease-associated germline nonsynonymous single nucleotide variants (nssnvs) in intrinsically disordered regions. Bioinformatics 36, 4977–4983. doi:10.1093/bioinformatics/btaa618
Keywords: variation, mutation, benchmark, method performance assessment, data sets, variation database
Citation: Shirvanizadeh N and Vihinen M (2023) VariBench, new variation benchmark categories and data sets. Front. Bioinform. 3:1248732. doi: 10.3389/fbinf.2023.1248732
Received: 27 June 2023; Accepted: 08 September 2023;
Published: 19 September 2023.
Edited by:
Marcelo Reis, State University of Campinas, BrazilReviewed by:
Castrense Savojardo, University of Bologna, ItalyCarlos Rodrigues, Baker Heart and Diabetes Institute, Australia
Seyed Jamalaldin Haddadi, State University of Campinas, Brazil
Copyright © 2023 Shirvanizadeh and Vihinen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mauno Vihinen, bWF1bm8udmloaW5lbkBtZWQubHUuc2U=
†Present address: Niloofar Shirvanizadeh, Cancer Genomics and Proteomics, Karolinska University Hospital, Huddinge, Sweden