 Roghayyeh Alipanahi1†
Roghayyeh Alipanahi1† Leila Safari
Leila Safari- 1Department of Computer Engineering, University of Zanjan, Zanjan, Iran
- 2Department of Neurozentrum, Universitätsklinikum Freiburg, Freiburg, Germany
Clustered regularly interspaced short palindromic repeats (CRISPR)-based gene editing has been widely used in various cell types and organisms. To make genome editing with Clustered regularly interspaced short palindromic repeats far more precise and practical, we must concentrate on the design of optimal gRNA and the selection of appropriate Cas enzymes. Numerous computational tools have been created in recent years to help researchers design the best gRNA for Clustered regularly interspaced short palindromic repeats researches. There are two approaches for designing an appropriate gRNA sequence (which targets our desired sites with high precision): experimental and predicting-based approaches. It is essential to reduce off-target sites when designing an optimal gRNA. Here we review both traditional and machine learning-based approaches for designing an appropriate gRNA sequence and predicting off-target sites. In this review, we summarize the key characteristics of all available tools (as far as possible) and compare them together. Machine learning-based tools and web servers are believed to become the most effective and reliable methods for predicting on-target and off-target activities of Clustered regularly interspaced short palindromic repeats in the future. However, these predictions are not so precise now and the performance of these algorithms -especially deep learning one’s-depends on the amount of data used during training phase. So, as more features are discovered and incorporated into these models, predictions become more in line with experimental observations. We must concentrate on the creation of ideal gRNA and the choice of suitable Cas enzymes in order to make genome editing with Clustered regularly interspaced short palindromic repeats far more accurate and feasible.
1 Introduction
Over the last decade, the Clustered regularly interspaced short palindromic repeats (CRISPR)/Cas system has become the dominant tool for genome editing due to its simplicity, high performance, accuracy, and programmability (Gaj et al., 2013; Jacquin et al., 2019; Afzal et al., 2020). In addition, other influential factors such as ease of use, low cost, high speed, multiplex potential, and higher specific DNA targeting ability have increased the success and popularity of CRISPR across the global scientific community (Mali et al., 2013). The unique characteristics of this technology have made it one of the broad topics in molecular biology, synthetic biology, and genetic engineering (Jinek et al., 2012). Gene activation (CRISPRa), gene repression, CRISPR interference (CRISPRi), and epigenome editing are popular tasks in genome engineering using CRISPER. The basic overflow of the CRISPR systems is illustrated in Figure 1.
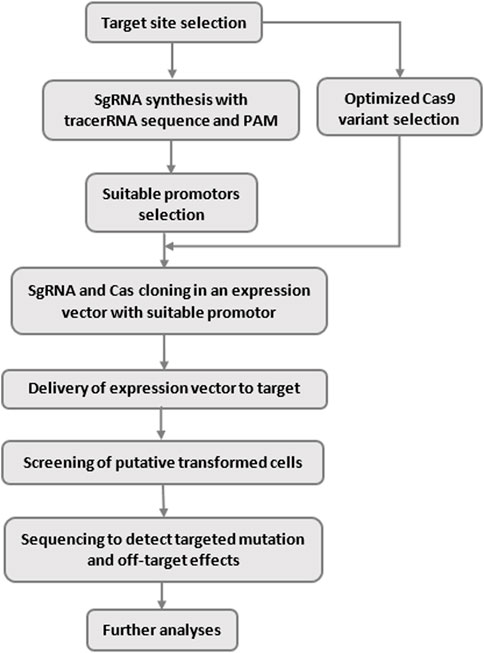
FIGURE1. Basic overflow of CRISPR systems.
As shown in Figure 2, CRISPR systems have three main components. The first one is a short synthetic guide RNA sequence (gRNA) necessary for Cas binding. The gRNA targets the Cas9 endonuclease (a protein which can cleave the DNA sequences) to define DNA. The gRNA can be supplied as a two-part system consisting of crRNA and tracrRNA, or as a single guide RNA (sgRNA), where the crRNA and tracrRNA are connected by a linker. The target’s recognition is facilitated by the protospacer-adjacent motif (PAM). Cleavage occurs on both strands 3 bp upstream of the PAM.
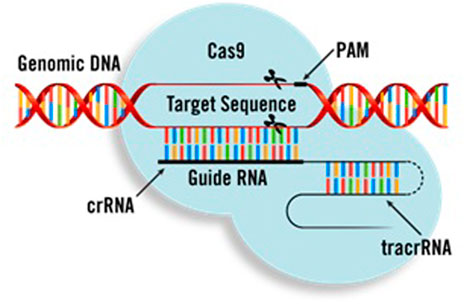
FIGURE 2. Main components of CRISPR (Duan et al., 2021).
To use CRISPR for genome engineering, we need to select two components: Cas9 and gRNA (Gasiunas et al., 2012; Cox et al., 2015). Once a genome modification is decided, the first step is to identify the best site/sites for targeting Cas-induced DSBs (Jinek et al., 2014). The second step is to design the appropriative gRNA (Cui et al., 2018).
After designing gRNA, the only requirement for cleaving a CRISPR target site is finding a 3-base pair (3 bp) PAM. The form of PAM varies depending on the bacterial species of the Cas9 gene. For example, the most commonly used Cas9 nuclease, derived from S.pyogenes, recognizes a PAM sequence of NGG (Rabinowitz et al., 2020). Using the frequency of “GG” = 5.21% in the reference human genome, there would be an expected 161,284,793 NGG PAM sites in the human genome, or roughly one “GG” dinucleotide every 42 bases. So, cleaving unwanted sites, called off-target sites, is very common (Duan et al., 2021). Therefore, CRISPR target sites should be selected in such a way that minimizes potential off-target cleavage (Herai, 2019; Rabinowitz et al., 2020). But this is not always straightforward as it is not guaranteed that the desired cleaves will appear on just the selected site. Unfortunately, the existence of these unwanted cleaves is possible in every experiment. Therefore, activity (on-target) and specificity (off-target) are two critical factors considered when designing a genomic edition with CRISPR (Herai, 2019).
According to research, the accuracy of CRISPR-based genomic edition depends on two issues: 1) the choice of Cas enzyme with suitable cutting power, 2) the choice of the appropriate cutting site, which relies on the performance of the gRNA. To achieve this, in the first step, we must select the optimal gRNAs contains high on-target activity and low (no) off-target efficiency (Moreno-Mateos et al., 2015; Luo et al., 2019; Manibalan et al., 2020). We will discuss this issue later. In the second step, we must select a suitable Cas enzyme [15]. In recent years, different variants of the Cas enzyme have been discovered. We can proceed according to Figure 3 to choose the proper Cas, depending on the type of editing. The choice of the Cas enzyme is effective on the PAM and the gRNA design.
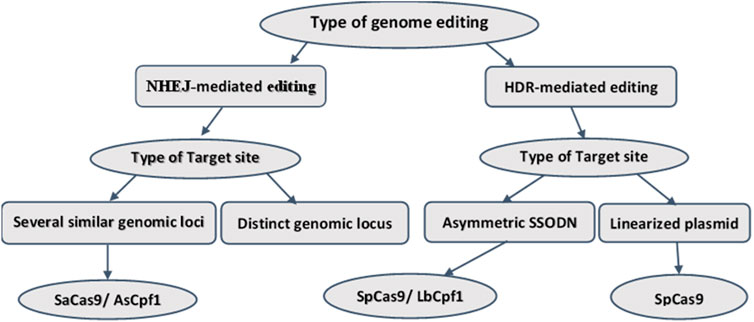
FIGURE 3. Selection of Cas enzyme.
In recent years, researchers have taken two main approaches for designing gRNAs, including experimental and machine learning-based methods (ML) (Lin and Luo, 2019). ML-based methods utilize the results of computational algorithms trained with real data to predict the effects of gRNAs instead of designing an actual experiment. Experimental methods are very costly and time-consuming (Chuai et al., 2017; Lin and Luo, 2019). In contrast, ML models are inexpensive and manageable. However, in terms of accuracy, they are still very different from experimental methods (Höijer et al., 2020). The accuracy of ML methods is highly dependent on the training process and the availability of adequate training data. Recent advances in the genome-wide analyses help researchers to discover all off-target sites, while the detection methods like Polymerase Chain Reaction (PCR) based methods, cannot find all of these sites. Using new sequencing technology, such as next-generation sequencing (NGS), and third generation sequencing which based on long-reads, can help us to detect more off-target sites. Mainly, single-molecule real-time sequencing (SMRT), has shown promising performance in genome sequencing. Researchers use these techniques to find more accurate information about off-target sites and use them in training their computational models (Lin and Wong, 2018; Höijer et al., 2020). Also, there are some repetitive, low complexity, AT/GC-rich regions, known as dark, in which ML-based tools cannot predict on-target and off-target sites in these areas. But amplification-free long-read sequencing technology helps to reveal Cas9 target sites even in these dark regions (Höijer et al., 2020). As the number of available features about on-target and off-target sites and the creation of large databases in this field increases, the predictions of ML-based methods become closer to experimental observations (Jiang et al., 2016; Abadi et al., 2017).
Some recent research has shown that ML-based methods can determine the extent of effective interactions and side-effects (changing unwanted sites) of each gRNA precisely (Abadi et al., 2017; Lin and Wong, 2018). Such a process can significantly accelerate the process of gRNA design for any part of human DNA, thus allowing us to edit anywhere in DNA (Jiang et al., 2016). However, existing models still have challenging issues, such as data imbalance, data heterogeneity, insufficient training data, generalizability, and cross-species inefficiency (Chuai et al., 2017).
We described the basic concepts of CRISPR systems and introduced activity and specificity as two main challenges in this area (Moreno-Mateos et al., 2015; Herai, 2019). In the rest of the paper, we provide an overview of computational approaches, especially machine and deep learning (MDL) algorithms, which we believe are the most effective and reliable methods for predicting gRNAs effects. The summary of our review is presented in Tables 1–Tables3, only for tools with active access link. Table 1 illustrates computational tools and software packages related to CRISPR systems; Table 2 summarizes tools and software packages related to finding off-target sites; Table 3 shows those related to gRNA design; and finally, Table 4 reports MDL-based tools and software packages related to CRISPR systems.
