 Kangsan Kim
Kangsan Kim Minjeong Kang
Minjeong Kang Byung-Kwan Cho
Byung-Kwan Cho- 1Department of Biological Sciences, Korea Advanced Institute of Science and Technology, Daejeon, Republic of Korea
- 2KAIST Institute for the BioCentury, Korea Advanced Institute of Science and Technology, Daejeon, Republic of Korea
- 3Graduate School of Engineering Biology, Korea Advanced Institute of Science and Technology, Daejeon, Republic of Korea
The past decade has seen growing interest in bacterial engineering for therapeutically relevant applications. While early efforts focused on repurposing genetically tractable model strains, such as Escherichia coli, engineering gut commensals is gaining traction owing to their innate capacity to survive and stably propagate in the intestine for an extended duration. Although limited genetic tractability has been a major roadblock, recent advances in systems and synthetic biology have unlocked our ability to effectively harness native gut commensals for therapeutic and diagnostic purposes, ranging from the rational design of synthetic microbial consortia to the construction of synthetic cells that execute “sense-and-respond” logic operations that allow real-time detection and therapeutic payload delivery in response to specific signals in the intestine. In this review, we outline the current progress and latest updates on microbial therapeutics, with particular emphasis on gut commensal engineering driven by synthetic biology and systems understanding of their molecular phenotypes. Finally, the challenges and prospects of engineering gut commensals for therapeutic applications are discussed.
1 Introduction
The human intestine is densely populated with an estimated 1013–1014 native commensals and an exhaustive catalog of enzymes essential for host metabolism (Sender et al., 2016). The collective genetic pools of the gut microbiota are estimated to surpass those of humans by nearly three orders of magnitude (Tierney et al., 2019) and contribute as much as 10% of the metabolites in the host bloodstream (Wikoff et al., 2009). The gut microbiota cooperates to support important chemical reactions that are otherwise absent in host metabolism. For example, the saccharolytic species of Bacteroides harbor carbohydrate-active enzymes that catabolize complex glycans into simpler substrates and short-chain fatty acids, which serve as nutrients accessible to other commensals and the host (Luis et al., 2018). In addition, the conversion of primary bile acids into their secondary forms in mammals is mediated by microbiota-derived bile salt hydrolases, the lack of which interferes with cell cycle regulation, immunity, and insulin secretion (Collins et al., 2023).
Therefore, the human intestine represents an important interface of host-microbe interactions, which have broad implications in diverse facets of host biology, from simple nutrient metabolism to complex roles such as immunomodulation. Thus, any aberrant shift in the gut microbiota composition, termed dysbiosis, could inadvertently lead to disease; associations with infection (Buffie et al., 2015), gut inflammation (Franzosa et al., 2019), and metabolic disorders (Smith et al., 2013), among various others (Koeth et al., 2013) have been reported. With the increasing realization that host–microbe interactions play an integral role in host biology, many studies have focused on microbial therapeutics, a growing field that entails the use of engineered bacterial chassis or synthetic microbe consortia for disease treatment.
Traditional means of microbe-based therapeutic interventions largely entailed administration of individual microbial strains or their consortia that intrinsically harbor therapeutically-relevant properties (Schultz, 2008; Khan et al., 2022). The microbe-based therapeutic modalities are gaining more tractions thanks to recent advances in systems and synthetic biology, which enabled customization the microbial cells and synthetic consortia with an aim to address a range of diseases with a better efficiency, while minimizing the inherent limitations that undermine the utility microbial-based therapeutics. The rapid progress in the research and development of microbial therapeutics is exemplified by the rapid increase in the number of enrolments in clinical trials investigating microbiota-based therapeutics (Dronkers et al., 2020), with over 4,000 clinical trials using the term “gut microbiota” registered at ClinicalTrials.gov to date. Analytical tools that incorporate multi-omics analysis and in silico simulations allow for spatiotemporal analysis of host-microbe interactions at resolutions down to a single metabolite (Heinken et al., 2023). Importantly, the integrated systems analysis could facilitate elucidation of structural and functional roles of gut microbiome in association with disease manifestation, enabling exploration of potential targets for therapy. This effort could ultimately translate to rational design of natural or synthetic microbial consortia to reverse intestinal dysbiosis and its associated complications, as exemplified by the case studies of Clostridioides difficile infection (CDI) (Buffie et al., 2015). The emerging utility of synthetic microbial chassis in therapy is largely facilitated by modularized bioparts that enable the execution of user-programmed functions, such as site-specific therapeutic delivery, in a highly reproducible manner. In addition, expanding synthetic biology toolkits now cater to non-model gut microbes that were previously intractable to genetic engineering, providing ways to circumvent the inherent drawbacks associated with non-commensal model strains. In this review, we outline the latest advances in microbial therapeutics using systems and synthetic biology frameworks for novel discoveries and strain engineering, with an emphasis on the engineering of gut bacteria.
2 Systems biology approach to understanding microbe–host interactions
Decades of omics biology research on the human gut microbiome have uncovered the complex interplay of host–microbe interactions at the molecular level, allowing us to consider the implications of such dynamics on the health of the host. In the past decades, many efforts have been devoted to characterizing the composition and patterns of the gut microbiota that correlate with specific host status, such as diet and age (Claesson et al., 2012), obesity (Turnbaugh et al., 2009), and malnutrition (Smith et al., 2013). Considering the postulation that the gut microbiota contributes as much as one-tenth of the metabolites in the host bloodstream (Wikoff et al., 2009), high-throughput metabolomics have been actively used to elucidate the link between the gut microbiome and host phenotypes. Such endeavors have enabled the mapping of microbiota-derived biochemicals in association with a range of host physiologies, including disease manifestations (Franzosa et al., 2019), xenobiotic metabolism (Zimmermann et al., 2019), and behavioral changes (Dohnalova et al., 2022). Importantly, integrative analysis of systems biology datasets and the development of computational platforms that consolidate multiple datasets into coherent information have considerably expanded our capacity to investigate host-microbe interactions.
Integrative systems biology analysis that reconciles multi-omics datasets with in silico modeling frameworks serves as an effective approach for revealing the implicit dynamics of host-microbe interactions. Briefly, the collective understanding of microbial genetics, genomics, and biochemistry is put into a single computational framework, the genome-scale metabolic model (GEM), which is a mathematical framework featuring a matrix of biochemical reactions that allows the prediction of metabolic flux distribution using linear programming. The optimization technique, flux balance analysis (FBA), calculates the most feasible metabolic state that maximizes a given objective function such as biomass generation or target metabolite production (Orth et al., 2010; Kim et al., 2021). The GEM also contains gene-protein-reaction associations that can be extended to assimilate -omics datasets, such as metagenomic readouts on the strain-specific relative abundance of a microbiome community (Heinken et al., 2019), serving as a platform to integrate and analyze disparate datasets (Monk et al., 2017). In particular, the integrative analysis of host–microbe interactions provides important inferences on implicit factors such as profiles of metabolites that are mutually cross-fed or subject to competition between the host and microbes (Heinken et al., 2013), the weighted contribution of a specific microbiome to host metabolism (Heinken et al., 2019), and microbiome-specific drug catabolism that affects host drug response (Heinken et al., 2023), which can be extrapolated further for predictive modeling of host biological status (Yang et al., 2022). More importantly, the ability to identify causal factors linked to an observed outcome and their mechanisms can translate to pragmatic applications, such as novel therapeutic modalities for enteric infection and inflammation (Buffie et al., 2015; Zhu et al., 2018). The knowledge on metagenomics, genetics, and biochemical profiles accumulating over decades has culminated in the “big-data-based” platforms that allow scalable prediction and simulation of potential impact of microbial metabolism on host physiology. Previously, constraint-based metabolic models that predict optimal metabolic fluxes for a given objective function (Heinken et al., 2023) and homology-based predictions of metabolic gene clusters that provide inference on metabolic pathway abundance in a given metagenome pool have been used (Pascal Andreu et al., 2021; Pascal Andreu et al., 2023). However, because of their highly multifaceted nature, host–microbe interactions remain one of the most actively explored avenues in systems biology (The Integrative, 2019).
The decade-long research on host-microbe interactions is now being translated into therapeutic product development. This is evident from the increasing number of microbe-based therapeutic regimens enrolling in clinical trials each year (Dronkers et al., 2020). In the next section, we provide a brief overview of the systems biology-driven approach to understanding the gut microbiota and outline how omics biology-driven analysis facilitates the exploration of host–microbe interactions at the molecular level. We also outline the ways in which the host-microbe interface is harnessed for potential therapeutic applications (Figure 1A).
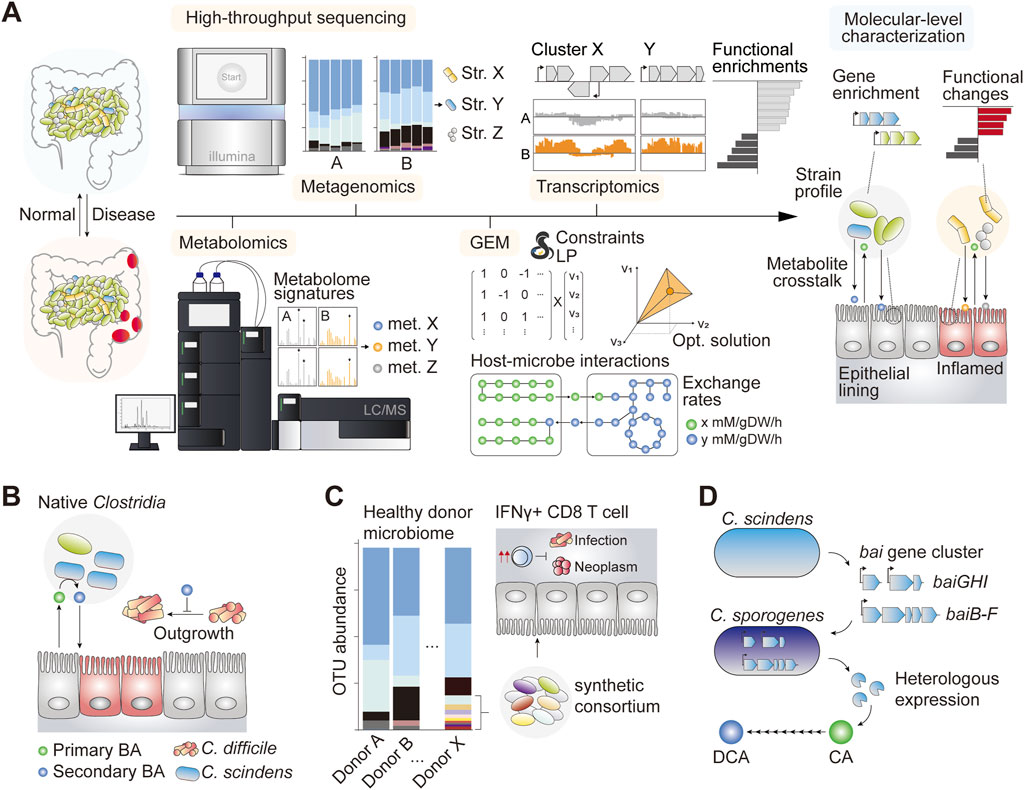
FIGURE 1. (A) Schematic of some notable systems biology approaches to understand the gut ecology and host–microbe interactions in the normal and inflamed gut (B) Introduction of Clostridium scindens and other causative species of Clostridium conferred C. difficile resistance phenotype primarily through the bioconversion of the luminal primary bile acid (BA) into the secondary form, which is inhibitory against C. difficile (Buffie et al., 2015) (C) A synthetically assembled consortium of 11 gut commensal bacteria induced proliferation of IFNγ CD8 T cells which showed anti-infective and anti-neoplastic effects in pre-clinical mouse models (Tanoue et al., 2019) (D) The bai gene cluster encoding primary BA deconjugation enzymes in C. scindens is heterologously expressed in genetically tractable species C. sporogenes, which functionally converts cholic acid (CA) into deoxycholic acid (DCA) in vitro (Funabashi et al., 2020).
