 Felix Hamburger1
Felix Hamburger1 Niels Schlichting1
Niels Schlichting1 Michael Eichenlaub2
Michael Eichenlaub2 Paul Igor Costea2
Paul Igor Costea2 Christopher Sauer2
Christopher Sauer2 Stefan Jenewein2*
Stefan Jenewein2* Johannes Kabisch1,3*
Johannes Kabisch1,3*- 1Computer-aided Synthetic Biology, TU Darmstadt, Darmstadt, Germany
- 2BASF SE, Ludwigshafen, Germany
- 3Institute for Biotechnology and Food Science, Norwegian University of Science and Technology (NTNU), Trondheim, Norway
Proteins face an obstacle race on their way to successful folding. Chaperones facilitate the proper folding of proteins by ensuring they remain on the correct path toward their final tertiary structure. In bacilli, the PrsA chaperone is essential for the correct folding and stabilization of proteins within the cell wall. Overexpression of the PrsA chaperone has been shown to improve the successful folding and secretion of many biotechnologically relevant secreted enzymes. This resulted in a double benefit: firstly, it promotes the efficient release of properly folded enzymes from the cell wall, and second, it reduces the folding stress for the cell, thereby enhancing the overall fitness of the production organism. This paper presents a workflow in which different wild-type PrsA molecules in Bacillus subtilis are co-expressed with different amylases having different signal peptides and promoters. To achieve this, six genome-reduced strains and nine PrsA proteins were systematically selected based on their cultivation performance and the production of two reference amylases. Following strain selection and deletion of major extracellular proteases, several hundred individual strains were created and screened using a stepwise and modular automation approach combined with amplicon sequencing. In addition to providing the key learnings from the workflow, it was revealed that no single PrsA molecule consistently improved amylase production, but genetic constructs combining different elements showed up to a 10-fold variation in yield. Among the screened constructs, the signal peptides YdjM and YvcE demonstrated the best performance.
1 Introduction
Bacillus subtilis is one of the most widely used organisms in biotechnology. It is the most studied Gram-positive organism with a genome sequenced in 1997 (Kunst et al., 1997; Earl et al., 2008). Its emergence as a model organism in biotechnology is supported by numerous processes that have received GRAS rating by the FDA and QPS assessment by EFSA (Leuschner et al., 2010; de Boer Sietske and Diderichsen, 1991). This includes its application in food production, such as historically in soy bean fermentation for thousands of years (Zweers et al., 2008). Because of their natural ability to secrete large amounts of homologous proteins like amylases and proteases into the medium and its good and prototrophic growth on cheap carbon sources, bacilli are generally used for the production of hydrolases (Gu et al., 2018). Secretion of the product is a major advantage in the industrial production of enzymes because it simplifies the purification process considerably in comparison with organisms that require lysis and separation from cytosolic host cell protein (Burdette et al., 2018) or possibly harbor undesired substances such as endotoxins in Escherichia coli (Petsch and Anspach, 2000). B. subtilis is readily genetically manipulable due to its ability to voluntarily absorb DNA through natural competence and integrate it into the genome. B. subtilis is now also used a heterologous expression system, although its ability to secrete non-homologous proteins has been described as being poor (Bolhuis et al., 1999). Yet some non-homologous amylase genes have been generated and secreted successfully since 1982 (Palva, 1982).
Amylases have a wide range of applications in the food, paper, detergent, biofuel, textile, pharma, and waste industries (Singh et al., 2016). Hence, they are among the most commercially interesting industrial enzyme classes.
B. subtilis produces a native
Secretion was shown to be a bottleneck in amylase production (Bolhuis et al., 1999; Yan and Wu, 2017). It can be generally split into three steps, namely, the cytosolic step, the membrane translocation, and the cell wall release and folding. In B. subtilis, proteins are generally secreted as pre-proteins with an amino-terminal signal peptide through the Sec pathway, aided by the signal recognition particle (SRP) and SecA (Kakeshtia et al., 2010), with the signal peptide likely acting as both a simple sorting signal for secretion and a folding factor, as described for E. coli (Park et al., 1988). Therefore, modifying the signal peptides is the most straightforward method that has been tried to optimize secretion (Freudl, 2018), but each step in the pathway harbors potential for improvement (Li et al., 2004). Although the cytosolic pathways (such as transcription, translation, and recognition by the secretion apparatus) are similar to those of other microbial hosts, the post-secretional processes in bacilli are unique, and the specific requirements have become obvious over the last few decades. It is now understood that post-secretional folding is important for successful cell wall release and that misfolding results in degradation at a cell wall-associated site, possibly before or during post-translocation (van Wely et al., 2001). In contrast, folding within the cytosol must be slow or even suppressed, with quick post-secretional folding being crucial to avoid aggregation and the stringent quality control found within the cell wall of bacilli. This kinetic partitioning has been initially described for E. coli with SecB as a holdase (Hardy and Randall, 1991). However, no functional SecB homolog has been described in bacilli. Therefore, alternative methods to enable kinetic partitioning are used. One example is the use of calcium ions to induce rapid folding: secreted enzymes fold efficiently with calcium addition into a protease-resistance form (Haddaoui et al., 1997), indicating that a low concentration of calcium in the cytosol might suppress folding, while a high level in the cell wall induce folding. Remodeling the cell wall has also been shown to improve post-secretional folding and impede with degradation (Vitikainen et al., 2005). PrsA, a cell wall-resident, membrane-bound chaperone, was found to increase amylase secretion in particular (Vitikainen et al., 2001; Vitikainen et al., 2004). It is the only chaperone in the cell wall of B. subtilis (Vitikainen et al., 2001). It has been described as the rate-limiting part of the secretion mechanism of
Modern screening techniques might, therefore, be used to identify the optimal PrsA chaperone with the appropriate level in the cell wall to ensure that the secretion and folding processes are smooth and efficient. In this publication, we present the automation-aided construction of hundreds of B. subtilis strains with different combinations of the PrsA chaperones (varying the chaperone and the promoter used for its expression) and the expression cassette for amylases (varying signal peptides, promoters, and the enzyme). We describe the process from strain selection to the construction and analysis of different PrsA strains with model amylases, the construction of a multi-protease knock-out strain, and the automation-aided construction of amylase expression cassettes, as well as share the practical experience when using a robotic platform for cultivation and strain generation.
Using amylase activity as a readout, we found that some PrsA molecules are generally more efficient than others, hinting that neither the homologous PrsA of the host nor the homologous PrsA to the amylase will be the superior and right choice. We also discovered that modifying the signal peptide does not replace the action of PrsA in bacilli, implying that signal peptides might contribute to enhance folding but might not replace the foldase.
2 Results
2.1 Selection of expression strains
2.1.1 Growth of parent strains
A suitable expression strain should have robust molecular biology traits such as transformation efficiency, good growth with low lysis in expression media, and finally an efficient expression of the target molecule. Six different B. subtilis strain options (S19002 through S19007; see Supplementary Table S1) were contributed to the project. To select the best amylase production chassis, the growth profile of the respective strains was monitored over a cultivation of 96 h. The strains were cultivated in microtiter plates (MTPs) for continuous growth measurements using a robotic platform. Additionally, they were cultivated in deepwell plates (DWPs) to produce a model amylase. The DWP-cultivations have higher comparability when it comes to upscaling for industrial processes (Habicher et al., 2021).
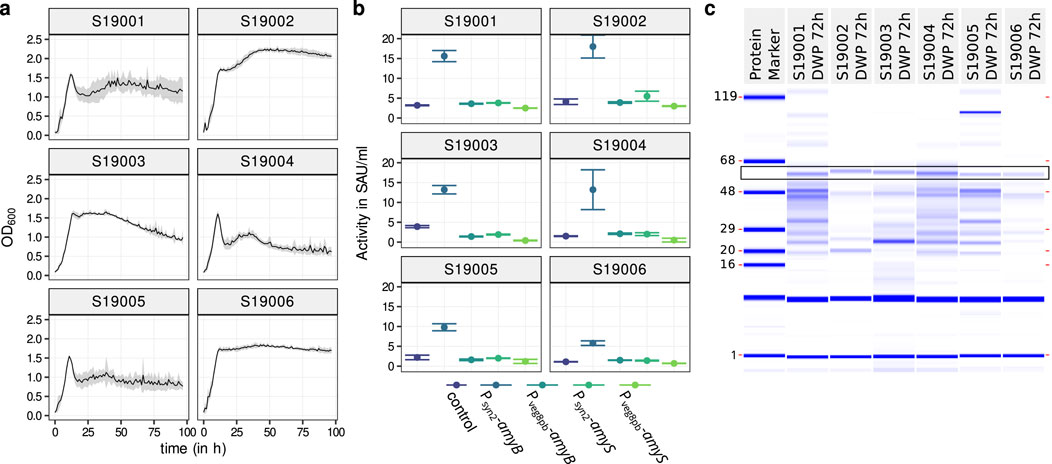
Figure 1. Selection of the initial strain. (A) Growth analysis of initial B. subtilis expression strains considered in this work (Supplementary Table S1). Cultivation was realized in 200-
2.1.2 Amylase activity
To determine the optimal strain for amylase expression, we integrated four different promoter–amylase combinations into the amyE locus of each strain’s genome.