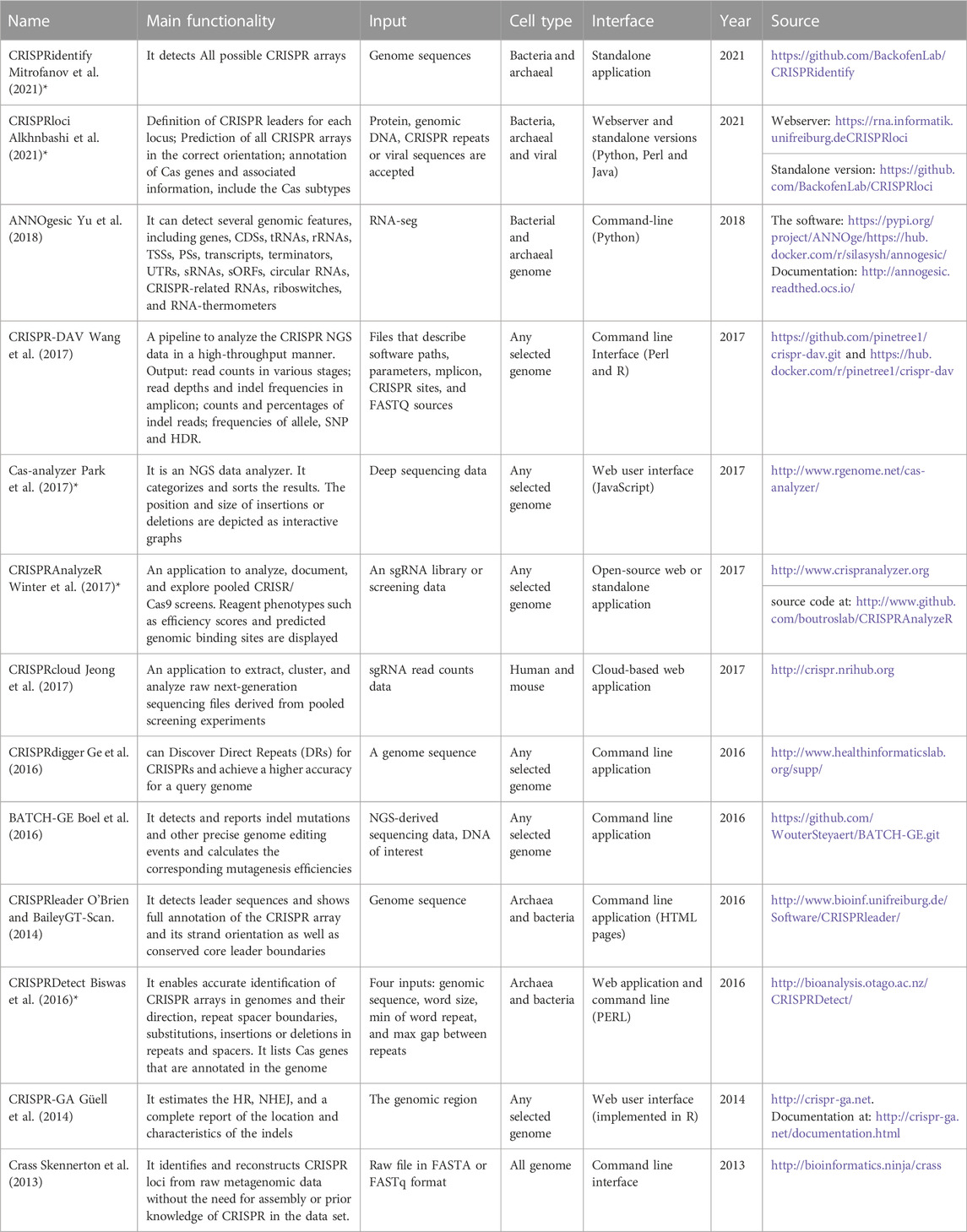
TABLE 1. Tools and software packages related to CRISPR systems.
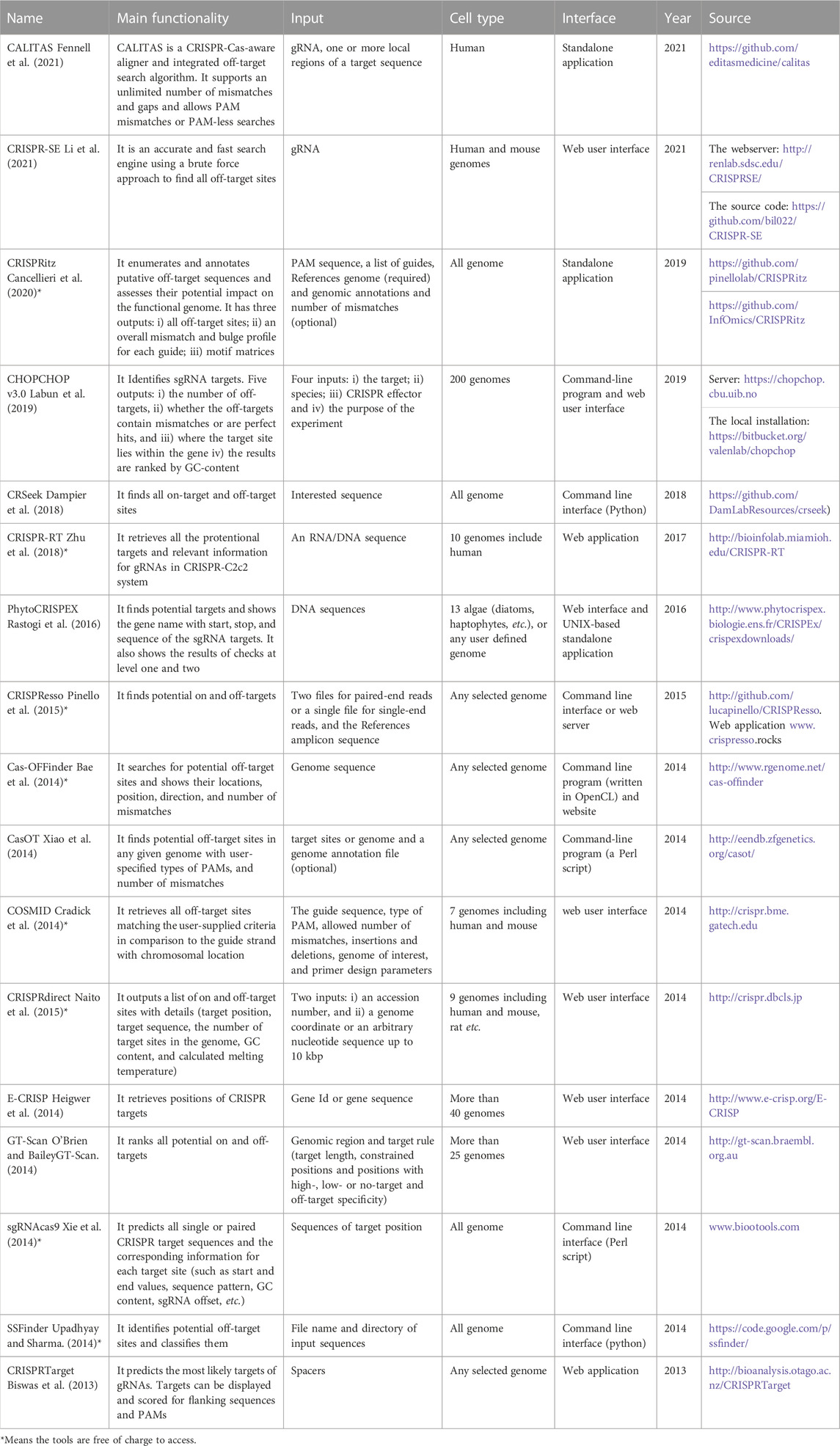
TABLE 2. Tools and software packages related to finding off-target sites.
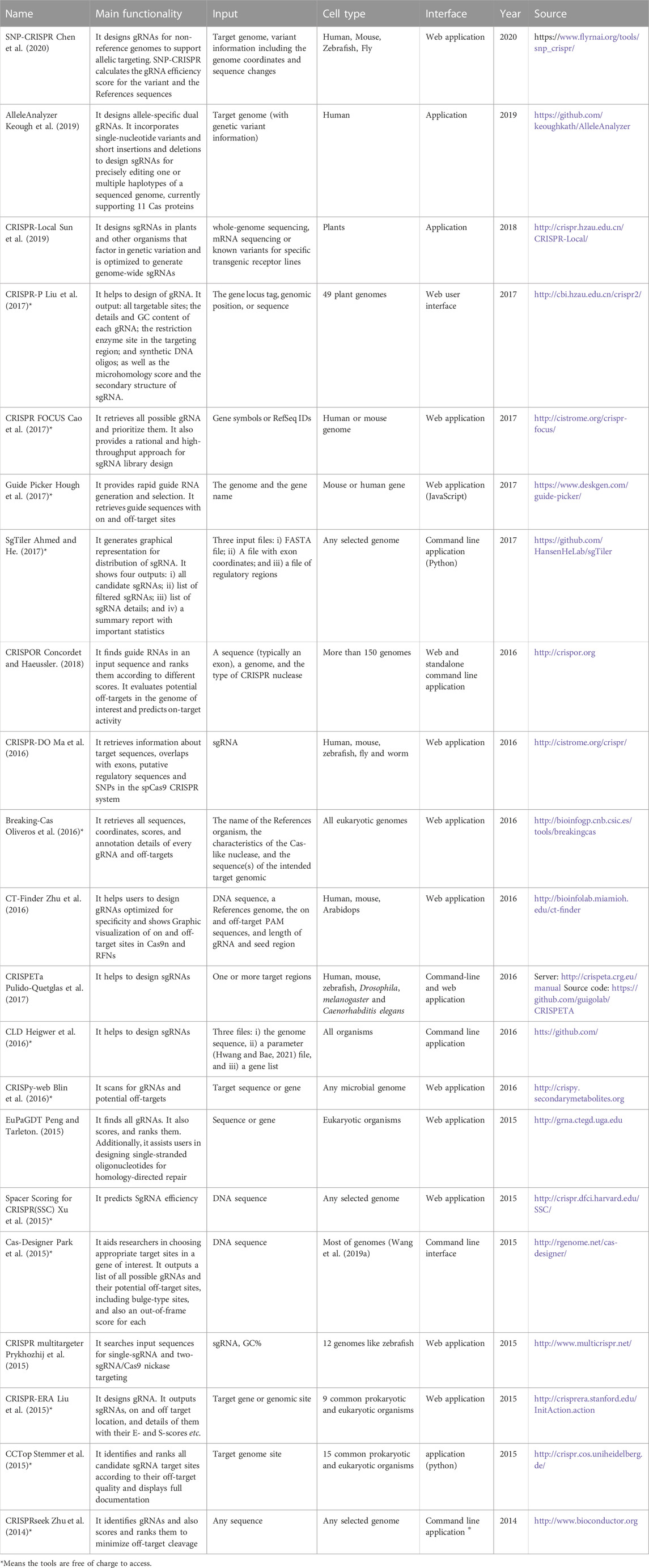
TABLE 3. Tools and software packages related to gRNA design.
