 Martina Dori
Martina Dori Jimmy Caroli
Jimmy Caroli Mattia Forcato
Mattia Forcato- 1Department of Life Sciences, University of Modena and Reggio Emilia, Modena, Italy
- 2Department of Drug Design and Pharmacology, University of Copenhagen, Copenhagen, Denmark
Circular RNAs (circRNAs) are known to act as important regulators of the microRNA (miRNA) activity. Yet, computational resources to identify miRNA:circRNA interactions are mostly limited to already annotated circRNAs or affected by high rates of false positive predictions. To overcome these limitations, we developed Circr, a computational tool for the prediction of associations between circRNAs and miRNAs. Circr combines three publicly available algorithms for de novo prediction of miRNA binding sites on target sequences (miRanda, RNAhybrid, and TargetScan) and annotates each identified miRNA:target pairs with experimentally validated miRNA:RNA interactions and binding sites for Argonaute proteins derived from either ChIPseq or CLIPseq data. The combination of multiple tools for the identification of a single miRNA recognition site with experimental data allows to efficiently prioritize candidate miRNA:circRNA interactions for functional studies in different organisms. Circr can use its internal annotation database or custom annotation tables to enhance the identification of novel and not previously annotated miRNA:circRNA sites in virtually any species. Circr is written in Python 3.6 and is released under the GNU GPL3.0 License at https://github.com/bicciatolab/Circr.
Introduction
Circular RNAs (circRNAs) (Salzman et al., 2012; Hansen et al., 2013a; Jeck et al., 2013; Memczak et al., 2013) are covalently closed RNA molecules characterized by a non-linear 3′-5′ junction resulting from an unusual splicing event denominated backsplice junction (Ashwal-Fluss et al., 2014; Lasda and Parker, 2014; Conn et al., 2015; Ivanov et al., 2015; Starke et al., 2015; Wang and Wang, 2015; Zhang et al., 2016). This particular splicing event causes circRNAs to lack both 3′ poly(A) tail and 5′ capping, conferring resistance to exonuclease activity [e.g., RNaseR (Suzuki et al., 2006; Vincent and Deutscher, 2006)] and therefore determining a general longer half-life as compared to linear RNAs (Jeck et al., 2013). Despite the great interest towards this class of non-coding RNA especially for their potential as disease biomarkers (Li et al., 2015; Memczak et al., 2015; Lyu and Huang, 2017; Jahani et al., 2020; Wang et al., 2021), only few of them have been functionally characterized to date [a few examples are reported in (Memczak et al., 2013; Ashwal-Fluss et al., 2014; Barrett and Salzman, 2016; Suenkel et al., 2020)]. Several biological functions have been proposed for circRNAs, ranging from the regulation of pluripotency to early lineage differentiation (Yu et al., 2017; Izuogu et al., 2018) to the control of unique functions in specialized cells (Berta et al., 1990; Capel et al., 1993). However, in terms of research interest, the most prominent function attributed to circRNAs is their action as miRNA sponge to regulate target gene expression by inhibiting miRNA activity through competition with the RISC complex, a function commonly defined as miRNA binding (Hansen et al., 2013a; Hansen et al., 2013b; Memczak et al., 2013; Huang et al., 2017; Panda, 2018; Verduci et al., 2019).
The computational resources that have been recently developed to investigate circRNA function and expression substantially consist of databases collecting all available information relative to already known circRNAs in different species [e.g., circBase (Glažar et al., 2014)]. Although these databases have been expanded to account for circRNA expression patterns, functional predictions (e.g., miRNAs binding sites) and disease associations (Yang et al., 2011; Ghosal et al., 2013; Li et al., 2014; Dudekula et al., 2016; Liu et al., 2016), still they are limited to previously identified circRNAs in human and mouse species only. Moreover, most of the available tools to predict miRNA:circRNA interactions fall short when dealing with previously uncharacterized circRNAs. For instance, some resources, requiring a Gene ID as input and not permitting sequence browsing, can predict miRNA targets only for annotated genes and are therefore informative only if circRNAs include a 3′ UTR. On the other hand, tools that allow the analysis of sequences are restricted to a fixed nucleotide sequence length limiting the number of sequences that can be investigated at once (Kertesz et al., 2007; Betel et al., 2008; Rennie et al., 2014; Agarwal et al., 2015; Rennie et al., 2016; Karagkouni et al., 2018). In an attempt to bypass these limitations, some prediction tools provide offline stand-alone versions of their algorithms (Enright et al., 2003; Rehmsmeier et al., 2004; Thadani and Tammi, 2006; Bandyopadhyay and Mitra, 2009; Hsu et al., 2011; Coronnello and Benos, 2013; Agarwal et al., 2015). This solution, while allowing a full customization of the analysis, comes with the drawback of a high rate of false positives as these approaches easily return thousands of putative miRNA:RNA interactions. A common solution to reduce these lists is to perform a systematic validation of the targets (e.g., with RIP assays) or the integration with known interactions. In particular, since the binding of the RISC complex is mediated by the interaction of the miRNA with members of the Argonaute protein family (Hammond et al., 2000; Hutvágner and Zamore, 2002), it is fundamental, for a predicted miRNA:circRNA site to be functional, that also Ago proteins are binding in the same positions. Currently, the development of various CLIP-Seq (Cross-linking and Immunoprecipitation followed by sequencing) protocols provides an extremely valuable source of high-throughput data for Ago binding sites (Chi et al., 2009; Hafner et al., 2010; Jungkamp et al., 2011; Leung et al., 2011; Helwak et al., 2013; Clark et al., 2014; Grosswendt et al., 2014). In addition, the availability of already validated miRNA:RNA sites obtained through RIP assays (or other experimental approaches) provides a further layer of valuable information for filtering candidate miRNA:circRNA pairs. Following the approach proposed in (Dori and Bicciato, 2019), we developed Circr, a tool that integrates miRNA:target predictions from three commonly used algorithms, i.e., miRanda (Agarwal et al., 2015), TargetScan (Enright et al., 2003), and RNAhybrid (Rehmsmeier et al., 2004) with validated miRNA:RNA interactions and AGO peaks data. The combination of multiple tools for the identification of a single miRNA recognition site together with experimental data allows to efficiently reduce the pool of candidate miRNA:circRNA interactions for functional studies.