All these results point to either S19002 or S19006 being the best choice of strain for further experiments because of good growth, high amylase activity, and weak protein background due to limited cell lysis. Although S19002 showed the overall best performance, it had the drawback that in contrast to S19006, the main extracellular proteases are not eliminated. In response to this strain, S19034 with deletions of these proteases was generated from S19002 in parallel to testing different promoter–prsA combinations in S19006, as described below. Full genotypes are listed in Supplementary Table S1.
2.2 PrsA tests
2.2.1 Generation of prsA expression strains
First, the selected amylase cassettes
2.2.2 Cultivation of amyB-expressing strains
During the cultivation, growth curves were recorded for every well in the MTP. These resembled the growth of the parent strain shown in Figure 1A and were very uniform (Supplementary Figure S4 top). Although MTPs show roughly the same results as DWPs, generally, DWP results are more pronounced. In DWP, the highest
2.2.3 Cultivation of amyS-expressing strains
As with amyB strains, growth curves recorded in amyS strains also closely resembled those of the parent strain S19006 (Figure 1A), with relatively minimal standard deviation (Supplementary Figure S4). Strains cultivated in DWP generally reached a higher
2.2.4
For measuring the amylase activity, MTP and DWP culture plates from the cultivation experiments were frozen after the end of the experiment and tested for
2.2.5
The activity data on amyS strains, on the other hand, are much more consistent (Supplementary Figure S5), but amyS, as previously observed (Figure 1B), shows a much lower amylase activity. DWP and MTP cultures have a nearly identical profile for the same PrsA in terms of final optical density. Contrary to the amyB strains, promoter 5 appears to be the one with the lowest activity results compared to promoters 3 and 4. When looking at the distance between control and PrsA strains, all PrsA except 457 provide an increase in amyS activity. PrsA 448, 452, and 453 have the highest activities, which are also among the top amyB strains. Peak activity measured in amyS samples with promoter 3 and PrsA 453 only amounts to 3.5 SAU/mL, while amyB activity values went up to 7.6 SAU/mL in DWP5 PrsA 453. In these experiments, promoter 3 provided the highest amylase activity increase compared to the controls in amyB and amyS strains, particularly in combination with PrsA 453, as well as PrsA 448, 449, 452, and 454. The PrsA conferring the lowest activity in combination with each promoter was 457, and its expression sometimes results in lower activity than in the controls (amyS strains and amyB MTP).
2.2.6 Protease knockouts in S19002
It was determined that S19002 was the strain of choice for the final experiment of combinations of amylase, PrsAs, signal peptides, and different promoters because its growth test, amylase expression, and transformation efficiency test results were better than those of S19006. As the lack of protease knockouts was the main argument against using S19002, the strain was modified accordingly. Deletion plasmids for the sequential knockouts of aprE, nprE, mpr, nprB, vpr, bpr, and epr were constructed. Homology regions were used for the replacement of the respective protease gene with a spectinomycin resistance gene flanked by lox-sites (lox-SSS cassette) via homologous recombination. Following successful integration, the lox-SSS cassette could be activated by inducing the genomically integrated Cre-recombinase, which removes the specR gene via site-specific recombination, leaving a lox72 scar, where the protease coding region was before. Each knockout was confirmed by sequencing the respective site. The resulting final strain was named S19034 and verified via whole-genome sequencing, confirming that no genomic rearrangement as a result of the 20 consecutive Cre-Lox reactions and lox72 scars occurred in the generation of this strain. The sequencing revealed 33 point mutations leading to frame-shifts described in Supplementary Table S5. Several of these are annotated with functions relating to sporulation, including the sporulation essential sigma factor E, as well as proteins involved in cell wall functions. S19034 was then transformed with different promoter–prsA PCR cassettes described before. Transformation success was determined via colony PCR and sequencing. Successfully transformed strains were stored as cryostock. The generated strains with the origin of PrsA and strain names are summarized in Table 1.
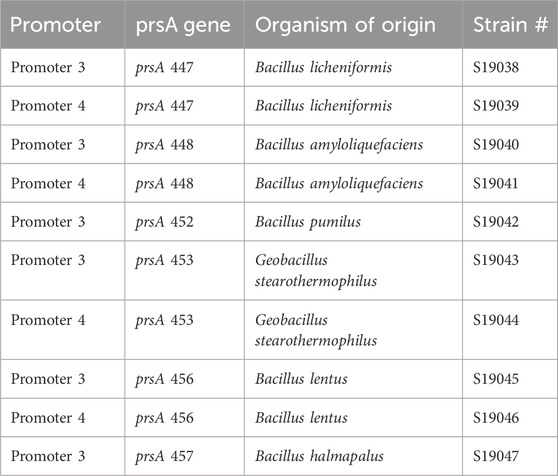
Table 1. Promoter–prsA combinations generated in strain 19034.
2.3 Preparation of automatic cloning
2.3.1 Construct selection
In total, 28 different amylases have been chosen from the “Termamyl-like” amylases of the GH13 family as the target sequences (Drula et al., 2022). Members of this family are used commercially in the food, biofuel, or detergent industries and are named after the first commercial product from this family (Termamyl L; introduced by Novo Nordisk in 1973). Members of this class are within a sequence range of 60% and display a core three-domain structure composed out of an interwoven A/B domain with an adjacent C-domain. Common is an unusual Ca2+–Na+–Ca2+ triad at the A/B-domain interface next to the substrate-binding site (Machius et al., 1995).
It was planned to transform a set of nine prsA with either promoter 3 or promoter 4 into the base strain S19034. These strains should then be transformed with the 28 different amylases, coupled with two promoters and four signal proteins. In total, 2,240 different combinations of PrsA, amylase, amylase promoter, and signal peptide were tested in a single run. As automation-supported strain generation of this scope needs careful preparation, we took into account the following aspects before starting with actual strain generation.
