 Nadine Ihle
Nadine Ihle Laura Grüßner1
Laura Grüßner1 Ceren Alkim
Ceren Alkim T. A. Stefanie Nguyen
T. A. Stefanie Nguyen Thomas Walther
Thomas Walther Cláudio J. R. Frazão
Cláudio J. R. Frazão
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Bioeng. Biotechnol., 20 February 2025
Sec. Synthetic Biology
Volume 13 - 2025 | https://doi.org/10.3389/fbioe.2025.1504785
(L)-2,4-dihydroxybutyrate (DHB) is a versatile compound that can serve as a precursor for the synthesis of the methionine analog 2-hydroxy-4-(methylthio)butyrate and new advanced polymers. We previously implemented in Escherichia coli an artificial biosynthetic pathway for the aerobic production of DHB from glucose, which relies on the deamination of (L)-homoserine followed by the reduction of 2-oxo-4-hydroxybutyrate (OHB) and yields DHB by an enzyme-bearing NADH-dependent OHB reductase activity. Under aerobic conditions, using NADPH as a cofactor is more favorable for reduction processes. We report the construction of an NADPH-dependent OHB reductase and increased intracellular NADPH supply by metabolic engineering to improve DHB production. Key cofactor discriminating positions were identified in the previously engineered NADH-dependent OHB reductase (E. coli malate dehydrogenase I12V:R81A:M85Q:D86S:G179D) and tested by mutational scanning. The two point mutations D34G:I35R were found to increase the specificity for NADPH by more than three orders of magnitude. Using the new OHB reductase enzyme, replacing the homoserine transaminase with the improved variant Ec.AlaC A142P:Y275D and increasing the NADPH supply by overexpressing the pntAB gene encoding the membrane-bound transhydrogenase yielded a strain that produced DHB from glucose at a yield of 0.25 molDHB molGlucose−1 in shake-flask experiments, which corresponds to a 50% increase compared to previous producer strains. Upon 24 h of batch cultivation of the most advanced DHB producer strain constructed in this work, a volumetric productivity of 0.83 mmolDHB L−1 h−1 was reached.
(L)-2,4-dihydroxybutyrate (DHB) is a versatile compound of growing industrial relevance, as it can serve as a precursor for the chemical production of the methionine analog 2-hydroxy-4-(methylthio)butyrate (HMTB) used in animal nutrition (Walther et al., 2017a) or as a building block for new advanced biopolymers (François, 2023). Furthermore, DHB can serve as a precursor for the synthesis of 1,3-propanediol (Frazão et al., 2019) or 1,2,4-butanetriol (Li et al., 2014). Although the occurrence of DHB at trace levels in patients with succinic semialdehyde dehydrogenase deficiency has been described previously (Shinka et al., 2002), there is no annotated natural metabolic pathway for its biosynthesis.
Aided by synthetic biology and enzyme engineering, we and others have previously reported three artificial biosynthetic pathways for the aerobic, microbial production of DHB starting from the widely abundant and inexpensive sugar glucose (Walther et al., 2017b; Walther et al., 2015; Walther et al., 2017a). It is of note, however, that all the new routes are fully compatible with the use of other sugars (e.g., xylose, mannose, sucrose) or alcohols (e.g., methanol, ethylene glycol) as starting carbon sources, as all DHB pathways start from naturally occurring metabolites (homoserine, malate, or glyoxylate/acetyl-CoA). The different metabolic routes were tested in Escherichia coli. The highest reported titers (7.9 ± 0.01 mM) and yields (0.10 ± 0.01 molDHB molGlucose−1) in shake-flask cultivations with glucose as carbon source have been achieved with the DHB pathway proceeding through the characteristic natural intermediate (L)-homoserine (Frazão et al., 2018). This route enables DHB synthesis via sequential deamination of (L)-homoserine by homoserine transaminase activity and reduction of 2-oxo-4-hydroxybutyrate (OHB) by an OHB reductase activity (Figure 1). In a previous study, we reported the construction of the highly active NADH-dependent OHB reductase Ec.Mdh5Q (Frazão et al., 2018). The mutant variant descends from the parent NAD+-dependent (L)-malate dehydrogenase from E. coli (Ec.Mdh) and incorporates five mutations (I12V:R81A:M85Q:D86S:G179D) to yield the desired synthetic activity. However, the typical intracellular ratios of [NADH]/[NAD+] and [NADPH]/[NADP+] in E. coli cells cultivated under aerobic conditions are 0.03 and 60, respectively (Bennett et al., 2009), which suggests that the utilization of NADPH as a cofactor in aerobic reduction processes may be more favorable. Therefore, employing an enzyme bearing NADPH-dependent OHB reductase activity could provide a strong advantage in terms of pathway performance.
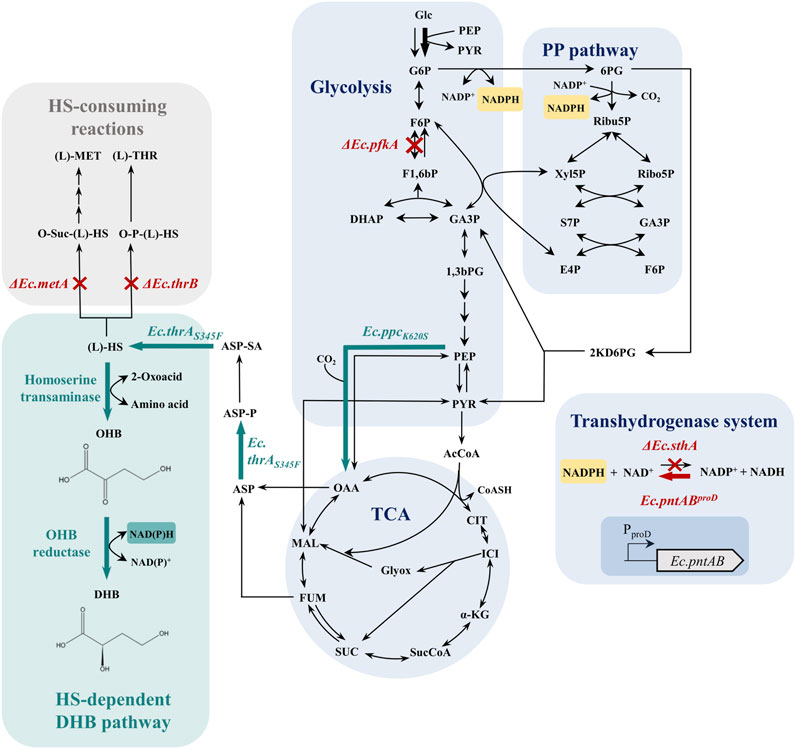
Figure 1. Aerobic synthesis of (L)-2,4-dihydroxybutyric acid (DHB) from glucose via the artificial homoserine pathway (bluish-green) and applied strategies for increased NADPH availability in Escherichia coli. Abbreviations: 1,3bPG, 1,3-bisphospho-glycerate; AcCoA, acetyl-CoA; ASP, aspartate; ASP-P, aspartyl phosphate; ASP-SA, aspartate semialdehyde; CIT, citrate; DHAP, dihydroxyacetone phosphate; F1,6bP, fructose-1,6-bisphosphate; F6P, fructose-6-phosphate; FUM, fumarate; G3P, glyceraldehyde-3-phosphate; G6P, glucose-6-phosphate; Glc, glucose; ICI, isocitrate; (L)-DHB, (L)-2,4-dihydroxybutyrate; (L)-HS, (L)-homoserine; (L)-MET, (L)-methionine; (L)-THR, (L)-threonine; MAL, malate; OAA, oxaloacetate; OHB, 2-oxo-4-hydroxybutyrate; O-P-(L)-HS, O-phospho-L-homoserine; O-Suc-(L)-HS, O-succinyl-(L)-homoserine; PEP, phosphoenolpyruvate; PP pathway, pentose phosphate pathway; PYR, pyruvate; SUC, succinate; SucCoA, succinyl-CoA; TCA, tricarboxylic acid cycle; α-KG, α-ketoglutarate; Gene names are written in italics. Ec.pfkA–gene encoding 6-phosphofructokinase; Ec.sthA–gene encoding soluble pyridine nucleotide transhydrogenase; Ec.pntAB–gene encoding membrane-bound pyridine nucleotide transhydrogenase; Ec.ppcK620S–gene encoding aspartate/malate insensitive phosphoenolpyruvate carboxylase mutant (Walther et al., 2017a); Ec.thrAS345F–gene encoding threonine-insensitive bifunctional aspartate kinase/HMS dehydrogenase mutant (Walther et al., 2017a). The main glucose uptake pathway in Escherichia coli via the phosphoenolpyruvate-dependent glucose-specific phosphotransferase system (Liang et al., 2015) is marked by a bold arrow. Red crosses mark chromosomal gene deletion, and the red arrow indicates chromosomal overexpression. Bluish-green arrows mark the enzyme activities of synthetic homoserine-dependent pathways provided by plasmid-based expression.
In the absence of annotated naturally occurring enzymes with NADPH-dependent OHB reductase activity, we chose NADH-dependent Ec.Mdh5Q as the template enzyme to engineer the required activity. Previous studies have focused on engineering the nicotinamide cofactor specificity of oxidoreductases following rational approaches [as reviewed by Chánique and Parra (2018)] and typically rely on introducing amino acid substitutions in the co-enzyme binding site. Following this logic, we used comparative sequence and structural analyses supported by the use of a structure-guided web tool (Cahn et al., 2017) to engineer NADPH-dependent OHB reductase activity. The best-performing mutant, assessed by in vitro analysis, was subsequently introduced into a strain that produced DHB via the synthetic homoserine pathway. To demonstrate the full potential of our engineered NADPH-dependent OHB reductase variant and to prevent NADPH becoming a limiting factor in product synthesis, we transferred the DHB pathway into NADPH-overproducing E. coli strains (Figure 1) expressing the improved transaminase variant Ec.alaCA142P:Y275D and showed a 50% increased DHB yield in shake-flask cultivations with glucose as the only carbon source for the most advanced strain.
