
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
BRIEF RESEARCH REPORT article
Front. Bioeng. Biotechnol. , 20 March 2023
Sec. Biomaterials
Volume 11 - 2023 | https://doi.org/10.3389/fbioe.2023.1101513
This article is part of the Research Topic Biofunctional Materials and Coatings for Orthopeadic and Dental Applications View all 8 articles
 Caroline Richard1*
Caroline Richard1* Mélissa Alfred-Arulrasa1
Mélissa Alfred-Arulrasa1 Harikrishnan Ramadas2Pubudu Tharanga Mahagamage1
Harikrishnan Ramadas2Pubudu Tharanga Mahagamage1 Thomas Defforge1Gael Gaultier1Cécile Autret-Lambert1
Thomas Defforge1Gael Gaultier1Cécile Autret-Lambert1 Nathalie Poirot1Eric Champion3
Nathalie Poirot1Eric Champion3 Amandine Magnaudeix3
Amandine Magnaudeix3The purpose of the study was to investigate the synthesis of economic calcium phosphate powders from recycled oyster shells, using a ball milling method. The oyster shell powder and a calcium pyrophosphate powder were used as starting materials and ball milled, then heat treated at 1,050°C for 5 h to produce calcium phosphate powders through a solid-state reaction. Electrochemically synthesized mesoporous silicon microparticles were then added to the prepared phosphate powders by mechanical mixer. The final powders were characterized using X-ray diffraction, Fourier transform infrared spectroscopy, and scanning electron microscopy to analyze their chemical composition and determine the most suitable process conditions. The biocompatibility of the produced powders was also tested in vitro using murine cells and the results showed good biocompatibility.
Every year millions patients worldwide sustain bone grafting procedures to repair bone defects stemming from diseases (cancer, infections, osteoporosis …) or traumatic events. Mammalian bone tissue is composed of about 70 wt% inorganic mineralized calcium phosphate in the form of biological hydroxyapatite (HAp). A first experiment of the use of calcium phosphate for a fracture treatment dates back a century (Habraken et al., 2016; Bobbio, 1972). Bone-like chemical composition, microporosity, specific surface area, crystallinity, crystal size, and surface roughness are important factors to consider, as are the properties of autologous and allogenic bone grafts. Thus, the ideal bone substitute (Jordana et al., 2017) should be biocompatible, bioresorbable, easy to use, inexpensive, while exhibiting i) a structural integrity similar to bone, ii) an osteoconductive matrix, and iii) contain osteoinductive factors, capable of recruiting and stimulating osteoforming cells for bone repair and regeneration. Bone tissue engineering is based on the synergy between an appropriate substitution biomaterial and cells (Jordana et al., 2017). Hydroxyapatite (HAp–Ca10(PO4)6(OH)2), Tricalcium Phosphate (TCP–Ca3(PO4)2) and Biphasic Calcium Phosphates (BCP, HAp + TCP mixtures) are the commonly used biomaterials in bone surgery. Most of the time, these compounds are of synthetic origin. However, biological or organic HAp can be found from heat-treated bovine cancellous bone. Recent studies have managed to create lower size nano hydroxyapatite by sol-gel method. Nano hydroxyapatite generated more interest in bone tissue engineering because of its size, similar to bone cells (Habraken et al., 2016). Mainly used for filling, HAp is osteoconductive by providing a support for bone cells and bone morphogenetic proteins. In contact with healthy bone, osteoid tissue is formed and then mineralized, with the progressive acquisition of a mechanical strength similar to that of bone. HAp can be also employed in various applications in medical industry: HA coated titanium and stainless-steel parts often used in clinical applications. These HA coated metal parts show low rejection rate and higher biocompatibility. Tooth Enamel contains 97%wt of nano-hydroxyapatite. A recent study shows that nano-hydroxyapatite was successfully used in enamel repair (Habraken et al., 2016).
TCP (Ca/P = 1.5) has two crystalline forms: ɑ-TCP and β-TCP. TCP is considered as the first resorbable bioceramic. β- TCP is the most used and can be converted to biological HAp in vivo. Wu et al., 2011a reported the order of the relative solubility of the bioceramics as ɑ-TCP > β-TCP >> HAp; Dorozhkin, (2009) pointed out TCP cannot be precipitated from aqueous solutions. The formation of β-TCP takes place at about 1,050°C in the solid route process for example from an equimolar blend of CaCO3 and Ca2P2O7. TCP can be also formed by thermal decomposition above 700°C of a precursor Ca-deficient hydroxyapatite obtained by aqueous precipitation with proper Ca/P of 1.5, also called apatitic tricalcium phosphate (Destainville et al., 2003).
The BCPs combine the physicochemical properties of each component. This allows a controlled bioactivity by modulating the HAp/TCP ratio.
Since the beginning of the 2000s, numerous studies demonstrated the capacity of the nacre from molluscan shells as an interesting natural substitution biomaterials versus the usually used synthetic biomaterials (Chen et al., 2016; Brundavanam et al., 2017; Jordana et al., 2017).
The Pacific oyster or Japanese oyster, Magallana Gigas or. Crassostrea Gigas (Thurnberg, 1793) was introduced in France at the end of the 1960s. This oyster is distinguished from those of most bivalves by a very flaky appearance, and by its material. It is made of almost pure calcite: 91–95%wt CaCO3 (containing about 40% of Calcium), 4%wt organic matter and small amount of oxides (Ho et al., 2021; Rujitanapanich et al. (2014)). Ramakrishna et al., 2018 stated that the oyster shell is usually composed of 90% calcite and 10% aragonite (aragonite transforms to calcite very slowly at room temperature). This oyster is widely farmed, especially in France, to such an extent that it represents 75% of the European production but only 2% of the world farming production (more than 4 million tons per year). This makes it the most commercially important tasting oyster species, with a world market share of 93.7% (Silva et al., 2019). Each year, oyster breeding in France generates about 140,000 tons of waste products. The major French oyster farming regions (Normandy, Brittany, Occitania.) have quite recently started to organise the collection of this biogenic waste with high diversified recycling potential.