2.3.2 Positional effects in the MTP incubator
During the previous experiments, observations showed that cultivations in MTP, which were performed in the Cytomat2 tower shakers, yielded less precise results than the cultivations in DWP, which were incubated in custom-designed humidity boxes (Bruder et al., 2019) in regular incubator shakers. Evaporation, particularly in wells at the edge of the MTP, appeared to be a cause of variation. This observation led to the question how big the positional effects already observed in other experiments actually are. To investigate this problem, a set of MTPs was grown together with the amyS strain with different prsA genes in various positions on the incubator shaking towers. A measure of 200 mL medium was inoculated with 2 mL of S19006 pre-culture and distributed into six MTPs with 200
It was observed in all positions that the higher column number H row always shows higher
2.3.3 Transformation tests
Since the B. subtilis transformation protocol described in the methods cannot be readily automated in MTPs, a new method of transforming it exclusively in liquid media had to be developed. A particular problem of downscaling the transformation in a fully automated manner is the plating onto antibiotic-containing agar plates for a selection of transformants. This could not be readily accomplished by the robotic platform, especially not in the required scale. The solution found is to carry out the selection process by continuous cultivation in liquid selective media, as shown in the scheme in Figure 2. In order to determine all the parameters necessary to reduce the transformation to MTP size and make it automation-compatible, manual tests were carried out in MTP. First, it had to be determined whether the transformation will work in such small volumes with different shaking parameters and therefore changing oxygen supply. At the same time, we wanted to test whether amplifying the Golden Gate product with PCR would yield better results. To determine this, an experiment was set up with the genetic material from the initial amylases test (
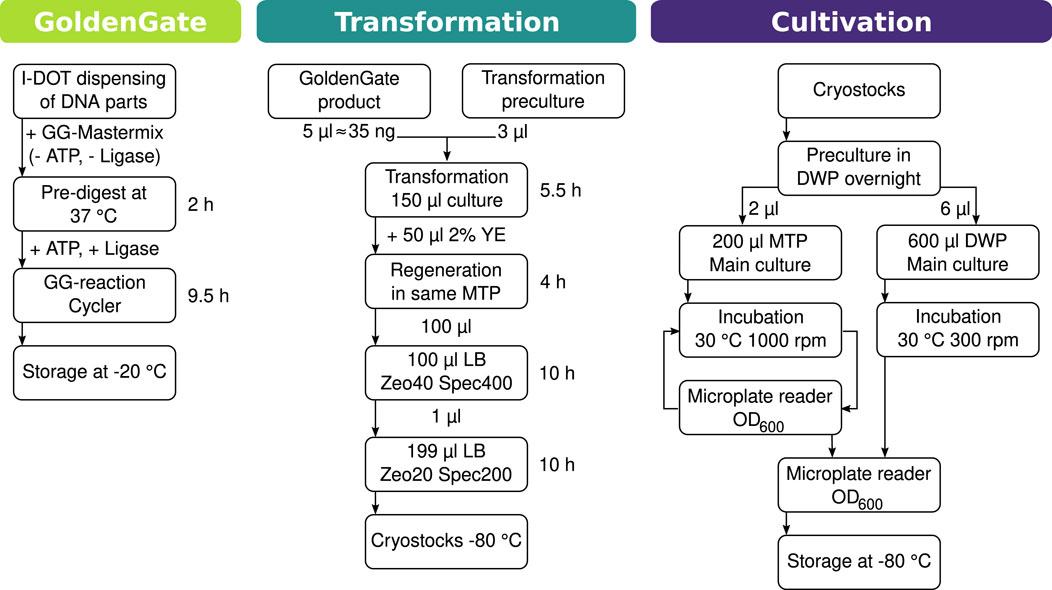
Figure 2. Chart of the workflow for automated cloning, transformation, and cultivation. The workflow is divided into three parts, after any of which pausing the process is possible. I.DOT is a contactless nanoliter dispenser. A more extensive version of this workflow can be found in Supplementary Figure S9.
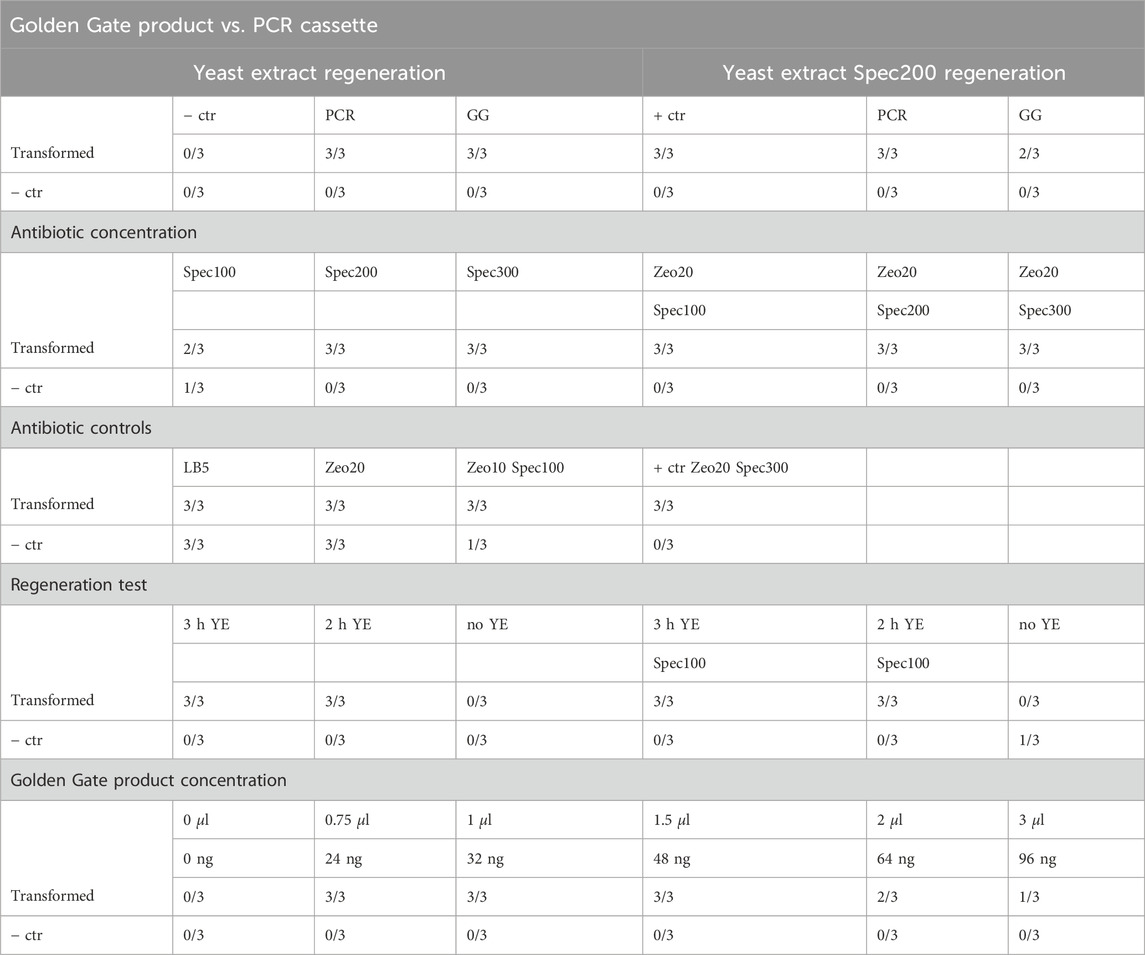
Table 2. Tests for determining transformation mix and liquid selection variables. Growth in MTP wells. −ctr describes transformation mixes without DNA, and + ctr represents positive controls transformed with 85 ng p19009.
The following transformation tests were conducted with a few adaptations. To facilitate pipetting on the robotic platform, the addition of 50
2.3.4 Workflow for automated cloning and cultivation
A workflow was designed to set up the automated cloning of strains on the robotic platform (see Figure 2). The procedure was split into three parts: creation of gene construction, transformation, and cultivation. Following each of these, the workflow can be suspended. For the cloning of the necessary gene constructs, it was decided to use the Golden Gate method because of its modularity and flexibility. To use as little DNA parts as possible, a nanoliter dispenser (I.DOT, Dispendix) was used for dispensing the Golden Gate parts into a 96-well PCR plate. Based on the data gathered in transformation experiments (Table 2), the workflow for the automatic transformation of prsA-transformed B. subtilis strains was designed. Transformation parameters were set to 1.5
2.4 Automated strain generation
The above mentioned workflow failed to yield the full, rational combinatorial of 28 different amylases with two different promoters and four different signal peptides that transform into 11 different prsA background strains. After automated transformation, a quality check was performed by first replicating the liquid-selected strains onto an LB selective plate and a starch plate, which was, in turn, stained using Lugol’s iodine. As can be seen in exemplary Supplementary Figure S10, the transformation of 67 out of 84 strains was successful, and only eight showed amylase activity. Analysis via sequencing of the negative clones revealed that often only one homology arm and the selection marker were integrated (data not shown). As a consequence, a less complex cloning and strain generation approach was chosen. Golden Gate reactions (500 ng total DNA) were set up by combining the Golden Gate fragments for the two different promoters
2.5 Cultivation of generated clones and amylase measurements
Eight individual clones per amylase were inoculated from cryocultures and cultivated in 96-deep well plates. Amylase activities were measured, and the genetic composition of the individual clones was determined using amplicon sequencing, with results elaborated below.
As shown in Figure 3A, the signal peptide from YdjM (a putative cell wall hydrolase, BSU_06250), closely followed by the signal peptide from YvcE (D,L-endopeptidase-type autolysin, BSU_34800), outperformed the other signal peptides overall. When resolved by the individual PrsA molecules present in the hosts, the AmyL signal peptide from B. licheniformis does not perform best with its native PrsA but instead with the chaperone from B. amyloliquefaciens, which also appears to support the secretion of the YdjM and YvcE signal peptides. On the amylase level (see Figure 3B), Amy0355 closely followed by Amy0365 performed best, while AmyS and 707 perform worse than the controls. When viewing the overall performance of the PrsA chaperones (see Supplementary Figure S11, middle), no clear best-suited candidate can be identified, but consistent with previous experiments, PrsA from B. halmapalus performs worst.