Unless stated otherwise, chemicals and solvents were purchased from Sigma-Aldrich (Darmstadt, Germany). Restriction enzymes and kits for plasmid DNA isolation, gel DNA extraction, and PCR clean-up were purchased from NEB (Frankfurt am Main, Germany) and used according to the manufacturer’s instructions. Primers were purchased from Sigma-Aldrich. Sanger sequencing was carried out by Genewiz (Leipzig, Germany).
All strains and plasmids used in this study are listed in Tables 1, 2.
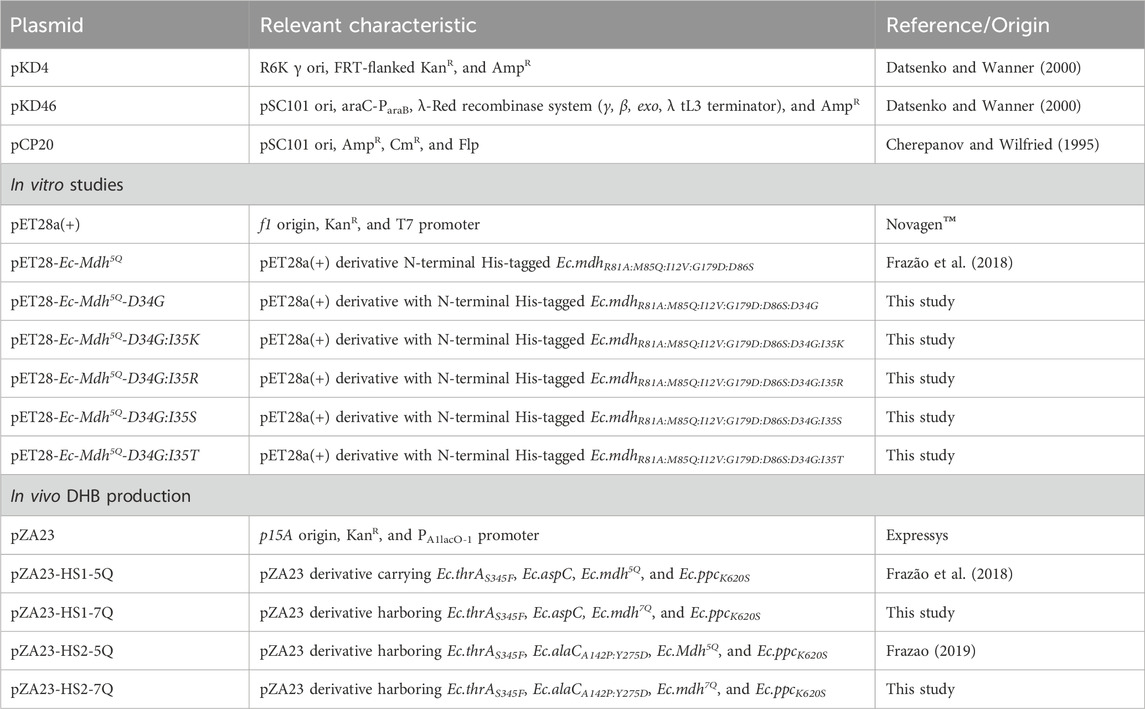
Table 1. Plasmids used in this study.
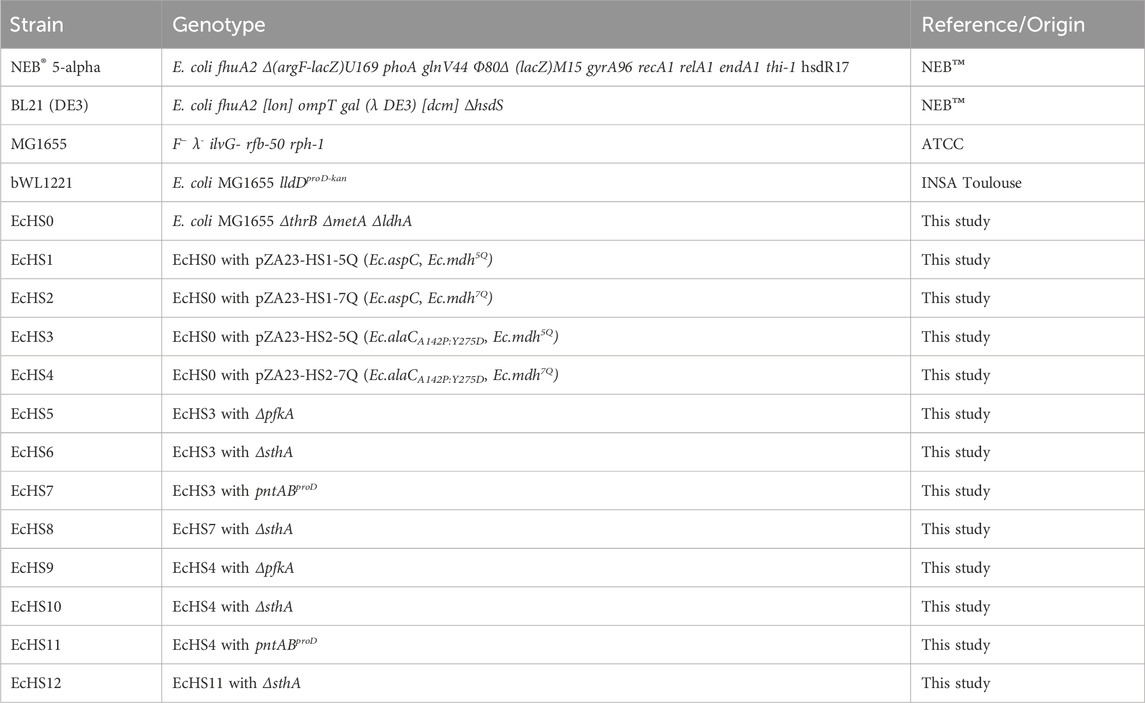
Table 2. Escherichia coli strains used in this study.
For cloning procedures, protein production, and cell recovery from glycerol stocks (30% v/v) kept at −80°C, cells were cultivated in lysogeny broth (LB) medium (10 g L−1 tryptone, 5 g L−1 yeast extract and 10 g L−1 NaCl). LB agar plates were prepared by adding 20 g L−1 agar-agar to liquid LB.
For DHB production studies, cells were cultivated in M9 mineral medium (Walther et al., 2017b), containing 20 g L−1 glucose, 100 mM MOPS (pH adjusted to 7 with KOH), 18 g L−1 Na2HPO4·12H2O, 3 g L−1 KH2PO4, 0.5 g L−1 NaCl, 2 g L−1 NH4Cl, 0.5 g L−1 MgSO4·7 H2O, 0.015 g L−1 CaCl2·2H2O, 0.010 g L−1 FeCl3, 0.012 g L−1 thiamine HCl, and trace elements (0.4 mg L−1 Na2EDTA·2H2O, 1.8 mg L−1 CoCl2·6H2O, 1.8 mg L−1 ZnCl2SO4·7H2O, 0.4 mg L−1 Na2MoO4·2H2O, 0.1 mg L−1 H3BO3, 1.2 mg L−1 MnSO4·H2O, and 1.2 mg L−1 CuCl2·2H2O). (L)-methionine and (L)-threonine were added at a final concentration of 0.2 g L−1 each to compensate for auxotrophies of the production strains. Where required, antibiotics were added at the following final concentrations: carbenicillin, 100 mg L−1, and kanamycin sulfate, 50 mg L−1.
Site-directed mutagenesis was performed via inverse PCR (Zheng et al., 2004) with primer pairs listed in Supplementary Table S1 using 6 ng of vector pET28-Ec.mdh5Q as a template (Frazão et al., 2018). The PCR reaction was performed with Q5 polymerase (NEB). Compared to the manufacturer’s instructions, the PCR cycle number was reduced to 19. After treatment with DpnI enzyme (NEB) to remove residual template DNA, plasmids were transformed into E. coli NEB® 5-alpha, and respective mutations were verified by DNA Sanger sequencing. The constructed plasmids are listed in Table 1.
N-terminally 6x-His-tagged enzymes were produced in E. coli BL21 (DE3) cells harboring respective pET28a expression vectors. A volume of 50 mL of lysogeny broth (LB) medium supplemented with kanamycin in a 250 mL unbaffled shake flask was inoculated at an initial optical density at 600 nm (OD600) of 0.2 from an overnight LB culture. The culture was incubated at 37°C and 220 rpm. Heterologous protein expression was induced when OD600 = 0.6 by the addition of 1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) to the medium. When an OD600 of 2 was reached, cells were harvested by centrifugation (10 min, 4,000 ×g, 4°C), and the cell pellets were stored at −20°C until further analysis. To purify the His-tagged protein, frozen cell pellets were thawed on ice, resuspended in 50 mM HEPES buffer containing 300 mM NaCl (pH = 7.5), and then disrupted using a sonicator (UDS 751, TOPAS, Germany, 4 × 30 s, 30% amplitude). Cell debris was removed from the soluble protein fraction by centrifugation (17,500 ×g, 15 min, 4°C). Upon washing of the Talon™ Cobalt affinity resin (Clontech, United States) according to the supplier’s instructions, the crude extract was added to the resin and incubated in a tube rotator (VWR) at room temperature for 20 min. After binding, a washing step with 50 mM HEPES buffer containing 300 mM NaCl (pH = 7.5) and a subsequent washing step with the same buffer but additionally containing 15 mM imidazole (pH = 7.5) were carried out. Afterward, the protein bound to the resin was eluted with 200 mM imidazole in 50 mM HEPES buffer containing 300 mM NaCl (pH = 7.5) in a final volume of 500 µL. The concentration of purified protein was determined using the Bradford assay (Rotiquant®, Carl Roth), usually yielding a protein concentration of 3–4 mg mL−1 after purification.
