 Sankar Basu
Sankar Basu Simon S. Assaf1
Simon S. Assaf1 Marianne Rooman
Marianne Rooman Fabrizio Pucci
Fabrizio Pucci- 1Computational Biology and Bioinformatics, Université Libre de Bruxelles, Brussels, Belgium
- 2Department of Microbiology, Austosh College, Under University of Calcutta, Kolkata, India
- 3Interuniversity Institute of Bioinformatics in Brussels, Brussels, Belgium
Understanding the role of stability strengths and weaknesses in proteins is a key objective for rationalizing their dynamical and functional properties such as conformational changes, catalytic activity, and protein-protein and protein-ligand interactions. We present BRANEart, a new, fast and accurate method to evaluate the per-residue contributions to the overall stability of membrane proteins. It is based on an extended set of recently introduced statistical potentials derived from membrane protein structures, which better describe the stability properties of this class of proteins than standard potentials derived from globular proteins. We defined a per-residue membrane propensity index from combinations of these potentials, which can be used to identify residues which strongly contribute to the stability of the transmembrane region or which would, on the contrary, be more stable in extramembrane regions, or vice versa. Large-scale application to membrane and globular proteins sets and application to tests cases show excellent agreement with experimental data. BRANEart thus appears as a useful instrument to analyze in detail the overall stability properties of a target membrane protein, to position it relative to the lipid bilayer, and to rationally modify its biophysical characteristics and function. BRANEart can be freely accessed from http://babylone.3bio.ulb.ac.be/BRANEart.
1 Introduction
The broad family of integral membrane proteins provides indispensable components of living cells. Being embedded in biological lipid membranes, these proteins include important scaffolds and functional sites, which bind targeted molecules floating around in the cytosol or in the extracellular medium. They therefore serve as attractive drug targets (Engel and Gaub, 2008; Cournia et al., 2015).
Stability and physico-chemical characteristics greatly vary between transmembrane (TM) and extramembrane (EM) regions of an integral membrane protein due to the difference in their surrounding chemical environment. EM domains in the cytosolic or extracellular medium resemble globular proteins as their surfaces are exposed to water, while residues embedded within the membrane are exposed to lipids and are thus characterized by an elevated hydrophobicity (Leman et al., 2018; Mbaye et al., 2019). Because of this mixed environment, the study of folding and stability of membrane proteins is very challenging and only few tools have been dedicated to investigate them (Lomize et al., 2012; Alford et al., 2015; Postic et al., 2015; Lu et al., 2018). Another reason why membrane proteins are less studied than globular proteins is their lower number of experimental three-dimensional (3D) structures (Shimizu et al., 2018).
Just as for globular proteins, the native structure of a membrane protein corresponds to the global minimum of the free energy landscape in physiological conditions. In general, however, some protein residues or regions, taken individually, are not in their global free energy minimum and correspond to what we call stability weaknesses (Kwasigroch and Rooman, 2006; De Laet et al., 2016; Hou et al., 2021). Indeed, not all residues can simultaneously adopt their lowest free energy conformations, because of the polypeptide chain which constraints the relative motions of the residues. Native conformations can be viewed as the best compromise between conflicting interactions. We define strong and weak residues as residues that are, or are not, optimized for stability properties, respectively. For example, stability weaknesses occur when a contact between two residues in the native structure does not have a favorable free energy contribution and can thus easily break, change conformation, and/or remain flexible. They often play a key role in protein function, interactions, and conformational changes. The concept of stability weaknesses is close to the notion of frustration except that the latter also includes kinetic constraints on fast folding (Ferreiro et al., 2014).
To further advance these issues, we implemented and expanded a series of new statistical mean-force potentials designed to specifically describe the stability properties of membrane proteins, which were first introduced in Mbaye et al. (2019). Based on these potentials, we defined here a per-residue membrane propensity index which predicts whether residues situated in the membrane have a stabilizing contribution or would prefer to be in EM regions, and similarly for residues outside the membrane. This index thus identifies strong and weak residues in EM and TM regions, and can also be used to predict how a protein is, or is not, inserted in the membrane.
It is to be noted that we are studying here relative, EM/TM, strengths and weaknesses, defined from the difference in folding free energy according to whether a residue is in one or the other region. They are different from the strengths and weaknesses defined as residues of which the folding free energy contribution is either highly optimal or not optimal at all in a given environment, as for example computed by the SWOTein web server for globular proteins (Hou et al., 2021). These quantities are related but nevertheless different and give complementary information.
We made our computational tool freely available in the form of a web server called BRANEart, designed to help the scientific community to explore stability strengths and weaknesses in membrane proteins, which is a key element in the study of their stability and function.
2 Materials and Methods
2.1 Protein Structure Data Sets
The membrane protein data set
The set
All selected protein chains in
Finally, we also set up a second data set
The list of all proteins belonging to the data sets
2.2 Statistical Potentials
Statistical potentials are coarse-grained mean-force energy functions derived from frequencies of associations of sequence and structure motifs in a data set of known protein structures. These frequencies are transformed into free energies using the inverse Boltzmann law (Sippl, 1990; Kocher et al., 1994; Dehouck et al., 2006). These potentials depend on the characteristics of the data set from which they are derived. For example, temperature-dependent statistical potentials are obtained from protein data sets of different melting temperatures (Folch et al., 2010; Pucci et al., 2014), and solubility-dependent potentials from proteins of different solubility (Hou et al., 2018).
Here we derived a series of membrane protein potentials from the sets
where kB is the Boltzmann constant, T, the absolute temperature conventionally taken to be room temperature, and F, the relative frequencies computed from a given data set of protein structures. The variables x, y, z stand for any of the four elementary structure or sequence descriptors s, d, t and a: s is an amino acid type, d, the spatial distance between the average side chain geometric centers of two residues separated by at least one residue along the polypeptide chain (Kocher et al., 1994), t, a (ϕ, ψ, ω) backbone torsion angle domain (Rooman et al., 1991), and a, a solvent accessibility bin where the solvent accessibility is defined as the ratio (in %) between the solvent accessible surface area (ASA) of a residue in the structure and in an extended Gly-X-Gly conformation (Kocher et al., 1994).
We constructed two versions of each of the potentials defined by Eqs. 1, 2. In the first, all frequencies F were computed from the structure set
The full list of 19 membrane statistical potentials derived here and their characteristics are given in Supplementary Material S1.
