
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Bioeng. Biotechnol. , 01 November 2023
Sec. Industrial Biotechnology
Volume 11 - 2023 | https://doi.org/10.3389/fbioe.2023.1275036
This article is part of the Research Topic Proceedings of the 18th International Symposium on BioPolymers (ISBP2022) View all 12 articles
 Maria-Tsampika Manoli1,2
Maria-Tsampika Manoli1,2 Francisco G. Blanco1,2
Francisco G. Blanco1,2 Virginia Rivero-Buceta1,2Ryan Kniewel1,2Sandra Herrera Alarcon1,2
Virginia Rivero-Buceta1,2Ryan Kniewel1,2Sandra Herrera Alarcon1,2 Sergio Salgado1,2
Sergio Salgado1,2 M. Auxiliadora Prieto1,2*
M. Auxiliadora Prieto1,2*Designing cell factories for the production of novel polyhydroxyalkanoates (PHAs) via smart metabolic engineering is key to obtain à la carte materials with tailored physicochemical properties. To this end, we used the model medium-chain-length-PHA producing bacterium, P. putida KT2440 as a chassis, which is characterized by its metabolic versatility and stress tolerance. Different PHA biosynthetic modules were assembled in expression plasmids using the Golden gate/MoClo modular assembly technique to implement an orthogonal short-chain-lengh-PHA (scl-PHA) switch in a “deaf” PHA mutant. This was specifically constructed to override endogenous multilevel regulation of PHA synthesis in the native strain. We generated a panel of engineered approaches carrying the genes from Rhodospirillum rubrum, Cupriavidus necator and Pseudomonas pseudoalcaligenes, demonstrating that diverse scl-PHAs can be constitutively produced in the chassis strain to varying yields from 23% to 84% PHA/CDW. Co-feeding assays of the most promising engineered strain harboring the PHA machinery from C. necator resulted to a panel of PHBV from 0.6% to 19% C5 monomeric incorporation. Chromosomally integrated PHA machineries with high PhaCCn synthase dosage successfully resulted in 68% PHA/CDW production. Interestingly, an inverse relationship between PhaC synthase dosage and granule size distribution was demonstrated in the heterologous host. In this vein, it is proposed the key involvement of inclusion body protein IbpA to the heterologous production of tailored PHA in P. putida KT2440.
Polyhydroxyalkanoates (PHAs) are biotechnologically useful natural polyesters produced in many microorganisms. PHAs function as an intracellular carbon and energy storage reservoir with physical and mechanical properties that make them promising bioplastics with many possible applications (Zheng et al., 2019). Optimal PHA production generally occurs when a deficit exists in the nutritional conditions of the cell, with the deprivation of nitrogen being the most widely applied in bioproduction strategies (Kim and Lenz, 2001; Mahato et al., 2021; Ronďošová et al., 2022). Hydrophobic PHA accumulates in roughly spherical cellular inclusions called PHA granules that are segregated from the cytoplasm by a surface layer of amphipathic phasin proteins and other granule-associated proteins (Jendrossek, 2009). The Gram-negative P. putida KT2440 has been extensively studied as archetypal producer of medium-chain-length PHA (mcl-PHA) containing monomers of 6–14 carbon atoms in length (C6-C14) (Mezzina et al., 2021). The P. putida pha genomic locus contains the genetic machinery (Supplementary Figure S1, panel A) which along with its metabolic flexibility, allows for the production of mcl-PHA from a variety of substrates, including both PHA-like aliphatic fatty acids as well as PHA-unrelated carbon sources (Figure 1) (Cheng and Charles, 2016; Koller et al., 2017).
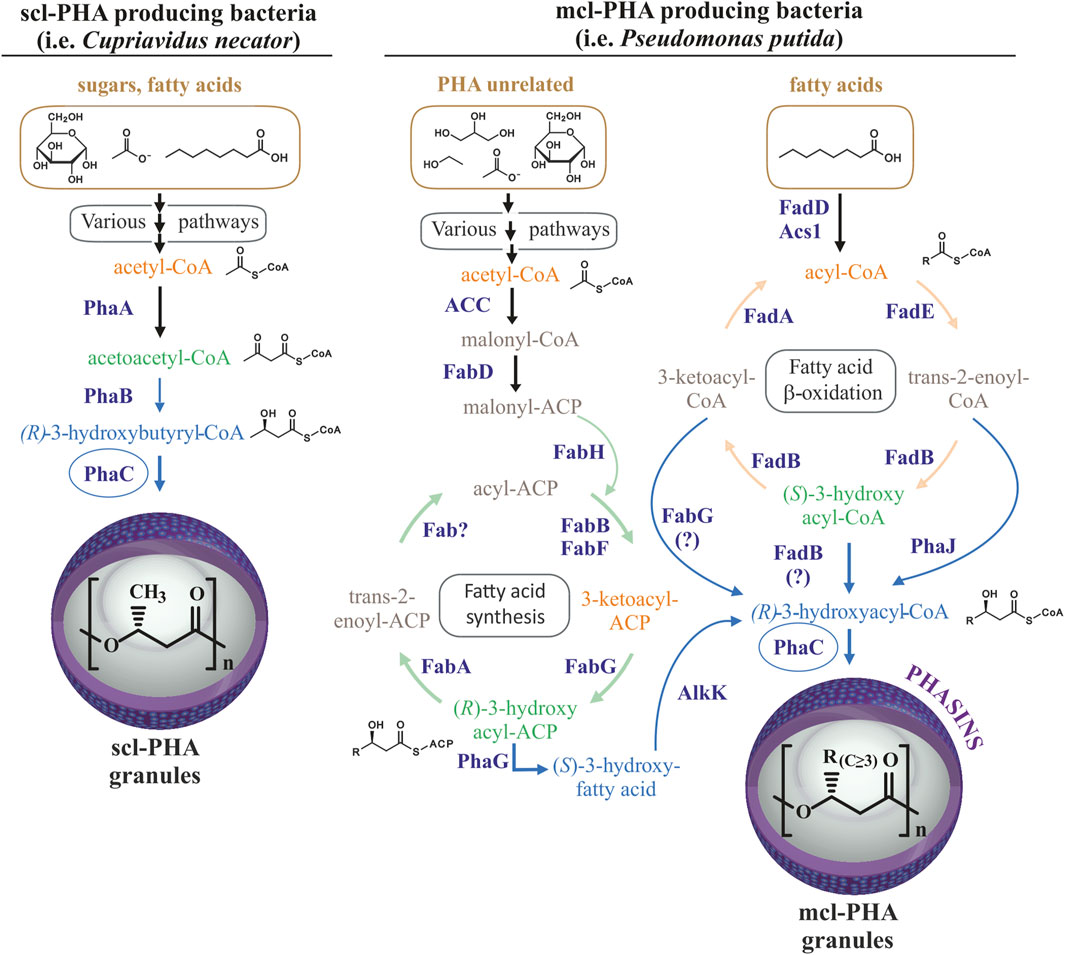
FIGURE 1. Routes for PHA production. Comparison of routes for scl-PHA production in many bacteria (left) and mcl-PHA production in Pseudomonas putida (right). Scl-PHA production requires the PhaA 3-ketothiolase and PhaB acetoacetyl-CoA reductase in addition to the PhaC synthase. Mcl-PHA 3-hydroxyacyl monomers are produced in Pseudomonas putida by de novo synthesis of fatty acids from PHA-unrelated substrates and ß-oxidation of fatty acids, allowing for the production of mcl-PHA from diverse carbon sources from diverse carbon sources (shown with brownish color).
In contrast to P. putida, many bacterial species are capable of producing short-chain-length PHA (scl-PHA) with monomers of 3–5 carbon atoms in length (C3-C5) from PHA-unrelated carbon sources (Figure 1). For example, the production of polyhydroxybutyrate (PHB) from sugars in C. necator requires the 3-ketothiolase PhaA to condense two glycolysis-derived acetyl-CoA molecules into acetoacetyl-CoA, and the acetoacetyl-CoA reductase PhaB to reduce acetoacetyl-CoA to four carbon 3-hydroxybutyryl-CoA, the substrate for PhaC synthase (Figure 1). Acetyl-CoA for PHB synthesis can also be provided by other carbon sources, such as the degradation of amino acids or ß-oxidation of fatty acids (Jendrossek and Pfeiffer, 2014). Some bacteria, such as Rhodospirillum rubrum, are capable of producing heteropolymers of both, scl-PHA with monomers of 4 and 5 carbon atoms, as well as mcl-PHA by incorporating 3-hydroxyhexanoate-CoA or 3-hydroxyheptanoate-CoA monomers when provided with medium-chain-length fatty acids (hexanoate) as the carbon source (Brandl et al., 1989; Jin and Nikolau, 2012; Godoy et al., 2023). Importantly, in addition to the supplied substrate and metabolic determinants, organism-specific PhaC synthase(s) determine the length monomers that can be incorporated into the polymer due to differences in substrate specificity (Kim et al., 2017). Thus, the class I PHA synthases, PhaC1 from Cupriavidus necator and PhaC2 from R. rubrum are generally able to incorporate monomers containing up to 5 or 7 carbons in length, respectively (Brandl et al., 1989). In contrast, the class II PHA synthases from P. putida can incorporate a wide variety of monomers of lengths between 6 and 14 carbons to produce mcl-PHAs. As mentioned above, wild type P. putida does not naturally produce scl-PHAs, which is likely due to the substrate specificity of its PHA synthases along with a metabolic propensity to produce longer chain 3-hydroxyacyl-CoA monomers (Huisman et al., 1989; Kim et al., 2017).
Pseudomonads gained special interest due to their metabolic versatility, adaptability to endogenous and exogenous stresses. Specifically, P. putida KT2440 has become a model organism for biotechnological, environmental and industrial applications due to the presence of different genome-scale metabolic models and high advances in synthetic biology and metabolic engineering fields (Mezzina et al., 2021). In fact, several studies have demonstrated the use of P. putida as a chassis for the heterologous expression of scl- or mcl-PHA machinery from other bacteria (Matsusaki et al., 1998; Clemente et al., 2000; Ouyang et al., 2007; Cha et al., 2020). For orthogonal scl-PHA production in this strain apart from the deletion of the native pha locus, the expression of phb genes is required (Figure 1) (Prieto et al., 2016). However, the majority of these studies were based on the heterologous inducible expression systems of phb gene clusters, which are not useful for scaling up processes. Digging into the optimization of the constitutive scl-PHA production in P. putida, in this work we have used as microbial chassis the PP05_01 strain with the entire native pha genomic locus deleted (Mato et al., 2020). To enhance the capacity for designing, building and testing of heterologous constitutive expression systems with different strengths, we adapted our Golden gate/MoClo modular assembly cloning method for obtaining plasmids suited for constitutive PHA production at different rates in P. putida (Blázquez et al., 2023). These approaches allowed us not only to control the monomer composition, but to identify the inclusion body protein IbpA as an important partner for heterologous production of scl-PHA in P. putida KT2440. We demonstrated an inverse relationship between PhaC synthase dosage and granule size/number distribution in the heterologous host driven by IbpA.
Bacterial strains and plasmids used in this work are listed in Table 1 and Supplementary Table S1 respectively. Unless otherwise indicated, E. coli and P. putida strains pre-cultures were grown in lysogeny broth (LB) at 37°C and 30°C respectively, at 200 rpm. Streptomycin (75 μg/mL), ampicillin (100 μg/mL), kanamycin (50 μg/mL), gentamycin (10 μg/mL), chloramphenicol (34 μg/mL), IPTG (0.5–1 mM) and Xgal (40 μg/mL) were added as required. Solid media were made by the addition of 1.5% (w/v) agar.

TABLE 1. Strains used in this study.
For PHA production standard laboratory methods were performed as previously described (Manoli et al., 2022). Briefly, P. putida strains were grown overnight in LB, the cells were washed twice with 0.85% saline solution and adjusted to an optical density of 600 nm of 0.3. Then, P. putida cells were grown for 24 h at 30°C and 200 rpm in 0.1 N M63, a nitrogen-limited minimal medium (13.6 g/L KH2PO4, 0.2 g/L (NH4)2SO4, 0.5 mg/L FeSO4·7H2O, adjusted to pH 7.0 with KOH). This medium was supplemented with 1 mM MgSO4, a solution of trace elements/Goodies (composition 1000X: 2.78 g FeSO4·7H2O, 1.98 g MnCl2·4H2O, 2.81 g CoSO4·7H2O, 1.47 g CaCl2·2H2O, 0.17 g CuCl2·2H2O, 0.29 g ZnSO4·7H2O dissolved in 1 L water), 15 mM sodium octanoate or 20 mM glucose as the carbon source (C/N ratio was maintained at 40 mol/mol). Concerning the inducible Ptrc cultures 1 mM IPTG was used from the beginning of the assays. Culture growth was monitored in shaking Erlenmeyer flasks of 250 mL (maintaining a volume to air ratio of 1/5) by measuring optical density at 600 nm (OD600) using a portable spectrophotometer (ThermoFisher Scientific).
