 Zhi-Hao Guo
Zhi-Hao Guo Li Yuan1,2
Li Yuan1,2 Ya-Zhou Shi
Ya-Zhou Shi
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Bioinform., 11 January 2022
Sec. Drug Discovery in Bioinformatics
Volume 1 - 2021 | https://doi.org/10.3389/fbinf.2021.809082
This article is part of the Research TopicA Balanced View Of Nucleic Acid Structural ModelingView all 4 articles
The 3D architectures of RNAs are essential for understanding their cellular functions. While an accurate scoring function based on the statistics of known RNA structures is a key component for successful RNA structure prediction or evaluation, there are few tools or web servers that can be directly used to make comprehensive statistical analysis for RNA 3D structures. In this work, we developed RNAStat, an integrated tool for making statistics on RNA 3D structures. For given RNA structures, RNAStat automatically calculates RNA structural properties such as size and shape, and shows their distributions. Based on the RNA structure annotation from DSSR, RNAStat provides statistical information of RNA secondary structure motifs including canonical/non-canonical base pairs, stems, and various loops. In particular, the geometry of base-pairing/stacking can be calculated in RNAStat by constructing a local coordinate system for each base. In addition, RNAStat also supplies the distribution of distance between any atoms to the users to help build distance-based RNA statistical potentials. To test the usability of the tool, we established a non-redundant RNA 3D structure dataset, and based on the dataset, we made a comprehensive statistical analysis on RNA structures, which could have the guiding significance for RNA structure modeling. The python code of RNAStat, the dataset used in this work, and corresponding statistical data files are freely available at GitHub (https://github.com/RNA-folding-lab/RNAStat).
RNA molecules play important roles in various biological processes, ranging from carrying genetic information, participating in protein synthesis, catalyzing biochemical reactions, and regulating gene expressions, to acting as a structural molecule in cellular organelles (Doherty and Doudna, 2001; Dethoff et al., 2012; Cech and Steitz, 2014). Generally, to perform functions, RNAs need to form special tertiary structures, which typically can be determined by experimental methods such as cryo-electron microscopy, X-ray crystallography, and nuclear magnetic resonance spectroscopy (NMR) (Fernandez-Leiro and Scheres, 2016; Rose et al., 2017; Westhof and Leontis, 2021). However, the structures deposited in Protein Data Bank (PDB) are still limited, since it is expensive and time-consuming to experimentally derive high-resolution RNA 3D structures (Rose et al., 2017; Westhof and Leontis, 2021). This situation has led to a great demand in structural biology to envisage the RNA structures using prediction methods (Hajdin et al., 2010; Shi Y.-Z. et al., 2014; Miao et al., 2017; Schlick and Pyle, 2017).
In the last decade, there are some computational models have been developed for predicting RNA 3D structures, among which the knowledge-based fragment assembly methods (Gan et al., 2004; Das and Baker, 2007; Parisien and Major, 2008; Das et al., 2010; Flores et al., 2010; Cao and Chen, 2011; Rother et al., 2011; Popenda et al., 2012; Zhao et al., 2012; Jian et al., 2019; Zhang et al., 2021) and the physics-based coarse-grained (CG) models have gained more attention (Jonikas et al., 2009; Flores and Altman, 2010; Pasquali and Derreumaux, 2010; Flores et al., 2012; Denesyuk and Thirumalai, 2013; Xia et al., 2013; Shi YZ. et al., 2014; Šulc et al., 2014; Krokhotin et al., 2015; Boniecki et al., 2016). For example, the FARNA/FARFAR can assemble trinucleotide fragments into 3D structures corresponding to an RNA sequence with the use of the Monte Carlo algorithm and a knowledge-based energy function, and the parameters of energy function were determined from the statistical analysis of known RNA 3D structures (Das and Baker, 2007; Das et al., 2010). The SimRNA with a CG representation, which employs a statistical potential derived from PDB structures, and can fold RNAs using only sequence information (Boniecki et al., 2016). Recently, we have also provided a new CG model to predict 3D structures and stability of an RNA in ion solutions from sequence alone (Shi Y.-Z. et al., 2014, 2015, 2018; Jin et al., 2019). Although the potential energy of our model is mainly physics-based, the potentials, especially bonded potentials, were also parameterized by the statistical analysis on the available 3D structures of RNAs in PDB (Shi YZ. et al., 2014; Jin et al., 2019).
Furthermore, the existing knowledge-based methods usually produce an ensemble of candidate structures, which should be further evaluated to recognize the best candidates as close to native structures as possible (Huang and Zou, 2011; Miao and Westhof, 2017; Yan et al., 2018; Tan et al., 2019; Magnus et al., 2020). To address this issue, several statistical potentials have been developed to evaluate RNA 3D structures (Bernauer et al., 2011; Capriotti et al., 2011; Wang et al., 2015; Li et al., 2016; Li et al., 2018; Masso, 2018; Yu et al., 2019; Zhang et al., 2020), such as RASP (Capriotti et al., 2011), RNA KB potentials (Bernauer et al., 2011), 3dRNAscore (Wang et al., 2015), and DFIRE (Zhang et al., 2020). Generally, these potentials are proportional to the frequencies of occurrence of atom pairs, angles, or dihedral angles in PDB structures based on Boltzmann or Bayesian formulations (Huang and Zou, 2011; Yan et al., 2018; Tan et al., 2019). For example, Capriotti et al. have built the RASP by calculating the density distribution of distance between any two atoms in all the known RNA structures (Capriotti et al., 2011). The 3dRNAscore introduced by Wang et al. uses seven typical RNA dihedral angles as well as distance-dependent geometrical descriptions for atom pairs to construct the statistical potentials (Wang et al., 2015). In addition to structure evaluation, very recently, Xiong et al. have proposed a fully knowledge-based function (BRiQ) based on statistics of orientation distribution of one base around another base from the PDB structures for improving RNA model refinement (Xiong et al., 2021).
Obviously, all these advances on RNA structure modeling indicate that to gather various statistics of RNA 3D structures is generally essential to predict RNA tertiary structures. However, there are few tools or web servers that can be used to make comprehensive statistical analysis for RNA 3D structures (Andronescu et al., 2008; Cock et al., 2009; Baulin et al., 2016; Danaee et al., 2018; Magnus et al., 2020). Recently, Baulin et al. have proposed a database URSDB (the Universe of RNA structures database) to store information (e.g., annotations of main structural elements) obtained all RNA-containing PDBs (Baulin et al., 2016). Although the URSDB can allow the user to get statistics on structural motifs (base pairs, stems, and loops) based on the information provided by the software of DSSR (dissecting the spatial structure of RNA) (Lu et al., 2015; Lu, 2020), these statistics on RNA secondary structure motifs could be far from enough to help RNA 3D structure modeling (Miao and Westhof, 2017; Tan et al., 2019). Fortunately, several works have provided statistics of RNA structures from different aspects. For example, both the RNA 3D Motif Atlas and bpRNA can provide a statistical summary of the hairpin and internal loop motifs (Parlea et al., 2016; Danaee et al., 2018). The RNA STRAND can also provide information on structural features such as types and sizes for stems and loops (Andronescu et al., 2008). To build scoring function for RNA structure prediction, Bottaro et al. as well as Das and Baker have developed methods to calculate the geometrical properties of RNA base-pairing and base-stacking (Bottaro et al., 2014; Das and Baker, 2007). Despite all this progress, with the rapidly increasing number of RNA structures deposited in PDB (Supplementary Figure S1 in the Supplementary Material) (Rose et al., 2017; Westhof and Leontis, 2021), an available tool to convenient access comprehensive statistical information of RNA 3D structures is still necessary.