2.3 Per-Residue Folding Free Energies
The statistical potentials
• If the structure descriptors involved in χ are localized on a single residue i (which is the case when e.g., χ = st, sa, sst, ssa), the total contribution is assigned to that residue. For example, the per-residue folding free energy contribution
where sj denotes the amino acid type at position j and ti, the backbone torsion angle domain at position i; L is the sequence length. Note that the torsion and solvent accessibility descriptors t and a are required to be in a sequence windows of maximum 17 residues centered on the sequence descriptor s, thus |i − j| ≤ 8 (Supplementary Material S1). Outside this window,
• If the structure descriptor is localized on two residues i and j (e.g., χ = sd, tt, aa, sds, saa, stt), half of the energy contribution is assigned to each of the two residues. For example, the per-residue folding free energy
where si and sj denote the amino acid types at positions i and j, respectively, and dij is the distance between residues i and j (Supplementary Figure S1A). Similarly, we have for the per-residue folding free energy
where ai and aj denote the solvent accessibility bin of residue i and j, respectively, and sk is the amino acid type of residue k. Both descriptors ai and aj are required to be in a sequence window of maximum 17 residues centered on the sequence descriptor sk, thus |i − k| ≤ 8 and |j − k| ≤ 8 (Supplementary Material S1).
• For the potential χ = ss which does not contain any structure descriptor, an equal amount was allocated to each of the two residues carrying a sequence descriptor s (Supplementary Figure S1B):
For further details, we refer the reader to Supplementary Material S1, S2 and to previous studies (De Laet et al., 2016; Mbaye et al., 2019; Hou et al., 2021).
In a last step, the folding free energy values so obtained for each residue i were smoothed by taking a weighted average over the 5-residue sequence window [i − 2, i + 2] (Mbaye et al., 2019):
The weighting parameters γ and β were chosen to minimize the level of weaknesses in the membrane protein data set (see Section 2.5). For the N- and C-terminal residues, the smoothing was done on the residues present in the [i − 2, i + 2] sequence interval.
In this way, each residue i was tagged with two folding free energy values
2.4 Membrane Propensity Index
We introduced a per-residue membrane propensity index MPri to predict to what extent a residue i in a folded protein corresponds to a stability strength or to a weakness when placed in a given, lipid or aqueous, environment. From a physico-chemical perspective, MPri estimates whether a residue shows a preference for the EM or TM environments, and can be interpreted as an index of stability of a residue within its structural context. It is defined as a linear combination of all folding free energy terms derived in the previous subsection:
where
2.5 Model Training and Parameter Optimization
To estimate the membrane propensity index MPri introduced in Eq. 8, we needed to identify 42 free parameters introduced in Eqs 7, 8: γ, β, αL, αN and 38
To perform the parameter identification on the
We defined the cost function
where the sum is over all residues in the
The performance of the predictor was evaluated using a strict leave-one-out cross validation procedure, in which each protein, in turn, was excluded from
The BRANEart model outputs a continuous membrane propensity score MPr varying approximately between −0.5 and 1.5. Values that are close to one identify residues predicted to be stable in lipid environment, with the most stable having the highest MPr score. In contrast, MPr values close to zero represent residues that are predicted to be stable in aqueous environment, with the most stable having the lowest MPr score. Residues that strongly contribute to the stability of the region to which they belong are considered as stability strengths, and residues that would be more stable elsewhere in the protein are called stability weaknesses.
2.6 EM/TM Residue Classification
The membrane propensity score MPr was also used to set up a binary classifier that predicts whether residues in a membrane protein belong to TM or to EM regions. For that purpose, we transformed the continuous MPr scores into a discrete binary function, where 0 means EM and 1 TM, using an appropriate cutoff value ϕ0 such that a residue i is predicted to be in the TM region if MPri ≥ ϕ0 and in the EM region otherwise. The value of ϕ0 was identified to minimize the difference with OPM assignments.
To evaluate the performance of this classifier, we used the balanced accuracy (BACC) defined as:
where TP and TN mean true positives and true negatives, respectively, and P an N positives and negatives.
In order to check our predictions using a threshold-independent metric, we also computed the AUC, i.e. the area under the receiver operating characteristic (ROC) curve, which plots sensitivity as a function of specificity for different threshold values.
2.7 Normalization of Crystallographic B-Factors
We considered crystallographic B-factors to get information about protein structural flexibility. However, B-factors of different protein structures cannot be compared without proper normalization (Smith et al., 2003). Two types of normalization were considered here.
The B-factors were extracted from the PDB files of all protein structures from
1. The zero-mean-unit-variance technique:
2. The min-max scaling technique:
3 Results and Discussion
3.1 Setting up BRANEart
We set up the BRANEart predictor, which predicts for each residue in a protein structure a membrane protein index MPr, defined as a linear combination of several types of statistical potential values, derived from either EM or TM regions of a set of membrane protein structures (
The MPr index yields a quantitative measure of the stabilizing or destabilizing contribution that a residue has in a specific environment, in other words, whether it acts as a weakness or a strength in that environment. Note that we compared here aqueous and lipid environments, but that this approach can be generalized to the comparison of other environments.
3.2 Large-Scale Application of BRANEart
We applied BRANEart to the membrane protein data set
As seen in Table 1, the mean ⟨MPr⟩ value is close to zero in globular proteins, ⟨MPr⟩ = 0.13, with a low standard deviation of σ = 0.13. This means that the large majority of the residues are stabler in an aqueous than in a lipid environment, which is obviously the case. Residues in membrane proteins have intermediate preferences (⟨MPr⟩ = 0.40) with a much larger σ of 0.37. However, splitting membrane proteins into TM and EM regions clarifies these findings: residues in EM regions clearly prefer to be in an aqueous environment (⟨MPr⟩ = 0.18), while residues inside the membrane prefer a lipid environment (⟨MPr⟩ = 0.74). These excellent results are a first validation of BRANEart.
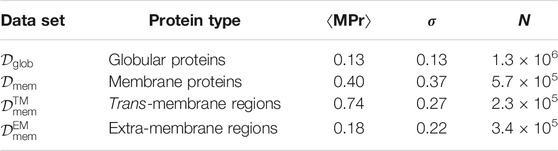
TABLE 1. Mean per-residue index ⟨MPr⟩ and standard deviation σ of the distributions computed for the four protein structure sets considered in this paper; N is the number of residues in each data set.
