 Chang Li
Chang Li Ian Hou2
Ian Hou2 Yongsheng Bai
Yongsheng Bai Xiaoming Liu
Xiaoming Liu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Bioinform. , 06 April 2023
Sec. Protein Bioinformatics
Volume 3 - 2023 | https://doi.org/10.3389/fbinf.2023.1122559
This article is part of the Research Topic Revisiting the Protein Universe by Deep Learning View all 4 articles
Introduction: Alzheimer’s disease (AD) is one of the most prominent medical conditions in the world. Understanding the genetic component of the disease can greatly advance our knowledge regarding its progression, treatment and prognosis. Single amino-acid variants (SAVs) in the APOE gene have been widely investigated as a risk factor for AD Studies, including genome-wide association studies, meta-analysis based studies, and in-vivo animal studies, were carried out to investigate the functional importance and pathogenesis potential of APOE SAVs. However, given the high cost of such large-scale or experimental studies, there are only a handful of variants being reported that have definite explanations. The recent development of in-silico analytical approaches, especially large-scale deep learning models, has opened new opportunities for us to probe the structural and functional importance of APOE variants extensively.
Method: In this study, we are taking an ensemble approach that simultaneously uses large-scale protein sequence-based models, including Evolutionary Scale Model and AlphaFold, together with a few in-silico functional prediction web services to investigate the known and possibly disease-causing SAVs in APOE and evaluate their likelihood of being functional and structurally disruptive.
Results: As a result, using an ensemble approach with little to no prior field-specific knowledge, we reported 5 SAVs in APOE gene to be potentially disruptive, one of which (C112R) was classificed by previous studies as a key risk factor for AD.
Discussion: Our study provided a novel framework to analyze and prioritize the functional and structural importance of SAVs for future experimental and functional validation.
Alzheimer’s disease (AD), a complex disease with a known genetic basis, is the most prominent cause of dementia in the elderly (Bettens et al., 2013). Understanding the genetic component of AD can be of great importance in its early diagnosis, effective treatment and improved prognosis. It has been widely studied and reported that the apolipoprotein E (APOE) gene, which is a key gene for lipid transportation, is closely associated with the risk of AD (Kamboh et al., 1995; Yamazaki et al., 2019; Martens et al., 2022). APOE gene has 3 different protein isoforms, namely, APOE2, APOE3, and APOE4 (Husain et al., 2021). These isoforms differ by two amino acids, APOE2 with Cys112 and Cys158, APOE3 with Cys112 and Arg158, and APOE4 with Arg112 and Arg158. APOE3 was considered the reference isoform and the APOE4 Cys112Arg variant was a strong risk factor for AD, while APOE2 Arg158Cys variant was reported to be protective (Bojanowski et al., 2006; Dolai et al., 2020). Given the functional importance and pathogenesis potential of APOE variants, many experimental studies using animal models, genome-wide association studies, and other meta-analyses have been performed to interrogate the impact of variants residing in the APOE gene (Bertram et al., 2008; Liu et al., 2014; Lewandowski et al., 2020). However, given the high cost of such large-scale or experimental studies, there are only a handful of variants being reported that have definite explanations.
The recent development of in silico analytical approaches, especially large-scale deep learning models, has opened new opportunities for us to probe the structural and functional importance of APOE variants extensively. Specifically, AlphaFold (Jumper et al., 2021), which exploited attention mechanisms from language modeling and multiple sequence alignment (MSA) data of protein homologs, has provided substantially increased coverage of high-confidence protein structure predictions. Additionally, the Evolutionary Scale Model (ESM) (Lin et al., 2022), which was pre-trained on 250 million protein sequences, has proven to be able to extract key functional domains and evaluate the functional importance of amino acid variants (Brandes et al., 2022) even in the absence of multiple sequence alignment (MSA) data which were required in AlphaFold modeling. Recent studies have tried to examine the ability of these tools individually to evaluate the impact of single amino-acid variants (SAVs), but reported conflicting results (Pak et al., 2021; Caswell et al., 2022). In this study, instead of using these tools separately, we are taking an ensemble approach that simultaneously uses these two large-scale protein sequence-based models together with a few in silico functional prediction web services to investigate the known and possibly disease-causing variants in APOE and evaluate their likelihood of being functional and structurally disruptive.
The protein sequence of the APOE gene was retrieved from Ensembl genome browser v107 in FASTA format (https://useast.ensembl.org/index.html). Python package Biopython was used to load and process the retrieved sequence. Only the reference isoform (APOE3) and the precursor APOE (pre-APOE) sequences were used in this study. The difference between pre-APOE and mature APOE was the addition of an 18-residue signal peptide at the beginning of the sequence. As a result, previously reported variants with respect to mature APOE, such as C112R and R158C, were reported as C130R and R176C, respectively, in this study.