Here, we present a novel tool, named as RNAStat, special for the statistical analysis of RNA 3D structures. It can be used to calculate structural information of RNA 3D structure(s) at different levels: global 3D structural level, secondary structure level, and atomic level. We first introduced the function and principle of the RNAStat. Afterward, based on a non-redundant RNA structure dataset established by us, we utilized the RNAStat to perform statistical analysis for RNA 3D structures, and provided various statistical data of RNA structural properties (e.g., size/shape, geometry of base-pairing/stacking, secondary structure motifs, and atom-atom distance). Throughout the article, we also discussed the potential value of these statistics on RNA 3D structure prediction and evaluation.
The RNAStat provided in this work can be used to make calculation (or statistics) for given RNA structure(s) in the following aspects: 1) the radius of gyration (i.e., size): and shape; 2) the secondary structure motifs; 3) the geometry of base-pairing and base-stacking; 4) the distances between atoms; see Figure 1.
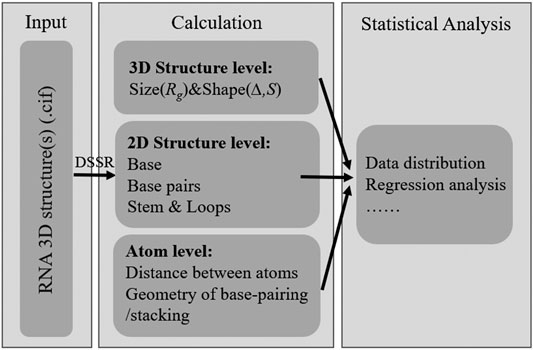
FIGURE 1. The basic functions of RNAStat for RNA 3D structure calculation and statistical analysis.
The mean radius of gyration
where N is the number of heavy atoms (C, P, N, and O) in the RNA molecule,
Since the shape of RNAs is rather important in determining the overall motion of RNA and their interaction with other biomolecules, two rotationally invariant quantities, the asphericity parameter
where
where
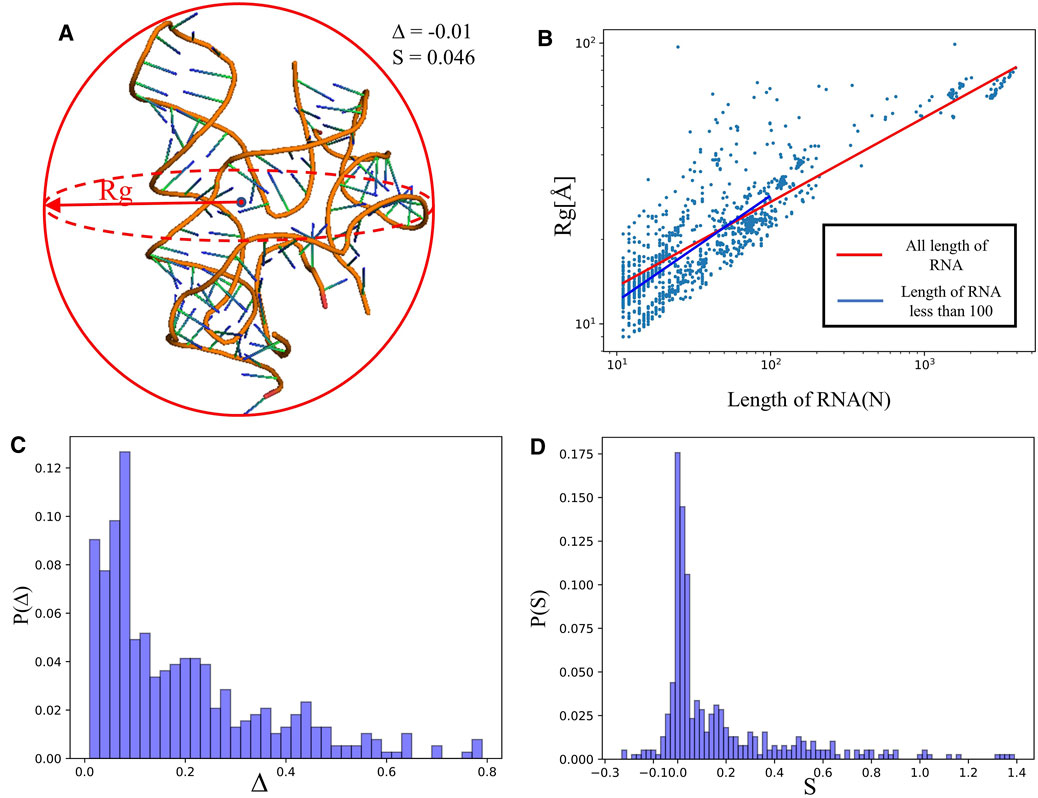
FIGURE 2. (A) The schematic diagram of size and shape for an RNA 3D structure (PDB ID: 4QLM).
To obtain the secondary structure motifs for an RNA PDB structure, the RNAStat can directly call the DSSR through the corresponding python command (e.g., x3dna-dssr.exe--json “+ ”-o = file); The DSSR is an integrated and automated command-line tool for analysis and annotation of RNA tertiary structures, and it can characterize nucleotides, base pairs, pseudoknots, loops, stems, and coaxially stacked helices (Lu et al., 2015; Lu, 2020); see an example in Figure 3. Based on the information extracted from DSSR, for an RNA structure set, the RNAStat can further provide the statistics of secondary structural elements, including base-pairs, stems, and various loops. In this work, we considered all C-G, A-U and G-U pairs to be canonical base pairs, and all other base pairs to be non-canonical ones, and the definitions of the secondary structural motifs can be found everywhere (Leontis and Westhof, 2001) and the simple illustration of them are also shown in Figure 3.
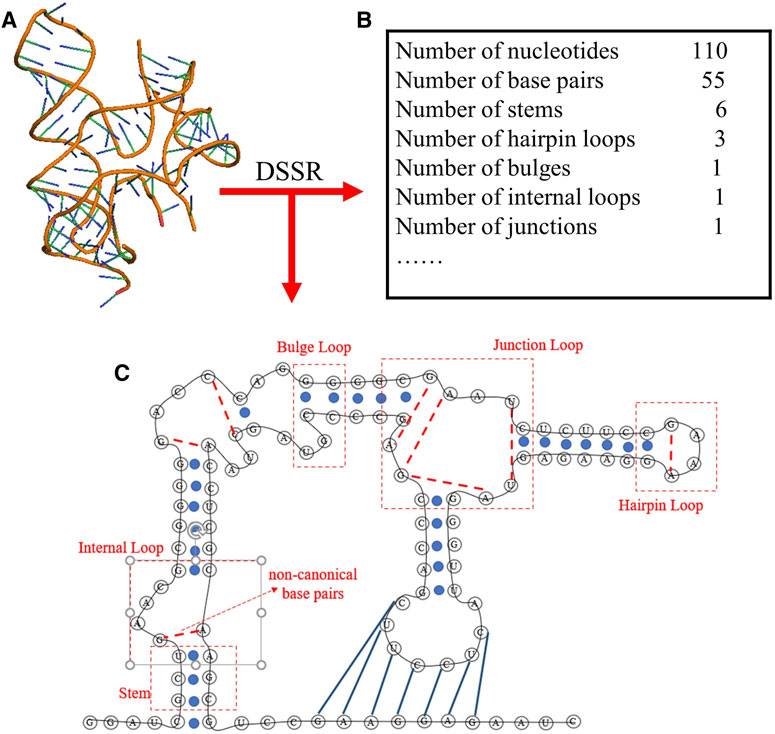
FIGURE 3. The schematic diagram of the secondary structure information extracted from an RNA 3D structure (ydaO riboswitch, PDB ID: 4QLM) using DSSR software in RNAStat. (A) 3D structure shown with the PyMol (http://www.pymol.org) for the RNA. (B) The DSSR software is called to analyze the RNA 3D structures, e.g., for the RNA, the secondary structure information including the details of canonical/noncanonical base pairs, stems, and various loops. (C) The secondary structure drawn based on the secondary structure information from (B) for the RNA. Black lines: backbone. Blue solid circles and blue solid lines: canonical base pairs (A-U, G-C, and G-U). Red dotted lines: non-canonical base pairs. Dashed boxes: samples of secondary structural motifs.