Pseudomonas putida strains were transformed following the protocol described by Choi et al. with some adaptations (Choi et al., 2006). Briefly, the strains of P. putida KT2440 and PP05_01 were grown overnight in 5 mL of LB at 30°C and 200 rpm and subcultured into 50 mL of LB to an OD600 between 0.5–1.0. These cultures were pelleted at 3,200 ×g for 8 min, washed five times with 300 mM sucrose and resuspended in 500 μL of 300 mM sucrose. 100 μL of cell suspension were mixed with 100 ng of desired plasmid and transferred to a 2 mm gap electroporation cuvette. After a pulse of 25 μF, 2.5 kV and 200 Ω; 900 μL of room temperature LB was added and transferred to a 100 × 16 mm round-bottom polypropylene tube and incubated for 1 h at 30°C, 200 rpm. 5 μL and 100 μL of the transformation cultures were plated on LB agar plates containing the corresponding antibiotic for the plasmid maintenance and grown at 30°C.
Plasmid DNA minipreps were made using the High Pure Plasmid Isolation Kit (Roche) following the manufacturer’s protocol. Genomic extractions of P. putida KT2440, R. rubrum ATCC11170, C. necator H16 and P. pseudoalcaligenes CECT5344 were performed with the illustra bacteria genomicPrep Mini Spin Kit (GE Healthcare). DNA agarose gel bands, PCR products and digestion products were purified with illustraTM GFX PCR DNA and Gel Band Purification Kit (GE Healthcare). DNA concentration was measured using a NanoDrop 2000 Spectrophotometer (ThermoFisher Scientific). The Golden gate restriction enzymes, BbsI (BpiI) and BsaI (Eco31I) were from ThermoFisher Scientific. Phusion DNA polymerase, T4 DNA ligase and all other restriction enzymes were from New England Biolabs. The MoClo toolkit was a gift from Sylvestre Marillonnet via Addgene (Kit #1000000044) (Weber et al., 2011; Werner et al., 2012).
Golden gate/MoClo assembly is a hierarchical method requiring the establishment of a library of promoter + RBS, CDS and terminator parts (level 0) that are assembled into higher order promoter-CDS-terminator transcription units (TU, level 1) and finally into assemblies containing 1 to 7 transcription units (level 2, Figure 2). The parts comprised the essential units needed to assemble the heterologous expression constructs for PHA production (i.e., phaC synthase, phaA 3-ketothiolase, phaB acetoacetyl-CoA, phaP phasin) from C. necator, P. pseudoalcaligenes and R. rubrum. Non-CDS parts included synthetic constitutive promoters of varying strengths. These promoters contained RBS sequences by the inclusion of the two Shine-Dalgarno sequences/RBSs in the bicistronic translational coupler BCD2 for the low expression promoter BG28/14a from Zobel et al. (level 0 plasmid pSS15) or the AGGGGG RBS for the moderate expression promoter SynPro16 from Tiso et al. (level 0 plasmid pRK154) (Zobel et al., 2015; Tiso et al., 2016). Following the design of SynPro16 validated by Tiso et al., the presence of the upstream insulating terminator λT0 was maintained in this promoter part. These two promoters were chosen for the assemblies used in the proof-of-concept experiments described below as they were expected to confer low and moderate constitutive expression in P. putida due to the published descriptions of their activities (Zobel et al., 2015; Tiso et al., 2016). Finally, parts were made for five rho-independent terminators with efficiencies of ≥98%, including the natural terminators λT0, rrnB_T1, rnpB_T1, rpoC and the synthetic T500 (Stueber and Bujard, 1982; Lutz and Bujard, 1997; Yarnell and Roberts, 1999; Larson et al., 2008; Cambray et al., 2013).
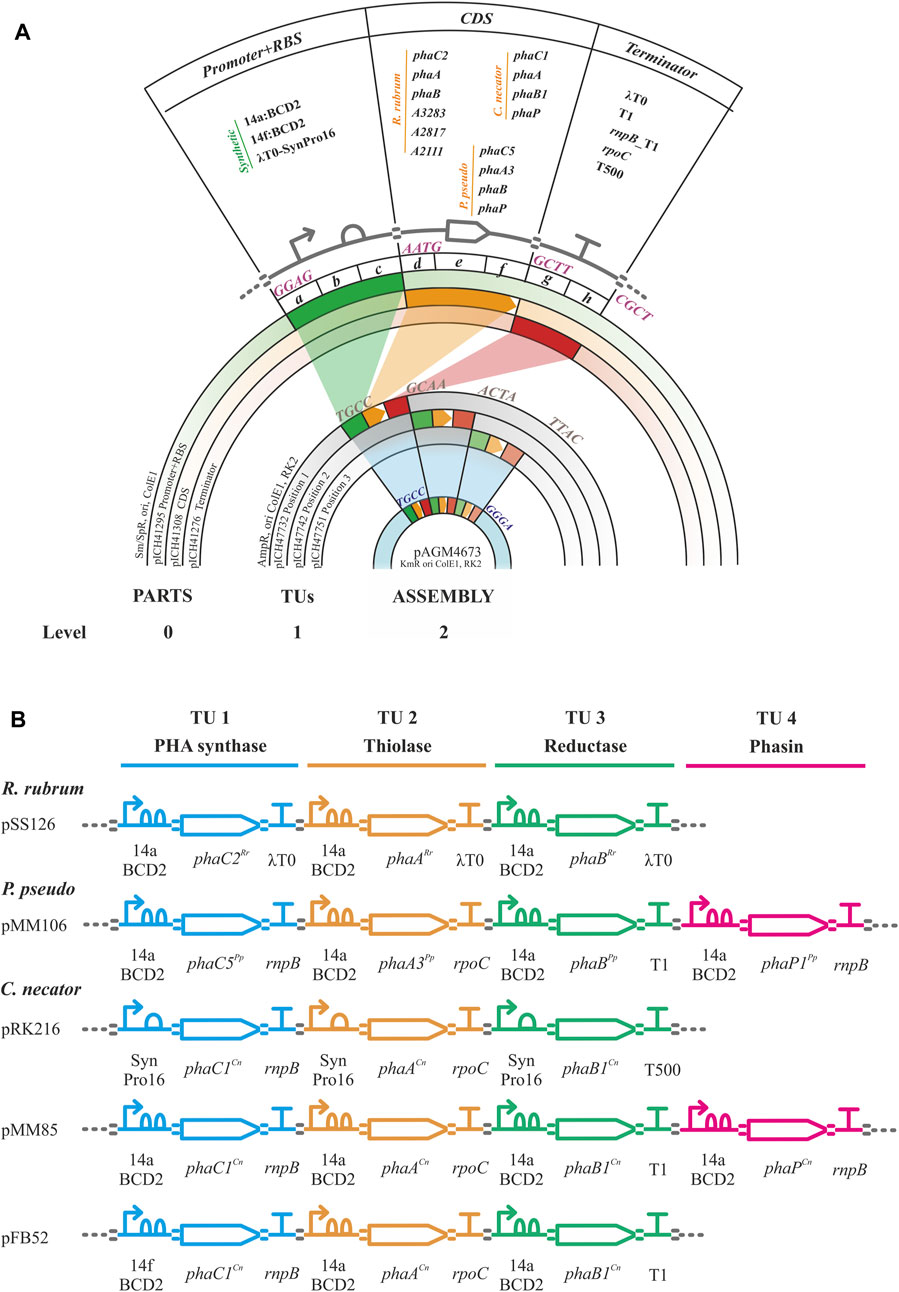
FIGURE 2. Golden gate/MoClo schema. (A). Golden gate/MoClo assembly cloning schema of levels 0, 1 and 2 as visualized by concentric plasmid constructs for the creation of a typical assembly containing three transcription units (TUs). Level 0: Promoter, CDS, and terminator parts cloned into level 0 plasmids pICH41295, pICH41308 and pICH41276, respectively. The Level 0 parts used are shown. The four nucleotide 5′ overhang BbsI ligation fusion sites are indicated for each level 0 part. Level 1: Three TU constructs in level 1 plasmids for positions 1, 2 and 3 by plasmids pICH4732, pICH47741 and pICH47751, respectively. Four nucleotide 5′ overhang BsaI ligation fusion sites for linking each of the TUs are indicated. Level 2: Final assembly of three TUs into plasmid pAGM4673 along with the inclusion of the end-linker from plasmid pICH41780 (not shown, provides fusion sites for ligation of 5′ TTAC and 5′ GGGA) to occupy positions 4–7. Four nucleotide 5′ overhang BbsI ligation fusion sites flanking the three TUs are indicated. (B). Example genetic design schematics of Golden gate/MoClo level 2 plasmid assemblies for the expression of PHB machinery from R. rubrum (pSS126), Pseudomonas pseudoalcaligenes (pMM106) and Cupriavidus necator (pRK216, pMM85, pFB52). The TU assemblies contain the PHA synthase, thiolase, reductase, and optional phasin genes with their promoters and terminators indicated. Genetic design glyphs follow the Synthetic Biology Open Language (SBOL) visual standard for elements: promoter, RBS, CDS, terminator and assembly scar.
Golden gate/MoClo plasmids were constructed following Weber et al. with some modifications and recently updated by Weber et al. (2011), Blázquez et al. (2023). For Level 0 plasmid construction, every part was PCR amplified with DNA oligos designed using Benchling (www.benchling.com) with the following characteristics: a tail containing the BbsI recognition site followed by the corresponding four nucleotide fusion site, 21 bp of minimal length for target complementarity, 50°C of minimal Tm for that region and a maximal Tm difference of ±1.5°C between both oligos. Promoter and CDS sequences were PCR amplified from C. necator H16, R. rubrum ATCC11170, P. pseudoalcaligenes CECT5344 and P. putida KT2440 genomic DNA or plasmid templates. Terminator sequences were PCR amplified from E. coli DH5α genomic DNA or plasmid templates. Oligo primers, templates and references for all Golden gate level 0 PCR amplifications are listed in Supplementary Table S2. Where needed DNA domestication (removal of naturally present internal BbsI or BsaI restriction sites was carried out using the strategy of Engler et al. (2008). This strategy generated PCR subparts with 5’ cohesive end fusion sites overlapping the internal restriction sites to be eliminated. The primers used for PCR contained synonymous substitution point mutations in the fusion nucleotides of a BbsI site that eliminate the restriction site upon ligation in the golden gate level 0 reaction. PCR products were purified using a gel purification kit following manufacturer instructions. Golden gate digestion-ligation reactions were set up with 100 ng of level 0 acceptor plasmid (position abc, pICH41295; position def, pICH41308; or position gh, pICH41276; Supplementary Table S1) and the corresponding amount of gel purified PCR product to achieve a 2:1 insert-vector molar ratio; 10 U BbsI, 400 U T4 DNA ligase and 1 mM ATP in Buffer G (10 mM Tris-HCl pH 7.5, 10 mM MgCl2, 50 mM NaCl and 0.1 mg/mL bovine serum albumin; ThermoFisher Scientific) in a reaction volume of 20 μL. The reaction was incubated in a thermocycler with four cycles of 37°C for 10 min and 16°C for 10 min followed by 65°C for 20 min. A 100 μL aliquot of chemically competent E. coli DH5α was then transformed by heat shock with 5 μL of the Golden gate reaction. Transformed cells were plated on LB-agar supplementedwith 75 μg/mL streptomycin, 0.5 mM IPTG and 40 μL/mL X-gal and grown overnight at 37°C for selection of the disruption of a-complementation ß-galactosidase activity. Several white colonies per transformation were transferred to 4 mL LB medium with 75 μg/mL streptomycin and grown overnight at 37°C for plasmid purification. The extracted plasmids were digested with BsaI to check for the presence of the correct insert size and confirmed by sequencing with primers RK81 and RK82 (Supplementary Table S2).
For Level 1 construction of TUs, the reaction mix contained 100 ng of acceptor plasmid (depending on the TU position, Supplementary Table S1), the three level 0 plasmids containing a promoter plasmid part, a CDS plasmid part and a terminator plasmid part in a 2:1 donor plasmid:acceptor plasmid molar ratio; 10 U BsaI, 400 U T4 DNA ligase and 1 mM ATP in Buffer G in a reaction volume of 20 μL. The reaction was incubated in a thermocycler for four cycles of 40°C for 10 min and 16°C for 10 min, then at 50°C for 10 min and 80°C for 20 min. Reactions were transformed into E. coli DH5α and plasmids prepared as for level 0 golden gate reactions except that 100 μg/mL ampicillin, 0.5 mM IPTG and 40 μL/mL X-gal were present for selection. The extracted plasmids were digested with BbsI to check for the presence of the correct insert size and sequenced with primers RK155 and RK156 (Supplementary Table S2).