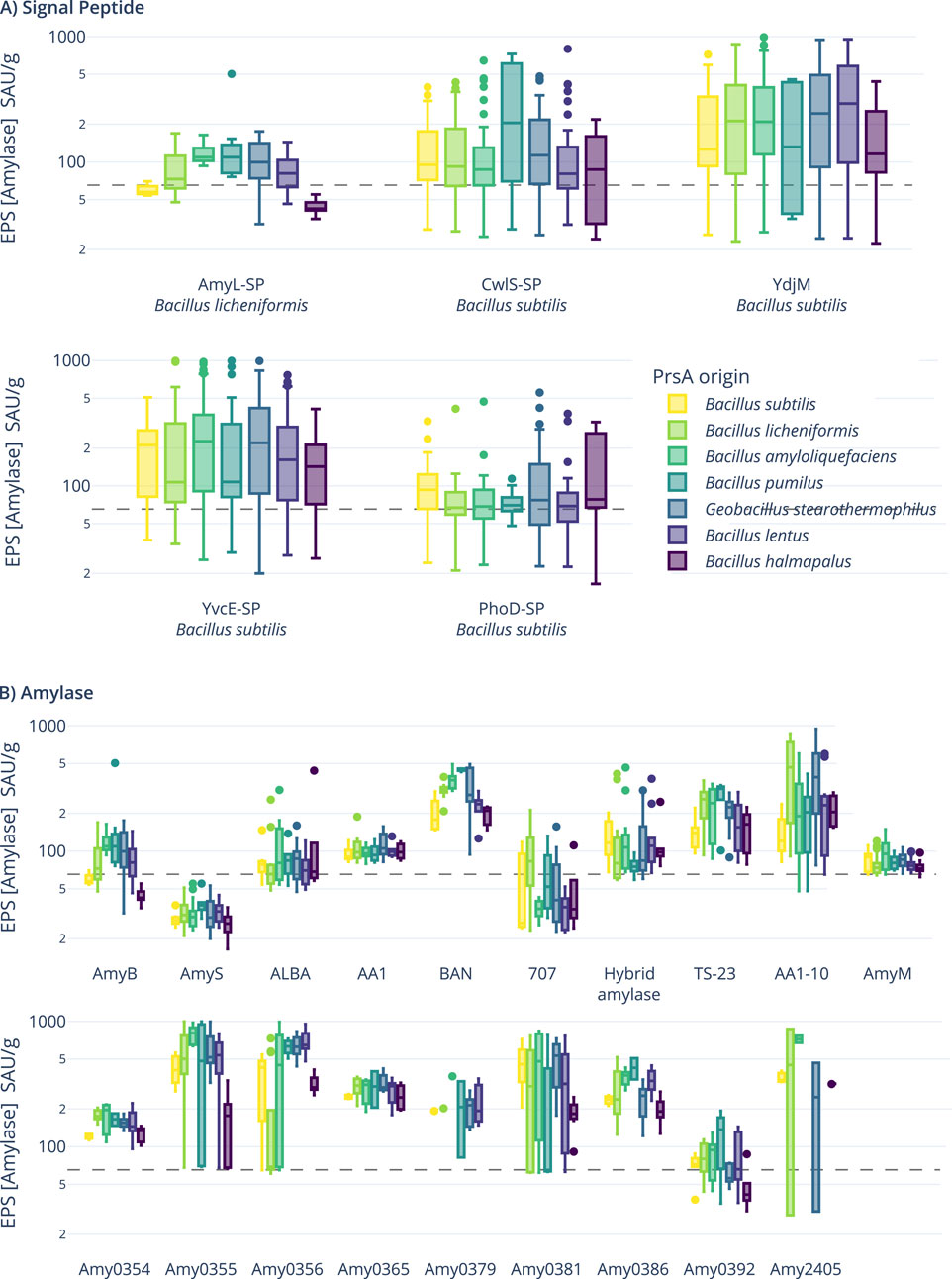
Figure 3. Boxplots of end-point determination of amylase activity (SAU/g: standardized amylase units per Gram). (A) Amylase activity plotted per signal peptide resolved by PrsA chaperones present in the strains. (B) Activity plotted per used amylase and resolved by the PrsA molecule. The gray dashed lines are the background activities of the negative controls, which, as all strains, contain the native amylase (AmyE) from B. subtilis.
2.6 Amplicon sequencing of cultivated clones
Cultivation was performed in a structured manner, with one MTP cultivation performed for each amylase molecule. This allows, after pooling of respective MTP wells, the use of the amylase sequence as a preset barcode to identify the construct in each clone via PCR amplicon sequencing (Mamanova et al., 2010; Head et al., 2014). Full genotypes of the expression cassette (promoter, signal peptide, and amylase) could be assigned to roughly two-thirds of the clones, while for the remaining clones, only parts of the genotype could be resolved.
3 Discussion
Genetic optimization of production strains necessitates the tuning of multiple genetic elements, as demonstrated by previous examples with B. subtilis (Westers et al., 2004). The six potential parent strains analyzed at the beginning of this work served as the foundation for such strain optimization. Resistance to lysis was of paramount importance since
It is established from the literature that the overexpression of PrsA (Vitikainen et al., 2001) and choosing a different PrsA (Quesada-Ganuza et al., 2019) can be beneficial for amylase secretion. The investigation of effects that different additional prsA genes have on the production of recombinant amylases in this study showed that PrsA in most cases has a positive effect on amylase activity (Supplementary Figure S5). This observation is consistent with the results obtained by Quesada-Ganuza et al. (2019), which increased amylase production by optimizing PrsA chaperones. However, it is worth mentioning that all strains still had the native B. subtilis prsA gene. It was expected that introducing an additional copy of a prsA gene would increase the amylase production and secretion since that is the case even for the overexpression of the native chaperone (Chen et al., 2015; Kontinen and Sarvas, 1993; Vitikainen et al., 2005). prsA 457 from B. halmapalus was the only one that had a negative effect on amylase activity in both amyB and amyS strains (Supplementary Figure S5). This implies an inhibitory function in either secretion or folding specific to the tested amylases, as well as a negative effect on the host caused by an unsuitable PrsA. Further research would be required to confirm this theory. prsA 457 was nevertheless continued to be examined in the following experiment as a negatively influencing gene. In addition to the chaperone gene, the promoter driving prsA expression appeared to have an impact on the growth of the strains, with promoter 4 showing the highest
The efficacy of PrsA appears to be highly expression- and amylase-dependent because the activity of cultures transformed with the same PrsA varied significantly depending on the used promoter and amylase. Hence, there was no single PrsA that could be selected as the best option for all or most amylases and/or promoters. The diversity of activities in different amylase–PrsA promoter combinations is striking and indicates that balancing all components might be the key to success.
Efficient screening requires parallelized cultivation preferably in MTP plates. Several issues stemming from cultivation in the Cytomat2 MTP shaker had to be identified and addressed. One of these was plate abrasion and increased evaporation on the plate edges caused by insufficient fixation of the MTP lids during shaking, which was solved through an engineering student project (see Supplementary Figure S12). MTP results generally had more outliers than DWP results. With an evaporation test (Supplementary Figure S6), one of the causes was confirmed to be positional effects in the MTP incubator. The shape of
A workflow has been devised for rapid cloning and the generation of many different amylase-integrated strains in one run. It was planned to, all in all, create 2,240 strains, which should be cultivated in MTP and DWP and screened for their amylase activity. The cloning and cultivation process was split into three functional, automated modules, each with a save point that enables pausing the process without losing progress. Designing automation processes in modules is extremely useful as it makes the automation process less complex and, therefore, less prone to errors. Such errors are mostly caused by the automated handling of labware, such as stuck lids, tips falling off, robot arm dropping labware, or missing labware because the operator forgot to load it or misplaced it. Furthermore, once a module has been well established, it can be reused for other processes.