Since base-pairing and base-stacking are critical interactions that stabilize RNA 3D structures (Butcher and Pyle, 2011; Bottaro et al., 2014; Wang et al., 2016; Wang et al., 2020), the RNAStat can calculate the geometry between two bases in base-pairing/stacking. First, the whole nucleobase (i.e., A, U, G, and C) is treated as a single rigid group, and a coordinate system is set up on each base, with the origin (O) at the geometric center of all the heavy atoms. Similar to the local referential of a nucleotide introduced by Gendron et al. (2001), for pyrimidines (or purines), the two unit vectors,
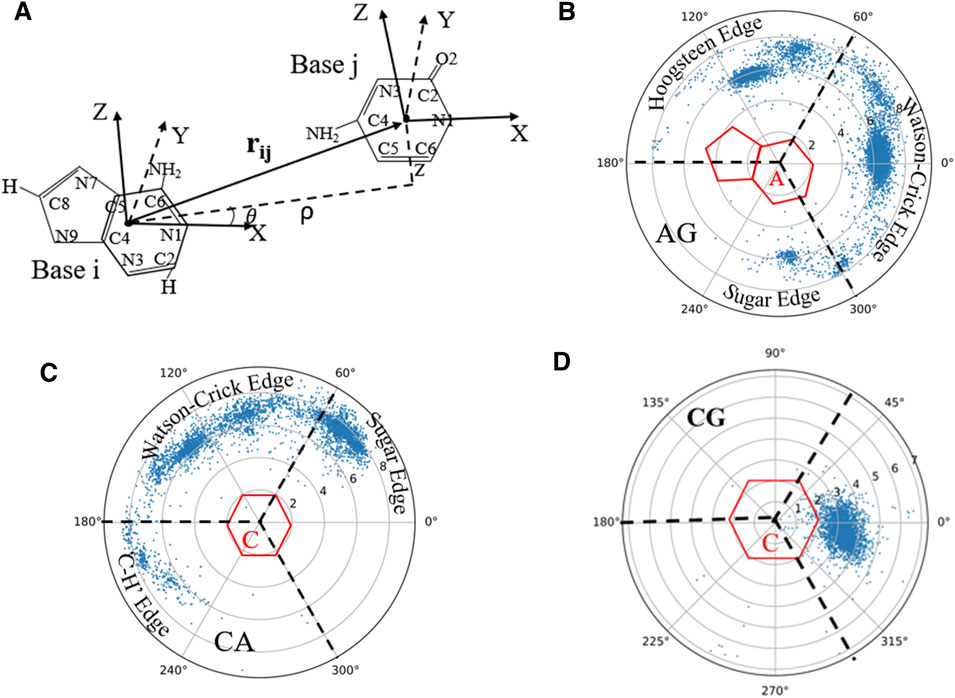
FIGURE 4. (A) The definition of the local coordinate system for bases, and in the coordinate system of one base (e.g., i), the position of another base (e.g., j) can be described by the vectors
As described in the Introduction, the most existing statistical potentials for RNA structure evaluation are based on the distances between various type atoms (Miao and Westhof, 2017; Tan et al., 2019). Based on the coordinates of all the heavy atoms in an RNA structure (i.e.,.cif file), the distance
where
To test the RNAStat, we established a non-redundant dataset based on the RNA 3D Hub set (Release nrlist_3.157_4.0 Å), in which the sequence identity between any two chains in the set is less than 95% (Leontis and Zirbel, 2012). Firstly, we collected 1,245 representative RNAs of all the different clusters with a resolution <4.0 Å from RNA 3D Hub list, which can be downloaded from http://rna.bgsu.edu/rna3dhub/nrlist. Then, we deleted the structure of non-RNA strands in the dataset. Afterwards, we removed the RNA structures with sequence identity ˃ 80% using the BLASTN program (Camacho et al., 2009). Finally, through the prior operation steps, 748 RNA structures were retained and their 3D structure files were downloaded from the PDB. The final RNA structure dataset used in this work can be found in the Supplementary Material as well as at GitHub (https://github.com/RNA-folding-lab/RNAStat), including PDB IDs, and PDB CIF files.
In this work, we present the RNAStat, an integrated tool for making comprehensive statistics on RNA 3D structures. As shown in Figure 1, the RNAStat can be used to do statistical analysis for RNA 3D structures at different levels, such as global 3D structure level, secondary structure level, and atom level. The code of the RNAStat in python can be found at GitHub through https://github.com/RNA-folding-lab/RNAStat. In the following, we will give a brief introduction of the usage method of the tool.
The input to RNAStat is the coordinate file(s) of RNA 3D structure(s) in CIF format. Based on the needs of users, the input can be a single PDB file of an RNA structure or the PDB files for a given RNA structure set. For each PDB file, the RNAStat can calculate the size and shape of the RNA through Eqs 1–4 (in section of Materials and Methods), and call the DSSR to obtain its secondary structure motifs, e.g., the information of base-pairs, stems and various loops; see Figure 3. In the RNAStat, the distance between any heavy atom pair can also be calculated by Eq. 5, and the atom pair types can be specified by the user or default to all kinds of atom types, where 85 heavy atom types in four nucleotides (A, U, G, and C) are considered (Wang et al., 2015; Tan et al., 2019); see Supplementary Table S2 in the Supplementary Material. In addition, based on the information of base-pairing and the coordinates of atoms in two paired bases, the geometrical properties of base-pairing and base-stacking can also be calculated.
More importantly, for RNA structure set, the RNAstat can provide statistical information for all the above structural properties as well as the frequency distribution of various base pairs, which could be directly used to build statistical potentials for RNA structure evaluation or refinement (Miao et al., 2017; Tan et al., 2019; Xiong et al., 2021). The details of the methods for the calculations and statistical analysis can be found in section of Materials and Methods.
To show the applicability of the RNAStat tool, we established a non-redundant RNA 3D structure dataset (see Materials and Methods), and took it as an example for RNA 3D structure analysis and statistic. Simultaneously, based on the RNA structure set, we also provided various statistical results of RNA structures, and which could contribute to building RNA statistical potentials or energy function of RNA CG models.
We calculated the radius of gyration
indicating that
Figure 2C depicts the distribution of asphericity parameter ∆ of RNA structures in the dataset, where ∆ spans over the whole range from 0 to 0.8, and ∼60% has ∆<0.2, suggesting that RNAs are mostly spherical in nature (Hyeon et al., 2006; Tan et al., 2015). The distribution of the shape parameter S for RNA structures is displayed in Figure 2D. The plot exhibits that almost all RNAs have S > 0, and the distribution has a significant peak around S = 0, implying that RNAs do not deviate much from the spherical symmetry. Our statistics on ∆ and S are very close to the results from RNA complexes reported in Ref. (Hyeon et al., 2006), while are with the different from those of single-chain RNAs.