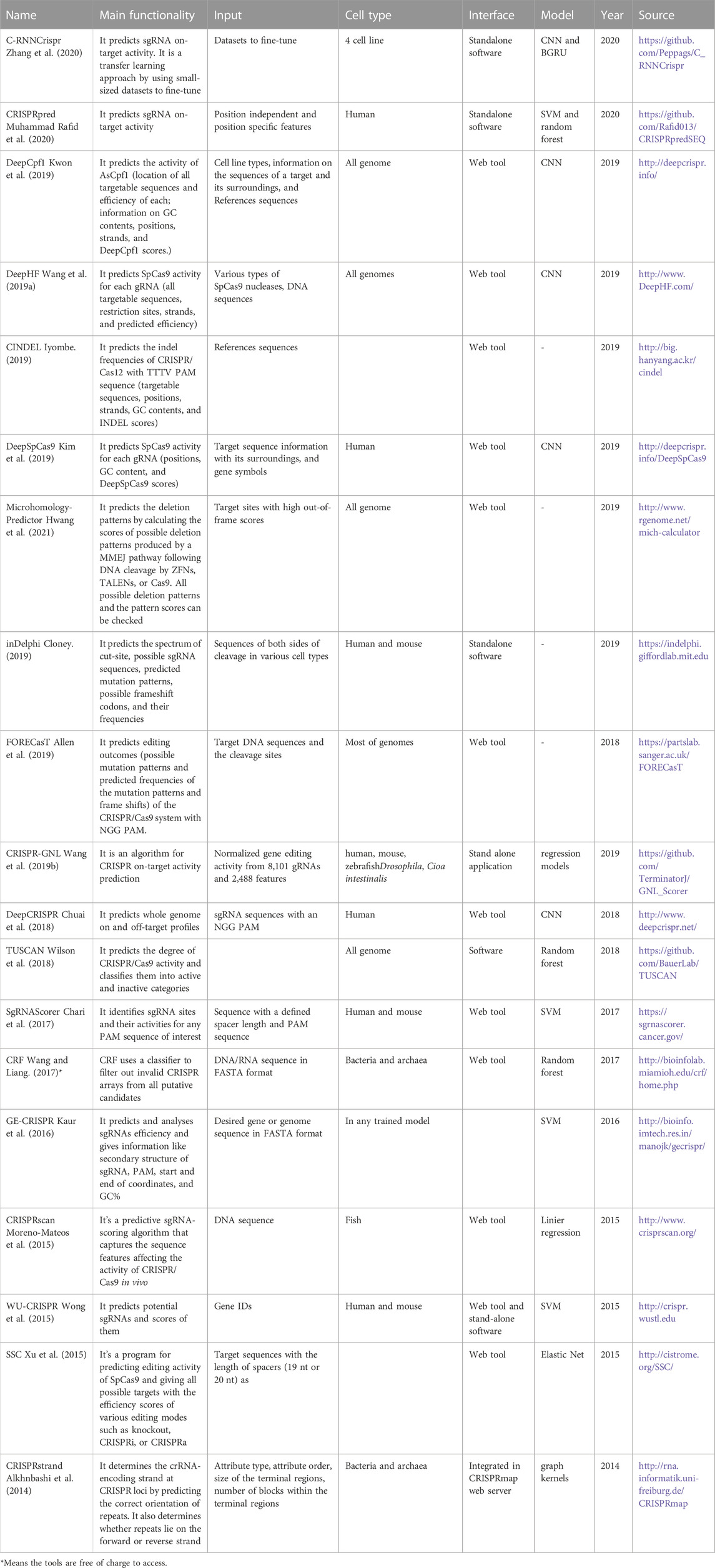
TABLE 4. MDL-based tools and software packages related to CRISPR systems.
2 Computational approaches in CRISPR
Computational approaches are an essential part of CRISPR research. The bioinformatics studies have made significant contributions to the initial discovery of CRISPR (Alkhnbashi et al., 2014; Makarova et al., 2015). We summarize some of them in Table 1. Bioinformatics tools play a significant role in these fields: 1) determination of the specific differences between the CRISPR/Cas systems from archaeal and bacterial sources; 2) determination of required repeat spacer sequences for processing the mature CRISPR RNA (crRNA); 3) prediction of the transcribed strand of CRISPR arrays; 4) determination of CRISPR leader sequences; 5) classification of Cas proteins; 6) prediction of proper gRNA; 7) prediction of on-target and off-target effects; and so on (Listgarten et al., 2016; Lin and Wong, 2018; Listgarten et al., 2018; Herai, 2019; Alkhnbashi et al., 2020; Smith et al., 2020).
According to our review, low cleavage efficiency and off-target effects hamper CRISPR development and application. So, prediction of proper gRNA and prediction of on-target and off-target effects is so critical. In the rest of the paper, we will focus on the tools that have been developed for designing optimal gRNA with low off-target effects.
2.1 gRNA design
There are two fundamental questions in CRISPR researches. The first question is: what are the targets of the given gRNA? Some methods, such as CRISPResso (Pinello et al., 2016) and CRISPRTarget (Biswas et al., 2013), try to calculate potential targets by taking a gRNA as input and using computational algorithms (more details are described in Table 3). Tools like CRISPRTarget (Biswas et al., 2013) offer a way to answer this question using a ML-based approach (Table 4 shows more details). The second important question is how to be confident about the accuracy of CRISPR edits. Most of the tools or methods in CRISPR’s field have been developed to answer these two questions. In Tables 2, 3, we tried to collect all of them and describe their details.
Also, we realized that most of researches in CRISPR area mainly focus on increasing cleavage activity (more on-targets) and cleavage efficiency (low off-target sites). As known, low efficiency makes CRISPR editing unreliable and also hampers CRISPR development and application (Wang et al., 2019a). Unfortunately, the high focus on more activity induces more off-target cleavage, which can be toxic. Therefore, we must maintain a balance between these two criteria. These issues can be resolved by designing successful CRISPR gRNA and choosing an appropriate Cas protein (Kuscu et al., 2014; Shen et al., 2018).
As mentioned earlier, cleavage efficiency varies significantly among different target sites and cell lines (Yan et al., 2018). Several features can influence the gRNA binding ability and the Cas enzyme cutting efficacy. Sequence composite features (nucleotide position, GC content), genetic and epigenetic features (chromatin accessibility, gene expression), and energetic properties (RNA secondary structure, melting temperature, free energy) are the most important influential features on cleavage efficiency (Pallarès Masmitjà et al., 2019; Wang et al., 2020). Based on these features, many computational tools have been developed for designing highly efficient gRNAs. In the rest of this section, we will discuss the most popular ones.
Rule set 1 (Liu et al., 2020) is a ML-based model that uses a support vector machine (SVM), a supervised ML method, and contains a linear regression method for classifying gRNAs. Rule set 1 uses sequence-based features, and its predictive data is highly correlated with experimental results (Xu et al., 2015). Rule set 2 (Liu et al., 2020) is an improved version of Rule set 1 and counts the nucleotides independent location of the gRNA target site within the gene to improve results (Doench et al., 2016). It is a powerful model, used for both CRISPR Knock Out (CRISPR KO) and CRISPR activation/interference (CRISPRa/i) experiments. Another powerful model-based package has been developed and implemented at the Broad Institute to predict gRNA efficiency, named sgRNA Designer (Pallarès Masmitjà et al., 2019).
Elastic Net is another ML-based and regularized regression-based method (Li and Lin, 2010). Although there are significant differences in nucleotide preference between CRISPR KO and CRISPRa/I, the Elastic Net algorithm is used to construct models for both CRISPR KO and CRISPRa/i. Also, this practical algorithm has been applied in Spacer Scoring for CRISPR (SSC) software to predict the gRNA efficiency (Qin et al., 2019). Additionally, well-known platforms such as E-CRISP (Heigwer et al., 2014), CHOPCHOP (Labun et al., 2019), and CRISPRFOCUS (Cao et al., 2017) have applied this method.
Moreno and his colleagues designed another logistic regression-based method and integrated it into CRISPRscan to predict the gRNA precision (Moreno-Mateos et al., 2015). Additionally, they have applied extra features such as guanine enrichment and adenine depletion, which increase the gRNA activity (Cui et al., 2018).
Another ML-based method is WU-CRISPR (Wong et al., 2015) which uses sequence composite features like guanine enrichment and adenine depletion, and some other novel features to build a higher precision model. The CRISPR/Cas9 target online predictor (CCTop) (Stemmer et al., 2015), a platform for CRISPR target prediction, takes advantage of this model. The SgRNAScorer is another software that uses SVM to calculate gRNA on-target scores. The new version of this software can predict other Cas systems such as SaCas9 (Qin et al., 2019) and AsCpf1 [94].
To avoid unwanted effects in other sites except for desired target sites (off-target), researchers try to modify a spacer sequence that does not adopt other sites in the genome. Tools such as CRISPRpred (Hwang and Bae, 2021), DeepSpCas9, and SgRNAScorer are usually limited to the set of preprocessed genomes used when training ML models. To build good gRNAs in genomes other than those used in the training process, researchers can use web-based tools such as CRISPy (Blin et al., 2016). Looking at Tables 1–Tables 4, we have listed the genome in which the editing takes place (named target genome) as a significant feature for all tools. The existence of target genome is even more critical for deep learning-based (DL) methods, because they are usually unpractical in genomes other than the ones from which training data was extracted. Basically, being used in all genomes is a significant strength for ML-based tools. But one tool may not have the same accuracy over all genomes or even all regions of a genome (see Figure 7) (Kim et al., 2021). Furthermore, structural correctness and base-level accuracy of the target genome are important. The accuracy of a genome differs not only between genome sequencing technologies but also across genomic regions, as some stretches of the genome are inherently more difficult to read (Kim et al., 2021). It is commonly known that certain genomic regions are more difficult for sequencing and extracting features. AT-rich or GC-rich regions, which are important for detecting off-target sites, are tough because they respond poorly to the amplification protocols required by some platforms. Palindromic sequences or hairpin structures similar to gRNA structures are difficult to denature, making such regions challenging for sequencing tools (Selvakumar et al., 2022).
2.1.1 Selecting the best gRNA
There may be several gRNAs for an experiment, in which case we have to pick the best one. Many computational approaches have been developed for scoring and selecting the best gRNAs. Some of them use experimental data to score a gRNA. According to the different criteria, these methods consider a specific score for each gRNA. The criteria and final score calculation are different in each algorithm. CHOPCHOP (Labun et al., 2019) provides multiple scores for users, such as Rule Set 1 and Rule set 2, SSC (Xu et al., 2015), CRISPRscan [13], and deepCpf1 (Kim et al., 2018). E-CRISP (Heigwer et al., 2014) uses a particular score to determine the quality of each gRNA, named SAE, which combines three scores: specificity, annotation and efficacy. E-CRISP uses Rule Set 1 and SSC too. CCTop (Stemmer et al., 2015) calculates the CRISPRater score to predict the efficiency of gRNAs. CCTop also calculates off-target scores for each sequence. The CRISPOR (Concordet and Haeussler, 2018) ranks gRNAs according to different scores, such as on-target activity and protentional off-targets scores.
To score a gRNA or determine whether it is suitable for the desired genome editing or not, we need to determine potential targets of a gRNA in the selected genome and determine which of these potential targets are desirable. Hence, the number of on-target and off-target sites is critical in gRNA evaluation. In other words, since genomic edits are permanent and very sensitive, it is crucial to determine potential targets before the main editing occurs and then remove or reduce them (Yan et al., 2018). Therefore, many researchers have focused on this issue. Furthermore, many developers have attempted to develop practical tools for this purpose. We will discuss these tools in the next section.
2.2 Prediction of CRISPR specificity (off-target sites)
The prediction of off-target mutations in CRISPR/Cas9 is a hot topic owing to its relevance to gene-editing research. Cas nucleases may cleave unintended genomic sites and cause unexpected mutations called off-target cleavage (Listgarten et al., 2018). Even though the CRISPR/Cas9 system is routinely used in a large variety of tasks, there is also a significant concern that off-target effects may reduce its effectiveness of CRISPR. In response to this concern, researchers have concluded that the best way to mitigate off-target effects is to know when and where they occur and then design guides to avoid them while balancing for on-target efficiency. By predicting CRISPR cutting specificity and designing optimal gRNAs, off-target effects can be effectively relieved. As noted earlier, careful CRISPR target selection and low concentrations of CRISPR components can reduce off-target cleavage (Zetsche et al., 2020).
The off-target predictive modelling problem can be broken down into three main tasks. Given a gRNA to evaluate off-target activity, one needs to (Afzal et al., 2020) search the whole genome for potential targets; in other words, search those regions of the genome matching the guide sequence with up to X number of mismatches (Gaj et al., 2013); score each potential target found in step 1 according to its activity (Jacquin et al., 2019); collect the second stage scores and evaluate the final score of a gRNA. Several solutions have been presented for these tasks, including Cas-OFFinder (Bae et al., 2014), CRISPOR (Concordet and Haeussler, 2018), CHOPCHOP (Labun et al., 2019), and e-CRISPR (Tarasava et al., 2018). These models differ in their search algorithms and the completeness of the search process. Completeness is dictated by options such as the maximum number of mismatches, allowed PAMs, and the search algorithm used.