The use of bone substitutes is constantly increasing thanks to the recent development of injectable forms. Several bone substitutes for surgical indications, combining growth factors and osteogenic cells to induce true bone tissue regeneration have been investigated or are proposed. It is often a question of adding a certain number of organic or inorganic compounds to boost osteoinduction (as PRP—Platelet Rich Plasma and PFR—platelet Rich Fibrin), or to limit the risks of sepsis (with Silver or Strontium) (Faiz Mohd Yusoff et al., 2021). Innovative bioceramics can be developed with association of Silicon for example. Silicon is a trace element found in the body. Although it is not an essential trace element, it is important for the immune system and for maintaining good bone health. Indeed, Silicon plays a role in the binding of calcium or bone calcification and is found in the osteoid border where bone is built (Camaioni et al., 2015). The release of silicon ions contributes to increase the multiplication and the differentiation of bone cells for a healing of shorter duration (Salonen and Lehto, 2008). Chadwick et al. (2010) indicated the first studies in 1995 concerning the dissolution of porous silicon (PS) in SBF (Simulated Body Fluid). They showed and confirmed the growth of HAp on the nanoporous silicon particles and proved that PS can be a real bioactive material. In the same way, Bowditch et al. (1998); Low and Voelcker. (2014); Dalilottojari, et al. (2015) reported the biocompatible, biodegradable and bioactive properties of PS with size pore tunability, high surface area, and opened outlooks in different fields as tissue engineering, drug delivery, oncology, orthopedics, ophthalmology and even in vivo/in vitro biosensors or biomarkers (Hernández-Montelongo et al., 2015).
As HAp is the most widely used biomaterial for bone repairs, various techniques have been developed to produce HAp powders. Two main routes are possible: wet route and solid-route. Different processes are possible as: solid-state, mechanochemical, conventional chemical precipitation, hydrolysis, sol-gel, hydrothermal method, emulsion method, sonochemical method, combustion and even pyrolysis. Zaman et al., 2020 have identified the advantages and disadvantages of each method. Nevertheless, the solid-state method is less frequently reported although it is relatively simple and inexpensive compared to the wet treatment methods. The major advantage of solid state reaction method is that the reactions are faster and there is no need to adjust the pH value as in the case of precipitation and other wet methods (Ho et al., 2013). It could be an effective route to obtained nanosized HAp and control of the size and morphology of the particles. The disadvantages mainly concern the control of the purity (contaminations) and, the formation of agglomerates. Rhee (2002) and Wu et al., 2011b demonstrated the feasibility of producing HAp from oyster shell powder by a detailed mechanochemical technique but without biocompatibility tests.
Our investigation focused on various synthesis conditions of HAp from recycled oyster shells (calcination temperature, heat treatment duration, milling time) using a mechanochemical way and solid state reaction to produce BCP powders from oyster shells. In order to improve the biological behaviour and extend the potential applications, addition of mesoporous silicon particles as source of silicon and possible drug inclusion in their pores, was also investigated. In vitro assays using a MC3T3-E1 pre-osteoblast cell line were carried out to investigate the biological behaviour of the materials.
Oyster shell powders (OSP) were supplied from Ovive SA (France), an industrial actor committed to the environment both through its actions of recycling shellfish waste. The origin of oyster shells is the Atlantic coast of France. The powders were specially conditioned (cleaned, dried and then crushed by Ovive SA) to produce the final product for medical applications. The particle size was in the range of 300–400 μm. This narrow size distribution was selected for more uniformity.
SEM and XRD analysis were conducted for OSP and for the produced calcium phosphate powders. X-ray diffraction (XRD) analysis was carried out using a Bruker D8 Advance Diffractometer to examine the phase purity, crystallinity and lattice parameters of calcium phosphate powders. XRD patterns were collected from 2θ: 20°–60° with steps of 0.02° using a Cu- Kɑ radiation (λ= 0.154 nm). Powder diffraction files (PDF, detailed in Figures 2, 5) from the International Centre for Diffraction Data (ICDD) were employed as the reference patterns to identify the crystalline phases. Microstructure observations of the initial powders and treated samples were done using Scanning Electron Microscope (SEM) (ZEISS Gemini series, Germany). The powders were coated with a 5–8 nm layer of gold-palladium using a Precision Etching System (Gatan Ametek, United States) to improve imaging. The elemental chemical compositions of OSP and also of the sintered powders were determined by Energy Dispersive X-ray spectroscopy (EDS) analysis.
FT-IR analysis was conducted on raw oyster shell powder samples using a Jasco model 4,600 (Japan) apparatus with the HATR (Horizontal Attenuated Total Reflectance) technique and a compression cell. The samples were analyzed in the range of 500–4,000 cm−1. The powders were mixed with KBr and pressed into pellets for analysis. A thermal analysis TGA/DTA was performed using a STA regulus 2,500 (Netzsch, Germany) facility under a nitrogen atmosphere with a heating rate of 10°C/min from 20°C to 1,000°C.
Granulometry analysis was performed with a Zetasizer Nano-ZS (Malvern Panalytical, Netherlands, red Laser) by Dynamic Light Scattering (DLS) in alcoholic medium (isopropanol) at room temperature. Hydrodynamic diameters of the particles were determined by Stokes-Einstein equation. Refractive index was 1.44 for porous silicon (Astrova and Tolmachev, 2000) and 1.67 for HAp.