Methods
Implementation
Circr is written in Python3 and is compatible with Linux, Mac OS, and the MS Windows subsystem for Linux. The tools is freely available from Github (https://github.com/bicciatolab/Circr) and depends on several modules (pandas, pybedtools, collections, multiprocessing, functools, itertools, operator) that can be installed via pip. Circr runs three different software for miRNA target prediction, namely miRanda (Agarwal et al., 2015), TargetScan (Enright et al., 2003), and RNAhybrid (Rehmsmeier et al., 2004).
miRanda (http://www.microrna.org/microrna/getDownloads.do) and RNAhybrid (https://bibiserv.cebitec.uni-bielefeld.de/rnahybrid/) must be downloaded and installed by the user from their respective official source; TargetScan is included in the Circr package as a standalone perl script. Circr supports single core and multithreading calculations. We suggest running Circr on a machine with at least 8 GB of RAM.
Circr comprises an internal annotation database that can be downloaded from the following folder https://drive.google.com/drive/folders/1zJVyzEFAMtvZTTueWRocxXs63jUxsl-U?usp=sharing (refer to the README file for details on the database content). This database includes the following information for H. sapiens, M. musculus, D. melanogaster, and C. elegans: 1) the genome sequence in FASTA format; 2) the ENSEMBL (Howe et al., 2021) gene annotation in GTF format and the rRNA coordinates; 3) the miRNA sequences from the latest version (v22) of miRBase (Kozomara et al., 2019); 4) the annotation files in BED format with the experimentally validated miRNA:RNA pairs (Grosswendt et al., 2014; Li et al., 2014; Luna et al., 2015; Moore et al., 2015; Luna et al., 2017; Karagkouni et al., 2018; Kobayashi et al., 2019); and 5) the Argonaute peaks coordinates (Moshkovich et al., 2011; Consortium, 2012; Huang et al., 2013; Balakrishnan et al., 2014; Boudreau et al., 2014; Erhard et al., 2014; Pillai et al., 2014; Rybak-Wolf et al., 2014; Moore et al., 2015; Gillen et al., 2016; Rayon-Estrada et al., 2017; Benway and Iacomini, 2018; Nowakowski et al., 2018; Sarshad et al., 2018; Xiao et al., 2019; Xu et al., 2019; Fernandes et al., 2021); see Supplementary Table S1 for the complete list of organism and genome versions provided. miRNA:RNA interactions and AGO peaks were retrieved from TarBase (Karagkouni et al., 2018) and ENCORI (Li et al., 2014) databases or they were available as supplementary data files in the Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/) series associated to the referenced publications. Then, we merged the files according to organism and genome assembly and finally used the UCSC Genome Browser tool LiftOver (Kent et al., 2002) to obtain the coordinates for the different genome versions (necessary files were downloaded from the Genome Browser Download Page http://hgdownload.soe.ucsc.edu/downloads.html).
Pipeline Overview
The core of Circr consists of three main steps: 1) sequence extraction from circRNA genomic coordinates; 2) prediction of miRNA:circRNA interactions with third party software; and 3) comparison of the predicted sites with a database of validated interactions and AGO peaks.
circRNAs Sequence Extraction
Circr requires in input a BED file containing the circRNAs genomic coordinates. Starting from this file, Circr generates a data frame of nucleotide sequences in FASTA format (Figure 1). By default, Circr assumes that the input coordinates are the coordinates of the circRNAs full sequences (i.e., containing both introns and exons) that need to be processed prior to sequence extraction. For this purpose, Circr compares circRNAs coordinates with gene annotation. If circRNAs overlap genes annotated on the same strand, they are split into intron/exon coordinates and only the coordinates of the latter are retained for sequence retrieval. Conversely, antisense and intergenic circRNAs are considered as a single exon. By providing the organism and genome build required for the analysis, the tool automatically retrieves the necessary files from its internal database. Circr also accepts custom reference files provided by the user to maximize the applicability of the method to different organisms or genome builds (for more details and examples on running Circr with custom files, see https://github.com/bicciatolab/Circr#running-circr-with-custom-annotation-files). As for linear mRNA, circRNAs that overlap genes can undergo alternative splicing, implying that they might not include all exons within the transcript start/end coordinates or might retain introns. Therefore, the assumption of an exon-only structure could lead to an erroneous analysis and loss of information. To avoid this pitfall, Circr can accept as input a set of coordinates of all the features (exon/intron) known to be included in each circRNA of interest (circRNA features). In this case, Circr skips the initial intron/exon splitting. This option can also be used to avoid exon/intron splitting when there is no prior knowledge of the actual circRNA sequence structure, thus considering each circRNA as a single exon. Once all the genomic coordinates of the provided circRNAs have been computed, Circr proceeds to extract their sequences in FASTA files.
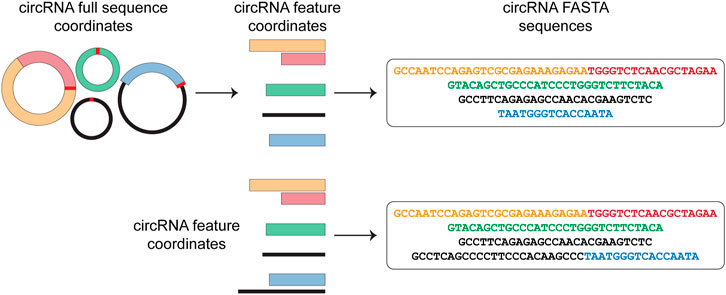
FIGURE 1. circRNAs sequence extraction. As a first step, Circr takes a set of coordinates in BED format as input. By default, it assumes that the transcripts overlapping genes on the same strand include only exons, therefore it retrieves the coordinates of the included features and uses them to construct the FASTA sequence of each circRNA. If the input file includes the exact composition of the circRNA (or if users want to avoid the automatic exon-only selection), Circr skips the initial exon/intron splitting and uses the supplied coordinates to retrieve the FASTA sequence.
Prediction of miRNA:circRNA Interactions
The sequences obtained from the sequence extraction step are used as input to three algorithms for the prediction of the miRNA binding sites (Figure 2). Among the various available tools for miRNA binding site prediction, we focused on the most commonly used, i.e., miRanda (Agarwal et al., 2015), RNAhybrid (Rehmsmeier et al., 2004), and TargetScan (Enright et al., 2003), as they all provide a standalone version that can be easily downloaded and installed on any Unix-based system. All three tools work primarily on sequence complementarity (seed match). Specifically, miRanda accounts for the free energy of the duplex together with seed sequence conservation and position and calculates a final score based on base match and gap penalties. RNAhybrid evaluates only the free energy from each possible match between the short (miRNA) and long (circRNA) sequences, retaining only the most stable duplexes. TargetScan searches for 8mer, 7mer, and 6mer on the target RNA that match the miRNA seed sequence. Circr runs each tool with default parameters and without applying any filter on the resulting output. To optimize the computation time, the three algorithms are run as parallel sub processes. Subsequently, the output files of each tool are first collected and then the predicted interactions are converted back to genomic coordinates and annotated using the miRNA:RNA seed category described in (Bartel, 2009) (e.g., 8mer or 7mer seed sequence). Finally, all processed outputs are collected into a table reporting also the number of tools that were able to identify that specific miRNA binding site.