2.1 Applications of systems biology in microbiome therapeutics
In April 2023, an investigational gut microbiota therapeutic tailored to address CDI, SER-109 (trade name VOWST), was approved by the Food and Drug Administration for clinical use (Mullard, 2023). This was the second FDA-approved microbiome therapeutic followed by a rectally administered microbiota cocktail, RBX2660 (trade name Rebyota). These two biotherapeutics have been tailored specifically for the prevention of CDI, which is a leading cause of antibiotic-associated diarrhea that results in 15,000–30,000 deaths annually (McDonald et al., 2018). Although the exact microbial compositions isolated from the therapeutic cocktails were not fully defined or disclosed (except that VOWST comprises selectively purified Firmicutes spores (Sims et al., 2023)), systems analysis of C. difficile etiology provides a glimpse into candidate species with potential therapeutic relevance and the underlying mechanisms of gut microbe-based modulation of the infection.
Gut microbiota dysbiosis is correlated with the depletion of luminal secondary bile acids (BAs) such as deoxycholic and lithocholic acids, which render the intestine highly prone to CDI (Theriot et al., 2016). The human gut microbiome, particularly the phyla Firmicutes and Bacteroidetes, have been predicted to harbor the biosynthetic capacity to transform host-derived primary BAs into 12 of the 13 types of secondary BAs in vivo (Heinken et al., 2019). With regard to infection susceptibility, primary bile acids promote spore germination in C. difficile, whereas secondary BAs are inhibitory towards its vegetative growth and the TcdB toxin (Icho et al., 2023). Thus, the depletion of BA-converting species and other antipeptide-producing native gut flora during an antibiotic sweep creates an environment highly conducive to spore germination, overgrowth, and infection. In addition, secondary BAs activate orthogonal receptors that antagonize pro-inflammatory nuclear factor-κB (NF-κB) signaling (Collins et al., 2023). Therefore, the reduction in luminal secondary BA levels is thought to exacerbate gut inflammation and increase susceptibility towards spore-forming pathogenic agents, and antibiotic treatment frequently leads to relapses.
Systems biology has shed light on the therapeutic potential of gut microbiota editing for thwarting recurring CDI. In one study, authors identified native species of gut microbiota that contributed significantly to CDI resistance (Buffie et al., 2015). Importantly, species-level characterization of microbial abundance together with time-course modeling of microbial dynamics in response to antibiotic perturbation identified a group of bacteria that ameliorated infection upon engraftment (Figure 1B). Among these, the adoptive transfer of Clostridium scindens, a species that retains a complete secondary BA biosynthetic pathway (Funabashi et al., 2020), confers infection resistance (Buffie et al., 2015). Interestingly, subsequent analysis revealed that resistance to the C. difficile gene family was strongly linked to the enrichment of genes associated with secondary BA biosynthesis. The results were reproducible in a constraint-based microbial community modeling framework, where the simulation suggested that patients with inflammatory bowel disease (IBD) varied considerably in their metabolic capacity to produce secondary BAs compared with healthy controls (Heinken et al., 2019). Taken together, the systems analysis of C. difficile pathobiology helped identify a potent probiotic candidate as well as the underlying mechanism of action, ultimately allowing novel therapeutic options for difficult-to-treat diseases such as CDI (with the FDA-approved therapeutics Rebyota and VOWST).
Recent progress in etiological characterization of celiac disease (CD) driven by-multi-omics analysis is also worth notable mentions (Leonard et al., 2020; Leonard et al., 2021). CD is an autoimmune condition triggered by gluten consumption. While it has been proposed that genetic predisposition and environmental stimuli play key role in CD development, its exact pathogenesis and etiology remains incompletely understood. Recently the Celiac Disease Genomic, Environmental, Microbiome, and Metabolomic (CDGEMM) study was launched in an attempt to investigate potential link between gut microbiota and CD development. The metagenomic and metabolomic features in stools collected from a longitudinal cohort of infants at high risks of CD were analyzed to explore multivariate risk factors encompassing genetics, mode of delivery and feeding, antibiotic use to CD development, in the venture point of gut microbiome (Leonard et al., 2020). Interestingly, the study reported major taxonomic and functional shifts in the gut microbiota of CD-prone infants, providing insights on how environmental factors that shape gut microbiota composition, in turn, may adversely affect immunomodulation of the host (Leonard et al., 2020). More recently, a longitudinal study that enrolled 10 CD and 10 non-CD infants reported changes in the gut metagenomes, pathway enrichments and associated metabolomes that are specific to the onset of CD (Leonard et al., 2021). As such, the multi-omics driven investigation of the longitudinal changes in gut microbiome has yielded valuable insights into the emerging role of gut microbiota in the development the chronic autoimmune disease. The data-driven findings shed light onto potentially novel therapeutic targets for the treatment and prevention of CD. The efforts are currently ongoing, with more than 550 subjects enrolled in the study as of May 2023. The large-scale longitudinal investigations hold the promise of unveiling undercharacterized molecular mechanisms and bring forth innovative therapeutic modalities for addressing CD (Leonard et al., 2023).
Synthetic microbial consortia designed with careful consideration of strain-specific metabolism, potential impact on host physiology, and safety profiles have been demonstrated to be a viable and effective therapeutic modality for different disease types (Table 1). In a study investigating CDI treatment, researchers developed a formula of synthetic microbial consortia with a defined species composition, using microbial isolates obtained from a healthy donor stool. This synthetic regimen was effective in preventing recurrent CDI in affected patients for up to 6 months (Petrof et al., 2013). In another study, a group of bacterial species that stimulated the proliferation of interferon-γ-secretory CD8 T cells demonstrated a potent therapeutic effect against infectious and neoplastic diseases in murine models (Figure 1C) (Tanoue et al., 2019). In this study, synthetic consortia comprising low-abundance species of healthy human microbiota exhibited enhanced clearance of the infectious agent Listeria monocytogenes and potent anti-tumor effects without observable adverse effects (Tanoue et al., 2019). The study underscored the importance of underrepresented species in omics data, which nonetheless have broad implications in host physiology, emphasizing the need to revisit sequence-oriented microbiome analysis weighted heavily on the quantitative evaluation of individual components.
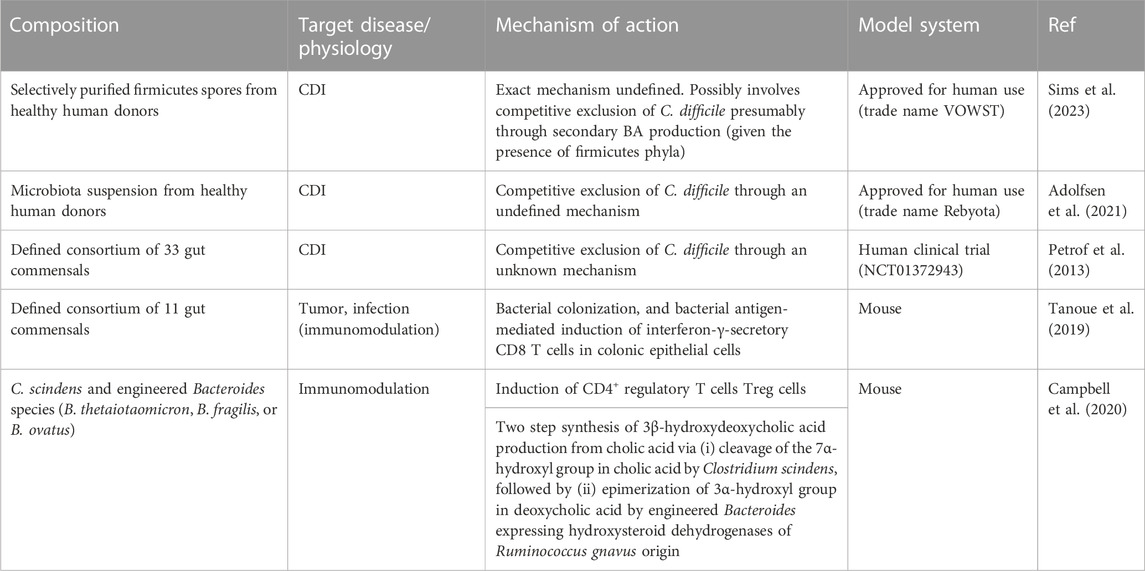
TABLE 1. Notable examples of native or synthetic microbe consortia in therapy.
It is also worth noting that the application of systems biology extends to functional genetics, enabling the characterization of the aforementioned causal factors at nucleotide-level. For example, functional coding sequences of the complete secondary BA biosynthetic pathway of intractable gut microbes have been published recently (Funabashi et al., 2020; Sato et al., 2021), paving the way for functional expression and control of secondary BA biosynthetic pools for therapeutic applications (Figure 1D) (Funabashi et al., 2020; Koh et al., 2022). While the multitudes of systems biology toolkits have drastically expanded our ability to characterize host-microbe interactions in unprecedented detail, our ability to effectively manipulate and engineer gut commensals remains a challenge. Genetic manipulation of gut commensal species is important for validating data-driven hypotheses and serves as an enabling technology in the growing field of live microbial therapeutics. In this section, we describe a novel therapeutic modality driven by synthetic biology.
3 Engineering microbes for therapeutic applications
Repurposing individual microbes for therapeutic applications, referred to as live bacterial therapeutics (LBTs), requires a robust chassis strain with the ability to survive, thrive, and drive the production and delivery of the desired payloads in vivo (Inda et al., 2019). Furthermore, the need for Generally Recognized as Safe (GRAS) makes probiotic bacteria that are harmless, beneficial to human health, capable of thriving in the host, best suited for therapeutic applications. The earliest and most prominent example was the use of the probiotic strain Escherichia coli Nissle 1917 (EcN), which has clinically relevant antimicrobial and immunomodulatory properties (Schultz, 2008; Sassone-Corsi et al., 2016). Since then, the administration of various probiotics with beneficial properties has been the treatment of choice for gut inflammation and dysbiosis (Fukuda et al., 2011; Sassone-Corsi et al., 2016).
Despite the reported efficacy and valid conceptual basis for its therapeutic effects (Schultz, 2008), probiotic treatment often leads to conflicting prognoses and is not recommended for therapeutic use by some medical organizations (Dickson, 2019; Guandalini and Sansotta, 2019). This calls for genetic engineering interventions to augment the beneficial properties of probiotics and confer novel clinically relevant phenotypes. The expanding availability of novel bioparts and genetic toolkits has allowed the programming of a microbial therapeutic chassis for IBD, cancer therapy, and synthetic metabolism (Lai et al., 2022; Lynch et al., 2022). Although the efficacy of most biotherapeutic projects has been limited to laboratory settings, increasing numbers of biotherapeutics are being evaluated for clinical applications (Dronkers et al., 2020). In this section, we describe the conceptual basis of LBT engineering, latest updates, and emerging use of gut commensal bacteria as a next-generation biotherapeutic chassis.
3.1 Synthetic biology approach to LBT construction
Assured safety, advanced genetic tractability, and relative ease of manipulation have led to the expanding use of probiotics as LBTs, which are live microbes engineered to deliver therapeutic payloads in vivo (Steidler et al., 2000). The development of titratable substrate-specific promoter arrays (Meyer et al., 2019) that facilitate complex logic operations (Nielsen et al., 2016; Jones et al., 2022) has streamlined the design and engineering of LBTs that perform real-time detection and therapeutic payload delivery in response to specific signals in vivo (Triassi et al., 2023). An LBT chassis can simultaneously diagnose, record, and self-regulate therapeutic payload delivery in response to host disease in vivo (Figure 2) (Zou et al., 2023). Such ‘smart LBT,’ for lack of a better term, is powered by versatile genetic circuitry that constitutes independent genetic components, each of which enables sensing of environmental cues, signal relay and processing, and actuation (Figure 2). Each genetic component is modularized as composite bioparts to enable reconfiguration of cellular functions in a “plug-and-play” manner; that is, as far as chassis strain is concerned, cells can be reprogrammed to execute various user-defined operations by re-wiring different input signals to output modules.