The MPr distributions are plotted in Figure 1 for each of the four data sets. They all show a unimodal bell-shaped distribution, except the one computed from
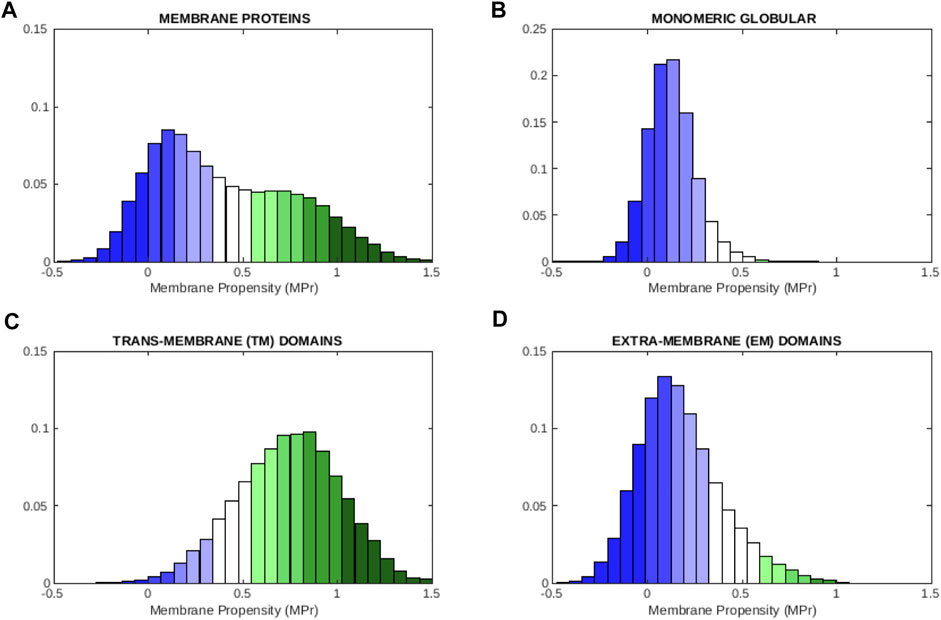
FIGURE 1. Distribution of MPr scores computed from different of protein data sets: (A) membrane proteins
To graphically visualize the level of stability of a residue, we defined stability classes in terms of the ϕ0 threshold value defined in section 2.6 and the standard deviation of the MPr distribution and we colored the residues accordingly, as explained in Table 2. Residues that are strong in aqueous environments but weak in lipid environments are colored in blue. Residues that are strong in the phosholipid bilayer and weak in aqueous solvent are colored in green. Different color graduations were defined to further differentiate between highly stable, stable, moderately stable and mildly stable residues in EM regions, and equivalently in TM regions.

TABLE 2. Classes of MPr scores defined in terms of the classification threshold ϕ0 defined in section 2.6 and the standard deviations σ™ and σEM of the MPr distributions computed from
Interestingly, the amount of strengths and weaknesses in TM and EM regions are almost identical, as computed from the MPr distributions: 7% of the residues are weak in both regions, 75% are strong and the remaining ones are neutral. Without surprise, most residues thus contribute to the stabilization of the regions to which they belong; note that the functional residues are usually among the weak residues.
We also examined how the MPr values change as a function of the solvent accessibility in globular proteins. Therefore, we divided all the residues in
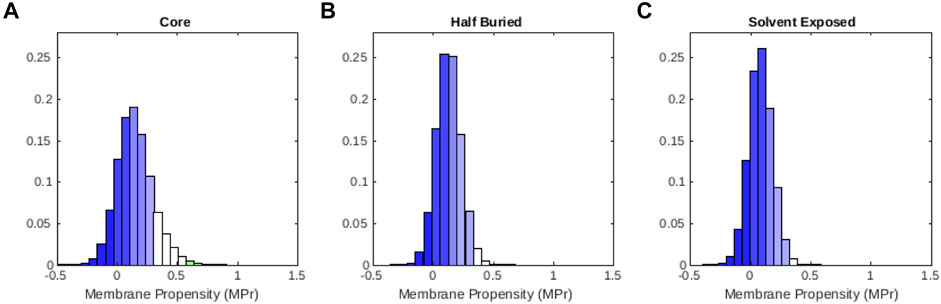
FIGURE 2. Distribution of MPr scores computed from the globular protein set
3.3 Application to α-helical and β-barrel Membrane Proteins
Membrane proteins of which the TM region has an α-helical or a β-barrel conformation exhibit different folding and stability properties (Leman et al., 2018; Mbaye et al., 2019). Indeed, the former type of proteins are essentially localized in the cytoplasmic membranes of eukaryotic and prokaryotic cells and quite rarely in outer membranes, whereas the latter type of proteins is found in outer membranes of Gram-negative bacteria, mitochondria or chloroplasts.
We observe from the distributions depicted in Figures 3A,B that residues pertaining to α-helical folds have a bigger preference for lipid environments than those pertaining to β-barrel folds. Indeed, average ⟨MPr⟩ values are equal to 0.44 (σ = 0.40) and 0.34 (σ = 0.31) for α and β proteins, respectively. If we focus on the TM regions, this tendency is even more marked (Figures 3C,D): ⟨MPr⟩ = 0.78 (σ = 0.28) and 0.63 (σ = 0.23).
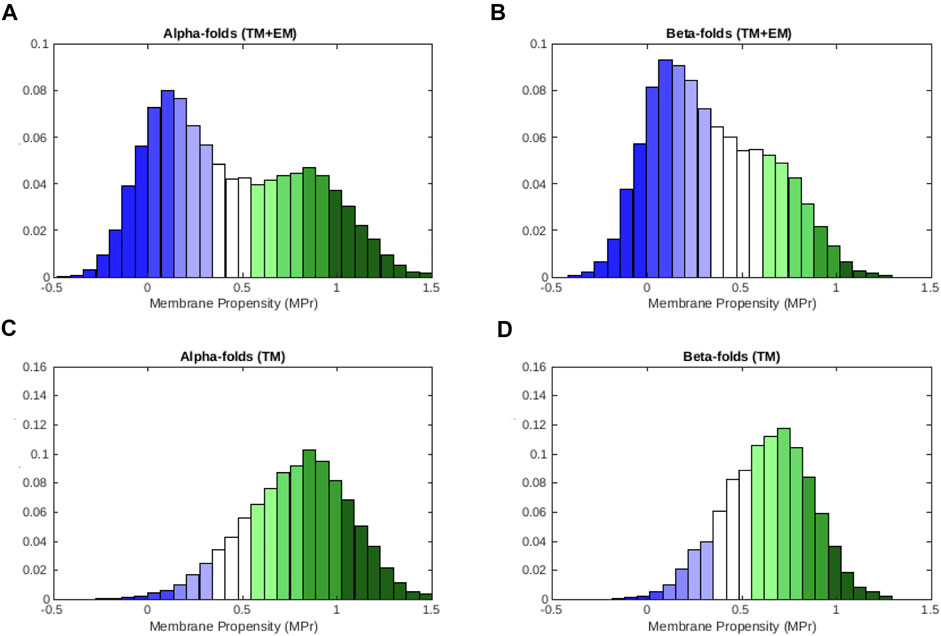
FIGURE 3. MPr distribution computed from the membrane proteins in
These results are in line with a series of facts. First, α-helices are coiled structures stabilized by regularly spaced hydrogen bonds between residues at positions i and i + 4 along the polypeptide chain. In contrast, β-sheets are stabilized by hydrogen bonds between residues from different β-strands, which are usually not close along the chain (Berg et al., 2002). They thus harbour greater geometric variability than α-helices and display greater deviations from ideal backbone bond angles (Touw and Vriend, 2010; Basu et al., 2014). Secondly, β-fold membrane proteins mainly have channel or porin conformations, through which molecules can cross the membrane. The internal faces of these β-barrels are therefore more hydrophilic even though they are in TM regions. They thus often correspond to weaknesses when computed in lipid environments. Finally, the asymmetrical bilayer of phospholipids in which β-barrel proteins are usually inserted have different characteristics than standard phospholipid bilayers. This implies that our statistical potentials generated from TM regions of the whole set of membrane proteins could be less accurate for this kind of proteins (see Mbaye et al. (2019) for further details).