Transformation cassettes were cloned via Golden Gate (Engler et al., 2008; 2009) reactions. The contactless nanoliter dispenser (I.DOT, Dispendix) enabled high-frequency, low volume dispensing of Golden Gate parts, making these types of devices essential for automation setups working with DNA assemblies (Holowko et al., 2021). The transformation of the obtained Golden Gate constructs into the 10 B. subtilis strains containing different prsA–promoter combinations was established to be fully automated by using a liquid selection process instead of plating and colony picking, which is much harder to implement as work moves away the structured plate/well format. The developed transformation method in MTP has been tested successfully in experiments conducted manually. Since we did not include colony PCR and our testing relied on starch-based amylase plate assays, the method depends on the reliability of the Golden Gate reaction and transformation. Engler et al. (2009) reported an efficiency of 97% for nine different modules in E. coli, while the aim of this work is just combining three of them. The amylase assay proved to be sufficient to detect that a full combinatorial DNA assembly generating 2,240 strains was not feasible without substantial optimization. For the presented workflow, manual picking was a task readily performed within 1 hour including replica plating using a 96-spike picker. The automated transformation worked as we received spectinomycin-resistant colonies that were negative for native amylase activity (derived from AmyE), indicating successful uptake of DNA and integration into the target locus; however, higher transformation rates might have solved the problem of many clones integrating only the antibiotic resistance gene and one homology arm. Fixing the frame shift in comP, which is involved in the development of natural competence (Maier, 2020), might have provided that increase. The fact that the cells can still become competent indicates that the point mutations that cause early stop codons are to some degree suppressed (Belinky et al., 2021). In order to overcome this construct generation bottleneck, we opted to proceed with a reduced combinatorial approach of one-pot reactions for the 19 amylase genes. In this study, the fragment for the signal peptide and the promoter was shuffled with a single amylase gene and transformed in a defined PrsA strain. The genotypes were assigned to the individual clones via amplicon sequencing of pooled clones, with the known amylase representing the barcode. This resolved the full genotype in two-thirds of the cases. For one-third of clones, only parts of the genotype could be determined due to the application of short-read sequencing with short average fragment sizes of the sequencing libraries, which did not span all construct element boundaries. For future approaches, long-read sequencing could be used to improve the identification of genotypes (Currin et al., 2019).
The screening results for optimal PrsA–amylase combinations can be easily summarized: no single, optimal PrsA molecule that always improved amylase production could be identified; however, within a group, different combinations of genetic elements and PrsA molecules could lead to a 10-fold variation in amylase production. For the screened genetic constructs and strains, PrsA from B. halmapalus, which is consistent with the results from the pre-experiments, tended to perform worst. Interestingly, growth was not affected, so one might exclude a negative effect on cellular functions such as the folding of penicillin-binding protein 2B (PBP2B), an essential protein for cell wall synthesis depending on PrsA for correct folding (Hyyryläinen et al., 2010). Sequence analysis revealed that the NC domain of the PrsA of B. halmapalus is strongly negatively charged with an overall charge of −17 at pH 7. This is consistent with the NC domain of, for example, the PrsA of G. stearothermophilus with a net charge of only −2 at pH 7. In a noteworthy publication, Quesada-Ganuza et al. (2019) created modifications in B. lentus PrsA associated with reduced hydrophobicity with an improved effect on secretion. The negative impact of B. halmapalus PrsA emphasizes the importance of overall charge, at least for the class of amylases. Evidently, further work such as testing hybrids might increase the efficiency of this foldase class to their client amylases.
For the signal peptides, YdjM and YvcE (synonym CwlO) performed best. YdjM was previously identified to be a well-performing signal peptide in protease secretion screening (Degering et al., 2010), and both sequences (YdjM and YvcE) are described to be suitable but not outperforming signal peptides for an alkaline xylanase (Zhang et al., 2016). Recent studies have not reached a clear conclusion about which sequence parameter is essential (Grasso et al., 2023; Freudl, 2018; Zhang et al., 2020; Brockmeier et al., 2006), and no overarching canonical signal peptide is known. As the signal peptide might not only present a marker within the cellular environment for secretion but may also affect mRNA stability, folding of the cognate protein, and cleavage of the pre-protein, more studies, such as by Grasso et al. (2023), are required to shed light on the requirements for efficient, predictable heterologous of secretion.
4 Conclusion
The presented automation setup allowed us to automatically generate and transform B. subtilis strain-producing amylases in various genetic contexts. Toward this goal, the genome-reduced production strain S19034 was generated with many favorable traits such as the absence of major extracellular proteases and robust growth with strongly reduced lysis. Further shortcomings on cultivation in tower shakers in automation setups were identified and addressed. Advantages of automated strain construction include higher throughput, avoidance of human errors while handling many samples, and reproducible data. Challenges include a high effort for the verification of constructs and high time requirements to develop robust workflows.
5 Methods
5.1 Strains and cultivation
5.1.1 General cultivation
All B. subtilis strains are listed in Supplementary Table S1 with their respective relevant genotypes. Parent strains S19001, S19002, and S19003 were kindly provided by BASF, while S19004, S19005, and S19006 were derived from the strain 6051HGW (Kabisch et al., 2013) and constructed previously in the Kabisch-lab. Cells were cultivated in Luria-Bertani medium with either 10 g/L (LB, Carl Roth, X968.3) or 5 g/L
5.1.2 Cultivation experiments
Cultivation experiments were carried out at 30°C using MTP (Greiner, 96-well plate, clear, flat bottom, M4811-40 EA) incubated in a Cytomat2 incubator (Thermo Fisher Scientific, US-MA) with 1,000 rpm (amplitude 1.5 mm) and plastic lids (Greiner, 656101). DWPs (Whatman, 96 deep-well plates, round bottom, 734–2,559) were incubated at 300 rpm (New Brunswick Innova 44, 51 mm orbit) and covered with adhesive gas permeable seals (Thermo Scientific, AB0718) in previously described 3D printed cultivation boxes (Bruder et al., 2019). A measure of 600
5.2 Cloning
5.2.1 DNA manipulations
Polymerase chain reaction (PCR) was performed as described in Kuslich et al. (2019), with hybrid polymerase (Roboklon, E2950-02) relying on the polymerase manual for annealing temperature calculation. Oligonucleotides were obtained from Sigma-Aldrich (Supplementary Table S4). Colony PCR was conducted according to Woodman et al.’s (2016) protocol, with an initial denaturation time of 10 min using either Taq (Roboklon, E2600-02) or OptiTaq polymerase (Roboklon, E2600-02). Agarose gel electrophoresis was performed as described in Armstrong and Schulz (2015), with ROTI GelStain (Carl Roth, 3,865.1) and 1 kbp DNA ladder (Carl Roth, Y014.2) or 1 kb Plus DNA ladder (New England Biolabs, N3200L) for DNA quantification. Genomic DNA was extracted using the High Pure PCR Template Preparation Kit (Roche, 11796828001). The innuPREP Plasmid Mini Kit 2.0 (Analytik Jena, 845-KS-5041250) was used for plasmid preparation and NucleoSpin Gel and PCR Clean-up kit (MACHEREY-NAGEL, 740609.50) for PCR cleanup. All DNA extractions were followed by NanoDrop (Mettler Toledo, UV5Nano) concentration measurements and an agarose gel. DNA was sequenced by Eurofins Genomics using the Mix2Seq Service.