FIGURE 2. Schematic of engineered live bacterial therapeutics. An LBT can simultaneously sense surrounding input signals, and process the signals using elaborate Boolean logic gates, which are relayed to guide response outpuslt. By tethering the output signals with appropriate actuator genes, an LBT can execute user-programmed functionalities such as genetic memory (with base editor; BE), and therapeutic delivery, in vivo. Abbreviations–TEA: terminal electron acceptor, BE: base editor.
Biosensor modules, such as endogenous transcription factors (TFs), enzymes, or nucleic acids, regulate gene expression. The mode of action of a genetic biosensor typically entails a conformational change upon ligand binding, followed by the activation of orthogonal transcription machinery. Using a two-component signal transduction system (TCS) as an example, the membrane-bound sensor histidine kinase ThsS phosphorelays thiosulfate (S2O32−)-induced signals at the receiving end of the DNA-binding regulator ThsR. Phosphorylated ThsR, in turn, activates its cognate promoter PphsA and expression of the downstream reporter gene superfolder GFP (sfGFP) to transform the ligand-binding signal into luminescence readouts in a dose-dependent manner (Daeffler et al., 2017). To date, an array of genetic biosensors specific to the host and/or disease-specific biomarkers has been reported. These include biosensors for inflammation, infection, bleeding, cancer, diabetes, and availability of abiotic factors such as pH and O2 (Table 2). The sensing modules confer stringent control over LBT phenotypes and augment the performance. In one instance, a refactoring of K5-type capsular polysaccharide expression with the lac promoter in EcN allowed programmable surface antigen display using isopropyl-b-D-thiogalactopyranoside (IPTG) induction. This rather simple tweak in transcription regulation led to prolonged in vivo survival by allowing programmable evasion of the host immune response, consequently augmenting the antitumor efficacy of LBT by ten-fold (Harimoto et al., 2022). The integration of more than one transcriptional regulator in a complex cellular response is characterized by multimodal transgene expression in response to diverse environmental cues (Taketani et al., 2020).
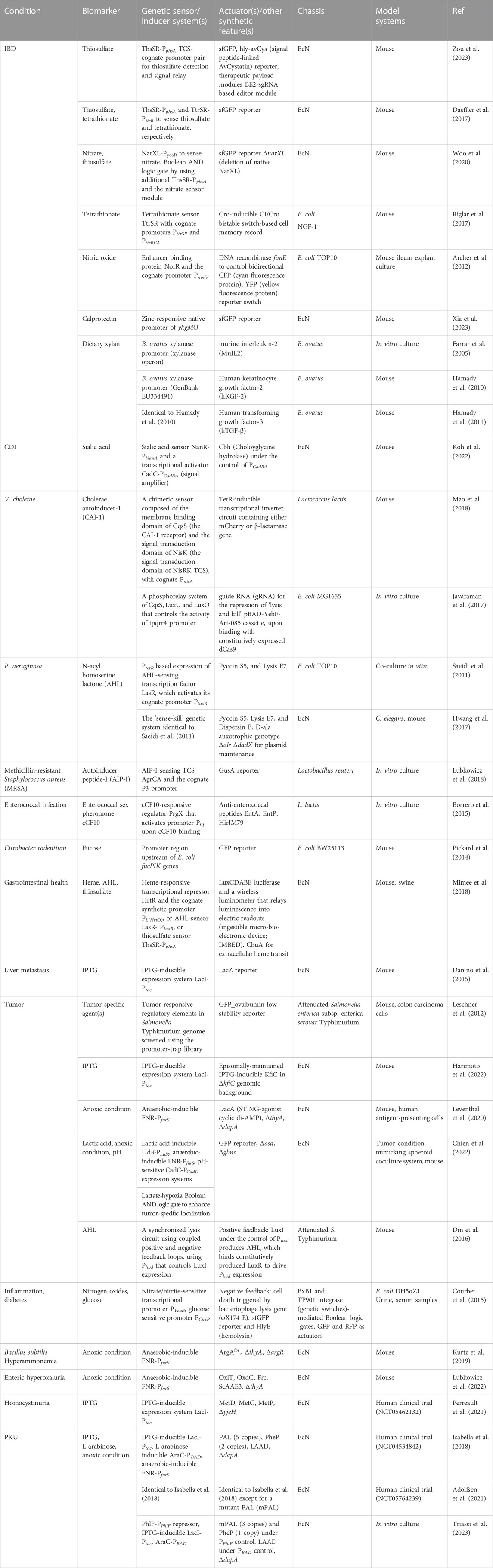
TABLE 2. Examples of LBTs for disease diagnosis and/or treatment.
Complex cell programming is achieved through signal processing modules, which are important for informing LBTs about specific locations and timings to produce therapeutic responses at the required dosage. To this end, Boolean logic gates that compute input signals to guide appropriate responses have been implemented in genetic circuit designs. For instance, the biomarkers of gut inflammation thiosulfate/tetrathionate (S4O62−) and nitrate (NO3−) are proxies for two distinct etiologies of IBD (Winter et al., 2010; Winter et al., 2013), and the means to detect each biomarker alone may be insufficient for IBD diagnosis. To address this issue, a genetic circuit incorporating an AND Boolean logic gate was constructed based on two TCSs and cognate promoter pairs, ThsSR with PphsA, and NarXL with PyeaR; creating a system that requires the presence of two IBD biomarkers, thiosulfate and nitrate, to elicit an actuator response (Woo et al., 2020). The use of complex logic operations further enhances the specificity of genetic circuits by imparting multilevel control to the system output. However, designing genetic circuits with an increasing number of control elements is difficult and time consuming, which limits the implementation of complex cell programming (Kwok, 2010). Automated genetic circuit design software (Cello) streamlines the entire workflow (Nielsen et al., 2016; Jones et al., 2022). The software allowed the functional design of a genetic circuit using a library of NOR/NOT gates as complex as 10 regulators and 55 parts. In addition, the response functions of the sensors and gates can be extrapolated to quantitatively predict the behavior of the designed genetic sensor. A reliable circuit design algorithm that boasts 92% circuit output prediction accuracy was applied in the design of a gut commensal chassis for multimodal transgene expression (Taketani et al., 2020) as well as in an LBT to enable timely, predictable expression of therapeutic payloads in a condition-specific manner (Triassi et al., 2023).
3.2 Enhancing LBT performance for clinical applications
Fine-tuning of transgene expression and the activity of encoded products are commonly sought to optimize strain performance. It should be noted that the functional expression of transgenes per the intended design may require extensive troubleshooting and ad hoc optimizations. For instance, owing to the highly subjective nature of composite elements within a genetic circuit, determining the optimal parameters for an expression system is confounded by multiple variables, such as the strength of the expression machinery (Chien et al., 2022; Choe et al., 2022; Song et al., 2022) and genetic and environmental contexts (Lou et al., 2012). In many instances, an optimal system output can be achieved by shuffling genetic parts (Choe et al., 2022) or their brute-force combinations (Song et al., 2022) and copy number variations (Isabella et al., 2018) to fine-tune the target transgene expression. Enhancing the activity of the encoded functions is also an integral part of the optimization process, particularly when transgene expression is not a bottleneck. To this end, the evolutionary (directed) engineering of target proteins at a specific scale, followed by biosensor-assisted screening, is effective in identifying target proteins with improved functionalities (Adolfsen et al., 2021). Furthermore, the characterization of highly expressed genomic regions within the chassis strains can be repurposed as a ‘landing pad’ for transgene expression cassettes, which confers higher genetic stability over the plasmid-based expression regimes (Park et al., 2020).
SYNB1618 and its derivatives are LBTs designed for the treatment of phenylketonuria (PKU) and assimilate the aforementioned engineering and optimization principles, which are worthy of a detailed discussion (Figure 3). The original strain, SYNB1618, is an EcN-based LBT engineered to complement a disabling mutation in the host phenylalanine hydroxylase (PAH), which is the cause of the debilitating metabolic disease. SYNB1618 carries two synthetic phenylalanine degradation pathways. First, PheP and PAL, encoded by phenylalanine transporter, pheP and phenylalanine ammonia-lyase, stlA, respectively. Second, LAAD, encoded by membrane-associated L-amino acid deaminase pma (Figure 3A) (Isabella et al., 2018). To achieve high expression levels and impart transcriptional control in vivo, multiple copies of pheP/stlA were genome-integrated and expression-controlled in an IPTG- (Ptac), L-arabinose (PBAD) or anaerobic-inducible (PfnrS) manner (Figure 3A). The in vivo analysis showed that SYNB1618 was effective in lowering blood phenylalanine levels in a mouse model of PKU, stabilized phenylalanine levels in healthy non-human primates (Isabella et al., 2018), and was safely tolerated by healthy individuals (Puurunen et al., 2021).
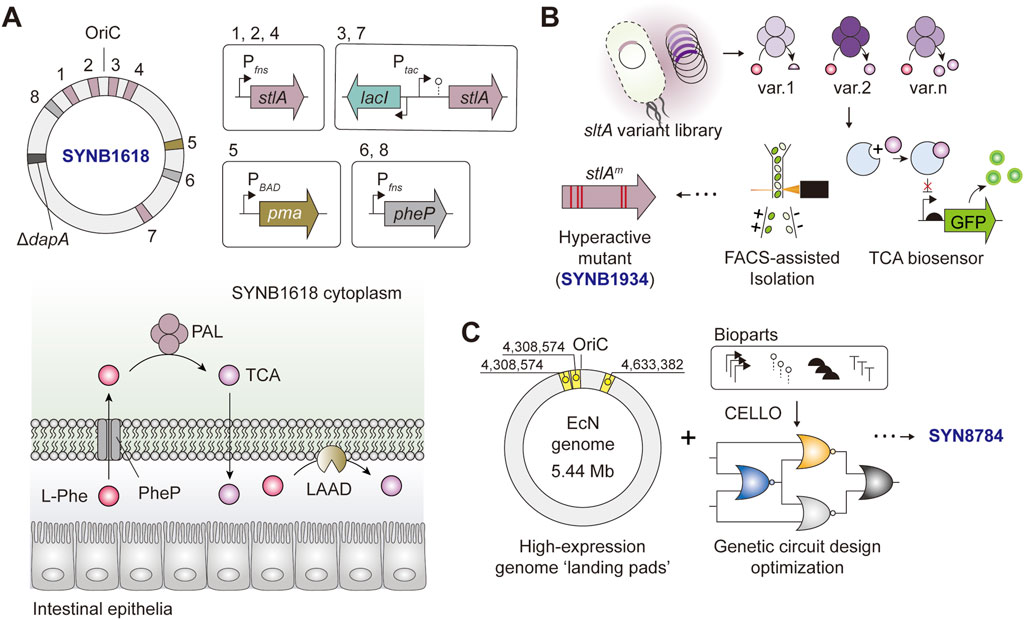
FIGURE 3. Simplified schematics of Escherichia coli Nissle 1917-based LBTs tailored for the treatment of PKU (A) SYNB1617 harbors five copies of stlA, two copies o f pheP, and a single copy of pma under the expression of Ptac, PBAD, or PfnrS. The deletion of 4-hydroxy-tetrahydrodipicolinate synthase (dapA) renders the bacteria auxotrophic to diaminopimelate (DAP) (B) A simplified workflow for screening and isolation of sltAm encoding “hyperactive” PAL mutants, the key component of SYNB1934 (C) Engineering logic behind SYN8784 construction.
In a follow-up study aimed at further augmenting the phenylalanine-degrading capacity of LBT, PAL was no longer rate-limiting, hence calling for efforts to optimize the performance of PAL (Adolfsen et al., 2021). In this study, the authors used a biosensor-assisted screen based on an allosteric TF activated in the presence of trans-cinnamate (TCA), a product of PAL, in a concentration-dependent manner. The stlA mutant library was designed considering the enzyme phylogeny, co-evolution, and structural attributes to infer enzyme residues for targeted engineering. To avoid cross-talk between different TCA producer strains (which also house the TF-based biosensor) in the pooled library, an oil-emulsion-assisted microculture followed by a fluorescence-activated cell sorting (FACS) strategy was adopted to effectively isolate high TCA-producing mutants (Figure 3B) (Adolfsen et al., 2021). The authors identified a PAL mutant (stlA* or mPAL) carrying five mutations (S92G, H133M, I167K, L432I, and V470A) with Vmax and KM two to three folds higher compared to that of wild-type PAL. The SYNB1618-derivative that expresses stlA*, designated SYNB 1934, showed an approximately 2-fold increase in in vivo phenylalanine conversion capacity compared to SYNB1618 (Adolfsen et al., 2021). The PKU-targeting LBT series SYNB1618 and SYNB1934 have undergone phase I and II clinical trials (ClinicalTrials.gov ID: NCT04984525 and NCT04534842, respectively), and a phase III trial of SYNB 1934 (NCT05764239) is underway in June 2023.