The C130R variant was manually introduced to create a separate sequence representing APOE4, and R176C was manually introduced to create a separate sequence representing APOE2.
ESM-1b model was retrieved from GitHub (https://github.com/facebookresearch/esm) using PyTorch Hub. The same tokenizer as the original ESM model was used to encode input protein sequences. The variant effect for each amino acid variant (ESM score) was calculated as the log-likelihood ratio between the variant and the corresponding reference amino acid. To show a positive score, we multiplied each prediction score by −1.
The variant was predicted to be more damaging if it had a higher ESM score.
AlphaFold v2 model was run locally using a third-party implementation, namely, LocalColabFold (https://github.com/YoshitakaMo/localcolabfold) (Jumper et al., 2021; Mirdita et al., 2022). The algorithm first implements MMseqs2 (Steinegger and Söding, 2017) to retrieve MSA for the target protein. Then, it predicts the 3D protein conformation for the given sequence.
Due to the high computational cost of running AlphaFold, it was extremely time-consuming to run predictions (in silico mutagenesis) for all possible SAVs in APOE, which would require running AlphaFold 6,023 times (Supplementary Table S1). As a workaround, we retrieved all SAVs in APOE reported in ClinVar (Landrum et al., 2015). First, ClinVar database version 20220507 was downloaded from https://ftp.ncbi.nlm.nih.gov/pub/clinvar/. Second, only variants annotated as inside the APOE gene were kept (n = 69). Third, only non-synonymous single nucleotide variants were kept, and all insertions and deletions were excluded (n = 38). As a result, a total of 38 SAVs were retrieved, and a separate protein sequence was created for each SAV. The predicted 3D protein structure for the wild-type and each mutant sequence was compared using the root-mean-square deviation (RMSD) of atomic positions, which was commonly used as a distance measurement between two protein structures. A variant with a higher RMSD score was expected to have a greater impact on the protein structure. Therefore, the RMSD score was used as a surrogate for AlphaFold’s prediction of the variant’s impact.
Besides the two computational tools described previously, we used two additional web services to measure/predict the stability of the protein with and without the variants. First, the Missense3D database for APOE was retrieved from http://missense3d.bc.ic.ac.uk:8080 (Khanna et al., 2021), which contains 307 pre-calculated predictions in APOE. Second, DynaMut2 was used to predict user supply variants (Rodrigues et al., 2020). The same SAVs retrieved from ClinVar were used and submitted to the DynaMut2 web service at: https://biosig.lab.uq.edu.au/dynamut2/.
To evaluate the performance of the main predictor (ESM-1b model), we retrieved population allele frequencies from gnomAD (https://gnomad.broadinstitute.org/news/2020-10-gnomad-v3-1/). Maximum population frequencies were retrieved for the same SAVs retrieved from ClinVar as described previously.
An Evolutionary conservation score, GERP++ (Davydov et al., 2010), was retrieved from the dbNSFP v4.3a database (Liu et al., 2011; Liu et al., 2020), available at https://sites.google.com/site/jpopgen/dbNSFP.
Additionally, we have retrieved 3 popular tools for predicting protein stability change upon mutation, namely, FoldX (Schymkowitz et al., 2005), DDGun (Montanucci et al., 2019) and Maestro (Laimer et al., 2015). First, FoldX was downloaded from https://foldxsuite.crg.eu/ using the academic license. The “Stability” command was used to calculate the Gibbs energy of protein folding for all 38 potential SAVs. The difference in folding energy between wild-type and mutant sequences was calculated and their absolute values were used to represent each SAV’s impact predicted by FoldX, since both stabilizing and destabilizing mutations may all have substantial impacts on the function of the protein. Second, the DDGun web service, available at: https://folding.biofold.org/ddgun/index.html, was used to make predictions on protein stability change given a list of mutations. Specifically, the wild-type sequence of APOE with a list of IDs for all 38 SAVs was uploaded. A global Delta Delta G (DDG) value was predicted for each of the SAVs, and its absolute value was used to represent each SAV’s impact predicted by DDGun. Third, Maestro v1.2.35 Linux executable file was downloaded from https://pbwww.services.came.sbg.ac.at/?page_id=477. All 38 SAVs were submitted as input for the Maestro program with the wild-type 3D structure of APOE obtained using AlphaFold2. Similarly, the DDG values were obtained from the prediction and their absolute values were used to represent each SAV’s impact predicted by Maestro.