5.2.2 Cloning of amylase and prsA expression cassettes
The Golden Gate protocol was adapted from Engler et al. (2008) and Engler et al. (2009), with the reaction mix containing 200 ng of each Golden Gate part: 1.5
5.2.3 Cloning of deletion plasmids using SLiCE
Deletion plasmids were constructed using SLiCE, according to Zhang et al. (2012). Homology regions were amplified from isolated genomic DNA of S19002 via PCR using hybrid polymerase. The SpecR cassette with lox and six sites [lox-SSS (Kumpfmüller et al., 2013)] was amplified in two parts from the chromosomal DNA of strain BsFLN040, a pJet backbone which was obtained via amplification from plasmid DNA p19012. After DNA cleanup of all components (New England Biolabs Sequencing Monarch PCR and DNA Clean Up), the SLiCE reaction was started. Electrocompetent E. coli NEB 10
5.2.4 Bacillus subtilis strain generation
Transformations of B. subtilis were accomplished by adding 50
5.2.5 Downscaling of B. subtilis transformation to microplates
The Golden Gate reaction and its PCR-amplified products were run on a 1% agarose gel and quantified using a DNA marker and free GelAnalyzer software to measure the amount of the relevant construct in each sample and calculate the amount of DNA required for a scaled-down transformation mix. The concentrations of the relevant bands were 40 ng/
5.3
Amylase activity was measured with an assay described by Lorentz (2000). For the calibration curve, a dilution series of 1:2 starting at 1:16 and ending at 1:512 of the amylase standard (Termamyl 120L, Sigma 3,403 Lot: SLBJ0544V with a given concentration of 18400 SAU/g; SAU/g = sigma amylase units/g) in MOPS buffer (50 mM of 3-morpholinopropane-1-sulfonic acid, 50 mM of NaCL, and 3 mM of
5.4 Automation
Automation was applied to several degrees. Generation of cryostocks, for example, was significantly more robust when using a semi-automated 96-well pipette (CyBio SELMA, Analytik Jena) as the operator had visible control over potential cross-contamination, such as bubbles at the tip-ends popping and spreading cells through various wells. All automation processes in the robotic platform were designed with as few steps as necessary to reach a stop and back-up point (see Figure 2. Pipetting of the Golden Gate reaction, for example, was done manually loading the nanoliter dispenser (I-Dot, Dispendix) with source and target plates. After successful liquid transfer, the plates were manually transferred to a thermocycler for the thermal reaction, which proved to be faster and more robust than starting from several hotel positions and using the built-in thermocycler for the required throughput. To characterize the growth of the strains, an automated setup using a microplate tower shaker with controlled humidity (Cytomat2, Thermo Fisher Scientific) was used for incubation. For OD measurements, plates were transferred between the Cytomat and a plate reader (BMG Labtech PHERAstar FSX) using a robotic arm (PreciseFlex 750, Brooks Automation). A representation of the complete robotic platform without the housing is shown in Supplementary Figure S1. Because the MTP cultivation used by Cytomat2 was prone to massive abrasion from the plastic lids, the tower shaker was modified by attaching 2-mm ethylene–propylene–diene cellular rubber to the back of the holders using superglue (see Supplementary Figure S12).
5.5 Amplicon sequencing of pooled MTP cultivations
For sequencing, the MTP plates were re-grown from a backup glycerol stock in LB media supplemented by 20 ng/
5.6 Data analysis
Data were analyzed using RStudio 3.6 with R version 3.6.1 on x86_64-pc-linux-gnu (64-bit) running under Ubuntu 19.10 with the following packages: growthcurver 0.3.0, viridis 0.5.1, viridisLite 0.3.0, gridExtra 2.3, cowplot 1.0.0, forcats 0.4.0, stringr 1.4.0, purrr 0.3.3, readr 1.3.1, tidyr 1.0.2, tibble 2.1.3, ggplot2 3.2.1, tidyverse 1.3.0, dplyr 0.8.3, and magrittr 1.5. Interactive plots were generated using plotly 5.18.0 and pandas 2.1.3. Geneious 11.1.5 was used for genetics. GelAnalyzer 19.1 was used for the gel analysis of DNA concentrations. For the calculation of significance, a Shapiro–Wilk test for normal distribution was done before two-way ANOVA, and then, Tukey’s honest significant difference test was performed.
5.7 LabChip measurements
Measurements were conducted with a PerkinElmer LabChip device according to manufacturer’s instructions using the Protein Express Assay Reagent Kit (PerkinElmer, CLS960008) with an AA560 amylase variant (55 kDa, 483aa) as a reference.
Data availability statement
The datasets presented in this article are not readily available because sequence data for proprietary sequences cannot be provided. Requests to access the datasets should be directed to am9oYW5uZXMua2FiaXNjaEBudG51Lm5v.
Author contributions
FH: Investigation, Methodology, Writing–original draft. NS: Investigation, Methodology, Writing–original draft. ME: Conceptualization, Methodology, Writing–review and editing. PC: Conceptualization, Methodology, Writing–review and editing. CS: Conceptualization, Methodology, Writing–review and editing. SJ: Conceptualization, Supervision, Visualization, Writing–original draft. JK: Conceptualization, Supervision, Visualization, Writing–original draft.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This study was supported by the Hessen State Ministry of Higher Education, Research and the Arts (HMWK) via the LOEWE CompuGene project for the purchase of the robotic platform.
Acknowledgments
SJ much appreciates internal discussions with Max Felle and Mathis Appelbaum and is also grateful for technical assistance to Frank Woellert.
Conflict of interest
Authors ME, PC, CS, and SJ were employed by BASF SE.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fbioe.2024.1479626/full#supplementary-material
References
Altenbuchner, J. (2016). Editing of the Bacillus subtilis genome by the CRISPR-cas9 system. Appl. Environ. Microbiol. 82, 5421–5427. doi:10.1128/AEM.01453-16
Armstrong, J. A., and Schulz, J. R. (2015). Agarose gel Electrophoresis. Curr. Protoc. Essent. Lab. Tech. 10 (7), 1. doi:10.1002/9780470089941.et0702s10
Bareither, R., and Pollard, D. (2011). A review of advanced small-scale parallel bioreactor technology for accelerated process development: current state and future need. Biotechnol. Prog. 27, 2–14. doi:10.1002/btpr.522
Belinky, F., Ganguly, I., Poliakov, E., Yurchenko, V., and Rogozin, I. B. (2021). Analysis of stop codons within prokaryotic protein-coding genes suggests frequent readthrough events. Int. J. Mol. Sci. 22, 1876. doi:10.3390/ijms22041876
Betts, J. I., and Baganz, F. (2006). Miniature bioreactors: current practices and future opportunities. Microb. Cell Factories 5, 21. doi:10.1186/1475-2859-5-21
Blackman, S., Smith, T., and Foster, S. (1998). The role of autolysins during vegetative growth of Bacillus subtilis 168. Microbiology 144, 73–82. doi:10.1099/00221287-144-1-73
Bolhuis, A., Tjalsma, H., Smith, H. E., de Jong, A., Meima, R., Venema, G., et al. (1999). Evaluation of bottlenecks in the late stages of protein secretion in Bacillus subtilis. Appl. Environ. Microbiol. 65, 2934–2941. doi:10.1128/AEM.65.7.2934-2941.1999
Brockmeier, U., Caspers, M., Freudl, R., Jockwer, A., Noll, T., and Eggert, T. (2006). Systematic screening of all signal peptides from Bacillus subtilis: a powerful strategy in optimizing heterologous protein secretion in Gram-positive bacteria. J. Mol. Biol. 362, 393–402. doi:10.1016/j.jmb.2006.07.034
Bruder, S., Moldenhauer, E. J., Lemke, R. D., Ledesma-Amaro, R., and Kabisch, J. (2019). Drop-in biofuel production using fatty acid photodecarboxylase from Chlorella variabilis in the oleaginous yeast Yarrowia lipolytica. Biotechnol. Biofuels 12, 202. doi:10.1186/s13068-019-1542-4
