 Yang Liu
Yang Liu You Wu
You Wu Xiaoke Shen1
Xiaoke Shen1 Lei Xie
Lei Xie- 1Department of Computer Science, Hunter College, The City University of New York, New York, NY, United States
- 2The Graduate Center, The City University of New York, New York, NY, United States
- 3Feil Family Brain and Mind Research Institute, Weill Cornell Medicine, Cornell University, New York, NY, United States
The life-threatening disease COVID-19 has inspired significant efforts to discover novel therapeutic agents through repurposing of existing drugs. Although multi-targeted (polypharmacological) therapies are recognized as the most efficient approach to system diseases such as COVID-19, computational multi-targeted compound screening has been limited by the scarcity of high-quality experimental data and difficulties in extracting information from molecules. This study introduces MolGNN, a new deep learning model for molecular property prediction. MolGNN applies a graph neural network to computational learning of chemical molecule embedding. Comparing to state-of-the-art approaches heavily relying on labeled experimental data, our method achieves equivalent or superior prediction performance without manual labels in the pretraining stage, and excellent performance on data with only a few labels. Our results indicate that MolGNN is robust to scarce training data, and hence a powerful few-shot learning tool. MolGNN predicted several multi-targeted molecules against both human Janus kinases and the SARS-CoV-2 main protease, which are preferential targets for drugs aiming, respectively, at alleviating cytokine storm COVID-19 symptoms and suppressing viral replication. We also predicted molecules potentially inhibiting cell death induced by SARS-CoV-2. Several of MolGNN top predictions are supported by existing experimental and clinical evidence, demonstrating the potential value of our method.
Introduction
The global COVID-19 pandemic caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) resulted in over 120 million infected patients and 2.6 million deaths worldwide by March 2021, mostly due to severe acute respiratory syndrome. Although COVID-19 vaccines offer a path to control the spread of coronavirus, there remains the challenge of creating widely available vaccines and rapidly developing updates to match fast-emerging new SARS-CoV-2 strains. Meanwhile, the discovery of novel drugs and therapies against the SARS-CoV-2 infection is critical for tackling the disease. However, discovery and development of effective antiviral therapies can be costly and time-consuming. For this reason, significant efforts have been made toward repurposing drugs for COVID-19 treatment (Beigel et al., 2020; Zhou et al., 2020; Galindez et al., 2021) as a time- and resource-saving alternative to de novo drug discovery (Chong and Sullivan, 2007; Jin and Wong, 2014).
Conventional target-based drug repurposing approaches have focused on the reuse of an existing drug against another single target. However, COVID-19 is a systemic disease caused by the direct effect of the viral infection and overreacted host inflammatory response. Thus, polypharmacological therapies are arguably more efficient by targeting multiple disease-associated viral genes (Paolini et al., 2006; Apsel et al., 2008; Hopkins, 2008; Hopkins, 2009). Identifying proper target combinations and designing effective multi-targeting agents require approaches such as in silico drug design, which provides a powerful tool to speed up chemical compound screening (Chaudhari et al., 2017; Peng et al., 2018; Balasubramaniam and Reis, 2020; González-Durruthy et al., 2020).
Machine learning techniques have been applied to various tasks in drug discovery, such as molecular property prediction (Duvenaud et al., 2015; Wu et al., 2018; Ayed et al., 2019) and drug–target interaction prediction (Yamanishi et al., 2010; Chen et al., 2012; Cai et al., 2021). One challenge for computational drug discovery is to effectively learn accurate and informative representation of molecules. Most traditional machine learning methods focus on feature engineering for molecular representation. However, recent advances in machine learning, especially deep neural networks, have played a significant role in virtual screening and fast development of new approaches to representation learning of molecular properties (Gilmer et al., 2017; Hu et al., 2019; Zheng et al., 2019). Among the new deep learning architectures, the graph neural network (GNN) has become a powerful tool for modeling molecule-related tasks. Although various studies have reported promising results (Kipf and Welling, 2017; Xu et al., 2019; Ying et al., 2019), computational drug discovery still faces the problem of insufficient labeled data precluding generalized predictive models. For example, the state-of-the-art GNN method ContextPred trains the model in a supervised manner based on experimentally determined labels, which are not available for many machine learning tasks (Hu et al., 2019).
To address the above issues, we have developed MolGNN as a novel method that is able to 1) leverage the power of the graph neural network with a pretraining process to learn molecular embedding, with molecules represented by a heterogeneous graph structure, with atoms as nodes and bonds as edges, and 2) employ a motif self-learning mechanism to encode information extracted from frequent subgraph structures, such as functional groups. In the following, we present evidence that our method represents the molecular structure more efficiently than the traditional sequence model (Wu et al., 2021), in addition to being completely independent from extra-labeled data. All data used in model pretraining are label free, and data preprocessing is easy and fast. Furthermore, node- and graph-level pretraining makes the pretrained model robust to scarce training data. As a result, the performance of our model satisfies the criteria of few-shot learning, which typically refers to machine learning problems, where the training set contains limited information (Garcia and Bruna, 2018). In the field of drug discovery, the outcome of few-shot learning is the prediction of molecular properties based on a small number of training samples. It is particularly important for the drug discovery of new diseases such as COVID-19 since few active compounds related to these diseases have been discovered.
In this study, we applied MolGNN to predict drug-like molecules potentially effective for COVID-19 treatment. We first screened polypharmacological compounds to target the Janus kinases (JAK) 1/2/3 and the main protease (MPro). JAK is a family of intracellular tyrosine kinases (Wilks, 1989) playing a major role in transmitting cytokine signals through receptor phosphorylation. The primary lethal syndrome associated with COVID-19 is the cytokine storm, an acute immune response that results in overdosed cytokine release into the blood in a short range of time (Fajgenbaum and June 2020; Hojyo et al., 2020). Inhibiting the activity of JAKs may therefore alleviate body responses to cytokine storms. MPro is a key enzyme initiating SARS-CoV-2 replication, and its inhibition may also slow down viral replication (Hilgenfeld, 2014; Pillaiyar et al., 2016; Zhang et al., 2020). In addition, we also predicted drug candidates derived from antiviral experiments lacking specific molecular targets. Both strategies produced several hits supported by existing experimental and clinical evidence, and hence they may represent relevant candidates for COVID-19 clinical trials.
