 Chris J. Radoux
Chris J. Radoux Francesca Vianello
Francesca Vianello Jake McGreig†
Jake McGreig† Nikita Desai
Nikita Desai Anthony R. Bradley
Anthony R. Bradley- Exscientia plc, Oxford, United Kingdom
The concept of the druggable genome has been with us for 20 years. During this time, researchers have developed several methods and resources to help assess a target’s druggability. In parallel, evidence for target-disease associations has been collated at scale by Open Targets. More recently, the Protein Data Bank in Europe (PDBe) have built a knowledge base matching per-residue annotations with available protein structure. While each resource is useful in isolation, we believe there is enormous potential in bringing all relevant data into a single knowledge graph, from gene-level to protein residue. Automation is vital for the processing and assessment of all available structures. We have developed scalable, automated workflows that provide hotspot-based druggability assessments for all available structures across large numbers of targets. Ultimately, we will run our method at a proteome scale, an ambition made more realistic by the arrival of AlphaFold 2. Bringing together annotations from the residue up to the gene level and building connections within the graph to represent pathways or protein-protein interactions will create complexity that mirrors the biological systems they represent. Such complexity is difficult for the human mind to utilise effectively, particularly at scale. We believe that graph-based AI methods will be able to expertly navigate such a knowledge graph, selecting the targets of the future.
Introduction
Twenty years ago Hopkins and Groom (2002) published “The Druggable Genome.” This seminal paper recognised that only a subset of the newly published human genome (Lander et al., 2001) encodes proteins capable of binding orally bioavailable (Lipinski et al., 1997) molecules: the druggable genome. Over the last 20 years, many druggable genome variations have been published, focussing on either a specific disease area (Kumar et al., 2013), or including targets of biologics and more recent medicinal chemistry efforts (Russ and Lampel, 2005; Finan et al., 2017). Now, we believe that a data-rich knowledge graph of target-based annotations down to the level of individual residues will lead to the most complete description of drug target space—the landscape of all potential drug targets described by the many factors that determine a drug target’s quality. While this amount of data would be overwhelming for human user exploration, AI algorithms can expertly navigate these knowledge graphs to select the drug targets of the future.
Hopkins and Groom stated “druggable does not equal drug target”. Their original definition of “druggable” focused on proteins that can bind orally bioavailable drug-like molecules; however, this would now be thought of as drug-like ligandability. Contemporary definitions of druggability (Leach and Radoux, 2021) expand upon the original definition to include additional requirements, addressing the far more complicated question of “can this target yield a successful drug?” An ideal small-molecule drug target is disease modifying, capable of binding a selective, orally bioavailable molecule at a site that elicits a functional effect, has no on-target toxicity and is expressed in disease-relevant tissue. This multi-parameter problem is typically tackled by a multidisciplinary team, gathering information from literature, publicly available resources, and computational prediction on a per-target basis. First, we discuss what we believe is the state of the art and offer the next steps to provide the most complete description of target space to date. Second, we explore critical considerations for performing structure-based druggability at scale. We show how we are leveraging automation and cloud computing to expand our internal knowledge graph with residue-level annotations. Finally, we discuss how we think the arrival of AlphaFold 2 (AF2) will affect target assessment.
Computer-readable annotation from gene to residue
Manually assessing all the factors that contribute to a suitable drug target is a time-consuming exercise. Fortunately, public resources such as Open Targets (Koscielny et al., 2017; Carvalho-Silva et al., 2018) and canSAR (Mitsopoulos et al., 2015; Mitsopoulos et al., 2020; Chau et al., 2016; Coker et al., 2018) bring together key data for target selection (Table 1). Open Targets focuses on linking targets to disease, but includes tractability data for small molecules, antibodies (Brown et al., 2018; Leach and Radoux, 2021) and PROTACs (proteolysis targeting chimeras) (Schneider et al., 2021). canSAR collates data from multiple sources and calculates structure-based, ligand-based, and network-based druggability scores, allowing users to assess the ligandability of specific cavities on each individual structure.

TABLE 1. Overview of data resources useful in the assessment of targets.
These platforms provide function-rich user interfaces (UIs), which are powerful tools for scientists looking to discover future drug targets, but they do not allow exploration with AI approaches. Incorporating this data into a knowledge graph would allow additional data sources to be layered on top, allowing more complex queries and graph-based algorithms for data interrogation.
A good example of using knowledge graphs to annotate proteins with data is the PDBe Knowledge Base (PDBe-KB) (Consortium et al., 2021), which provides a neo4j graph database that maps data from several partner providers at the residue level. The growth of the PDB and improved protein structure prediction (Kryshtafovych et al., 2021) has increased opportunities for structure-based assessment. Additional considerations are necessary, however, to correctly identify therapeutically relevant pockets beyond simple ligandability prediction. Predicting whether pockets are orthosterically or allosterically functional, which offers opportunities for selectivity (Smilova et al., 2022), or are conserved across species (where required), provides key insights into a protein’s drug target suitability.
Structure-based druggability assessments typically focus on a single static protein structure (Hendlich et al., 1997; Hajduk et al., 2005; Cheng et al., 2007; Halgren, 2009; Huang, 2009; Kawabata, 2010; Volkamer et al., 2010; Volkamer et al., 2012a; Volkamer et al., 2012b; Krasowski et al., 2011; Desaphy et al., 2012; Borrel et al., 2015; Aggarwal et al., 2021), providing a score at the pocket level. Hotspot-based approaches, using either molecular dynamics (Young et al., 2007; Seco et al., 2009; Yang and Wang, 2010; Huang and Caflisch, 2011; Lexa and Carlson, 2011; Schmidtke et al., 2011; Bakan et al., 2012; Alvarez-Garcia and Barril, 2014; Ichihara et al., 2014; Vukovic et al., 2016; Arcon et al., 2017; Uehara and Tanaka, 2017; Vajda et al., 2018; Zariquiey et al., 2019; Yuan et al., 2020; Evans et al., 2021a) or static structures (Kozakov et al., 2015; Radoux et al., 2016; Curran et al., 2020), are capable of providing residue-level scoring. A hotspot-based assessment run at scale would provide residue level tractability annotations to be added to a knowledge graph such as the PDBe-KB, with all available structures for a given target used to calculate these scores.