To evaluate the correlation between allele frequencies of the variants and predictions made by computational tools, Pearson’s correlation coefficient was calculated using the Python library SciPy (https://scipy.org/). We calculated the area under the receiver operating characteristic curve (auROC) and average precision scores to evaluate each predictor’s ability in prioritizing potential clinically relevant variants. Specifically, an auROC was calculated by measuring the predictor’s true positive rate (TPR) and false positive rate (FPR) using different score cutoffs. Similarly, the average precision (AP) score was calculated by measuring the predictor’s precision and recall (same as TPR) using different score cutoffs. The formulas for calculating TPR, FPR, and precision are:
Where TP refers to the number of true positives (correctly predicted ClinVar pathogenic variants), FN refers to the number of false negatives (incorrectly predicted ClinVar pathogenic variants as benign), FP refers to the number of false positives (incorrectly predicted ClinVar benign variants as pathogenic), and TN refers to the number of true negatives (correctly predicted ClinVar benign variants). Both auROC and AP scores were calculated using the Python library sklearn with functions roc_auc_score and average_precision_score, respectively.
Additionally, PyTorch was used to calculate ESM model predictions, and Tensorflow was used to calculate AlphaFold model predictions.
As illustrated in Figure 1, the entire length of the APOE protein was predicted by the ESM-1b model, and all potential amino acid variants were evaluated as the log odds ratio between the mutant and wild-type predictions. Variants with lighter colors indicate a low predicted likelihood of the existence of a variant at this position, which implies their functional importance. Key functional domains, including a signal peptide, receptor binding domain and lipid binding domain showed higher importance, as illustrated by light color bands. Interestingly, these regions of high importance showed higher conservation scores (GERP++ score), as illustrated by the top panel. In contrast, amino acids from positions 18–45 showed both low conservation and low predicted functional importance. This observed concordance of the ESM prediction with annotated functional domains and evolutionary conservation demonstrated the model’s ability to capture important regions in the APOE gene, given that the gene is only moderately conserved and is quite tolerant to missense variants (Lek et al., 2016).
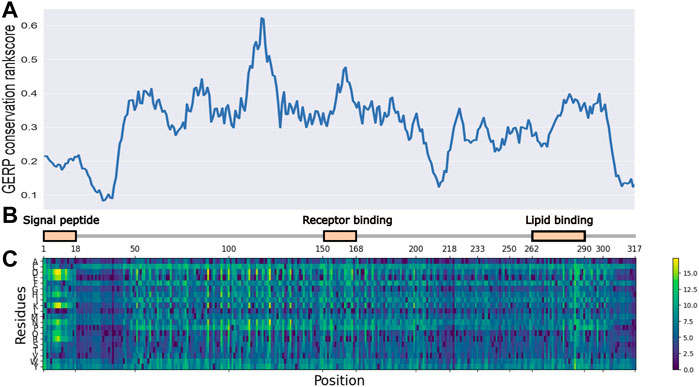
FIGURE 1. ESM-1b and GERP predicted functional importance scores for all potential SAVs in APOE gene. (A) GERP conservation scores for APOE gene. (B) functional domains for APOE gene. (C) ESM-1b in silico mutagenesis predictions for APOE gene.
Additionally, multiple clustering patterns were observed in the prediction heatmap, as illustrated by regions with high predicted values. One of these regions was amino acids 1–18, representing the signal peptide region. While few studies have tried to evaluate the functional importance of variants residing in this region, it is clear that multiple variants can be extremely harmful to the protein’s function.
To illustrate if the scores predicted by ESM-1b can truly reflect function importance at the variant level, we next evaluated allele frequencies observed in a large-scale population cohort, namely, gnomAD, and see if the model’s predictions show correlations with allele frequencies (AFs) of the variants in general populations. Due to purifying selection, variants with lower AFs are more likely to be deleterious, whereas variants with higher AFs are more likely to be tolerated (benign). As illustrated in Figure 2, predictions by ESM-1 showed statistically significant positive correlations with −log10(AFs) in the general population, which indicates its capability of identifying truly functional variants that have undergone purifying selection.
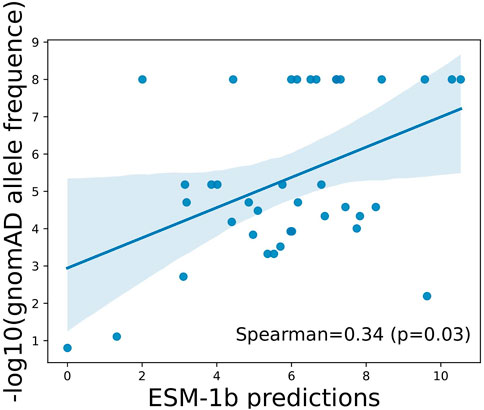
FIGURE 2. Correlation between ESM-1b predictions and population allele frequencies among ClinVar reported variants in APOE. Pearson correlation coefficient and associated p-value were reported.
Next, we investigated the predictions made by AlphaFold. AlphaFold gives a per-residue confidence metric called pLDDT (predicted Local Distance Difference Test) score for all alpha-carbon atoms (Mariani et al., 2013). Regions with high pLDDT scores usually have fewer clashes and structural violations. As shown in Figure 3, pLDDT scores correlate with conservation scores (Spearman correlation coefficient = 0.43, p-value = 1.94 × 10−15), which was expected, as AlphaFold prediction relies on MSA data as input, which primarily utilizes conservation data. Additionally, in regions with high pLDDT scores (pLDDT >70), for example, the amino acid’s approximate position from 45–170, only pathogenic variants and no benign variants were reported. Their potential structural importance could explain this observed pattern.