Burdette, L. A., Leach, S. A., Wong, H. T., and Tullman-Ercek, D. (2018). Developing gram-negative bacteria for the secretion of heterologous proteins. Microb. Cell Factories 17, 196. doi:10.1186/s12934-018-1041-5
Chen, J., Fu, G., Gai, Y., Zheng, P., Zhang, D., and Wen, J. (2015). Combinatorial Sec pathway analysis for improved heterologous protein secretion in Bacillus subtilis: identification of bottlenecks by systematic gene overexpression. Microb. Cell Factories 14, 92. doi:10.1186/s12934-015-0282-9
Currin, A., Swainston, N., Dunstan, M. S., Jervis, A. J., Mulherin, P., Robinson, C. J., et al. (2019). Highly multiplexed, fast and accurate nanopore sequencing for verification of synthetic DNA constructs and sequence libraries. Synth. Biol. 4, ysz025. doi:10.1093/synbio/ysz025
de Boer Sietske, A., and Diderichsen, B. (1991). On the safety of Bacillus subtilis and B. amyloliquefaciens: a review. Appl. Microbiol. Biotechnol. 36, 1–4. doi:10.1007/BF00164689
Degering, C., Eggert, T., Puls, M., Bongaerts, J., Evers, S., Maurer, K.-H., et al. (2010). Optimization of protease secretion in Bacillus subtilis and Bacillus licheniformis by screening of homologous and heterologous signal peptides. Appl. Environ. Microbiol. 76, 6370–6376. doi:10.1128/AEM.01146-10
de Souza, P. M., and e Magalhães, P. d. O. (2010). Application of microbial α-amylase in industry - a review. Braz. J. Microbiol. 41, 850–861. doi:10.1590/S1517-83822010000400004
Drula, E., Garron, M.-L., Dogan, S., Lombard, V., Henrissat, B., and Terrapon, N. (2022). The carbohydrate-active enzyme database: functions and literature. Nucleic Acids Res. 50, D571–D577. doi:10.1093/nar/gkab1045
Duetz, W. A., Rüedi, L., Hermann, R., O’Connor, K., Büchs, J., and Witholt, B. (2000). Methods for intense aeration, growth, storage, and replication of bacterial strains in microtiter plates. Appl. Environ. Microbiol. 66, 2641–2646. doi:10.1128/AEM.66.6.2641-2646.2000
Earl, A. M., Losick, R., and Kolter, R. (2008). Ecology and genomics of Bacillus subtilis. Trends Microbiol. 16, 269–275. doi:10.1016/j.tim.2008.03.004
Elyasi Far, B., Ahmadi, Y., Yari Khosroshahi, A., and Dilmaghani, A. (2020). Microbial alpha-amylase production: progress, challenges and perspectives. Adv. Pharm. Bull. 10, 350–358. doi:10.34172/apb.2020.043
Engler, C., Gruetzner, R., Kandzia, R., and Marillonnet, S. (2009). Golden gate shuffling: a one-pot DNA shuffling method based on type IIs restriction enzymes. PLOS ONE 4, e5553. doi:10.1371/journal.pone.0005553
Engler, C., Kandzia, R., and Marillonnet, S. (2008). A one pot, one step, precision cloning method with high throughput capability. PLOS ONE 3, e3647. doi:10.1371/journal.pone.0003647
Freudl, R. (2018). Signal peptides for recombinant protein secretion in bacterial expression systems. Microb. Cell Factories 17, 52. doi:10.1186/s12934-018-0901-3
Grasso, S., Dabene, V., Hendriks, M. M. W. B., Zwartjens, P., Pellaux, R., Held, M., et al. (2023). Signal peptide efficiency: from high-throughput data to prediction and explanation. ACS Synth. Biol. 12, 390–404. doi:10.1021/acssynbio.2c00328
Gu, Y., Xu, X., Wu, Y., Niu, T., Liu, Y., Li, J., et al. (2018). Advances and prospects of Bacillus subtilis cellular factories: from rational design to industrial applications. Metab. Eng. 50, 109–121. doi:10.1016/j.ymben.2018.05.006
Gupta, M., and Rao, K. K. (2014). Phosphorylation of DegU is essential for activation of amyE expression in Bacillus subtilis. J. Biosci. 39, 747–752. doi:10.1007/s12038-014-9481-5
Habicher, T., Klein, T., Becker, J., Daub, A., and Büchs, J. (2021). Screening for optimal protease producing Bacillus licheniformis strains with polymer-based controlled-release fed-batch microtiter plates. Microb. Cell Factories 20, 51. doi:10.1186/s12934-021-01541-2
Habicher, T., Rauls, E. K. A., Egidi, F., Keil, T., Klein, T., Daub, A., et al. (2020). Establishing a fed-batch process for protease expression with Bacillus licheniformis in polymer-based controlled-release microtiter plates. Biotechnol. J. 15, 1900088. doi:10.1002/biot.201900088
Haddaoui, E. A., Leloup, L., Petit-Glatron, M.-F., and Chambert, R. (1997). Characterization of A Stable intermediate trapped during reversible refolding of Bacillus subtilis α-amylase. Eur. J. Biochem. 249, 505–509. doi:10.1111/j.1432-1033.1997.00505.x
Hardy, S. J., and Randall, L. L. (1991). A kinetic partitioning model of selective binding of nonnative proteins by the bacterial chaperone SecB. Sci. (New York, N.Y.) 251, 439–443. doi:10.1126/science.1989077
Head, S. R., Komori, H. K., LaMere, S. A., Whisenant, T., Van Nieuwerburgh, F., Salomon, D. R., et al. (2014). Library construction for next-generation sequencing: overviews and challenges. BioTechniques 56, 61–77. doi:10.2144/000114133
Holowko, M. B., Frow, E. K., Reid, J. C., Rourke, M., and Vickers, C. E. (2021). Building a biofoundry. Synth. Biol. 6, ysaa026. doi:10.1093/synbio/ysaa026
Hyyryläinen, H.-L., Marciniak, B. C., Dahncke, K., Pietiäinen, M., Courtin, P., Vitikainen, M., et al. (2010). Penicillin-binding protein folding is dependent on the PrsA peptidyl-prolyl cis-trans isomerase in Bacillus subtilis. Mol. Microbiol. 77, 108–127. doi:10.1111/j.1365-2958.2010.07188.x
Kabisch, J., Thürmer, A., Hübel, T., Popper, L., Daniel, R., and Schweder, T. (2013). Characterization and optimization of Bacillus subtilis ATCC 6051 as an expression host. J. Biotechnol. 163, 97–104. doi:10.1016/j.jbiotec.2012.06.034
Kakeshtia, H., Kageyama, Y., Ara, K., Ozaki, K., and Nakamura, K. (2010). Enhanced extracellular production of heterologous proteins in Bacillus subtilis by deleting the C-terminal region of the SecA secretory machinery. Mol. Biotechnol. 46, 250–257. doi:10.1007/s12033-010-9295-0
Kontinen, V. P., and Sarvas, M. (1993). The PrsA lipoprotein is essential for protein secretion in Bacillus subtilis and sets a limit for high-level secretion. Mol. Microbiol. 8, 727–737. doi:10.1111/j.1365-2958.1993.tb01616.x
Krishnappa, L., Monteferrante, C. G., Neef, J., Dreisbach, A., and van Dijl, J. M. (2014). Degradation of extracytoplasmic catalysts for protein folding in Bacillus subtilis. Appl. Environ. Microbiol. 80, 1463–1468. doi:10.1128/AEM.02799-13
Kumpfmüller, J., Kabisch, J., and Schweder, T. (2013). An optimized technique for rapid genome modifications of Bacillus subtilis. J. Microbiol. Methods 95, 350–352. doi:10.1016/j.mimet.2013.10.003
Kunst, F., Ogasawara, N., Moszer, I., Albertini, A. M., Alloni, G., Azevedo, V., et al. (1997). The complete genome sequence of the gram-positive bacterium Bacillus subtilis. Nature 390, 249–256. doi:10.1038/36786
Kuslich, C. D., Chui, B., and Yamashiro, C. T. (2019). Overview of PCR. Curr. Protoc. Essent. Lab. Tech. 18, e27. doi:10.1002/cpet.27
Leuschner, R. G. K., Robinson, T. P., Hugas, M., Cocconcelli, P. S., Richard-Forget, F., Klein, G., et al. (2010). Qualified presumption of safety (QPS): a generic risk assessment approach for biological agents notified to the European food safety authority (EFSA). Trends Food Sci. and Technol. 21, 425–435. doi:10.1016/j.tifs.2010.07.003
Li, W., Zhou, X., and Lu, P. (2004). Bottlenecks in the expression and secretion of heterologous proteins in Bacillus subtilis. Res. Microbiol. 155, 605–610. doi:10.1016/j.resmic.2004.05.002
Liu, Y., Lu, F., Chen, G., Snyder, C. L., Sun, J., Li, Y., et al. (2009). High-level expression, purification and characterization of a recombinant medium-temperature α-amylase from Bacillus subtilis. Biotechnol. Lett. 32, 119–124. doi:10.1007/s10529-009-0112-4