The as received OSP was mixed with calcium pyrophosphate (Ca2P2O7) supplied by Santacruz Biotechnology (France) in the molar ratio 4:3 (Ca/P ratio in stoichiometric HAp). For the synthesis of approximatively 10 g of sample, 7.59 g of Ca2P2O7 and 3.98 g of OSP were mixed with 8 mL of deionized water to avoid wear and debris of stainless steel balls and surface of stainless steel container during milling process. A Fritsch pulverisette 6 (classic line) planetary ball milling machine was used for milling the samples. 100 g of Fritsch stainless steel balls (5 mm diameter) were employed so that the weight ratio of powder to that of balls remains at 1:10 (capacity of container: 80 mL). The samples were milled for 18 h at 3 RCF (g). The obtained pastes were then dried in an oven at 150°C for 3 h. The dried pastes were heat treated, at a rate of 10°C/min, at 1,100°C during 5 h. Then they were milled at 4.5 RCF (g) during 1 h to pulverize them. The obtained powder was coded as M18H5. In addition, to facilitate the mechanical milling process and eliminate aggregates, the dried pastes were ground by hand in an agate mortar both before and after the milling step.
The choice of the temperature and duration of heat treatment were determined by literature review and previous studies. Increasing the temperature of the heat treatment increases generally the amount of HAp. The ideal temperature for the synthesis is set between 1,000°C and 1100°C (the formation of β-TCP takes place at about 1,050°C). For the experiments, the temperature was chosen to optimize the formation of HAp and β-TCP. A long heating time allows the complete transformation of CaCO3 and Ca2O7P2 for a final product without impurities. According to literature, the heating time can vary between 3 and 5 h depending on the source. Therefore, the longer time of 5 h was chosen.
Mesoporous silicon was produced by using the electrochemical etching technique. The cell was made of two platinum electrodes placed on each side of a p-type silicon wafer (10–20 mΩ·cm) that was oriented in the (100) direction and immersed in an aqueous solution containing hydrofluoric acid (HF). The polarization of the electrodes enabled the one of the wafer, defining the anodic (porous) and cathodic sides. The electrolyte consisted of 30% HF, 25% acetic acid, and 45% deionized water (in weight). The anodizing treatment was performed at a constant current density of 30 mA/cm2 for 90–120 min with the goal of creating mesoporous silicon layers with average pore sizes ranging from 5 to 50 nm (Uhlir, 1956; Herino et al., 1987; Lehmann et al., 2000). After the etching process, the wafer was rinsed with water and then isopropanol (IPA) to remove any residual electrolyte from the porous structure. It was then placed in an ultrasonic container for 12 min to separate the porous layer from the parent substrate. The resulting millimeter-scale particles were removed from the container, placed in a paper filter, and rinsed with IPA for 20 min. The particles were stored in IPA and then ball-milled for 30 min in a Fritsch Pulverisette 6 ball miller using 4.5 RCF (g) of stainless steel balls with a diameter of 10 mm. The micro- and nanoparticles were then separated by size using a Hettich Universal 320 centrifuge at two different rotation speeds (40 RCF (g) and 3985 RCF (g) for 10 min each) to remove the largest and smallest particles, respectively. The final step was the evaporation of the IPA.
M18H5 powder and MePS particles were mixed manually with a spatula for 1-2 min, in a ratio of 70% M18H5 to 30% MePS in accordance with the considerations for organic glasses (Lao and Nedelec. 2014). The mixture was preserved in alcoholic solution. Then, this mixture was dried at 150°C for 3 h.
The MC3T3-E1 subclone 14 cell line (CRL2594®, ATCC, United States, then referred as MC3T3-E1) was used as cell model. These cells are murine pre-osteoblasts derived from newborn calvaria and were selected for their ability to differentiate homogeneously in mature osteoblasts able to produce a mineralized extracellular matrix. These cells were routinely cultured in a non-differentiating culture medium in an incubator heated at 37°C under 5% CO2 and wet atmosphere. The culture medium named after “complete culture medium” was composed of alpha modified Eagle’s medium, without ascorbic acid containing L-glutamine (ɑMEM, A1049001, Gibco, United States) and supplemented with 10% fetal calf serum (FBS, 10270106) and 100 μg/mL streptomycin, 100 U/mL penicillin (BP2959-50, Fisher Scientific, United Kingdom).
The medium was replaced every 2-3 days and cells were passaged when the cell confluence reached 70%–80% of the culture area. After having discarded the cell culture medium, the cell layer was rinsed in 1X phosphate buffer saline (PBS, 141900094, Gibco, United States). It was then covered by TrypLE select (12563029, Gibco, United States) for 3 min to allow cells to detach from culture plasticware. The cells were harvested in complete culture medium and centrifuged at 300 g for 5 min at room temperature. The cell pellet was resuspended in complete culture medium and the concentration of viable cells was established by numeration of an aliquot in a Mallassez’ hematocytometer for further seedings.
Three powders extracts were prepared in complete culture medium as described hereafter, according to the ISO EN 100993-5: 2010 standard. For standardization of the exchange surface with the solution, an amount of powder was weighed using a microbalance (Sartorius, Germany) to get an Extraction Ratio (ER) of 6 cm2/mL (<0.5 mm thick) in a final amount of solution of 100 mL. Powder extracts obtained from M18H5 powder were prepared according to its specific surface area of 45 m2/g. Regarding the M18H5/MePS mixture, two powder extracts, respectively named M18H5/MePS_1 and M18H5/MePS_2 were prepared according to the specific surface of M18H5 powder (45 m2/g—powder extracts named M18H5/MePS_1) or MePS (300 m2/g—powder extracts named M18H5/MePS_2—Table 1). The powders were conditioned in glass tubes closed by an aluminum foil and heated at 200°C for 2 h for sterilization. Under laminar flow hood, powders were resuspended in 1 mL of complete culture medium and thoroughly mixed. 250 μL of this suspension were immediately mixed into 24.75 mL of complete culture medium in 50 mL sterile conic centrifuge tube (Corning, United States). The solutions were incubated for 24 h at 37°C under 300 rpm agitation.