Since RNA structure formation is generally hierarchical (Brion and Westhof, 1997), the information of RNA secondary structures could be the key to evaluate or predict RNA tertiary structures. The DSSR software can be called by the RNAStat to analyze all the RNA tertiary structures in the dataset; see Figure 3. Based on the results from DSSR, various statistics on RNA secondary motifs can be showed.
As shown in Figure 5; Supplementary Tables S3–S5 in the Supplementary Material, the guanine nucleotide (i.e., G) and the base pairs of G-C/C-G are the most common in the RNA dataset, e.g., the probability of occurrence of G (∼34%) is apparently higher than that of the other bases. Using the dataset of RNA structures, we found that the number of base pairs
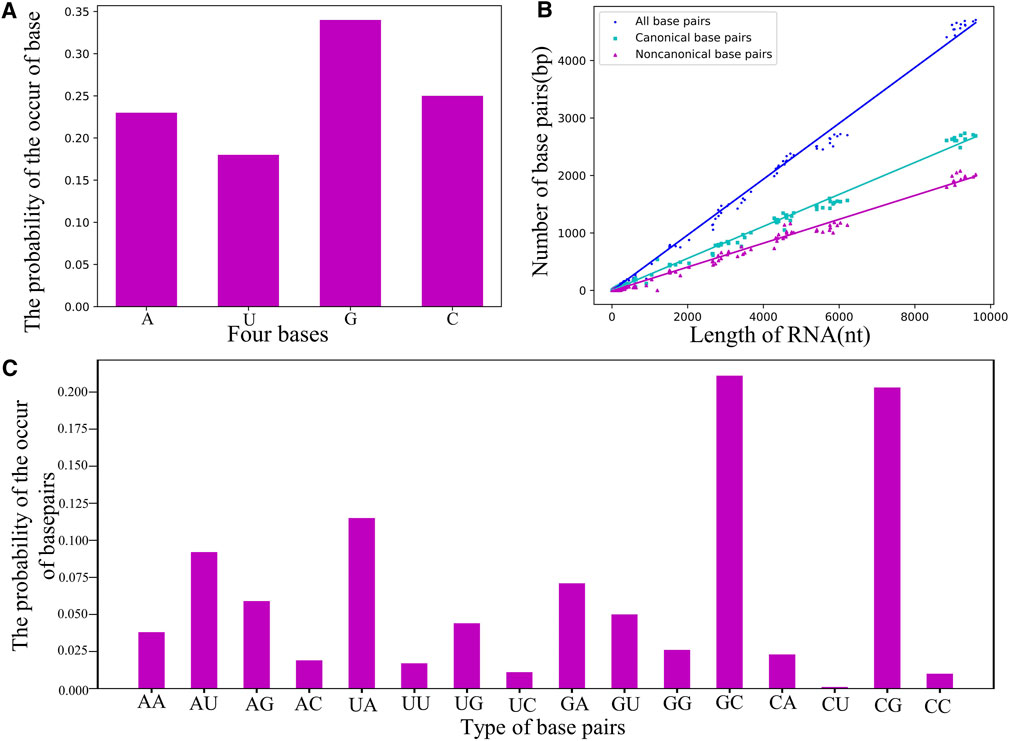
FIGURE 5. (A) The probability of the occurrence of nucleotides in the non-redundant dataset. (B) The counts of base-pairs as a function of length N for RNA structures in the dataset. Green squares: canonical base pairs. Purple triangle: non-canonical base pairs. Blue circle: all canonical and non-canonical base pairs. (C) The probability of the occurrence of base pairs including canonical and non-canonical ones.
Figure 5C shows the probability of the occurrence of base pairs including canonical and non-canonical base pairs; seen also in Supplementary Table S4 in the Supplementary Material, and due to the proportional relation between base-pairing strength and their relative probability, this statistic of base pairs can be directly used to parameterize the base-pairing energy function for RNA models. For example, based on the relative probability between G-C/C-G (∼40%) and A-U/U-A (∼20%), we have set that the energy of G-C is twice the strength of the A-U in our CG model (Shi Y.-Z. et al., 2014; Jin et al., 2019), and the common non-canonical base pairs (e.g., A-G, A-A, and G-G) will be further taken into account. In addition, base-pair stacking make a significant contribution to the stability of an RNA structure (Schlick and Pyle, 2017; Miao and Westhof, 2017; Brion and Westhof, 1997; Laing and Schlick, 2009), and the stacking interaction parameters can also be obtained from the statistical frequency of base-pair stack (Supplementary Table S5 in the Supplementary Material), which could improve the predictions of RNA secondary (or 3D) structures and their thermodynamic stability (Dima et al., 2005; Gardner et al., 2011; Sloma and Mathews, 2017).
Furthermore, the distribution of length of RNA secondary structure motifs (e.g., stem and loops) could be helpful in the evaluation of structures predicted by ab initio models (Brion and Westhof, 1997; Danaee et al., 2018). Figure 6A displays the distribution of the length of stem, which is defined by the number of continuous canonical base pairs (Lu et al., 2015). Although the distribution of stem length for the RNAs in dataset is very broad, there is a prominent peak around ∼2 bp and the length of stem greater than 10 bp occur much less frequently; see Figure 6A, suggesting that stems are constantly interrupted by loops (Figure 6B) (Danaee et al., 2018). For hairpin loops shown in Figure 6C, we found that hairpin loops are most likely to have a length of 4 nt, i.e., tetraloops, which have been proved to be extremely stable by thermodynamic experiments (Butcher and Pyle, 2011), and the heptaloops (i.e., hairpin loops of length 7 nt) are the second most frequent, in line with the results from bpRNA, and RNA 3D Motif Atlas (Danaee et al., 2018; Parlea et al., 2016). On the contrary, the distribution of the bulge loop length only has one very significant peak at 1 nt, and almost all the bulge loops are with length less than 5 nt; seen in Figure 6D. The reasons could be that one stem interrupted by short bulge loops (e.g., 1 nt) is generally as stable as continuous helix with same sequence due to the coaxial-stacking interaction between two stems (Shi et al., 2015; Butcher and Pyle, 2011), while the stability of RNAs is reduced with the increase of the length of bulge loop (Zhang et al., 2019). As shown in Figures 6E,F, the distributions of internal/junction loop lengths are more complex, with more than one broad peak. For example, there are about four visible peaks observed for internal loop at 2, 4, 6, and 9 nt, respectively. Since the bases in two sides (5′ and 3′) of an internal loop often pairing together in non-canonical way, the internal loops often tend to be symmetric in order to keep a more stable structure (Laing and Schlick, 2009; Butcher and Pyle, 2011; Gardner et al., 2011). However, we only calculated the length of the entire loop without distinguishing 5′ and 3’ loop sequences, for simplicity in the present version of the RNAStat. More detailed statistics of internal/multi-loops should be taken into account in the future to help improve their energy parameters calculation.