Materials and Methods
Graph Neural Network Representation of Chemical Structure
We used a graph neural network to model the ability of small molecules to activate or inhibit potential drug targets. Let
Network Motif
Network motifs are recurrent substructures or subgraphs within a larger graph. In a chemical compound, chemical functional groups or fragments such as benzene rings are endogenous motifs. We applied PubChem fingerprints encoding molecular fragments with binary digits to represent motifs (Kim et al., 2021). PubChem fingerprints used in pretraining were calculated with the Chemistry Development Kit (Willighagen et al., 2017). The original fingerprint had 881 digits. Since the chemical molecules in the data sets used in this study are mostly organic drugs, we reduced the number of digits in the fingerprint by removing digits associated with atoms rarely appearing in drugs. Specifically, we only kept the digits related to C, H, O, N, S, F, Cl, and Br atoms. That results in a filtered fingerprint with 740 digits.
Model Architecture
The model was built on a multitask learning framework with three tasks: node- and edge-level embedding learning, self-supervised motif learning, and supervised fine-tuning/graph classification (Figure 1). We followed the “context” method from Hu et al. (2019) to perform node and edge embedding learning. Briefly, a subgraph that contains the central node is chosen and the central node embedding is generated with a GNN model. This embedding is trained to be similar with the embedding generated with nodes within
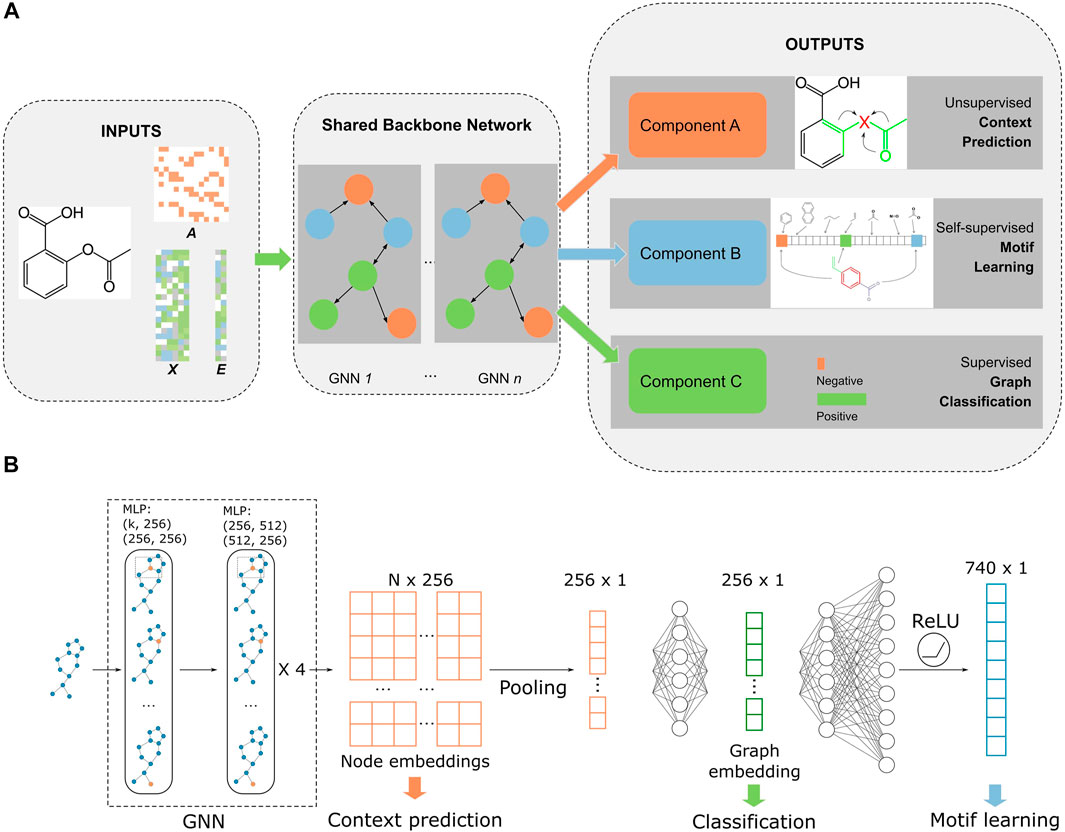
FIGURE 1. Overall workflow of MolGNN. (A). Inputs were edges represented by the adjacency matrix
The architecture of the backbone GNN Figure 1B is a five-layer graph isomorphism network (GIN) with 512 and 256 hidden units for MLPs in each layer (Hu et al., 2019; Xu et al., 2019). The GNN outputs a latent representation of all nodes in each graph. To make the model permutational invariant, a pooling function symmetric to permutations was applied to node representations to generate graph-level embeddings. We chose a mean pooling function that outperformed sum or max functions in our experiments. The fingerprint branch readout MLP had 370 hidden units (half the size of the filtered PubChem fingerprint dimension), while the classification MLP had 256 hidden units (same as the dimensions of node embeddings). Our model was built with PyTorch and PyTorch Geometric (Fey and Lenssen, 2019; Paszke et al., 2019). All pretraining and fine-tuning was performed on a single Tesla V100 GPU.
Input Data
Our model inputs consisted of chemical molecules as graphs represented by adjacency matrices
For node and edge context prediction pretraining, we used two million unlabeled chemical molecules sampled from the ZINC15 database (Sterling and Irwin, 2015). For graph-level self-supervised pretraining, we used a data set of 456 K molecules sampled from ChEMBL (Gaulton et al., 2012; Mayr et al., 2018). For downstream classification tasks testing model efficiency in drug development, we applied our method to chemical molecules related to COVID-19. We derived our JAK data set from ChEMBL with kinases JAK1, JAK2, and JAK3 binding affinity as labels. The original data set included experimental IC50 values of thousands of chemical molecules against JAK1, JAK2, and JAK3. We labeled all molecules with the IC50 value under
Data set Splitting
The benchmark data sets were split with the scaffold splitting method (Ramsundar et al., 2019). The Murcko scaffold of each chemical was captured with RDKit (Landrum, 2006), and only chemicals with the same scaffold were grouped together. Groups were randomly permutated and added into training, validating, or the testing set. This procedure made sure that the testing set only contained chemicals with scaffolds differing from those in the training and validating sets. Scaffold splitting also causes chemical properties to differ between training and testing sets and impairs prediction performance of a model trained exclusively with labeled training data. As a result, the splitting method allows for a better assessment of how the model benefits from self-supervised pretraining with unlabeled data. Furthermore, since new drug scaffolds often differ from existing drugs, scaffold splitting was expected to provide superior insights into the potential for drug discovery of our trained model.