3.4 Protein Embedding in the Membrane
The MPr score can be used not only to identify weak and strong regions but also to predict the protein embedding in the lipid bilayer membrane and more specifically, whether residues are inside or outside the membrane. For that purpose, we transformed the per-residue MPr scores into a binary function, where 0 and 1 mean EM and TM, respectively, using the ϕ0 threshold value defined in section 2.6. We computed the BACC and AUC scores (Eq. 10 and Supplementary Material S4) between the so predicted membrane assignments and OPM annotations, in a strict protein-level cross validation (Section 2.5). The BACC score is equal to 0.87 and the AUC score reaches 0.94.
These scores are very high and we did not expect better results. Indeed, weak residues in TM and EM regions fall in the class of wrong predictions, even though some of them are truly weak and are essential for membrane protein functioning as we will see in the next two subsections.
Note that we used annotations of OPM (Lomize et al., 2012) to train the predictor, and that these annotations are actually also computational predictions. Some discrepancies between the results of our prediction and OPM could therefore be due to errors of OPM rather than of our predictor. Moreover, the discrepancies between OPM and BRANEart predictions are essentially located close to the membrane borders and highlight some structural flexibility in these regions as illustrated in Supplementary Figure S5. We could use our predictor to yield new EM/TM assignments, which would differ from OPM assignments in some places. Alternatively, the MPr score could be used as a feature, in combination with other features, to improve the accuracy of the membrane protein embedding in the membrane (Lomize and Pogozheva, 2013; Postic et al., 2016).
3.5 Probing Membrane Helix Associations
Experimental data about the association energy
where LA and LB are the number of residues in A and B. In the case the complex is a tetramer, the MPr score of the four separate monomers are subtracted from that of the complex, and divided by four. We computed the Pearson correlation coefficient r between
For comparison, we computed the correlation coefficient between
Note that BRANEart was not designed for predicting stability inside the membrane, but rather to detect the protein regions that prefer to be inside or outside the membrane. This result comes thus as an additional, unexpected, application of BRANEart. Note also that the predictions were completely blind as no experimental association energies were used in the model construction.
The list of all proteins with their experimental
3.6 Relating the MPr Score to Biophysical Quantities
This section explores the relationships between the MPr score that identifies stability strength and weakness regions in membrane proteins and several quantities describing structural and biophysical residue properties. We started by computing the correlation between MPr and different hydrophobicity scales for all residues belonging to
Our BRANEart MPr score is only weakly correlated with these various hydrophobicity scales: the Pearson correlation coefficient is always around r =−0.20 (see Supplementary Material S3.1 for details). This is not surprising as the MPr score contains much more information than simple hydrophobicity values and takes into account structural data and their complexity. It classifies TM and EM residues with much higher accuracy and can properly describe the local stability of different conformations of the same membrane protein, as shown in the next subsection.
We also explored the relationship between the BRANEart MPr score and the depth in the lipid bilayer. Our membrane-dependent potentials are based on a series of conformational descriptors and their combinations, but do not use depth in contrast to some other statistical potentials (Senes et al., 2007; Schramm et al., 2012). Despite this, we found a good correlation between the per-residue MPr scores and the absolute values of the Z-depth, defined as the distance between the residue side chain centroids and the plane parallel to the membranes cutting the bilayer into two equal parts. The Pearson correlation coefficient is indeed r =−0.55. This means that the larger the MPr score and thus the larger the preference of a residue for a lipid environment, the deeper it is embedded in the lipid membrane. Note that the relationship is non-linear as can be seen from Supplementary Figure S3. This suggests the use of non-linear functions of our statistical potentials to predict the Z-depth more precisely.
Finally, we investigated whether MPr scores are correlated with residue flexibility, estimated from crystallographic B-factors that indicate the relative vibrational motion of atoms in different parts of the structure. To be able to compare the B-factors of different proteins, they need to be properly normalized. We used two normalization techniques, the zero-mean-unit-variance and min-max-scaling techniques, described in Methods subsection 2.7. We computed the Pearson correlation coefficient between the MPr scores and the per-residue normalized B-factors of all residues in
This characteristic anti-correlation can be explained by the opposite trends of MPr scores and B-factors. For the B-factor, the higher the value, the higher the degree of flexibility. For the MPr score, a higher value means a higher preference for lipid environments that restrict atomic movements. The anti-correlation thus means that EM regions are more flexible than TM regions. The low value of the correlation is due to the fact that the MPr score contains information about stability across different environments, which is much more than information about flexibility. Note, moreover, that stability and MPr are related to free energy in which flexibility is entropically favorable.
3.7 Application to Leucine Transporters
In general, membrane proteins undergo conformational changes to accomplish their biological functions (Latorraca et al., 2017; Chataigner et al., 2020). Residues that are at the basis of these large-scale movements are often stability weaknesses (Hou et al., 2021) or in other words, frustrated (Ferreiro et al., 2014). Indeed, functional constraints prevent them to adopt conformations with high stability contributions. Here we show how we can use the MPr score to gain insight into the functional role of these residues. Note that, to our knowledge, no other method employs dedicated energy functions to study frustration in membrane proteins.
As an example, we analyzed leucine transporter (LeuT), a bacterial homodimeric protein containing twelve transmembrane helices, which uses the electrochemical potential of sodium ions to transport leucine from outside to inside the cell (Penmatsa and Gouaux, 2014). Both the substrate-free outward-open LeuT structure and the inward-open apo conformation have been resolved via X-ray crystallography (Krishnamurthy and Gouaux, 2012) (PDB codes: 3tt1 and 3tt3, respectively).
We analyzed the difference in membrane propensity index (ΔMPr) between the two conformations in Figure 4A. We can easily identify the two regions with highest ΔMPr in absolute value: the transmembrane helix 1 (TM1) and the extracellular helix 4 (EL4), which are key elements that allow the opening and closing of the intracellular and extracellular gates, respectively (Krishnamurthy and Gouaux, 2012). We thus focused on these two regions and analyzed their MPr values in both outward and inward conformations (Figures 4B–E).