In addition to structure-based assessment, drug discovery precedence for a target can be searched. This could mean identifying crystal structures in the PDB corresponding to drug-like compounds, or active drug-like compounds in ChEMBL (Mendez et al., 2018). The presence of multiple distinct chemical series further increases the chances that the target is tractable. If active compounds and protein crystal structures are not available, the target can be cross-referenced with published druggable genome sets (Hopkins and Groom, 2002; Russ and Lampel, 2005; Finan et al., 2017) to see if it is predicted to be tractable based on similarity to known drug targets.
Automation and scalability are essential in confidently expanding the druggable genome into novel and overlooked areas. Capturing all relevant data for target selection, from target-level evidence to per-residue data, in a single knowledge graph is a daunting but important task. Doing so will enable AI methods to undertake the work normally performed by large multidisciplinary teams and identify the very best novel targets.
Structure-based druggability at scale
The human proteome comprises the protein sequences of all coding genes, including splice variants, from the human reference genome (Breuza et al., 2016). There are currently 20,360 human proteins in Swiss-Prot (Boutet et al., 2007), of which approximately 4,600 are implicated in disease according to the OMIM database (Hamosh et al., 2005), representing around 22% of human proteins with roles in disease. These proteins are the obvious subset of the human proteome likely to contain viable drug targets. An estimated 70% of the human proteome is covered by homologous protein structures (Somody et al., 2017), which can be exploited to characterise druggable pockets.
Where protein structures are available, they are often missing atoms, contain alternate atom placements, and are missing hydrogens. To obtain consistent high-quality structures, a method of automating the preparation of structures for computational experiments is required. Once a prepared set of structures is available, large-scale analysis requires a robust automation platform to locate target-binding sites across multiple structures per target.
It has long been known that considering proteins as rigid structures fails to consider energetic fluctuations that lead to proteins exploring a multitude of complex conformational states (Elber and Karplus, 1987). Moreover, important conformational changes in proteins are often associated with ligand binding; therefore, incorporating target flexibility into drug discovery pipelines will improve a project’s likelihood of success (Amaro et al., 2018). Molecular dynamics is one approach to incorporate protein flexibility; however, this is too computationally expensive to run at the proteome scale. Therefore, a rational approach to structure-based assessment would need to assess druggability across all structural data available rather than picking one representative structure. This is particularly important when data exist for multiple conformational states (e.g., active vs. inactive structures). Such an approach would necessarily yield a large amount of data and require careful analysis, especially in the context of automated pocket detection.
Exscientia’s approach to structure-based assessment
Exscientia’s pipeline for automated target druggability assessments, summarised in Figure 1, has been designed to fulfil the above requirements. This pipeline captures a profile of druggability for each target that retains essential details such as single structures with non-conserved druggable binding pockets, while providing a global overview of the chosen target. Our workflows are run using scalable cloud computing infrastructure to facilitate the expansion of assessments to whole proteomes.

FIGURE 1. High-level schematic showing the overall structure-based assessment workflow.
The first step leverages PDBe information relative to the structural coverage of each full-length protein characterised by a UniProt ID. The PDBe provides crucial information on the protein segments that are structurally enabled. Structural studies of larger proteins tend to yield multiple structures of shorter, non-overlapping segments. Our pipeline treats each of these segments separately, identifying the best representative structures available for each based on sequence coverage, resolution, and data quality.
For each individual target, publicly available structures are combined with in-house simulated and generated structures, and then aligned to the previously determined reference structure. All structural files undergo the same processing steps (related to rebuilding sidechains, protonating the protein and ligand, and assigning partial charges) to ensure data compatibility for downstream modelling tasks such as docking and molecular dynamics. The result of this step is a standard dataset of all structural data available for each target. Tractable binding pockets on each prepared structure are then assessed using Fragment Hotspot Maps with 3D grids, which highlight the areas most attractive to small molecules (Radoux et al., 2016; Curran et al., 2020). The centre of mass of each of these drug-like tractable volumes (which correspond to putative binding pockets on an individual structure) is extracted and stored.
The centres of mass from all structures are clustered, shown in Figure 2, with each cluster taken to correspond to a distinct binding pocket, referred to as a global pocket. This is vital for referencing each binding pocket consistently across multiple structures, allowing tractability scores to be collated for each global pocket. This enables researchers to evaluate a range of scores for a given global pocket, and determine whether a target needs to adopt a particular conformation for effective binding.