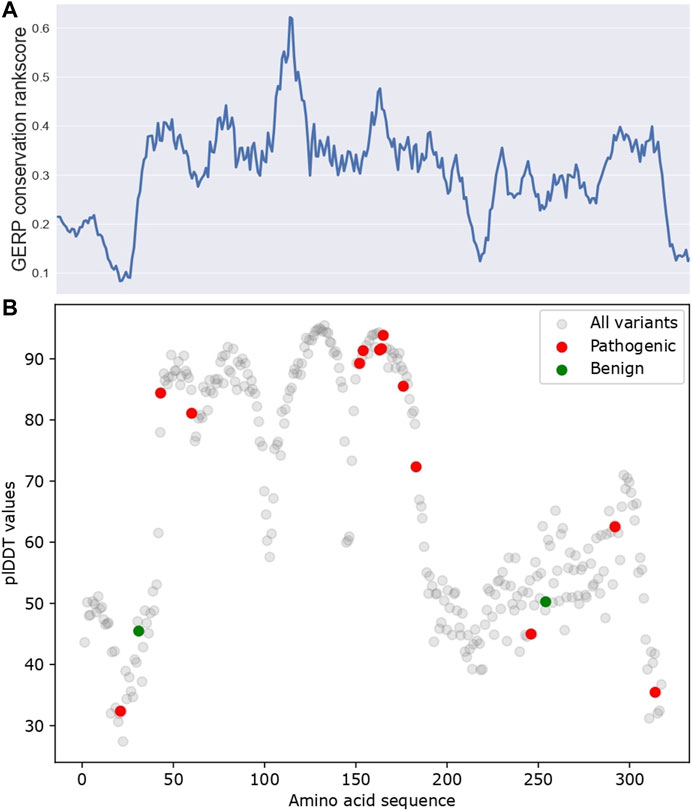
FIGURE 3. The pLDDT scores for AlphaFold predicted APOE structure. (A) GERP conservation score. (B) pLDDT scores along APOE protein sequence. ClinVar pathogenic/benign variants were highlighted in red and green, respectively. All variants are referred to as all amino acids in APOE, which reflect the pLDDT distribution for all amino acids of APOE.
We compared the correlation of the predictions made by four popular computational frameworks, namely, ESM-1b, AlphaFold, Missense3D, and DynaMut2, which measure protein properties from different perspectives (Figure 4). Specifically, ESM model studies comprehensive protein sequence features from millions of protein sequences using a language model. AlphaFold model studies protein sequences and tries to predict 3D protein structures using sequences and available templates. Missense3D adopts a bioinformatics pipeline and evaluates a wide range of structural impacts of an SAV. DynaMut2 model predicts protein stability by learning a series of biochemical and biophysical features from the target proteins. We have examined additional popular in silico tools that can predict protein stability (Caldararu et al., 2020; Pan et al., 2022), including FoldX (Schymkowitz et al., 2005), DDGun (Montanucci et al., 2019) and Maestro (Laimer et al., 2015), but all of them showed inferior performance compared to DynaMut2 in APOE (Supplementary Figures S1, S2; Supplementary Table S2). Therefore, we chose only DynaMut2 as the representative tool for protein stability prediction. Interestingly, benchmarked using ClinVar labels, the DynaMut2, as the best individual predictor among protein stability methods, outperformed predictors from other categories, including ESM and AlphaFold. Therefore, we provided predictions from DynaMut2 for all possible SAVs in APOE in Supplementary Table S3.