More recently, an independent study revisited the transgene expression architecture of SYNB1618 with the final goal of optimizing its therapeutic gene expression and alleviating the potential genetic burdens incurred during the process. By leveraging the knowledge of genomic ‘hot spots’ that are conducive to strong gene expression and fine-tuning transgene expression using automated genetic circuit design, the authors constructed the final strain, SYN8784 (Figure 3C). With fewer genomic copies of stlA* and PheP, SYN8784 maintained higher TCA activity while also exhibiting enhanced strain fitness compared to SYNB1618, underscoring the importance of rationalized genetic circuit design informed by computer-aided design tools and augmentation of strain functions to reinforce system performance (Park et al., 2020; Triassi et al., 2023).
Together, these studies show that a synthetic-biology-powered chassis platform can be readily translated into real-life applications. The use of modularized bioparts with quantitatively measured and standardized characteristics confers enormous flexibility and predictability to the biological function design, facilitating the reconfiguration of cellular phenotypes in a user-defined manner. However, our insufficient understanding of complex biology limits our capacity to overcome certain biological constraints. The inherent biological constraints of probiotic strains, such as rapid clearance, low in vivo cell density, and inability to colonize the intestinal niche for an extended duration, represent hurdles in probiotic-based therapeutics (Zmora et al., 2018), especially for addressing chronic diseases (Inda et al., 2019). As a promising alternative, repurposing native gut microbes for therapeutic applications has gained traction in recent years. In the next section, we discuss systems and synthetic biology approaches for repurposing native gut resident species for therapeutic purposes.
4 Engineering gut commensal species for therapeutic applications
Repurposing the gut microbiota for various applications follows the same engineering principles. However, such attempts have been hampered by the incompatibility of existing molecular toolkits and the relative scarcity of relevant resources to manipulate genomes in the past (Salyers et al., 2000). In recent years, a suite of multi-omics and bioinformatics analytic pipelines has led to the discovery of unique transcription architectures as well as regulatory components that can be repurposed as transgene expression tools. In addition, continued efforts to harness the gut microbiota will expand the genetic toolbox for engineering individual commensal species.
In particular, Bacteroides species represent some of the most genetically amenable and extensively engineered gut commensals. Recent reviews/proceedings on microbiome (in particular, Bacteroides) engineering highlights the “technological readiness level” of engineering gut commensals that is nearly on par with that of other model species (Lai et al., 2022; McClure et al., 2022). For example, the availability of validated bioparts enables functional design and implementation of artificial genetic circuits to elicit multimodal actuator responses in individual gut commensals and their consortia, demonstrating programmed sense-and-respond operations in vitro and in vivo (Taketani et al., 2020; Huang et al., 2022). Remarkably, an artificially assembled porphyran PUL gene segment measuring 60 kb was transplanted into the Bacteroides thetaiotaomicron genome to enable the utilization of the polysaccharide porphyran, a carbohydrate found in seaweed that is inaccessible to the native gut microbiota (Shepherd et al., 2018). The colonization efficiency and in vivo abundance of engineered B. thetaiotaomicron can be controlled exogenously by porphyran supplementation, which is a promising development for stable engraftment and control of LBTs in vivo (Shepherd et al., 2018). Owing to this superior genetic tractability and numerical predominance of the species across individuals, Bacteroides in their native and engineered forms have been used in a host of gut microbiome studies, which ultimately led them to earn the status of, as Wexler and Goodman put it, “a window into the microbiome” (Wexler and Goodman, 2017).
Bacteroides possess an ensemble of adaptive features that are advantageous for in vivo survival. First, it encodes a broad class of carbohydrate-active enzymes that allow foraging on different forms of carbohydrates, from complex dietary glycans to host-derived glycoproteins (Martens et al., 2008; Luis et al., 2018), which confer survival advantages through the feast and famine cycles. Specifically, B. thetaiotaomicron expresses an elaborate repertoire of saccharolytic enzymes via specific gene clusters known as polysaccharide utilization loci (PUL), which together account for nearly 18% of the whole genome (Xu et al., 2003). There are as many as 88 such loci, each embedded with at least one homolog of susC and susD, which enable B. thetaiotaomicron to utilize a broad range of substrates (Martens et al., 2008) (Figure 4). In addition, phase-variable systems controlling surface antigen expression enable the evasion of host immunity and phage infection (Cullen et al., 2015; Taketani et al., 2015; Porter et al., 2020), and promote phenotypic diversity that mediates colonization in the intestine (Jiang et al., 2019) (Figure 4). Such intrinsic features enable native members of the gut microbiota to stably colonize the gut ecosystem, occupy specific luminal niches such as glands and crypts, and propagate autonomously at high density for as long as a decade (Faith et al., 2013; Lee et al., 2013). Therefore, gut commensal-based LBTs may be better suited than their probiotic counterparts to address chronic diseases or those that require long-term interventions (Inda et al., 2019).
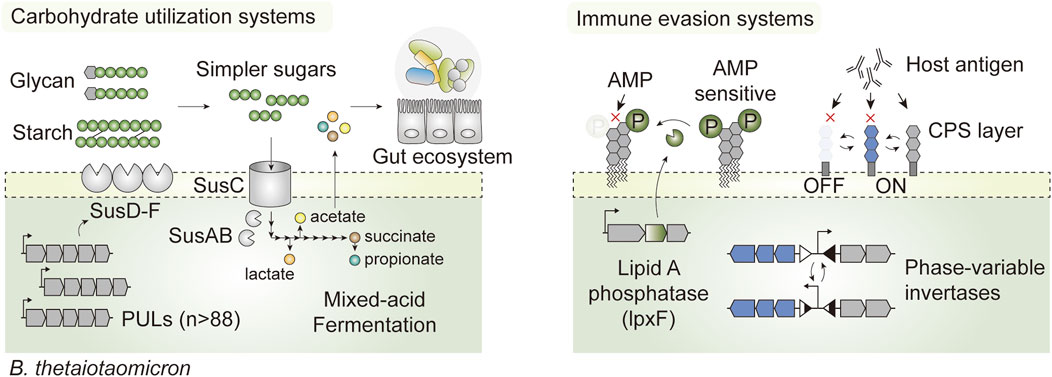
FIGURE 4. Hallmark features that promote in vivo survival in the prominent gut commensal Bacteroides thetaiotaomicron. The presence of exhaustive carbohydrate-active enzymes encoded by PULs enables foraging on complex dietary carbohydrates and host glycans (Starch utilization systems; SusA-F for illustration purposes). This not only confers a competitive edge for survival through the feast and famine cycles in the intestine, but it also shapes the luminal nutrient (or metabolic) landscape by converting otherwise inaccessible carbohydrates into simpler oligosaccharides and short chain fatty acids that can be utilized by the native gut flora and host epithelia. B. thetaiotaomicron also houses phase variable invertases and phosphatases that dynamically shift cell surface architectures in response to environmental perturbation, promoting evasion of the host immune system and exogenous antimicrobial peptides (AMP).
In addition to the adaptive features that favor gut colonization, mounting evidence highlights the therapeutic relevance of native Bacteroides and other commensals in human health and disease, including beneficial systemic effects on host metabolism and immunomodulation in inflammation, infection, and cancer therapy (Buffie et al., 2015; Plovier et al., 2017; Tanoue et al., 2019). Notably, B. thetaiotaomicron alleviated gut inflammation in a human Caco-2 infection model by selectively antagonizing pro-inflammatory NF-κB signaling through the nuclear export of the transcriptionally active NF-κB subunit RelA into the cytoplasm, thereby preventing downstream inflammatory cascades (Kelly et al., 2004). The protective effects of B. thetaiotaomicron extend to murine models of colitis, characterized by reduced weight loss, histopathological damage, and inflammation (Delday et al., 2019). Interestingly, treatment with purified pirin-like protein (BT0187) in the Caco-2 cell lines led to reduced NF-κB levels and reproduced several hallmarks of therapeutic effects in live cells, suggesting its potential therapeutic use for inflammatory diseases (Delday et al., 2019). Recently, an assessment of the safety and tolerability status of B. thetaiotaomicron suggested that the strain was well tolerated by patients with Crohn’s disease (NCT02704728) (Hansen et al., 2021), further underscoring the applicability of beneficial gut commensals in LBTs. In this section, we describe the systems and synthetic biology-guided design and engineering of LBTs using the prominent native gut commensal Bacteroides as a model. A host of other gut inhabitants and probiotics species are also being employed for LBT applications, interested readers may refer to selected review articles (Inda et al., 2019; Aggarwal et al., 2020).
4.1 Systems biology approach to understanding gut commensal biology
An a priori understanding of the cellular systems precedes their rational design and engineering. In the case of gut commensals, factors such as the genetic determinants of strain fitness within the intestinal niche provide important engineering considerations for in vivo applications. However, conventional approaches to characterize genetic functions, such as gene knockout, are highly effective but limited in scale and require considerable labor and resources (Baba et al., 2006). Therefore, the vast majority of genetic elements in species belong to hypothetical genes that lack validation in literature, even in model gut commensal species.
Attempts to functionally characterize the genetic functions of gut commensal have accelerated using high-throughput screening systems. These include large-scale expression libraries for gain-of-function analyses (Yaung et al., 2015), genome-wide transposon mutagenesis and sequencing (Tn-Seq) (Goodman et al., 2009; Wu et al., 2015; Liu et al., 2021), and CRISPR interference screening platforms for loss-of-function studies (Shin et al., 2023). Such high-throughput sequencing methodologies are an effective means to build and validate novel hypotheses on genotype-phenotype relationships and streamline the investigation of genotype-phenotype relationships in a context-specific manner. For example, a mutant population of B. thetaiotaomicron containing a 3.5
High-throughput screening of genome-wide transcriptome changes, such as transcriptome sequencing (RNA-Seq), is a common strategy for analyzing condition-specific changes in mRNA transcript abundance. Variations in transcriptome sequencing technologies allow the characterization of distinct transcriptome landscapes across species that reflect unique ecological features and provide an important system-level inference that translates into engineering applications. For example, investigation of B. thetaiotaomicron transcriptome response in the presence of sub-components of porcine mucin, a glycoprotein that makes up the mucus lining of the mammalian intestine, has revealed a synchronized action of its PULs along with the regulatory extracytoplasmic sigma factors (ECF-σ). The deletion of genes that encode ECF-σs embedded within O-glycan-responsive PULs led to a significant in vivo deficit, elucidating yet another essential genetic determinant of in vivo fitness, along with the regulatory elements that impart transcriptional control (Martens et al., 2008). Transcriptome analysis has also identified two hybrid TCSs, BT3334 and BT0267, that activate cognate promoters in response to specific carbon substrates (Martens et al., 2008; Martens et al., 2011), which have been repurposed as inducible promoter bioparts in B. thetaiotaomicron (Mimee et al., 2015).