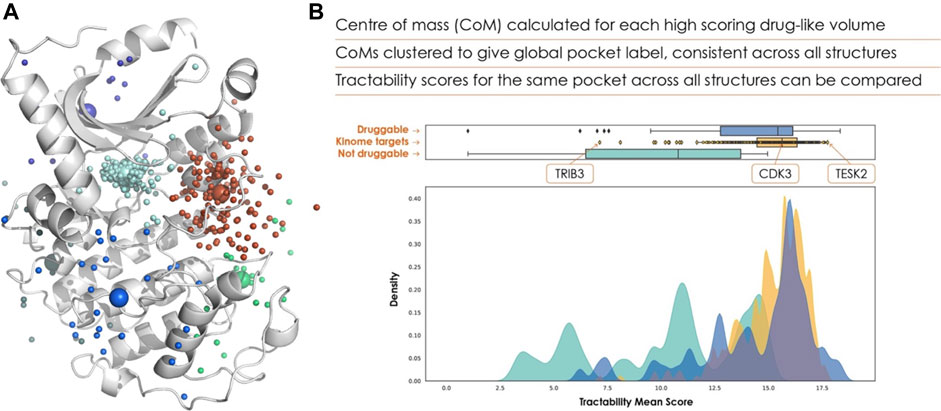
FIGURE 2. (A) Clustered pockets across all AlphaFold 2 structures of the human kinome, with the AlphaFold 2 model of cyclin-dependent kinase 7 (UniProt ID P50613). The cyan points show the ATP binding site of each kinase, allowing us to rank the kinome purely based on ATP-pocket druggability. (B) The frequency distribution of ATP site scores is shown in yellow, with known druggable (blue) and non-druggable (green) targets for comparison.
Assessing viral targets
With the ongoing COVID-19 pandemic, target selection for pandemic preparedness is of particular importance. The conservation of a drug target is assessed to capture the robustness of the protein and its binding pockets and used to provide valuable insights into the longevity of the drug in the face of resistance and pathogen emergence. These insights are obtained by computing a range of conservation metrics that rely on sequence alignments generated by MUSCLE (Madeira et al., 2019) for each target. The sequences that make up these alignments are identified using a BLAST (Altschul et al., 1990) search of the reference protein against a database of non-redundant protein sequences, restricted by taxonomies of interest. This enables a plethora of sequences of the target in the organism of interest to be captured, as well as orthologs for identifying drug activity against similar proteins.
At each residue in these alignments, we identify variants, the amino acid feature frequency across variants, and the most common amino acid frequency, which are mapped onto the reference PDB numbering. Additionally, we use Jensen-Shannon divergence (Capra and Singh, 2007) (JSD) to identify the conservation at each alignment position. JSD predicts functionally important residues using an estimator for sequence conservation. Each alignment position and its neighbour residues are compared to a background set of amino acids under no evolutionary pressure, and positions that differ substantially from this set are predicted as functionally important or constrained.
Functional insights from UniProt and FunPDBe are collated to identify amino acids that have interactions with small molecules or play a role in the target’s function. By extracting residue-level hotspot scores and mapping the conservation scores to them, we can quickly identify the most tractable and robust positions that can be used to inform design.
Papain-like protease case study
Our recent assessment of the papain-like protease (PLpro), an attractive target for the treatment of COVID-19 (Shin et al., 2020), highlighted the importance of each step of the target assessment workflow. PLpro has a β-turn/loop formed by residues Gly266-Gly271 next to the active site (Osipiuk et al., 2021). Upon binding of a substrate or inhibitor, this loop closes, creating a more buried and druggable pocket. Assessment of only a single structure may have resulted in missing this difference, causing the PLpro site to be deemed not druggable. Consideration of all structures captures a range of druggability scores (Figure 3). Structures with a closed loop conformation are well within the druggable range, and the active site, Cys111, provides the additional opportunity for pursuing covalent strategies.
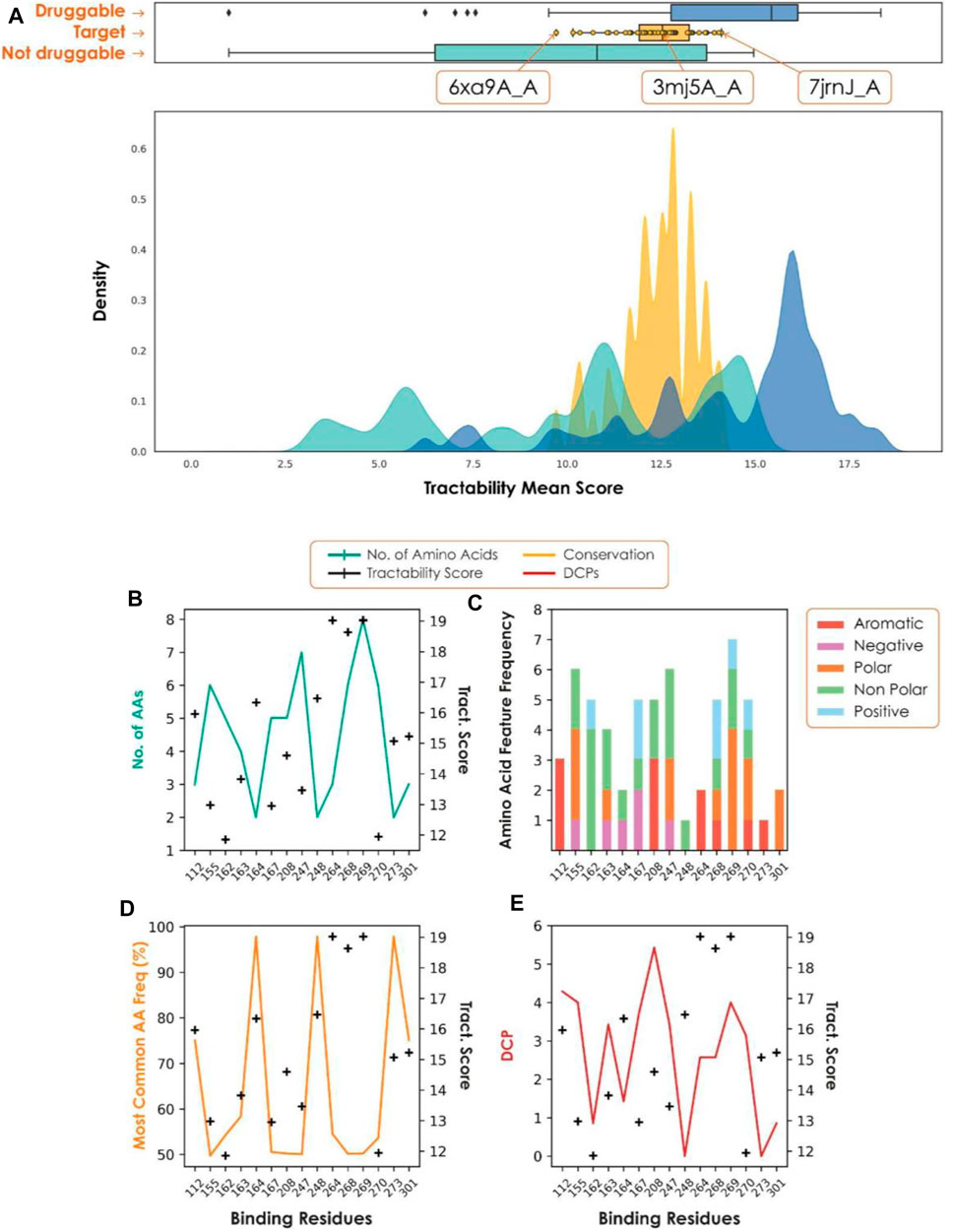
FIGURE 3. (A) Tractability for PLpro (yellow) vs. known druggable (blue) and non-druggable (green) targets. (B–E) Plots to show conservation metrics across coronavirus species HKU1, 229E, OC43, NL63, MERS, SARS-CoV-2 and SARS-CoV. In each plot, the hotspot tractability score for a given residue is shown as a black cross. (B) Number of different amino acids. (C) Number of amino acid properties. (D) Frequency of the most common amino acid observed. (E) Frequency with which an amino acid is differentially conserved between species for each predicted hotspot residue. A position is considered differentially conserved when it is conserved within a given species (JS-divergence score >0.8) and different from a corresponding conserved position in a different species. As we consider multiple species, this plot shows the average number of times a given position is labelled as differentially conserved.
A fundamental requirement of viral target selection in the context of pandemic preparedness is that future variants and species will not develop drug resistance. Here we used conservation across multiple coronavirus species, combined with per-residue tractability scores, to assess the risk of drug resistance. In the case of PLpro, most of the residues with high tractability scores, and therefore essential for binding, were highly variable across a set of coronaviruses (SARS, MERS, 229E, NL63, HKU1, OC43). As a result, there is a high risk of future variants and novel coronaviruses being resistant to drugs developed against SARS-CoV-2 PLpro, making it unsuitable for pandemic preparedness.