There are two basic methods to predict the specificity of CRISPR gRNAs: the alignment-based and the scoring-based methods. In the following, we will explain these approaches and give successful examples of each one. Also, the overview of these approaches is depicted in Figure 4.
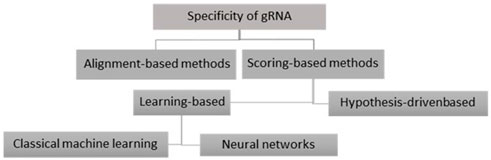
FIGURE 4. Basic methods to predict the specificity of CRISPR gRNAs.
2.2.1 Alignment-based methods
In the alignment-based method, gRNAs are aligned to a given genome, and off-target sequences and sites are returned. These methods are mainly used to find out all potential off-target sites in silico. Choosing a search engine and setting search parameters plays an important role in evaluating these tools (Liu et al., 2020). For example, if we set the maximum number of mismatches to a large number, like four or more, we will probably find all possible off-targets. The observed rate of off-target activity is about 59% when there is one mismatch between the target DNA and gRNA sequences and decreases toward 0% when four or more mismatches exist (Kim et al., 2021). So, it can be concluded that an increased number of mismatches decreases the likelihood of off-target activity.
Common sequence alignment tools use BLAST, BLAT, Bowtie, Bowtie2, BWA or customized search engines. Table 5 summarizes the search engine of famous alignment-based tools in CRISPR.
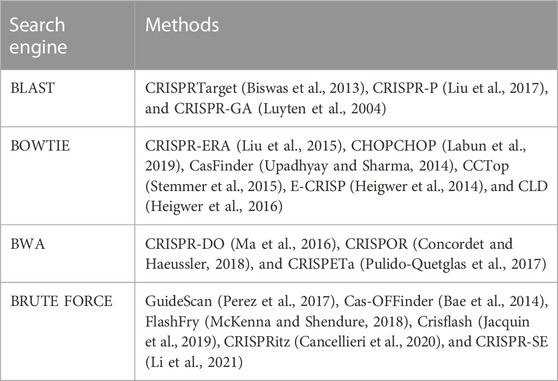
TABLE 5. The most popular alignment-based methods and related search engines.
Compared to methods which use BLAST, Bowtie and BWA as search engine, methods like GuideScan (Perez et al., 2017), Cas-OFFinder (Bae et al., 2014), FlashFry (McKenna and Shendure, 2018), Crisflash (Jacquin et al., 2019), CRISPRitz (Cancellieri et al., 2020), and finally, CRISPR-SE (Li et al., 2021)are faster due to the use of Brute force search engine. In addition, unlike most methods that support only a limited number of mismatches (mostly 3 or 4), Cas-OFFinder, CRISPRitz and CRISPR-SE have more preference due to their support of any number of mismatches.
The Bowtie and BWA are traditional tools for short sequence alignment that can be used for off-target sites detection (de Ruijter and Guldenmund, 2016). However, they cannot identify small PAMs since they were developed for NGS read alignment. Moreover, these tools allow very limited mismatches with default parameters, so they cannot identify all potential off-target sites.
Most tools, like CCTop (Stemmer et al., 2015), modify default algorithms and parameters and utilize Bowtie (de Ruijter and Guldenmund, 2016) to find off-target sites. CCTop follows three main steps. In the first step, CCTop identifies PAM sites; In the second step, it modifies default parameters (up to five mismatches against one in default) of Bowtie, and uses them to search for matches and mismatches in protospacer sequences. In the third step, it evaluates the off-target score for each candidate gRNA.
SeqMAp (Jiang and Wong, 2008) is an ultrafast short sequence mapping tool used in sgRNAcas9 (Xie et al., 2014) to find off-target sites. The sgRNAcas9 classifies all off-target sites into three categories and scores them to choose the best gRNA.
CasOT (Xiao et al., 2014) is another tool that can find Cas9 on-target and off-target sites with up to six mismatches in the seed region (12 nucleotides adjacent to the PAM). This tool can also determine whether off-targets are within a coding exon (Listgarten et al., 2016) or not. FlashFry (McKenna and Shendure, 2018) is another alignment-based method that defines off-targets with high speed. Additionally, it chooses the best gRNA and provides useful information such as annotating off-target sites, on and off-target scores, GC content, etc. FlashFry is a good choice for many applications because of its high speed and comprehensive output. Crisflash (Jacquin et al., 2019) is another one that belongs to the alignment-based approaches group. Crisflash designs gRNAs with a tree-based algorithm and uses user-supplied variant data to optimizes gRNA accuracy. It uses an N-ary tree structure, which searches up to four mismatches. CRISPRitz (Cancellieri et al., 2020) used a four-bit-based encoding to represent each nucleotide to allow for efficient bitwise operations. CRISPRitz supports off-targets with both mismatches and indels.
CALITAS (Fennell et al., 2021) is a new CRISPR-Cas-aware aligner tool which uses a modified and CRISPR-tuned version of the Needleman–Wunsch algorithm, supports an unlimited number of mismatches and gaps, and allows PAM mismatches or PAM-less searches. CALITAS returns a single best alignment for a given off-target site and it enables off-targets to be referenced directly using alignment coordinate.
CHOPCHOP v3.0 (Labun et al., 2019), a well-known model, is another tool that uses Bowtie with parameters–V and–L to detect off-target sites [90]. But, CRISPOR uses BWA to find all potential off-target sites iteratively and can find all validated off-targets as well as Cas-OFFinder (Bae et al., 2014).
Sequence alignment tools like CRISPy (Qin et al., 2019) and CRISPRdirect (Heigwer et al., 2016) rely on a minimum of one K-mer exact match. They are likely to miss some off-targets, spatially with a high number of mismatches and ultra-short gRNAs (20-mer). So, the accuracy of these methods cannot be very high.
In recent years, some tools like GuideScan (Perez et al., 2017), Cas-OfFinder (Bae et al., 2014), and CRISPR-SE (Li et al., 2021) have been developed with Brute force algorithm as their search engine. GuideScan uses a “tree” data structure with a brute-force algorithm that guarantees the search accuracy. Another tool in this category is Cas-OFFinder. Cas-OFFinder is one of the most popular tools for detecting potential off-target sites, with no limit to the number of mismatches, PAM types, or gRNA length. In our opinion, the most significant advantage of Cas-OFFinder is its high running speed due to using GPUs. It can also predict off-target sites with one-bp deletions or insertions.
OffScan (Cui et al., 2020) is the last one we considered in this study that is, belongs to the alignment-based approaches group. OffScan is not limited by the number of mismatches and allows custom PAM. Besides, OffScan adopts the FM-index, which efficiently improves query speed and reduce memory consumption.
Here, we discussed several alignment-based methods for the prediction of the gRNA output and realized that Cas-OFFinder may be the best option for identifying all potential off-targets with any Cas nucleases among these tools. Although users can reduce the number of outputs by restricting the maximum mismatches while exploring off-target cleavage, there are always redundant outputs; many are false positives.
On the whole, all nucleotide positions containing mismatches do not have the same decisive effect on off-target cleavage, but this issue is not considered in alignment-based methods. Because of this problem, and in order to increase the accuracy of the off-target detection methods, adding the features that influence the non-specific binding of CRISPR gRNAs to the methods is essential. As a result, another group of approaches emerged called scoring-based methods, which are discussed in the following sub-section.
2.2.2 Scoring-based methods
In the scoring-based method, the gRNAs identified in the alignment process are scored and ranked, and the sgRNA with the highest score is selected. There are two groups of scoring-based approaches: 1) hypothesis-driven-based approaches, where off-targets are scored based on the contribution of specific genome context factors to gRNA specificity; 2) learning-based approaches, where gRNAs are scored and predicted from a training model that considers the different features affecting specificity.
MIT (Hsu et al., 2013) is the first popular score-based tool for CRISPR off-target evaluation. To score the off-target efficiency of each gRNA, it counts and evaluates the contributions made by different mismatch positions. It also calculates a weight matrix to determine off-target efficiency for each gRNA (Chuai et al., 2017). The MIT score has been integrated into many CRISPR gRNA design tools, such as CHOPCHOP v3.0 CHOP (Labun et al., 2019) and CRISPOR (Concordet and Haeussler, 2018).
Another popular score-based tool for off-target evaluation is CFD (Cutting Frequency Determination). It is noticeable that gRNA can bind genome loci with non-canonical PAMs such as NAG, NCG, and NGA. So, CFD has added PAM features to their scoring metrics (Abadi et al., 2017). Also, for examining correlations between RNAs and off-targets, gRNAs with mismatches and indels in target sequences are added. GUIDE-seq (Tsai et al., 2015) validated the CFD score and proved that it performs better than the MIT score. The CFD score has been integrated into CRISPRscan (Moreno-Mateos et al., 2015), GuideScan (Perez et al., 2017), CRISPOR (Concordet and Haeussler, 2018), and others. CRISPRoff (Carlson-Stevermer et al., 2020) and uCRISPR (Carlson-Stevermer et al., 2020) integrated energetic properties into their scoring metrics. They both yielded better accuracy than MIT and CFD in off-target prediction.
Scoring-based methods consider only a few features, and unfortunately, all practical features cannot be considered. Also, most features are not understood yet, while learning-based methods use combinations of multiple features to build complex models for better prediction of off-target sites. These models are based on ML and, more recently, DL methods.
DL-based methods are attractive for CRISPR gRNA target efficacy prediction. They are mainly based on CNNs. Table 4 introduces some famous models that use MDL models for gRNA on-target prediction. These models used neural networks to extract features from the input genomic sequence. Generally, they are superior to models that use classical ML tools in prediction accuracy.
DeepCRISPR (Chuai et al., 2018) is a DL-based platform that combines gRNA on-target and off-target site predictions. As mentioned, in DL-based models, we do not need to identify all effective features, as they are detected automatically using the deep neural network. DeepCRISPR learns all possible sequence and epigenetic features that may affect gRNA Knock Out (KO) efficacy (Hana et al., 2021) in its learning process with a large dataset that is, gathered for it.
CRISPR-Cpf1 (Kim et al., 2017) is a ML-based model that achieved high efficiency, although it suffers minor off-target effects. DeepCpf1 (Kwon et al., 2019) is another highly used DL-based algorithm, mainly used in predicting Cpf1 activity. It uses chromatin accessibility data. It showed a significant improvement in the accuracy of Cpf1 activity prediction. CRISPR-DT (Zhu and LiangCRISPR-, 2019) is a recently developed platform for predicting the Cpf1 target efficiency. This model has been implemented with the SVM algorithm and displays better performance than the DL-based models such as DeepCpf1.
CRISPOR (Concordet and Haeussler, 2018) may be the best tool for designing gRNAs. CRISPOR combines multiple tools and gathers a large dataset to develop a highly efficient CRISPR gRNA design. CRISPOR contains 417 genomes and 19 PAM types, making it useful in almost all genomes. CRISPOR calculates two specificity scores: MIT and CFD. Additionally, it calculates ten efficiency scores, including Rule Set 2, CRISPRscan, microhomology, Lindel scores (Chen et al., 2019) and others for outcome prediction. CRISPOR designs primers for each gRNA as well as off-target sites. These primers are helpful when conducting on and off-target validation. CRISPOR enables the filtering of gRNAs with genomic variants based on well-known variant databases.