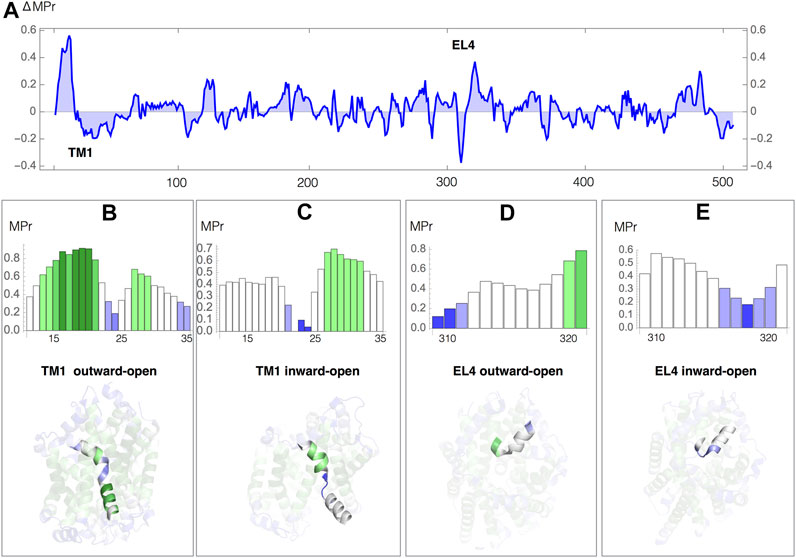
FIGURE 4. Leu transporter: MPr score in the outward-open (PDB code 3tt1) and inward-open (PDB code 3tt3) conformations (A) Difference in MPr score (ΔMpr) between the two conformations as a function of the sequence position; the positions of the helices TM1 and EL4 are indicated. (B–E) MPr of the TM1 and EL4 helices as a function of the sequence position, and TM1 and EL4 helices embedded in the 3D structure, colored according to the code defined in Table 2; (B) TM1, outward-open conformation; (C) TM1, inward-open conformation; (D) EL4, outward-open conformation; (D) EL4, inward-open conformation.
The transmembrane segment TM1 (residues 11–35) is a kinked helix formed by two helical regions, TM1a (residues 11–21) and TM1b (residues 25–35), separated by a hinge fragment (residues 22–24). TM1a undergoes a large movement upon opening of the intracellular gate (Figures 4B,C). Despite the fact that the whole TM1 is inside the lipid membrane, the hinge residues have a very low MPr score in both conformations. They are thus stability weaknesses, relating them to their functional role in facilitating the opening and closing of the gate, as confirmed by experiments (Krishnamurthy and Gouaux, 2012). The MPr of TM1a changes significantly during the (closed → open) conformational transition. In the inward-open conformation, when the intracellular gate is open, TM1 is only found weakly stable (Figure 4C), whereas in the outward-open conformation, when the intracellular gate is closed, TM1a establishes stabilizing contacts with TM5 and TM7 which is reflected by high MPr values (Figure 4B).
In a similar way, we identified the strong and weak residues in the outer membrane environment related to the conformational movement that EL4 undergoes while opening and closing of the extracellular gate. EL4 (residues 309–321) is a helix of which the last residues (319–321) are unwound and called hinge; its position appears displaced in the outward-open and inward-open conformations. In the outward-open conformation, when the extracellular gate is open, the N-terminal EL4 residues 309–311 are stable in aqueous environment but the hinge residues are weak (Figure 4D). While closing the gate, EL4 changes in terms of its stability profile, with its N-terminus becoming weak and its C-terminus and the hinge getting more stable (Figure 4C) due to extensive contacts including hydrophobic interactions and a hydrogen bond between Ala 319 and Asp 401 in TM10.
This example illustrates how we can use BRANEart to identify in a simple way residues in membrane proteins that are not optimized for their stability, which is particularly relevant for functional residues.
3.8 Application to Human Phospholamban
As a second test case, we studied the stability of human phospholamban (PLB), a protein anchored into the cardiac sarcoplasmic reticulum membrane, which is essential to myocardial contractility (Koss and Kranias, 1996). Let us start with the analysis of the MPr index as a function of the sequence, computed from the standard pentameric form of PLB (PDB code 1zll). As shown in Figure 5A, the two domains of the protein are easy to identify: the EM part (residues 1–22) has low MPr values and thus the propensity to be stable in water, and the TM part (residues 23–52) has overall much higher MPr values indicating its stability in lipids. A closer look shows that the TM region must be divided in two: the C-terminal part (residues 33–52) is apolar, contains the well known motif LxxIxxx (Mravic et al., 2019) and has high MPr values indicating its stability in lipids, whereas the N-terminus (residues 23–32) is a TM weakness.
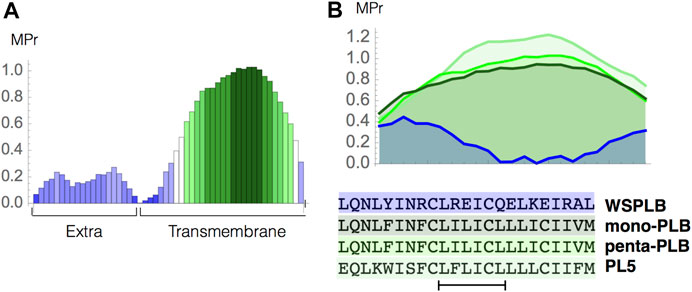
FIGURE 5. Phospholamban case study. (A) MPr values computed for the pentameric structure of the full-length phospholambam (PDB code 1zll); (B) amino acid sequences and MPr values for the TM fragment (residues 28–50) of four different PLB structures: PLB monomer (light green, PDB code 1zll), PLB pentamer (green, PDB code 1zll), redesigned pentamer PL5 (dark green, PDB code 6mqu) and water soluble PLB tetramer (blue, PDB code 1yod).
Note that the latter part connects the EM and TM regions and is very close to the water-membrane interface. It thus probably changes dynamically from the lipid TM to the aqueous EM environment and vice versa. This prediction is in perfect agreement with both NMR data (Oxenoid and Chou, 2005), molecular dynamics simulations (Kim et al., 2009; Mravic et al., 2019) and mutagenesis data (Fujii et al., 1989; Simmerman et al., 1996), which identified this region as highly dynamical, unimportant for the stabilization of the structure, but crucial for modulating functional interactions of PLB.
We also analyzed the MPr index of the different types of PLB structures. More specifically, we compared the stability of the wild-type monomeric and pentameric forms (PDB code 1zll) (Oxenoid and Chou, 2005), of a highly stable designed pentameric variant (PL5, PDB code 6mqu) (Mravic et al., 2019) and of a water-soluble tetrameric variant (WSPLB, PDB code 1yod) (Slovic et al., 2005). The MPr values of the TM region (residues 28–50) in all these structures are reported in Figure 5B. The TM region of the monomeric form shows a clear preference for the lipid environment with all residues having an MPr value bigger than ∼ 0.5. The pentamer form is predicted to be more stable than the monomer form since inter-chain interactions between the TM fragments strongly stabilize this structural assembly. Note that both the monomer and pentamer forms do exist in phospholipid bilayers even though the latter is dominant. Such oligomer equilibrium is dynamic and can change due to mutations or phosphorylation (Cornea et al., 1997).