Loss Function and Metrics
Binary cross-entropy loss was used in the pretraining step of context prediction. We treated PubChem fingerprints in the motif learning network as a multi-label prediction problem, and a binary cross-entropy loss was used for this network. For graph classification, we used cross-entropy loss for multi-class classification, and binary cross-entropy loss for binary or multi-label classification.
Because of the label imbalance in the data sets, accuracy was not a good metric to evaluate our experiments. Instead, we selected the area under the receiver operating characteristic curve (ROC-AUC), average precision (AP), and F1 score as metrics. All metrics were calculated with the scikit-learn package (Pedregosa et al., 2011).
Results and Discussion
Label-Independent Self-Supervised Pretraining is Critical to Model Performance
Our MolGNN included a two-stage pretraining method derived from ContextPred (Hu et al., 2019). The first stage was an atom- and edge-level pretraining stage with context prediction, which is the same as in ContextPred. The second stage was a graph-level self-supervised and label-independent pretraining step, different from ContextPred that relies on experimental data (see Methods for details). To demonstrate that MolGNN benefited from both pretraining stages, we performed an ablation study (Table 1). For all six data sets, MolGNN outperformed both models without pretraining and models pretrained only with context prediction. Ellinger was the most imbalanced data set with an 84:1 negative to positive ratio and was associated with the highest improvement due to graph-level pretraining. The ROC-AUC showed significant improvements of 17.9 and 29.3% compared to the model pretrained with context prediction only and the model with no pretraining, respectively. The AP showed nearly eightfold relative improvement compared to the model with no pretraining, although the absolute value of improvement was low.
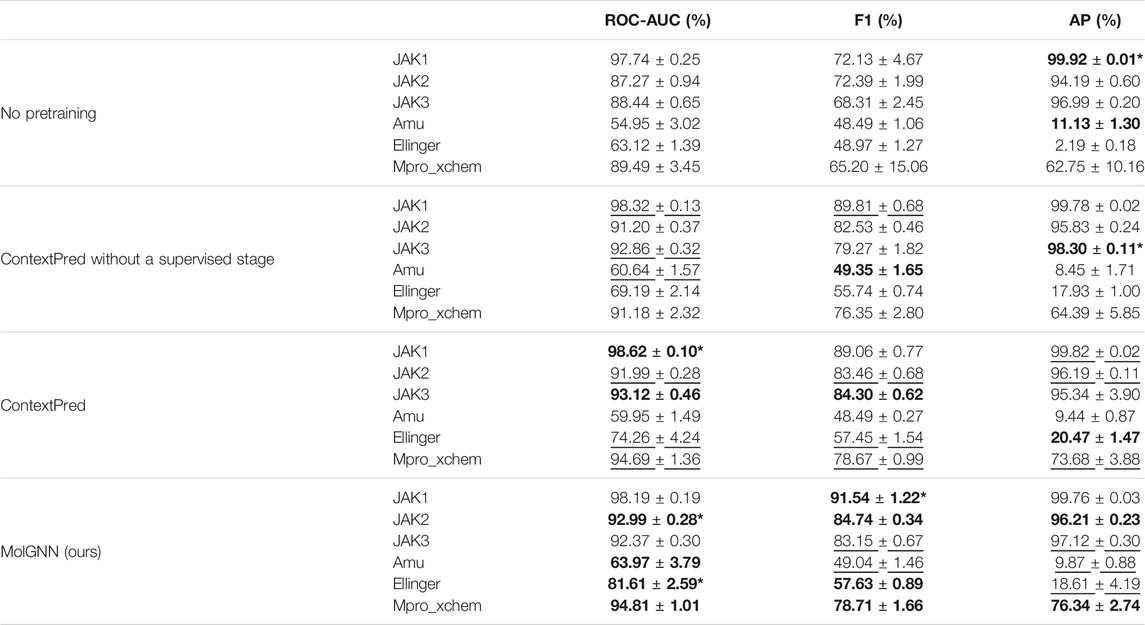
TABLE 1. Performance comparisons between MolGNN, ContextPred, ContextPred without supervised stage, and GNN models without pretraining. The best results are highlighted in bold. The second-best performance is underscored. * Indicates the statistically significant differences (p < 0.05) between the best and second-best performer.
Label-Independent Self-Supervised Pretraining Outperforms Label-Dependent Pretraining
Next, we compared MolGNN to the experimental label-based, supervised pretraining model from ContextPred. We applied the same scaffold-based splitting method from ContextPred to our JAK and SARS-CoV-2 data sets. Both methods improved model performance compared to the model without pretraining (Table 1). MolGNN performance was superior or equivalent to supervised pretraining. In JAK2 and Ellinger, our method was significantly better than the supervised pretraining with p-values of 0.0051 and 0.0107, respectively. There is no statistically significant difference in other data sets. Given the supervised pretraining needed a large number of experimentally labeled data, the pretraining data set used in MolGNN was less costly, easier, and faster to acquire. Our motif network pretraining could be a complete replacement for the experimental label-based supervised pretraining.
MolGNN Significantly Improves Few-Shot Learning Performance
A challenge to machine learning when applied to chemical molecules has been the scarcity of labeled data. We therefore tested the performance of MolGNN with reduced labeled fine-tuning data (Figure 2; Table 2). Compared to the GIN model without pretraining, MolGNN benefitted from pretraining even with very little training data. As a rule, the most significant improvements occurred, when the ratio of training to testing data was 1:8. When using the JAK1 data set, MolGNN showed a relative improvement in the F1 score of 55.8% over the model with no pretraining and 25.9% when the training to testing ratio was 8:1. This was also the case for JAK2 and JAK3 with improvements of 34.8 and 34.8% with the 1:8 ratio and 10.0 and 21.7% with the 8:1 ratio, respectively.