Scaling up to proteome-wide assessment
Our pipeline focuses on automating the process for single targets and runs using only a UniProt ID as input, which facilitates running the pipeline on a proteome-wide scale. Once this large-scale calculation is complete, and the results are captured in a knowledge graph, it can be layered together with public data from the PDBe and Open Targets, and our own internal knowledge graphs. Such a complete and data-rich description of the drug target space will enable far more precise search queries.
The more data included in the knowledge graph, the more complex these queries can become. We can consider further target annotations such as the subcellular location or tissue expression, or create edges between targets based on homology thresholds, pathway information or protein-protein interactions, mirroring the biological systems they represent. The benefits of introducing this structure and complexity can be exemplified by synthetic lethality, a promising area for novel cancer therapeutics. The first approved synthetic lethal therapy, for indications including breast and ovarian cancer, targets poly(ADP-ribose) polymerase (PARP). PARP is essential in cancer cells with BRCA1/2 mutations due to their defective homologous recombination (HR) pathway for DNA damage repair (Gutmanas et al., 2014). Healthy cells can survive PARP inhibition due to compensation by the HR pathway, allowing the BRCA-mutated cancer cells to be selectively killed. Using a knowledge graph as described above, pathways with similar function to the one containing an oncogene of interest, in this case DNA damage repair pathways for BRCA1/2 mutations, can be identified. These can then be searched for tractable functional binding sites to identify novel opportunities for synthetic lethality therapy targets. This could be done in a targeted way for a specific mutation or expanded to consider all identified oncogenic mutations and their respective pathways. Eventually, manually curated queries will likely become unwieldy, and graph-based algorithms will be the most effective approach for navigating this data-rich drug target space.
Many of the annotations do not rely on protein structure, but ultimately protein structures are vital for a pocket-centric target view. As well as helping the assessment of target tractability, a protein structure makes a project more doable by enabling structure-based design. Increased structural coverage of the proteome will have huge consequences on how much of the target space can be assessed and pursued.
Impact of AlphaFold 2
The last 20 years have seen significant advances in the power and accuracy of homology modelling and de novo protein structure prediction methods (Pereira et al., 2021). These advances have recently culminated in the remarkable performance of AF2, developed by DeepMind, as demonstrated in the CASP14 experiment (Jumper et al., 2021a; Jumper et al., 2021b; Tunyasuvunakool et al., 2021). AF2 showed significant improvement from previous CASP experiments, with several structure predictions almost indistinguishable from experimental structures (Jumper et al., 2021a). Recently, more than 200 million AF2 structures have been released across more than 1 million species (Callaway 2022), greatly increasing the scope for structure-based assessment; however, care must be taken.
One of the most significant aspects of the AF2 method for structural bioinformatics and structure-based computational prediction has been the development of informative confidence metrics for local (pLDDT) and global (PAE) structural accuracy (Jumper et al., 2021a; Jumper et al., 2021b; Akdel et al., 2021). These metrics have been shown to estimate the local accuracy of AF2 predictions with remarkable reliability (Jumper et al., 2021a; Jumper et al., 2021b; Akdel et al., 2021; Pereira et al., 2021; Tunyasuvunakool et al., 2021). Specifically, at pLDDT scores >90 (high confidence), one estimation shows AF2 χ1 rotamers are 80% correct (Jumper et al., 2021b; Tunyasuvunakool et al., 2021). At pLDDT >70 (confident), AF2 has generally correct backbone predictions, although side chain conformations may be less accurate in these regions (Jumper et al., 2021b; Tunyasuvunakool et al., 2021). In addition, PAE scores may indicate domain orientation in multi-domain chains and possibly of proteins in multi-chain complexes (Akdel et al., 2021; Evans et al., 2021b; Tunyasuvunakool et al., 2021).