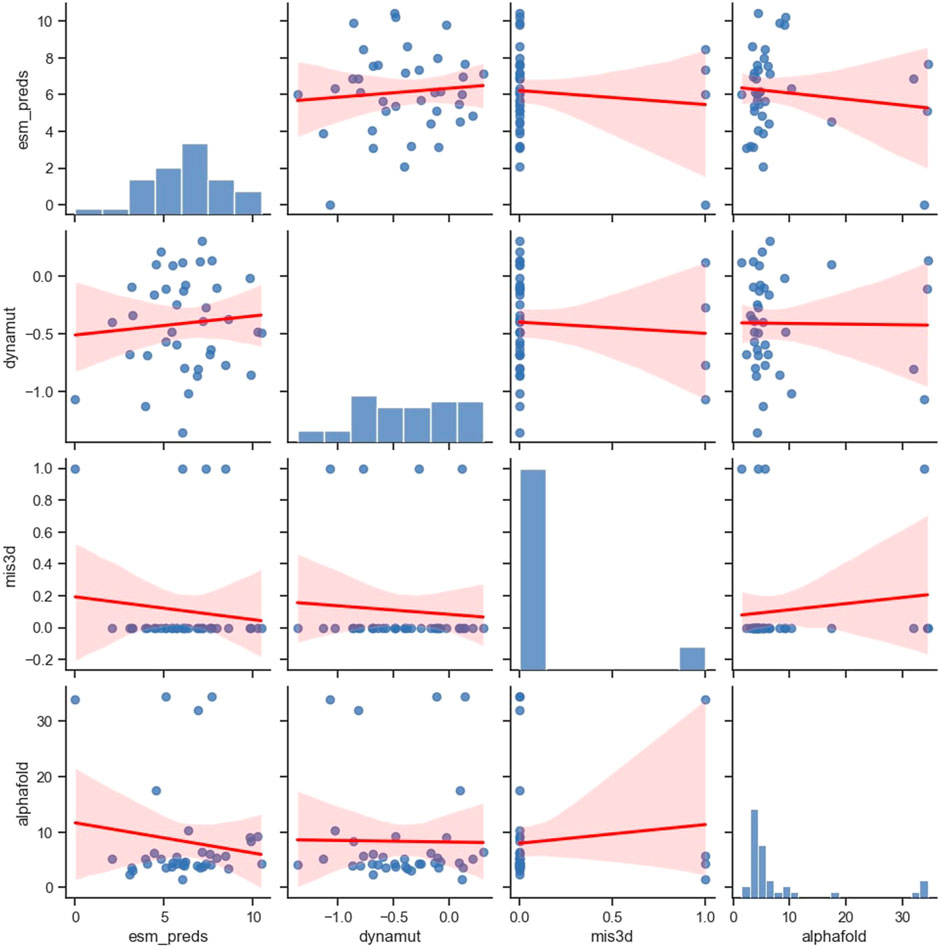
FIGURE 4. Pairwise correlation of the variants between the four in silico predictors.
All these four selected tools showed no or very weak correlations with each other, which can be both concerning and useful. On one hand, if the inconsistency arises from methodological flaws, their ability to capture useful information is extremely limited. Users should be cautious when adopting these tools in their workflow. On the other hand, if this inconsistency arises from the differences in methodological preferences and their ability to capture different aspects of protein functions, then these tools can provide valuable orthogonal information.
To evaluate the usefulness of the previously described tools and illustrate if their low correlation can be beneficial to explaining variant effects, we obtained top candidates from multiple predictions and examined their biological relevance as a means of validation. The top candidates were obtained based on the predictions made by each of the four tools. We consider variants to be potentially pathogenic if predictions from two or more tools showed indicative of a disruptive effect. Using this ensemble approach (majority vote), five candidate variants were obtained. As shown in Table 1, variants C130R, R163C, and R132C are most likely to be functional. Importantly, DynaMut2, which predicts the stability of the mutant protein sequence, showed destabilizing effects for all these 3 variants. AlphaFold models also predict the top 2 variants to disrupt the key functional domains. Interestingly, the most promising variant, C130R, is the variant that separates the transcript that carries the variant gene (APOE4) from the wild-type transcript (APOE3). The variant replaces Cysteine with Arginine, which was predicted to change a residue state from buried to exposed (Figure 5). The functional importance of the C130R variant was validated by previous studies, which reported the variant to be associated with an elevated risk of AD (Husain et al., 2021; Martens et al., 2022). This observation highlighted the ability to combine multiple functional prediction tools in finding key functional variants.

TABLE 1. Five candidate variants that affect APOE function.
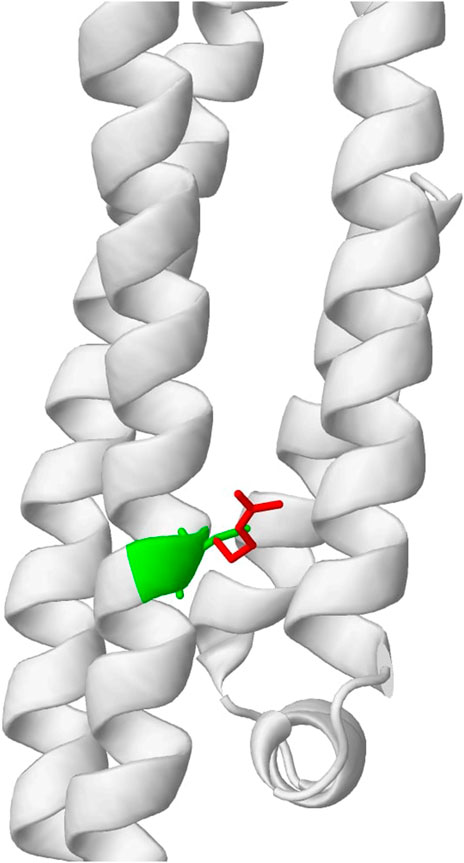
FIGURE 5. Predicted change in residue properties between Cysteine (green) and Arginine (red) in C130R variant by Missense3D.