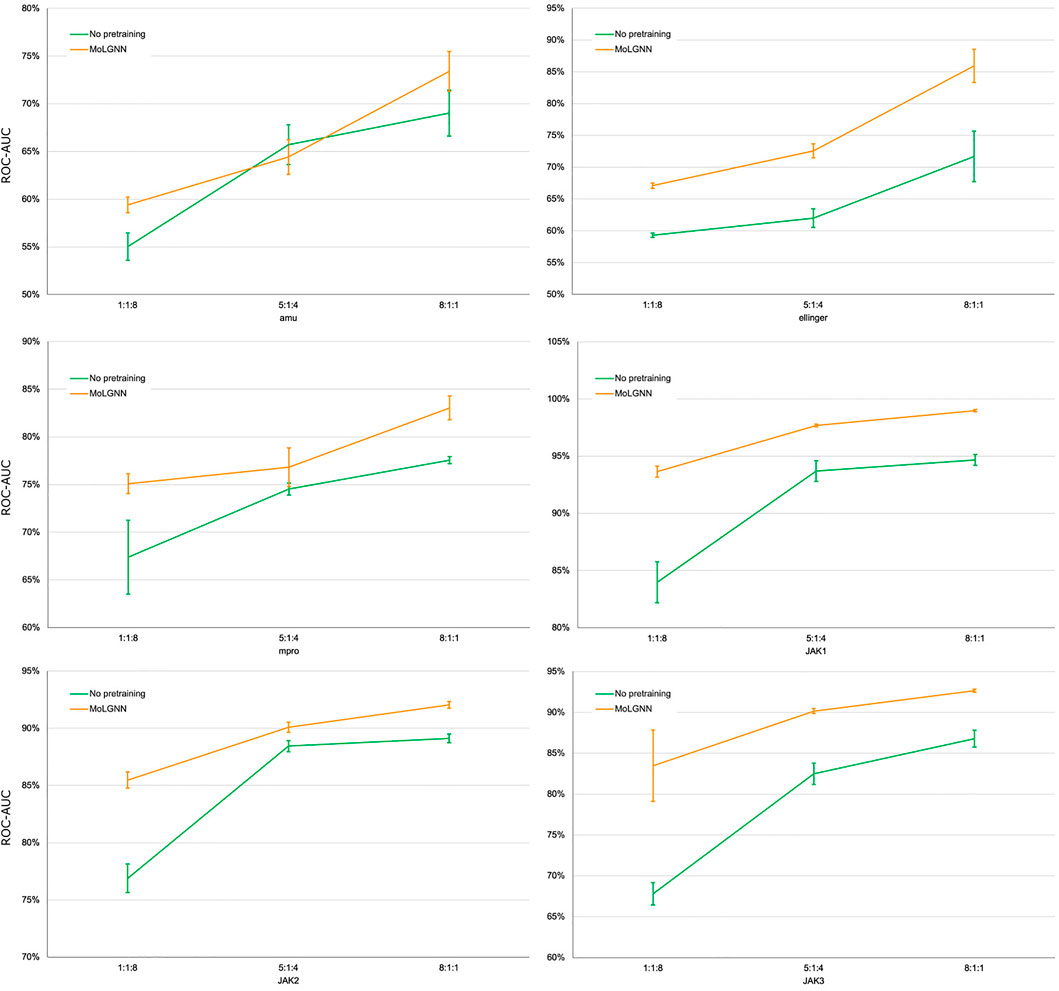
FIGURE 2. ROC-AUC scores of few-shot learning experiments. The ratio number shown under the plots were the ratio of training, validation, and testing sets. Error bars represent the standard deviation of five replicates.
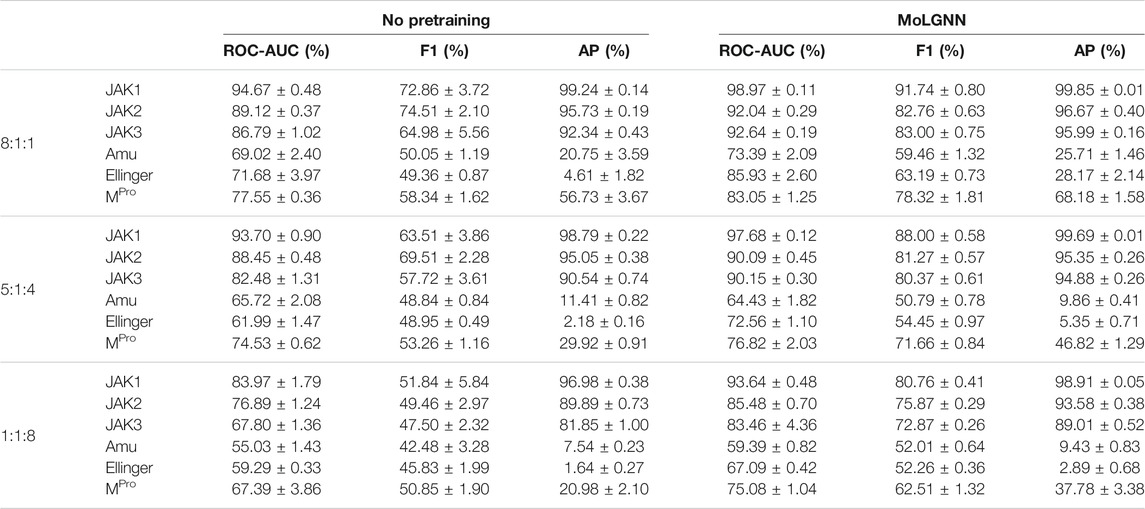
TABLE 2. Experimental results with reduced training data. Data ratio in training, validation, and testing sets are labeled in the first column.