Since CASP14, DeepMind has released the AF2 code, model parameters and a database with AF2 model predictions (Akdel et al., 2021; Tunyasuvunakool et al., 2021). The AF2 database provides almost complete (98.5%) coverage of the human proteome, with structural predictions for all 20,000 proteins of the human proteome (Akdel et al., 2021; Tunyasuvunakool et al., 2021). Of the residues modelled, 36% were predicted with confidence (pLDDT >70), and another 22% were predicted with high confidence (pLDDT >90) (Akdel et al., 2021; Tunyasuvunakool et al., 2021). In terms of proteins, AF2 has confident predictions for >75% of protein sequence for 44% of human protein targets (Mullard, 2021). One analysis (Porta-Pardo et al., 2021) showed that the AF2 database increased the proportion of the human proteome with valuable structural insights from 47% to 75%, and reduced the number of proteins with no structural information from 4,832 to between 29 and 1,336 proteins (depending on confidence thresholds).
Having structural annotation and prediction for unannotated proteins and regions will benefit ligand binding-pocket predictions on those regions. Work by Beltrao (Akdel et al., 2021) indicates that while using low-confidence regions for binding-pocket detection can result in many false positives and negatives, predictions made on confident regions become comparable to using experimental crystal structures (Akdel et al., 2021). These preliminary results, however, will likely need further robust benchmarking, with more stringent filtering of homologs at both sequence and structure levels. Additionally, large-scale validation experiments will be required (Pak et al., 2021; Jones and Thornton, 2022).
While AF2 models can be good starting points for ligand-binding or pocket prediction, especially where there are few or no homologous structures available, it is essential to account for both confidence metrics and homology of proteins or regions of interest to the PDB (Akdel et al., 2021; Mullard, 2021; Tunyasuvunakool et al., 2021; Jones and Thornton, 2022). Understanding AF2 model limitations will be vital to ensure their impact on large-scale druggability predictions. With the correct preparation and consideration of confidence metrics, AF2 models will allow the predicted druggable genome to expand into areas previously not considered.
Conclusion
In the 20 years since the publication of the druggable genome, tremendous advances have been made in multiple areas. These include improved structure prediction, structure-based assessment, public resources of collated data, and new architecture for data storage and methods for data interrogation. This provides the opportunity to build a detailed description of drug target space, an opportunity we must seize to select the very best future drug targets. The original druggable genome was represented as a simple Venn diagram, with “drug targets” at the intersection of “druggable genes” and “disease-modifying genes.” It is increasingly becoming a multi-dimensional problem, difficult to represent for human minds, particularly at large scales.
Graph-based AI algorithms can effectively work with data of this scale and complexity. Identifying the best method for selecting targets from a knowledge graph of this scale will be the subject of future research. Databases such as the Cambridge Structural Database (Groom et al., 2016) (CSD), ChEMBL (Mendez et al., 2018) and the PDB have all shown that bringing data together in an organised fashion allows far greater insights than from each individual data point.
Data availability statement
The original contributions presented in the study are included in the article’s supplementary materials, and any further inquiries can be directed to the corresponding author.
Author contributions
AB and CR contributed to the conception of the manuscript. CR wrote the first draft of the manuscript. FV, JM, and ND wrote sections of the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
Conflict of interest
Authors CR, FV, JM, ND, and AB were employed by the company Exscientia.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Aggarwal, R., Gupta, A., Chelur, V., Jawahar, C. V., and Priyakumar, U. D. (2021). DeepPocket: Ligand binding site detection and segmentation using 3D convolutional neural networks. J. Chem. Inf. Model.. 10.1021/acs.jcim.1c00799. doi:10.1021/acs.jcim.1c00799
Akdel, M., Pires, D. E. V., Pardo, E. P., Jänes, J., Zalevsky, A. O., Mészáros, B., et al. (2021). A structural biology community assessment of AlphaFold 2 applications. Biorxiv 2021, 461876. doi:10.1101/2021.09.26.461876
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi:10.1016/s0022-2836(05)80360-2
Alvarez-Garcia, D., and Barril, X. (2014). Molecular simulations with solvent competition quantify water displaceability and provide accurate interaction maps of protein binding sites. J. Med. Chem. 57, 8530–8539. doi:10.1021/jm5010418