Some computational tools use CNNs for feature extraction or classification of CRISPR Cas. For instance, Seq-deepCpf1 (Kim et al., 2018; Kwon et al., 2019) has used CNN to extract features from the input gRNA sequence. And DeepCRISPR incorporates a CNN for predicting CRISPR/Cas9 gRNA on-target knockout efficiency and whole-genome off-target profiles. Also, DeepCas9 uses CNN to automatically learn the sequence determinants and predict the activities of gRNAs across multiple species genomes (Bhagwat and Khuri, 2021). Deeper-Bind (Hassanzadeh and Wang, 2016) used a LSTM layer to learn the dependencies between sequence features; this helps improve the prediction of protein binding specificity (Zhang et al., 2020). C-RNNCrispr (Zhang et al., 2020) has used a hybrid architecture combining CNN with bidirectional GRU (BGRU) to predict sgRNA cleavage efficacy (Sledzinski et al., 2020).
The performance of these tools is quantitatively assessed with two commonly used evaluation metrics, including accuracy and Spearman Correlation Coefficient (SCC) between predicted and real detected off-target activity. However, other evaluation metrics like Precision and Sensitivity (Eqs 2, 3) are used in some research as well. Spearman correlation seems to be a more reliable criterion. Most of these tools achieve promising accuracy in off-target prediction. Figures 5, 6 compare the off-target prediction efficacy of some popular tools. Due to their importance, we compare the accuracy of DL-based tools in separate diagram. The average accuracy of these tools is illustrated in the figures, as their accuracy differs among different genomes. For example, DeepCRISPR was the most accurate tool in the HEL cell line but performed poorly in the others. More details can be found in (Wang et al., 2019a; Zhu and LiangCRISPR-, 2019). Also, as a ML method, the accuracy differs between the train and test datasets. Unfortunately, for DeepCas9 and DeepSpCas9 (Chen et al., 2019), there is no report in their primary reference for the training dataset and the test dataset in CRISPRLearner (Bhagwat and Khuri, 2021). Accuracy, Precision, and Sensitivity are defined as follows, where TP, FP, TN, and FN represent true positive, false positive, true negative, and false negative, respectively.
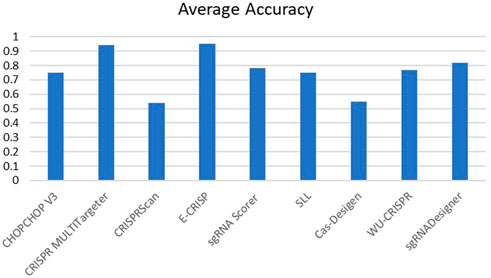
FIGURE 5. Average accuracy of off-target prediction.
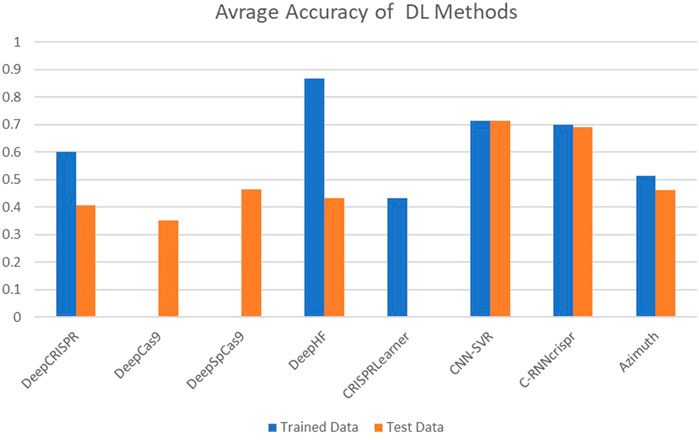
FIGURE 6. Average accuracy of off-target prediction in DL-based methods.
SCC evaluates the ability of the models to predict the actual efficiency of each gRNA sequence (Konstantakos et al., 2022). While some models are trained to minimize the mean squared error (MSE), the comparison between models on different datasets is necessarily made using Spearman correlation. Figure 7 compares the predictive ability of off-target sites in some ML-based tools over five datasets named Zebrafish_G, Zebrafish_S, HEL, A375, and mESC. In general, the larger the polygon area, the better the overall performance of the tool. Figure 7 clearly illustrates the better and more robust performance of the DeepHF, DeepSpCas9, and DeepCas9 models. As shown, classic ML-based tools such as Azimuth 2.0 achieve comparable performance to DL-based tools. Also, even though E-CRISP is more accurate than some learning-based tools, it does not achieve high enough correlations. However, E-CRISP has stable performance across all datasets. In addition, as it is clear from Figure 7, DeepCRISPR outperforms the other tools on the HEL dataset, and E-CRISP and CRISPRLearner achieve better results based on this metric.
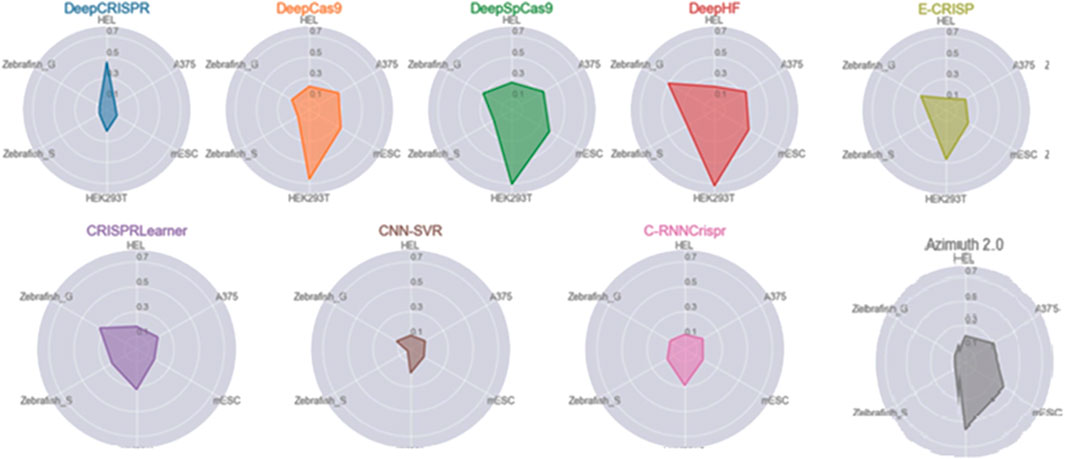
FIGURE 7. Spearman correlation for ML-based tools over the different datasets. Each polygon represents a tool, and the edges illustrate the obtained correlation over the respective dataset.
As mentioned, gRNAs are typically designed by computational tools which compare gRNA sequence with a reference genome to predict the activity of on-target and potential off-targets. However, these tools can yield false-positive (FP) or false-negative (FN) results. Furthermore, the DNA in clinical experiments can differ from the reference genome used in the computational modeling, which means they would be more false predictions. Therefore, the accuracy is less than the values shown in Figure 7 in the actual experiment. To resolve this problem, in-vitro based tools have been developed for the experimental detection of off-target sites in a particular DNA sample. Tools like SMRT-OTS and Nano-OTS (Höijer et al., 2020) use long-read single-molecule sequencing.
In this article, we review both traditional and ML-based approaches for gRNA designing and predicting off-target sites. As mentioned before, experimental methods which use third-generation sequencing technology, have a better performance in Cas9 target detection on dark genomic regions (Höijer et al., 2020). This new technology helps us to detect more on-target and off-target sites and to design optimal gRNA. Furthermore, collected data in experimental methods, could improve the accuracy of DL-based tools.
Also, we have presented a comprehensive list of available tools. Each tool has merits and demerits, and the performance of different tools differs in different situations. According to our studies, some tools can be a better choice in some situations; However, others may be more popular in the scientific community. So, choosing the right tool depends on the conditions and limitations of an application.
Among the alignment-based methods, tools like CRISPR-P, Flycrispr, CRISPRseek, Cas-OFFinder, CasOT, sgRNACas9, and Flashfly have high accuracy and efficiency; however, CRISPR-P and Flycrispr are only used in specific genomes. Other tools such as CRISPRseek, Cas-Offinder, and CasOT, are used in almost all genomes. Moreover, they support only particular types of PAMs, while methods such as sgRNACas9 and Flashfly are compatible with all types of PAMs and seem to be a better option for designing gRNAs.
Among the learning-based methods, DL-based methods, including C-RNNCrispr, DeepCpf1, DeepHF, DeepSpCas9, and DeepCRISP, have drawn much interest recently. However, learning-based methods such as CLD, CRISPR-ERA, sgRNA-design, E-CRISP are significant due to their high accuracy and use in all genomes. Finally, based on our study, methods such as CRISPR-SE and E-CRISP are the best options to be used in all genomes with high accuracy.
3 Conclusion
CRISPR systems have been developed for accurate genome editing. Since genomic modifications are permanent (Ding et al., 2018), it is crucial to make precise edits. Most of the tools or methods in CRISPR’s field have been developed to help users design proper gRNA with fewer off-target effects. It is considered that the efficiency of one gRNA may differ among different models and databases. Users must evaluate several gRNAs using multiple models and select the best one for their experiments.
The previous successes of CNN and RNN architectures in bioinformatics motivated other researchers to extend their applications with a DL platform, which we believe is the best solution for predicting off-target effects. DL methods are inexpensive and fast compared to experimental methods. However, their accuracy depends on the amount of available data for a model’s training. Additionally, most of existing methods have three big problems, which means their predictions are not exact. First, they calculate scores based on mismatches to the guide sequence. However, DL-based methods can extract more efficient features hidden in the input data. In other words, DL-based methods can capture features other than gRNA sequence-based features. These features can be utilized and encoded in the input sequence to improve the performance of the existing DL architectures. In addition, most proposed DL-based methods use a one-hot vector representation to encode the input data. (Charlier et al., 2021). The use of newer encoding and embedding methods proposed in the field of DL can enhance the efficiency of existing DL-based methods. Also, the use of gRNA-DNA pair encoding can be helpful. Second, there is a rapid expansion in experimental data in CRISPR research. Most methods cannot scale and improve their performance with this new data. As known, DL-based methods achieve better performance by training on large datasets, but they require a pre-processing step to prepare and aggregate data obtained from diverse sources based on different experimental methods. This step requires enough knowledge about the type of input data, the operation mechanism of CRISPR, and the architecture of the deep neural network. Finally, the most severe issue is that existing DL-based methods still need to be improved in providing sufficient precision for clinical practice usage. NGS-based whole-genome sequencing technologies help to discover almost all off-target sites in the target genome and create a large and more accurate train dataset. As the number of instances in a train dataset increases, the predictions of DL-based methods become closer to experimental observations.