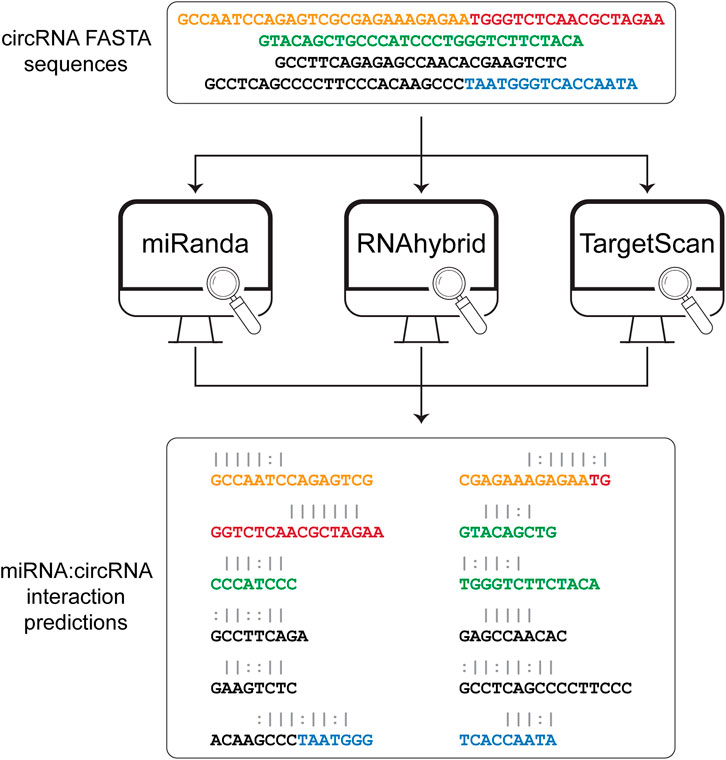
FIGURE 2. Prediction of miRNA:circRNA interactions. The list of FASTA sequences generated in the first step is then given as input to each of the 3 tools that are included in Circr, i.e., miRanda, RNAhybrid, and TargetScan. The result for each tool is then parsed and collected in a comprehensive result table where only unique miRNA:circRNA interacting sites are retained. White spaces indicate a mismatch, pipe represents a match while colon represent a G-U pair.
Comparison of Predicted Interaction Against Validated Databases
In the last step, Circr compares the predicted interactions against the collection of publicly available and experimentally validated miRNA:RNA pairs as well as AGO peaks coordinates contained in the internal database. This comparison is central to select potentially relevant miRNA:circRNA pairs, as it focuses on seed regions that were either validated (and therefore there is a known direct interaction among the two RNAs) or overlap an Argonaute binding site (meaning that the RISC complex is potentially recruited to that specific site). The final table reports whether there is an overlap between the coordinates of validated seed regions or AGO peaks and the predicted ones (Figure 3). Although Circr comprises an internal database of validated interactions and AGO peaks derived from publicly available data, the user can provide custom sets of interactions to focus the analysis on specific sequences of interest.
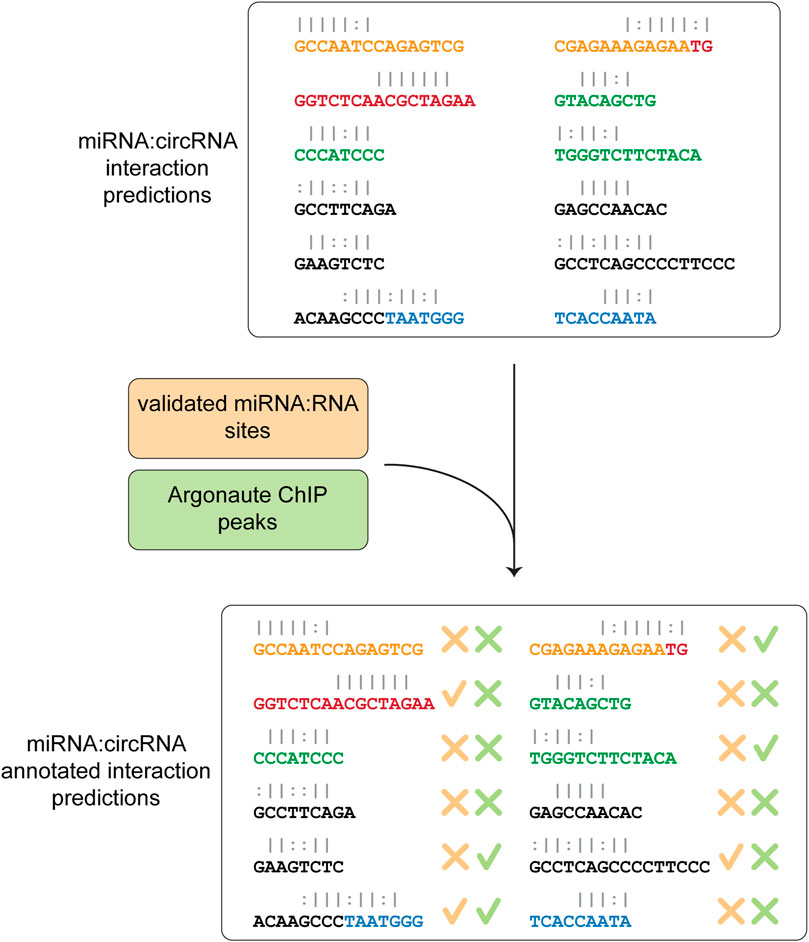
FIGURE 3. Comparison of predicted interaction against validated databases. The final step consists in the annotation of the predicted miRNA:circRNA interactions with experimentally validated miRNA-RNA sites and peaks of Argonaute binding. The final table is then saved as a CSV format file.
Output Format
As output, Circr generates a table that allows the user to easily explore and filter out the seed sequences of interest. The output table consists of 12 columns containing the genomic coordinates of the miRNA binding site, the miRNA name, the investigated circRNA, and its strand, the inferred seed category, a unique interaction identifier, the number of software that have predicted the interaction, flags specifying whether the predicted interaction is experimentally validated or overlaps an Argonaute peak and, if the circRNA coordinates are reported in circBase (http://www.circbase.org/), the corresponding circBase ID (Table 1).