Potential Drugs Predicted From the Repurposing Data set
To test our method in drug candidate prediction, we applied MolGNN on JAK data sets to screen molecules, with the potential to alleviate COVID-19 symptoms from the repurposing data set. We then applied MolGNN to the MPro data set to search for molecule candidates possibly inhibiting viral replication. The top-ranked candidates from JAK and MPro data sets were selected, and their intersection is listed in Table 3.
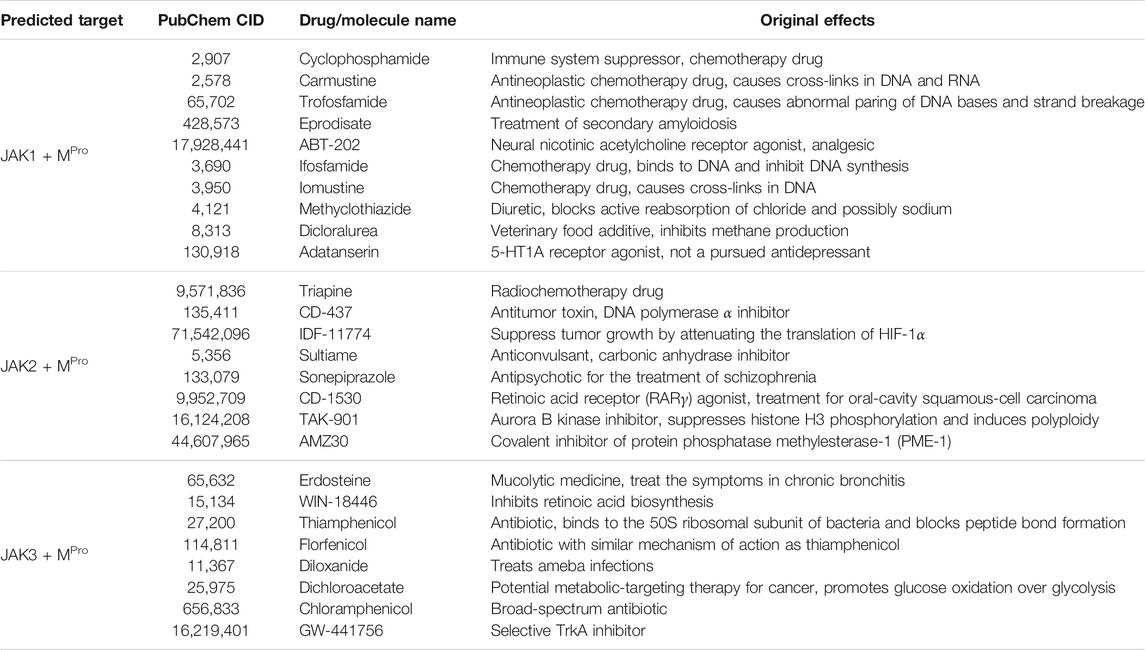
TABLE 3. Top-ranked drugs according to predicted potential to bind JAK and MPro.
We also predicted potential COVID-19 drugs by applying MolGNN to data sets based on in vitro assays not specifying drug targets. The top hits with their original effects are listed in Table 4. Attempts to obtain intersections between these two groups, or with JAK and MPro top-ranked, molecules failed, suggesting that molecular structures effective against SARS-CoV-2 in vitro were different from molecules specifically able to bind to JAK or MPro.
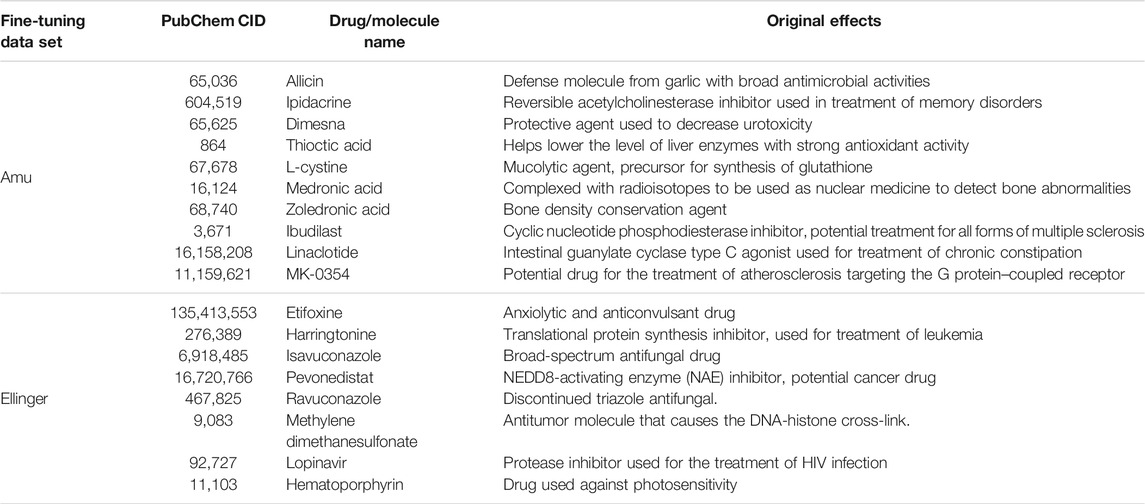
TABLE 4. Top-ranked drugs/molecules according to potential anti-COVID effects.
Among predicted drug candidates, several molecules have already been under study due to their anti–COVID-19 effects, providing a validation to our predictions. For example, we predicted that cyclophosphamide, recently shown to mitigate acute respiratory distress syndrome among COVID-19 patients (Revannasiddaiah et al., 2020), could inhibit JAK1 and MPro. We also predicted that erdosteine, which has shown promising results in improving the condition of COVID-19 patients (Santus et al., 2020), may be a co-inhibitor of JAK3 and MPro. Among other examples, allicin (an organosulfur molecule found in garlic) is believed to decrease the rate of SARS-CoV-2 viral infection (Donma and Donma, 2020; Khubber et al., 2020; Shekh et al., 2020). Ipidacrine, a reversible acetylcholinesterase inhibitor originally used for the treatment of memory disorders, was found in X-ray crystallographic screening studies to exhibit MPro binding activity (Günther et al., 2020). Thioctic (alpha-lipoic) acid may protect diabetic patients against COVID-19 (Cure and Cumhur Cure, 2020). Harringtonine used in leukemia treatment has also been included in COVID-19 clinical trials (Wen et al., 2021). Lopinavir, a protease inhibitor used in HIV treatment, was under the clinical trial for the treatment of adults with severe COVID-19 symptoms (Cao et al., 2020).