Author contributions
RA: supplied acquisition of data, analysis, interpretation of data and drafting the paper. LS: provided the conception and design of the study, analysis and interpretation of data, revised it critically for important intellectual content, and final approval of the version to be submitted. AK: provided the conception and design of the study, analysis and interpretation of data, revised it critically for important intellectual content, and final approval of the version to be submitted. RA has the first authorship right. LS and AK contributed equally to this work and share senior authorship.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abadi, S., Yan, W. X., Amar, D., and Mayrose, I. (2017). A machine learning approach for predicting CRISPR-Cas9 cleavage efficiencies and patterns underlying its mechanism of action. PLoS Comput. Biol. 13 (10), e1005807. doi:10.1371/journal.pcbi.1005807
Afzal, S., Sirohi, P., and Singh, N. K. (2020). A review of CRISPR associated genome engineering: Application, advances and future prospects of genome targeting tool for crop improvement. Biotechnol. Lett. 42, 1611–1632. doi:10.1007/s10529-020-02950-w
Ahmed, M., and He, H. H. (2017). SgTiler: A fast method to design tiling sgRNAs for CRISPR/cas9 mediated screening, BioRxiv. 217166.
Alkhnbashi, O. S., Costa, F., Shah, S. A., Garrett, R. A., Saunders, S. J., and Backofen, R. (2014). CRISPRstrand: Predicting repeat orientations to determine the crRNA-encoding strand at CRISPR loci. Bioinformatics 30 (17), i489–i496. doi:10.1093/bioinformatics/btu459
Alkhnbashi, O. S., Meier, T., Mitrofanov, A., Backofen, R., and Voß, B. (2020). CRISPR-Cas bioinformatics. Methods 172, 3–11. doi:10.1016/j.ymeth.2019.07.013
Alkhnbashi, O. S., Mitrofanov, A., Bonidia, R., Raden, M., Tran, V. D., Eggenhofer, F., et al. (2021). CRISPRloci: Comprehensive and accurate annotation of CRISPR–cas systems. Nucleic Acids Res. 49, W125–W130. doi:10.1093/nar/gkab456
Allen, F., Crepaldi, L., Alsinet, C., Strong, A. J., Kleshchevnikov, V., De Angeli, P., et al. (2019). Predicting the mutations generated by repair of Cas9-induced double-strand breaks. Nat. Biotechnol. 37 (1), 64–72. doi:10.1038/nbt.4317
Bae, S., Park, J., and Kim, J-S. (2014). Cas-OFFinder: A fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics 30 (10), 1473–1475. doi:10.1093/bioinformatics/btu048
Bhagwat, N., and Khuri, N. (2021). “Predicting targets for genome editing with long short term memory networks,” in Advances in computer vision and computational biology (Berlin, Germany: Springer), 657–670.
Biswas, A., Gagnon, J. N., Brouns, S. J., Fineran, P. C., and Brown, C. M. (2013). CRISPRTarget: Bioinformatic prediction and analysis of crRNA targets. RNA Biol. 10 (5), 817–827. doi:10.4161/rna.24046
Biswas, A., Staals, R. H., Morales, S. E., Fineran, P. C., and Brown, C. M. (2016). CRISPRDetect: A flexible algorithm to define CRISPR arrays. BMC genomics 17 (1), 356–370. doi:10.1186/s12864-016-2627-0
Blin, K., Pedersen, L. E., Weber, T., and Lee, S. Y. (2016). CRISPy-web: An online resource to design sgRNAs for CRISPR applications. Synthetic Syst. Biotechnol. 1 (2), 118–121. doi:10.1016/j.synbio.2016.01.003
Boel, A., Steyaert, W., De Rocker, N., Menten, B., Callewaert, B., De Paepe, A., et al. (2016). BATCH-GE: Batch analysis of Next-Generation Sequencing data for genome editing assessment. Sci. Rep. 6 (1). doi:10.1038/srep30330
Cancellieri, S., Canver, M. C., Bombieri, N., Giugno, R., and Pinello, L. (2020). CRISPRitz: Rapid, high-throughput and variant-aware in silico off-target site identification for CRISPR genome editing. Bioinformatics 36 (7), 2001–2008. doi:10.1093/bioinformatics/btz867
Cao, Q., Ma, J., Chen, C-H., Xu, H., Chen, Z., Li, W., et al. (2017). CRISPR-FOCUS: A web server for designing focused CRISPR screening experiments. PLoS One 12 (9), e0184281. doi:10.1371/journal.pone.0184281
Carlson-Stevermer, J., Kelso, R., Kadina, A., Joshi, S., Rossi, N., Walker, J., et al. (2020). CRISPRoff enables spatio-temporal control of CRISPR editing. Nat. Commun. 11 (1), 5041–5047. doi:10.1038/s41467-020-18853-3
Chari, R., Yeo, N. C., Chavez, A., and Church, G. M. (2017). sgRNA Scorer 2.0: a species-independent model to predict CRISPR/Cas9 activity. ACS Synth. Biol. 6 (5), 902–904. doi:10.1021/acssynbio.6b00343
Charlier, J., Nadon, R., and Makarenkov, V. (2021). Accurate deep learning off-target prediction with novel sgRNA-DNA sequence encoding in CRISPR-Cas9 gene editing. Bioinformatics 37 (16), 2299–2307. doi:10.1093/bioinformatics/btab112
Chen, C-L., Rodiger, J., Chung, V., Viswanatha, R., Mohr, S. E., Hu, Y., et al. (2020). SNP-CRISPR: A web tool for SNP-specific genome editing. G3 Genes, Genomes, Genet. 10 (2), 489–494. doi:10.1534/g3.119.400904
Chen, W., McKenna, A., Schreiber, J., Haeussler, M., Yin, Y., Agarwal, V., et al. (2019). Massively parallel profiling and predictive modeling of the outcomes of CRISPR/Cas9-mediated double-strand break repair. Nucleic acids Res. 47 (15), 7989–8003. doi:10.1093/nar/gkz487
Chuai, G., Ma, H., Yan, J., Chen, M., Hong, N., Xue, D., et al. (2018). DeepCRISPR: Optimized CRISPR guide RNA design by deep learning. Genome Biol. 19 (1), 80–18. doi:10.1186/s13059-018-1459-4
Chuai, G-h., Wang, Q-L., and Liu, Q. (2017). In silico meets in vivo: Towards computational CRISPR-based sgRNA design. Trends Biotechnol. 35 (1), 12–21. doi:10.1016/j.tibtech.2016.06.008
Cloney, R. (2019). The oracle of inDelphi predicts Cas9 repair outcomes. Nat. Rev. Genet. 20 (1), 4–5. doi:10.1038/s41576-018-0077-z
Concordet, J-P., and Haeussler, M. (2018). Crispor: Intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic acids Res. 46 (W1), W242–W245. doi:10.1093/nar/gky354
Cox, D. B. T., Platt, R. J., and Zhang, F. (2015). Therapeutic genome editing: Prospects and challenges. Nat. Med. 21 (2), 121–131. doi:10.1038/nm.3793
Cradick, T. J., Qiu, P., Lee, C. M., Fine, E. J., and Bao, G. (2014). COSMID: A web-based tool for identifying and validating CRISPR/cas off-target sites. Mol. Therapy-Nucleic Acids. 3, e214. doi:10.1038/mtna.2014.64
Cui, Y., Liao, X., Peng, S., Tang, T., Huang, C., and Yang, C. (2020). OffScan: A universal and fast CRISPR off-target sites detection tool. BMC genomics 21 (1), 872–876. doi:10.1186/s12864-019-6241-9
Cui, Y., Xu, J., Cheng, M., Liao, X., and Peng, S. (2018). Review of CRISPR/Cas9 sgRNA design tools. Interdiscip. Sci. Comput. Life Sci. 10 (2), 455–465. doi:10.1007/s12539-018-0298-z
Dampier, W., Chung, C-H., Sullivan, N. T., Atkins, A. J., Nonnemacher, M. R., and Wigdahl, B. (2018). CRSeek: A Python module for facilitating complicated CRISPR design strategies, PeerJ Prepr. Report No, 2167–9843.
de Ruijter, A., and Guldenmund, F. (2016). The bowtie method: A review. Saf. Sci. 88, 211–218. doi:10.1016/j.ssci.2016.03.001
Ding, W., Mao, W., Shao, D., Zhang, W., and Gong, H. (2018). DeepConPred2: An improved method for the prediction of protein residue contacts. Comput. Struct. Biotechnol. J. 16, 503–510. doi:10.1016/j.csbj.2018.10.009
Doench, J. G., Fusi, N., Sullender, M., Hegde, M., Vaimberg, E. W., Donovan, K. F., et al. (2016). Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 34 (2), 184–191. doi:10.1038/nbt.3437
Duan, L., Ouyang, K., Xu, X., Xu, L., Wen, C., Zhou, X., et al. (2021). Nanoparticle delivery of CRISPR/Cas9 for genome editing. Front. Genet. 12, 673286. doi:10.3389/fgene.2021.673286
Fennell, T., Zhang, D., Isik, M., Wang, T., Gotta, G., Wilson, C. J., et al. (2021). CALITAS: A CRISPR-cas-aware ALigner for in silico off-TArget search. CRISPR J. 4 (2), 264–274. doi:10.1089/crispr.2020.0036
Gaj, T., Gersbach, C. A., and Barbas, C. F. (2013). ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 31 (7), 397–405. doi:10.1016/j.tibtech.2013.04.004
Gasiunas, G., Barrangou, R., Horvath, P., and Siksnys, V. (2012). Cas9–crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl. Acad. Sci. 109 (39), E2579–E2586. doi:10.1073/pnas.1208507109
Ge, R., Mai, G., Wang, P., Zhou, M., Luo, Y., Cai, Y., et al. (2016). CRISPRdigger: Detecting CRISPRs with better direct repeat annotations. Sci. Rep. 6 (1). doi:10.1038/srep32942
Güell, M., Yang, L., and Church, G. M. (2014). Genome editing assessment using CRISPR Genome Analyzer (CRISPR-GA). Bioinformatics 30 (20), 2968–2970. doi:10.1093/bioinformatics/btu427
Hana, S., Peterson, M., McLaughlin, H., Marshall, E., Fabian, A. J., McKissick, O., et al. (2021). Highly efficient neuronal gene knockout in vivo by CRISPR-Cas9 via neonatal intracerebroventricular injection of AAV in mice. Gene Ther. 28, 646–658. doi:10.1038/s41434-021-00224-2
Hassanzadeh, H. R., and Wang, M. D. (2016). “DeeperBind: Enhancing prediction of sequence specificities of DNA binding proteins,” in 2016 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), Shenzhen, China, 15-18 December 2016 (IEEE).
Heigwer, F., Kerr, G., and Boutros, M. E-C. R. I. S. P. (2014). E-CRISP: Fast CRISPR target site identification. Nat. methods 11 (2), 122–123. doi:10.1038/nmeth.2812
Heigwer, F., Zhan, T., Breinig, M., Winter, J., Brügemann, D., Leible, S., et al. (2016). CRISPR library designer (CLD): Software for multispecies design of single guide RNA libraries. Genome Biol. 17 (1), 55–10. doi:10.1186/s13059-016-0915-2
Herai, R. H. (2019). Avoiding the off-target effects of CRISPR/cas9 system is still a challenging accomplishment for genetic transformation. Gene 700, 176–178. doi:10.1016/j.gene.2019.03.019
Höijer, I., Johansson, J., Gudmundsson, S., Chin, C-S., Bunikis, I., Häggqvist, S., et al. (2020). Amplification-free long-read sequencing reveals unforeseen CRISPR-Cas9 off-target activity. Genome Biol. 21 (1), 290. doi:10.1186/s13059-020-02206-w
Hough, S. H., Kancleris, K., Brody, L., Humphryes-Kirilov, N., Wolanski, J., Dunaway, K., et al. (2017). Guide Picker is a comprehensive design tool for visualizing and selecting guides for CRISPR experiments. BMC Bioinforma. 18 (1), 167–210. doi:10.1186/s12859-017-1581-4
Hsu, P. D., Scott, D. A., Weinstein, J. A., Ran, F. A., Konermann, S., Agarwala, V., et al. (2013). DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 31 (9), 827–832. doi:10.1038/nbt.2647
Hwang, G-H., and Bae, S. (2021). Computational methods in synthetic biology. Berlin, Germany: Springer, 81–88.Web-based base editing toolkits: BE-Designer and BE-analyzer
Hwang, G-H., Song, B., and Bae, S. (2021). Current widely-used web-based tools for CRISPR nucleases, base editors, and prime editors. Gene Genome Ed. 1, 100004. doi:10.1016/j.ggedit.2021.100004
Iyombe, J-P. (2019). Correction du gène de la dystrophine avec la méthode CRISPR induced deletion. Québec: CinDel.