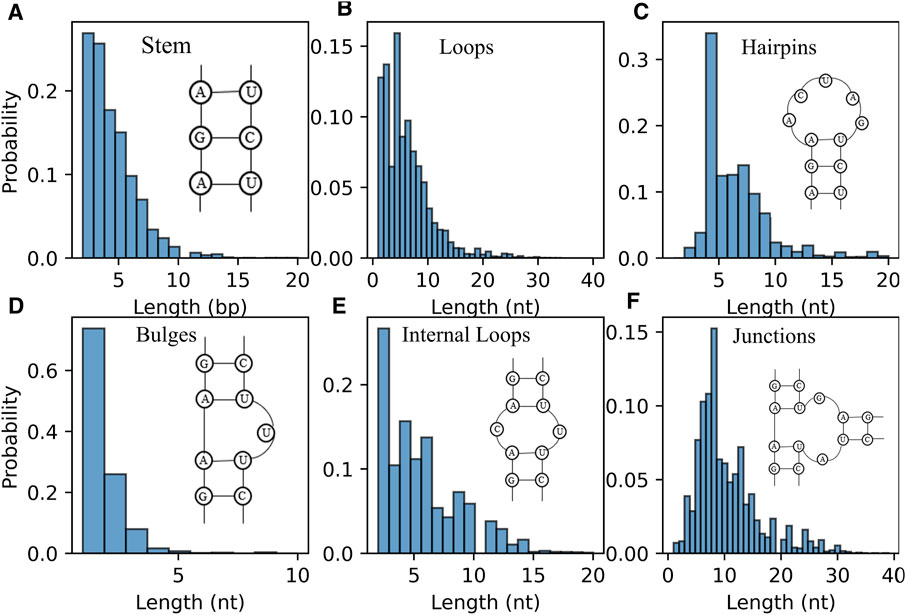
FIGURE 6. The distribution of length of RNA secondary structure motifs in the dataset. (A) Histogram of the occurrence for the length of stems. (B–F) Histogram of the occurrence for the length of loops (B) all loops; (C) hairpin loops; (D) bulge loops; (E) internal loops; (F) junction loops.
On account of the importance of the geometrical configuration of base-pair/stacking in RNA 3D modeling (Das and Baker, 2007; Bottaro et al., 2014), the RNAStat provides the calculation or statistic of geometry of base-pairing/stacking for RNA structures; see the section of Materials and Methods. For the RNA structure dataset used in this work, the statistical results of base pairs including canonical and non-canonical ones are shown in Figure 4 and Supplementary Figure S4 in the Supplementary Material. For example, Figure 4B shows the geometric position
Supplementary Figures S4–S8 in the Supplementary Material show the distributions for all the base-pairing and stacking, and the corresponding data files as well as fitting parameters (
In view of the fact that most of the knowledge-based statistical potentials for RNA structure evaluation are based on the distances between atoms (Bernauer et al., 2011; Capriotti et al., 2011; Huang and Zou, 2011; Tan et al., 2019). The RNAStat can also be used to calculate the distance between any two non-bonded heavy atoms located at different nucleotides in RNA. For example, the distribution of distance between two atoms with type of P is shown in Figure 7A. In addition to a very broad peak at ∼70 Å, there are three noteworthy peaks at ∼5.7 Å, ∼11.2 Å, and ∼18.4 Å, respectively. The first two peaks are corresponding to the distances of two P atoms in the nearest neighbor nucleotides and next-nearest neighbor nucleotides, respectively, and the third peak represents distance between two P atoms in paired nucleotides; see Figures 7B,C. More distance distributions of atoms with various types can also be found in Supplementary Figure S9 in the Supplementary Material as well as data files at GitHub. Besides, the RNAStat tool also allows the users to input the atoms or atom types to perform statistical analysis for their distances; see in the section of Materials and Methods.
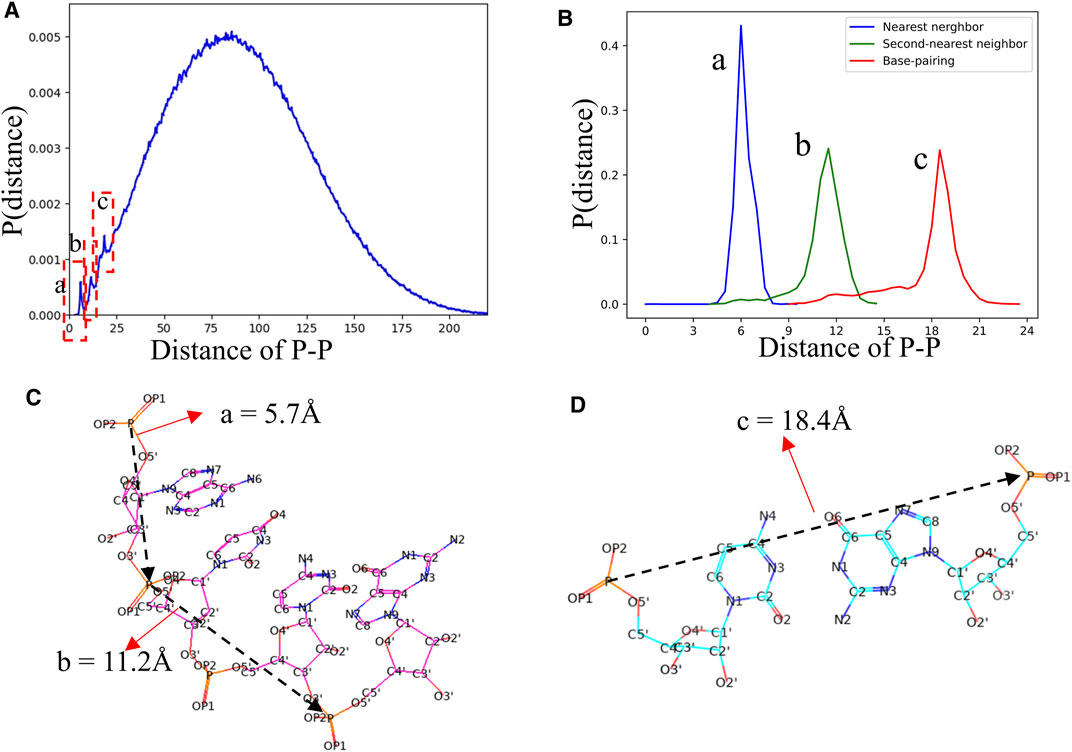
FIGURE 7. (A) The distance distribution between P atoms in our dataset. Three significant peaks are marked by dashed boxes. (B) The distance distributions between two P atoms in the nearest neighbor nucleotides (a, blue line), second-nearest neighbor nucleotides (c, green line), and paired nucleotides (c, red line), respectively. (C, D) Schematic diagram of the distances between P atoms in the nearest neighbor nucleotide, second-nearest neighbor nucleotides, and paired nucleotides. The a, b, and c in (B–D) are corresponding to the three peaks in (A).
In summary, RNAStat is an integrated computational tool to perform comprehensive statistical analysis for the RNA 3D structures given by the users. The tool cannot only automatically calculate RNA global structural properties such as size and shape, but also analyze atom-atom distance distributions at atomic level. Furthermore, the tool can provide statistics of RNA secondary structure elements (e.g., canonical/non-canonical base pairs, stems and various loops) and geometric properties of base-pairing and base-stacking. In this work, we have established and utilized a non-redundant RNA 3D structure dataset to test the usability of the tool, and the statistical data could be directly used to build statistical potentials or energy functions for RNA 3D structure evaluation and prediction.