The PL5 variant has the largest MPr values and is the most stable form in the lipid environment. This is again in perfect agreement with experiments; in redesigning this protein fragment, the polar residues have been substituted by apolar residues to get stronger interchain interactions driven by apolar sidechain packing and an increased stability of the overall fold (Mravic et al., 2019). Finally, the redesigned, truncated, water-soluble variant has extremely low propensities to be in a lipid environment, as can easily be seen from our predictions.
3.9 Application in the Evaluation of Docked Complexes
BRANEart has recently been applied in the context of COVID-19 research to cross-check the correctness of docked peptide-protein complexes between angiotensin II and its cognate receptor, angiotensin converting enzyme 2 (ACE2) (Basu et al., 2021). The survey was performed by comparing the MPr profiles of the peptide ligand and the docking sites in the top-ranked docked poses returned by ClusPro 2.0 (Desta et al., 2020). BRANEart unanimously selected peptides mapping to ACE2’s deep pocket buried in the hydrophobic core. This result shows the usefulness of BRANEart to analyze hydrophobicity compatibility between protein and/or peptide partners.
3.10 BRANEart Web Server: Features and Functionalities
We implemented our structure-based prediction method into the easy-to-use BRANEart web server. To run a query, the user first chooses the structure of a membrane protein by either providing its 4-letter code which is then automatically retrieved if available in the PDB, or by uploading a protein structure file in PDB format. The user is then asked to select the relevant chain(s) on which BRANEart has to perform the predictions, taking into account the other chains present in the structure file. Finally, the computation starts.
The main BRANEart output is the MPr score for each amino acid residue in the selected chain(s) of the target protein structure. In addition to returning these values as a downloadable text file, the web server also displays a table with the MPr of each residue of the targeted protein chain(s), colored according to the code defined in Table 2. In addition, BRANEart provides a multi-featured browser tool to visualize the protein 3D structure, where each residue is colored according to its MPr score. It has several advanced visualization functionalities. More precisely, it is possible to:
• hide/show specific chain(s) in the displayed structure, and zoom in and out by left dragging the mouse.
• switch to a full screen visualization mode and take a “.png” snapshot of the visualized structures.
• select a residue of interest by double clicking on it. The neighboring residues are then displayed in “ball and sticks”.
• download a PyMol session file (.pml) (The PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC) to switch to the PyMol representation of the 3D structure.
For further technical details and information about the web server, we refer to the BRANEart help page.
4 Conclusion
We presented BRANEart, a computational method to identify the stability strengths and weaknesses in membrane proteins. Extending and combining the newly developed membrane statistical potentials introduced in Mbaye et al. (2019), we defined a MPr score that quantifies whether a residue is stable in a lipid or aqueous environment. Large-scale predictions and applications to test cases show BRANEart’s ability to correctly identify regions in their respective environment that strongly contribute to the stabilization of their host protein, and residues that have instead low impact on stabilization but have functional roles.
Note that our approach can be extended to identify strengths and weaknesses in other environments than the membrane, for example in hot and cold environments using temperature-dependent potentials (Folch et al., 2010).
We additionally provided a user friendly web server that, on the basis of the 3D structure of the target membrane protein, computes the MPr score for each residue in the input structure. Visualization tools are provided which simplify the understanding and interpretation of BRANEart results.
To our knowledge, BRANEart is the first accessible, fast and accurate tool that use dedicated membrane protein potentials to identify stability strengths and weaknesses in membrane proteins. The use of such mean force potentials drastically simplifies the analysis of membrane protein stability, since the effect of the lipid environment is considered in an implicit manner. BRANEart can be used in a wide series of applications that range from the analysis of the conformational changes in membrane proteins, the embedding of proteins in the lipid membrane, to the identification of residues to target for the rational modification of biophysical characteristics of membrane proteins.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Author Contributions
Conceptualization, FP and MR; formal analysis and investigation SB and FP; methodology and validation, SB, SA, FT, FP, and MR; writing—original draft preparation, SB, MR, and FP; writing—review and editing SB, MR, and FP. All authors have read and agreed to the published version of the manuscript.
Funding
SB, SA, and FP are or were Postdoctoral researcher at the Belgian F.R.S.-FNRS Fund for Scientific Research. MR is Research Director at the F.R.S.-FNRS. This work is financially supported by PDR and PER projects of the same fund.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
SB wants to convey his sincere gratitude to Dr. Abhirup Bandyopadhyay, Institute De Neurosciences Des Systems, Aix-Marseille University, France for his constant motivation and helpful discussions during the work.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fbinf.2021.742843/full#supplementary-material
References
Alford, R. F., Koehler Leman, J., Weitzner, B. D., Duran, A. M., Tilley, D. C., Elazar, A., et al. (2015). An Integrated Framework Advancing Membrane Protein Modeling and Design. Plos Comput. Biol. 11, e1004398. doi:10.1371/journal.pcbi.1004398
Basu, S., Bhattacharyya, D., and Banerjee, R. (2014). Applications of Complementarity Plot in Error Detection and Structure Validation of Proteins. Indian J. Biochem. Biophys. 51, 188–200.
Basu, S., Chakravarty, D., Bhattacharyya, D., Saha, P., and Patra, H. K. (2021). Plausible Blockers of Spike Rbd in Sars-Cov2-Molecular Design and Underlying Interaction Dynamics from High-Level Structural Descriptors. J. Mol. Model. 27, 191. doi:10.1007/s00894-021-04779-0
Berg, J., Tymoczko, J., and Stryer, L. (2002). Secondary Structure: Polypeptide Chains Can Fold into Regular Structures Such as the Alpha Helix, the Beta Sheet, and Turns and Loops. Biochemistry, W.H. Freeman 5th edn, New York, United States.