TABLE 1. Field description of the output file generated by Circr.
Testing on Example Dataset
To assess Circr performance and running time, we analyzed 100 circRNAs identified in the developing mouse brain, selected from Supplementary Data S1 of (Dori et al., 2019). These analyses were performed on both a server running Ubuntu 18.04.4 and a computer running macOS 10.15, using 8 CPUs.
Results
To test the performances of Circr, we analyzed a set of circRNAs predicted to be expressed in the developing mouse brain (Dori et al., 2019). Data were obtained from RNAseR treated RNA of the three main cell population of the lateral cortex (proliferating and differentiating progenitors and newborn neurons). From this cohort of sequences, we selected 100 circRNAs of different lengths overlapping genes in the sense and antisense strand, and intergenic ones to cover all possible genomic features of circRNAs. As described in the Methods section, it is possible to provide in input the coordinates of the full sequence of the circRNA and let Circr calculate the exon coordinates or a final set of coordinates with all the features included in each circRNA. To test Circr, we used the 100 mouse sequences and analyzed them using both types of input (i.e., full sequence and final set of coordinates), with the default set of support files relative to the mm9 mouse genome. When providing the genomic coordinates of circRNAs full sequences, Circr completed the analysis in 46 min and retrieved more than 190,000 miRNA:circRNA binding sites (Figure 4). Conversely, providing the input file as a set of final exon/intron coordinates, Circr took 227 min for the prediction of 263,581 sites (Figure 4). Starting from Circr output, we can either focus on interactions identified by all 3 algorithms, resulting in 12,142 hits for the full sequence analysis and 53,539 for the feature coordinates one, or we can select only those overlapping either validated seed regions or an AGO peak, obtaining a list of 25,529 sites (32 if we account for the presence of both an AGO peak and a validated interaction) from the full sequence analysis and 16,991 (20 with both AGO/validated interaction) for the feature coordinates set up. By combining all criteria (3 tools and either an AGO peak or a validated interaction), the list goes down to 1,198 and 1,413 interaction sites for the full sequence and feature coordinates respectively, providing the user with a more manageable number of putative interacting miRNA:circRNA pairs for further validation. The analyses were carried out on an Ubuntu server using 8 threads. The same analyses performed on a personal computer with MacOS 10.15 and 8 threads required 132 min for the full sequence and 384 min for the final set input.
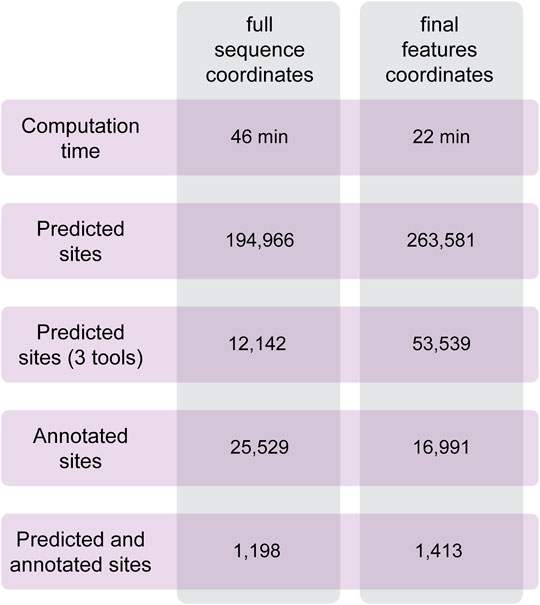
FIGURE 4. Summary of the results obtained with Circr on a server with Ubuntu 18.04. The analysis was performed on 100 circRNAs selected from the list of transcripts expressed in the lateral cortex of the mouse developing brain. The BED file including the coordinates of the chosen circRNAs was supplied both as the full sequence and as the set of final features.
Discussion
Circular RNAs have emerged as a significant player in both physiological and pathological conditions and have become an invaluable source of information as prognostic markers in cancer and several other diseases. However, despite a large number of circRNA databases is currently available, their limitation to already known and annotated sequences in specific organisms hampers the identification of novel circRNAs and the investigation of their potential role as miRNA regulators. As an example, CircNet 2.0 (Liu et al., 2016) focuses on circRNAs found in human cancers whereas ENCORI (Li et al., 2014) collects circRNAs only from human and mouse. Other limitations reside in the nature of miRNA:circRNA interactions reported and their annotation: CircNet 2.0 reports only interaction predicted by at least one tool among PITA, miRanda and TargetScan, but provides only the miRNA ID without specifying the number and genomic location of binding sites. The reported interactions are all supported by miRTarBase (Huang et al., 2020) but these validations involve the miRNA and the gene hosting the circRNA, irrespectively of the site of the interaction. ENCORI reports interactions predicted by just a single tool (miRanda) and only if supported by Ago CLIP-seq Data. From a usability point of view, ENCORI is miRNA-centered and does not allow searching by circRNA ID or coordinates. To cope with these needs, we developed Circr, a tool that combines the output of different miRNA binding site prediction algorithms with validated miRNA:RNA interactions (through direct approaches as luciferase assays or RNA-IP) and Argonaute binding site data. The output of Circr returns a comprehensive view of the circRNAs interactome, it has been structured to be easily browsed and investigated, thus helping researchers to narrow down the list of potential targets for further validation and functional characterization. We applied Circr to the analysis of a set of circRNAs predicted to be expressed in the developing mouse brain and, among the interactions supported by a validated seed region or an AGO peak, we focused our attention to those involving miRNAs known to be expressed in the same tissue (as, for instance, miR-9, miR-124, miR92a, miR92b). As a confirmation, we found a binding site identified by all 3 algorithms for miR-9 on CiCo_mm9_circ_000349, a circRNA already annotated in circBase (mmu_circ_0000044) and identified in mouse brain by (Memczak et al., 2013; Rybak-Wolf et al., 2015). In addition, we also found a binding site, predicted by 2 tools, for miR-92a/miR-92b on CiCo_mm9_circ_002328 (mmu_circ_0006166). To compare Circr performance on a circRNA reported in existing databases, we interrogated CircNet 2.0 and ENCORI for miRNA bindings of hsa_circ_0001946 (CDR1as), a well characterized human circRNA. Circr results reproduced the findings of the two databases and was able to identify two validated interactions with mir-7 and mir-671 (Hansen et al., 2011; Memczak et al., 2013).