Level 2 assembly reactions were carried out with 100 ng of acceptor plasmid (pAGM4673, RK2 origin of replication providing some range in copy number in pseudomonads estimated to be maintained at 30 ± 10 copies per genome equivalent in P. putida, Supplementary Table S1), the corresponding end-linker plasmid depending on the number of TUs to be inserted, each level 1 plasmid in a 2:1 donor plasmid:acceptor plasmid molar ratio, 10 U BbsI, 400 U T4 DNA ligase and 1 mM ATP in Buffer G in a reaction volume of 20 μL. The reaction was incubated in a thermocycler with four cycles of 37°C for 10 min and 16°C for 10 min followed by 65°C for 20 min. Reactions were transformed into E. coli DH5α as for level 0 golden gate reactions except that 50 μg/mL kanamycin was added for selection. Red-white color selection was carried out (red color canthaxanthin produced by its operon in the cloning site of pAGM4673) and several white colonies were transferred to 4 mL of liquid LB medium with 50 μg/mL kanamycin for plasmid minipreps. The extracted plasmids were confirmed by digestion with DraIII or EcoRI and sequenced using primers RK157 and RK158 (Supplementary Table S2).
For ibpA gene deletion, the pEMG knockout system was used, generating the PP05_15 strain. ibpA encodes a small heat shock protein, located in PP_1982 locus. Two pairs of primers (MM388-MM389, MM390-MM391, Supplementary Table S2) were designed to amplify the flanking fragments of the ibpA locus. An overlap PCR was carried out giving a product of 1.6 kb and was cloned into pEMG plasmid using EcoRI and BamHI restriction enzymes. For the first and second recombination steps, we followed the protocol described above. In this case for the confirmation of the second recombination event, external pair of primers (i.e., MM392-MM393) of to be deleted region were used.
For IbpA complementation assays, the PP05_15 strain was transformed with pMM194 plasmid (obtained using Golden gate/MoClo strategy) that contains the ibpA gene under the low strength 14a promoter and the BCD2 element. PHA accumulation assays were then performed as stated below.
For the co-localization experiments one multipurpose vector was constructed, with a phasin fusion position tags and under the control of the inducible Pm promoter. For the in vivo co-localization experiments the pBDN2 vector was used, since it contains the cloning site for the phasin between two SapI restriction sites (Blanco et al., 2023). Then, PP01_02 strain was transformed with the pBDN2 derived plasmids (i.e., empty, including only the msf-GFP, as negative control and harboring the wild type PhaP1Cn). The resulting strains were inoculated at OD600 nm of 0.3 under PHA accumulating conditions and were allowed to grow until the polymer accumulation was visible (i.e., OD600 = 0.7). At this point the cultures were induced with 1 mM 3 MB for the expression of the fusion protein phasin-GFP (Mato et al., 2020).
To facilitate the single-copy genes into bacterial chromosome (i.e., for the generation of PP05_12 and PP01_02 strains), an adapted for Golden gate broad host range mini-Tn7 vector (pRK99) was used. The genome integration is based on a neutral and naturally evolved attTn7 site, located downstream of a highly conserved glmS gene (Zobel et al., 2015). A four parental mating process was carried out from overnight LB precultures of E. coli CC118λpir bearing pMM175 and pFB52 plasmids (donor strains), E. coli HB101 (pRK600) (helper strain), E. coli DH5aλpir (pTnS-1) (leading transposase strain), and P. putida PP05_01 (recipient strain). The transconjugants were selected on cetrimide agar containing 10 μg/mL gentamycin plates and incubated at 30°C for 18 h. The next day few colonies were picked on LB kanamycin, to verify the loss of the plasmid and LB gentamycin and incubated at 30°C for 18 h. Few gentamycin resistant and kanamycin sensitive clones were selected to verify the correct insertion of the transposon into the attTn7 stie and checked via colony PCR and sequencing (Zobel et al., 2015).
For PHA quantification, 20 mL of each P. putida strain grown in PHA production conditions (see 2.1 section) for 24 h were centrifuged for 30 min at 3,200 ×g. Cells were washed once with 0.85% NaCl and lyophilized for 24 h. Lyophilized pellets were weighed to obtain the cell dry weight (CDW) of total biomass. PHA monomer composition and PHA content were determined by Gas Chromatography-Mass Spectrometry (GC-MS) of the methanolysed polyester (Braunegg et al., 1978; Revelles et al., 2016; Manoli et al., 2022). In each condition, at least two independent biological replicates were performed. Where the statistical error was higher of 10%, four biological replicates were performed. During the methanolysis process, two technical replicates were included for each biological sample. 2–5 mg of lyophilized cells were resuspended in 2 mL of methanol acidified with 3% (v/v) H2SO4 for scl-PHA analysis, and resuspended in 2 mL of methanol containing 15% (v/v) H2SO4 for mcl-PHA analysis. 2 mL of chloroform containing 0.5 mg/mL 3-methylbenzoic acid was added to the samples as an internal standard. Samples were boiled in a screw-capped tube at 100°C for 4 or 5 h to assay scl-PHA or mcl-PHA, respectively. After cooling, the mixture was washed twice by adding 1 mL of distilled water, centrifuged for 10 min, and followed by the removal of the aqueous phase. The organic layer containing the resulting methyl ester of each monomer was analyzed by GC-MS using an Agilent 7890A GC equipped with a DB-5HT capillary column (30 m length, 0.25 mm internal diameter, 0.1 µm film thickness) and mass data were acquired and processed with an Agilent 5975C mass spectrometer. Samples (1 μL) of the organic phase were injected with helium as carrier gas at a ratio of 1:10 with 1 part sample to 10 parts helium, and the oven temperature was programmed to remain at 80°C for 2 min and then increased at a rate of 5°C/min up to 115°C for the efficient separation of peaks. The temperature of the injector was 250°C. Spectra were obtained as electron impacts with an ionizing energy for MS operation of 70 eV. Standard curves with known quantities of PHB (Sigma-Aldrich) or poly (3-hydroxyhexanoate-co-3-hydroxyoctanoate) (Bioplastech, Ltd.) dissolved in chloroform were used to calculate the monomer composition of the extracted polymers.
Monomer composition was also analysed by NMR. For these experiments, the used solvent was deuterated chloroform (chloroform-d 99.8%) that contains 0.03% (v/v) tetramethylsilane (TMS) (CDCl3) (ref. 225,789 from Sigma-Aldrich). Proton NMR spectra (1H-NMR) were recorded using a 90° pulse experiment under the following acquisition parameters: 128 scans with a fixed receiver gain value of 287, spectral width of 12.0164 ppm, 32,768 points in the time domain, and acquisition time of 2.27 s. COSY spectra were recorded using the standard Bruker sequence cosygpqf. Spectra were recorded under the following acquisition parameters: fixed receiver gain value of 1290, 128 scans and a spectral width of 13.0177 × 13.0177 ppm.
The 1D (1H), 2D Correlated Spectroscopy (COSY) NMR spectra of the extracted polymers were recorded on Bruker AV III 600 MHz spectrometer (Bruker, Rheinstetten, Germany) using a XI 600 MHz S3 5 mm probe with Z-gradient in CDCl3. The resulting NMR spectra were processed by Mestrelab MNova software (Version 14.2.3–29241). Phasing and baseline correction were manually completed.
According to 1H-NMR and COSY assays (Supplementary Figure S3), the signals assigned to the protons of the HB monomer are methyl at 1.27 ppm (4), methylene at 2.50 ppm (2) and methine at 5.25 ppm (3), and the protons assigned to the mcl-HA monomer are methyl at 0.90 ppm (9), methylene groups of the side chain at 1.28 and 1.59 ppm (8), methylene group at 2.50 ppm (6) and methine at 5.25 ppm (7).
For granule extraction, cultures grown under PHA-producing conditions for 24 h as mentioned above were harvested by centrifugation for 20 min at 10,000 ×g. Cells were resuspended in 15 mM Tris-HCl pH 8.0 buffer and then disrupted twice using French Press (at ≈ 1 000 psi). PHB granules from pellets (resuspended in 15 mM Tris-HCl pH 8.0) were purified by two subsequent glycerol density gradient centrifugations at 18,000 ×g for 40 min. The first gradient consisted of 6 mL of sample layered over 3 mL of 85% glycerol and 6 mL of 50% glycerol. Granules were isolated after centrifugation with a Pasteur pipette at the glycerol 50%–85% interface. The second gradient consisted of 3 mL of granule fraction from the first gradient over 4 layers of 3 mL of glycerol at 85%, 80%, 60%, and 40% glycerol. Granules were isolated after centrifugation from the glycerol 40%–60% interface.
For granule-associated protein detection, independent granule isolations were normalized to the same PHB content by PHA quantification as explained above. Aliquots containing 22.5 μg of PHB from each extraction were run onto 12.5% SDS-PAGE gels and stained using BlueSafe (Nzytech).
For N-terminal sequencing of granule-associated proteins, these SDS-PAGE (12.5%) gels were transferred onto methanol activated-polyvinylidene fluoride (PVDF) membranes in a semidry transfer device (Biorad) soaked in transfer buffer (25 mM Tris, 192 mM glycine, 20% methanol, pH 8.3) for 1 h 15 min at 15 mV. The resulting transferred membranes were stained with Ponceau S stain (ThermoFisher), and the visible protein bands of selected proteins were subjected to N-terminal sequencing by Edman degradation in a protein sequencer (Applied Biosystems, Procise 494).
Cultures were routinely visualized with a 100× phase-contrast objective using an epifluorescence microscope Leica DM4B (Wetzlar, Germany) and images were taken with an attached camera (Leica DFC345 FX). Where needed a filter system L5 was used for GFP observation. In order to fix cells and achieve a correct superposition of images from the different channels, microscope slides were covered by a thin layer of 0.1% poly-L-lysine. Then, 5 µL of the cell suspension was deposited on the covered slide and immediately observed under microscopy.
For Transmission electron microscopy (TEM) experiments, P. putida cells previously grown during 24 h under M63 0.1 N minimal medium supplemented with 15 mM octanoate, were harvested and washed twice with 1 X PBS. Then, the cells were fixed for 1 h in 3% glutaraldehyde in PBS and washed twice with PBS. Samples were post-fixed in 1% osmium tetroxide and 0.8% potassium ferricyanide for 1 h at 4°C. Samples were washed with PBS prior to dehydration with an increasing gradient of ethanol (30%, 50%, 70%, 80%, 90% and 100%) of 10 min per step. Samples were embedded in LX112 resin and were polymerized for 48 h at 60°C. 60–80 nm sections were placed in copper grids of 75 mesh and stained with 5% uranyl acetate for 15 min and lead citrate for 3 min. Samples were viewed in a JEOL 1230 TEM and images were taken with a CMOS TVIPS 16 mp camera.
To determine the size of PHA granules from TEM micrographs, 50 cells of each engineered strain were selected, in which PHA granule diameters were measured using ImageJ software. In each case, 100 granules with sharp boundaries were selected and analyzed. Thus, to avoid measuring cells in different section plans, only granules from cells with a size of 0.9-1 x 2–2.2 µm (width x length) were considered (Galán et al., 2011).
The chassis applied in this work was the PHA-deficient strain (P. putida KT2440 Δpha, named as PP05_01). This lacks the native pha gene cluster, including phaC1ZC2DFI that encodes the two PHA synthases, the depolymerase, the transcriptional activator PhaD and the two phasins (including promoters and regulatory regions that drive the expression of pha genes) (Supplementary Figure S1 and Supplementary Table S3).
Phenotypic evaluation of the PP05_01 (Δpha) strain confirmed that it does not produce PHA as determined microscopically by the complete lack of PHA granules when cultured in LB or 0.1 N M63 supplemented with 15 mM octanoate (Supplementary Figure S1) and by the lack of detectable PHA by GC-MS (Table 2). Growth of the PP05_01 strain was highly similar to wild type. Comparison of the two strains revealed lower relative CFUs in the wild type (Supplementary Figure S1). This was expected due to the allocation of metabolic resources towards PHA production in the wild type that were instead dedicated to biomass accumulation in the Δpha strain (De Eugenio et al., 2010a; Manoli et al., 2022).