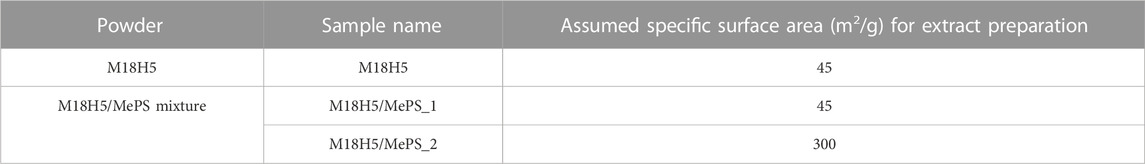
TABLE 1. Powders used and their assumed surface specific area for the preparation of extract used for biological assays.
The extracts were then characterized by their silicon content released from powders determined by Inductive Coupled Plasma—Optical Emission Spectrometry (ICP-OES)—OptimaDV8300, PerkinElmer, United States). The extract solutions were used either pure (100%) or diluted in complete culture medium at 25%, 50%, and 70%.
Cells were seeded at a density of 20,000 cell/cm2 in 100 µL of complete culture medium in 96-well plates (Sarstedt, Germany) for MTT assay, 24-well containing 12 mm borosilicate coverslip (ThermoFisher Scientific, United States) for assays using microscopy. The cells were then incubated at 37°C for 24 h to let the cells to adhere. The culture medium was then discarded and replaced by powder extracts or their dilutions for 24 h and 48 h.
After 24 h and 48 h of culture, 10 µL of 12 mM MTT 3-[4,5-dimethylthiazol-2-yl] −2,5 diphenyl tetrazolium bromide (Sigma-Aldrich, United States) were added to the wells and the cells were incubated for 2 h at 37°C to let the cellular dehydrogenases to metabolize the soluble yellow MTT into deep-purple insoluble crystals of formazan. The culture medium containing MTT was discarded and 50 µL of dimethyl sulfoxide (Sigma-Aldrich, United States) were pipetted onto the cells to lyse them and dissolve the formazan crystals. After 15 min of incubation at room temperature in the dark, under agitation, the optical density or absorbance was measured at 595 nm using a microplate reader (Fluostar Optima, BMG Labtech, Germany).
For cells cultured on borosilicate glass coverslips, after 20 h of incubation, EdU (5-ethynyl-2′-deoxyuridine, thymidine ThermoFisher Scientific, United States) was added to the culture media for a final concentration of 10 µM per well. After 4 h of incubation at 37°C, cells were rinsed 3 times in PBS 1X and fixed in paraformaldehyde (PFA, Sigma Aldrich, Germany) 4%w/v in PBS 1X for 10 min at room temperature. After 3 washes in PBS 1X, cells were immersed in Tris 100 mM, pH 7.6 for 10 min at room temperature and permeabilized in 0,5%v/v Triton-X100 (ThermoFisher, United States) in PBS 1X for 15 min at RT. A click-chemistry reaction solution was prepared by adding one by one as follows 887,5 µL PBS 1X, 10 µL 200 mM CuSO4 (Sigma Aldrich, United States), 2,5 µL AlexaFluor 488 azide (ThermoFisher Scientific, United States) and 100 µL 200 mg/mL ascorbic acid (Acros organics, United States), and incubated on cells in the dark for 30 min at RT. A counterstaining of nuclear DNA was carried out with 20 µM Hoechst 33,342 (ThermoFisher Scientific, United States) in PBS 1X for 5 min at RT in the dark.
After 24 h, cells cultured on borosilicate glass coverslips were rinsed 3 times in PBS 1X and fixed in PFA 4% for 10 min at room temperature. They were rinsed 3 times in PBS 1X and permeabilized for 15 min at room temperature in 0.1%v/v Triton-X100 (ThermoFisher, United States) in PBS 1X. Aspecific antigenic sites were saturated by immersing the cells for 1 h at room temperature in Bovine Serum Albumin (BSA, Sigma Aldrich, United States) 3%w/v in PBS 1X. Actin cytoskeleton was stained by incubated the cells in presence of Phalloidin conjugated to Dylight 488 (ThermoFisher Scientific, United States) for 1 h at room temperature in the dark. An immunolabelling of ɑ-tubulin was performed by hybridizing the cells first, with a monoclonal mouse anti ɑ-tubulin antibody (SantaCruz Biotechnology, United States) for 1 h at room temperature and, secondly, with a goat-anti-mouse antibody conjugated to Dylight 550 (ThermoFisher Scientific) for 1 h at room temperature. Nuclei were stained with Hoechst 33,342 as described above.
After 24 h of cell culture in the powder extracts, the culture supernatant was collected and the calcium was dosed using a commercial fluorimetric dosage assay (Abcam, United Kingdom) according to the manufacturer’s instructions in black 96-well plates (Corning, United States). The fluorescence was measured in a plate reader (Fluostar Optima, BMG Labtech, Germany) with an excitation at 544 nm and emission at 590 nm.
For each experiment needing to make microscopy observation, at least 10 fields were imaged using an upright AxioImager M2 fluorescence microscope coupled to an AxioCam MRm camera (Zeiss, Germany). Images were then analyzed using the ImageJ software. Cell density was measured by counting the Hoechst 33,342 positive nuclei reflecting the cell number and reported as number of cell/cm2. The proliferation rate was calculated as the percentage of EdU positive nuclei on Hoechst 33,432 positive nuclei.
For each test, at least 3 independent experiments were performed with freshly prepared powder extracts. Statistical analyses were done using GraphPad Prism 9. Data were assayed for normality using the Shapiro-Wilk test. According to the data set and the results of the normality test, either a Kruskal–Wallis test followed by a Dunn’s post hoc test (non-parametric) or an ANOVA one-way followed by a Tukey post hoc test was performed. Differences were considered as significant for p ≤ 0.05.