Berman, H. M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T. N., Weissig, H., et al. (2000). The Protein Data Bank. Nucleic Acids Res. 28, 235–242. doi:10.1093/nar/28.1.235
Chataigner, L. M. P., Leloup, N., and Janssen, B. J. C. (2020). Structural Perspectives on Extracellular Recognition and Conformational Changes of Several Type-I Transmembrane Receptors. Front. Mol. Biosci. 7, 129. doi:10.3389/fmolb.2020.00129
Cornea, R. L., Jones, L. R., Autry, J. M., and Thomas, D. D. (1997). Mutation and Phosphorylation Change the Oligomeric Structure of Phospholamban in Lipid Bilayers. Biochemistry 36, 2960–2967. doi:10.1021/bi961955q
Cournia, Z., Allen, T. W., Andricioaei, I., Antonny, B., Baum, D., Brannigan, G., et al. (2015). Membrane Protein Structure, Function, and Dynamics: a Perspective from Experiments and Theory. J. Membr. Biol. 248, 611–640. doi:10.1007/s00232-015-9802-0
De Laet, M., Gilis, D., and Rooman, M. (2016). Stability Strengths and Weaknesses in Protein Structures Detected by Statistical Potentials: Application to Bovine Seminal Ribonuclease. Proteins 84, 143–158. doi:10.1002/prot.24962
Deber, C. M., Wang, C., Liu, L. P., Prior, A. S., Agrawal, S., Muskat, B. L., et al. (2001). TM Finder: a Prediction Program for Transmembrane Protein Segments Using a Combination of Hydrophobicity and Nonpolar Phase Helicity Scales. Protein Sci. 10, 212–219. doi:10.1110/ps.30301
Dehouck, Y., Gilis, D., and Rooman, M. (2006). A New Generation of Statistical Potentials for Proteins. Biophys. J. 90, 4010–4017. doi:10.1529/biophysj.105.079434
Desta, I. T., Porter, K. A., Xia, B., Kozakov, D., and Vajda, S. (2020). Performance and its Limits in Rigid Body Protein-Protein Docking. Structure 28, 1071. doi:10.1016/j.str.2020.06.006
Engel, A., and Gaub, H. E. (2008). Structure and Mechanics of Membrane Proteins. Annu. Rev. Biochem. 77, 127–148. doi:10.1146/annurev.biochem.77.062706.154450
Engelman, D. M., Steitz, T. A., and Goldman, A. (1986). Identifying Nonpolar Transbilayer Helices in Amino Acid Sequences of Membrane Proteins. Annu. Rev. Biophys. Biophys. Chem. 15, 321–353. doi:10.1146/annurev.bb.15.060186.001541
Ferreiro, D. U., Komives, E. A., and Wolynes, P. G. (2014). Frustration in Biomolecules. Q. Rev. Biophys. 47, 285–363. doi:10.1017/S0033583514000092
Folch, B., Dehouck, Y., and Rooman, M. (2010). Thermo- and Mesostabilizing Protein Interactions Identified by Temperature-dependent Statistical Potentials. Biophys. J. 98, 667–677. doi:10.1016/j.bpj.2009.10.050
Fujii, J., Maruyama, K., Tada, M., and MacLennan, D. H. (1989). Expression and Site-specific Mutagenesis of Phospholamban. Studies of Residues Involved in Phosphorylation and Pentamer Formation. J. Biol. Chem. 264, 12950–12955. doi:10.1016/s0021-9258(18)51579-9
Hessa, T., Kim, H., Bihlmaier, K., Lundin, C., Boekel, J., Andersson, H., et al. (2005). Recognition of Transmembrane Helices by the Endoplasmic Reticulum Translocon. Nature 433, 377–381. doi:10.1038/nature03216
Hou, Q., Bourgeas, R., Pucci, F., and Rooman, M. (2018). Computational Analysis of the Amino Acid Interactions that Promote or Decrease Protein Solubility. Sci. Rep. 8, 14661. doi:10.1038/s41598-018-32988-w
Hou, Q., Pucci, F., Ancien, F., Kwasigroch, J. M., Bourgeas, R., and Rooman, M. (2021). SWOTein: A Structure-Based Approach to Predict Stability Strengths and Weaknesses of prOTEINs. Bioinformatics 37, 1963–1971. doi:10.1093/bioinformatics/btab034
Kim, T., Lee, J., and Im, W. (2009). Molecular Dynamics Studies on Structure and Dynamics of Phospholamban Monomer and Pentamer in Membranes. Proteins 76, 86–98. doi:10.1002/prot.22322
Kocher, J. P., Rooman, M. J., and Wodak, S. J. (1994). Factors Influencing the Ability of Knowledge-Based Potentials to Identify Native Sequence-Structure Matches. J. Mol. Biol. 235, 1598–1613. doi:10.1006/jmbi.1994.1109
Koehler, J., Woetzel, N., Staritzbichler, R., Sanders, C. R., and Meiler, J. (2009). A Unified Hydrophobicity Scale for Multispan Membrane Proteins. Proteins 76, 13–29. doi:10.1002/prot.22315
Koehler Leman, J., Bonneau, R., and Ulmschneider, M. B. (2018). Statistically Derived Asymmetric Membrane Potentials from α-helical and β-barrel Membrane Proteins. Sci. Rep. 8, 4446. doi:10.1038/s41598-018-22476-6
Koss, K. L., and Kranias, E. G. (1996). Phospholamban: a Prominent Regulator of Myocardial Contractility. Circ. Res. 79, 1059–1063. doi:10.1161/01.res.79.6.1059
Krishnamurthy, H., and Gouaux, E. (2012). X-ray Structures of LeuT in Substrate-free Outward-Open and Apo Inward-Open States. Nature 481, 469–474. doi:10.1038/nature10737
Krissinel, E., and Henrick, K. (2007). Inference of Macromolecular Assemblies from Crystalline State. J. Mol. Biol. 372, 774–797. doi:10.1016/j.jmb.2007.05.022
Kwasigroch, J. M., and Rooman, M. (2006). Prelude and Fugue, Predicting Local Protein Structure, Early Folding Regions and Structural Weaknesses. Bioinformatics 22, 1800–1802. doi:10.1093/bioinformatics/btl176
Latorraca, N. R., Venkatakrishnan, A. J., and Dror, R. O. (2017). GPCR Dynamics: Structures in Motion. Chem. Rev. 117, 139–155. doi:10.1021/acs.chemrev.6b00177
Lomize, A. L., Schnitzer, K. A., and Pogozheva, I. D. (2020). TMPfold: A Web Tool for Predicting Stability of Transmembrane α-Helix Association. J. Mol. Biol. 432, 3388–3394. doi:10.1016/j.jmb.2019.10.024
Lomize, A. L., and Pogozheva, I. D. (2013). “Solvation Models and Computational Prediction of Orientations of Peptides and Proteins in Membranes,” in Membrane Proteins (Clifton, NJ: Springer), 125–142. doi:10.1007/978-1-62703-583-5_7
Lomize, M. A., Pogozheva, I. D., Joo, H., Mosberg, H. I., and Lomize, A. L. (2012). OPM Database and PPM Web Server: Resources for Positioning of Proteins in Membranes. Nucleic Acids Res. 40, D370–D376. doi:10.1093/nar/gkr703