Conclusion
We developed a new GNN-based and self-supervised learning method MolGNN to facilitate drug discovery. Compared to state-of-the-art techniques, our implementation showed the following advantages:
1. In the pretraining step, MolGNN was fully self-supervised. It did not require any extra-labeled data to obtain graph-level embedding as in Hu et al. (2019), while achieving equivalent performance.
2. Specifically designed for chemicals, MolGNN not only captured atom- and bond-level information but also substructure information, which was critical for its superior performance in chemical-related tasks.
3. MolGNN can successfully handle sparse labeled data. The graph-level label we used in self-supervised pretraining could be much more easily acquired than labels derived from specific experiments, providing our method with a wider range of use.
The GNN model trained with MolGNN showed robustness, when applied to a small labeled fine-tuning data set, suggesting a potentially powerful few-shot learning method. Even with very little fine-tuning data, pretraining was able to improve final performance by a large margin. This confirms that substructure-based labels can assist neural networks in capturing intrinsic chemical attributes of molecules in their latent space.
Our method provides a powerful tool for new drug development, especially in the case of new and poorly known diseases. Our fine-tuned model successfully identified various molecules exhibiting anti–COVID-19 activity from a large set of chemical compounds. Some of our proposed candidates have already shown potential to contribute to COVID-19 treatment and have been included in the clinical trial. We suggest that such compounds should be tested both in vitro and in vivo. The experimental validation of our testable hypotheses should also be the subject of future collaborative work. Our method may contribute to polypharmacology by predicting candidate molecules for multiple targets, based on various models pretrained and fine-tuned with MolGNN. Finally, we believe that our method may speed up drug development both in the specific case of COVID-19 and of other diseases for which few effective therapies are currently available.
Data Availability Statement
Publicly available data sets were analyzed in this study. This data can be found here: https://github.com/AdorableYoyo/MolGNN_fewshot.
Author Contributions
LX was the principal investigator, designed the methods, supervised all experiments, and contributed to writing the manuscript. YL contributed to data processing, experiments running, results analysis, and manuscript writing. YW assisted in running the experiments and contributed to manuscript writing and reviewing. XS assisted in running experiments and reviewing the manuscript.
Funding
This work was supported by the National Institute of General Medical Sciences (NIGMS) (R01GM122845) and National Institute on Aging (NIA) (R01AD057555) of the National Institute of Health (NIH).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Apsel, B., Blair, J. A., Gonzalez, B., Nazif, T. M., Feldman, M. E., Aizenstein, B., et al. (2008). Targeted Polypharmacology: Discovery of Dual Inhibitors of Tyrosine and Phosphoinositide Kinases. Nat. Chem. Biol. 4, 691–699. doi:10.1038/nchembio.117
Ayed, M., Lim, H., and Xie, L. (2019). Biological Representation of Chemicals Using Latent Target Interaction Profile. BMC Bioinformatics 20, 674. doi:10.1186/s12859-019-3241-3
Balasubramaniam, M., and Shmookler Reis, R. (2020). Computational Target-Based Drug Repurposing of Elbasvir, an Antiviral Drug Predicted to Bind Multiple SARS-CoV-2 Proteins. New York, NY: ChemRxiv Prepr. Serv. Chem. doi:10.26434/chemrxiv.12084822
Beigel, J. H., Tomashek, K. M., Dodd, L. E., Mehta, A. K., Zingman, B. S., Kalil, A. C., et al. (2020). Remdesivir for the Treatment of Covid-19—Preliminary Report. N. Engl. J. Med. 383 (19), 1813–1826. doi:10.1056/NEJMoa2007764
Cai, T., Lim, H., Abbu, K. A., Qiu, Y., Nussinov, R., and Xie, L. (2021). MSA-Regularized Protein Sequence Transformer toward Predicting Genome-Wide Chemical-Protein Interactions: Application to GPCRome Deorphanization. J. Chem. Inf. Model. 61, 1570–1582. doi:10.1021/acs.jcim.0c01285
Cao, B., Wang, Y., Wen, D., Liu, W., Wang, J., Fan, G., et al. (2020). A Trial of Lopinavir-Ritonavir in Adults Hospitalized with Severe Covid-19. N. Engl. J. Med. 382, 1787–1799. doi:10.1056/NEJMoa2001282
Chaudhari, R., Tan, Z., Huang, B., and Zhang, S. (2017). Computational Polypharmacology: a New Paradigm for Drug Discovery. Expert Opin. Drug Discov. 12, 279–291. doi:10.1080/17460441.2017.1280024
Chen, X., Liu, M.-X., and Yan, G.-Y. (2012). Drug-target Interaction Prediction by Random Walk on the Heterogeneous Network. Mol. Biosyst. 8, 1970–1978. doi:10.1039/C2MB00002D
Chong, C. R., and Sullivan, D. J. (2007). New Uses for Old Drugs. Nature 448, 645–646. doi:10.1038/448645a
Corsello, S. M., Bittker, J. A., Liu, Z., Gould, J., McCarren, P., Hirschman, J. E., et al. (2017). The Drug Repurposing Hub: a Next-Generation Drug Library and Information Resource. Nat. Med. 23, 405–408. doi:10.1038/nm.4306
Cure, E., and Cumhur Cure, M. (2020). Alpha-lipoic Acid May Protect Patients with Diabetes against COVID-19 Infection. Med. Hypotheses 143, 110185. doi:10.1016/j.mehy.2020.110185
Donma, M. M., and Donma, O. (2020). The Effects of Allium Sativum on Immunity within the Scope of COVID-19 Infection. Med. Hypotheses 144, 109934. doi:10.1016/j.mehy.2020.109934
Duvenaud, D., Maclaurin, D., Aguilera-Iparraguirre, J., Gómez-Bombarelli, R., Hirzel, T., Aspuru-Guzik, A., et al. (2015). Convolutional Networks on Graphs for Learning Molecular Fingerprints. Ithaca, NY: arXivAvailable at: http://papers.nips.cc/paper/5954-convolutional-networks-on-graphs-for-learning-molecular-fingerprints.pdf (Accessed April 19, 2019).