The SEM analysis provided the micrographies of the OSP particles. Figure 1A shows at high magnification details of the surface of a particle with typical outer prismatic layers or sheet layers of the shell. They are generally oriented to the growth direction of the oyster shell (Yoon et al., 2003). The surface is irregular with a lot of debris as short mineral fibers and Figure 1B. Shows the global morphology and size of the particles of the selected powder.

FIGURE 1. SEM images (SE mode) of selected raw oyster shell powder for the synthesis. (A) Detail of the surface of a particle (bar: 1 µm). (B) Global view and morphology of the particles (bar: 200 µm).
The Energy Dispersive X ray analysis (EDS) was also performed on the samples of OSP. The presence of Calcium (36.09%), Oxygen (47.25%), Carbon (14.43%) was evident. Elements like Magnesium (0.71%), Sodium, Silicon (1.53%) were also present in trace amounts. These elements could replace Calcium in the carbonate.
The Figure 2A. Shows the X ray pattern of raw OSP. Calcite is the major mineral component of the powder (JCPDS N° 47-1743)). No aragonite phase, other polymorphic biocarbonate, was detected. The presence of sharp diffraction peaks in the raw sample indicates high crystallinity (rhomboedric microstructure—R
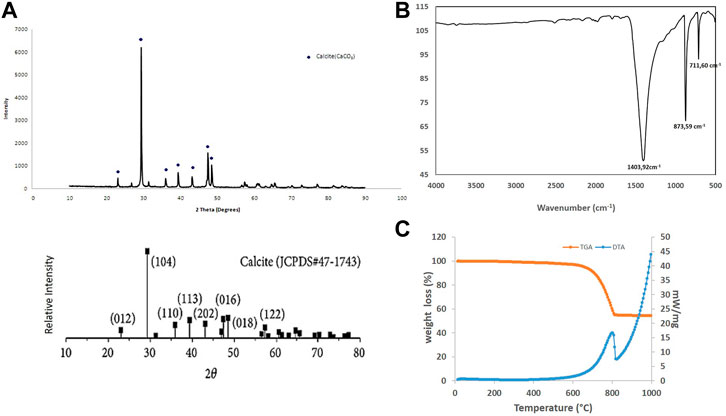
FIGURE 2. Physicochemical characterization of oyster shell powder. (A) X ray diffractogram compared with JCPDS standard with the diffracting crystal planes and (B) FTIR pattern with the characteristic carbonate vibrational bands. (C) TGA/DTA of raw oyster shell powder.
Characteristic FT-IR bands at 1,403.92 cm−1, 873.59 cm−1, 711.60 cm−1were noted (Figure 2B). 1,403.92 cm−1 and 873.59 cm−1 correspond to ν3-asymmetric CO3 stretching and ν1 asymmetric CO3 deformation respectively. 711.73 cm−1 is attributed to ν4 symmetric CO3 deformation. The 873.59 cm−1 and 711.60 cm-1 bands match closely with the calcite reference vibrational bands (Gunasekaran et al., 2006; Rujitanapanich et al., 2014) and confirm the composition of the raw oyster shell powder in calcite. The slight shifts may be attributed to impurities and/or distortion of carbonate groups.
Figure 2C presents the results of a TGA/DTA performed on raw OSP. The organic matter and humidity present in the sample were removed between 150°C and 200°C. An endothermic reaction, as shown by the DTA curve is recorded between around 700°C and 820°C. The calcination of CaCO3 was observed to decompose into CaO following the reaction CaCO3→CaO+CO2↑ (Choi et al., 2011; Hasan Mahmud et al., 2015; Cudrado and Rica, 2017). This measurement confirmed the chemical composition of the oyster shell powder.
Figure 3 shows the crushed OSP (duration of milling: 18 h). The grain shape is rather uniform, the surface smooth, and few aggregates are present. The grain size is more or less homogeneous. By image analysis, the average diameter is 2.06 µm.

FIGURE 3. SEM images of crushed OSP sample (Milling duration: 18 h).
After heat treatment, the grains agglomerate and their shapes are less regular than previously. The crystals are white and opaque. In addition, the surface of the grains looks rougher (Figure 4).

FIGURE 4. SEM Images of M18H5 sample.
The X-ray diffraction analysis of the M18H5 powder is presented in Figure 5. The XRD pattern reveals the presence of a mixture of calcium phosphates, including β-tricalcium phosphate (TCP; PDF 00-055-0898) and calcium phosphate apatites that can be indexed as hydroxyapatite (HAp; PDF 01-074-9775), as illustrated in Figure 5.
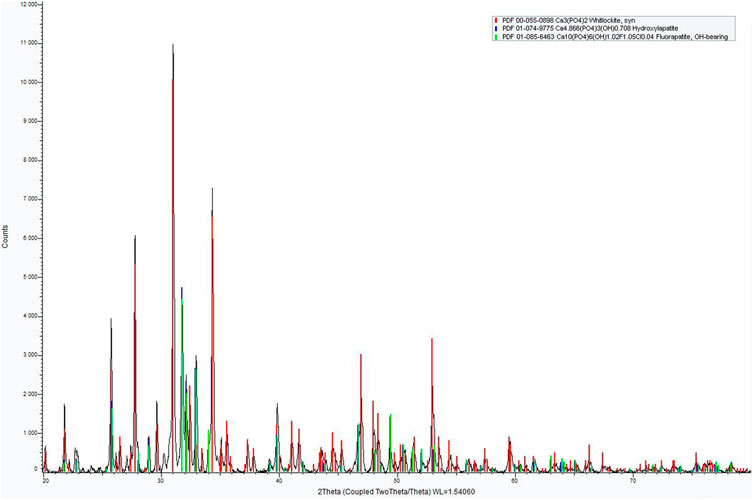
FIGURE 5. XRD pattern of M18H5 powder.