More recently, exploration of the primary transcriptome landscape of B. thetaiotaomicron genome-wide led to the discovery of more than 4
4.2 Engineering gut commensal-based LBTs
Gut commensals equipped with different features in synthetic biology have been shown to be viable means for therapeutic payload delivery. For example, pioneering research groups engineered Bacteroides ovatus to produce anti-inflammatory peptides such as murine interleukin-2 (MuIL2), human keratinocyte growth factor-2 (hKGF-2), and human transforming growth factor-β (hTGF-β) using a xylanase-inducible promoter in pre-clinical models of gut inflammation (Table 2) (Farrar et al., 2005; Hamady et al., 2010; Hamady et al., 2011). The robust heterologous expression system of Bacteroides species is often used as a “surrogate cell” to produce potent proteins of intractable commensal origin in the gut for the evaluation of host physiology. For example, a gene encoding tryptophan decarboxylase in the native gut resident R. gnavus was heterologously expressed in B. thetaiotaomicron to assess the biological implications of this relatively rare pathway that converts tryptophan into tryptamine in vivo setting. Gnotobiotic mice colonized with tryptamine-producing B. thetaiotaomicron (TrpD +) showed an increase in colonic secretion and a shortened gut transit time (Bhattarai et al., 2018). In a follow-up study, the introduction of B. thetaiotaomicron TrpD+ decreased weight loss in a colitis mouse model, primarily through tryptamine-induced activation of host serotonin receptor 4 (5-HT4R), which promoted goblet cell differentiation (Bhattarai et al., 2020). Similarly, to evaluate the impact of microbial bile acid metabolism in the colon, genes from Ruminococcus gnavus encoding a subset of the 3β-hydroxydeoxycholic acid (isoDCA) biosynthetic pathway, hydroxysteroid dehydrogenases, were heterologously expressed in model Bacteroides species (Campbell et al., 2020). Synthetic bile acid metabolism was performed by co-culturing engineered Bacteroides strains with C. scindens, which encodes the remaining subset of the isoDCA pathway. The introduction of synthetic consortia into the mouse model increased the colonic isoDCA concentration, with a concomitant elevation in the levels of colonic CD4+ regulatory T cells (Table 1).
Another avenue of strain engineering is based on a mathematical approach, first to understand biological phenotypes, and second to simulate optimal strain engineering designs that yield the desired outcomes. As mentioned in Section 2, in silico simulations using the GEM provide important inferences regarding the implicit dynamics of metabolic interactions within or between the host and microbes. For example, the first GEM reconstruction of B. thetaiotaomicron iAH991 was built with the extension of a mouse GEM compartment to simulate host-microbe interactions in response to different dietary regimes. While the multi-species GEM captured metabolite exchanges that were comparable with published metabolomics data, a stand-alone simulation of iAH991 for B. thetaiotaomicron monoculture phenotypes was incongruent with the experimental values (Heinken et al., 2013). Accumulating knowledge on gut commensal genetics and biochemistry has streamlined the reconstruction of strain-specific GEMs with improved prediction accuracy. Nearly a decade after its reconstruction, iAH991 was revisited by incorporating up-to-date strain-specific data and underwent extensive curation of model properties, resulting in an expanded GEM iKS1119 that showed remarkably improved in silico prediction accuracy (Kim et al., 2021). Subsequently, GEM-guided metabolic engineering was applied to B. thetaiotaomicron to produce non-native butyrate, a short-chain fatty acid (SCFA) with proposed immunomodulatory properties. While the strain failed to produce butyrate in the wild-type background, a round of metabolic engineering informed by the Optknock simulation redirected the cellular metabolic flux toward non-native butyrate biosynthesis, albeit at a much lower concentration than the native SCFA products (Kim et al., 2021).
Taken together, these studies demonstrate that prominent gut commensal species can be engineered to functionally express exogenous compounds at a sufficient dosage to elicit the desired changes in vivo. Furthermore, the development of in silico simulation platforms aids in the rational design and engineering to optimize the production of non-native metabolites. Leveraging this capacity, Novome Biotechnologies Inc. developed a B. thetaiotaomicron-based proprietary strain, NB1000S, designed to degrade oxalate for the treatment of enteric hyperoxaluria, and a porphyrin-based prebiotic, NB 2000P, that facilitates engraftment and tunes in vivo abundance of NB1000S. The combination product NOV-001 yielded promising results in phase 1 clinical trial assessing safety, tolerability, and strain colonization pharmacodynamics (NCT04909723), paving the way toward the clinical use of commensal-based LBTs that have hitherto been prospective.
5 Safety considerations
However, safety concerns regarding the use of genetically engineered bacteria as therapeutic agents remain unresolved. The primary concern is the dissemination of genetically modified bacteria into the environment, posing potential risks, such as horizontal gene transfer (HGT) of artificial genetic elements into native ecosystems. In addition, aberrant behaviors of chassis strains such as B. thetaiotaomicron in the expansion of enteric pathogens in genetically-predisposed hosts (Curtis et al., 2014) and invoking pro-inflammatory responses (Brown et al., 2019) call for an effective measure to contain the bioactivity of engineered microbes in a controllable manner. As part of the biocontainment strategy, LBTs are rendered auxotrophic by deleting biosynthetic genes associated with cell survival. In the Synlogic LBT series, dapA encoding 4-hydroxy-tetrahydrodipicolinate synthase was inactivated and required exogenous diaminopimelate (DAP) supplementation for survival (Table 2). Other proactive options involve the use of genetic circuits that trigger an actuator response, such as self-lysis effectors, inducer treatment, or exposure to the environment (Chan et al., 2016). Similarly, a “single-use” LBT in which a lysis factor Lysis E7 was integrated into the actuator to induce programmed lysis of the chassis, thereby facilitating the dissemination of anti-pathogenic effector upon sensing pathogen-specific molecule (Table 2) (Saeidi et al., 2011; Hwang et al., 2017). More sophisticated containment strategies are being developed, such as CRISPRi-assisted maintenance of synthetic auxotrophy (article deposited in bioRxiv, refer to Lai et al., 2022 for more details) (Lai et al., 2022). With novel progress in synthetic biology, we expect to see creative endeavors to achieve safer and more efficient LBT biocontainment.
6 Conclusion and perspectives
The rapid progress in synthetic biology, accompanied by efforts to decipher complex biology, has culminated in the ability to harness the native gut microbiota to our benefit, which has unveiled a host of novel therapeutic modalities. First, the existing paradigm of microbiome-based therapeutics explores the untapped reservoir of the genetic and biochemical repertoire of the native gut flora, which has been demonstrated to be efficacious, as exemplified by the recent success of VOWST and Rebyota (Adolfsen et al., 2021; Sims et al., 2023), and many more that await clinical assessment.
This marks a promising advancement in the once-controversial fecal microbiota transplant (FMT) therapy, which raised safety flags and was haunted by the weak reproducibility of its therapeutic efficacy. The rational design of synthetic microbial consortia based on a systems biology-driven understanding can selectively screen candidate microbial species and their combinations for maximum therapeutic potency (Tanoue et al., 2019; Campbell et al., 2020). Potentially, the capacity to simulate xenobiotic metabolism in the context of microbiome metagenomics that can account for inter-personal variations (Heinken et al., 2023), can be fully harnessed in challenging areas such as immunocompromised pediatric hosts by informing the most effective therapeutic effects based on predictive analysis of host-specific metabolic features (Flerlage et al., 2020). To this end, the implementation of iterative cycles of design-build-test-learn (DBTL) to systematically assess the outcome of the built designs, learn the system function, and install novel features to obtain an optimally functioning microbiome therapeutic product (Lawson et al., 2019).
Specific avenues for microbiome therapeutics require genetic engineering to augment strain performance and endow nonnative functionalities. In addition to direct microbe-to-host delivery, LBTs equipped with reliable biosensors can now execute diagnoses and site-specific dose-dependent payload delivery (production) in vivo. One prospect of the LBT lies in its ability to administer the correct drug dosage at the right time and place, which is expected to significantly improve treatment efficiency while minimizing side effects that are not uncommon in current therapeutic regimens (Inda et al., 2019). Furthermore, the emerging utility of gut commensals as chassis strains is expected to overcome several obstacles in probiotic-based in vivo therapeutic applications such as engraftment efficiency, rapid clearance, and low cell density. The rapid pace of development in the synthetic biology of gut commensals has culminated in NOV-001, the first gut commensal-based LBT that recently passed a phase 1 clinical trial (NCT04909723), paving the way toward real-life applications of commensal-based therapeutics that have long remained prospective. However, a recent report showed a distinct approach to circumvent the existing hurdles in probiotic (E. coli)-based LBTs to isolate and engineer native gut-resident E. coli which engrafts perpetually upon reintroduction (Russell et al., 2022). This is an interesting development in native gut commensal engineering and is worth considering in future LBT applications.
Living cells constantly undergo evolution, similar to that of LBTs. SYNB1618, the original Phe-degrading LBT that passed a phase 1 trial (NCT03516487), was upgraded using a catalytically superior mutant PAL enzyme discovered through high-throughput screening (Adolfsen et al., 2021). In another independent study, the transgene expression architecture of SYNB1618 was remodeled using an AutoCAD (Cello)-assisted genetic circuit design that reduced cellular burden (Triassi et al., 2023). The progress in the Phe-degrading LBT series implies that the ‘rooms for development’ in the existing chassis may be considerably wider than we previously thought. Machine learning algorithms expedite the interpretation of large agglomerates of high-throughput datasets, facilitating the discovery of hidden layers of regulation that can be translated into pragmatic applications (Sastry et al., 2019; Rychel et al., 2021). High-throughput automated screening platforms combine a wealth of (un)validated bioparts to streamline the exploration of solution spaces for optimal strain function (Yu et al., 2023). Unparalleled advances in systems and synthetic biology will accelerate the pace of chassis development and expedite the evolution of LBT performance.
Author contributions
KK: Visualization, Writing–original draft, Writing–review and editing. MK: Writing–review and editing. B-KC: Conceptualization, Supervision, Writing–review and editing.