OHB was prepared enzymatically starting from (L)-homoserine in a reaction catalyzed by the (L)-amino acid oxidase from Crotalus adamanteus (Sigma-Aldrich, A9253) as described by Wellner and Lichtenberg (1971). The reaction mix contained 125 mM (L)-homoserine, 100 mM Tris-HCl (pH = 7.8), and 4,374 U mL−1 catalase from Aspergillus niger (Sigma-Aldrich, C3515), and 4.7 U mL−1 (L)-amino acid oxidase. Control reactions were performed with the same reaction mix but in the absence of (L)-homoserine or (L)-amino acid oxidase, respectively. The reaction was performed for 4.5 h at 37°C and 220 rpm. After incubation on ice for 1 h, enzymes were removed using Amicon® Ultra Centrifugal filters (cut-off, <10 kDa; Merck, Germany) for 45 min at 4°C. Quantification of OHB was performed based on a ketone calibration curve (0–300 mM pyruvate). A volume of 100 µL of standard/sample was mixed with 1 mL of a solution containing 1 M sodium arsenate and 1 M boric acid (pH = 6.5). After incubation at room temperature for 30 min, the absorbance at 325 nm was measured. The absence of homoserine was further confirmed by high-performance liquid chromatography (HPLC) analysis.
Enzymatic assays with purified enzyme were conducted at 37°C in 96-well flat-bottomed microtiter plates with a final reaction volume of 250 µL per well. The reaction kinetics were monitored in a microplate reader (NanoQuant Plate™, Infinite M200 PRO, TECAN) by following the characteristic absorption of NAD(P)H at 340 nm. The reaction mixture contained 0.25 mM NAD(P)H, 60 mM Hepes (pH 7.0, adjusted with 5 M KOH), 5 mM MgCl2, 50 mM KCl, and appropriate amounts of the purified enzyme. Reactions were started by adding 2 mM OHB to assess the cofactor preference of the constructed enzyme variants. To determine the kinetic constants on the substrate of Ec.Mdh5Q and top-performing NADPH-dependent OHB reductase, specific activities were determined at variable OHB concentrations (0.005–10 mM) and 0.25 mM of the preferred co-substrate. In order to estimate the kinetic constants on the cofactors, specific activities were determined at fixed amounts of substrate (OHB, 2 mM) and variable amounts of NAD(P)H (0.03–1 mM). Experimental data were fitted to the Michaelis–Menten model or to the substrate inhibition model using non-linear regression (Curve fitting tool, MATLAB R2021a). One unit (U) is defined as the amount of enzyme that catalyzes the conversion of 1 µmol of NAD(P)H per minute at pH 7.0 and 37°C. Km,app [mM] is defined as the apparent Michaelis–Menten constant, Ki [mM] is the substrate inhibition constant, vmax,app [U mg−1] is the apparent maximum reaction speed, and kcat,app is the apparent catalytic constant [s−1]. The catalytic efficiency is described by kcat/Km [mM s−1] (Chmiel, 2018). The specificity for NADPH is described as (kcat,app/Km,app)NADPH/(kcat,app/Km,app)NADH, calculated from the mean values of kcat and Km (Cahn et al., 2017). We furthermore define the overall catalytic efficiency with respect to substrate conversion in the presence of the preferred cofactor as (kcat,app/Km,app)OHB × (kcat,app/Km,app)NAD(P)H.
All plasmids constructed and used for in vivo DHB synthesis are listed in Table 1. The vectors are based on the pZA23 backbone of the pZ expression system (Expressys). Plasmids pZA23-HS1-5Q (Ec.aspC, Ec.mdh5Q) and pZA23-HS2-5Q (Ec.alaCA142P:Y275D, Ec.mdh5Q) were a kind gift of Prof. J. M. François from Toulouse Biotechnology Institute, INSA Toulouse, France, and served as the basis for gene replacements. To replace OHB reductase-encoding gene Ec.mdh5Q, the gene Ec.mdh7Q was first amplified by PCR from the corresponding pET28 vector using the primers TW2949 and TW2427 listed in Supplementary Table S1, thereby introducing 5′ overhangs containing NotI and XbaI restriction sites. Backbone vectors pZA23-HS1-5Q and pZA23-HS2-5Q and insert were digested with the restriction enzymes NotI and XbaI (NEB). The digested backbone was further treated with Antarctic phosphatase (NEB). After DNA purification by gel extraction (Gel Extraction Kit, NEB), the backbone and insert were ligated using T4 DNA ligase (NEB) according to the provider’s protocol. After verification via sequencing, plasmids were transformed into appropriate host strains.
All E. coli strains constructed and used are listed in Table 2. Chromosomal gene deletions in E. coli MG1655 were introduced by P1vir phage transduction (Lennox, 1955) using single-gene knockout mutants from the Keio collection (Baba et al., 2006) as donor strains. After transduction, the kanamycin resistance cassettes were removed using flippase (FLP) recombinase-catalyzed excision. The FLP recombinase was expressed from pCP20 (Cherepanov and Wilfried, 1995). Gene deletions and successful removal of resistance cassettes were confirmed by diagnostic PCR (Primers listed in Supplementary Table S1) using DreamTaq polymerase (ThermoFisher Scientific), following the protocol provided by the manufacturer. Afterward, a new round of chromosomal modification was initiated, and the procedure was repeated until all target deletions were introduced.
For chromosomal overexpression of Ec.pntAB, the native chromosomal 5′-untranslated region of the gene was replaced by the insulated constitutive promoter proD (Davis et al., 2011) via PCR-mediated λ-Red recombination following the protocol of Datsenko and Wanner (2000). The FRT-kan-FRT cassette fused to proD promoter sequence with 50 bp 5′-extensions homologous to the target genomic locus was amplified by PCR (primers are listed in Supplementary Table S1) from the genomic DNA of strain bWL1221 in our lab collection (kindly provided by Prof. J. M. François from Toulouse Biotechnology Institute, INSA Toulouse, France). Recipient cells were transformed with the helper plasmid pKD46 (Datsenko and Wanner, 2000) for expression of λ-Red recombination genes and with linearized, gel-purified PCR product. Successful integration was confirmed by colony PCR, as described above. After kan-cassette removal using FLP recombinase expressed from pCP20, the chromosomal promoter exchange was furthermore confirmed via DNA sequencing.
All cell cultivations were performed at 37°C, 220 rpm in an orbital shaker (Ecotron, Infors). First, pre-cultures were inoculated with a single colony picked from an LB agar plate and cultivated in 3 mL LB media (15 mL Falcon tube lying flat), and 50 μg mL−1 kanamycin was added to the strains harboring pZA23 plasmids. After 8 h, a volume of 0.5 mL of the first pre-culture was transferred to 10 mL of M9 mineral medium, supplemented with (L)-methionine (0.2 g L−1), (L)-threonine (0.2 g L−1), and kanamycin. After 16 h of cultivation, cells were harvested by centrifugation in a table-top centrifuge (5 min, 6,000 ×g, room temperature). The main cultures were carried out in 25 mL M9 media supplemented with (L)-methionine (0.2 g L−1), (L)-threonine (0.2 g L−1), and kanamycin in 250 mL baffled shake flasks. The main cultures were inoculated with the harvested cells at a starting OD600 of 0.2. When an OD600 of ∼0.6 was reached, 1 mM IPTG was added to induce the expression of pathway genes. Samples were regularly withdrawn and centrifuged (2 min, 16,000 ×g, room temperature), and the supernatant was kept at −20°C until further analysis.
Extracellular metabolites (glucose, DHB, acetate, and lactate) in supernatant samples from cultivations were analyzed using HPLC. The samples (1 mL) were filter sterilized with 0.2 µm filters, transferred into 2 mL HPLC sample vials, and subsequently analyzed with the Dionex UltiMate 3000 UHPLC system (Thermo Scientific). The device was equipped with an RI and UV/Vis detector. For separation, a Rezex™ ROA-Organic Acid H+ (8%) column (Phenomenex) with a size of 300 mm × 7.8 mm was used, protected by a SecurityGuard™ Carbo H+ pre-column (4 mm × 3 mm, Phenomenex). A sample volume of 20 µL was injected, and analytes were eluted using 0.5 mM H2SO4 as mobile phase, with a flow rate of 0.5 mL min−1. The column oven temperature was set to 80°C, and the temperature of the autosampler was set to 6°C.
Depending on the concentration range, the presence of DHB was verified by LC/MS analyses using our previously described method (Frazão et al., 2023). The LC/MS platform consists of a Vanquish and a Thermo Scientific™ Q Exactive™ Focus (ThermoFisher Scientific), controlled by Xcalibur software (version 2.1, ThermoFisher Scientific). Separation by liquid chromatography was achieved using a Rezex RoA-organic acid H+ (8%) resin-based column preceded by a SecurityGuard guard cartridge (Phenomenex) held at 80°C with 0.1% formic acid as the mobile phase. The temperature of the autosampler was kept at 6°C, the injection volume was 20 µL, and an isocratic flow of 0.4 mL min−1 was adjusted. Peak areas were corrected for the contribution of all naturally abundant isotopes using the software IsoCor (version 2.2.0) (Millard et al., 2012).
Multiple sequence alignment of NAD(P)H-dependent malate dehydrogenases and lactate dehydrogenases was performed using MAFFT (v 7.525) provided by EMBL-EBI (Madeira et al., 2024). UniProt identifiers of all proteins used in the alignment are listed in the supplementary material. Structure alignments were performed in PyMol v2.5.1 (http://www.pymol.org/pymol). Three-dimensional structures were retrieved from the Protein Data Bank (PDB) or predicted with AlphaFold Colab (Jumper et al., 2021).
We have previously engineered a highly active OHB reductase using the NAD+-dependent (L)-malate dehydrogenase from E. coli (Ec.Mdh; UniProtKB code P61889) as a template enzyme. The best-performing variant, Ec.Mdh5Q, contains five point mutations (I12V:R81A:M85Q:D86S:G179D) and displays a 108-fold higher catalytic efficiency with NADH than with NADPH (Figure 2A). Because the typical intracellular ratios of [NAD(P)H]/[NAD(P)] in E. coli (Bennett et al., 2009) indicate that NADPH should be preferentially used as a cofactor in reduction processes under aerobic conditions, we set out to engineer an NADPH-dependent OHB reductase using Ec.Mdh5Q as the template enzyme.