Ellinger, B., Bojkova, D., Zaliani, A., Cinatl, J., Claussen, C., Westhaus, S., et al. (2021). A SARS-CoV-2 Cytopathicity Dataset Generated by High-Content Screening of a Large Drug Repurposing Collection. Sci. Data 8, 70. doi:10.1038/s41597-021-00848-4
Fajgenbaum, D. C., and June, C. H. (2020). Cytokine Storm. N. Engl. J. Med. 383, 2255–2273. doi:10.1056/NEJMra2026131
Fey, M., and Lenssen, J. E. (2019). “Fast Graph Representation Learning with PyTorch Geometric,” in ICLR Workshop on Representation Learning on Graphs and Manifolds.
Galindez, G., Matschinske, J., Rose, T. D., Sadegh, S., Salgado-Albarrán, M., Späth, J., et al. (2021). Lessons from the COVID-19 Pandemic for Advancing Computational Drug Repurposing Strategies. Nat. Comput. Sci. 1, 33–41. doi:10.1038/s43588-020-00007-6
Gaulton, A., Bellis, L. J., Bento, A. P., Chambers, J., Davies, M., Hersey, A., et al. (2012). ChEMBL: a Large-Scale Bioactivity Database for Drug Discovery. Nucleic Acids Res. 40, D1100–D1107. doi:10.1093/nar/gkr777
Gilmer, J., Schoenholz, S. S., Riley, P. F., Vinyals, O., and Dahl, G. E. (2017). “Neural Message Passing for Quantum Chemistry,” in Proceedings Of the 34th International Conference On Machine Learning Proceedings of Machine Learning Research. Editors D. Precup, and Y. W. Teh (PMLR), 1263–1272. Available at: http://proceedings.mlr.press/v70/gilmer17a.html.
González-Durruthy, M., Concu, R., Vendrame, L. F. O., Zanella, I., Ruso, J. M., and Cordeiro, M. N. D. S. (2020). Targeting Beta-Blocker Drug-Drug Interactions with Fibrinogen Blood Plasma Protein: A Computational and Experimental Study. Molecules 25, 5425. doi:10.3390/molecules25225425
Günther, S., Reinke, P. Y. A., Fernández-García, Y., Lieske, J., Lane, T. J., Ginn, H. M., et al. (2020). Inhibition of SARS-CoV-2 Main Protease by Allosteric Drug-Binding. New York City: bioRxiv. doi:10.1101/2020.11.12.378422
Hilgenfeld, R. (2014). From SARS to MERS: Crystallographic Studies on Coronaviral Proteases Enable Antiviral Drug Design. FEBS J. 281, 4085–4096. doi:10.1111/febs.12936
Hojyo, S., Uchida, M., Tanaka, K., Hasebe, R., Tanaka, Y., Murakami, M., et al. (2020). How COVID-19 Induces Cytokine Storm with High Mortality. Inflamm. Regener 40, 37. doi:10.1186/s41232-020-00146-3
Hopkins, A. L. (2008). Network Pharmacology: the Next Paradigm in Drug Discovery. Nat. Chem. Biol. 4, 682–690. doi:10.1038/nchembio.118
Hu, W., Liu, B., Gomes, J., Zitnik, M., Liang, P., Pande, V., et al. (2019). Strategies for Pre-training Graph Neural Networks. Available at: http://arxiv.org/abs/1905.12265 [Accessed February 15, 2021].
Jin, G., and Wong, S. T. C. (2014). Toward Better Drug Repositioning: Prioritizing and Integrating Existing Methods into Efficient Pipelines. Drug Discov. Today 19, 637–644. doi:10.1016/j.drudis.2013.11.005
Khubber, S., Hashemifesharaki, R., Mohammadi, M., and Gharibzahedi, S. M. T. (2020). Garlic (Allium Sativum L.): a Potential Unique Therapeutic Food Rich in Organosulfur and Flavonoid Compounds to Fight with COVID-19. Nutr. J. 19, 124. doi:10.1186/s12937-020-00643-8
Kim, S., Chen, J., Cheng, T., Gindulyte, A., He, J., He, S., et al. (2021). PubChem in 2021: New Data Content and Improved Web Interfaces. Nucleic Acids Res. 49, D1388–D1395. doi:10.1093/nar/gkaa971
Kipf, T. N., and Welling, M. (2017). Semi-Supervised Classification with Graph Convolutional Networks. Ithaca, NY: arXiv. doi:10.1515/9783110527230-021
Landrum, G. (2006). RDKit: Open-Source Cheminformatics. Ithaca, NY: arXivAvailable at: http://www.rdkit.org.
Mayr, A., Klambauer, G., Unterthiner, T., Steijaert, M., Wegner, J. K., Ceulemans, H., et al. (2018). Large-scale Comparison of Machine Learning Methods for Drug Target Prediction on ChEMBL. Chem. Sci. 9, 5441–5451. doi:10.1039/C8SC00148K
Paolini, G. V., Shapland, R. H. B., van Hoorn, W. P., Mason, J. S., and Hopkins, A. L. (2006). Global Mapping of Pharmacological Space. Nat. Biotechnol. 24, 805–815. doi:10.1038/nbt1228
Paszke, A., Gross, S., Massa, F., Lerer, A., Bradbury, J., Chanan, G., et al. (2019). “PyTorch: An Imperative Style, High-Performance Deep Learning Library,” in Advances in Neural Information Processing Systems 32. Editors H. Wallach, H. Larochelle, A. Beygelzimer, F. d\textquotesingle Alché-Buc, E. Fox, and R. Garnett (Curran Associates, Inc.)), 8024–8035. Available at: http://papers.neurips.cc/paper/9015-pytorch-an-imperative-style-high-performance-deep-learning-library.pdf.
Pedregosa, F., Varoquaux, G., Gramfort, A., Michel, V., Thirion, B., Grisel, O., et al. (2011). Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 12, 2825–2830.