Amaro, R. E., Baudry, J., Chodera, J., Demir, Ö., McCammon, J. A., Miao, Y., et al. (2018). Ensemble docking in drug discovery. Biophys. J. 114, 2271–2278. doi:10.1016/j.bpj.2018.02.038
Arcon, J. P., Defelipe, L. A., Modenutti, C. P., López, E. D., Alvarez-Garcia, D., Barril, X., et al. (2017). Molecular dynamics in mixed solvents reveals protein–ligand interactions, improves docking, and allows accurate binding free energy predictions. J. Chem. Inf. Model. 57, 846–863. doi:10.1021/acs.jcim.6b00678
Bakan, A., Nevins, N., Lakdawala, A. S., and Bahar, I. (2012). Druggability assessment of allosteric proteins by dynamics simulations in the presence of probe molecules. J. Chem. Theory Comput. 8, 2435–2447. doi:10.1021/ct300117j
Borrel, A., Regad, L., Xhaard, H., Petitjean, M., and Camproux, A.-C. (2015). PockDrug: A model for predicting pocket druggability that overcomes pocket estimation uncertainties. J. Chem. Inf. Model. 55, 882–895. doi:10.1021/ci5006004
Boutet, E., Lieberherr, D., Tognolli, M., Schneider, M., and Bairoch, A. (2007). UniProtKB/Swiss-Prot. Methods Mol. Biol. 406, 89–112. doi:10.1007/978-1-59745-535-0_4
Breuza, L., Poux, S., Estreicher, A., Famiglietti, M. L., Magrane, M., Tognolli, M., et al. (2016). The UniProtKB guide to the human proteome. Database (Oxford). 2016, bav120. doi:10.1093/database/bav120
Brown, K. K., Hann, M. M., Lakdawala, A. S., Santos, R., Thomas, P. J., and Todd, K. (2018). Approaches to target tractability assessment – A practical perspective. MedChemComm 9, 606–613. doi:10.1039/c7md00633k
Callaway, E. (2022). ‘The entire protein universe’: AI predicts shape of nearly every known protein. Nature 608, 15–16. doi:10.1038/d41586-022-02083-2
Capra, J. A., and Singh, M. (2007). Predicting functionally important residues from sequence conservation. Bioinform Oxf Engl. 23, 1875–1882. doi:10.1093/bioinformatics/btm270
Carvalho-Silva, D., Pierleoni, A., Pignatelli, M., Ong, C., Fumis, L., Karamanis, N., et al. (2018). Open targets platform: New developments and updates two years on. Nucleic Acids Res. 47, D1056–D1065. doi:10.1093/nar/gky1133
Chau, C. H., O’Keefe, B. R., and Figg, W. D. (2016). The canSAR data hub for drug discovery. Lancet Oncol. 17, 286. doi:10.1016/s1470-2045(16)00095-4
Cheng, A. C., Coleman, R. G., Smyth, K. T., Cao, Q., Soulard, P., Caffrey, D. R., et al. (2007). Structure-based maximal affinity model predicts small-molecule druggability. Nat. Biotechnol. 25, 71–75. doi:10.1038/nbt1273
Coker, E. A., Mitsopoulos, C., Tym, J. E., Komianou, A., Kannas, C., Di Micco, P., et al. (2018). canSAR: update to the cancer translational research and drug discovery knowledgebase. Nucleic Acids Res. 47, D917–D922. doi:10.1093/nar/gky1129
Consortium, T. U., Bateman, A., Martin, M.-J., Orchard, S., Magrane, M., Agivetova, R., et al. (2020). UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 49, D480–D489. doi:10.1093/nar/gkaa1100
Consortium, Pdb.-K., Varadi, M., Anyango, S., Armstrong, D., Berrisford, J., Choudhary, P., et al. (2021). PDBe-KB: Collaboratively defining the biological context of structural data. Nucleic Acids Res. 50, D534–D542. doi:10.1093/nar/gkab988
Curran, P. R., Radoux, C., Smilova, M. D., Sykes, R., Higueruelo, A., Bradley, A., et al. (2020). Hotspots api: A Python package for the detection of small molecule binding hotspots and application to structure-based drug design. J. Chem. Inf. Model. 60, 1911–1916. doi:10.1021/acs.jcim.9b00996
Desaphy, J., Azdimousa, K., Kellenberger, E., and Rognan, D. (2012). Comparison and druggability prediction of protein–ligand binding sites from pharmacophore-annotated cavity shapes. J. Chem. Inf. Model. 52, 2287–2299. doi:10.1021/ci300184x
Elber, R., and Karplus, M. (1987). Multiple conformational states of proteins: A molecular dynamics analysis of myoglobin. Science 235, 318–321. doi:10.1126/science.3798113
Evans, D. J., Yovanno, R. A., Rahman, S., Cao, D. W., Beckett, M. Q., Patel, M. H., et al. (2021a). Finding druggable sites in proteins using TACTICS. J. Chem. Inf. Model. 61, 2897–2910. doi:10.1021/acs.jcim.1c00204
Evans, R., O’Neill, M., Pritzel, A., Antropova, N., Senior, A., Green, T., et al. (2021b). Protein complex prediction with AlphaFold-Multimer. Biorxiv 2021, 463034. doi:10.1101/2021.10.04.463034
Finan, C., Gaulton, A., Kruger, F. A., Lumbers, R. T., Shah, T., Engmann, J., et al. (2017). The druggable genome and support for target identification and validation in drug development. Sci. Transl. Med. 9, eaag1166. doi:10.1126/scitranslmed.aag1166
Groom, C. R., Bruno, I. J., Lightfoot, M. P., and Ward, S. C. (2016). The Cambridge structural database. Acta Crystallogr. B Struct. Sci. Cryst. Eng. Mat. 72, 171–179. doi:10.1107/s2052520616003954
Gutmanas, A., Alhroub, Y., Battle, G. M., Berrisford, J. M., Bochet, E., Conroy, M. J., et al. (2014). PDBe: Protein Data Bank in Europe. Nucleic Acids Res. 42, D285–D291. doi:10.1093/nar/gkt1180
Hajduk, P. J., Huth, J. R., and Fesik, S. W. (2005). Druggability indices for protein targets derived from NMR-based screening data. J. Med. Chem. 48, 2518–2525. doi:10.1021/jm049131r