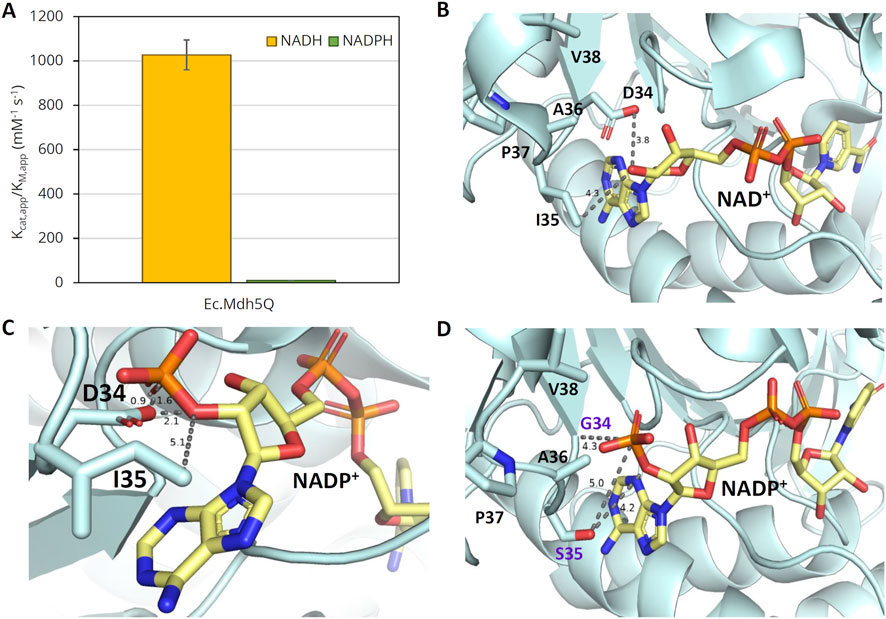
Figure 2. Specific activity of Ec.Mdh5Q on OHB with NAD(P)H and 3D-protein structures with NAD+ or NADP+. (A) Catalytic efficiency (kcat,app/KM,app) of Ec.Mdh5Q with NAD(P)H. Enzymatic activity was measured with purified enzyme at 37°C and pH 7.0 in 96-well flat-bottomed microtiter plates with 2 mM OHB and 0.25 mM NAD(P)H. The reactions were followed by monitoring the NAD(P)H absorption at 340 nm. Error bars indicate the standard deviation of the mean (n = 2). (B) X-ray structure of Ec.Mdh with bound NAD+ (Pdb code: 1emd). Side chains of amino acids in the selectivity control loop are shown (positions 34–38). (C) X-ray structure of Ec.Mdh with superimposed NADP+. The figure was generated by the structural alignment of Ec.Mdh with bound NAD (Pdb code: 1emd) and the malate dehydrogenase from Flaveria bidentis (Fb.Mdh) with bound NADP (Pdb code: 1civ). The structures of Fb-Mdh and NAD were omitted from the representation. (D) Model structure of Ec.Mdh D34G:I35S with superimposed NADP+. The figure was generated by structural alignment of the mutant enzyme model [generated with AlphaFold Colab (Jumper et al., 2021); mutations relative to wt enzyme are marked in violet] and the crystal structure of Fb.Mdh with bound NADP. Subfigures (B–D) were generated in PyMol v2.5.1 (http://www.pymol.org/pymol). The distance between selected residues is shown in Å. The carbon ribbon of cofactors NAD(P)+ is shown in pale yellow. The color scheme of NAD(P)+ and relevant amino acid side chains (position 34–38) is oxygen – red, nitrogen – blue, and sulfur – orange.
NAD+-dependent (L)-malate dehydrogenases (cytosolic; Mdh type 1 family) belong to the large superfamily of (L)-Mdh/(L)-Ldh enzymes, which further includes NADP+-dependent enzymes of identical function (chloroplastic; Mdh type 2 family) and NAD+-dependent (L)-lactate dehydrogenases (Ldh). Multiple sequence alignments between several members of the superfamily of Mdh and Ldh enzymes revealed a strong conservation of the primary protein structure (Supplementary Figure S1). Previous studies elucidated the crucial role of a loop region in a conserved cofactor binding motif of the Rossman fold for cofactor discrimination (Holmberg et al., 1999; Nishiyama et al., 1993; Feeney et al., 1990; Rossmann et al., 1974). Multiple sequence alignments showed that this region corresponds to the amino acid residues at positions 34 and 35 in Ec.Mdh (Supplementary Figure S1). Of crucial interest in Ec.Mdh is the active site residue Asp34, which is pivotal for conferring cofactor specificity because its negative charge has previously been shown to result in electrostatic repulsion of the 2′-phosphate group of NADP(H) (Nishiyama et al., 1993; Feeney et al., 1990). Visual inspection of the X-ray crystal structure of Ec.Mdh (PDB code with bound NAD+: 1emd, Figure 2B) bound with superimposed NADPH (Figure 2C) further revealed a potential steric clash between the aspartate residue at position 34 and the 2′-phosphate moiety of NADPH. Multiple sequence alignment analyses showed strict conservation at the corresponding position in NADP+-dependent malate dehydrogenases with a glycine residue (Supplementary Figure S1). Replacement of Asp34 residue in Ec.Mdh by the smaller and uncharged residue glycine, therefore, seemed crucial to accommodate the larger NADP(H) and to remove the unfavorable electrostatic interaction with NADP(H). In addition, residue Ile35 is of interest for switching the cofactor preference of Ec.Mdh. Due to its close proximity to the 2′-phosphate group of NADPH (Hall and Banaszak, 1993), the amino acid residue located at position 35 is prone to form an electrostatic interaction or a hydrogen bond with the cofactor. Multiple sequence alignment of natural NADPH-dependent Mdh enzymes reveals complete conservation of a potentially hydrogen-bond donating serine residue at the position corresponding to Ile35 in Ec.Mdh. Therefore, we hypothesized that the isoleucine residue at position 35 should be exchanged for serine. This idea was complemented by the structure-guided web tool CSR-salad (Cahn et al., 2017), which suggested an exchange of Ile35 by positively charged residues (Lys, Arg) or other polar uncharged amino acids (Thr) to allow for an electrostatic or polar interaction with the 2′-phosphate group of NADPH. Visual inspection of the predicted 3D structure of a respective Ec.Mdh D34G:I35S double mutant bound with NADP+ confirms the predicted absence of steric clashes in the selectivity control loop (Figure 2D).
Point mutations to switch cofactor preference were stepwise introduced into the Ec.mdh5Q gene by site-directed mutagenesis. The constructed N-terminally 6x-His-tagged variants were expressed from pET28a vectors transformed in E. coli BL21(DE3) cells. After purification, the specific activity of the purified variants was first quantified in the presence of OHB (2 mM) and either NADH or NADPH (0.25 mM) as cofactor. As shown in Figure 3, the template enzyme Ec.Mdh5Q was highly active on OHB with NADH as a co-substrate (68 ± 4 U mg−1) but displayed low activity in the presence of NADPH (4 ± 0.2 U mg−1). For all of the engineered mutants, the cofactor preference (here defined as v(NADPH)/v(NADH)) was found to be altered. Replacement of the Asp34 residue by glycine resulted in comparable OHB reductase activity in the presence of both cofactors (v(NADH) = 23 ± 2 U mg−1, v(NADPH) = 35 ± 3 U mg−1), possibly indicating dual cofactor preference. The additional substitution of isoleucine at position 35 by serine, threonine, lysine, or arginine resulted in at least 2.5-fold higher activities in the presence of NADPH than NADH. With 64 ± 4.5 U mg−1, the specific OHB reductase activity of Ec.Mdh5Q D34G:I35R (hereafter abbreviated as Ec.Mdh7Q) with NADPH as a cofactor was nearly six-fold higher than with NADH (12 ± 0.02 U mg−1). The mutant variant showed a similar specific activity on OHB when compared to that of Ec.Mdh5Q. Thus, Ec.Mdh7Q was identified as the most promising NADPH-dependent OHB reductase enzyme, and its kinetic parameters on both cofactors and OHB were determined and compared to those of Ec.Mdh5Q.
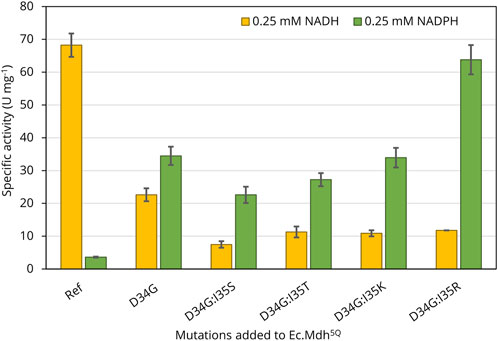
Figure 3. Specific NAD(P)H-dependent OHB reductase activity of engineered variants derived from Ec.Mdh5Q. The enzymes were produced from E. coli BL21 (DE3) harboring the respective pET28a expression vectors cultivated in 50 mL LB. Heterologous expression was induced at OD600 of 0.6 with 1 mM IPTG. Protein expression was carried out until OD600 = 2 was reached. Enzymatic assays were performed with purified enzyme at 37°C and pH 7.0 in 96-well flat-bottomed microtiter plates with 2 mM OHB and 0.25 mM NAD(P)H. The reactions were followed by monitoring the NAD(P)H absorption at 340 nm. Error bars indicate the standard deviation of the mean (n = 2).