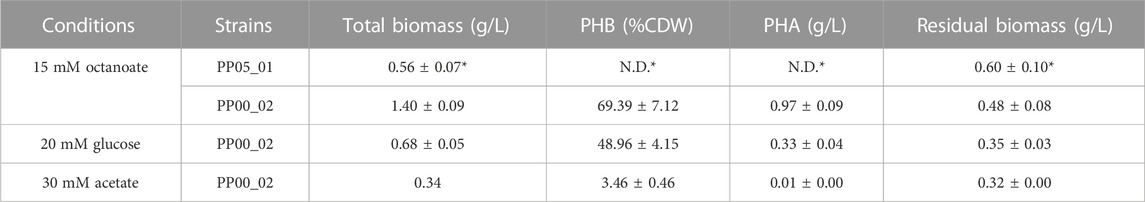
TABLE 2. PHA yield following 24 h of growth under the corresponding conditions using 0.1 N M63 minimal medium. The data correspond to the mean values and standard deviations of four biological replicates (with two technical replicates for the methanolysis analysis). Residual biomass indicates the biomass free of PHA. N.D.: not detected *data obtained from (Manoli et al., 2022).
Since PP05_01 strain will be the foundation for implanting an orthogonal, synthetic phb gene cluster designed for constitutive scl-PHA production, we firstly validated the scl-PHA production capacity when carrying an inducible monocopy system that allows the production of the PHA machinery in the presence of an inducer. Thus, we chromosomally inserted via mini Tn5 transposon the Ptrc:phaCCn-phaACn-phaBCn cluster from C. necator driven by the Ptrc promoter inducible by IPTG. Two strains were obtained on wild type background and pha null, PP00_03 and PP00_02, respectively. By initial screening of PP00_03 using octanoate as carbon and energy source, we obtained 60% PHA/CDW, composed of 43% C4, 4% C6 and 53% C8 (data not shown). For the purpose of this study, the influence of different carbon sources under nitrogen limited conditions on PHA accumulation was evaluated in the resulting pha null background strain PP00_02 (Table 2). The best PHA accumulation was observed when a fatty acid precursor such as octanoic acid was used compared to glucose or acetate. In fact, PP00_02, lacking the pha cluster, reached high amounts of PHA accumulation with octanoate (e.g., nearly 70% PHA/CDW) and 50% of PHA/CDW under glucose conditions. Taking into account the toxicity effect of acetate and the low growth performance under these conditions (e.g., reaching up to 0.3 g/L total biomass), PP00_02 produced slight amounts of 3% PHA/CDW. Taken together, these experiments validated the PP05_01 chassis as modifiable to enable orthogonal constitutive PHA production.
For the installation of efficient constitutive PHA production machinery, we aimed to develop a modular, extensible system for the creation of PHA gene expression constructs that would be easy to deploy with the chassis strain. To this end, we adapted the Golden gate/MoClo assembly cloning technique to rapidly generate gene expression constructs organized in synthetic operons containing multiple transcription units with the minimum phb genes needed (Weber et al., 2011; Blázquez et al., 2023). The resulting synthetic phb clusters followed the modular structure shown in the synthetic phb orthogonalization pathway (Figure 2), which enabled interchangeability of genetic parts across modules as needed.
Since dosage of the different synthetic phb modules is crucial for proper functioning of the PHA machinery (Hiroe et al., 2012; Li et al., 2016; Li et al., 2017), we varied the strength of synthetic promoters driving the expression of these genes using low (14a) and medium (SynPro16 or SP16) strength constitutive promoters, previously validated in P. putida (Zobel et al., 2015; Tiso et al., 2016; Blázquez et al., 2023). During the strains’ construction, we considered the specificity of the phb module as a potential tool for diversification of the monomeric content and possibly different catalytic capacities of these enzymes in a heterologous chassis. For this, the wild type genes were obtained from bacteria able to produce different types of scl-PHA (i.e., R. rubrum, P. pseudoalcaligenes and C. necator). Numerous plasmid-based synthetic phb modules were generated and tested (Figure 2, Supplementary Table S1), each module contained the three minimal phb transcriptional units necessary for PHB production (i.e., PHA synthase, phaC; 3-ketoacyl-CoA thiolase, phaA; 3-ketoacyl-CoA reductase, phaB).
We assessed the production of scl-PHA (i.e., consisting of C4 or C5 monomers) and mcl-PHA (i.e., consisting of predominantly C6 and C8 monomers) following 24 h of growth in 0.1 N M63 minimal medium supplemented with 15 mM octanoate as the sole carbon source. The strain PP05_01 (pSS126), expressing the PHA machinery from R. rubrum under the low strength 14a constitutive promoter, yielded ∼25% CDW of PHB (Table 3). TEM images of PP05_01 (pSS126) strain revealed that most cells contained a single PHB granule occupying a large proportion of the cytoplasm (Figure 3). To improve the granule stability, in the same construct the three phasins from R. rubrum (i.e., A3283Rr, A2817Rr, A2111Rr) were additionally expressed under the SynPro16 promoter generating the PP05_01 (pRK182) strain. However, no major effect on the overall PHA production properties were observed by yielding 25% PHB/CDW (Table 3).
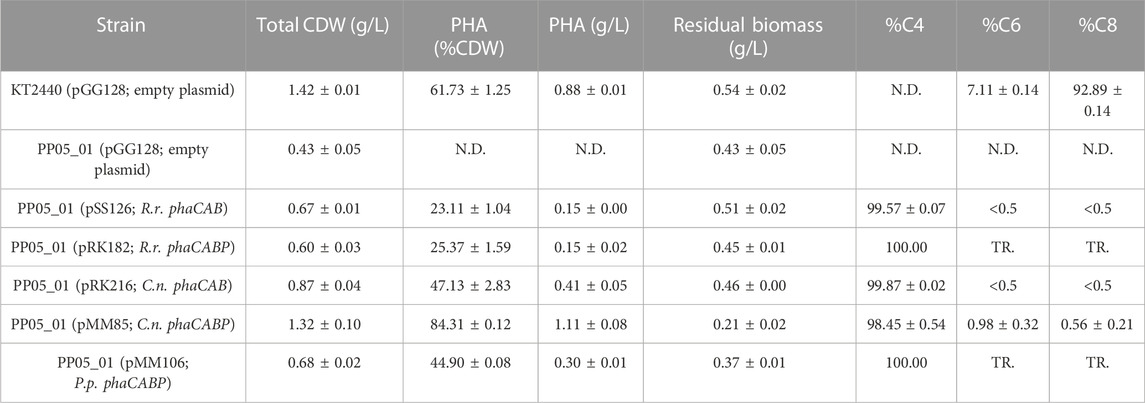
TABLE 3. Properties of heterologous PHA production in Pseudomonas putida chassis strain. GC-MS analysis of PHA content in 0.1 N M63 minimal medium supplemented with 15 mM octanoate for 24 h. The data correspond to the mean values and standard deviations of at least two independent biological replicates. TR.: Traces; N.D.: not detected.
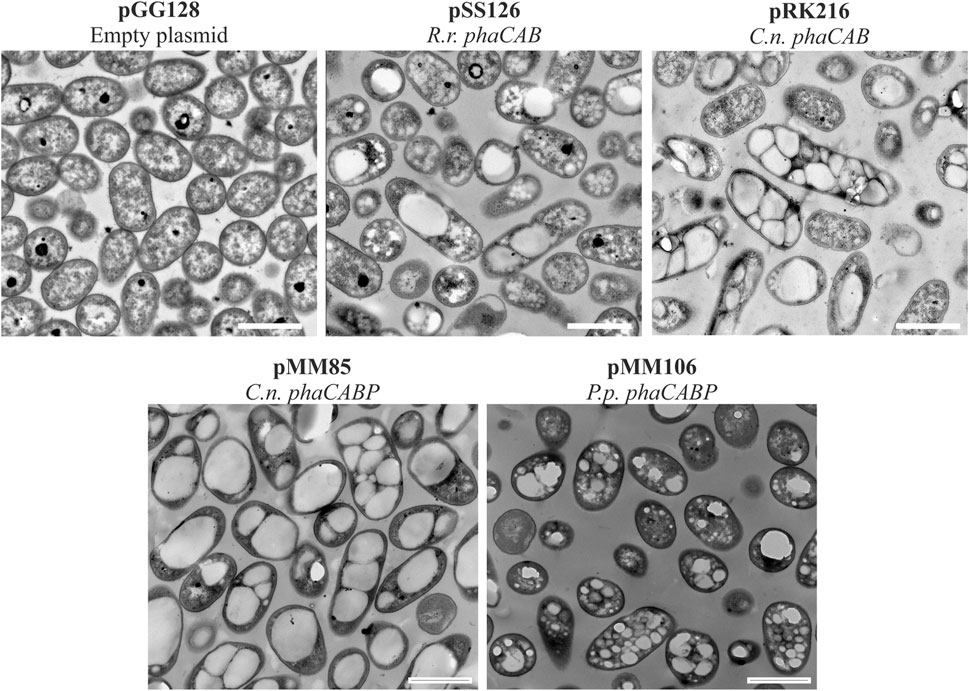
FIGURE 3. Transmission electron microscopy photos of heterologous PHA production. Pseudomonas putida PP05_01 strains expressing heterologous PHA machinery grown for 24 h in PHA production conditions with 15 mMoctanoate. Scale bars represent 1 μm. Abbreviations: R. r.: Rhodospirillum rubrum, C. n.: Cupriavidus necator, P. p.: Pseudomonas pseudoalcaligenes.
Similarly, the PHA machinery of P. pseudoalcaligenes (pMM106) and C. necator (pMM85) were expressed under the 14a constitutive promoter reaching 45%–84% PHA/CDW, respectively (Table 3; Figure 3). These were encouraging results, since we were able to obtain similar PHA content from C. necator genes expressed in a constitutive multicopy system compared to the inducible monocopy strain PP00_02 (Table 2). From TEM images, we could confirm that cells producing PHB generally contained multiple PHB granules that occupied the majority of the intracellular space (Figure 3). We also tested the influence of expressing the PHA machinery of C. necator under the moderate SynPro16 promoter (PP05_01 (pRK216)). However, assessing PP05_01 (pRK216) under the same growth scenario, there was no apparent improvement in the PHA production capabilities compared to the pMM85 plasmid (47% PHB/CDW, Table 3). In general, the PHA machinery constructs with SynPro16 promoter showed a more variable phenotype compared to 14a constitutive promoter. This can be also observed by the TEM images, where several cells harboring pRK216 did not produce PHA, leading to an overall decrease of the %PHA/CDW quantified by GC-MS analyses (Figure 3). Considering the unstableness issues raised by the SynPro16 promoter, in this study, we did not pursue the combination of the different genetic parts with this expression system.
As a product of a synthetic pathway involving four enzymes, PHA content relies heavily on the activity and relative ratios of these enzymes and the applied growth conditions. Thus, the best candidate strain harboring C. necator PHA machinery was tested for the feasibility to produce tailored PHA towards diverse personalized applications. For this purpose, several nutritional scenarios were planned for the production of copolymers (i.e., PHBV) in PP05_01 harboring pMM85 with the wild type phbCn cassette. As listed in Table 4, a panel of PHBV copolymers was successfully obtained with varying C4:C5 compositions. In fact, co-feeding with 5 mM propionic acid yielded 35% of PHA/CDW with 3% C5 monomeric composition while 1 mM undecenoic acid co-feeding yielded 44% PHA/CDW with 19% of C5. Overall, we demonstrated the successful deployment of custom assembled plasmids to produce à la carte PHB/PHBV polymers in the engineered P. putida strains.
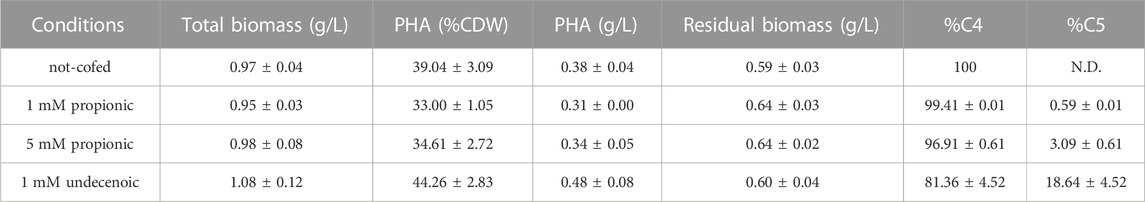
TABLE 4. PHA yield of PP05_01 harboring pMM85 (C.n. phaCABP) following 24 h growth with 0.1 N M63 supplemented with 20 mM glucose and co-fed with the indicated odd length fatty acids. The data correspond to the mean values and standard deviations of two independent biological replicates.