Halgren, T. A. (2009). Identifying and characterizing binding sites and assessing druggability. J. Chem. Inf. Model. 49, 377–389. doi:10.1021/ci800324m
Hamosh, A., Scott, A. F., Amberger, J. S., Bocchini, C. A., and McKusick, V. A. (2005). Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res. 33, D514–D517. doi:10.1093/nar/gki033
Hendlich, M., Rippmann, F., and Barnickel, G. (1997). Ligsite: Automatic and efficient detection of potential small molecule-binding sites in proteins. J. Mol. Graph. Model. 15, 359–363. doi:10.1016/s1093-3263(98)00002-3
Hopkins, A. L., and Groom, C. R. (2002). The druggable genome. Nat. Rev. Drug Discov. 1, 727–730. doi:10.1038/nrd892
Huang, B. (2009). MetaPocket: A meta approach to improve protein ligand binding site prediction. OMICS A J. Integr. Biol. 13, 325–330. doi:10.1089/omi.2009.0045
Huang, D., and Caflisch, A. (2011). Small molecule binding to proteins: Affinity and binding/unbinding dynamics from atomistic simulations. ChemMedChem 6, 1578–1580. doi:10.1002/cmdc.201100237
Ichihara, O., Shimada, Y., and Yoshidome, D. (2014). The importance of hydration thermodynamics in fragment-to-lead optimization. ChemMedChem 9, 2708–2717. doi:10.1002/cmdc.201402207
Jones, D. T., and Thornton, J. M. (2022). The impact of AlphaFold2 one year on. Nat. Methods 19, 15–20. doi:10.1038/s41592-021-01365-3
Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., et al. (2021a). Applying and improving AlphaFold at CASP14. Proteins. 89, 1711–1721. doi:10.1002/prot.26257
Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., et al. (2021b). Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589. doi:10.1038/s41586-021-03819-2
Kawabata, T. (2010). Detection of multiscale pockets on protein surfaces using mathematical morphology. Proteins. 78, 1195–1211. doi:10.1002/prot.22639
Koscielny, G., An, P., Carvalho-Silva, D., Cham, J. A., Fumis, L., Gasparyan, R., et al. (2017). Open targets: A platform for therapeutic target identification and validation. Nucleic Acids Res. 45, D985–D994. doi:10.1093/nar/gkw1055
Kozakov, D., Hall, D. R., Napoleon, R. L., Yueh, C., Whitty, A., and Vajda, S. (2015). New Frontiers in druggability. J. Med. Chem. 58, 9063–9088. doi:10.1021/acs.jmedchem.5b00586
Krasowski, A., Muthas, D., Sarkar, A., Schmitt, S., and Brenk, R. (2011). DrugPred: A structure-based approach to predict protein druggability developed using an extensive nonredundant data set. J. Chem. Inf. Model. 51, 2829–2842. doi:10.1021/ci200266d
Kryshtafovych, A., Schwede, T., Topf, M., Fidelis, K., and Moult, J. (2021). Critical assessment of methods of protein structure prediction (CASP)—round XIV. Proteins. 89, 1607–1617. doi:10.1002/prot.26237
Kumar, R. D., Chang, L.-W., Ellis, M. J., and Bose, R. (2013). Prioritizing potentially druggable mutations with dGene: An annotation tool for cancer genome sequencing data. Plos One 8, e67980. doi:10.1371/journal.pone.0067980
Lander, E. S., Linton, L. M., Birren, B., and Nusbaum, C. (2001). Initial sequencing and analysis of the human genome. Nature 409, 860–921. doi:10.1038/35057062
Leach, A. R., and Radoux, C. (2021). Computational drug target tractability analysis. Syst. Med., 4, 145–153. doi:10.1016/b978-0-12-801238-3.11531-4
Lexa, K. W., and Carlson, H. A. (2011). Full protein flexibility is essential for proper hot-spot mapping. J. Am. Chem. Soc. 133, 200–202. doi:10.1021/ja1079332
Lipinski, C. A., Lombardo, F., Dominy, B. W., and Feeney, P. J. (1997). Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 23, 3–25. doi:10.1016/s0169-409x(96)00423-1
Madeira, F., Park, Y., Lee, J., Buso, N., Gur, T., Madhusoodanan, N., et al. (2019). The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 47, W636–W641. doi:10.1093/nar/gkz268
Mendez, D., Gaulton, A., Bento, A. P., Chambers, J., De Veij, M., Félix, E., et al. (2018). ChEMBL: Towards direct deposition of bioassay data. Nucleic Acids Res. 47, D930–D940. doi:10.1093/nar/gky1075
Mitsopoulos, C., Schierz, A. C., Workman, P., and Al-Lazikani, B. (2015). Distinctive behaviors of druggable proteins in cellular networks. PLoS Comput. Biol. 11, e1004597. doi:10.1371/journal.pcbi.1004597
Mitsopoulos, C., Di Micco, P., Fernandez, E. V., Dolciami, D., Holt, E., Mica, I. L., et al. (2020). canSAR: update to the cancer translational research and drug discovery knowledgebase. Nucleic Acids Res. 49, D1074–D1082. doi:10.1093/nar/gkaa1059
Mullard, A. (2021). What does AlphaFold mean for drug discovery? Nat. Rev. Drug Discov. 20, 725–727. doi:10.1038/d41573-021-00161-0
Osipiuk, J., Azizi, S.-A., Dvorkin, S., Endres, M., Jedrzejczak, R., Jones, K. A., et al. (2021). Structure of papain-like protease from SARS-CoV-2 and its complexes with non-covalent inhibitors. Nat. Commun. 12, 743. doi:10.1038/s41467-021-21060-3
Pak, M. A., Markhieva, K. A., Novikova, M. S., Petrov, D. S., Vorobyev, I. S., Maksimova, E. S., et al. (2021). Using AlphaFold to predict the impact of single mutations on protein stability and function. Biorxiv 2021, 460937. doi:10.1101/2021.09.19.460937