The kinetic parameters of both enzyme variants are summarized in Table 3. The engineered variant Ec.Mdh7Q exhibited more than three orders of magnitude higher specificity (defined as (kcat/Km(NADPH))/(kcat/Km(NADH)) for NADPH than Ec.Mdh5Q (16 and 0.01, respectively). No loss in OHB affinity or impairment of the overall catalytic efficiency ((kcat/Km)OHB × (kcat/Km)NAD(P)H) was observed. We observed uncompetitive substrate inhibition of Ec.Mdh7Q by OHB (Ki = 5.5 ± 0.5 mM). Substrate inhibition was found to be stronger than for Ec.Mdh5Q (Ki = 31.9 mM ± 5.6 mM) (Frazão et al., 2018).
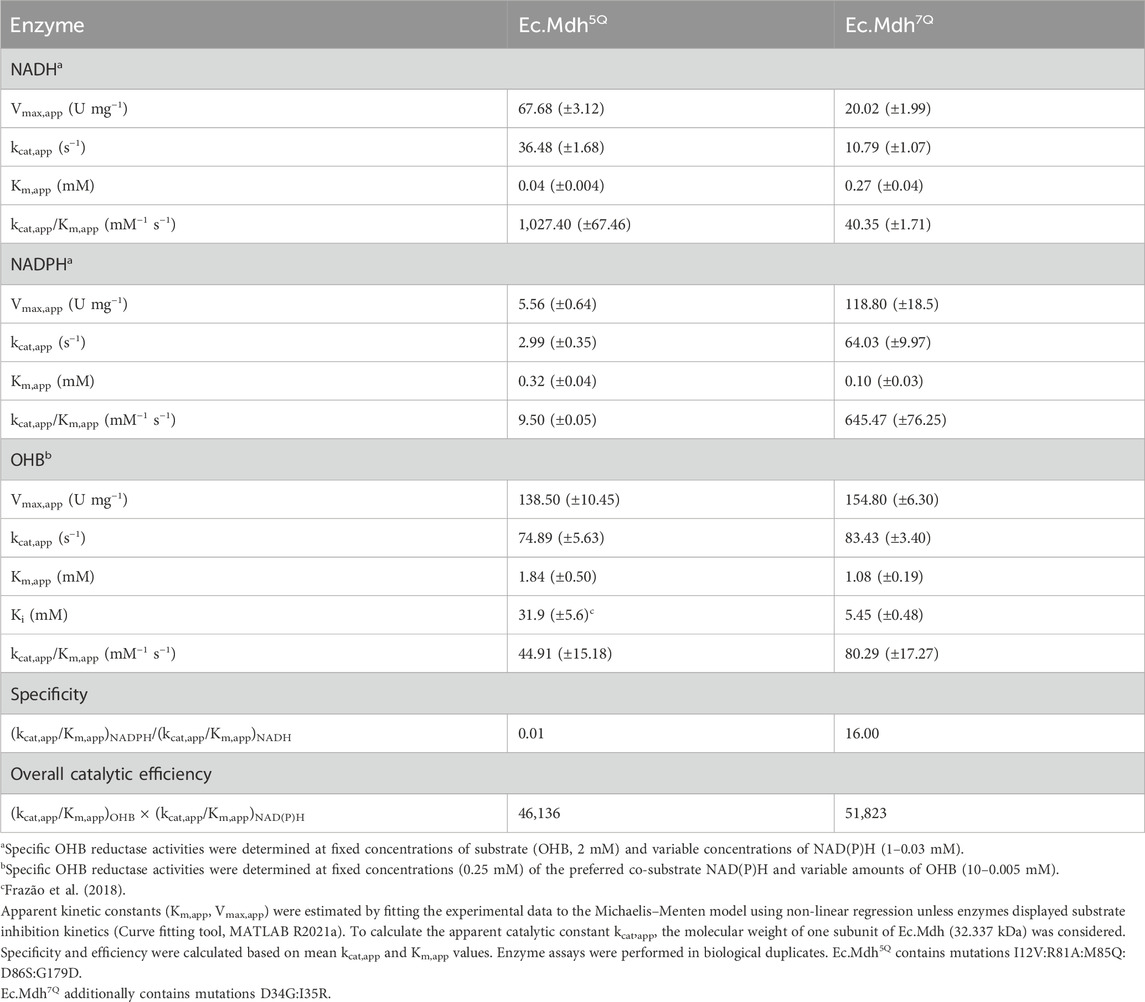
Table 3. Kinetic analysis of OHB reductases Ec.Mdh5Q and Ec.Mdh7Q.
Next, the in vivo performance of the NADPH-dependent OHB reductase Ec.Mdh7Q for DHB biosynthesis was investigated. To achieve DHB production from glucose, we selected E. coli MG1655 ΔthrB ΔmetA ΔldhA as a production host to ensure a sufficient supply of the homoserine precursor. The host was then equipped with a DHB pathway consisting of homoserine transaminase activity (catalyzed by Ec.AspC from E. coli), and NAD(P)H-dependent OHB reductase activity (either Ec.Mdh5Q or Ec.Mdh7Q) expressed from a medium-copy plasmid pZA23 under the control of the ITPG-inducible PA1lacO-1 promoter. Constructed plasmids additionally carried threonine-insensitive bifunctional aspartate kinase/homoserine dehydrogenase (Ec.ThrAS345F) and the aspartate/malate insensitive phosphoenolpyruvate carboxylase variant Ec.PpcK620S (Frazão et al., 2018) (see the metabolic setting in Figure 1).
Producer strains were cultivated in 25 mL M9 mineral medium containing 20 g L−1 glucose. (L)-methionine and (L)-threonine were added to the medium (at a final concentration of 0.2 g L−1 each) to compensate for the host strain’s auxotrophies. Expression of pathway genes was induced by IPTG (1 mM) at the mid-exponential phase, and DHB production was quantified after 24 h of cell cultivation. Upon expression of Ec.Mdh5Q, we found the culture supernatant to contain 7.0 ± 0.5 mM DHB (EcHS1, Figure 4), reaching a product yield of 0.10 ± 0.004 molDHB molGlucose−1 and a volumetric productivity of 0.29 ± 0.02 mmolDHB L−1 h−1. Strain EcHS2 expressing Ec.Mdh7Q was able to produce 7.5 ± 0.6 mM DHB. Because only a modest increase in DHB production was observed with NADPH-dependent OHB reductase, we speculated at this stage that homoserine transaminase activity was limiting. Indeed, purified Ec.AspC enzyme has previously been shown to display only low in vitro activity on (L)-homoserine (0.082 U mg−1), and it could not be saturated at substrate concentrations of up to 50 mM (L)-homoserine (Walther et al., 2017a; Walther et al., 2017b). Crucially, however, Bouzon and co-workers (2017) previously disclosed a homoserine transaminase (Ec.AlaCA142P:Y275D) with a much higher affinity for homoserine (Km = 1.7 mM). Therefore, we reanalyzed the effect of the different OHB reductases when co-expressing Ec.AlaCA142P:Y275D in the producer strains. Indeed, co-expression of Ec.Mdh5Q and Ec.AlaCA142P:Y275D (EcHS3) led to a DHB titer of 13.8 ± 0.5 mM, which constitutes roughly a 2-fold improvement over the use of Ec.AspC. This result confirms the homoserine transaminase activity of Ec.AspC as a rate-limiting step in the pathway, and we retained the transaminase Ec.AlaCA142P:Y275D for all subsequent experiments. Replacement of Ec.Mdh5Q by NADPH-dependent OHB reductase led to the production of 17.2 ± 0.2 mM (EcHS4), which represents a 25% improvement compared to the corresponding strain with the NADH-dependent variant. By co-expressing Ec.alaCA142P:Y275D and Ec.mdh7Q genes, a maximum DHB product yield of 0.20 ± 0.005 molDHB molGlucose−1 and a volumetric productivity of 0.72 ± 0.01 mmolDHB L−1 h−1 were achieved. Analysis of the enantiomeric purity of DHB revealed the presence of only the L-form of the organic acid (data not shown).
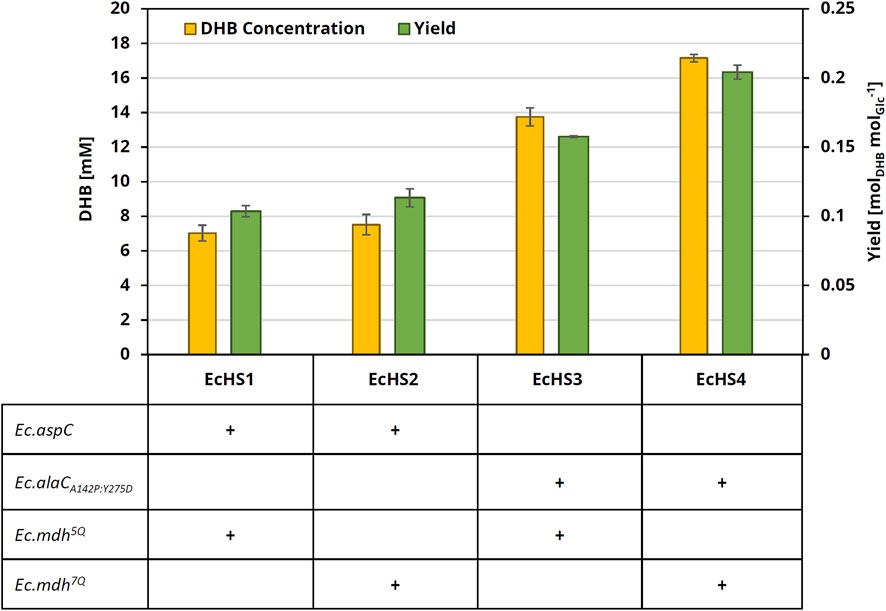
Figure 4. DHB concentration and yield after 24 h cultivation of engineered production strains. All strains are derived from the parental strain E. coli MG1655 ΔthrB ΔmetA ΔldhA, which was transformed with medium-copy DHB production plasmids expressing homoserine transaminase (Ec.aspC or Ec.alaCA142P:Y275D) and OHB reductase (Ec.Mdh5Q or Ec.Mdh7Q). All plasmids further carry Ec.thrAS345F and Ec.ppcK620S. Cultivation was performed at 37°C and 220 rpm in 250 mL baffled flasks containing 25 mL M9 medium with 20 g L−1 glucose, supplemented with 0.2 g L−1 (L)-methionine and 0.2 g L−1 (L)-threonine. The expression of pathway genes was induced at an OD600 of 0.6 with 1 mM IPTG. DHB titers and yields after 24 h of cultivation are shown. The experiments were performed in biological duplicates. Error bars indicate the standard deviation of the mean.