Jacquin, A. L., Odom, D. T., and Lukk, M. (2019). Crisflash: Open-source software to generate CRISPR guide RNAs against genomes annotated with individual variation. Bioinformatics 35 (17), 3146–3147. doi:10.1093/bioinformatics/btz019
Jeong, H-H., Kim, S. Y., Rousseaux, M. W., Zoghbi, H. Y., and Liu, Z. (2017). CRISPRcloud: A secure cloud-based pipeline for CRISPR pooled screen deconvolution. Bioinformatics 33 (18), 2963–2965. doi:10.1093/bioinformatics/btx335
Jiang, F., Taylor, D. W., Chen, J. S., Kornfeld, J. E., Zhou, K., Thompson, A. J., et al. (2016). Structures of a CRISPR-Cas9 R-loop complex primed for DNA cleavage. Science 351 (6275), 867–871. doi:10.1126/science.aad8282
Jiang, H., and Wong, W. H. (2008). SeqMap: Mapping massive amount of oligonucleotides to the genome. Bioinformatics 24 (20), 2395–2396. doi:10.1093/bioinformatics/btn429
Jinek, M., Chylinski, K., Fonfara, I., Hauer, M., Doudna, J. A., and Charpentier, E. (2012). A programmable dual-RNA–guided DNA endonuclease in adaptive bacterial immunity. science 337 (6096), 816–821. doi:10.1126/science.1225829
Jinek, M., Jiang, F., Taylor, D. W., Sternberg, S. H., Kaya, E., Ma, E., et al. (2014). Structures of Cas9 endonucleases reveal RNA-mediated conformational activation. Science 343 (6176), 1247997. doi:10.1126/science.1247997
Kaur, K., Gupta, A. K., Rajput, A., and Kumar, M. (2016). ge-CRISPR-An integrated pipeline for the prediction and analysis of sgRNAs genome editing efficiency for CRISPR/Cas system. Sci. Rep. 6 (1). doi:10.1038/srep30870
Keough, K. C., Lyalina, S., Olvera, M. P., Whalen, S., Conklin, B. R., and Pollard, K. S. (2019). AlleleAnalyzer: A tool for personalized and allele-specific sgRNA design. Genome Biol. 20 (1), 167–169. doi:10.1186/s13059-019-1783-3
Kim, D., Kang, B. C., and Kim, J. S. (2021). Identifying genome-wide off-target sites of CRISPR RNA-guided nucleases and deaminases with Digenome-seq. Nat. Protoc. 16 (2), 1170–1192. doi:10.1038/s41596-020-00453-6
Kim, H., Kim, S-T., Ryu, J., Kang, B-C., Kim, J-S., and Kim, S-G. (2017). CRISPR/Cpf1-mediated DNA-free plant genome editing. Nat. Commun. 8 (1), 14406–14407. doi:10.1038/ncomms14406
Kim, H. K., Kim, Y., Lee, S., Min, S., Bae, J. Y., Choi, J. W., et al. (2019). SpCas9 activity prediction by DeepSpCas9, a deep learning–based model with high generalization performance. Sci. Adv. 5 (11), eaax9249. doi:10.1126/sciadv.aax9249
Kim, H. K., Min, S., Song, M., Jung, S., Choi, J. W., Kim, Y., et al. (2018). Deep learning improves prediction of CRISPR–Cpf1 guide RNA activity. Nat. Biotechnol. 36 (3), 239–241. doi:10.1038/nbt.4061
Konstantakos, V., Nentidis, A., Krithara, A., and Paliouras, G. (2022). CRISPR-Cas9 gRNA efficiency prediction: An overview of predictive tools and the role of deep learning. Nucleic acids Res. 50 (7), 3616–3637. doi:10.1093/nar/gkac192
Kuscu, C., Arslan, S., Singh, R., Thorpe, J., and Adli, M. (2014). Genome-wide analysis reveals characteristics of off-target sites bound by the Cas9 endonuclease. Nat. Biotechnol. 32 (7), 677–683. doi:10.1038/nbt.2916
Kwon, K. H., Seonwoo, M., Myungjae, S., Soobin, J., Woo, C. J., Younggwang, K., et al. (2019). DeepCpf1: Deep learning-based prediction of CRISPR-Cpf1 activity atendogenous sites. Proc. Annu. Meet. Jpn. Pharmacol. Soc. 92, JKL-05.
Labun, K., Montague, T. G., Krause, M., Torres Cleuren, Y. N., Tjeldnes, H., and Valen, E. (2019). CHOPCHOP v3: Expanding the CRISPR web toolbox beyond genome editing. Nucleic acids Res. 47 (W1), W171–W174. doi:10.1093/nar/gkz365
Li, B., Chen, P. B., and DiaoCRISPR-, Y. S. E. (2021). CRISPR-SE: A brute force search engine for CRISPR design. NAR genomics Bioinforma. 3 (1), lqab013. doi:10.1093/nargab/lqab013
Li, Q., and Lin, N. (2010). The Bayesian elastic net. Bayesian anal. 5 (1), 151–170. doi:10.1214/10-ba506
Lin, J., and Wong, K-C. (2018). Off-target predictions in CRISPR-Cas9 gene editing using deep learning. Bioinformatics 34 (17), i656–i663. doi:10.1093/bioinformatics/bty554
Lin, L., and Luo, Y. (2019). Tracking CRISPR’s footprints. CRISPR Gene Ed. 1961, 13–28. doi:10.1007/978-1-4939-9170-9_2
Listgarten, J., Weinstein, M., Elibol, M., Hoang, L., Doench, J., and Fusi, N. (2016) Predicting off-target effects for end-to-end CRISPR guide design. bioRxiv.:078253.
Listgarten, J., Weinstein, M., Kleinstiver, B. P., Sousa, A. A., Joung, J. K., Crawford, J., et al. (2018). Prediction of off-target activities for the end-to-end design of CRISPR guide RNAs. Nat. Biomed. Eng. 2 (1), 38–47. doi:10.1038/s41551-017-0178-6
Liu, G., Zhang, Y., and Zhang, T. (2020). Computational approaches for effective CRISPR guide RNA design and evaluation. Comput. Struct. Biotechnol. J. 18, 35–44. doi:10.1016/j.csbj.2019.11.006
Liu, H., Ding, Y., Zhou, Y., Jin, W., Xie, K., and Chen, L-L. (2017). CRISPR-P 2.0: An improved CRISPR-cas9 tool for genome editing in plants. Mol. plant 10 (3), 530–532. doi:10.1016/j.molp.2017.01.003
Liu, H., Wei, Z., Dominguez, A., Li, Y., Wang, X., and Qi, L. S. (2015). CRISPR-ERA: A comprehensive design tool for CRISPR-mediated gene editing, repression and activation: Fig. 1. Bioinformatics 31 (22), 3676–3678. doi:10.1093/bioinformatics/btv423
Luo, J., Chen, W., Xue, L., and Tang, B. (2019). Prediction of activity and specificity of CRISPR-Cpf1 using convolutional deep learning neural networks. BMC Bioinforma. 20 (1). doi:10.1186/s12859-019-2939-6
Luyten, H., Plijter, J. J., and Van Vliet, T. (2004). Crispy/crunchy crusts of cellular solid foods: A literature review with discussion. J. texture Stud. 35 (5), 445–492. doi:10.1111/j.1745-4603.2004.35501.x
Ma, J., Köster, J., Qin, Q., Hu, S., Li, W., Chen, C., et al. (2016). CRISPR-DO for genome-wide CRISPR design and optimization. Bioinformatics 32 (21), 3336–3338. doi:10.1093/bioinformatics/btw476
Makarova, K. S., Wolf, Y. I., Alkhnbashi, O. S., Costa, F., Shah, S. A., Saunders, S. J., et al. (2015). An updated evolutionary classification of CRISPR–Cas systems. Nat. Rev. Microbiol. 13 (11), 722–736. doi:10.1038/nrmicro3569
Mali, P., Yang, L., Esvelt, K. M., Aach, J., Guell, M., DiCarlo, J. E., et al. (2013). RNA-guided human genome engineering via Cas9. Science 339 (6121), 823–826. doi:10.1126/science.1232033
Manibalan, S., Thirukumaran, K., Varshni, M., Shobana, A., and Achary, A. (2020). Report on biopharmaceutical profile of recent biotherapeutics and insilco docking studies on target bindings of known aptamer biotherapeutics. Biotechnol. Genet. Eng. Rev. 36 (2), 57–80. doi:10.1080/02648725.2020.1858395
McKenna, A., and Shendure, J. (2018). FlashFry: A fast and flexible tool for large-scale CRISPR target design. BMC Biol. 16 (1), 74–76. doi:10.1186/s12915-018-0545-0
Mitrofanov, A., Alkhnbashi, O. S., Shmakov, S. A., Makarova, K. S., Koonin, E. V., and Backofen, R. (2021). CRISPRidentify: Identification of CRISPR arrays using machine learning approach. Nucleic acids Res. 49 (4), e20–e. doi:10.1093/nar/gkaa1158
Moreno-Mateos, M. A., Vejnar, C. E., Beaudoin, J-D., Fernandez, J. P., Mis, E. K., Khokha, M. K., et al. (2015). CRISPRscan: Designing highly efficient sgRNAs for CRISPR-cas9 targeting in vivo. Nat. methods 12 (10), 982–988. doi:10.1038/nmeth.3543
Muhammad Rafid, A. H., Toufikuzzaman, M., Rahman, M. S., and Rahman, M. S. (2020). CRISPRpred (SEQ): A sequence-based method for sgRNA on target activity prediction using traditional machine learning. BMC Bioinforma. 21. doi:10.1186/s12859-020-3531-9
Naito, Y., Hino, K., Bono, H., and Ui-Tei, K. (2015). CRISPRdirect: Software for designing CRISPR/cas guide RNA with reduced off-target sites. Bioinformatics 31 (7), 1120–1123. doi:10.1093/bioinformatics/btu743
O’Brien, A., and BaileyGT-Scan, T. L. (2014). GT-scan: Identifying unique genomic targets. Bioinformatics 30 (18), 2673–2675. doi:10.1093/bioinformatics/btu354
Oliveros, J. C., Franch, M., Tabas-Madrid, D., San-León, D., Montoliu, L., Cubas, P., et al. (2016). Breaking-Cas—Interactive design of guide RNAs for CRISPR-cas experiments for ENSEMBL genomes. Nucleic acids Res. 44 (W1), W267–W271. doi:10.1093/nar/gkw407
Pallarès Masmitjà, M., Knödlseder, N., and Güell, M. (2019). CRISPR gene editing. Berlin, Germany: Springer, 3–11.CRISPR-gRNA design
Park, J., Bae, S., and Kim, J-S. (2015). Cas-Designer: A web-based tool for choice of CRISPR-cas9 target sites. Bioinformatics 31 (24), 4014–4016. doi:10.1093/bioinformatics/btv537
Park, J., Lim, K., Kim, J-S., and Bae, S. (2017). Cas-analyzer: An online tool for assessing genome editing results using NGS data. Bioinformatics 33 (2), 286–288. doi:10.1093/bioinformatics/btw561
Peng, D., and Tarleton, R. (2015). EuPaGDT: A web tool tailored to design CRISPR guide RNAs for eukaryotic pathogens. Microb. genomics 1 (4), e000033. doi:10.1099/mgen.0.000033
Perez, A. R., Pritykin, Y., Vidigal, J. A., Chhangawala, S., Zamparo, L., Leslie, C. S., et al. (2017). GuideScan software for improved single and paired CRISPR guide RNA design. Nat. Biotechnol. 35 (4), 347–349. doi:10.1038/nbt.3804
Pinello, L., Canver, M. C., Hoban, M. D., Orkin, S. H., Kohn, D. B., Bauer, D. E., et al. CRISPResso: Sequencing analysis toolbox for CRISPR genome editing. bioRxiv. 2016:031203.