Peng, Y., McCorvy, J. D., Harpsøe, K., Lansu, K., Yuan, S., Popov, P., et al. (2018). 5-HT2C Receptor Structures Reveal the Structural Basis of GPCR Polypharmacology. Cell 172, 719–730. e14. doi:10.1016/j.cell.2018.01.001
Pillaiyar, T., Manickam, M., Namasivayam, V., Hayashi, Y., and Jung, S.-H. (2016). An Overview of Severe Acute Respiratory Syndrome-Coronavirus (SARS-CoV) 3CL Protease Inhibitors: Peptidomimetics and Small Molecule Chemotherapy. J. Med. Chem. 59, 6595–6628. doi:10.1021/acs.jmedchem.5b01461
Ramsundar, B., Eastman, P., Walters, P., and Pande, V. (2019). Deep Learning for the Life Sciences: Applying Deep Learning to Genomics, Microscopy, Drug Discovery, and More. Sebastopol, CA: O’Reilly MediaAvailable at: https://books.google.com/books?id=tYFKuwEACAAJ.
Revannasiddaiah, S., Kumar Devadas, S., Palassery, R., Kumar Pant, N., and Maka, V. V. (2020). A Potential Role for Cyclophosphamide in the Mitigation of Acute Respiratory Distress Syndrome Among Patients with SARS-CoV-2. Med. Hypotheses 144, 109850. doi:10.1016/j.mehy.2020.109850
Santus, P., Tursi, F., Croce, G., Di Simone, C., Frassanito, F., Gaboardi, P., et al. (2020). Changes in Quality of Life and Dyspnoea after Hospitalization in COVID-19 Patients Discharged at home. Multidis Res. Med. 15, 713. doi:10.4081/mrm.2020.713
Shekh, S., Reddy, K. K. A., and Gowd, K. H. (2020). In Silico allicin Induced S-Thioallylation of SARS-CoV-2 Main Protease. J. Sulfur Chem. 42, 109–120. doi:10.1080/17415993.2020.1817457
Sterling, T., and Irwin, J. J. (2015). ZINC 15 - Ligand Discovery for Everyone. J. Chem. Inf. Model. 55, 2324–2337. doi:10.1021/acs.jcim.5b00559
Touret, F., Gilles, M., Barral, K., Nougairède, A., Decroly, E., de Lamballerie, X., et al. (2020). Vitro Screening of a FDA Approved Chemical Library Reveals Potential Inhibitors of SARS-CoV-2 Replication. New York, NY: bioRxiv. doi:10.1101/2020.04.03.023846
Wen, H.-J., Liu, F.-L., Huang, M.-X., Luo, R.-H., He, W.-B., Feng, J., et al. (2021). A Proposal for Clinical Trials of COVID-19 Treatment Using Homo-Harringtonine. Natl. Sci. Rev. 8, nwaa257. doi:10.1093/nsr/nwaa257
Wilks, A. F. (1989). Two Putative Protein-Tyrosine Kinases Identified by Application of the Polymerase Chain Reaction. Proc. Natl. Acad. Sci. 86, 1603–1607. doi:10.1073/pnas.86.5.1603
Willighagen, E. L., Mayfield, J. W., Alvarsson, J., Berg, A., Carlsson, L., Jeliazkova, N., et al. (2017). The Chemistry Development Kit (CDK) v2.0: Atom Typing, Depiction, Molecular Formulas, and Substructure Searching. J. Cheminform. 9, 33. doi:10.1186/s13321-017-0220-4
Wu, Z., Pan, S., Chen, F., Long, G., Zhang, C., and Yu, P. S. (2021). A Comprehensive Survey on Graph Neural Networks. IEEE Trans. Neural Netw. Learn. Syst. 32, 4–24. doi:10.1109/TNNLS.2020.2978386
Wu, Z., Ramsundar, B., Feinberg, E. N., Gomes, J., Geniesse, C., Pappu, A. S., et al. (2018). MoleculeNet: a Benchmark for Molecular Machine Learning. Chem. Sci. 9, 513–530. doi:10.1039/C7SC02664A
Xu, K., Jegelka, S., Hu, W., and Leskovec, J. (2019). “How Powerful Are Graph Neural Networks?,” in 7th International Conference on Learning Representations (ICLR 2019). doi:10.1109/ijcnn.2019.8852173
Yamanishi, Y., Kotera, M., Kanehisa, M., and Goto, S. (2010). Drug-target Interaction Prediction from Chemical, Genomic and Pharmacological Data in an Integrated Framework. Bioinformatics 26, i246–i254. doi:10.1093/bioinformatics/btq176
Ying, R., You, J., Morris, C., Ren, X., Hamilton, W. L., and Leskovec, J. (2019). Hierarchical Graph Representation Learning with Differentiable Pooling. Ithaca, NY: arXiv.
Zhang, L., Lin, D., Sun, X., Curth, U., Drosten, C., Sauerhering, L., et al. (2020). Crystal Structure of SARS-CoV-2 Main Protease Provides a Basis for Design of Improved α-ketoamide Inhibitors. Science 368, 409–412. doi:10.1126/science.abb3405
Zheng, S., Yan, X., Yang, Y., and Xu, J. (2019). Identifying Structure-Property Relationships through SMILES Syntax Analysis with Self-Attention Mechanism. J. Chem. Inf. Model. 59, 914–923. doi:10.1021/acs.jcim.8b00803
Keywords: SARS-CoV-2, deep learning, graph neural network, self-supervised learning, polypharmacology, virtual screening
Citation: Liu Y, Wu Y, Shen X and Xie L (2021) COVID-19 Multi-Targeted Drug Repurposing Using Few-Shot Learning. Front. Bioinform. 1:693177. doi: 10.3389/fbinf.2021.693177
Received: 10 April 2021; Accepted: 25 May 2021;
Published: 15 June 2021.
Edited by:
Liam James McGuffin, University of Reading, United KingdomReviewed by:
Rodrigo Ligabue-Braun, Federal University of Health Sciences of Porto Alegre, BrazilJan Zaucha, Technical University of Munich, Germany
Copyright © 2021 Liu, Wu, Shen and Xie. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lei Xie, bHhpZUBpc2NiLm9yZw==