Still and all, further improvements need to be made on the tool to perform more detailed statistical analysis and to make it easier to use. For example, most of the available RNA statistical potentials generally adopt a distance-dependent scheme, however for proteins, the orientation-dependent statistical potentials, which consider the many-body interactions by statistically describing both distance and relative orientation between interacting atom groups, and have been proved to have better performance than the traditional distance-dependent potentials (Masso, 2018; Yu et al., 2019; Zhang et al., 2020). Thus, in the further development of RNAStat, the distribution of orientation (e.g., angle and torsion angle) between atoms as well as the joint probability at the given relative distance and orientation of observing two atoms should be taken into account. In addition, although the RNAStat is free-installation and convenient to use through command lines, it is still required the python installation or corresponding environment configuration. Thus, a user-friendly webserver could be further built after the deepened improvement for the tool. Very recent studies have shown that RNA scoring functions derived from deep learning of RNA 3D structures performed well in identification of accurate structural models (Kurgan and Zhou, 2011; Li et al., 2018; Wang et al., 2018; Huang et al., 2020; Townshend et al., 2021), which suggests that more potential structural features of RNAs should be further mined with the aid of deep neural networks.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Z-HG, Y-ZS, and B-GZ designed the research; Z-HG and LY performed the experiments. Z-HG and Y-ZS analyzed the data. Y-ZS, Z-HG, and Y-LT wrote the manuscript. All authors discussed the results and reviewed the manuscript.
This work was supported by the Grants from the National Science Foundation of China (11971367 and 11605125).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We are grateful to Professors Zhi-Jie Tan (Wuhan University), and Jie Liu (Wuhan Textile University) for valuable discussions.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fbinf.2021.809082/full#supplementary-material
Andronescu, M., Bereg, V., Hoos, H. H., and Condon, A. (2008). RNA STRAND: the RNA Secondary Structure and Statistical Analysis Database. BMC Bioinformatics 9, 340. doi:10.1186/1471-2105-9-340
Baulin, E., Yacovlev, V., Khachko, D., Spirin, S., and Roytberg, M. (2016). URS DataBase: Universe of RNA Structures and Their Motifs. Database (Oxford) 2016, baw085. doi:10.1093/database/baw085
Bernauer, J., Huang, X., Sim, A. Y., and Levitt, M. (2011). Fully Differentiable Coarse-Grained and All-Atom Knowledge-Based Potentials for RNA Structure Evaluation. RNA 17 (6), 1066–1075. doi:10.1261/rna.2543711
Boniecki, M. J., Lach, G., Dawson, W. K., Tomala, K., Lukasz, P., Soltysinski, T., et al. (2016). SimRNA: a Coarse-Grained Method for RNA Folding Simulations and 3D Structure Prediction. Nucleic Acids Res. 44 (7), e63. doi:10.1093/nar/gkv1479
Bottaro, S., Di Palma, F., and Bussi, G. (2014). The Role of Nucleobase Interactions in RNA Structure and Dynamics. Nucleic Acids Res. 42 (21), 13306–13314. doi:10.1093/nar/gku972
Brion, P., and Westhof, E. (1997). Hierarchy and Dynamics of RNA Folding. Annu. Rev. Biophys. Biomol. Struct. 26, 113–137. doi:10.1146/annurev.biophys.26.1.113
Butcher, S. E., and Pyle, A. M. (2011). The Molecular Interactions that Stabilize RNA Tertiary Structure: RNA Motifs, Patterns, and Networks. Acc. Chem. Res. 44 (12), 1302–1311. doi:10.1021/ar200098t
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., et al. (2009). BLAST+: Architecture and Applications. BMC Bioinformatics 10, 421. doi:10.1186/1471-2105-10-421
Cao, S., and Chen, S. J. (2011). Physics-based De Novo Prediction of RNA 3D Structures. J. Phys. Chem. B 115, 4216–4226. doi:10.1021/jp112059y
Capriotti, E., Norambuena, T., Marti-Renom, M. A., and Melo, F. (2011). All-atom Knowledge-Based Potential for RNA Structure Prediction and Assessment. Bioinformatics 27 (8), 1086–1093. doi:10.1093/bioinformatics/btr093
Cech, T. R., and Steitz, J. A. (2014). The Noncoding RNA Revolution-Trashing Old Rules to Forge New Ones. Cell 157, 77–94. doi:10.1016/j.cell.2014.03.008
Cock, P. J., Antao, T., Chang, J. T., Chapman, B. A., Cox, C. J., Dalke, A., et al. (2009). Biopython: Freely Available Python Tools for Computational Molecular Biology and Bioinformatics. Bioinformatics 25 (11), 1422–1423. doi:10.1093/bioinformatics/btp163
Danaee, P., Rouches, M., Wiley, M., Deng, D., Huang, L., and Hendrix, D. (2018). bpRNA: Large-Scale Automated Annotation and Analysis of RNA Secondary Structure. Nucleic Acids Res. 46, 5381–5394. doi:10.1093/nar/gky285
Das, R., and Baker, D. (2007). Automated De Novo Prediction of Native-like RNA Tertiary Structures. Proc. Natl. Acad. Sci. U S A. 104, 14664–14669. doi:10.1073/pnas.0703836104
Das, R., Karanicolas, J., and Baker, D. (2010). Atomic Accuracy in Predicting and Designing Noncanonical RNA Structure. Nat. Methods 7 (4), 291–294. doi:10.1038/nmeth.1433
Denesyuk, N. A., and Thirumalai, D. (2013). Coarse-grained Model for Predicting RNA Folding Thermodynamics. J. Phys. Chem. B 117, 4901–4911. doi:10.1021/jp401087x
Dethoff, E. A., Chugh, J., Mustoe, A. M., and Al-Hashimi, H. M. (2012). Functional Complexity and Regulation through RNA Dynamics. Nature 482, 322–330. doi:10.1038/nature10885
Dima, R. I., Hyeon, C., and Thirumalai, D. (2005). Extracting Stacking Interaction Parameters for RNA from the Data Set of Native Structures. J. Mol. Biol. 347, 53–69. doi:10.1016/j.jmb.2004.12.012
Doherty, E. A., and Doudna, J. A. (2001). Ribozyme Structures and Mechanisms. Annu. Rev. Biophys. Biomol. Struct. 30, 457–475. doi:10.1146/annurev.biophys.30.1.457
Fernandez-Leiro, R., and Scheres, S. H. (2016). Unravelling Biological Macromolecules with Cryo-Electron Microscopy. Nature 537, 339–346. doi:10.1038/nature19948
Flores, S. C., and Altman, R. B. (2010). Turning Limited Experimental Information into 3D Models of RNA. RNA 16 (9), 1769–1778. doi:10.1261/rna.2112110