The powder is a Biphasic Calcium Phosphate (BCP). The XRD pattern indicated a majority of β-TCP. The quantification of the amount of each calcium phosphate is roughly estimated by a RIR (Reference Intensity Ratio) method with the two higher peak intensities of β-TCP and HAp as proposed by Raynaud et al., 2001 and in the works of Destainville et al., 2003. The results indicated 75 wt% for β-TCP, 25 wt% for HAp. To obtain a more precise determination, a Rietveld refinement was conducted using the Fullprof software, and the results are presented in Figure 6.
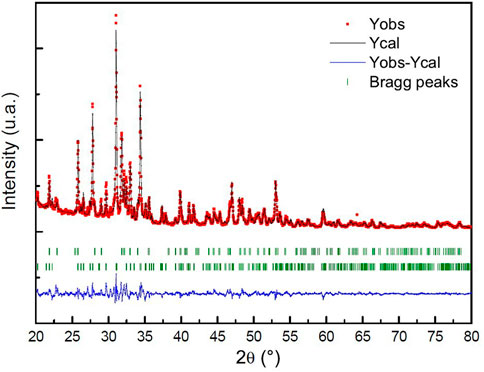
FIGURE 6. Rietveld analysis pattern of obtained powder. The black solid line concerns the calculated intensities and the red asterisk line, the observed intensities. The short vertical lines show the position of possible Bragg reflections. The difference between the observed and calculated intensities is plotted below the profile.
The Rietveld refinement method shows that 24.1% of the sample is composed of hydroxyapatite (HAp) and 75.9% is composed of β-tricalcium phosphate (β-TCP). The reliability factors for this structural refinement of the X-ray diagram, Rp and Rwp, are close to 7.32% and 9.42%, respectively. It should be noted that these values are somewhat high because minor phases present in the sample were not taken into account in the refinement due to their low proportion. Some lines in the X-ray diagram are not indexed, including broad lines around 24° and two peaks at 27.12° and 30.13°. It is possible that these minor phases, which could include oxides such as CaO and MgO, calcite (CaCO3), dicalcium phosphates (α-Ca2P2O7 and β-Ca2P2O7), or even α-TCP, contribute to the lack of intensity of these lines (Gomes et al., 2010).
The molar ratio of calcium to phosphorus in the M18H5 powder was estimated to be 1.53 using the following Eq. 1 (Raynaud et al., 2001):
Where MHAP and Mβ-TCP are the molar weights of HAP and β-TCP (1004.64 and 310.18 g.mol−1, respectively).
The granulometric analysis (Table 2) shows that the average hydrodynamic diameter of the particles of M18H5 powder has the same order of magnitude as the average hydrodynamic diameter of porous silicon. Figure 7 displays a SEM image of the mixture M18H5 and MeSP. MeSP can be identified by its elongated shape and is marked with white stars. The protocol used to produce the MePS is the same as that described in Melhem et al. (2017), resulting in mesoporous silicon with columnar pores growing along the <100> directions, well-aligned, with an estimated average diameter of around 50 nm. Visible striations along the length of the marked particles correspond to columnar pores with relatively smooth walls and small branches. As the p-wafer is moderately doped (around 1017 cm−3), only one pore distribution is observed (Zhang, 2004). The porosity of the wafer is around 60%. MePS particles are generally darker in contrast than hydroxyapatite particles. Table 3 shows that the size of each type of particle is equivalent. Karbassian (2018) reported that mesoporous silicon particles can be resorbed after several days.
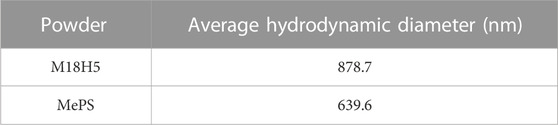
TABLE 2. Average hydrodynamic diameter of the particles of the different powders.

FIGURE 7. (A) SEM images of the mixture M18H5 and MePS. (B) Detail of particles of MePS and M18H5 particles.
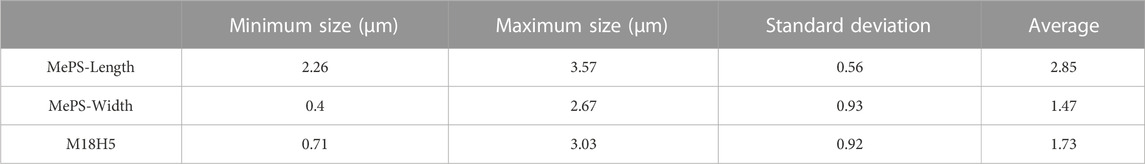
TABLE 3. Size of the different particles (powder M18H5 and MePS).
The biocompatibility of powders was assayed from powder extracts prepared as described above according guidelines of the ISO standard 10093-5. As detailed above, in order to follow these guidelines regarding mixture of M18H5 and MePS, two extracts were prepared according to the specific area of the powder mixture components. As the concentration of calcium ions in cells environment influences their behavior and due to the solubility of β-TCP and Hap at 37°C (−log (Ks) = 29.5 and 117.2, respectively (Elliott, 1994)), calcium as Ca2+ was dosed in the powder extracts and compared with the concentration in the complete culture medium alone. Average Ca2+ concentration in the complete medium was 349 ± 7.5 µM. No significant difference was found with the Ca2+ dosed in the powder extracts, indicating a low release of Ca2+ from HAp. Consequently, it can be assumed that there was a chemical equilibrium regarding Ca2+in these solutions.