For scale up processing the ideal strain would not rely on antibiotic resistance for plasmid maintenance nor the use of an inducer for expression of the heterologous cassette. Additionally, the chromosomic PHA machinery integration would result to a more stable and homogeneous phenotype compared to plasmid. For this reason, a strain was generated to efficiently produce PHB in a monocopy constitutive expression system inserted in the P. putida chromosome. As a starting point, the phb cassette from C. necator was specifically inserted into the attTn7 loci of P. putida PP05_01, generating the PP05_16 and PP05_12 strains (Table 1). PP05_16 strain contained the four genes necessary to drive PHB synthesis under the low strength constitutive promoter 14a and PP05_12 did not contain the PhaPCn phasin (please refer to Table 1 and (Supplementary Table S1) for strains, plasmids and genomic information).
As expected, under the same growth conditions supplemented with 15 mM octanoate, the monocopy expression of the phbCn cassette led to a decrease in percent PHA/CDW accumulation (Table 5) that was also reflected in a decrease in total biomass (12%–15% by PP05_12 and PP05_16 versus 84% with pMM85 plasmid). To improve on PHB productivity observed in PP05_12 and PP05_16, the PP01_02 strain was generated. PP01_02 harbored the same phb cassette as PP05_12 in the attTn7 chromosomic locus but with the phaCCn synthase under control of a stronger 14f promoter (Table 5; Zobel et al., 2015). PP01_02 resulted in 69% PHA/CDW accumulation, which by NMR quantification we confirmed that 92% of the produced polymer was C4 and 8% C6 monomer (Supplementary Figure S3). Altogether, we successfully obtained a battery of chromosomic integrated PHB constructs that resulted in tuned PHA productivities (e.g., from 12%–69% PHA/CDW). These observations strongly suggested the impact of pha synthase dosage on PHA yield and monomeric composition.
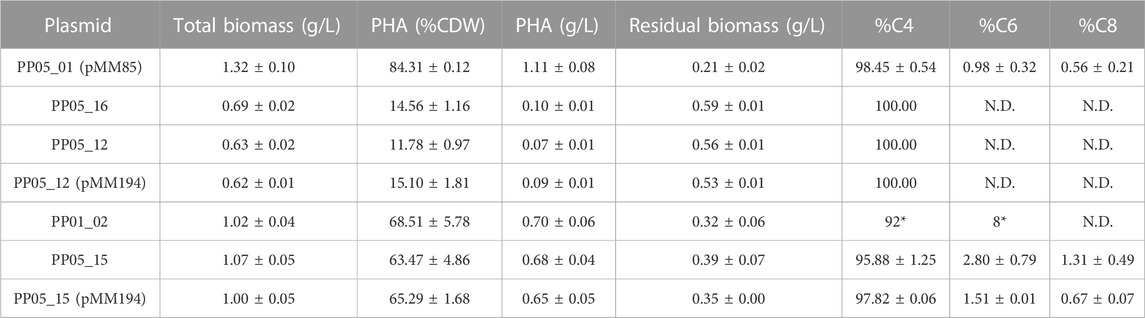
TABLE 5. PHA yield in PP05_01 modified strain following 24 h growth with 0.1 N M63 supplemented with 15 mM octanoate as the sole carbon source. N.D.: not detected. Mean values and standard deviations of at least two independent biological replicates are shown. *Data derived from NMR quantification.
It is well known that PHA synthesis is tightly controlled by a number of regulatory networks that govern PHA content, granule size and distribution in cells (recently reviewed by (Mitra et al., 2022)). However, the heterologously produced phasins in engineered strains did not show an obvious influence over PHA production properties or granule size (Table 3; Figure 3).
To elucidate other factors that might impact granule number and size (i.e., synthase dosage), TEM microscopic photos were taken after 24 h of growth using 15 mM octanoate as the sole carbon and energy source. For this, PP05_12 and PP01_02 strains were used, expressing the pha synthase under the control of 14a (low strength) and 14f (high strength) promoters, respectively. Figure 4 shows that low synthase dosage in the PP05_12 strain, generated a heterogeneous cell population, with several cells without PHB inclusions and others with few, but large PHB granules. However, high synthase doses in the PP01_02 strain resulted in a more homogeneous population of cells that contained numerous smaller granules. Granule size analysis of the PP05_12 strain showed a broader size distribution with an average size of 300 nm, while PP01_02 strain had a narrower size distribution with a smaller average size of 180 nm (Figure 4C).
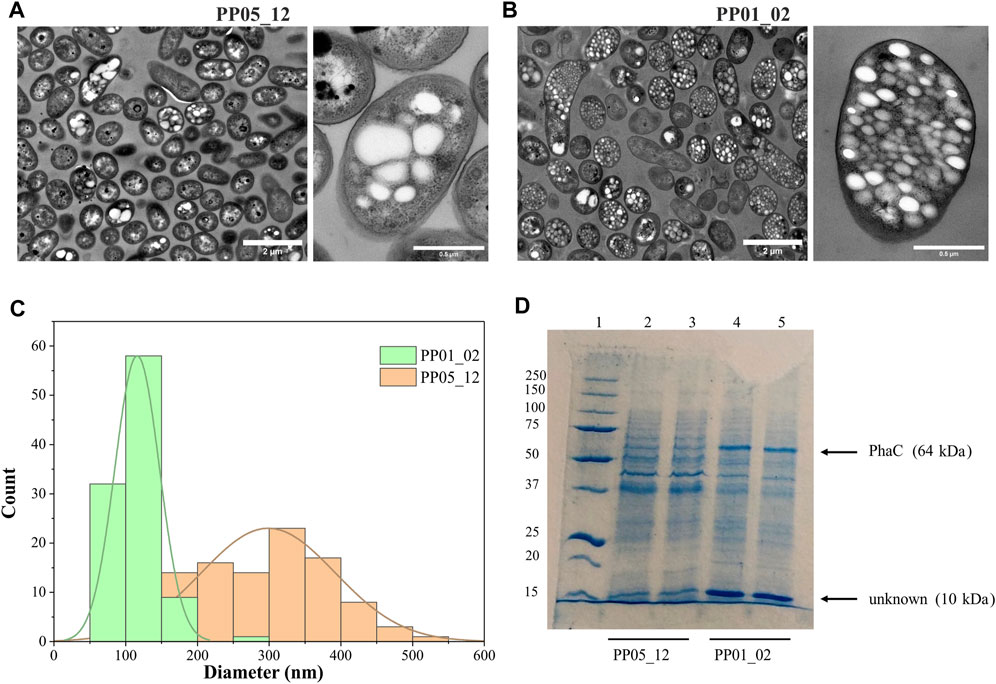
FIGURE 4. Impact of synthase dosage on granule number and size. (A, B). TEM images of PP01_02 and PP05_12, respectively under standard PHA accumulation conditions supplemented with 15 mM octanoate. (C). Granule size distribution obtained from 100 granules measured from TEM images using ImageJ software, PP01_02 (green) and PP05_12 (orange). (D). SDS - PAGE gels of granule extractions. Lane 1 MW marker; lanes 2–3: PP05_12 independently extracted PHB granules; lanes 4–5: PP01_02 independently extracted PHB granules. MW of the corresponding GAPs are indicated. Precision Plus Protein Standards (Biorad) was used as a molecular weight marker using Tris-Glycine 4%–20% conditions.
To further confirm the relative increase in PhaC synthase expression, granule preparations were extracted and run on SDS-PAGE to estimate GAPs present on the granule surface (Figure 4D). To ascertain that the same granule quantity was run on the gels, methanolysis of granule preparations were performed to quantify the actual amount of PHB in each preparation. The control experiments with the empty plasmid may be found in the Supplementary Figure S2. As expected, PP05_01 (pGG128) strain did not produce any detectable PHA and, thus, no granule formation was visible in the SDS-PAGE gels (Supplementary Figure S2). Comparing with the control band pattern, the presence of acetoacetyl-CoA-reductase PhaB (26 kDa) and acetyl-CoA-transferase PhaA (40 kDa) in both strains’ granule preparation resulted challenging. This could be explained to some extent by their low abundance in the granule surface since they were expressed under the low 14a promoter’s strength. However, as expected, the PP01_02 strain with a higher strength promoter for PhaC synthase (64 kDa) showed higher levels of PhaC protein than PP05_12 (Figure 4D). N-terminal sequencing of the 64 kDa band confirmed that this corresponded to PhaC.
In this granule preparations, we could observe a repeated pattern of a co-increased protein dosage of approximately 10 kDa protein together with the increase in PhaCCn. Interestingly, this low molecular weight protein was also observed in the granules’ extraction harboring the PHA machinery from R. rubrum (Supplementary Figure S2). To elucidate the identity of this unknown protein, we performed N-terminal sequencing. A protein BLAST of “TTAFSLAPLF” against the P. putida KT2440 proteome revealed the presence of the small heat shock protein, lbpA (PP_1982).
Small heat-shock proteins (sHSP), are characterized by a molecular mass of 12–43 kDa, and function as ubiquitous and diverse molecular chaperones that prevent protein aggregation under heat shock conditions. Two sHSP from E. coli, IbpA/B have been previously reported to bind to the inclusion bodies of recombinant proteins (Han et al., 2006). IbpA is also considered a stress-related chaperone with an intrinsic holdase activity. This ATP-independent holding function allows them to bind to denatured and partly unfolded proteins under stress conditions. The proteins bound to sHSPs are maintained in a refolding-competent state and are thereby protected from irreversible aggregation (Han et al., 2006; Roy et al., 2014).
To assess the involvement of IbpA on bacterial PHA machinery, an ibpA deletion mutant was constructed in the PP01_02 background, generating strain PP05_15. Phenotypic evaluation of PP05_15 showed no major impact on the growth capacity at 30°C compared to the parental PP01_02 strain (e.g., similar residual biomass, Table 5). These results are in agreement with previous observations to ibpA deletion mutant in KT2440, where the growth was only significantly affected at 40°C (Krajewski et al., 2013). Looking at the PHA profile properties, PP05_15 revealed no major differences concerning the PHA accumulation profile compared with the parental strain, PP01_02; 63.5% PHA/CDW versus 68.5% PHA/CDW, respectively (Table 5). These observations are in accordance with the recombinant E. coli IbpA/B null strain behavior, which also slightly affected PHA production, when carried the C. necator PHA machinery (Han et al., 2006). To look closer on the involvement of the IbpA deletion on granule size distribution, TEM analyses were performed (Figure 5A). In fact, PP05_15 strain showed a broader granule size distribution with an average size of about 300 nm compared with 180 nm of PP01_02 (Figure 5C). Finally, the influence of IbpA over other granule-associated proteins was analyzed by granule extraction assay (Figure 5D). As expected for granules extracted from the ibpA null (PP05_15) strain, the 10 kDa band corresponding to IbpA disappeared, while showing little change in the other bands corresponding to GAPs involved in PHA production (i.e., PhaC). It is worth mentioning that during the extraction procedure granules from the PP05_15 strain appeared to be more aggregated and adherent compared to granules from PP01_02. This aspect could be explained, at least to some extent, from the deletion of IbpA, which could lead to higher protein aggregation.
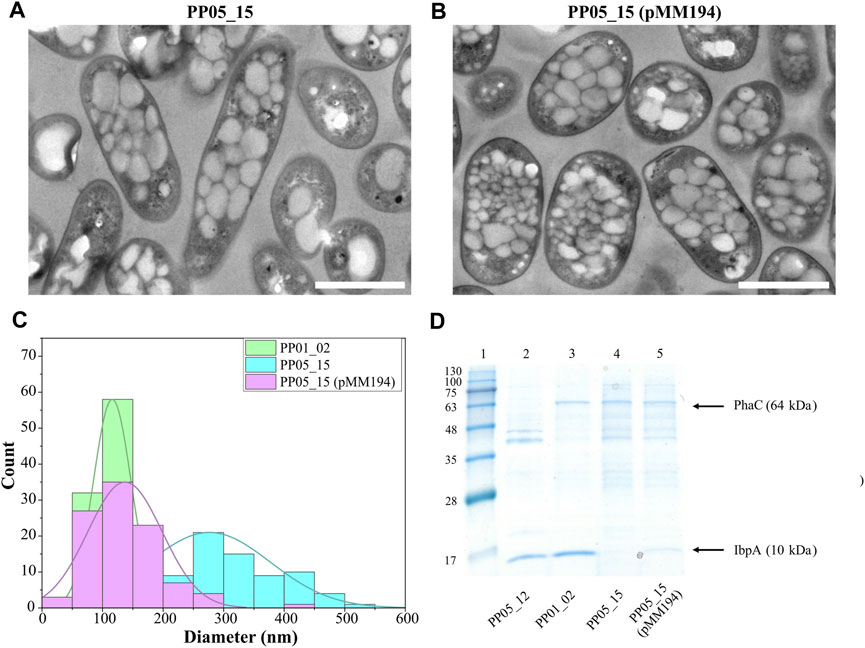
FIGURE 5. Impact of IbpA deletion on granule number and size. (A, B). TEM images of PP05_15 and PP05_15 (pMM194), respectively under standard PHA accumulation conditions supplemented with 15 mM octanoate. (C). Granule size distribution obtained from 100 granules measured from TEM images using ImageJ software, in green PP01_02, blue PP05_15 and purple PP05_15 (pMM194). Scale bars represent 1 µm. (D). SDS - PAGE gels of granule extractions. Lane 1 MW marker; lanes 2: PP05_12; lane 3: PP01_02; lane 4: PP05_15; lane 5: PP05_15 (pMM194). The MW of the corresponding GAPs are indicated. BlueStar Prestained protein marker (Nippon Genetics) was used as a molecular weight marker using Tris-Glycine 4%–20% conditions.