Lu, W., Schafer, N. P., and Wolynes, P. G. (2018). Energy Landscape Underlying Spontaneous Insertion and Folding of an Alpha-Helical Transmembrane Protein into a Bilayer. Nat. Commun. 9, 4949. doi:10.1038/s41467-018-07320-9
Mbaye, M. N., Hou, Q., Basu, S., Teheux, F., Pucci, F., and Rooman, M. (2019). A Comprehensive Computational Study of Amino Acid Interactions in Membrane Proteins. Sci. Rep. 9, 12043. doi:10.1038/s41598-019-48541-2
Moon, C. P., and Fleming, K. G. (2011). Side-chain Hydrophobicity Scale Derived from Transmembrane Protein Folding into Lipid Bilayers. Proc. Natl. Acad. Sci. U S A. 108, 10174–10177. doi:10.1073/pnas.1103979108
Mravic, M., Thomaston, J. L., Tucker, M., Solomon, P. E., Liu, L., and DeGrado, W. F. (2019). Packing of Apolar Side Chains Enables Accurate Design of Highly Stable Membrane Proteins. Science 363, 1418–1423. doi:10.1126/science.aav7541
Oxenoid, K., and Chou, J. J. (2005). The Structure of Phospholamban Pentamer Reveals a Channel-like Architecture in Membranes. Proc. Natl. Acad. Sci. U S A. 102, 10870–10875. doi:10.1073/pnas.0504920102
Penmatsa, A., and Gouaux, E. (2014). How LeuT Shapes Our Understanding of the Mechanisms of Sodium-Coupled Neurotransmitter Transporters. J. Physiol. 592, 863–869. doi:10.1113/jphysiol.2013.259051
Postic, G., Ghouzam, Y., and Gelly, J. C. (2015). An Empirical Energy Function for Structural Assessment of Protein Transmembrane Domains. Biochimie 115, 155–161. doi:10.1016/j.biochi.2015.05.018
Postic, G., Ghouzam, Y., and Gelly, J. C. (2016). OREMPRO Web Server: Orientation and Assessment of Atomistic and Coarse-Grained Structures of Membrane Proteins. Bioinformatics 32, 2548–2550. doi:10.1093/bioinformatics/btw208
Pucci, F., Dhanani, M., Dehouck, Y., and Rooman, M. (2014). Protein Thermostability Prediction within Homologous Families Using Temperature-dependent Statistical Potentials. PloS one 9, e91659. doi:10.1371/journal.pone.0091659
Rooman, M. J., Kocher, J. P., and Wodak, S. J. (1991). Prediction of Protein Backbone Conformation Based on Seven Structure Assignments. Influence of Local Interactions. J. Mol. Biol. 221, 961–979. doi:10.1016/0022-2836(91)80186-x
Schramm, C. A., Hannigan, B. T., Donald, J. E., Keasar, C., Saven, J. G., DeGrado, W. F., et al. (2012). Knowledge-based Potential for Positioning Membrane-Associated Structures and Assessing Residue-specific Energetic Contributions. Structure 20, 924–935. doi:10.1016/j.str.2012.03.016
Senes, A., Chadi, D. C., Law, P. B., Walters, R. F., Nanda, V., and DeGrado, W. F. (2007). E(z), a Depth-dependent Potential for Assessing the Energies of Insertion of Amino Acid Side-Chains into Membranes: Derivation and Applications to Determining the Orientation of Transmembrane and Interfacial Helices. J. Mol. Biol. 366, 436–448. doi:10.1016/j.jmb.2006.09.020
Shimizu, K., Cao, W., Saad, G., Shoji, M., and Terada, T. (2018). Comparative Analysis of Membrane Protein Structure Databases. Biochim. Biophys. Acta Biomembr 1860, 1077–1091. doi:10.1016/j.bbamem.2018.01.005
Simmerman, H. K., Kobayashi, Y. M., Autry, J. M., and Jones, L. R. (1996). A Leucine Zipper Stabilizes the Pentameric Membrane Domain of Phospholamban and Forms a Coiled-Coil Pore Structure. J. Biol. Chem. 271, 5941–5946. doi:10.1074/jbc.271.10.5941
Sippl, M. J. (1990). Calculation of Conformational Ensembles from Potentials of Mean Force. An Approach to the Knowledge-Based Prediction of Local Structures in Globular Proteins. J. Mol. Biol. 213, 859–883. doi:10.1016/s0022-2836(05)80269-4
Slovic, A. M., Stayrook, S. E., North, B., and DeGrado, W. F. (2005). X-ray Structure of a Water-Soluble Analog of the Membrane Protein Phospholamban: Sequence Determinants Defining the Topology of Tetrameric and Pentameric Coiled Coils. J. Mol. Biol. 348, 777–787. doi:10.1016/j.jmb.2005.02.040
Smith, D. K., Radivojac, P., Obradovic, Z., Dunker, A. K., and Zhu, G. (2003). Improved Amino Acid Flexibility Parameters. Protein Sci. 12, 1060–1072. doi:10.1110/ps.0236203
Tian, W., Naveed, H., Lin, M., and Liang, J. (2020). GeTFEP: A General Transfer Free Energy Profile of Transmembrane Proteins. Protein Sci. 29, 469–479. doi:10.1002/pro.3763
Touw, W. G., and Vriend, G. (2014). BDB: Databank of Pdb Files with Consistent B-Factors. Protein Eng. Des. Sel 27, 457–462. doi:10.1093/protein/gzu044
Touw, W. G., and Vriend, G. (2010). On the Complexity of Engh and Huber Refinement Restraints: the Angle τ as Example. Acta Crystallogr. D Biol. Crystallogr. 66, 1341–1350. doi:10.1107/S0907444910040928
Wang, G., and Dunbrack, R. L. (2003). PISCES: a Protein Sequence Culling Server. Bioinformatics 19, 1589–1591. doi:10.1093/bioinformatics/btg224
Keywords: computational prediction, membrane protein structure and stability, folding free energy, frustration, statistical potentials, Leucine Transporters, Human Phospholamban
Citation: Basu S, Assaf SS, Teheux F, Rooman M and Pucci F (2021) BRANEart: Identify Stability Strength and Weakness Regions in Membrane Proteins. Front. Bioinform. 1:742843. doi: 10.3389/fbinf.2021.742843
Received: 16 July 2021; Accepted: 03 November 2021;
Published: 02 December 2021.
Edited by:
Igor N. Berezovsky, Bioinformatics Institute (A∗STAR), SingaporeReviewed by:
Sai Ganesan, University of California, San Francisco, United StatesToni Giorgino, National Research Council (CNR), Italy
Copyright © 2021 Basu, Assaf, Teheux, Rooman and Pucci. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marianne Rooman, TWFyaWFubmUuUm9vbWFuQHVsYi5iZQ==; Fabrizio Pucci, RmFicml6aW8uUHVjY2lAdWxiLmJl
†These authors have contributed equally to this work