The MTT assay measuring the metabolic activity of the cells cultured for 24 h and 48 h in presence of the three powder extracts either pure (100%) or diluted in the complete culture medium at 1/4 (75%), 1/2 (50%) and 3/4 (25%)) is given in Figure 8. In order to evaluate the cytocompatibility (absence of cytotoxicity) of the powder extracts, the results were expressed as a percentage of activity with regards to the control, i.e. the cells cultured in the complete culture medium (Figures 8A, B). For M18H5 powders, whatever the concentration of the powder extract might be, no difference with the control was observed at both time points of 24 and 48 h.
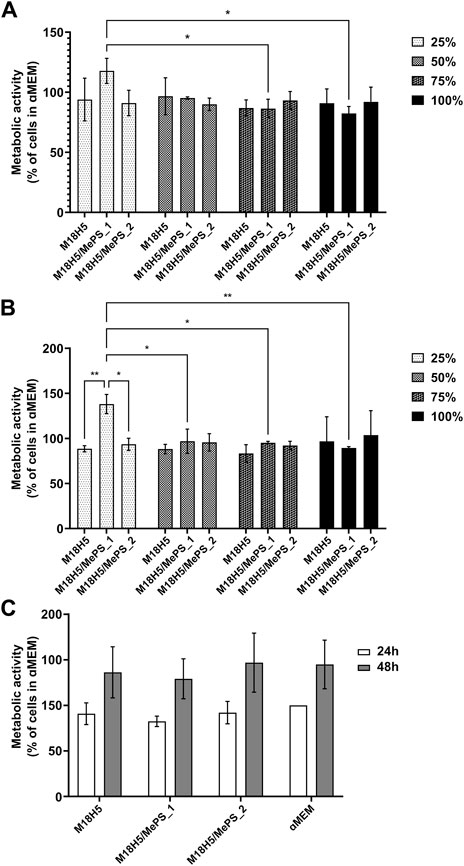
FIGURE 8. Evaluation of the metabolic activity of MC3T3-E1 cells cultured in presence of M18H5, M18H5/MePS_1 and M18H5/MePS_2 powder extracts, pure (100%) or diluted at d = 1/4 (75%), d = 1/2 (50%) and d = 3/4 (25%) for (A): 24 h and (B): 48 h. The metabolic activity is expressed as a percentage of the control, i.e. the value obtained for the cells cultured in complete culture medium (ɑMEM). (C): Metabolic activity of the cell measured after 24 h and 48 h of cultivation and expressed as a percentage of the value obtained for the cell cultured in complete culture medium (ɑMEM) after 24 h as a control. Statistical analysis: ANOVA one-way followed by a Tukey post hoc test. *: p ≤ 0.05, **: p < 0.01. n = 3 independent experiments.
Regarding the extracts obtained from M18H5/MePS powders (M18H5/MePS_1 and M18H5/MePS_2), no difference with M18H5 powder extract was detected at the exception of the diluted extract from the M18H5/MePS_1 powder. When the cells were cultured with 25% of the M18H5/MePS_1 powder extract in the complete culture medium, a significant increase of the cell metabolic activity was observed at both time points, 24 h (Figure 8A) and 48 h (Figure 8B). This variation can be explained either by a stimulation of the cell proliferation induced by dissolution products from M18H5/MePS powder or by an elevation of the cell metabolism (for example to meet a growing need of energy). On the Figure 8C, the results were normalized to the cells population at 24 h in order to evaluate the growth of the cell population overtime. Whatever the conditions, the cellular metabolic activity was multiplied by 1.5 over time. No significant difference was detected even regarding the condition M18H5/MePS_1 at 25% in the complete culture medium. This implies that the significant increase of the cell metabolic activity in comparison with the other conditions is not due to a stimulation of the cell proliferation leading to a higher number of metabolically active cells in the well but to a direct or indirect stimulation of the cell metabolism. The fact that only the lowest dilution induces a cell stimulation suggests that a chemical element present in the dissolution product from this sample induces a biological activity at low concentration, and that there is a threshold beyond which this stimulating effect is lost. In this respect, silicon would be a good candidate. The silicon content in 100% M18H5/MePS_1 was 53 µM (1.49 mg/L). Silicon was not detected for 100% M18H5/MePS_2 likely because the silicon concentration in this powder extract was below the detection threshold of ICP-OES. Assuming that the quantity of the released silicon is proportional to the powder amount in the culture medium, in 25% M18H5/MePS_1 the silicon content should be 1.33 µM and 0.8 µM (0.23 mg/L) for 100% M18H5/MePS_2. For this 100% M18H5/MePS_2, there was not any detected effect. Silicon substitution is widely investigated to improve the biological properties of hydroxyapatite ceramics as bone substitutes (Szurkowska and Kolmas, 2017). As a dopant in biomaterial composition, silicon was assessed to be able to induce osteogenesis and angiogenesis (Bose et al., 2013). While different responses to silicon were observed according to the cell type (Zou et al., 2008), silicon affects the in vitro cell behavior in a concentration range around 10 times higher than the silicon concentration measured in the 100% M18H5/MePS_1 samples (i.e. Wiens et al., 2010; Zhou et al., 2016). For example, Zhou et al., have tested concentrations in silicate of 10 and 50 mg/L on both mesenchymal stem cells and MC3T3-E1 cell line with a detectable effect on metabolic activity only after 7 days. Thus, the silicon release in the case of M18H5/MePS_2 extracts was likely too low to induce a cell response and the effect seen for diluted at 25% M18H5/MePS_1 is due to another phenomenon.
The main goal of the biological assays here was to check the biocompatibility of the extracts. According to the ISO standard 10093-5, the cytotoxicity of a compound is assessed when the metabolic activity falls down under 70% of those of the control. The metabolic activity is not altered in comparison to the control, the powder extracts are considered as not cytotoxic. Thus, while keeping in mind that a deeper insight about the effect of the more diluted M18H5/MePS_1 powder extract should be done, in the following experiments only the pure powder extracts were used.