To demonstrate the full potential of NADPH-dependent OHB reductase toward DHB production, we next engineered the host strain E. coli MG1655 ΔthrB ΔmetA ΔldhA toward increased NADPH supply. We selected multiple chromosomal targets previously shown to increase the intracellular availability of NADPH, including the deletion of the Ec.pfkA gene (encoding 6-phosphofructokinase I) to enhance flux through the pentose phosphate pathway (Chin and Cirino, 2011). We further altered the expression of the transhydrogenase system toward the formation of NADPH by deleting soluble pyridine nucleotide transhydrogenase Ec.SthA (NAD+ + NADPH → NADH + NADP+; Boonstra et al., 1999; Sauer et al., 2004) and/or overexpressing membrane-bound pyridine nucleotide transhydrogenase Ec.PntAB (NADH + NADP+ → NAD+ + NADPH; Clarke et al., 1986; Sauer et al., 2004). The resulting host strains were then used to characterize the impact of increased NADPH availability on DHB production with the NADPH-dependent OHB reductase. To investigate potential effects on DHB production that may be caused by altered homoserine availability, we additionally tested Ec.Mdh5Q.
After 24 h of cultivation in glucose-containing mineral medium, the producer strains with the chromosomal deletion of the Ec.pfkA gene and expressing the NADH-dependent Ec.mdh5Q (EcHS5) exhibited a severe drop of both DHB concentration and yield when compared to the reference strain EcHS3 (Figure 5). This may be related to the observed growth defect caused by Ec.pfkA deletion (data not shown). With EcHS6 deleted for Ec.sthA, the DHB yield was equal to 0.19 ± 0.03 molDHB molGlucose−1, which corresponds to a 19% increase when compared to reference strain EcHS3 with a yield of 0.16 mol ± 0.001 molDHB molGlucose−1. Upon chromosomal overexpression of Ec.pntAB (EcHS7), the product yield was further increased to 0.21 ± 0.007 molDHB molGlucose−1, which corresponds to a total increase of 30% compared to the reference strain (EcHS3). The additional deletion of Ec.sthA did not provide an advantage and indeed caused the DHB yield to drop to 0.18 ± 0.008 molDHB molGlucose−1.
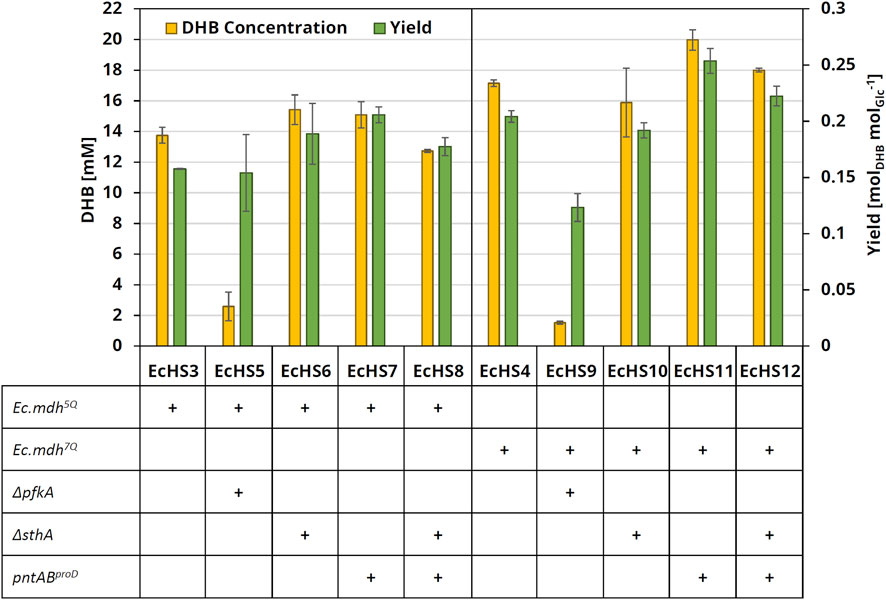
Figure 5. DHB concentration and yield after 24 h cultivation of engineered production strains with increased NADPH availability. All strains were derived from the parental strain E. coli MG1655 ΔthrB ΔmetA ΔldhA, which was transformed with medium-copy DHB production plasmids expressing homoserine transaminase (Ec.alaCA142P:Y275D) and OHB reductase (Ec.mdh5Q or Ec.mdh7Q). All plasmids further carried Ec.thrAS345F and Ec.ppcK620S. Cultivation was performed at 37°C and 220 rpm in 250-mL baffled flasks containing 25 mL M9 medium with 20 g L−1 glucose, supplemented with 0.2 g L−1 (L)-methionine and 0.2 g L−1 (L)-threonine. Expression of pathway genes was induced at an OD600 of 0.6 with 1 mM IPTG. DHB titers and yields after 24 h of cultivation are shown. The experiments were performed in biological duplicates. Error bars indicate the standard deviation of the mean.
The production strains expressing Ec.mdh7Q displayed similar tendencies as the strains expressing Ec.mdh5Q. However, the positive effect on DHB production upon chromosomal overexpression of Ec.pntAB was enhanced, yielding 0.25 ± 0.01 molDHB molGlucose−1 after 24 h cultivation of EcHS11. Thus, increased NADPH availability via chromosomal overexpression of pntAB promoted DHB synthesis, particularly in the presence of the engineered NADPH-dependent enzyme variant.
To sum up, by co-expression of the improved homoserine transaminase (Ec.alaCA142P:Y275D) with the NADPH-dependent OHB reductase Ec.mdh7Q and by further modification of the production strain (Ec.pntABproD), it was possible to increase the DHB yield by 50% to 0.25 ± 0.01 molDHB molGlucose−1 and reach a volumetric productivity of 0.83 ± 0.03 mmolDHB L−1 h−1 within 24 h of batch cultivation of EcHS11.
In previous work, we used a rational engineering approach to construct a highly active OHB reductase enzyme, catalyzing the last reaction step of the artificial homoserine-dependent DHB synthesis route (Frazão et al., 2018). Because we used the NAD+-dependent E. coli malate dehydrogenase (Ec.Mdh; UniProtKB code P61889) as a template to construct Ec.Mdh5Q (I12V:R81A:M85Q:D86S:G179D), the reduction of OHB in the synthetic pathway relied on NADH as a cofactor. However, typical intracellular ratios of [NAD(P)H]/[NAD(P)] in E. coli (Bennett et al., 2009) indicate that the use of a NADPH-dependent OHB reductase provides a thermodynamic advantage. In this study, we aimed to replace the currently used NADH-dependent OHB reductase by an enzyme with NADPH-dependent activity.
Oxidoreductases with nicotinamide cofactor dependency exhibit a strong preference for either NAD(H) or NADP(H) (Chánique and Parra, 2018). In the large and complex (L)-malate/(L)-lactate dehydrogenase superfamily, most enzyme candidates of microbial origin rely on the NAD(H) cofactor system, typically showing only low residual activities with NADP(H) (Takahashi-Íñiguez et al., 2016; Brochier-Armanet and Madern, 2021). Whereas naturally existing NADP(H)-dependent Ldhs have not yet been identified (Richter et al., 2011), NADP(H)-preferring Mdhs exist in chloroplasts (Lemaire et al., 1996). Indeed, we initially envisaged the construction of a NADPH-dependent OHB reductase using the NADP+-dependent chloroplast malate dehydrogenase originating from Sorghum bicolor (Sb.chMdhP) as a template enzyme (Crétin et al., 1990). Because we did not succeed in the production of a soluble, active enzyme variant in E. coli, we altered our strategy and instead focused on switching the cofactor specificity of previously reported NADH-dependent OHB reductase variant Ec.Mdh5Q toward NADPH.
Previous studies investigating the cofactor specificity of enzymes belonging to the (L)-malate/(L)-lactate dehydrogenase superfamily revealed that only a few amino acid residues are responsible for conferring the preference for either NAD(H) or NADP(H). In attempts to reverse the enzyme cofactor dependency, identifying crucial cofactor-discriminating key positions is usually followed by site-directed mutagenesis of the respective amino acid residues or a loop exchange approach (Chánique and Parra, 2018). Our present study confirms the crucial importance of the amino acid residues at positions 34 and 35 in Ec.Mdh in conferring cofactor preference. Upon replacing Asp34 and Ile35 with glycine and arginine, respectively, the cofactor specificity of the mutant Ec.Mdh7Q was successfully altered from NADH to NADPH. Switching the cofactor specificity while maintaining comparable catalytic efficiency to that of the wild-type enzyme represents a significant challenge. In the majority of previous attempts of switching the cofactor specificity of oxidoreductases from NAD(H) to NADP(H), the catalytic efficiency of the mutant with NADP(H) was severely reduced, reaching less than 50% of the wild type’s catalytic efficiency with NAD(H) (Chánique and Parra, 2018). Although the catalytic efficiency of Ec.Mdh7Q with NADPH was also reduced in the context of this study, it still reached more than 60% of that of the template enzymes with NADH. Further engineering efforts should be made to fully recover the activity of Ec.Mdh7Q with NADPH compared to the template enzyme Ec.Mdh5Q with NADH. More relevant for in vivo DHB pathway operation using the NADPH-dependent variant, the overall catalytic efficiency for OHB reduction in the presence of the preferred cofactor defined as (kcat,app/Km,app)(OHB) × (kcat,app/Km,app)(NAD(P)H) of the mutant Ec.Mdh7Q (on OHB with NADPH) was comparable to the template enzyme Ec.Mdh5Q (on OHB with NADH). Thus, the engineered Ec.Mdh7Q was a promising candidate for in vivo application.