Pinello, L., Canver, M. C., Hoban, M. D., Orkin, S. H., Kohn, D. B., Bauer, D. E., et al. (2015). CRISPResso: Sequencing analysis toolbox for CRISPR-cas9 genome editing, bioRxiv. 031203.
Prykhozhij, S. V., Rajan, V., Gaston, D., and Berman, J. N. (2015). CRISPR multitargeter: A web tool to find common and unique CRISPR single guide RNA targets in a set of similar sequences. PloS onee0119372 10 (3). doi:10.1371/journal.pone.0119372
Pulido-Quetglas, C., Aparicio-Prat, E., Arnan, C., Polidori, T., Hermoso, T., Palumbo, E., et al. (2017). Scalable design of paired CRISPR guide RNAs for genomic deletion. PLoS Comput. Biol. 13 (3), e1005341. doi:10.1371/journal.pcbi.1005341
Qin, R., Li, J., Li, H., Zhang, Y., Liu, X., Miao, Y., et al. (2019). Developing a highly efficient and wildly adaptive CRISPR-SaCas9 toolset for plant genome editing. Plant Biotechnol. J. 17 (4), 706–708. doi:10.1111/pbi.13047
Rabinowitz, R., Almog, S., Darnell, R., and Offen, D. (2020). CrisPam: SNP-Derived PAM analysis tool for allele-specific targeting of genetic variants using CRISPR-cas systems. Front. Genet. 11, 851. doi:10.3389/fgene.2020.00851
Rastogi, A., Murik, O., Bowler, C., and Tirichine, L. (2016). PhytoCRISP-ex: A web-based and stand-alone application to find specific target sequences for CRISPR/CAS editing. BMC Bioinforma. 17 (1), 261–264. doi:10.1186/s12859-016-1143-1
Selvakumar, S. C., Preethi, K. A., Ross, K., Tusubira, D., Khan, M. W. A., Mani, P., et al. (2022). CRISPR/Cas9 and next generation sequencing in the personalized treatment of Cancer. Mol. Cancer 21 (1), 83. doi:10.1186/s12943-022-01565-1
Shen, M. W., Arbab, M., Hsu, J. Y., Worstell, D., Culbertson, S. J., Krabbe, O., et al. (2018). Predictable and precise template-free CRISPR editing of pathogenic variants. Nature 563 (7733), 646–651. doi:10.1038/s41586-018-0686-x
Skennerton, C. T., Imelfort, M., and Tyson, G. W. (2013). Crass: Identification and reconstruction of CRISPR from unassembled metagenomic data. Nucleic acids Res. 41 (10), e105–e. doi:10.1093/nar/gkt183
Sledzinski, P., Nowaczyk, M., and Olejniczak, M. (2020). Computational tools and resources supporting CRISPR-Cas experiments. Cells 9 (5), 1288. doi:10.3390/cells9051288
Smith, R. H., Chen, Y-C., Seifuddin, F., Hupalo, D., Alba, C., Reger, R., et al. (2020). Genome-wide analysis of off-target CRISPR/Cas9 activity in single-cell-derived human hematopoietic stem and progenitor cell clones. Genes 11 (12), 1501. doi:10.3390/genes11121501
Stemmer, M., Thumberger, T., del Sol Keyer, M., Wittbrodt, J., and Mateo, J. L. (2015). CCTop: An intuitive, flexible and reliable CRISPR/Cas9 target prediction tool. PloS one 10 (4), e0124633. doi:10.1371/journal.pone.0124633
Sun, J., Liu, H., Liu, J., Cheng, S., Peng, Y., Zhang, Q., et al. (2019). CRISPR-local: A local single-guide RNA (sgRNA) design tool for non-reference plant genomes. Bioinformatics 35 (14), 2501–2503. doi:10.1093/bioinformatics/bty970
Tarasava, K., Liu, R., Garst, A., and Gill, R. T. (2018). Combinatorial pathway engineering using type I-E CRISPR interference. Biotechnol. Bioeng. 115 (7), 1878–1883. doi:10.1002/bit.26589
Tsai, S. Q., Zheng, Z., Nguyen, N. T., Liebers, M., Topkar, V. V., Thapar, V., et al. (2015). GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol. 33 (2), 187–197. doi:10.1038/nbt.3117
Upadhyay, S. K., and Sharma, S. (2014). SSFinder: High throughput CRISPR-cas target sites prediction tool. BioMed Res. Int. 2014, 1–4. doi:10.1155/2014/742482
Wang, D., Zhang, C., Wang, B., Li, B., Wang, Q., Liu, D., et al. (2019). Optimized CRISPR guide RNA design for two high-fidelity Cas9 variants by deep learning. Nat. Commun. 10 (1). doi:10.1038/s41467-019-12281-8
Wang, J., Xiang, X., Cheng, L., Zhang, X., and Luo, Y. (2020). CRISPR-GNL: An improved model for predicting CRISPR activity by machine learning and featurization. bioRxiv. 2019:605790.
Wang, J., Zhang, X., Cheng, L., and Luo, Y. (2020). An overview and metanalysis of machine and deep learning-based CRISPR gRNA design tools. RNA Biol. 17 (1), 13–22. doi:10.1080/15476286.2019.1669406
Wang, K., and Liang, C. C. R. F. (2017). CRF: Detection of CRISPR arrays using random forest. PeerJ 5, e3219. doi:10.7717/peerj.3219
Wang, X., Tilford, C., Neuhaus, I., Mintier, G., Guo, Q., Feder, J. N., et al. (2017). CRISPR-DAV: CRISPR NGS data analysis and visualization pipeline. Bioinformatics 33 (23), 3811–3812. doi:10.1093/bioinformatics/btx518
Wilson, L. O., Reti, D., O'Brien, A. R., Dunne, R. A., and Bauer, D. C. (2018). High activity target-site identification using phenotypic independent CRISPR-Cas9 core functionality. CRISPR J. 1 (2), 182–190. doi:10.1089/crispr.2017.0021
Winter, J., Schwering, M., Pelz, O., Rauscher, B., Zhan, T., Heigwer, F., et al. CRISPRAnalyzeR: Interactive analysis, annotation and documentation of pooled CRISPR screens. BioRxiv. 2017:109967.
Wong, N., Liu, W., and WangWU-Crispr, X. (2015). Wu-CRISPR: Characteristics of functional guide RNAs for the CRISPR/Cas9 system. Genome Biol. 16 (1), 218–8. doi:10.1186/s13059-015-0784-0
Xiao, A., Cheng, Z., Kong, L., Zhu, Z., Lin, S., Gao, G., et al. (2014). CasOT: A genome-wide cas9/gRNA off-target searching tool. Bioinformatics 30 (8), 1180–1182. doi:10.1093/bioinformatics/btt764
Xie, S., Shen, B., Zhang, C., Huang, X., and Zhang, Y. (2014). sgRNAcas9: a software package for designing CRISPR sgRNA and evaluating potential off-target cleavage sites. PloS one 9 (6), e100448. doi:10.1371/journal.pone.0100448
Xu, H., Xiao, T., Chen, C-H., Li, W., Meyer, C. A., Wu, Q., et al. (2015). Sequence determinants of improved CRISPR sgRNA design. Genome Res. 25 (8), 1147–1157. doi:10.1101/gr.191452.115
Yan, J., Chuai, G., Zhou, C., Zhu, C., Yang, J., Zhang, C., et al. (2018). Benchmarking CRISPR on-target sgRNA design. Briefings Bioinforma. 19 (4), 721–724. doi:10.1093/bib/bbx001
Yu, S-H., Vogel, J., and Förstner, K. U. (2018). ANNOgesic: A Swiss army knife for the RNA-seq based annotation of bacterial/archaeal genomes. GigaScience 7 (9), giy096. doi:10.1093/gigascience/giy096
Zetsche, B., Abudayyeh, O. O., Gootenberg, J. S., Scott, D. A., and Zhang, F. (2020). A survey of genome editing activity for 16 Cas12a orthologs. Keio J. Med. 69 (3), 59–65. doi:10.2302/kjm.2019-0009-oa
Zhang, G., Dai, Z., and Dai, X. (2020). C-RNNCrispr: Prediction of CRISPR/Cas9 sgRNA activity using convolutional and recurrent neural networks. Comput. Struct. Biotechnol. J. 18, 344–354. doi:10.1016/j.csbj.2020.01.013
Zhu, H., and LiangCRISPR-, C. D. T. (2019). CRISPR-DT: Designing gRNAs for the CRISPR-cpf1 system with improved target efficiency and specificity. Bioinformatics 35 (16), 2783–2789. doi:10.1093/bioinformatics/bty1061
Zhu, H., Misel, L., Graham, M., Robinson, M. L., and LiangCT-Finder, C. (2016). CT-finder: A web service for CRISPR optimal target prediction and visualization. Sci. Rep. 6 (1), 25516–25518. doi:10.1038/srep25516
Zhu, H., Richmond, E., and LiangCRISPR-Rt, C. (2018). CRISPR-RT: A web application for designing CRISPR-C2c2 crRNA with improved target specificity. Bioinformatics 34 (1), 117–119. doi:10.1093/bioinformatics/btx580
Keywords: CRiSPR/Cas, gRNA design, on-target, off-target, computational approach, machine learning
Citation: Alipanahi R, Safari L and Khanteymoori A (2023) CRISPR genome editing using computational approaches: A survey. Front. Bioinform. 2:1001131. doi: 10.3389/fbinf.2022.1001131
Received: 22 July 2022; Accepted: 19 December 2022;
Published: 11 January 2023.
Edited by:
Shizuka Uchida, Aalborg University Copenhagen, DenmarkReviewed by:
Arjun Ray, Indraprastha Institute of Information Technology Delhi, IndiaOlli-Pekka Smolander, Tallinn University of Technology, Estonia
Copyright © 2023 Alipanahi, Safari and Khanteymoori. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Leila Safari, bHNhZmFyaUB6bnUuYWMuaXI=
†This author share first authorship
‡These authors have contributed equally to this work and share senior authorship