One of the major features of Circr resides in its flexibility, as users can virtually investigate any given sequence in any given organism, as long as all the necessary files are available and provided. A possible limitation of our method could be represented by the fact that, in its current implementation, Circr adopts exclusively prediction tools based on seed matching, a feature that might affect the completeness of the information provided. However, we foresee that the growing interests toward this class of RNA will lead to the development of more refined and complete databases coupled with dedicated computational approaches that could be easily incorporated in Circr. In conclusion, although exploiting a previously described approach [e.g., (Chen et al., 2021)], to date Circr represents the first standalone tool that combines multiple prediction algorithms together with experimental data and we believe that it can provide a relevant contribution to the discovery of new and potentially functionally relevant miRNA:circRNA interactions.
Data Availability Statement
Publicly available datasets were analyzed in this study. This data can be found here: https://github.com/bicciatolab/Circr.
Author Contributions
MD conceived the project; MD and JC wrote the code; MD, JC, and MF wrote and revised the manuscript.
Funding
This work was supported by funds from the PRIN 2017 Project 2017HWTP2K of the Italian Ministry of Education, University and Research and the FAR 2019 (E54I19002000001), and GR-2016-02362451 of the Italian Ministry of Health to MF.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fbinf.2022.852834/full#supplementary-material
References
Agarwal, V., Bell, G. W., Nam, J. W., and Bartel, D. P. (2015). Predicting Effective microRNA Target Sites in Mammalian mRNAs. Elife 4. doi:10.7554/eLife.05005
Ashwal-Fluss, R., Meyer, M., Pamudurti, N. R., Ivanov, A., Bartok, O., Hanan, M., et al. (2014). circRNA Biogenesis Competes with Pre-mRNA Splicing. Mol. Cel 56 (1), 55–66. doi:10.1016/j.molcel.2014.08.019
Balakrishnan, I., Yang, X., Brown, J., Ramakrishnan, A., Torok-Storb, B., Kabos, P., et al. (2014). Genome-wide Analysis of miRNA-mRNA Interactions in Marrow Stromal Cells. Stem Cells 32 (3), 662–673. doi:10.1002/stem.1531
Bandyopadhyay, S., and Mitra, R. (2009). TargetMiner: microRNA Target Prediction with Systematic Identification of Tissue-specific Negative Examples. Bioinformatics 25 (20), 2625–2631. doi:10.1093/bioinformatics/btp503
Barrett, S. P., and Salzman, J. (2016). Circular RNAs: Analysis, Expression and Potential Functions. Development 143 (11), 1838–1847. doi:10.1242/dev.128074
Bartel, D. P. (2009). MicroRNAs: Target Recognition and Regulatory Functions. Cell 136 (2), 215–233. doi:10.1016/j.cell.2009.01.002
Benway, C. J., and Iacomini, J. (2018). Defining a microRNA-mRNA Interaction Map for Calcineurin Inhibitor Induced Nephrotoxicity. Am. J. Transplant 18 (4), 796–809. doi:10.1111/ajt.14503
Berta, P., Hawkins, J. R., Sinclair, A. H., Taylor, A., Griffiths, B. L., Goodfellow, P. N., et al. (1990). Genetic Evidence Equating SRY and the Testis-Determining Factor. Nature 348 (6300), 448–450. doi:10.1038/348448a0
Betel, D., Wilson, M., Gabow, A., Marks, D. S., and Sander, C. (2008). The microRNA.Org Resource: Targets and Expression. Nucleic Acids Res. 36, D149–D153. Database issue). doi:10.1093/nar/gkm995
Boudreau, R. L., Jiang, P., Gilmore, B. L., Spengler, R. M., Tirabassi, R., Nelson, J. A., et al. (2014). Transcriptome-wide Discovery of microRNA Binding Sites in Human Brain. Neuron 81 (2), 294–305. doi:10.1016/j.neuron.2013.10.062
Capel, B., Swain, A., Nicolis, S., Hacker, A., Walter, M., Koopman, P., et al. (1993). Circular Transcripts of the Testis-Determining Gene Sry in Adult Mouse Testis. Cell 73 (5), 1019–1030. doi:10.1016/0092-8674(93)90279-y
Chen, Y., Yao, L., Tang, Y., Jhong, J.-H., Wan, J., Chang, J., et al. (2021). CircNet 2.0: an Updated Database for Exploring Circular RNA Regulatory Networks in Cancers. Nucleic Acids Res. 50, D93–D101. doi:10.1093/nar/gkab1036
Chi, S. W., Zang, J. B., Mele, A., and Darnell, R. B. (2009). Argonaute HITS-CLIP Decodes microRNA-mRNA Interaction Maps. Nature 460 (7254), 479–486. doi:10.1038/nature08170
Clark, P. M., Loher, P., Quann, K., Brody, J., Londin, E. R., and Rigoutsos, I. (2014). Argonaute CLIP-Seq Reveals miRNA Targetome Diversity across Tissue Types. Sci. Rep. 4, 5947. doi:10.1038/srep05947
Conn, S. J., Pillman, K. A., Toubia, J., Conn, V. M., Salmanidis, M., Phillips, C. A., et al. (2015). The RNA Binding Protein Quaking Regulates Formation of circRNAs. Cell 160 (6), 1125–1134. doi:10.1016/j.cell.2015.02.014
Consortium, E. P. (2012). An Integrated Encyclopedia of DNA Elements in the Human Genome. Nature 489 (7414), 57–74. doi:10.1038/nature11247
Coronnello, C., and Benos, P. V. (2013). ComiR: Combinatorial microRNA Target Prediction Tool. Nucleic Acids Res. 41, W159–W164. Web Server issue). doi:10.1093/nar/gkt379
Dori, M., and Bicciato, S. (2019). Integration of Bioinformatic Predictions and Experimental Data to Identify circRNA-miRNA Associations. Genes (Basel) 10 (9). doi:10.3390/genes10090642