When we first replaced the NADH-dependent OHB reductase with our engineered NADPH-dependent variant for in vivo DHB production via the homoserine-dependent pathway, a positive effect could not be observed. Thus, we speculated that our current transaminase, Ec.AspC, was limiting the pathway flux (Figure 4). By exchanging Ec.AspC with the alanine aminotransferase double mutant Ec.AlaCA142P:Y275D (Bouzon et al., 2017) and co-expression with the NADH-dependent Ec.Mdh5Q, we could indeed show a two-fold improvement in DHB production, reaching a concentration of 13.8 ± 0.5 mM. Upon co-expression of Ec.AlaCA142P:Y275D and Ec.Mdh7Q, we could even reach up to 17.2 ± 0.2 mM DHB and a product yield of 0.20 ± 0.005 molDHB molGlucose−1 after 24 h of cultivation. The results show the superior in vivo performance of the engineered NADPH-dependent OHB reductase variant but also clearly indicate the crucial role of sufficient transaminase activity for efficient pathway operation. However, it must be considered that the affinity of the engineered OHB reductase variant for the synthetic substrate might still be insufficient (Km = 1.08 ± 0.19 mM), as intracellular OHB concentrations are expected to be in the range of sub-mM levels (Walther et al., 2017a). Due to the reversibility of the transaminase reaction and the high intracellular glutamate concentrations (Bennett et al., 2009), the availability of an effective OHB reductase with high affinity toward the synthetic substrate and high overall catalytic efficiency is crucial to shift the in vivo pathway flux in the direction of the target product DHB. Thus, further engineering might be required to reduce the Km of the engineered OHB reductase variant.
Several studies showed how increased NADPH availability resulted in higher yields and productivities of NADPH-dependent product formations. To show the full potential of our engineered NADPH-dependent OHB reductase, we engineered the host strain toward increased NADPH availability using common strategies, including chromosomal deletion of Ec.pfkA to increase the flux through the PPP, deletion of Ec.sthA, and chromosomal overexpression of Ec.pntAB (Kabus et al., 2007; Rathnasingh et al., 2012; Shi et al., 2013; Cui et al., 2014; Cabulong et al., 2019; Hao et al., 2020; Chen et al., 2024; Zhang et al., 2021; Chin and Cirino, 2011). To investigate the potential effects of increased precursor supply, we also evaluated DHB production in NADPH-overproducing host strains expressing the NADH-dependent OHB reductase Ec.mdh5Q. Noticeably, we observed a 19% increased DHB yield in our producer strain EcHS5 deleted for sthA, expressing Ec.alaCA142P:Y275D and Ec.mdh5Q and a 25% yield increase in the strain EcHS6 overexpressing pntAB compared to the respective reference strain EcHS3 (0.16 mol ±0.001 molDHB molGlucose−1). These improvements could possibly be ascribed to an enhancement of homoserine production, as two mol of NADPH are necessary to produce one mol of homoserine. However, the positive effect of engineering NADPH supply via pntAB overexpression was clearly strongest in the producer strain EcHS11 expressing the engineered NADPH-dependent Ec.mdh7Q, reaching the highest DHB yield on glucose (0.25 ± 0.01 molDHB molGlucose−1) reported so far. Although much higher DHB yields starting from homoserine have been reported (0.94 mol mol−1) with high-cell densities in recent work of Liu et al. (2022), such an approach requires the separate fermentation of L-homoserine from glucose, thereby increasing production costs. Furthermore, to the best of our knowledge, the authors did not report yields taking into account the use of the initial glucose substrate. In this study, we report a 50% increase in DHB yield with producer strain EcHS11, compared to the reference EcHS1 (0.10 ± 0.004 molDHB molGlucose−1). It would be worthwhile investigating whether DHB titers could be increased further through enhanced NADPH availability. In a previous study, Ng et al. (2015) presented a promising strategy to increase the NADPH regeneration rate in E. coli by 25-fold, based on the heterologous expression of a synthetic Entner–Doudoroff pathway.
In a larger, more general context, our study shows how streamlining NADPH cofactor preference of the biosynthetic pathway (by enzyme engineering) and NADPH cofactor supply (by metabolic engineering) can increase the efficiency of aerobic biosyntheses of reduced compounds.
The original contributions presented in the study are included in the article/Supplementary Material; further inquiries can be directed to the corresponding author.
NI: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Validation, Visualization, Writing – original draft, Writing – review & editing. LG: Investigation, Writing – review & editing. CA: Investigation, Writing – review & editing. TN: Investigation, Writing – review & editing. TW: Conceptualization, Funding acquisition, Methodology, Project administration, Resources, Supervision, Writing – original draft, Writing – review & editing. CF: Conceptualization, Investigation, Methodology, Project administration, Supervision, Writing – original draft, Writing – review & editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This study was supported by a grant from ERA-CoBioTech (ID: 20) awarded to TW. This project is co-funded by the European Union and co-financed from tax revenues on the basis of the budget adopted by the Saxon State Parliament (Project number 100549942). The LC/MS system was in part founded by the Deutsche Forschungsgemeinschaft (INST 269/792-1 FUGG).
The authors thank Prof. Jean-Marie François for the helpful discussions and for kindly providing the strain bWL1221.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fbioe.2025.1504785/full#supplementary-material
Baba, T., Ara, T., Hasegawa, M., Takai, Y., Okumura, Y., Baba, M., et al. (2006). Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2, 2006.0008. doi:10.1038/msb4100050
Bennett, B. D., Kimball, E. H., Gao, M., Osterhout, R., Van Dien, S. J., and Rabinowitz, J. D. (2009). Absolute metabolite concentrations and implied enzyme active site occupancy in Escherichia coli. Nat. Chem. Biol. 5 (8), 593–599. doi:10.1038/nchembio.186
Boonstra, B., French, C. E., Wainwright, I., and Bruce, N. C. (1999). The UdhA gene of Escherichia coli encodes a soluble pyridine nucleotide transhydrogenase. J. Bacteriol. 181 (3), 1030–1034. doi:10.1128/JB.181.3.1030-1034.1999
Bouzon, M., Perret, A., Loreau, O., Delmas, V., Perchat, N., Jean, W., et al. (2017). A synthetic alternative to canonical one-carbon metabolism. ACS Synth. Biol. 6 (8), 1520–1533. doi:10.1021/acssynbio.7b00029
Brochier-Armanet, C., and Madern, D. (2021). Phylogenetics and biochemistry elucidate the evolutionary link between L-malate and L-lactate dehydrogenases and disclose an intermediate group of sequences with mix functional properties. Biochimie 191, 140–153. doi:10.1016/j.biochi.2021.08.004
Cabulong, R. B., Valdehuesa, K. N. G., Bañares, A. B., Ramos, K. R. M., Nisola, G. M., Lee, W. K., et al. (2019). Improved cell growth and biosynthesis of glycolic acid by overexpression of membrane-bound pyridine nucleotide transhydrogenase. J. Industrial Microbiol. Biotechnol. 46 (2), 159–169. doi:10.1007/s10295-018-2117-2
Cahn, J. K. B., Werlang, C. A., Baumschlager, A., Brinkmann-Chen, S., Mayo, S. L., and Arnold, F. H. (2017). A general tool for engineering the NAD/NADP cofactor preference of oxidoreductases. ACS Synth. Biol. 6 (2), 326–333. doi:10.1021/acssynbio.6b00188
Chánique, A. M., and Parra, L. P. (2018). Protein engineering for nicotinamide coenzyme specificity in oxidoreductases: attempts and challenges. Front. Microbiol. 9, 194. doi:10.3389/fmicb.2018.00194
Chen, Y., Huang, L., Yu, T., Yao, Y., Zhao, M., Pang, A., et al. (2024). Balancing the AspC and AspA pathways of Escherichia coli by systematic metabolic engineering strategy for high-efficient L-homoserine production. ACS Synth. Biol. 13, 2457–2469. doi:10.1021/acssynbio.4c00208
Cherepanov, P. P., and Wilfried, W. (1995). Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of flp-catalyzed excision of the Antibiotic-resistance determinant. Gene 158 (1), 9–14. doi:10.1016/0378-1119(95)00193-A
Chin, J. W., and Cirino, P. C. (2011). Improved NADPH supply for xylitol production by engineered Escherichia coli with glycolytic mutations. Biotechnol. Prog. 27 (2), 333–341. doi:10.1002/btpr.559
Chmiel, H. (2018). Bioprozesstechnik. 4th edn, Editors H. Chmiel, R. Takors, and D. Weuster-Botz (Berlin, Heidelberg: Springer Berlin Heidelberg). doi:10.1007/978-3-662-54042-8
Clarke, D. M., Loo, T. W., Gilliam, S., and Bragg, P. D. (1986). Nucleotide sequence of the PntA and PntB genes encoding the pyridine nucleotide transhydrogenase of Escherichia coli. Eur. J. Biochem. 158 (3), 647–653. doi:10.1111/j.1432-1033.1986.tb09802.x