Next, using a similar approach, we identify one variant to be potentially benign. As shown in Table 2, all four tools predicted the variant to be non-functional. ClinVar reported a conflicting interpretation of pathogenicity for this SAV, meaning that multiple clinical laboratories reported contradictory interpretations for the same variant. Specifically, some studies reported it to be benign while others report it to be uncertain significance, according to the 2015 ACMG-AMP guidelines (Richards et al., 2015). Given its previous uncertain annotations and the fact that all orthogonal in silico methods showed concordant prediction, its function is worth investigating in future studies to confirm whether the SAV is truly benign. All calculated scores for all 38 SAVs analyzed in the study are provided in Supplementary Table S2.

TABLE 2. Candidate variant that predicted to be benign by all tools.
In this study, we explored the usefulness of various orthogonal in silico predictors in their ability to prioritize functionally and structurally disruptive SAVs in the APOE gene. Using little to no prior knowledge, we identified 5 potentially disrupting variants, one of which (C130R) was classified by previous studies as a key risk factor for AD (Holtzman et al., 2012).
As illustrated by our study, the ESM model, which utilized large-scale pretraining and state-of-the-art deep learning architectures, can efficiently identify highly important domains and functional SAVs. The N-terminal of the APOE protein consists of 4 helices, H1, H2, H3, and H4, which form a four-helix bundle that spans amino acids from 42 to 182 (Wilson et al., 1991). These helices contain some key functional domains, such as the LDL-receptor binding region (residues 154–168). As illustrated by the ESM prediction (Figure 1), this region indeed contains multiple highlighted bands, reflecting the potential functional importance of the variants. Moreover, the previously mentioned domain for the signal peptide (residues 1–18) represents another region of interest. It has been previously reported that variants located in signal-peptide-encoding sequences may severely impact protein transportation (Jarjanazi et al., 2007). For this under-investigated region, no variant was reported in ClinVar, including benign, pathogenic, or variant of unknown significance (VUS), which calls for future studies to perform functional validation of variants in this region that focus on the transportation and maturation of APOE.
However, the ESM model was imperfect, and it may fail to predict variants residing in regions with little homologous coverage. For example, in our study, the ESM model incorrectly predicted the C130R variant to be non-functional. On the other hand, the AlphaFold model has demonstrated potential in identifying such highly disruptive SAVs. While the C130R variant was predicted as non-functional by the ESM model, it showed the highest disruptive effect predicted by AlphaFold among the top 5 candidate SAVs. Based on our results from non-specific in silico predictions, this C130R variant may convey its functional impact through altered protein 3D structure rather than the function encoded in the underlying amino acid. Indeed, this C130R, or the equivalently C112R in mature APOE, was reported to destabilize the protein structure, which was considered to improve its ability to bind to lipid and amyloid-β surfaces, which may ultimately increase the risk of AD (Chetty et al., 2017).
Aside from the promising results of using a set of orthogonal in silico tools to help us understand the functional importance of APOE variants, we believe there are a few limitations in our study that future studies could improve upon. First, our illustration and analysis in this study were based only on a single gene APOE, and future studies may include other apolipoprotein genes to investigate the capability of these novel computational tools in assisting lipid research. Second, we only considered SAVs in this study, and we note that InDels (short insertions or deletions) may play a greater role in protein stability and function. It is still an open question regarding if and how these existing computational tools can help with this regard. Third, in this study, we performed validation across multiple data resources, including conservation score and population allele frequency, and future studies may be conducted to include additional in silico validations and even experimental validations, such as deep mutational scanning data, to further elucidate the functional importance of the reported variants.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
XL, YB, and CL conceived the idea. CL, IH, and MM performed the formal analysis. CL, IH, MM, and GW wrote the manuscript. XL and YB edited the manuscript and supervised the findings of this work.
This study was funded for article processing charge is provided by the University of South Florida to XL.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fbinf.2023.1122559/full#supplementary-material
SUPPLEMENTARY FIGURE S1 | Performances of all quantitative predictors analyzed in this study benchmarked using ClinVar labels. The dashed red line represents the ROC cut-off value. Tools with greater ROC values were chosen to construct the ensemble.
SUPPLEMENTARY FIGURE S2 | ROC curves for all quantitative predictors analyzed in this study benchmarked using ClinVar labels.