Lorentz, K. (2000). Routine α-amylase assay using protected 4-Nitrophenyl-1,4-α-d-Maltoheptaoside and a novel α-glucosidase. Clin. Chem. 46, 644–649. doi:10.1093/clinchem/46.5.644
Machius, M., Wiegand, G., and Huber, R. (1995). Crystal structure of calcium-depleted Bacillus licheniformis α-amylase at 2.2 Å resolution. J. Mol. Biol. 246, 545–559. doi:10.1006/jmbi.1994.0106
Maier, B. (2020). Competence and transformation in Bacillus subtilis. Curr. Issues Mol. Biol. 37, 57–76. doi:10.21775/cimb.037.057
Mamanova, L., Coffey, A. J., Scott, C. E., Kozarewa, I., Turner, E. H., Kumar, A., et al. (2010). Target-enrichment strategies for next-generation sequencing. Nat. Methods 7, 111–118. doi:10.1038/nmeth.1419
Morrison, S. L. (1997). Transformation of E. coli by electroporation. Curr. Protoc. Immunol. 21, Appendix 3N–A.3N.4. doi:10.1002/0471142735.ima03ns21
Nordholt, N., van Heerden, J., Kort, R., and Bruggeman, F. J. (2017). Effects of growth rate and promoter activity on single-cell protein expression. Sci. Rep. 7, 6299. doi:10.1038/s41598-017-05871-3
Palva, I. (1982). Molecular cloning of α-amylase gene from Bacillus amyloliquefaciens and its expression in Bacillus subtilis. Gene 19, 81–87. doi:10.1016/0378-1119(82)90191-3
Park, S., Liu, G., Topping, T. B., Cover, W. H., and Randall, L. L. (1988). Modulation of folding pathways of exported proteins by the leader sequence. Science 239, 1033–1035. doi:10.1126/science.3278378
Pedreira, T., Elfmann, C., and Stülke, J. (2022). The current state of SubtiWiki, the database for the model organism Bacillus subtilis. Nucleic Acids Res. 50, D875–D882. doi:10.1093/nar/gkab943
Petsch, D., and Anspach, F. B. (2000). Endotoxin removal from protein solutions. J. Biotechnol. 76, 97–119. doi:10.1016/S0168-1656(99)00185-6
Quesada-Ganuza, A., Antelo-Varela, M., Mouritzen, J. C., Bartel, J., Becher, D., Gjermansen, M., et al. (2019). Identification and optimization of PrsA in Bacillus subtilis for improved yield of amylase. Microb. Cell Factories 18, 158. doi:10.1186/s12934-019-1203-0
Sieben, M., Giese, H., Grosch, J.-H., Kauffmann, K., and Büchs, J. (2016). Permeability of currently available microtiter plate sealing tapes fail to fulfil the requirements for aerobic microbial cultivation. Biotechnol. J. 11, 1525–1538. doi:10.1002/biot.201600054
Singh, R., Kumar, M., Mittal, A., and Mehta, P. K. (2016). Microbial enzymes: industrial progress in 21st century. 3 Biotech. 6, 174. doi:10.1007/s13205-016-0485-8
Son, Y. J., Ryu, A. J., Li, L., Han, N. S., and Jeong, K. J. (2016). Development of a high-copy plasmid for enhanced production of recombinant proteins in leuconostoc citreum. Microb. Cell Factories 15, 12. doi:10.1186/s12934-015-0400-8
Stephenson, K., and Harwood, C. R. (1998). Influence of a cell-wall-associated protease on production of α-amylase by Bacillus subtilis. Appl. Environ. Microbiol. 64, 2875–2881. doi:10.1128/AEM.64.8.2875-2881.1998
Tännler, S., Zamboni, N., Kiraly, C., Aymerich, S., and Sauer, U. (2008). Screening of Bacillus subtilis transposon mutants with altered riboflavin production. Metab. Eng. 10, 216–226. doi:10.1016/j.ymben.2008.06.002
Tsuge, K., Ohata, Y., and Shoda, M. (2001). Gene yerP, involved in surfactin self-resistance in Bacillus subtilis. Antimicrob. Agents Chemother. 45, 3566–3573. doi:10.1128/AAC.45.12.3566-3573.2001
van Wely, K. H., Swaving, J., Freudl, R., and Driessen, A. J. (2001). Translocation of proteins across the cell envelope of Gram-positive bacteria. FEMS Microbiol. Rev. 25, 437–454. doi:10.1111/j.1574-6976.2001.tb00586.x
Vingataramin, L., and Frost, E. H. (2015). A single protocol for extraction of gDNA from bacteria and yeast. BioTechniques 58, 120–125. doi:10.2144/000114263
Vitikainen, M., Hyyryläinen, H.-L., Kivimäki, A., Kontinen, V. P., and Sarvas, M. (2005). Secretion of heterologous proteins in Bacillus subtilis can Be improved by engineering cell components affecting post-translocational protein folding and degradation. J. Appl. Microbiol. 99, 363–375. doi:10.1111/j.1365-2672.2005.02572.x
Vitikainen, M., Lappalainen, I., Seppala, R., Antelmann, H., Boer, H., Taira, S., et al. (2004). Structure-function analysis of PrsA reveals roles for the parvulin-like and flanking N- and C-terminal domains in protein folding and secretion in Bacillus subtilis. J. Biol. Chem. 279, 19302–19314. doi:10.1074/jbc.M400861200
Vitikainen, M., Pummi, T., Airaksinen, U., Wahlström, E., Wu, H., Sarvas, M., et al. (2001). Quantitation of the capacity of the secretion apparatus and requirement for PrsA in growth and secretion of α-amylase in Bacillus subtilis. J. Bacteriol. 183, 1881–1890. doi:10.1128/JB.183.6.1881-1890.2001
Wang, Y., Chen, Z., Zhao, R., Jin, T., Zhang, X., and Chen, X. (2014). Deleting multiple lytic genes enhances biomass yield and production of recombinant proteins by Bacillus subtilis. Microb. Cell Factories 13, 129. doi:10.1186/s12934-014-0129-9
Westers, L., Westers, H., and Quax, W. J. (2004). Bacillus subtilis as cell factory for pharmaceutical proteins: a biotechnological approach to optimize the host organism. Biochimica Biophysica Acta (BBA) - Mol. Cell Res. 1694, 299–310. doi:10.1016/j.bbamcr.2004.02.011
Wittmann, C., Kim, H. M., John, G., and Heinzle, E. (2003). Characterization and application of an optical sensor for quantification of dissolved O2 in shake-flasks. Biotechnol. Lett. 25, 377–380. doi:10.1023/A:1022402212537
Woodman, M. E., Savage, C. R., Arnold, W. K., and Stevenson, B. (2016). Direct PCR of intact bacteria (colony PCR). Curr. Protoc. Microbiol. 42, A.3D.1–A.3D.7. doi:10.1002/cpmc.14
Wu, S.-C., Ye, R., Wu, X.-C., Ng, S.-C., and Wong, S.-L. (1998). Enhanced secretory production of a single-chain antibody fragment from Bacillus subtilis by coproduction of molecular chaperones. J. Bacteriol. 180, 2830–2835. doi:10.1128/JB.180.11.2830-2835.1998
Yan, S., and Wu, G. (2017). Bottleneck in secretion of α-amylase in Bacillus subtilis. Microb. Cell Factories 16, 124. doi:10.1186/s12934-017-0738-1
Zhang, K., Su, L., and Wu, J. (2020). Recent advances in recombinant protein production by Bacillus subtilis. Annu. Rev. Food Sci. Technol. 11, 295–318. doi:10.1146/annurev-food-032519-051750
Zhang, W., Yang, M., Yang, Y., Zhan, J., Zhou, Y., and Zhao, X. (2016). Optimal secretion of alkali-tolerant xylanase in Bacillus subtilis by signal peptide screening. Appl. Microbiol. Biotechnol. 100, 8745–8756. doi:10.1007/s00253-016-7615-4
Zhang, Y., Werling, U., and Edelmann, W. (2012). SLiCE: a novel bacterial cell extract-based DNA cloning method. Nucleic Acids Res. 40, e55. doi:10.1093/nar/gkr1288
Zimmermann, H. F., John, G. T., Trauthwein, H., Dingerdissen, U., and Huthmacher, K. (2003). Rapid evaluation of oxygen and water permeation through microplate sealing Tapes. Biotechnol. Prog. 19, 1061–1063. doi:10.1021/bp025774t
Keywords: automation, B. subtilis, amylase, secretion, chaperone, PrsA
Citation: Hamburger F, Schlichting N, Eichenlaub M, Costea PI, Sauer C, Jenewein S and Kabisch J (2025) Automation-aided construction and characterization of Bacillus subtilis PrsA strains for the secretion of amylases. Front. Bioeng. Biotechnol. 12:1479626. doi: 10.3389/fbioe.2024.1479626
Received: 12 August 2024; Accepted: 30 December 2024;
Published: 23 January 2025.
Edited by:
Birgitta Elisabeth Ebert, University of Queensland, AustraliaReviewed by:
Daipayan Sarkar, National Institutes of Health (NIH), United StatesSong Liu, Jiangnan University, China
Copyright © 2025 Hamburger, Schlichting, Eichenlaub, Costea, Sauer, Jenewein and Kabisch. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stefan Jenewein, c3RlZmFuLmplbmV3ZWluQGJhc2YuY29t; Johannes Kabisch, am9oYW5uZXMua2FiaXNjaEBudG51Lm5v