Complementation assays with pMM194 plasmid were performed by introducing the ibpA gene expressed by the constitutive 14a promoter:BCD2 element. Similar PHA production properties were obtained by the complemented strains PP05_12 (pMM194) and PP05_15 (pMM194), reaching 15% and 65% PHA/CDW, similar to the parental strains (Table 5). This could be explained to some extent by the low dosage of complemented IbpA. We cannot discard that higher IbpA dosage could lead to higher PHA content to the engineered strains. Even though no major effect was observed by PHA production, TEM images revealed that PP05_15 (pMM194) successfully reverted the granule formation pattern, with a clear tendency towards smaller granule size compared to PP01_02 strain (Figures 5B, C). Additionally, granule extraction assays confirmed that the presence of IbpA by pMM194 resulted in less granule aggregation (data not shown) and a re-appearance of the IbpA band to the SDS-PAGE (Figure 5D).
IbpA/B seem to exhibit several different functions depending on physiological conditions, among them it has been ascribed that these phasin-like proteins might function as a phase stabilizer at the interface of hydrophilic cytoplasm and hydrophobic PHB granules when this polymer is heterologously produced in E. coli (Han et al., 2006). To elucidate if the presence of IbpA in the granule could inhibit the binding of other GAPs, we tested the binding of the PhaP phasin from C. necator to scl-PHA granules. For this, PhaP1Cn was fused to the green fluorescent protein (msf-GFP) and heterologously expressed in the PP01_02 strain (Figure 6A). For the co-localization experiments, as expected, the control PP01_02 strain harboring (pBDN2-GFP), empty plasmid including only the msf-GFP, showed a diffuse cytoplasmic fluorescence surrounding the scl-PHA granules (Figure 6B). This observation confirmed the non-binding affinity of msf-GFP towards scl-PHA. As anticipated, the scl-family phasin (e.g., PhaP1Cn in pBDN2-PhaP plasmid) showed a co-localized fluorescence to the polymer, thus, maintaining its ability to bind to scl-PHA even in the presence of IbpA (Figure 6B).
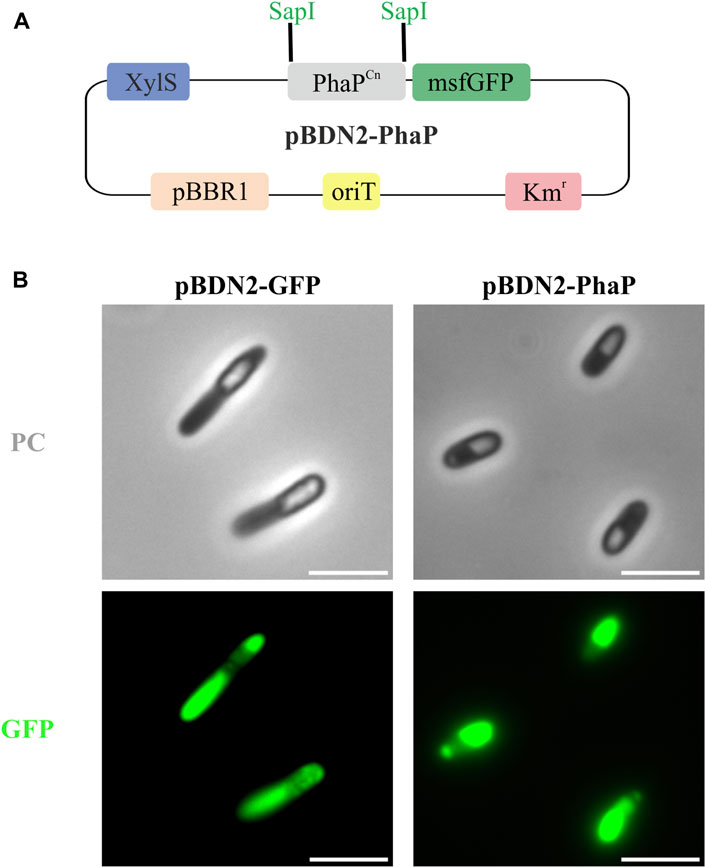
FIGURE 6. Co-localization experiments in the scl-PHA chassis (PP01_02). (A). Schematic representation of pBDN2-PhaP vector. (B). Fluorescence images with phase contrast (PC) amd GFP, left panel PP01_02 (pBDN2-GFP) negative control of binding, the strain that expresses the msf-GFP gene and right panel PP01_02 (pBDN2-PhaP) that expresses the PhaP1 from Cupriavidus necator. The scale bar corresponds to 3 µm.
Pseudomonas putida is a model bacterium for mcl-PHA production with a complex regulatory system driving the expression of genes encoding the PHA machinery. When grown on fatty acids, transcription of pha locus genes is augmented when compared to growth on simple carbon sources (De Eugenio et al., 2010b; Wang and Nomura, 2010; Manoli et al., 2020). Additionally, the Crc catabolite repression regulator influences transcription of phaC1 such that its transcription is inhibited in balanced carbon/nitrogen conditions (La Rosa et al., 2014). While other global transcriptional factors, including RpoS, PsrA and GacS/GacA also influence transcription of genes in the pha locus, though their precise roles remain to be fully elucidated (De Eugenio et al., 2010b; Mezzina et al., 2021). In fact, from previous works from our group, we showed a complex interactome among key components of PHA production (i.e., PhaF-PhaD) (Tarazona et al., 2020). However, a deletion of the entire pha locus disengages these regulatory functions from the native production of mcl-PHA, making orthogonal PHA production independent of cellular regulators of gene expression. Nevertheless, we cannot discard the involvement of other cellular components during the heterologously expressed scl-PHA machinery. A full pha locus deletion chassis strain can then serve to host heterologous pha expression constructs. This allows for à la carte production of bespoke PHAs in P. putida using custom expression constructs coupled with growth of the resulting strain on any number of carbon sources.
There have been numerous examples of the production of orthogonal PHA in pha null Pseudomonas through the heterologous expression of pha genes. These include the expression of phaC synthase genes from scl-/mcl-PHA producing C. necator, Rhodobacter sphaeroides, Nocardia corrallina, Thiocystis violacea, or various pseudomonads in Δpha mutants of P. putida to generate scl- and mcl-PHAs with unique compositions (Huisman et al., 1991; Preusting et al., 1993; Timm et al., 1994; Lee et al., 1995; Dennis et al., 1998; Matsusaki et al., 1998; Clemente et al., 2000; Ouyang et al., 2007). Another study created a P. putida KT2440 phaC1-, ΔphaZ, phaC2- Ω interposon mutant and used this Δpha strain as a host for the functional screening of soil metagenomic cosmid clones to identify novel PHA synthases (Cheng and Charles, 2016). Our work expands upon these previous investigations by generating a sequence-confirmed P. putida KT2440 Δpha locus chassis, PP05_01 strain coupled with the advantage of using a highly flexible assembly cloning system for the generation of custom, stable and constitutively produced scl- and mcl-PHAs.
Pseudomonads can utilize direct PHA precursor pathways to convert fatty acids and PHA unrelated carbon sources (i.e., acetate, ethanol, glycerol, sugars, etc.) via ß-oxidation and de novo synthesis of fatty acids, respectively, into various (R)-3-hydroxyacyl-CoAs (Figure 1). In contrast, R. rubrum, P. pseudoalcaligenes, and C. necator are limited in their ability to channel fatty acid metabolites into PHA and instead PHA production relies upon the availability of acetyl-CoA derived from the catabolism of carbon sources (Senior and Dawes, 1971; Budde et al., 2010). The creation of the PP05_01 chassis strain lacking the entire pha locus allows for the decoupling of the natural cycle of mcl-PHA production and consumption that is an integral part of energy apportionment and central carbon flux in P. putida (Escapa et al., 2012). As mentioned above, mcl-PHA production in P. putida is connected to central and peripheral metabolic pathways. The PP05_01 chassis is not subject to catabolite repression of PHA machinery expression and thus, avoids the dependence on certain carbon sources for PHA production. Generally, a nutritional imbalance (i.e., excess carbon and/or the limitation of nutrients such as nitrogen) favors PHA production in P. putida (Madison and Huisman, 1999). This imbalance is most significant for PHA production when substrates other than fatty acids are used as carbon sources (Sun et al., 2007; Wang and Nomura, 2010; Follonier et al., 2011). In the presence of fatty acids, nitrogen limitation is not necessary for PHA production, yet greatly improves PHA yields (Poblete-Castro et al., 2012). Notwithstanding, for our studies in the majority of cases, PHA production was carried out in nitrogen limited conditions with octanoate as the sole carbon source.
The modular and hierarchical nature of biological designs reveals new possibilities for the development of rational and standardized mechanisms in order to improve the engineering process of specific biological solutions. DNA assembly is a widely used method to build synthetic genetic circuits. Traditionally, these cloning strategies were performed by digestion and ligation of DNA fragments using BioBricks or Gibson assembly. However, these approaches require specific designs for each step that can hamper complete standardization and parts reuse. Modular Cloning (MoClo) methodology has emerged as a powerful tool for standardizing the assembly of genetic parts. MoClo is based on Golden gate cloning, which allows simultaneous and directional assembly of multiple DNA parts.
PHA polyesters can be derived from over 150 different (R)-3-hydroxyalkanoic acid monomers, giving rise to a huge variety of physical and mechanical properties in the resulting polymers (Steinbüchel and Valentin, 1995; Hazer and Steinbüchel, 2007). Production of orthogonal PHAs in P. putida greatly expands the envelope of polymer diversity. Due to its metabolic flexibility and ability to use a variety of substrates, such as fatty acids or aromatic compounds, it is expected that our P. putida toolkit can be exploited to generate new and useful PHAs (Linger et al., 2014; Blázquez et al., 2023).
By using Golden gate/MoClo technology, we demonstrated the successful production of PHB and PHBV copolymers in P. putida PP05_01 through the expression of heterologous PHB machinery from R. rubrum, P. pseudoalcaligenes or C. necator. Indeed, since the synthetic phb clusters used in this study followed the same brick structure, this could enable the interchangeability of the genetic parts across the modules, if needed. Thus, we could discover the best synthetic parts’ (i.e., promoter, RBS, CDS) combination for most optimal PHA production. Additionally, the involvement of phasins in PHA accumulation and granule stability was studied, suggesting that the presence of these proteins in a heterologous host is not crucial. The absence or presence of phasins did not demonstrate significant differences in PHA accumulation and granule formation. However, we observed that the dosage of PhaC in the chromosome integrated constructs significantly impacted PHA production in several ways. In this sense, we identified interesting patterns: i) higher scl-PHA production, ii) production of a panel of PHAs with increased mcl-PHA (predominantly C6) composition (i.e. 90% C4 and approximately 10% mcl-PHA, iii) more numerous and smaller size granules, and iv) that the presence of the small heat shock protein IbpA augments heterologous PHA production.
IbpA/B belong to the alpha-crystalline type small heat-shock proteins (sHSP) with a molecular mass of 12–43 kDa, and are known to act as holding chaperones (Krajewski et al., 2013; Roy et al., 2014). Most chaperones possess intrinsic holdase activity, where the ATP-independent holding function is used to bind unstable proteins and prevent the formation of dysfunctional aggregates. Then, misfolded proteins can be transferred from holdase chaperones to downstream ATP-dependent chaperones. These use energy from ATP hydrolysis to power conformational changes in the chaperone, which promotes unfolding, refolding, or translocation of bound substrate proteins as part of their processing. Therefore, the combined action of molecular chaperones may increase the cellular pool of native proteins while minimizing inactive proteins and potentially harmful protein aggregates (Jewett and Shea, 2006).