Dori, M., Haj Abdullah Alieh, L., Cavalli, D., Massalini, S., Lesche, M., Dahl, A., et al. (2019). Sequence and Expression Levels of Circular RNAs in Progenitor Cell Types during Mouse Corticogenesis. Life Sci. Alliance 2 (2). doi:10.26508/lsa.201900354
Dudekula, D. B., Panda, A. C., Grammatikakis, I., De, S., Abdelmohsen, K., and Gorospe, M. (2016). CircInteractome: A Web Tool for Exploring Circular RNAs and Their Interacting Proteins and microRNAs. RNA Biol. 13 (1), 34–42. doi:10.1080/15476286.2015.1128065
Enright, A. J., John, B., Gaul, U., Tuschl, T., Sander, C., and Marks, D. S. (2003). MicroRNA Targets in Drosophila. Genome Biol. 5 (1), R1. doi:10.1186/gb-2003-5-1-r1
Erhard, F., Haas, J., Lieber, D., Malterer, G., Jaskiewicz, L., Zavolan, M., et al. (2014). Widespread Context Dependency of microRNA-Mediated Regulation. Genome Res. 24 (6), 906–919. doi:10.1101/gr.166702.113
Fernandes, R. C., Toubia, J., Townley, S., Hanson, A. R., Dredge, B. K., Pillman, K. A., et al. (2021). Post-transcriptional Gene Regulation by MicroRNA-194 Promotes Neuroendocrine Transdifferentiation in Prostate Cancer. Cell Rep 34 (1), 108585. doi:10.1016/j.celrep.2020.108585
Ghosal, S., Das, S., Sen, R., Basak, P., and Chakrabarti, J. (2013). Circ2Traits: a Comprehensive Database for Circular RNA Potentially Associated with Disease and Traits. Front. Genet. 4, 283. doi:10.3389/fgene.2013.00283
Gillen, A. E., Yamamoto, T. M., Kline, E., Hesselberth, J. R., and Kabos, P. (2016). Improvements to the HITS-CLIP Protocol Eliminate Widespread Mispriming Artifacts. BMC Genomics 17, 338. doi:10.1186/s12864-016-2675-5
Glažar, P., Papavasileiou, P., and Rajewsky, N. (2014). circBase: a Database for Circular RNAs. Rna 20 (11), 1666–1670. doi:10.1261/rna.043687.113
Grosswendt, S., Filipchyk, A., Manzano, M., Klironomos, F., Schilling, M., Herzog, M., et al. (2014). Unambiguous Identification of miRNA:target Site Interactions by Different Types of Ligation Reactions. Mol. Cel 54 (6), 1042–1054. doi:10.1016/j.molcel.2014.03.049
Hafner, M., Landthaler, M., Burger, L., Khorshid, M., Hausser, J., Berninger, P., et al. (2010). Transcriptome-wide Identification of RNA-Binding Protein and microRNA Target Sites by PAR-CLIP. Cell 141 (1), 129–141. doi:10.1016/j.cell.2010.03.009
Hammond, S. M., Bernstein, E., Beach, D., and Hannon, G. J. (2000). An RNA-Directed Nuclease Mediates post-transcriptional Gene Silencing in Drosophila Cells. Nature 404 (6775), 293–296. doi:10.1038/35005107
Hansen, T. B., Jensen, T. I., Clausen, B. H., Bramsen, J. B., Finsen, B., Damgaard, C. K., et al. (2013a). Natural RNA Circles Function as Efficient microRNA Sponges. Nature 495 (7441), 384–388. doi:10.1038/nature11993
Hansen, T. B., Kjems, J., and Damgaard, C. K. (2013b). Circular RNA and miR-7 in Cancer. Cancer Res. 73 (18), 5609–5612. doi:10.1158/0008-5472.Can-13-1568
Hansen, T. B., Wiklund, E. D., Bramsen, J. B., Villadsen, S. B., Statham, A. L., Clark, S. J., et al. (2011). miRNA-Dependent Gene Silencing Involving Ago2-Mediated Cleavage of a Circular Antisense RNA. Embo j 30 (21), 4414–4422. doi:10.1038/emboj.2011.359
Helwak, A., Kudla, G., Dudnakova, T., and Tollervey, D. (2013). Mapping the Human miRNA Interactome by CLASH Reveals Frequent Noncanonical Binding. Cell 153 (3), 654–665. doi:10.1016/j.cell.2013.03.043
Howe, K. L., Achuthan, P., Allen, J., Allen, J., Alvarez-Jarreta, J., Amode, M. R., et al. (2021). Ensembl 2021. Nucleic Acids Res. 49 (D1), D884–d891. doi:10.1093/nar/gkaa942
Hsu, J. B., Chiu, C. M., Hsu, S. D., Huang, W. Y., Chien, C. H., Lee, T. Y., et al. (2011). miRTar: an Integrated System for Identifying miRNA-Target Interactions in Human. BMC Bioinformatics 12, 300. doi:10.1186/1471-2105-12-300
Huang, H. Y., Lin, Y. C., Li, J., Huang, K. Y., Shrestha, S., Hong, H. C., et al. (2020). miRTarBase 2020: Updates to the Experimentally Validated microRNA-Target Interaction Database. Nucleic Acids Res. 48 (D1), D148–d154. doi:10.1093/nar/gkz896
Huang, S., Yang, B., Chen, B. J., Bliim, N., Ueberham, U., Arendt, T., et al. (2017). The Emerging Role of Circular RNAs in Transcriptome Regulation. Genomics 109 (5-6), 401–407. doi:10.1016/j.ygeno.2017.06.005
Huang, V., Zheng, J., Qi, Z., Wang, J., Place, R. F., Yu, J., et al. (2013). Ago1 Interacts with RNA Polymerase II and Binds to the Promoters of Actively Transcribed Genes in Human Cancer Cells. Plos Genet. 9 (9), e1003821. doi:10.1371/journal.pgen.1003821
Hutvágner, G., and Zamore, P. D. (2002). A microRNA in a Multiple-Turnover RNAi Enzyme Complex. Science 297 (5589), 2056–2060. doi:10.1126/science.1073827
Ivanov, A., Memczak, S., Wyler, E., Torti, F., Porath, H. T., Orejuela, M. R., et al. (2015). Analysis of Intron Sequences Reveals Hallmarks of Circular RNA Biogenesis in Animals. Cel Rep 10 (2), 170–177. doi:10.1016/j.celrep.2014.12.019
Izuogu, O. G., Alhasan, A. A., Mellough, C., Collin, J., Gallon, R., Hyslop, J., et al. (2018). Analysis of Human ES Cell Differentiation Establishes that the Dominant Isoforms of the lncRNAs RMST and FIRRE Are Circular. BMC Genomics 19 (1), 276. doi:10.1186/s12864-018-4660-7