Bertram, L., Lange, C., Mullin, K., Parkinson, M., Hsiao, M., Hogan, M. F., et al. (2008). Genome-wide association analysis reveals putative alzheimer's disease susceptibility loci in addition to APOE. Am. J. Hum. Genet. 83 (5), 623–632. doi:10.1016/j.ajhg.2008.10.008
Bettens, K., Sleegers, K., and Van broeckhoven, C. (2013). Genetic insights in alzheimer's disease. Lancet Neurology 12 (1), 92–104. doi:10.1016/S1474-4422(12)70259-4
Bojanowski, C. M., Shen, D., Chew, E. Y., Ning, B., Csaky, K. G., Green, W. R., et al. (2006). Anapolipoprotein E variant may protect against age-related macular degeneration through cytokine regulation. Environ. Mol. Mutagen. 47 (8), 594–602. doi:10.1002/em.20233
Brandes, N., Goldman, G., Wang, C. H., Ye, C. J., and Ntranos, V. (2022). Genome-wide prediction of disease variants with a deep protein language model. bioRxiv. doi:10.1101/2022.08.25.505311
Caldararu, O., Mehra, R., Blundell, T. L., and Kepp, K. P. (2020). Systematic investigation of the data set dependency of protein stability predictors. J. Chem. Inf. Model. 60 (10), 4772–4784. doi:10.1021/acs.jcim.0c00591
Caswell, R. C., Gunning, A. C., Owens, M. M., Ellard, S., and Wright, C. F. (2022). Assessing the clinical utility of protein structural analysis in genomic variant classification: Experiences from a diagnostic laboratory. Genome Med. 14 (1), 77. doi:10.1186/s13073-022-01082-2
Chetty, P. S., Mayne, L., Lund-katz, S., Englander, S. W., and Phillips, M. C. (2017). Helical structure, stability, and dynamics in human apolipoprotein e3 and e4 by hydrogen exchange and mass spectrometry. Proc. Natl. Acad. Sci. 114 (5), 968–973. doi:10.1073/pnas.1617523114
Davydov, E. V., Goode, D. L., Sirota, M., Cooper, G. M., Sidow, A., and Batzoglou, S. (2010). Identifying a high fraction of the human genome to be under selective constraint using gerp++. PLoS Comput. Biol. 6 (12), e1001025. doi:10.1371/journal.pcbi.1001025
Dolai, S., Cherakara, S., and Garai, K. (2020). Apolipoprotein e4 exhibits intermediates with domain interaction. Biochimica Biophysica Acta (BBA) - Proteins Proteomics 1868 (12), 140535. doi:10.1016/j.bbapap.2020.140535
Holtzman, D. M., Herz, J., and Bu, G. (2012). Apolipoprotein E and apolipoprotein E receptors: Normal biology and roles in alzheimer disease. Cold Spring Harb. Perspect. Med. 2 (3), a006312. doi:10.1101/cshperspect.a006312
Husain, M. A., Laurent, B., and Plourde, M. (2021). APOE and alzheimer's disease: From lipid transport to physiopathology and therapeutics. Front. Neurosci. 15, 630502. doi:10.3389/fnins.2021.630502
Jarjanazi, H., Savas, S., Pabalan, N., Dennis, J. W., and Ozcelik, H. (2007). Biological implications of snps in signal peptide domains of human proteins. Proteins Struct. Funct. Bioinforma. 70 (2), 394–403. doi:10.1002/prot.21548
Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., et al. (2021). Highly accurate protein structure prediction with alphafold. Nature 596 (7873), 583–589. doi:10.1038/s41586-021-03819-2
Kamboh, M. I., Sanghera, D. K., Ferrell, R. E., and Dekosky, S. T. (1995). A4POE*4-associated alzheimer's disease risk is modified by α1–antichymotrypsin polymorphism. Nat. Genet. 10 (4), 486–488. doi:10.1038/ng0895-486
Khanna, T., Hanna, G., Sternberg, M. J. E., and David, A. (2021). Missense3D-DB web catalogue: An atom-based analysis and repository of 4M human protein-coding genetic variants. Hum. Genet. 140 (5), 805–812. doi:10.1007/s00439-020-02246-z
Laimer, J., Hofer, H., Fritz, M., Wegenkittl, S., and Lackner, P. (2015). Maestro - multi agent stability prediction upon point mutations. BMC Bioinforma. 16 (1), 116. doi:10.1186/s12859-015-0548-6
Landrum, M. J., Lee, J. M., Benson, M., Brown, G., Chao, C., Chitipiralla, S., et al. (2015). ClinVar: Public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 44 (D1), D862–D868. doi:10.1093/nar/gkv1222
Lek, M., Karczewski, K. J., Minikel, E. V., Samocha, K. E., Banks, E., Fennell, T., et al. (2016). Analysis of protein-coding genetic variation in 60,706 humans. Nature 536 (7616), 285–291. doi:10.1038/nature19057
Lewandowski, C. T., Maldonado weng, J., and Ladu, M. J. (2020). Alzheimer's disease pathology in APOE transgenic mouse models: The who, what, when, where, why, and how. Neurobiol. Dis. 139, 104811. doi:10.1016/j.nbd.2020.104811