Indeed, two sHSP from E. coli, IbpA/B have been previously reported to bind to the inclusion bodies of recombinant proteins. Han and collaborators demonstrated that a recombinant E. coli IbpA/B deletion mutant led to significant changes in PHB granule morphology, whereby they were shown to become distorted and wrinkled (Han et al., 2006). Therefore, it was suggested that in the absence of IbpA/B, PHB granules were expected to bind more cytosolic proteins in a non-specific manner compared to the parental strain. For this, IbpA/B in E. coli were proposed to act as phasin-like proteins that function as a phase stabilizer at the interface of the hydrophobic PHB granules and the hydrophilic cytoplasm (Han et al., 2006).
In this work, we constructed a viable IbpA null, PP05_15 strain. Even though the phenotypic evaluation revealed no major changes in PHA production, differences were observed in granule size distribution. Indeed, in the absence of IbpA, the tendency was towards overall larger granules compared to parental PP01_02 strain. Interestingly, this granule size distribution of PP05_15 is quite similar to PP05_12 (containing low PhaC synthase expression), suggesting that the deletion of IbpA may decrease the effective level or function of PhaC. We did not observe an obvious lower PhaC synthase level in the granule extracts of PP05_15 compared to PP01_02, indicating that the IbpA either modulates the function of PhaC by allowing its proper folding or operates on granule size in a PhaC-independent manner. Whether or not the PhaC/other granule-associated proteins could be partially aggregated in the PP05_15 strain was not determined, yet our findings confirmed the importance of IbpA protein in granule size determination and GAPs localization.
Altogether, in this study we demonstrated that P. putida optimized cell factories can be used for the production of tailored scl-PHA. Our results also suggest that native mcl-PHA regulatory network might be different to that of orthogonal scl-PHA system and we cannot discard the involvement of non-envisaged players such as IbpA.
The raw data supporting the conclusion of this article will be made available by the authors, without undue reservation.
M-TM: Conceptualization, Supervision, Writing–review and editing, Investigation, Methodology, Writing–original draft, Validation, Data curation. FB: Writing–review and editing, Investigation, Methodology. VR-B: Methodology, Writing–review and editing. RK: Conceptualization, Supervision, Writing–review and editing, Investigation, Methodology, Writing–original draft, Project administration. SA: Writing–review and editing, Methodology. S: Writing–review and editing, Investigation, Methodology. MP: Conceptualization, Funding acquisition, Resources, Supervision, Writing–review and editing, Investigation, Project administration, Writing–original draft.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research received funding from the European Union’s Horizon 2020 research and innovation program under grant agreement number 633962 (P4SB), 814418 (SinFonia) and 870294 (MIX-up). This work was supported by the CSIC Interdisciplinary Thematic Platform (PTI+) Sustainable Plastics towards a Circular Economy (PTI-Susplast+), the Community of Madrid (P2018/NMT4389) and the Spanish Ministry of Science and Innovation under the research grant BIOCIR (PID 2020-112766RB-C21).
We would like to thank Aranzazu Mato for helping with the construction of some strains used in this work. Also, we would like to acknowledge Carlos de Cerro for helping with the genome’s sequencing and Santiago Roque de Miguel Sanz with the methanolysis process of some of the samples used in this work. Ana Valencia’s technical work is also greatly appreciated. We thank CIB and CNB scientific facilities (i.e., gas chromatography, protein chemistry, nuclear magnetic resonance, and transmission electron microscopy services).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fbioe.2023.1275036/full#supplementary-material
Bagdasarian, M., Lurz, R., Rückert, B., Franklin, F. C., Bagdasarian, M. M., Frey, J., et al. (1981). Specific-purpose plasmid cloning vectors. II. Broad host range, high copy number, RSF1010-derived vectors, and a host-vector system for gene cloning in Pseudomonas. Gene 16, 237–247. doi:10.1016/0378-1119(81)90080-9
Jewett, A. I., and Shea, J.-E. (2006). Folding on the chaperone: yield enhancement through loose binding. J. Mol. Biol. 363 (5), 945–957. doi:10.1016/j.jmb.2006.08.040
Blanco, F. G., Machatschek, R., Keller, M., Hernández-Arriaga, A. M., Godoy, M. S., Tarazona, N. A., et al. (2023). Nature-inspired material binding peptides with versatile polyester affinities and binding strengths. Int. J. Biol. Macromol. 253, 126760. doi:10.1016/j.ijbiomac.2023.126760
Blázquez, B., León, D. S., Torres-Bacete, J., Gómez-Luengo, Á., Kniewel, R., Martínez, I., et al. (2023). Golden Standard: a complete standard, portable, and interoperative MoClo tool for model and non-model proteobacteria. Nucleic Acids Res. 23, gkad758. doi:10.1093/nar/gkad758
Boyer, H. W., and Roulland-Dussoix, D. (1969). A complementation analysis of the restriction and modification of DNA in Escherichia coli. J. Mol. Biol. 41, 459–472. doi:10.1016/0022-2836(69)90288-5
Brandl, H., Knee, E. J., Fuller, R. C., Gross, R. A., and Lenz, R. W. (1989). Ability of the phototrophic bacterium Rhodospirillum rubrum to produce various poly (β-hydroxyalkanoates): potential sources for biodegradable polyesters. Int. J. Biol. Macromol. 11, 49–55. doi:10.1016/0141-8130(89)90040-8
Braunegg, G., Sonnleitner, B., and Lafferty, R. M. (1978). A rapid gas chromatographic method for the determination of poly-β-hydroxybutyric acid in microbial biomass. Eur. J. Appl. Microbiol. Biotechnol. 6, 29–37. doi:10.1007/BF00500854
Budde, C. F., Mahan, A. E., Lu, J., Rha, C., and Sinskey, A. J. (2010). Roles of multiple acetoacetyl coenzyme A reductases in polyhydroxybutyrate biosynthesis in Ralstonia eutropha H16. J. Bacteriol. 192, 5319–5328. doi:10.1128/JB.00207-10
Cambray, G., Guimaraes, J. C., Mutalik, V. K., Lam, C., Mai, Q.-A., Thimmaiah, T., et al. (2013). Measurement and modeling of intrinsic transcription terminators. Nucleic Acids Res. 41, 5139–5148. doi:10.1093/nar/gkt163
Cha, D., Ha, H. S., and Lee, S. K. (2020). Metabolic engineering of Pseudomonas putida for the production of various types of short-chain-length polyhydroxyalkanoates from levulinic acid. Bioresour. Technol. 309, 123332. doi:10.1016/j.biortech.2020.123332
Cheng, J., and Charles, T. C. (2016). Novel polyhydroxyalkanoate copolymers produced in Pseudomonas putida by metagenomic polyhydroxyalkanoate synthases. Appl. Microbiol. Biotechnol. 100, 7611–7627. doi:10.1007/s00253-016-7666-6
Choi, K.-H., Kumar, A., and Schweizer, H. P. (2006). A 10-min method for preparation of highly electrocompetent Pseudomonas aeruginosa cells: application for DNA fragment transfer between chromosomes and plasmid transformation. J. Microbiol. Methods 64, 391–397. doi:10.1016/j.mimet.2005.06.001
Clemente, T., Shah, D., Tran, M., Stark, D., Padgette, S., Dennis, D., et al. (2000). Sequence of PHA synthase gene from two strains of Rhodospirillum rubrum and in vivo substrate specificity of four PHA synthases across two heterologous expression systems. Appl. Microbiol. Biotechnol. 53, 420–429. doi:10.1007/s002530051636
De Eugenio, L. I., Escapa, I. F., Morales, V., Dinjaski, N., Galán, B., García, J. L., et al. (2010a). The turnover of medium-chain-length polyhydroxyalkanoates in Pseudomonas putida KT2442 and the fundamental role of PhaZ depolymerase for the metabolic balance. Environ. Microbiol. 12, 207–221. doi:10.1111/j.1462-2920.2009.02061.x
De Eugenio, L. I., Galán, B., Escapa, I. F., Maestro, B., Sanz, J. M., García, J. L., et al. (2010b). The PhaD regulator controls the simultaneous expression of the pha genes involved in polyhydroxyalkanoate metabolism and turnover in Pseudomonas putida KT2442. Environ. Microbiol. 12, 1591–1603. doi:10.1111/j.1462-2920.2010.02199.x
Dennis, D., McCoy, M., Stangl, A., Valentin, H. E., and Wu, Z. (1998). Formation of poly(3-hydroxybutyrate-co-3-hydroxyhexanoate) by PHA synthase from Ralstonia eutropha. J. Biotechnol. 64, 177–186. doi:10.1016/S0168-1656(98)00110-2
Engler, C., Kandzia, R., and Marillonnet, S. (2008). A one pot, one step, precision cloning method with high throughput capability. PLOS ONE 3, e3647. doi:10.1371/journal.pone.0003647
Escapa, I. F., García, J. L., Bühler, B., Blank, L. M., and Prieto, M. A. (2012). The polyhydroxyalkanoate metabolism controls carbon and energy spillage in Pseudomonas putida. Environ. Microbiol. 14, 1049–1063. doi:10.1111/j.1462-2920.2011.02684.x
Follonier, S., Panke, S., and Zinn, M. (2011). A reduction in growth rate of Pseudomonas putida KT2442 counteracts productivity advances in medium-chain-length polyhydroxyalkanoate production from gluconate. Microb. Cell Factories 10, 25. doi:10.1186/1475-2859-10-25
Galán, B., Dinjaski, N., Maestro, B., De Eugenio, L. I., Escapa, I. F., Sanz, J. M., et al. (2011). Nucleoid-associated PhaF phasin drives intracellular location and segregation of polyhydroxyalkanoate granules in Pseudomonas putida KT2442. Mol. Microbiol. 79, 402–418. doi:10.1111/j.1365-2958.2010.07450.x
Godoy, M. S., de Miguel, S. R., and Prieto, M. A. (2023). Aerobic-anaerobic transition boosts poly(3-hydroxybutyrate-co-3-hydroxyvalerate) synthesis in Rhodospirillum rubrum: the key role of carbon dioxide. Microb. Cell Factories 22, 47. doi:10.1186/s12934-023-02045-x
Han, M.-J., Park, S.-J., Lee, J.-W., Min, B.-H., Lee, S.-Y., Kim, S.-J., et al. (2006). Analysis of poly (3-hydroxybutyrate) granule-associated proteome in recombinant Escherichia coli. J. Microbiol. Biotechnol. 16, 901–910.