Flores, S. C., Bernauer, J., Shin, S., Zhou, R., and Huang, X. (2012). Multiscale Modeling of Macromolecular Biosystems. Brief Bioinform 13 (4), 395–405. doi:10.1093/bib/bbr077
Flores, S. C., Sherman, M. A., Bruns, C. M., Eastman, P., and Altman, R. B. (2011). Fast Flexible Modeling of RNA Structure Using Internal Coordinates. Ieee/acm Trans. Comput. Biol. Bioinform. 8 (5), 1247–1257. doi:10.1109/TCBB.2010.104
Flores, S. C., Wan, Y., Russell, R., and Altman, R. B. (2010). Predicting RNA Structure by Multiple Template Homology Modeling. Pac. Symp. Biocomput 2010, 216–227. doi:10.1142/9789814295291_0024
Gan, H. H., Fera, D., Zorn, J., Shiffeldrim, N., Tang, M., Laserson, U., et al. (2004). RAG: RNA-As-Graphs Database-Cconcepts, Analysis, and Features. Bioinformatics 20 (8), 1285–1291. doi:10.1093/bioinformatics/bth084
Gardner, D. P., Ren, P., Ozer, S., and Gutell, R. R. (2011). Statistical Potentials for Hairpin and Internal Loops Improve the Accuracy of the Predicted RNA Structure. J. Mol. Biol. 413, 473–483. doi:10.1016/j.jmb.2011.08.033
Gendron, P., Lemieux, S., and Major, F. (2001). Quantitative Analysis of Nucleic Acid Three-Dimensional Structures. J. Mol. Biol. 308 (5), 919–936. doi:10.1006/jmbi.2001.4626
Hajdin, C. E., Ding, F., Dokholyan, N. V., and Weeks, K. M. (2010). On the Significance of an RNA Tertiary Structure Prediction. RNA 16, 1340–1349. doi:10.1261/rna.1837410
Huang, B., Du, Y., Zhang, S., Li, W., Wang, J., and Zhang, J. (2020). Computational Prediction of RNA Tertiary Structures Using Machine Learning Methods. Chin. Phys. B 29, 108704. doi:10.1088/1674-1056/abb303
Huang, S. Y., and Zou, X. (2011). Statistical Mechanics-Based Method to Extract Atomic Distance-dependent Potentials from Protein Structures. Proteins 79 (9), 2648–2661. doi:10.1002/prot.23086
Hyeon, C., Dima, R. I., and Thirumalai, D. (2006). Size, Shape, and Flexibility of RNA Structures. J. Chem. Phys. 125 (19), 194905. doi:10.1063/1.2364190
Jian, Y., Wang, X., Qiu, J., Wang, H., Liu, Z., Zhao, Y., et al. (2019). DIRECT: RNA Contact Predictions by Integrating Structural Patterns. BMC Bioinformatics 20 (1), 497. doi:10.1186/s12859-019-3099-4
Jin, L., Tan, Y. L., Wu, Y., Wang, X., Shi, Y. Z., and Tan, Z. J. (2019). Structure Folding of RNA Kissing Complexes in Salt Solutions: Predicting 3D Structure, Stability, and Folding Pathway. RNA 25, 1532–1548. doi:10.1261/rna.071662.119
Jonikas, M. A., Radmer, R. J., Laederach, A., Das, R., Pearlman, S., Herschlag, D., et al. (2009). Coarse-grained Modeling of Large RNA Molecules with Knowledge-Based Potentials and Structural Filters. RNA 15, 189–199. doi:10.1261/rna.1270809
Krokhotin, A., Houlihan, K., and Dokholyan, N. V. (2015). iFoldRNA V2: Folding RNA with Constraints. Bioinformatics 31 (17), 2891–2893. doi:10.1093/bioinformatics/btv221
Kurgan, L., and Zhou, Y. (2011). Machine Learning Models in Protein Bioinformatics. Curr. Protein Pept. Sci. 12 (6), 455. doi:10.2174/138920311796957621
Laing, C., and Schlick, T. (2009). Analysis of Four-Way Junctions in RNA Structures. J. Mol. Biol. 390 (3), 547–559. doi:10.1016/j.jmb.2009.04.084
Leontis, N. B., and Westhof, E. (2001). Geometric Nomenclature and Classification of RNA Base Pairs. RNA 7 (4), 499–512. doi:10.1017/s1355838201002515
Leontis, N. B., and Zirbel, C. L. (2012). “Nonredundant 3D Structure Datasets for RNA Knowledge Extraction and Benchmarking,”. RNA 3D Structure Analysis and Prediction. Editors N Leontis, and E Westhof (Berlin, Heidelberg: Springer), 27, 281–298. doi:10.1007/978-3-642-25740-7_13
Li, J., Zhang, J., Wang, J., Li, W., and Wang, W. (2016). Structure Prediction of RNA Loops with a Probabilistic Approach. Plos Comput. Biol. 12 (8), e1005032. doi:10.1371/journal.pcbi.1005032
Li, J., Zhu, W., Wang, J., Li, W., Gong, S., Zhang, J., et al. (2018). RNA3DCNN: Local and Global Quality Assessments of RNA 3D Structures Using 3D Deep Convolutional Neural Networks. Plos Comput. Biol. 14 (11), e1006514. doi:10.1371/journal.pcbi.1006514
Lu, X. J., Bussemaker, H. J., and Olson, W. K. (2015). DSSR: an Integrated Software Tool for Dissecting the Spatial Structure of RNA. Nucleic Acids Res. 43 (21), e142. doi:10.1093/nar/gkv716
Lu, X. J. (2020). DSSR-enabled Innovative Schematics of 3D Nucleic Acid Structures with PyMOL. Nucleic Acids Res. 48 (13), e74. doi:10.1093/nar/gkaa426
Magnus, M., Antczak, M., Zok, T., Wiedemann, J., Lukasiak, P., Cao, Y., et al. (2020). RNA-puzzles Toolkit: a Computational Resource of RNA 3D Structure Benchmark Datasets, Structure Manipulation, and Evaluation Tools. Nucleic Acids Res. 48 (2), 576–588. doi:10.1093/nar/gkz1108
Masso, M. (2018). All-atom Four-Body Knowledge-Based Statistical Potential to Distinguish Native Tertiary RNA Structures from Nonnative Folds. J. Theor. Biol. 453, 58–67. doi:10.1016/j.jtbi.2018.05.022
Miao, Z., Adamiak, R. W., Antczak, M., Batey, R. T., Becka, A. J., Biesiada, M., et al. (2017). RNA-puzzles Round III: 3D RNA Structure Prediction of Five Riboswitches and One Ribozyme. RNA 23, 655–672. doi:10.1261/rna.060368.116
Miao, Z., and Westhof, E. (2017). RNA Structure: Advances and Assessment of 3D Structure Prediction. Annu. Rev. Biophys. 46, 483–503. doi:10.1146/annurev-biophys-070816-034125
Parisien, M., and Major, F. (2008). The MC-fold and MC-Sym Pipeline Infers RNA Structure from Sequence Data. Nature 452, 51–55. doi:10.1038/nature06684
Parlea, L. G., Sweeney, B. A., Hosseini-Asanjan, M., Zirbel, C. L., and Leontis, N. B. (2016). The RNA 3D Motif Atlas: Computational Methods for Extraction, Organization and Evaluation of RNA Motifs. Methods 103, 99–119. doi:10.1016/j.ymeth.2016.04.025
Pasquali, S., and Derreumaux, P. (2010). HiRE-RNA: a High Resolution Coarse-Grained Energy Model for RNA. J. Phys. Chem. B 114 (37), 11957–11966. doi:10.1021/jp102497y
Popenda, M., Szachniuk, M., Antczak, M., Purzycka, K. J., Lukasiak, P., Bartol, N., et al. (2012). Automated 3D Structure Composition for Large RNAs. Nucleic Acids Res. 40, e112. doi:10.1093/nar/gks339
Rawat, N., and Biswas, P. (2009). Size, Shape, and Flexibility of Proteins and DNA. J. Chem. Phys. 131 (16), 165104. doi:10.1063/1.3251769