Funding
The authors declare financial support was received for the research, authorship, and/or publication of this article. This study was supported by the Korea Bio Grand Challenge (2018M3A9H3024759 to B-KC) through the National Research Foundation of Korea (NRF), funded by the Ministry of Science and ICT.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Adolfsen, K. J., Callihan, I., Monahan, C. E., Greisen, P. J., Spoonamore, J., Momin, M., et al. (2021). Improvement of a synthetic live bacterial therapeutic for phenylketonuria with biosensor-enabled enzyme engineering. Nat. Commun. 12, 6215. doi:10.1038/s41467-021-26524-0
Aggarwal, N., Breedon, A. M. E., Davis, C. M., Hwang, I. Y., and Chang, M. W. (2020). Engineering probiotics for therapeutic applications: recent examples and translational outlook. Curr. Opin. Biotechnol. 65, 171–179. doi:10.1016/j.copbio.2020.02.016
Archer, E. J., Robinson, A. B., and Suel, G. M. (2012). Engineered E. coli that detect and respond to gut inflammation through nitric oxide sensing. ACS Synth. Biol. 1, 451–457. doi:10.1021/sb3000595
Baba, T., Ara, T., Hasegawa, M., Takai, Y., Okumura, Y., Baba, M., et al. (2006). Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2, 2006.0008. doi:10.1038/msb4100050
Bhattarai, Y., Jie, S., Linden, D. R., Ghatak, S., Mars, R. a.T., Williams, B. B., et al. (2020). Bacterially derived tryptamine increases mucus release by activating a host receptor in a mouse model of inflammatory bowel disease. iScience 23, 101798. doi:10.1016/j.isci.2020.101798
Bhattarai, Y., Williams, B. B., Battaglioli, E. J., Whitaker, W. R., Till, L., Grover, M., et al. (2018). Gut microbiota-produced tryptamine activates an epithelial G-protein-coupled receptor to increase colonic secretion. Cell Host Microbe 23, 775–785.e5. doi:10.1016/j.chom.2018.05.004
Borrero, J., Chen, Y., Dunny, G. M., and Kaznessis, Y. N. (2015). Modified lactic acid bacteria detect and inhibit multiresistant enterococci. ACS Synth. Biol. 4, 299–306. doi:10.1021/sb500090b
Brown, E. M., Ke, X., Hitchcock, D., Jeanfavre, S., Avila-Pacheco, J., Nakata, T., et al. (2019). Bacteroides-derived sphingolipids are critical for maintaining intestinal homeostasis and symbiosis. Cell Host Microbe 25, 668–680.e7. doi:10.1016/j.chom.2019.04.002
Buffie, C. G., Bucci, V., Stein, R. R., Mckenney, P. T., Ling, L., Gobourne, A., et al. (2015). Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature 517, 205–208. doi:10.1038/nature13828
Campbell, C., Mckenney, P. T., Konstantinovsky, D., Isaeva, O. I., Schizas, M., Verter, J., et al. (2020). Bacterial metabolism of bile acids promotes generation of peripheral regulatory T cells. Nature 581, 475–479. doi:10.1038/s41586-020-2193-0
Chan, C. T., Lee, J. W., Cameron, D. E., Bashor, C. J., and Collins, J. J. (2016). 'Deadman' and 'Passcode' microbial kill switches for bacterial containment. Nat. Chem. Biol. 12, 82–86. doi:10.1038/nchembio.1979
Chien, T., Harimoto, T., Kepecs, B., Gray, K., Coker, C., Hou, N., et al. (2022). Enhancing the tropism of bacteria via genetically programmed biosensors. Nat. Biomed. Eng. 6, 94–104. doi:10.1038/s41551-021-00772-3
Choe, D., Kim, K., Kang, M., Lee, S. G., Cho, S., Palsson, B., et al. (2022). Synthetic 3'-UTR valves for optimal metabolic flux control in Escherichia coli. Nucleic Acids Res. 50, 4171–4186. doi:10.1093/nar/gkac206
Claesson, M. J., Jeffery, I. B., Conde, S., Power, S. E., O'connor, E. M., Cusack, S., et al. (2012). Gut microbiota composition correlates with diet and health in the elderly. Nature 488, 178–184. doi:10.1038/nature11319
Collins, S. L., Stine, J. G., Bisanz, J. E., Okafor, C. D., and Patterson, A. D. (2023). Bile acids and the gut microbiota: metabolic interactions and impacts on disease. Nat. Rev. Microbiol. 21, 236–247. doi:10.1038/s41579-022-00805-x
Courbet, A., Endy, D., Renard, E., Molina, F., and Bonnet, J. (2015). Detection of pathological biomarkers in human clinical samples via amplifying genetic switches and logic gates. Sci. Transl. Med. 7, 289ra83. 289ra283. doi:10.1126/scitranslmed.aaa3601
Cullen, T. W., Schofield, W. B., Barry, N. A., Putnam, E. E., Rundell, E. A., Trent, M. S., et al. (2015). Antimicrobial peptide resistance mediates resilience of prominent gut commensals during inflammation. Science 347, 170–175. doi:10.1126/science.1260580
Curtis, M. M., Hu, Z., Klimko, C., Narayanan, S., Deberardinis, R., and Sperandio, V. (2014). The gut commensal Bacteroides thetaiotaomicron exacerbates enteric infection through modification of the metabolic landscape. Cell Host Microbe 16, 759–769. doi:10.1016/j.chom.2014.11.005
Daeffler, K. N., Galley, J. D., Sheth, R. U., Ortiz-Velez, L. C., Bibb, C. O., Shroyer, N. F., et al. (2017). Engineering bacterial thiosulfate and tetrathionate sensors for detecting gut inflammation. Mol. Syst. Biol. 13, 923. doi:10.15252/msb.20167416
Danino, T., Prindle, A., Kwong, G. A., Skalak, M., Li, H., Allen, K., et al. (2015). Programmable probiotics for detection of cancer in urine. Sci. Transl. Med. 7, 289ra84. 289ra284. doi:10.1126/scitranslmed.aaa3519
Delday, M., Mulder, I., Logan, E. T., and Grant, G. (2019). Bacteroides thetaiotaomicron ameliorates colon inflammation in preclinical models of crohn's disease. Inflamm. Bowel Dis. 25, 85–96. doi:10.1093/ibd/izy281
Dickson, I. (2019). Probiotics fail to improve preschool gastroenteritis. Nat. Rev. Gastroenterol. Hepatol. 16, 76. doi:10.1038/s41575-019-0104-3
Din, M. O., Danino, T., Prindle, A., Skalak, M., Selimkhanov, J., Allen, K., et al. (2016). Synchronized cycles of bacterial lysis for in vivo delivery. Nature 536, 81–85. doi:10.1038/nature18930
Dohnalova, L., Lundgren, P., Carty, J. R. E., Goldstein, N., Wenski, S. L., Nanudorn, P., et al. (2022). A microbiome-dependent gut-brain pathway regulates motivation for exercise. Nature 612, 739–747. doi:10.1038/s41586-022-05525-z
Dronkers, T. M. G., Ouwehand, A. C., and Rijkers, G. T. (2020). Global analysis of clinical trials with probiotics. Heliyon 6, e04467. doi:10.1016/j.heliyon.2020.e04467
Faith, J. J., Guruge, J. L., Charbonneau, M., Subramanian, S., Seedorf, H., Goodman, A. L., et al. (2013). The long-term stability of the human gut microbiota. Science 341, 1237439. doi:10.1126/science.1237439
Farrar, M. D., Whitehead, T. R., Lan, J., Dilger, P., Thorpe, R., Holland, K. T., et al. (2005). Engineering of the gut commensal bacterium Bacteroides ovatus to produce and secrete biologically active murine interleukin-2 in response to xylan. J. Appl. Microbiol. 98, 1191–1197. doi:10.1111/j.1365-2672.2005.02565.x
Flerlage, T., Brazelton De Cardenas, J. N., Garner, C. D., Hasan, N. A., Karathia, H., Qudeimat, A., et al. (2020). Multiple NDM-5-expressing Escherichia coli isolates from an immunocompromised pediatric host. Open Forum Infect. Dis. 7, ofaa018. ofaa018. doi:10.1093/ofid/ofaa018
Franzosa, E. A., Sirota-Madi, A., Avila-Pacheco, J., Fornelos, N., Haiser, H. J., Reinker, S., et al. (2019). Gut microbiome structure and metabolic activity in inflammatory bowel disease. Nat. Microbiol. 4, 293–305. doi:10.1038/s41564-018-0306-4
Fukuda, S., Toh, H., Hase, K., Oshima, K., Nakanishi, Y., Yoshimura, K., et al. (2011). Bifidobacteria can protect from enteropathogenic infection through production of acetate. Nature 469, 543–547. doi:10.1038/nature09646
Funabashi, M., Grove, T. L., Wang, M., Varma, Y., Mcfadden, M. E., Brown, L. C., et al. (2020). A metabolic pathway for bile acid dehydroxylation by the gut microbiome. Nature 582, 566–570. doi:10.1038/s41586-020-2396-4
Goodman, A. L., Mcnulty, N. P., Zhao, Y., Leip, D., Mitra, R. D., Lozupone, C. A., et al. (2009). Identifying genetic determinants needed to establish a human gut symbiont in its habitat. Cell Host Microbe 6, 279–289. doi:10.1016/j.chom.2009.08.003
Guandalini, S., and Sansotta, N. (2019). “Probiotics in the treatment of inflammatory bowel disease,” in Probiotics and child gastrointestinal health: advances in microbiology, infectious diseases and public health. Editors S. GUANDALINI, and F. INDRIO (Cham: Springer International Publishing), Vol. 10. doi:10.1007/5584_2018_319
Hamady, Z. Z., Scott, N., Farrar, M. D., Wadhwa, M., Dilger, P., Whitehead, T. R., et al. (2011). Treatment of colitis with a commensal gut bacterium engineered to secrete human tgf-β1 under the control of dietary xylan. Inflamm. Bowel Dis. 17, 1925–1935. doi:10.1002/ibd.21565
Hamady, Z. Z., Scott, N., Farrar, M. D., Lodge, J. P., Holland, K. T., Whitehead, T., et al. (2010). Xylan-regulated delivery of human keratinocyte growth factor-2 to the inflamed colon by the human anaerobic commensal bacterium Bacteroides ovatus. Gut 59, 461–469. doi:10.1136/gut.2008.176131
Hansen, R., Sanderson, I. R., Muhammed, R., Allen, S., Tzivinikos, C., Henderson, P., et al. (2021). A double-blind, placebo-controlled trial to assess safety and tolerability of (thetanix) Bacteroides thetaiotaomicron in adolescent crohn's disease. Clin. Transl. Gastroenterol. 12, e00287. doi:10.14309/ctg.0000000000000287
Harimoto, T., Hahn, J., Chen, Y. Y., Im, J., Zhang, J., Hou, N., et al. (2022). A programmable encapsulation system improves delivery of therapeutic bacteria in mice. Nat. Biotechnol. 40, 1259–1269. doi:10.1038/s41587-022-01244-y
Heinken, A., Hertel, J., Acharya, G., Ravcheev, D. A., Nyga, M., Okpala, O. E., et al. (2023). Genome-scale metabolic reconstruction of 7,302 human microorganisms for personalized medicine. Nat. Biotechnol. 41, 1320–1331. doi:10.1038/s41587-022-01628-0
Heinken, A., Ravcheev, D. A., Baldini, F., Heirendt, L., Fleming, R. M. T., and Thiele, I. (2019). Systematic assessment of secondary bile acid metabolism in gut microbes reveals distinct metabolic capabilities in inflammatory bowel disease. Microbiome 7, 75. doi:10.1186/s40168-019-0689-3
Heinken, A., Sahoo, S., Fleming, R. M., and Thiele, I. (2013). Systems-level characterization of a host-microbe metabolic symbiosis in the mammalian gut. Gut Microbes 4, 28–40. doi:10.4161/gmic.22370
Huang, B. D., Groseclose, T. M., and Wilson, C. J. (2022). Transcriptional programming in a Bacteroides consortium. Nat. Commun. 13, 3901. doi:10.1038/s41467-022-31614-8
Hwang, I. Y., Koh, E., Wong, A., March, J. C., Bentley, W. E., Lee, Y. S., et al. (2017). Engineered probiotic Escherichia coli can eliminate and prevent Pseudomonas aeruginosa gut infection in animal models. Nat. Commun. 8, 15028. doi:10.1038/ncomms15028