Crétin, C., Luchetta, P., Joly, C., Decottignies, P., Lepiniec, L., Gadal, P., et al. (1990). Primary structure of Sorghum malate dehydrogenase (NADP) deduced from CDNA sequence. Eur. J. Biochem. 192 (2), 299–303. doi:10.1111/j.1432-1033.1990.tb19227.x
Cui, Y. Y., Chen, L., Yuan, Y. Z., Huang, J., and Liu, J. Z. (2014). Production of shikimic acid from Escherichia coli through chemically inducible chromosomal evolution and cofactor metabolic engineering. Microb. Cell Factories 13 (1), 1–11. doi:10.1186/1475-2859-13-21
Datsenko, K. A., and Wanner, B. L. (2000). One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. 97 (12), 6640–6645. doi:10.1073/pnas.120163297
Davis, J. H., Rubin, A. J., and Sauer, R. T. (2011). Design, construction and characterization of a set of insulated bacterial promoters. Nucleic Acids Res. 39 (3), 1131–1141. doi:10.1093/nar/gkq810
Feeney, R., Clarke, A. R., and Holbrook, J. J. (1990). A single amino acid substitution in lactate dehydrogenase improves the catalytic efficiency with an alternative coenzyme. Biochem. Biophysical Res. Commun. 166 (2), 667–672. doi:10.1016/0006-291X(90)90861-G
François, J. M. (2023). Progress advances in the production of bio-sourced methionine and its hydroxyl analogues. Biotechnol. Adv. 69, 108259. doi:10.1016/j.biotechadv.2023.108259
Frazao, C. J. R. (2019). Refactoring metabolic pathways for synthon production from renew-able carbon sources. Agricultural sciences. INSA de Toulouse
Frazão, C. J. R., Topham, C. M., Malbert, Y., François, J. M., and Walther, T. (2018). Rational engineering of a malate dehydrogenase for microbial production of 2,4-dihydroxybutyric acid via homoserine pathway. Biochem. J. 475 (23), 3887–3901. doi:10.1042/BCJ20180765
Frazão, C. J. R., Trichez, D., Serrano-Bataille, H., Dagkesamanskaia, A., Topham, C. M., Walther, T., et al. (2019). Construction of a synthetic pathway for the production of 1,3-propanediol from glucose. Sci. Rep. 9 (1), 11576. doi:10.1038/s41598-019-48091-7
Frazão, C. J. R., Wagner, N., Kenny, R., and Thomas, W. (2023). Construction of a synthetic metabolic pathway for biosynthesis of 2,4-dihydroxybutyric acid from ethylene glycol. Nat. Commun. 14 (1), 1931. doi:10.1038/s41467-023-37558-x
Hall, M. D., and Banaszak, L. J. (1993). Crystal structure of a ternary complex of Escherichia coli malate dehydrogenase citrate and NAD at 1·9 Å resolution. J. Mol. Biol. 232, 213–222. doi:10.1006/jmbi.1993.1377
Hao, Y., Ma, Q., Liu, X., Fan, X., Men, J., Wu, H., et al. (2020). High-yield production of L-valine in engineered Escherichia coli by a novel two-stage fermentation. Metab. Eng. 62, 198–206. doi:10.1016/j.ymben.2020.09.007
Holmberg, N., Ryde, U., and Bülow, L. (1999). Redesign of the coenzyme specificity in L-lactate dehydrogenase from Bacillus stearothermophilus using site-directed mutagenesis and media engineering. Protein Eng. 12 (10), 851–856. doi:10.1093/protein/12.10.851
Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., et al. (2021). Highly accurate protein structure prediction with AlphaFold. Nature 596 (7873), 583–589. doi:10.1038/s41586-021-03819-2
Kabus, A., Georgi, T., Wendisch, V. F., and Bott, M. (2007). Expression of the Escherichia coli PntAB genes encoding a membrane-bound transhydrogenase in corynebacterium glutamicum improves l-lysine formation. Appl. Microbiol. Biotechnol. 75 (1), 47–53. doi:10.1007/s00253-006-0804-9
Lemaire, M., Miginiac-Maslow, M., and Decottignies, P. (1996). The catalytic site of chloroplastic NADP-dependent malate dehydrogenase contains a his/Asp pair. Eur. J. Biochem. 236 (3), 947–952. doi:10.1111/j.1432-1033.1996.00947.x
Lennox, E. S. (1955). Transduction of linked genetic characters of the host by bacteriophage P1. Virology 1 (2), 190–206. doi:10.1016/0042-6822(55)90016-7
Li, X., Cai, Z., Li, Y., and Zhang, Y. (2014). Design and construction of a non-natural malate to 1,2,4-butanetriol pathway creates possibility to produce 1,2,4-butanetriol from glucose. Sci. Rep. 4 (1), 5541. doi:10.1038/srep05541
Liang, Q., Zhang, F., Li, Y., Zhang, Xu, Li, J., Yang, P., et al. (2015). Comparison of individual component deletions in a glucose-specific phosphotransferase system revealed their different applications. Sci. Rep. 5 (1), 13200. doi:10.1038/srep13200
Liu, Y., Zhang, J., Li, R., and Yu, B. (2022). Efficient production of 2,4-dihydroxybutyrate from l -homoserine by the designed cofactor self-sufficient route. ACS Sustain. Chem. Eng. 10, 14361–14369. doi:10.1021/acssuschemeng.2c05012
Madeira, F., Madhusoodanan, N., Lee, J., Eusebi, A., Niewielska, A., Tivey, A. R. N., et al. (2024). The EMBL-EBI job dispatcher sequence analysis tools framework in 2024. Nucleic Acids Res. 52, W521–W525. doi:10.1093/nar/gkae241
Millard, P., Letisse, F., Sokol, S., and Portais, J. C. (2012). IsoCor: correcting MS data in isotope labeling experiments. Bioinformatics 28 (9), 1294–1296. doi:10.1093/BIOINFORMATICS/BTS127
Ng, C.Yu, Farasat, I., Maranas, C. D., and Salis, H. M. (2015). Rational design of a synthetic entner-doudoroff pathway for improved and controllable NADPH regeneration. Metab. Eng. 29, 86–96. doi:10.1016/j.ymben.2015.03.001
Nishiyama, M., Birktoft, J. J., and Beppu, T. (1993). Alteration of coenzyme specificity of malate dehydrogenase from thermus flavus by site-directed mutagenesis. J. Biol. Chem. 268 (7), 4656–4660. doi:10.1016/s0021-9258(18)53446-3
Rathnasingh, C., Subramanian, M. R., Lee, Y., Catherine, C., Ashok, S., and Park, S. (2012). Production of 3-hydroxypropionic acid via malonyl-CoA pathway using recombinant Escherichia coli strains. J. Biotechnol. 157 (4), 633–640. doi:10.1016/j.jbiotec.2011.06.008
Richter, N., Zienert, A., and Werner, H. (2011). A single-point mutation enables lactate dehydrogenase from Bacillus subtilis to utilize NAD+ and NADP+ as cofactor. Eng. Life Sci. 11 (1), 26–36. doi:10.1002/elsc.201000151
Rossmann, M. G., Moras, D., and Olsen, K. W. (1974). Chemical and biological evolution of a nucleotide-binding protein. Nature 250 (5463), 194–199. doi:10.1038/250194a0
Sauer, U., Canonaco, F., Heri, S., Perrenoud, A., and Fischer, E. (2004). The soluble and membrane-bound transhydrogenases UdhA and PntAB have divergent functions in NADPH metabolism of Escherichia coli. J. Biol. Chem. 279 (8), 6613–6619. doi:10.1074/jbc.M311657200
Shi, A., Zhu, X., Lu, J., Zhang, X., and Ma, Y. (2013). Activating transhydrogenase and NAD kinase in combination for improving isobutanol production. Metab. Eng. 16 (1), 1–10. doi:10.1016/j.ymben.2012.11.008
Shinka, T., Inoue, Y., Ohse, M., Ito, A., Ohfu, M., Hirose, S., et al. (2002). Rapid and sensitive detection of urinary 4-hydroxybutyric acid and its related compounds by gas chromatography–mass spectrometry in a patient with succinic semialdehyde dehydrogenase deficiency. J. Chromatogr. B 776 (1), 57–63. doi:10.1016/S1570-0232(02)00126-5
Takahashi-Íñiguez, T., Aburto-Rodríguez, N., Vilchis-González, A. L., and Flores, M. E. (2016). Function, kinetic properties, crystallization, and regulation of microbial malate dehydrogenase. J. Zhejiang Univ. Sci. B 17 (4), 247–261. doi:10.1631/jzus.B1500219
Walther, T., Calvayrac, F., Malbert, Y., Alkim, C., Dressaire, C., Cordier, H., et al. (2017a). Construction of a synthetic metabolic pathway for the production of 2,4-dihydroxybutyric acid from homoserine. Metab. Eng. 45, 237–245. doi:10.1016/j.ymben.2017.12.005
Walther, T., Dressaire, C., Cordier, H., and Francois, J.-M. (2015). Method of production of 2,4-dihydroxybutyric acid. U. S. 2015/0159182 A1.
Walther, T., Topham, C. M., Irague, R., Auriol, C., Baylac, A., Cordier, H., et al. (2017b). Construction of a synthetic metabolic pathway for biosynthesis of the non-natural methionine precursor 2,4-dihydroxybutyric acid. Nat. Commun. 8 (1), 15828. doi:10.1038/ncomms15828
Wellner, D., and Lichtenberg, L. A. (1971). [218a] Assay of amino acid oxidase. Methods Enzym. 17, 593–596. doi:10.1016/0076-6879(71)17104-2
Zhang, Yu, Wei, M., Zhao, G., Zhang, W., Li, Y., Lin, B., et al. (2021). High-level production of L-homoserine using a non-induced, non-auxotrophic Escherichia coli chassis through metabolic engineering. Bioresour. Technol. 327 (29), 124814. doi:10.1016/j.biortech.2021.124814
Keywords: enzyme engineering, strain engineering, cofactor specificity, synthetic metabolic pathway, 2,4-dihydroxybutyric acid, homoserine, Escherichia coli
Citation: Ihle N, Grüßner L, Alkim C, Nguyen TAS, Walther T and Frazão CJR (2025) Cofactor engineering for improved production of 2,4-dihydroxybutyric acid via the synthetic homoserine pathway. Front. Bioeng. Biotechnol. 13:1504785. doi: 10.3389/fbioe.2025.1504785
Received: 01 October 2024; Accepted: 23 January 2025;
Published: 20 February 2025.
Edited by:
Ruud Weusthuis, Wageningen University and Research, NetherlandsReviewed by:
Zhiming Rao, Jiangnan University, ChinaCopyright © 2025 Ihle, Grüßner, Alkim, Nguyen, Walther and Frazão. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cláudio J. R. Frazão, Y2xhdWRpby5mcmF6YW9AdHUtZHJlc2Rlbi5kZQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.