Hazer, B., and Steinbüchel, A. (2007). Increased diversification of polyhydroxyalkanoates by modification reactions for industrial and medical applications. Appl. Microbiol. Biotechnol. 74, 1–12. doi:10.1007/s00253-006-0732-8
Hiroe, A., Tsuge, K., Nomura, C. T., Itaya, M., and Tsuge, T. (2012). Rearrangement of gene order in the phaCAB operon leads to effective production of ultrahigh-molecular-weight poly[(R)-3-Hydroxybutyrate] in genetically engineered Escherichia coli. Appl. Environ. Microbiol. 78, 3177–3184. doi:10.1128/AEM.07715-11
Huisman, G. W., de Leeuw, O., Eggink, G., and Witholt, B. (1989). Synthesis of poly-3-hydroxyalkanoates is a common feature of fluorescent pseudomonads. Appl. Environ. Microbiol. 55, 1949–1954. doi:10.1128/aem.55.8.1949-1954.1989
Huisman, G. W., Wonink, E., Meima, R., Kazemier, B., Terpstra, P., and Witholt, B. (1991). Metabolism of poly(3-hydroxyalkanoates) (PHAs) by Pseudomonas oleovorans. Identification and sequences of genes and function of the encoded proteins in the synthesis and degradation of PHA. J. Biol. Chem. 266, 2191–2198. doi:10.1016/s0021-9258(18)52227-4
Jendrossek, D. (2009). Polyhydroxyalkanoate granules are complex subcellular organelles (carbonosomes). J. Bacteriol. 191, 3195–3202. doi:10.1128/JB.01723-08
Jendrossek, D., and Pfeiffer, D. (2014). New insights in the formation of polyhydroxyalkanoate granules (carbonosomes) and novel functions of poly(3-hydroxybutyrate). Environ. Microbiol. 16, 2357–2373. doi:10.1111/1462-2920.12356
Jin, H., and Nikolau, B. J. (2012). Role of genetic redundancy in polyhydroxyalkanoate (PHA) polymerases in PHA biosynthesis in Rhodospirillum rubrum. J. Bacteriol. 194, 5522–5529. doi:10.1128/JB.01111-12
Kim, J., Kim, Y.-J., Choi, S. Y., Lee, S. Y., and Kim, K.-J. (2017). Crystal structure of Ralstonia eutropha polyhydroxyalkanoate synthase C-terminal domain and reaction mechanisms. Biotechnol. J. 12, 1600648. doi:10.1002/biot.201600648
Kim, Y. B., and Lenz, R. W. (2001). Polyesters from microorganisms. Adv. Biochem. Eng. Biotechnol. 71, 51–79. doi:10.1007/3-540-40021-4_2
Koller, M., Maršálek, L., de Sousa Dias, M. M., and Braunegg, G. (2017). Producing microbial polyhydroxyalkanoate (PHA) biopolyesters in a sustainable manner. N. Biotechnol. 37, 24–38. doi:10.1016/j.nbt.2016.05.001
Krajewski, S. S., Nagel, M., and Narberhaus, F. (2013). Short ROSE-like RNA thermometers control IbpA synthesis in Pseudomonas species. PLoS ONE 8, e65168. doi:10.1371/journal.pone.0065168
La Rosa, R., de la Peña, F., Prieto, M. A., and Rojo, F. (2014). The Crc protein inhibits the production of polyhydroxyalkanoates in Pseudomonas putida under balanced carbon/nitrogen growth conditions. Environ. Microbiol. 16, 278–290. doi:10.1111/1462-2920.12303
Larson, M. H., Greenleaf, W. J., Landick, R., and Block, S. M. (2008). Applied force reveals mechanistic and energetic details of transcription termination. Cell 132, 971–982. doi:10.1016/j.cell.2008.01.027
Lee, E. Y., Jendrossek, D., Schirmer, A., Choi, C. Y., and Steinbüchel, A. (1995). Biosynthesis of copolyesters consisting of 3-hydroxybutyric acid and medium-chain-length 3-hydroxyalkanoic acids from 1,3-butanediol or from 3-hydroxybutyrate by Pseudomonas. Appl. Microbiol. Biotechnol. 42, 901–909. doi:10.1007/BF00191189
Li, D., Lv, L., Chen, J.-C., and Chen, G.-Q. (2017). Controlling microbial PHB synthesis via CRISPRi. Appl. Microbiol. Biotechnol. 101, 5861–5867. doi:10.1007/s00253-017-8374-6
Li, T., Ye, J., Shen, R., Zong, Y., Zhao, X., Lou, C., et al. (2016). Semirational approach for ultrahigh poly(3-hydroxybutyrate) accumulation in Escherichia coli by combining one-step library construction and high-throughput screening. ACS Synth. Biol. 5, 1308–1317. doi:10.1021/acssynbio.6b00083
Linger, J. G., Vardon, D. R., Guarnieri, M. T., Karp, E. M., Hunsinger, G. B., Franden, M. A., et al. (2014). Lignin valorization through integrated biological funneling and chemical catalysis. Proc. Natl. Acad. Sci. U. S. A. 111, 12013–12018. doi:10.1073/pnas.1410657111
Lutz, R., and Bujard, H. (1997). Independent and tight regulation of transcriptional units in Escherichia coli via the LacR/O, the TetR/O and AraC/I1-I2 regulatory elements. Nucleic Acids Res. 25, 1203–1210. doi:10.1093/nar/25.6.1203
Madison, L. L., and Huisman, G. W. (1999). Metabolic engineering of poly(3-hydroxyalkanoates): from DNA to plastic. Microbiol. Mol. Biol. Rev. 63, 21–53. doi:10.1128/mmbr.63.1.21-53.1999
Mahato, R. P., Kumar, S., and Singh, P. (2021). Optimization of growth conditions to produce sustainable polyhydroxyalkanoate bioplastic by Pseudomonas aeruginosa EO1. Front. Microbiol. 12, 711588. doi:10.3389/fmicb.2021.711588
Makkar, N. S., and Casida, L. E. (1987). Technique for estimating low numbers of a bacterial strain(s) in soil. Appl. Environ. Microbiol. 53, 887–888. doi:10.1128/aem.53.4.887-888.1987
Manoli, M.-T., Nogales, J., and Prieto, A. (2022). Synthetic control of metabolic states in Pseudomonas putida by tuning polyhydroxyalkanoate cycle. mBio 13, 01794–e1821. doi:10.1128/mbio.01794-21
Manoli, M. T., Tarazona, N., Mato, A., Maestro, B., Sanz, J. M., Nogales, J., et al. (2020). “Molecular basis of medium-chain length-PHA metabolism of Pseudomonas putida,” in The handbook of polyhydroxyalkanoates (United States: CRC Press), 89–114.
Mato, A., Blanco, F. G., Maestro, B., Sanz, J. M., Pérez-Gil, J., and Prieto, M. A. (2020). Dissecting the polyhydroxyalkanoate-binding domain of the PhaF phasin: rational design of a minimized affinity tag. Appl. Environ. Microbiol. 86, 00570–e620. doi:10.1128/AEM.00570-20
Matsusaki, H., Manji, S., Taguchi, K., Kato, M., Fukui, T., and Doi, Y. (1998). Cloning and molecular analysis of the Poly(3-hydroxybutyrate) and Poly(3-hydroxybutyrate-co-3-hydroxyalkanoate) biosynthesis genes in Pseudomonas sp. strain 61-3. J. Bacteriol. 180, 6459–6467. doi:10.1128/jb.180.24.6459-6467.1998
Mezzina, M. P., Manoli, M. T., Prieto, M. A., and Nikel, P. I. (2021). Engineering native and synthetic pathways in Pseudomonas putida for the production of tailored polyhydroxyalkanoates. Biotechnol. J. 16, 2000165. doi:10.1002/biot.202000165
Mitra, R., Xu, T., Chen, G., Xiang, H., and Han, J. (2022). An updated overview on the regulatory circuits of polyhydroxyalkanoates synthesis. Microb. Biotechnol. 15, 1446–1470. doi:10.1111/1751-7915.13915
Ouyang, S.-P., Liu, Q., Fang, L., and Chen, G.-Q. (2007). Construction of pha-operon-defined knockout mutants of Pseudomonas putida KT2442 and their applications in poly(hydroxyalkanoate) production. Macromol. Biosci. 7, 227–233. doi:10.1002/mabi.200600187
Pfennig, N., and Trüper, H. G. (1971). Type and neotype strains of the species of phototrophic bacteria maintained in pure culture. Int. J. Syst. Evol. Microbiol. 21, 19–24. doi:10.1099/00207713-21-1-19
Poblete-Castro, I., Escapa, I. F., Jäger, C., Puchalka, J., Lam, C. M. C., Schomburg, D., et al. (2012). The metabolic response of P. putida KT2442 producing high levels of polyhydroxyalkanoate under single- and multiple-nutrient-limited growth: highlights from a multi-level omics approach. Microb. Cell Factories 11, 34. doi:10.1186/1475-2859-11-34
Preusting, H., Kingma, J., Huisman, G., Steinbüchel, A., and Witholt, B. (1993). Formation of polyester blends by a recombinant strain of Pseudomonas oleovorans: different poly(3-hydroxyalkanoates) are stored in separate granules. J. Environ. Polym. Degrad. 1, 11–21. doi:10.1007/BF01457649
Prieto, A., Escapa, I. F., Martínez, V., Dinjaski, N., Herencias, C., de la Peña, F., et al. (2016). A holistic view of polyhydroxyalkanoate metabolism in Pseudomonas putida. Environ. Microbiol. 18, 341–357. doi:10.1111/1462-2920.12760
Revelles, O., Tarazona, N., García, J. L., and Prieto, M. A. (2016). Carbon roadmap from syngas to polyhydroxyalkanoates in Rhodospirillum rubrum. Environ. Microbiol. 18, 708–720. doi:10.1111/1462-2920.13087
Ronďošová, S., Legerská, B., Chmelová, D., Ondrejovič, M., and Miertuš, S. (2022). Optimization of growth conditions to enhance PHA production by Cupriavidus necator. Fermentation 8, 451. doi:10.3390/fermentation8090451
Roy, S., Patra, M., Nandy, S., Banik, M., Dasgupta, R., and Basu, T. (2014). In vitro holdase activity of E. coli small heat-shock proteins IbpA, IbpB and IbpAB: a biophysical study with some unconventional techniques. Protein Pept. Lett. 21, 564–571. doi:10.2174/0929866521666131224094408
Senior, P. J., and Dawes, E. A. (1971). The role and regulation of poly-beta-hydroxybutyrate synthesis in Azotobacter beijerinckii. Biochem. J. 123, 29P. doi:10.1042/bj1230029pa
Steinbüchel, A., and Valentin, H. E. (1995). Diversity of bacterial polyhydroxyalkanoic acids. FEMS Microbiol. Lett. 128, 219–228. doi:10.1111/j.1574-6968.1995.tb07528.x
Stueber, D., and Bujard, H. (1982). Transcription from efficient promoters can interfere with plasmid replication and diminish expression of plasmid specified genes. EMBO J. 1, 1399–1404. doi:10.1002/j.1460-2075.1982.tb01329.x
Sun, Z., Ramsay, J. A., Guay, M., and Ramsay, B. A. (2007). Carbon-limited fed-batch production of medium-chain-length polyhydroxyalkanoates from nonanoic acid by Pseudomonas putida KT2440. Appl. Microbiol. Biotechnol. 74, 69–77. doi:10.1007/s00253-006-0655-4
Tarazona, N. A., Hernández-Arriaga, A. M., Kniewel, R., and Prieto, M. A. (2020). Phasin interactome reveals the interplay of PhaF with the polyhydroxyalkanoate transcriptional regulatory protein PhaD in Pseudomonas putida. Environ. Microbiol. 22, 3922–3936. doi:10.1111/1462-2920.15175
Timm, A., Wiese, S., and Steinbüchel, A. (1994). A general method for identification of polyhydroxyalkanoic acid synthase genes from pseudomonads belonging to the rRNA homology group I. Appl. Microbiol. Biotechnol. 40, 669–675. doi:10.1007/BF00173327
Tiso, T., Sabelhaus, P., Behrens, B., Wittgens, A., Rosenau, F., Hayen, H., et al. (2016). Creating metabolic demand as an engineering strategy in Pseudomonas putida – rhamnolipid synthesis as an example. Metab. Eng. Commun. 3, 234–244. doi:10.1016/j.meteno.2016.08.002
Wang, Q., and Nomura, C. T. (2010). Monitoring differences in gene expression levels and polyhydroxyalkanoate (PHA) production in Pseudomonas putida KT2440 grown on different carbon sources. J. Biosci. Bioeng. 110, 653–659. doi:10.1016/j.jbiosc.2010.08.001
Weber, E., Engler, C., Gruetzner, R., Werner, S., and Marillonnet, S. (2011). A modular cloning system for standardized assembly of multigene constructs. PLOS ONE 6, e16765. doi:10.1371/journal.pone.0016765
Werner, S., Engler, C., Weber, E., Gruetzner, R., and Marillonnet, S. (2012). Fast track assembly of multigene constructs using Golden Gate cloning and the MoClo system. Bioengineered 3, 38–43. doi:10.4161/bbug.3.1.18223
Yarnell, W. S., and Roberts, J. W. (1999). Mechanism of intrinsic transcription termination and antitermination. Science 284, 611–615. doi:10.1126/science.284.5414.611
Zheng, Y., Chen, J.-C., Ma, Y.-M., and Chen, G.-Q. (2019). Engineering biosynthesis of polyhydroxyalkanoates (PHA) for diversity and cost reduction. Metab. Eng. 58, 82–93. doi:10.1016/j.ymben.2019.07.004
Keywords: Pseudomonas putida, synthetic biology, polyhydroxyalkanoates, modular cloning assembly, IbpA inclusion body protein
Citation: Manoli M-T, Blanco FG, Rivero-Buceta V, Kniewel R, Alarcon SH, Salgado S and Prieto MA (2023) Heterologous constitutive production of short-chain-length polyhydroxyalkanoates in Pseudomonas putida KT2440: the involvement of IbpA inclusion body protein. Front. Bioeng. Biotechnol. 11:1275036. doi: 10.3389/fbioe.2023.1275036
Received: 09 August 2023; Accepted: 18 October 2023;
Published: 01 November 2023.
Edited by:
Jeong Chan Joo, Catholic University of Korea, Republic of KoreaReviewed by:
Sung Kuk Lee, Ulsan National Institute of Science and Technology, Republic of KoreaCopyright © 2023 Manoli, Blanco, Rivero-Buceta, Kniewel, Alarcon, Salgado and Prieto. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: M. Auxiliadora Prieto, YXV4aUBjaWIuY3NpYy5lcw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.