Jahani, S., Nazeri, E., Majidzadeh-A, K., Jahani, M., and Esmaeili, R. (2020). Circular RNA; a New Biomarker for Breast Cancer: A Systematic Review. J. Cel Physiol 235 (7-8), 5501–5510. doi:10.1002/jcp.29558
Jeck, W. R., Sorrentino, J. A., Wang, K., Slevin, M. K., Burd, C. E., Liu, J., et al. (2013). Circular RNAs Are Abundant, Conserved, and Associated with ALU Repeats. Rna 19 (2), 141–157. doi:10.1261/rna.035667.112
Jungkamp, A. C., Stoeckius, M., Mecenas, D., Grün, D., Mastrobuoni, G., Kempa, S., et al. (2011). In Vivo and Transcriptome-wide Identification of RNA Binding Protein Target Sites. Mol. Cel 44 (5), 828–840. doi:10.1016/j.molcel.2011.11.009
Karagkouni, D., Paraskevopoulou, M. D., Chatzopoulos, S., Vlachos, I. S., Tastsoglou, S., Kanellos, I., et al. (2018). DIANA-TarBase V8: a Decade-Long Collection of Experimentally Supported miRNA-Gene Interactions. Nucleic Acids Res. 46 (D1), D239–d245. doi:10.1093/nar/gkx1141
Kent, W. J., Sugnet, C. W., Furey, T. S., Roskin, K. M., Pringle, T. H., Zahler, A. M., et al. (2002). The Human Genome Browser at UCSC. Genome Res. 12 (6), 996–1006. doi:10.1101/gr.229102
Kertesz, M., Iovino, N., Unnerstall, U., Gaul, U., and Segal, E. (2007). The Role of Site Accessibility in microRNA Target Recognition. Nat. Genet. 39 (10), 1278–1284. doi:10.1038/ng2135
Kobayashi, M., Benakis, C., Anderson, C., Moore, M. J., Poon, C., Uekawa, K., et al. (2019). AGO CLIP Reveals an Activated Network for Acute Regulation of Brain Glutamate Homeostasis in Ischemic Stroke. Cel Rep 28 (4), 979–e6. e976. doi:10.1016/j.celrep.2019.06.075
Kozomara, A., Birgaoanu, M., and Griffiths-Jones, S. (2019). miRBase: from microRNA Sequences to Function. Nucleic Acids Res. 47 (D1), D155–d162. doi:10.1093/nar/gky1141
Lasda, E., and Parker, R. (2014). Circular RNAs: Diversity of Form and Function. Rna 20 (12), 1829–1842. doi:10.1261/rna.047126.114
Leung, A. K., Young, A. G., Bhutkar, A., Zheng, G. X., Bosson, A. D., Nielsen, C. B., et al. (2011). Genome-wide Identification of Ago2 Binding Sites from Mouse Embryonic Stem Cells with and without Mature microRNAs. Nat. Struct. Mol. Biol. 18 (2), 237–244. doi:10.1038/nsmb.1991
Li, J. H., Liu, S., Zhou, H., Qu, L. H., and Yang, J. H. (2014). starBase v2.0: Decoding miRNA-ceRNA, miRNA-ncRNA and Protein-RNA Interaction Networks from Large-Scale CLIP-Seq Data. Nucleic Acids Res. 42, D92–D97. Database issue). doi:10.1093/nar/gkt1248
Li, P., Chen, S., Chen, H., Mo, X., Li, T., Shao, Y., et al. (2015). Using Circular RNA as a Novel Type of Biomarker in the Screening of Gastric Cancer. Clin. Chim. Acta 444, 132–136. doi:10.1016/j.cca.2015.02.018
Liu, Y. C., Li, J. R., Sun, C. H., Andrews, E., Chao, R. F., Lin, F. M., et al. (2016). CircNet: a Database of Circular RNAs Derived from Transcriptome Sequencing Data. Nucleic Acids Res. 44 (D1), D209–D215. doi:10.1093/nar/gkv940
Luna, J. M., Barajas, J. M., Teng, K. Y., Sun, H. L., Moore, M. J., Rice, C. M., et al. (2017). Argonaute CLIP Defines a Deregulated miR-122-Bound Transcriptome that Correlates with Patient Survival in Human Liver Cancer. Mol. Cel 67 (3), 400–e7. e407. doi:10.1016/j.molcel.2017.06.025
Luna, J. M., Scheel, T. K., Danino, T., Shaw, K. S., Mele, A., Fak, J. J., et al. (2015). Hepatitis C Virus RNA Functionally Sequesters miR-122. Cell 160 (6), 1099–1110. doi:10.1016/j.cell.2015.02.025
Lyu, D., and Huang, S. (2017). The Emerging Role and Clinical Implication of Human Exonic Circular RNA. RNA Biol. 14 (8), 1000–1006. doi:10.1080/15476286.2016.1227904
Memczak, S., Jens, M., Elefsinioti, A., Torti, F., Krueger, J., Rybak, A., et al. (2013). Circular RNAs Are a Large Class of Animal RNAs with Regulatory Potency. Nature 495 (7441), 333–338. doi:10.1038/nature11928
Memczak, S., Papavasileiou, P., Peters, O., and Rajewsky, N. (2015). Identification and Characterization of Circular RNAs as a New Class of Putative Biomarkers in Human Blood. PLoS One 10 (10), e0141214. doi:10.1371/journal.pone.0141214
Moore, M. J., Scheel, T. K., Luna, J. M., Park, C. Y., Fak, J. J., Nishiuchi, E., et al. (2015). miRNA-target Chimeras Reveal miRNA 3'-end Pairing as a Major Determinant of Argonaute Target Specificity. Nat. Commun. 6, 8864. doi:10.1038/ncomms9864
Moshkovich, N., Nisha, P., Boyle, P. J., Thompson, B. A., Dale, R. K., and Lei, E. P. (2011). RNAi-Independent Role for Argonaute2 in CTCF/CP190 Chromatin Insulator Function. Genes Dev. 25 (16), 1686–1701. doi:10.1101/gad.16651211
Nowakowski, T. J., Rani, N., Golkaram, M., Zhou, H. R., Alvarado, B., Huch, K., et al. (2018). Regulation of Cell-type-specific Transcriptomes by microRNA Networks during Human Brain Development. Nat. Neurosci. 21 (12), 1784–1792. doi:10.1038/s41593-018-0265-3
Panda, A. C. (2018). Circular RNAs Act as miRNA Sponges. Adv. Exp. Med. Biol. 1087, 67–79. doi:10.1007/978-981-13-1426-1_6
Pillai, M. M., Gillen, A. E., Yamamoto, T. M., Kline, E., Brown, J., Flory, K., et al. (2014). HITS-CLIP Reveals Key Regulators of Nuclear Receptor Signaling in Breast Cancer. Breast Cancer Res. Treat. 146 (1), 85–97. doi:10.1007/s10549-014-3004-9