Icho, S., Ward, J. S., Tam, J., Kociolek, L. K., Theriot, C. M., and Melnyk, R. A. (2023). Intestinal bile acids provide a surmountable barrier against C. difficile TcdB-induced disease pathogenesis. Proc. Natl. Acad. Sci. U. S. A. 120, e2301252120. doi:10.1073/pnas.2301252120
Inda, M. E., Broset, E., Lu, T. K., and De La Fuente-Nunez, C. (2019). Emerging Frontiers in microbiome engineering. Trends Immunol. 40, 952–973. doi:10.1016/j.it.2019.08.007
Isabella, V. M., Ha, B. N., Castillo, M. J., Lubkowicz, D. J., Rowe, S. E., Millet, Y. A., et al. (2018). Development of a synthetic live bacterial therapeutic for the human metabolic disease phenylketonuria. Nat. Biotechnol. 36, 857–864. doi:10.1038/nbt.4222
Jayaraman, P., Holowko, M. B., Yeoh, J. W., Lim, S., and Poh, C. L. (2017). Repurposing a two-component system-based biosensor for the killing of Vibrio cholerae. ACS Synth. Biol. 6, 1403–1415. doi:10.1021/acssynbio.7b00058
Jiang, X., Hall, A. B., Arthur, T. D., Plichta, D. R., Covington, C. T., Poyet, M., et al. (2019). Invertible promoters mediate bacterial phase variation, antibiotic resistance, and host adaptation in the gut. Science 363, 181–187. doi:10.1126/science.aau5238
Jones, T. S., Oliveira, S. M. D., Myers, C. J., Voigt, C. A., and Densmore, D. (2022). Genetic circuit design automation with Cello 2.0. Nat. Protoc. 17, 1097–1113. doi:10.1038/s41596-021-00675-2
Kelly, D., Campbell, J. I., King, T. P., Grant, G., Jansson, E. A., Coutts, A. G., et al. (2004). Commensal anaerobic gut bacteria attenuate inflammation by regulating nuclear-cytoplasmic shuttling of PPAR-gamma and RelA. Nat. Immunol. 5, 104–112. doi:10.1038/ni1018
Khan, R., Roy, N., Ali, H., and Naeem, M. (2022). Fecal microbiota transplants for inflammatory bowel disease treatment: synthetic- and engineered communities-based microbiota transplants are the future. Gastroenterology Res. Pract. 2022, 1–9. doi:10.1155/2022/9999925
Kim, K., Choe, D., Song, Y., Kang, M., Lee, S. G., Lee, D. H., et al. (2021). Engineering Bacteroides thetaiotaomicron to produce non-native butyrate based on a genome-scale metabolic model-guided design. Metab. Eng. 68, 174–186. doi:10.1016/j.ymben.2021.10.005
Koeth, R. A., Wang, Z., Levison, B. S., Buffa, J. A., Org, E., Sheehy, B. T., et al. (2013). Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat. Med. 19, 576–585. doi:10.1038/nm.3145
Koh, E., Hwang, I. Y., Lee, H. L., De Sotto, R., Lee, J. W. J., Lee, Y. S., et al. (2022). Engineering probiotics to inhibit Clostridioides difficile infection by dynamic regulation of intestinal metabolism. Nat. Commun. 13, 3834. doi:10.1038/s41467-022-31334-z
Kurtz, C. B., Millet, Y. A., Puurunen, M. K., Perreault, M., Charbonneau, M. R., Isabella, V. M., et al. (2019). An engineered E. coli Nissle improves hyperammonemia and survival in mice and shows dose-dependent exposure in healthy humans. Sci. Transl. Med. 11, eaau7975. doi:10.1126/scitranslmed.aau7975
Lai, Y., Hayashi, N., and Lu, T. K. (2022). Engineering the human gut commensal Bacteroides thetaiotaomicron with synthetic biology. Curr. Opin. Chem. Biol. 70, 102178. doi:10.1016/j.cbpa.2022.102178
Lawson, C. E., Harcombe, W. R., Hatzenpichler, R., Lindemann, S. R., Loffler, F. E., O'malley, M. A., et al. (2019). Common principles and best practices for engineering microbiomes. Nat. Rev. Microbiol. 17, 725–741. doi:10.1038/s41579-019-0255-9
Lee, S. M., Donaldson, G. P., Mikulski, Z., Boyajian, S., Ley, K., and Mazmanian, S. K. (2013). Bacterial colonization factors control specificity and stability of the gut microbiota. Nature 501, 426–429. doi:10.1038/nature12447
Leonard, M. M., Karathia, H., Pujolassos, M., Troisi, J., Valitutti, F., Subramanian, P., et al. (2020). Multi-omics analysis reveals the influence of genetic and environmental risk factors on developing gut microbiota in infants at risk of celiac disease. Microbiome 8, 130. doi:10.1186/s40168-020-00906-w
Leonard, M. M., Kenyon, V., Valitutti, F., Pennacchio-Harrington, R., Piemontese, P., Francavilla, R., et al. (2023). Cohort profile: celiac disease genomic, environmental, microbiome and metabolome study; a prospective longitudinal birth cohort study of children at-risk for celiac disease. PLoS One 18, e0282739. doi:10.1371/journal.pone.0282739
Leonard, M. M., Valitutti, F., Karathia, H., Pujolassos, M., Kenyon, V., Fanelli, B., et al. (2021). Microbiome signatures of progression toward celiac disease onset in at-risk children in a longitudinal prospective cohort study. Proc. Natl. Acad. Sci. U. S. A. 118, e2020322118. doi:10.1073/pnas.2020322118
Leschner, S., Deyneko, I. V., Lienenklaus, S., Wolf, K., Bloecker, H., Bumann, D., et al. (2012). Identification of tumor-specific Salmonella Typhimurium promoters and their regulatory logic. Nucleic Acids Res. 40, 2984–2994. doi:10.1093/nar/gkr1041
Leventhal, D. S., Sokolovska, A., Li, N., Plescia, C., Kolodziej, S. A., Gallant, C. W., et al. (2020). Immunotherapy with engineered bacteria by targeting the STING pathway for anti-tumor immunity. Nat. Commun. 11, 2739. doi:10.1038/s41467-020-16602-0
Liu, H., Shiver, A. L., Price, M. N., Carlson, H. K., Trotter, V. V., Chen, Y., et al. (2021). Functional genetics of human gut commensal Bacteroides thetaiotaomicron reveals metabolic requirements for growth across environments. Cell Rep. 34, 108789. doi:10.1016/j.celrep.2021.108789
Lou, C., Stanton, B., Chen, Y. J., Munsky, B., and Voigt, C. A. (2012). Ribozyme-based insulator parts buffer synthetic circuits from genetic context. Nat. Biotechnol. 30, 1137–1142. doi:10.1038/nbt.2401
Lubkowicz, D., Ho, C. L., Hwang, I. Y., Yew, W. S., Lee, Y. S., and Chang, M. W. (2018). Reprogramming probiotic lactobacillus reuteri as a biosensor for Staphylococcus aureus derived AIP-I detection. ACS Synth. Biol. 7, 1229–1237. doi:10.1021/acssynbio.8b00063
Lubkowicz, D., Horvath, N. G., James, M. J., Cantarella, P., Renaud, L., Bergeron, C. G., et al. (2022). An engineered bacterial therapeutic lowers urinary oxalate in preclinical models and in silico simulations of enteric hyperoxaluria. Mol. Syst. Biol. 18, e10539. doi:10.15252/msb.202110539
Luis, A. S., Briggs, J., Zhang, X., Farnell, B., Ndeh, D., Labourel, A., et al. (2018). Dietary pectic glycans are degraded by coordinated enzyme pathways in human colonic Bacteroides. Nat. Microbiol. 3, 210–219. doi:10.1038/s41564-017-0079-1
Lynch, J. P., Goers, L., and Lesser, C. F. (2022). Emerging strategies for engineering Escherichia coli Nissle 1917-based therapeutics. Trends Pharmacol. Sci. 43, 772–786. doi:10.1016/j.tips.2022.02.002
Mao, N., Cubillos-Ruiz, A., Cameron, D. E., and Collins, J. J. (2018). Probiotic strains detect and suppress cholera in mice. Sci. Transl. Med. 10, eaao2586. doi:10.1126/scitranslmed.aao2586
Martens, E. C., Chiang, H. C., and Gordon, J. I. (2008). Mucosal glycan foraging enhances fitness and transmission of a saccharolytic human gut bacterial symbiont. Cell Host Microbe 4, 447–457. doi:10.1016/j.chom.2008.09.007
Martens, E. C., Lowe, E. C., Chiang, H., Pudlo, N. A., Wu, M., Mcnulty, N. P., et al. (2011). Recognition and degradation of plant cell wall polysaccharides by two human gut symbionts. PLoS Biol. 9, e1001221. doi:10.1371/journal.pbio.1001221
Mastropaolo, M. D., Thorson, M. L., and Stevens, A. M. (2009). Comparison of Bacteroides thetaiotaomicron and Escherichia coli 16S rRNA gene expression signals. Microbiol. Read. 155, 2683–2693. doi:10.1099/mic.0.027748-0
Mcclure, S., Enam, F., Arnold, J., and Mimee, M. (2022). Proceedings from the 3rd international conference on microbiome engineering. Biotechnol. Prog. 38, e3241. doi:10.1002/btpr.3241
Mcdonald, L. C., Gerding, D. N., Johnson, S., Bakken, J. S., Carroll, K. C., Coffin, S. E., et al. (2018). Clinical practice guidelines for Clostridium difficile infection in adults and children: 2017 update by the infectious diseases society of America (IDSA) and society for healthcare epidemiology of America (SHEA). Clin. Infect. Dis. 66, e1–e48. doi:10.1093/cid/cix1085
Meyer, A. J., Segall-Shapiro, T. H., Glassey, E., Zhang, J., and Voigt, C. A. (2019). Escherichia coli “Marionette” strains with 12 highly optimized small-molecule sensors. Nat. Chem. Biol. 15, 196–204. doi:10.1038/s41589-018-0168-3
Mimee, M., Nadeau, P., Hayward, A., Carim, S., Flanagan, S., Jerger, L., et al. (2018). An ingestible bacterial-electronic system to monitor gastrointestinal health. Science 360, 915–918. doi:10.1126/science.aas9315
Mimee, M., Tucker, A. C., Voigt, C. A., and Lu, T. K. (2015). Programming a human commensal bacterium, Bacteroides thetaiotaomicron, to sense and respond to stimuli in the murine gut microbiota. Cell Syst. 1, 62–71. doi:10.1016/j.cels.2015.06.001
Monk, J. M., Lloyd, C. J., Brunk, E., Mih, N., Sastry, A., King, Z., et al. (2017). iML1515, a knowledgebase that computes Escherichia coli traits. Nat. Biotechnol. 35, 904–908. doi:10.1038/nbt.3956
Mullard, A. (2023). FDA approves second microbiome-based C. difficile therapy. Nat. Rev. Drug Discov. 22, 436. doi:10.1038/d41573-023-00081-1
Nielsen, A. a.K., Der, B. S., Shin, J., Vaidyanathan, P., Paralanov, V., Strychalski, E. A., et al. (2016). Genetic circuit design automation. Science 352, aac7341. doi:10.1126/science.aac7341
Orth, J. D., Thiele, I., and Palsson, B. O. (2010). What is flux balance analysis? Nat. Biotechnol. 28, 245–248. doi:10.1038/nbt.1614
Park, Y., Espah Borujeni, A., Gorochowski, T. E., Shin, J., and Voigt, C. A. (2020). Precision design of stable genetic circuits carried in highly-insulated E. coli genomic landing pads. Mol. Syst. Biol. 16, e9584. doi:10.15252/msb.20209584
Pascal Andreu, V., Augustijn, H. E., Chen, L., Zhernakova, A., Fu, J., Fischbach, M. A., et al. (2023). gutSMASH predicts specialized primary metabolic pathways from the human gut microbiota. Nat. Biotechnol. doi:10.1038/s41587-023-01675-1
Pascal Andreu, V., Roel-Touris, J., Dodd, D., Fischbach, M. A., and Medema, M. H. (2021). The gutSMASH web server: automated identification of primary metabolic gene clusters from the gut microbiota. Nucleic Acids Res. 49, W263–W270. doi:10.1093/nar/gkab353
Perreault, M., Means, J., Gerson, E., Lee, D., Horvath, N., Rajasuriyar, A., et al. (2021). Development of an investigational methionine-consuming synthetic biotic medicine (SYNB1353) for the treatment of homocystinuria. Sydney, Australia: 14th International Congress of Inborn Errors of Metabolism.