Lin, Z., Akin, H., Rao, R., Hie, B., Zhu, Z., Lu, W., et al. (2022). Evolutionary-scale prediction of atomic level protein structure with a language model. bioRxiv. doi:10.1101/2022.07.20.500902
Liu, X., Jian, X., and Boerwinkle, E. (2011). DbNSFP: A lightweight database of human nonsynonymous snps and their functional predictions. Hum. Mutat. 32 (8), 894–899. doi:10.1002/humu.21517
Liu, X., Li, C., Mou, C., Dong, Y., and Tu, Y. (2020). DbNSFP v4: A comprehensive database of transcript-specific functional predictions and annotations for human nonsynonymous and splice-site snvs. Genome Med. 12 (1), 103. doi:10.1186/s13073-020-00803-9
Liu, Y., Yu, J.-T., Wang, H.-F., Han, P.-R., Tan, C.-C., Wang, C., et al. (2014). APOE genotype and neuroimaging markers of alzheimer's disease: Systematic review and meta-analysis. J. Neurology, Neurosurg. Psychiatry 86 (2), 127–134. doi:10.1136/jnnp-2014-307719
Mariani, V., Biasini, M., Barbato, A., and Schwede, T. (2013). Lddt: A local superposition-free score for comparing protein structures and models using distance difference tests. Bioinformatics 29 (21), 2722–2728. doi:10.1093/bioinformatics/btt473
Martens, Y. A., Zhao, N., Liu, C.-C., Kanekiyo, T., Yang, A. J., Goate, A. M., et al. (2022). ApoE cascade hypothesis in the pathogenesis of alzheimer's disease and related dementias. Neuron 110 (8), 1304–1317. doi:10.1016/j.neuron.2022.03.004
Mirdita, M., Schütze, K., Moriwaki, Y., Heo, L., Ovchinnikov, S., and Steinegger, M. (2022). ColabFold: Making protein folding accessible to all. Nat. Methods 19 (6), 679–682. doi:10.1038/s41592-022-01488-1
Montanucci, L., Capriotti, E., Frank, Y., Ben-Tal, N., and Fariselli, P. (2019). DDGun: An untrained method for the prediction of protein stability changes upon single and multiple point variations. BMC Bioinforma. 20 (S14), 335. doi:10.1186/s12859-019-2923-1
Pak, M. A., Markhieva, K. A., Novikova, M. S., Petrov, D. S., Vorobyev, I. S., Maksimova, E. S., et al. (2021). Using alphafold to predict the impact of single mutations on protein stability and function. bioRxiv. doi:10.1101/2021.09.19.460937
Pan, Q., Nguyen, T. B., Ascher, D. B., and Pires, D. E. V. (2022). Systematic evaluation of computational tools to predict the effects of mutations on protein stability in the absence of experimental structures. Briefings Bioinforma. 23 (2), bbac025. doi:10.1093/bib/bbac025
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet. Med. 17 (5), 405–424. doi:10.1038/gim.2015.30
Rodrigues, C. H. m., Pires, D. E. v., and Ascher, D. B. (2020). <DynaMut2: Assessing changes in stability and flexibility upon single and multiple point missense mutations. Protein Sci. 30 (1), 60–69. doi:10.1002/pro.3942
Schymkowitz, J., Borg, J., Stricher, F., Nys, R., Rousseau, F., and Serrano, L. (2005). The FoldX web server: An online force field. Nucleic Acids Res. 33, W382–W388. doi:10.1093/nar/gki387
Steinegger, M., and Söding, J. (2017). MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 35 (11), 1026–1028. doi:10.1038/nbt.3988
Wilson, C., Wardell, M. R., Weisgraber, K. H., Mahley, R. W., and Agard, D. A. (1991). Three-Dimensional structure of the LDL receptor-binding domain of human apolipoprotein E. Science 252 (5014), 1817–1822. doi:10.1126/science.2063194
Keywords: AlphaFold, missense variant, APOE, Alzheimer’s disease, deep learning, ensemble
Citation: Li C, Hou I, Ma M, Wang G, Bai Y and Liu X (2023) Orthogonal analysis of variants in APOE gene using in-silico approaches reveals novel disrupting variants. Front. Bioinform. 3:1122559. doi: 10.3389/fbinf.2023.1122559
Received: 14 December 2022; Accepted: 31 March 2023;
Published: 06 April 2023.
Edited by:
Andrzej Kloczkowski, The Research Institute at Nationwide Children’s Hospital, United StatesReviewed by:
Uday S. Evani, New York Genome Center, United StatesCopyright © 2023 Li, Hou, Ma, Wang, Bai and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chang Li, bGljQHVzZi5lZHU=; Yongsheng Bai, YmlvaW5mb3JtYXRpY3NyZXNlYXJjaHRvbW9ycm93QGdtYWlsLmNvbQ==; Xiaoming Liu, eGlhb21pbmdsaXVAdXNmLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.