Pereira, J., Simpkin, A. J., Hartmann, M. D., Rigden, D. J., Keegan, R. M., and Lupas, A. N. (2021). High-accuracy protein structure prediction in CASP14. Proteins. 89, 1687–1699. doi:10.1002/prot.26171
Porta-Pardo, E., Ruiz-Serra, V., Valentini, S., and Valencia, A. (2021). The structural coverage of the human proteome before and after AlphaFold. Biorxiv 2021, 454980. doi:10.1101/2021.08.03.454980
Radoux, C. J., Olsson, T. S. G., Pitt, W. R., Groom, C. R., and Blundell, T. L. (2016). Identifying interactions that determine fragment binding at protein hotspots. J. Med. Chem. 59, 4314–4325. doi:10.1021/acs.jmedchem.5b01980
Russ, A. P., and Lampel, S. (2005). The druggable genome: An update. Drug Discov. Today 10, 1607–1610. doi:10.1016/s1359-6446(05)03666-4
Schmidtke, P., Bidon-Chanal, A., Luque, F. J., and Barril, X. (2011). MDpocket: Open-source cavity detection and characterization on molecular dynamics trajectories. Bioinformatics 27, 3276–3285. doi:10.1093/bioinformatics/btr550
Schneider, M., Radoux, C. J., Hercules, A., Ochoa, D., Dunham, I., Zalmas, L.-P., et al. (2021). The PROTACtable genome. Nat. Rev. Drug Discov. 20, 789–797. doi:10.1038/s41573-021-00245-x
Seco, J., Luque, F. J., and Barril, X. (2009). Binding site detection and druggability index from first principles. J. Med. Chem. 52, 2363–2371. doi:10.1021/jm801385d
Shin, D., Mukherjee, R., Grewe, D., Bojkova, D., Baek, K., Bhattacharya, A., et al. (2020). Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature 1, 657–662. doi:10.1038/s41586-020-2601-5
Smilova, M. D., Curran, P. R., Radoux, C. J., Delft, F. V., Cole, J. C., Bradley, A. R., et al. (2022). Fragment hotspot mapping to identify selectivity-determining regions between related proteins. J. Chem. Inf. Model. 62, 284–294. doi:10.1021/acs.jcim.1c00823
Somody, J. C., MacKinnon, S. S., and Windemuth, A. (2017). Structural coverage of the proteome for pharmaceutical applications. Drug Discov. Today 22, 1792–1799. doi:10.1016/j.drudis.2017.08.004
Tunyasuvunakool, K., Adler, J., Wu, Z., Green, T., Zielinski, M., Žídek, A., et al. (2021). Highly accurate protein structure prediction for the human proteome. Nature 1, 590–596. doi:10.1038/s41586-021-03828-1
Uehara, S., and Tanaka, S. (2017). Cosolvent-based molecular dynamics for ensemble docking: Practical method for generating druggable protein conformations. J. Chem. Inf. Model. 57, 742–756. doi:10.1021/acs.jcim.6b00791
Vajda, S., Beglov, D., Wakefield, A. E., Egbert, M., and Whitty, A. (2018). Cryptic binding sites on proteins: Definition, detection, and druggability. Curr. Opin. Chem. Biol. 44, 1–8. doi:10.1016/j.cbpa.2018.05.003
Volkamer, A., Griewel, A., Grombacher, T., and Rarey, M. (2010). Analyzing the topology of active sites: On the prediction of pockets and subpockets. J. Chem. Inf. Model. 50, 2041–2052. doi:10.1021/ci100241y
Volkamer, A., Kuhn, D., Grombacher, T., Rippmann, F., and Rarey, M. (2012a). Combining global and local measures for structure-based druggability predictions. J. Chem. Inf. Model. 52, 360–372. doi:10.1021/ci200454v
Volkamer, A., Kuhn, D., Rippmann, F., and Rarey, M. (2012b). DoGSiteScorer: A web server for automatic binding site prediction, analysis and druggability assessment. Bioinformatics 28, 2074–2075. doi:10.1093/bioinformatics/bts310
Vukovic, S., Brennan, P. E., and Huggins, D. J. (2016). Exploring the role of water in molecular recognition: Predicting protein ligandability using a combinatorial search of surface hydration sites. J. Phys. Condens. Matter 28, 344007. doi:10.1088/0953-8984/28/34/344007
Yang, C.-Y., and Wang, S. (2010). Computational analysis of protein hotspots. ACS Med. Chem. Lett. 1, 125–129. doi:10.1021/ml100026a
Young, T., Abel, R., Kim, B., Berne, B. J., and Friesner, R. A. (2007). Motifs for molecular recognition exploiting hydrophobic enclosure in protein–ligand binding. Proc. Natl. Acad. Sci. U. S. A. 104, 808–813. doi:10.1073/pnas.0610202104
Yuan, J.-H., Han, S. B., Richter, S., Wade, R. C., and Kokh, D. B. (2020). Druggability assessment in TRAPP using machine learning approaches. J. Chem. Inf. Model. 60, 1685–1699. doi:10.1021/acs.jcim.9b01185
Keywords: druggability, druggable genome, tractability, knowledge graph, drug target identification, artificial intelligence
Citation: Radoux CJ, Vianello F, McGreig J, Desai N and Bradley AR (2022) The druggable genome: Twenty years later. Front. Bioinform. 2:958378. doi: 10.3389/fbinf.2022.958378
Received: 31 May 2022; Accepted: 15 August 2022;
Published: 30 September 2022.
Edited by:
Garrett M. Morris, University of Oxford, United KingdomReviewed by:
Jie Zheng, ShanghaiTech University, ChinaCopyright © 2022 Radoux, Vianello, McGreig, Desai and Bradley. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chris J. Radoux, Y3JhZG91eEBleHNjaWVudGlhLmFpLmNvLnVr
†These authors have contributed equally to this work