Petrof, E. O., Gloor, G. B., Vanner, S. J., Weese, S. J., Carter, D., Daigneault, M. C., et al. (2013). Stool substitute transplant therapy for the eradication of Clostridium difficile infection: 'RePOOPulating' the gut. Microbiome 1, 3. doi:10.1186/2049-2618-1-3
Pickard, J. M., Maurice, C. F., Kinnebrew, M. A., Abt, M. C., Schenten, D., Golovkina, T. V., et al. (2014). Rapid fucosylation of intestinal epithelium sustains host-commensal symbiosis in sickness. Nature 514, 638–641. doi:10.1038/nature13823
Plovier, H., Everard, A., Druart, C., Depommier, C., Van Hul, M., Geurts, L., et al. (2017). A purified membrane protein from Akkermansia muciniphila or the pasteurized bacterium improves metabolism in obese and diabetic mice. Nat. Med. 23, 107–113. doi:10.1038/nm.4236
Porter, N. T., Hryckowian, A. J., Merrill, B. D., Fuentes, J. J., Gardner, J. O., Glowacki, R. W. P., et al. (2020). Phase-variable capsular polysaccharides and lipoproteins modify bacteriophage susceptibility in Bacteroides thetaiotaomicron. Nat. Microbiol. 5, 1170–1181. doi:10.1038/s41564-020-0746-5
Puurunen, M. K., Vockley, J., Searle, S. L., Sacharow, S. J., Phillips, J. A., Denney, W. S., et al. (2021). Safety and pharmacodynamics of an engineered E. coli Nissle for the treatment of phenylketonuria: a first-in-human phase 1/2a study. Nat. Metab. 3, 1125–1132. doi:10.1038/s42255-021-00430-7
Riglar, D. T., Giessen, T. W., Baym, M., Kerns, S. J., Niederhuber, M. J., Bronson, R. T., et al. (2017). Engineered bacteria can function in the mammalian gut long-term as live diagnostics of inflammation. Nat. Biotechnol. 35, 653–658. doi:10.1038/nbt.3879
Russell, B. J., Brown, S. D., Siguenza, N., Mai, I., Saran, A. R., Lingaraju, A., et al. (2022). Intestinal transgene delivery with native E. coli chassis allows persistent physiological changes. Cell 185, 3263–3277.e15. doi:10.1016/j.cell.2022.06.050
Ryan, D., Jenniches, L., Reichardt, S., Barquist, L., and Westermann, A. J. (2020). A high-resolution transcriptome map identifies small RNA regulation of metabolism in the gut microbe Bacteroides thetaiotaomicron. Nat. Commun. 11, 3557. doi:10.1038/s41467-020-17348-5
Rychel, K., Decker, K., Sastry, A. V., Phaneuf, P. V., Poudel, S., and Palsson, B. O. (2021). iModulonDB: a knowledgebase of microbial transcriptional regulation derived from machine learning. Nucleic Acids Res. 49, D112–D120. doi:10.1093/nar/gkaa810
Saeidi, N., Wong, C. K., Lo, T. M., Nguyen, H. X., Ling, H., Leong, S. S., et al. (2011). Engineering microbes to sense and eradicate Pseudomonas aeruginosa, a human pathogen. Mol. Syst. Biol. 7, 521. doi:10.1038/msb.2011.55
Salyers, A. A., Bonheyo, G., and Shoemaker, N. B. (2000). Starting a new genetic system: lessons from bacteroides. Methods 20, 35–46. doi:10.1006/meth.1999.0903
Sassone-Corsi, M., Nuccio, S. P., Liu, H., Hernandez, D., Vu, C. T., Takahashi, A. A., et al. (2016). Microcins mediate competition among Enterobacteriaceae in the inflamed gut. Nature 540, 280–283. doi:10.1038/nature20557
Sastry, A. V., Gao, Y., Szubin, R., Hefner, Y., Xu, S., Kim, D., et al. (2019). The Escherichia coli transcriptome mostly consists of independently regulated modules. Nat. Commun. 10, 5536. doi:10.1038/s41467-019-13483-w
Sato, Y., Atarashi, K., Plichta, D. R., Arai, Y., Sasajima, S., Kearney, S. M., et al. (2021). Novel bile acid biosynthetic pathways are enriched in the microbiome of centenarians. Nature 599, 458–464. doi:10.1038/s41586-021-03832-5
Schultz, M. (2008). Clinical use of E. coli Nissle 1917 in inflammatory bowel disease. Inflamm. Bowel Dis. 14, 1012–1018. doi:10.1002/ibd.20377
Sender, R., Fuchs, S., and Milo, R. (2016). Revised estimates for the number of human and bacteria cells in the body. PLoS Biol. 14, e1002533. doi:10.1371/journal.pbio.1002533
Sharma, C. M., Hoffmann, S., Darfeuille, F., Reignier, J., Findeiss, S., Sittka, A., et al. (2010). The primary transcriptome of the major human pathogen Helicobacter pylori. Nature 464, 250–255. doi:10.1038/nature08756
Shepherd, E. S., Deloache, W. C., Pruss, K. M., Whitaker, W. R., and Sonnenburg, J. L. (2018). An exclusive metabolic niche enables strain engraftment in the gut microbiota. Nature 557, 434–438. doi:10.1038/s41586-018-0092-4
Shin, J., Bae, J., Lee, H., Kang, S., Jin, S., Song, Y., et al. (2023). Genome-wide CRISPRi screen identifies enhanced autolithotrophic phenotypes in acetogenic bacterium Eubacterium limosum. Proc. Natl. Acad. Sci. U. S. A. 120, e2216244120. doi:10.1073/pnas.2216244120
Sims, M. D., Khanna, S., Feuerstadt, P., Louie, T. J., Kelly, C. R., Huang, E. S., et al. (2023). Safety and tolerability of SER-109 as an investigational microbiome therapeutic in adults with recurrent Clostridioides difficile infection: a phase 3, open-label, single-arm trial. JAMA Netw. Open 6, e2255758. doi:10.1001/jamanetworkopen.2022.55758
Smith, M. I., Yatsunenko, T., Manary, M. J., Trehan, I., Mkakosya, R., Cheng, J., et al. (2013). Gut microbiomes of Malawian twin pairs discordant for kwashiorkor. Science 339, 548–554. doi:10.1126/science.1229000
Song, Y., Bae, J., Jin, S., Lee, H., Kang, S., Lee, J., et al. (2022). Development of highly characterized genetic bioparts for efficient gene expression in CO(2)-fixing Eubacterium limosum. Metab. Eng. 72, 215–226. doi:10.1016/j.ymben.2022.03.016
Steidler, L., Hans, W., Schotte, L., Neirynck, S., Obermeier, F., Falk, W., et al. (2000). Treatment of murine colitis by Lactococcus lactis secreting interleukin-10. Science 289, 1352–1355. doi:10.1126/science.289.5483.1352
Taketani, M., Donia, M. S., Jacobson, A. N., Lambris, J. D., and Fischbach, M. A. (2015). A phase-variable surface layer from the gut symbiont Bacteroides thetaiotaomicron. mBio 6, e01339–e01315. doi:10.1128/mBio.01339-15
Taketani, M., Zhang, J., Zhang, S., Triassi, A. J., Huang, Y. J., Griffith, L. G., et al. (2020). Genetic circuit design automation for the gut resident species Bacteroides thetaiotaomicron. Nat. Biotechnol. 38, 962–969. doi:10.1038/s41587-020-0468-5
Tanoue, T., Morita, S., Plichta, D. R., Skelly, A. N., Suda, W., Sugiura, Y., et al. (2019). A defined commensal consortium elicits CD8 T cells and anti-cancer immunity. Nature 565, 600–605. doi:10.1038/s41586-019-0878-z
The Integrative, H. M. P. R. N. C. (2019). The integrative human microbiome project. Nature 569, 641–648. doi:10.1038/s41586-019-1238-8
Theriot, C. M., Bowman, A. A., and Young, V. B. (2016). Antibiotic-induced alterations of the gut microbiota alter secondary bile acid production and allow for Clostridium difficile spore germination and outgrowth in the large intestine. mSphere 1, e00045-15. doi:10.1128/mSphere.00045-15
Tierney, B. T., Yang, Z., Luber, J. M., Beaudin, M., Wibowo, M. C., Baek, C., et al. (2019). The landscape of genetic content in the gut and oral human microbiome. Cell Host Microbe 26, 283–295.e8. doi:10.1016/j.chom.2019.07.008
Triassi, A. J., Fields, B. D., Monahan, C. E., Means, J. M., Park, Y., Doosthosseini, H., et al. (2023). Redesign of an Escherichia coli Nissle treatment for phenylketonuria using insulated genomic landing pads and genetic circuits to reduce burden. Cell Syst. 14, 512–524.e12. doi:10.1016/j.cels.2023.05.004
Turnbaugh, P. J., Hamady, M., Yatsunenko, T., Cantarel, B. L., Duncan, A., Ley, R. E., et al. (2009). A core gut microbiome in obese and lean twins. Nature 457, 480–484. doi:10.1038/nature07540
Wexler, A. G., and Goodman, A. L. (2017). An insider's perspective: bacteroides as a window into the microbiome. Nat. Microbiol. 2, 17026. doi:10.1038/nmicrobiol.2017.26
Wikoff, W. R., Anfora, A. T., Liu, J., Schultz, P. G., Lesley, S. A., Peters, E. C., et al. (2009). Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc. Natl. Acad. Sci. U. S. A. 106, 3698–3703. doi:10.1073/pnas.0812874106
Winter, S. E., Thiennimitr, P., Winter, M. G., Butler, B. P., Huseby, D. L., Crawford, R. W., et al. (2010). Gut inflammation provides a respiratory electron acceptor for Salmonella. Nature 467, 426–429. doi:10.1038/nature09415
Winter, S. E., Winter, M. G., Xavier, M. N., Thiennimitr, P., Poon, V., Keestra, A. M., et al. (2013). Host-derived nitrate boosts growth of E. coli in the inflamed gut. Science 339, 708–711. doi:10.1126/science.1232467
Woo, S. G., Moon, S. J., Kim, S. K., Kim, T. H., Lim, H. S., Yeon, G. H., et al. (2020). A designed whole-cell biosensor for live diagnosis of gut inflammation through nitrate sensing. Biosens. Bioelectron. 168, 112523. doi:10.1016/j.bios.2020.112523
Wu, M., Mcnulty, N. P., Rodionov, D. A., Khoroshkin, M. S., Griffin, N. W., Cheng, J., et al. (2015). Genetic determinants of in vivo fitness and diet responsiveness in multiple human gut Bacteroides. Science 350, aac5992. doi:10.1126/science.aac5992
Xia, J. Y., Hepler, C., Tran, P., Waldeck, N. J., Bass, J., and Prindle, A. (2023). Engineered calprotectin-sensing probiotics for IBD surveillance in humans. Proc. Natl. Acad. Sci. U. S. A. 120, e2221121120. doi:10.1073/pnas.2221121120
Xu, J., Bjursell, M. K., Himrod, J., Deng, S., Carmichael, L. K., Chiang, H. C., et al. (2003). A genomic view of the human-Bacteroides thetaiotaomicron symbiosis. Science 299, 2074–2076. doi:10.1126/science.1080029
Yang, S., Wang, S., Wang, Y., Rong, R., Kim, J., Li, B., et al. (2022). MB-SupCon: microbiome-based predictive models via supervised contrastive learning. J. Mol. Biol. 434, 167693. doi:10.1016/j.jmb.2022.167693
Yaung, S. J., Deng, L., Li, N., Braff, J. L., Church, G. M., Bry, L., et al. (2015). Improving microbial fitness in the mammalian gut by in vivo temporal functional metagenomics. Mol. Syst. Biol. 11, 788. doi:10.15252/msb.20145866
Yu, T., Boob, A. G., Singh, N., Su, Y., and Zhao, H. (2023). In vitro continuous protein evolution empowered by machine learning and automation. Cell Syst. 14, 633–644. doi:10.1016/j.cels.2023.04.006
Zhu, W., Winter, M. G., Byndloss, M. X., Spiga, L., Duerkop, B. A., Hughes, E. R., et al. (2018). Precision editing of the gut microbiota ameliorates colitis. Nature 553, 208–211. doi:10.1038/nature25172
Zimmermann, M., Zimmermann-Kogadeeva, M., Wegmann, R., and Goodman, A. L. (2019). Mapping human microbiome drug metabolism by gut bacteria and their genes. Nature 570, 462–467. doi:10.1038/s41586-019-1291-3
Zmora, N., Zilberman-Schapira, G., Suez, J., Mor, U., Dori-Bachash, M., Bashiardes, S., et al. (2018). Personalized gut mucosal colonization resistance to empiric probiotics is associated with unique host and microbiome features. Cell 174, 1388–1405.e21. doi:10.1016/j.cell.2018.08.041
Keywords: live biotherapeutics, probiotics, microbiome, synthetic biology, systems biology
Citation: Kim K, Kang M and Cho B-K (2023) Systems and synthetic biology-driven engineering of live bacterial therapeutics. Front. Bioeng. Biotechnol. 11:1267378. doi: 10.3389/fbioe.2023.1267378
Received: 26 July 2023; Accepted: 09 October 2023;
Published: 19 October 2023.
Edited by:
In Young Hwang, Singapore Institute of Technology, SingaporeReviewed by:
Adison Choonkit Wong, Singapore Institute of Technology, SingaporeHiren Karathia, Greenwood Genetic Center, United States
Ho Chun Loong, Southern University of Science and Technology, China
Copyright © 2023 Kim, Kang and Cho. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Byung-Kwan Cho, YmNob0BrYWlzdC5hYy5rcg==