Rose, P. W., Prlić, A., Altunkaya, A., Bi, C., Bradley, A. R., Christie, C. H., et al. (2017). The RCSB Protein Data Bank: Integrative View of Protein, Gene and 3D Structural Information. Nucleic Acids Res. 45, D271–D281. doi:10.1093/nar/gkw1000
Rother, M., Rother, K., Puton, T., and Bujnicki, J. M. (2011). ModeRNA: a Tool for Comparative Modeling of RNA 3D Structure. Nucleic Acids Res. 39 (10), 4007–4022. doi:10.1093/nar/gkq1320
Schlick, T., and Pyle, A. M. (2017). Opportunities and Challenges in RNA Structural Modeling and Design. Biophys. J. 113, 225–234. doi:10.1016/j.bpj.2016.12.037
Shi, Y.-Z., Wu, Y.-Y., Wang, F.-H., and Tan, Z.-J. (2014b). RNA Structure Prediction: Progress and Perspective. Chin. Phys. B 23, 078701. doi:10.1088/1674-1056/23/7/078701
Shi, Y. Z., Jin, L., Feng, C. J., Tan, Y. L., and Tan, Z. J. (2018). Predicting 3D Structure and Stability of RNA Pseudoknots in Monovalent and Divalent Ion Solutions. Plos Comput. Biol. 14 (6), e1006222. doi:10.1371/journal.pcbi.1006222
Shi, Y. Z., Jin, L., Wang, F. H., Zhu, X. L., and Tan, Z. J. (2015). Predicting 3D Structure, Flexibility, and Stability of RNA Hairpins in Monovalent and Divalent Ion Solutions. Biophys. J. 109, 2654–2665. doi:10.1016/j.bpj.2015.11.006
Shi, Y. Z., Wang, F. H., Wu, Y. Y., and Tan, Z. J. (2014a). A Coarse-Grained Model with Implicit Salt for RNAs: Predicting 3D Structure, Stability and Salt Effect. J. Chem. Phys. 141, 105102. doi:10.1063/1.4894752
Sloma, M. F., and Mathews, D. H. (2017). Base Pair Probability Estimates Improve the Prediction Accuracy of RNA Non-canonical Base Pairs. Plos Comput. Biol. 13 (11), e1005827. doi:10.1371/journal.pcbi.1005827
Šulc, P., Romano, F., Ouldridge, T. E., Doye, J. P., and Louis, A. A. (2014). A Nucleotide-Level Coarse-Grained Model of RNA. J. Chem. Phys. 140 (23), 235102. doi:10.1063/1.4881424
Tan, Y. L., Feng, C. J., Jin, L., Shi, Y. Z., Zhang, W., and Tan, Z. J. (2019). What Is the Best Reference State for Building Statistical Potentials in RNA 3D Structure Evaluation? RNA 25 (7), 793–812. doi:10.1261/rna.069872.118
Tan, Z., Zhang, W., Shi, Y., and Wang, F. (2015). RNA Folding: Structure Prediction, Folding Kinetics and Ion Electrostatics. Adv. Exp. Med. Biol. 827, 143–183. doi:10.1007/978-94-017-9245-5_11
Tan, Z. J., and Chen, S. J. (2006). Nucleic Acid helix Stability: Effects of Salt Concentration, Cation Valence and Size, and Chain Length. Biophys. J. 90, 1175–1190. doi:10.1529/biophysj.105.070904
Tanner, J. J. (2016). Empirical Power Laws for the Radii of Gyration of Protein Oligomers. Acta Crystallogr. D Struct. Biol. 72, 1119–1129. doi:10.1107/S2059798316013218
Townshend, R. J. L., Eismann, S., Watkins, A. M., Rangan, R., Karelina, M., Das, R., et al. (2021). Geometric Deep Learning of RNA Structure. Science 373 (6558), 1047–1051. doi:10.1126/science.abe5650
Wang, J., Zhao, Y., Zhu, C., and Xiao, Y. (2015). 3dRNAscore: a Distance and Torsion Angle Dependent Evaluation Function of 3D RNA Structures. Nucleic Acids Res. 43 (10), e63. doi:10.1093/nar/gkv141
Wang, K., Jian, Y., Wang, H., Zeng, C., and Zhao, Y. (2018). RBind: Computational Network Method to Predict RNA Binding Sites. Bioinformatics 34 (18), 3131–3136. doi:10.1093/bioinformatics/bty345
Wang, Y., Gong, S., Wang, Z., and Zhang, W. (2016). The Thermodynamics and Kinetics of a Nucleotide Base Pair. J. Chem. Phys. 144 (11), 115101. doi:10.1063/1.4944067
Wang, Y., Liu, T., Yu, T., Tan, Z. J., and Zhang, W. (2020). Salt Effect on Thermodynamics and Kinetics of a Single RNA Base Pair. RNA 26 (4), 470–480. doi:10.1261/rna.073882.119
Westhof, E., and Leontis, N. B. (2021). An RNA-Centric Historical Narrative Around the Protein Data Bank. J. Biol. Chem. 296, 100555. doi:10.1016/j.jbc.2021.100555
Woodson, S. A. (2005). Metal Ions and RNA Folding: a Highly Charged Topic with a Dynamic Future. Curr. Opin. Chem. Biol. 9, 104–109. doi:10.1016/j.cbpa.2005.02.004
Xia, Z., Bell, D. R., Shi, Y., and Ren, P. (2013). RNA 3D Structure Prediction by Using a Coarse-Grained Model and Experimental Data. J. Phys. Chem. B 117 (11), 3135–3144. doi:10.1021/jp400751w
Xiong, P., Wu, R., Zhan, J., and Zhou, Y. (2021). Pairing a High-Resolution Statistical Potential with a Nucleobase-Centric Sampling Algorithm for Improving RNA Model Refinement. Nat. Commun. 12 (1), 2777. doi:10.1038/s41467-021-23100-4
Yan, Y., Wen, Z., Zhang, D., and Huang, S. Y. (2018). Determination of an Effective Scoring Function for RNA-RNA Interactions with a Physics-Based Double-Iterative Method. Nucleic Acids Res. 46 (9), e56. doi:10.1093/nar/gky113
Yu, Z., Yao, Y., Deng, H., and Yi, M. (2019). ANDIS: an Atomic Angle- and Distance-dependent Statistical Potential for Protein Structure Quality Assessment. BMC Bioinformatics 20 (1), 299. doi:10.1186/s12859-019-2898-y
Zhang, B. G., Qiu, H. H., Jiang, J., Liu, J., and Shi, Y. Z. (2019). 3D Structure Stability of the HIV-1 TAR RNA in Ion Solutions: A Coarse-Grained Model Study. J. Chem. Phys. 151 (16), 165101. doi:10.1063/1.5126128
Zhang, D., Li, J., and Chen, S. J. (2021). IsRNA1: De Novo Prediction and Blind Screening of RNA 3D Structures. J. Chem. Theor. Comput 17, 1842–1857. doi:10.1021/acs.jctc.0c01148
Zhang, T., Hu, G., Yang, Y., Wang, J., and Zhou, Y. (2020). All-atom Knowledge-Based Potential for RNA Structure Discrimination Based on the Distance-Scaled Finite Ideal-Gas Reference State. J. Comput. Biol. 27, 856–867. doi:10.1089/cmb.2019.0251
Keywords: RNA 3D structure, statistical analysis, secondary structure motifs, non-canonical base pair, structure evaluation
Citation: Guo Z-H, Yuan L, Tan Y-L, Zhang B-G and Shi Y-Z (2022) RNAStat: An Integrated Tool for Statistical Analysis of RNA 3D Structures. Front. Bioinform. 1:809082. doi: 10.3389/fbinf.2021.809082
Received: 04 November 2021; Accepted: 17 December 2021;
Published: 11 January 2022.
Edited by:
Samuel Coulbourn Flores, Stockholm University, SwedenReviewed by:
Xiaolei Zhu, Anhui Agricultural University, ChinaCopyright © 2022 Guo, Yuan, Tan, Zhang and Shi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ya-Zhou Shi, eXpzaGlAd3R1LmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.