Rayon-Estrada, V., Harjanto, D., Hamilton, C. E., Berchiche, Y. A., Gantman, E. C., Sakmar, T. P., et al. (2017). Epitranscriptomic Profiling across Cell Types Reveals Associations between APOBEC1-Mediated RNA Editing, Gene Expression Outcomes, and Cellular Function. Proc. Natl. Acad. Sci. U S A. 114 (50), 13296–13301. doi:10.1073/pnas.1714227114
Rehmsmeier, M., Steffen, P., Hochsmann, M., and Giegerich, R. (2004). Fast and Effective Prediction of microRNA/target Duplexes. Rna 10 (10), 1507–1517. doi:10.1261/rna.5248604
Rennie, W., Kanoria, S., Liu, C., Mallick, B., Long, D., Wolenc, A., et al. (2016). STarMirDB: A Database of microRNA Binding Sites. RNA Biol. 13 (6), 554–560. doi:10.1080/15476286.2016.1182279
Rennie, W., Liu, C., Carmack, C. S., Wolenc, A., Kanoria, S., Lu, J., et al. (2014). STarMir: a Web Server for Prediction of microRNA Binding Sites. Nucleic Acids Res. 42, W114–W118. Web Server issue). doi:10.1093/nar/gku376
Rybak-Wolf, A., Jens, M., Murakawa, Y., Herzog, M., Landthaler, M., and Rajewsky, N. (2014). A Variety of Dicer Substrates in Human and C. elegans. Cell 159 (5), 1153–1167. doi:10.1016/j.cell.2014.10.040
Rybak-Wolf, A., Stottmeister, C., Glažar, P., Jens, M., Pino, N., Giusti, S., et al. (2015). Circular RNAs in the Mammalian Brain Are Highly Abundant, Conserved, and Dynamically Expressed. Mol. Cel 58 (5), 870–885. doi:10.1016/j.molcel.2015.03.027
Salzman, J., Gawad, C., Wang, P. L., Lacayo, N., and Brown, P. O. (2012). Circular RNAs Are the Predominant Transcript Isoform from Hundreds of Human Genes in Diverse Cell Types. PLoS One 7 (2), e30733. doi:10.1371/journal.pone.0030733
Sarshad, A. A., Juan, A. H., Muler, A. I. C., Anastasakis, D. G., Wang, X., Genzor, P., et al. (2018). Argonaute-miRNA Complexes Silence Target mRNAs in the Nucleus of Mammalian Stem Cells. Mol. Cel 71 (6), 1040-e8–e1048. doi:10.1016/j.molcel.2018.07.020
Starke, S., Jost, I., Rossbach, O., Schneider, T., Schreiner, S., Hung, L. H., et al. (2015). Exon Circularization Requires Canonical Splice Signals. Cel Rep 10 (1), 103–111. doi:10.1016/j.celrep.2014.12.002
Suenkel, C., Cavalli, D., Massalini, S., Calegari, F., and Rajewsky, N. (2020). A Highly Conserved Circular RNA Is Required to Keep Neural Cells in a Progenitor State in the Mammalian Brain. Cel Rep 30 (7), 2170–e5. e2175. doi:10.1016/j.celrep.2020.01.083
Suzuki, H., Zuo, Y., Wang, J., Zhang, M. Q., Malhotra, A., and Mayeda, A. (2006). Characterization of RNase R-Digested Cellular RNA Source that Consists of Lariat and Circular RNAs from Pre-mRNA Splicing. Nucleic Acids Res. 34 (8), e63. doi:10.1093/nar/gkl151
Thadani, R., and Tammi, M. T. (2006). MicroTar: Predicting microRNA Targets from RNA Duplexes. BMC Bioinformatics 7 Suppl 5 (Suppl. 5Suppl 5), S20. doi:10.1186/1471-2105-7-s5-s20
Verduci, L., Strano, S., Yarden, Y., and Blandino, G. (2019). The circRNA-microRNA Code: Emerging Implications for Cancer Diagnosis and Treatment. Mol. Oncol. 13 (4), 669–680. doi:10.1002/1878-0261.12468
Vincent, H. A., and Deutscher, M. P. (2006). Substrate Recognition and Catalysis by the Exoribonuclease RNase R. J. Biol. Chem. 281 (40), 29769–29775. doi:10.1074/jbc.M606744200
Wang, S., Zhang, K., Tan, S., Xin, J., Yuan, Q., Xu, H., et al. (2021). Circular RNAs in Body Fluids as Cancer Biomarkers: the New Frontier of Liquid Biopsies. Mol. Cancer 20 (1), 13. doi:10.1186/s12943-020-01298-z
Wang, Y., and Wang, Z. (2015). Efficient Backsplicing Produces Translatable Circular mRNAs. Rna 21 (2), 172–179. doi:10.1261/rna.048272.114
Xiao, R., Chen, J. Y., Liang, Z., Luo, D., Chen, G., Lu, Z. J., et al. (2019). Pervasive Chromatin-RNA Binding Protein Interactions Enable RNA-Based Regulation of Transcription. Cell 178 (1), 107–e18. e118. doi:10.1016/j.cell.2019.06.001
Xu, P., Palmer, L. E., Lechauve, C., Zhao, G., Yao, Y., Luan, J., et al. (2019). Regulation of Gene Expression by miR-144/451 during Mouse Erythropoiesis. Blood 133 (23), 2518–2528. doi:10.1182/blood.2018854604
Yang, J. H., Li, J. H., Shao, P., Zhou, H., Chen, Y. Q., and Qu, L. H. (2011). starBase: a Database for Exploring microRNA-mRNA Interaction Maps from Argonaute CLIP-Seq and Degradome-Seq Data. Nucleic Acids Res. 39 (Database issue), D202–D209. doi:10.1093/nar/gkq1056
Yu, C. Y., Li, T. C., Wu, Y. Y., Yeh, C. H., Chiang, W., Chuang, C. Y., et al. (2017). The Circular RNA circBIRC6 Participates in the Molecular Circuitry Controlling Human Pluripotency. Nat. Commun. 8 (1), 1149. doi:10.1038/s41467-017-01216-w
Keywords: circRNA, miRNA, interactions prediction, miRNA binding, prediction tools
Citation: Dori M, Caroli J and Forcato M (2022) Circr, a Computational Tool to Identify miRNA:circRNA Associations. Front. Bioinform. 2:852834. doi: 10.3389/fbinf.2022.852834
Received: 11 January 2022; Accepted: 21 February 2022;
Published: 11 March 2022.
Edited by:
Stefania Bortoluzzi, University of Padua, ItalyReviewed by:
Chiara Pastrello, University Health Network (UHN), CanadaQuan Zou, University of Electronic Science and Technology of China, China
Copyright © 2022 Dori, Caroli and Forcato. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mattia Forcato, bWF0dGlhLmZvcmNhdG9AdW5pbW9yZS5pdA==
†These authors have contributed equally to this work