The MTT assay showed that the MC3T3-E1 cell viability is not affected by the powder extracts and allowed to hypothesize that the proliferation rate is not affected. But the MTT assay do not test directly the proliferation rate so that it was evaluated by an EdU incorporation assay, in situ (Figure 9). The cells incorporated EdU during the duplication of their DNA content at the beginning of the mitosis process. Consequently, the percentage of cells having incorporated EdU (Figure 9C), obtained by image analysis after having imaged the cells in fluorescence microscopy (Figure 9A), directly reflected the proliferation rate of the cell population. Nuclei, representing the total cell number, were stained by the Hoechst 33,432. The cell populations (Figure 9A) confirmed the MTT results. The nuclei presented a healthy morphology, circular with a regular contour. The cell layers were homogeneous. Very few condensed nuclei, hallmarks of cell death, were found. The calculation of the cell density (Figure 9B) did not show any significant difference between the conditions, which confirmed the visual observations. The average density is about 27,400 cells/cm2 is above the cell seeding density (25,000 cells/cm2). After 4 h of EdU incorporation, about 40% of cells were positive (Figure 9C), and so entered the S phase of the cell cycle. The cells cultured in complete culture medium exhibited a percentage of EdU incorporation of 38.5% ± 2.0%, which cannot be considered as different from the conditions with powder extracts. The powder extracts did not alter the proliferation rate of the MC3T3-E1 cells.
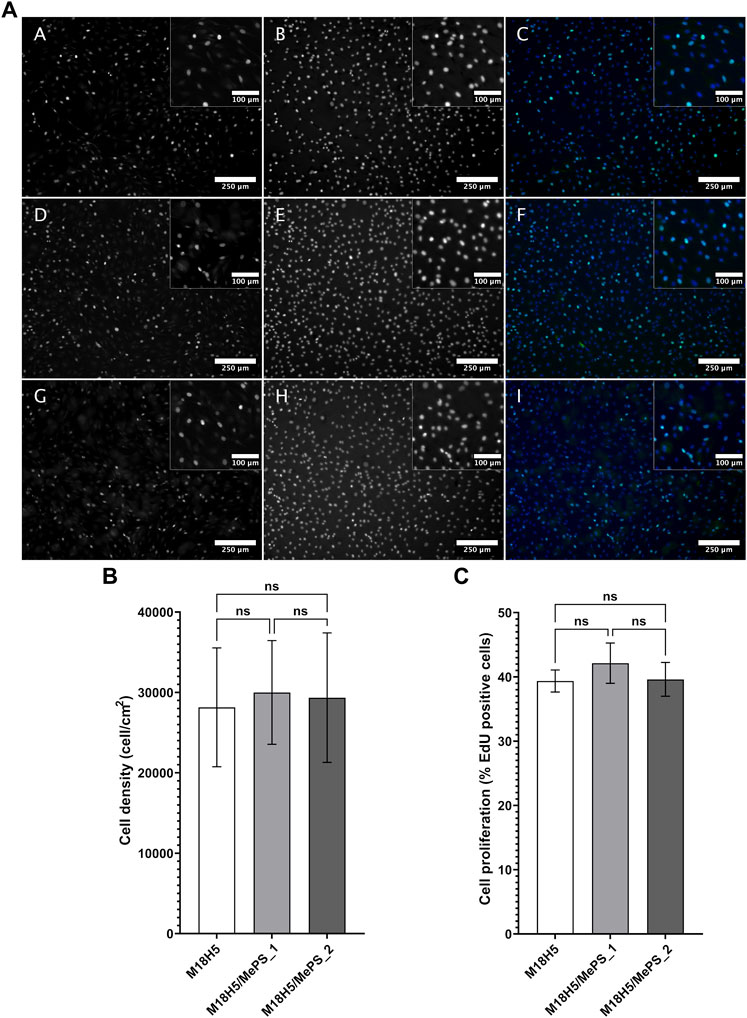
FIGURE 9. Density and proliferation rate of MC3T3-E1 cells cultured in presence of M18H5, M18H5/MePS_1 and M18H5/MePS_2 powder extracts. (A) Micrographs of cells cultured in presence of M18H5 (A–C), M18H5/MePS _1 (D–F) and M18H5/MePS_2 (G–I) powder extracts stained after EdU incorporation assay. A, D and (G) EdU after conjugation with AlexaFluor488; B, E and (F) nuclei stained by Hoechst 33,342; C, F and (I) merge. Scale bar: 250 µm (insets: 100 µm). Upper right: insets showing the magnification of areas in the main image (B) Cell density, determined by image analysis and expressed as cells/cm2. Statistical analysis: ANOVA one way followed by a Tukey post hoc test; ns: p > 0.05. (C) Cell proliferation, expressed in % of cells positive for EdU in total cell population (Hoechst 3,342 positive nuclei) determined by image analysis. Statistical analysis: Kruskall-Wallis followed by a Dunn’s post hoc test; ns: p > 0.05. n = 3 independent experiments.
The MC3T3-E1 cell morphology in presence of powder extracts in comparison with the complete culture growth medium alone was evaluated in situ by fluorescence microscopy after cytoskeleton staining (Figure 10). In complete culture medium (lower row, M, N, O, P in Figure 10) the cells were well spread. Actin staining (M and green staining on P images in Figure 10) is arranged as stress fibers. At the top end of these stress fibers, the actin staining is more intense in punctual areas that might correspond to the anchoring points of the cell on the glass coverslip surface. Microtubules (N and red staining on P images in Figure 10), were distributed from the microtubule organizing center (where all microtubules converge) which is close to the nucleus, towards the cell periphery. When considering the conditions with the powder extracts, whatever the nature of the powder, the cell morphology and cytoskeleton distributions are not visually different. The cell morphology was not altered suggesting that the cell physiological behavior in terms of adhesion, orientation and related cell-cell adhesion as well as migratory aspects should not be dramatically different between these conditions.
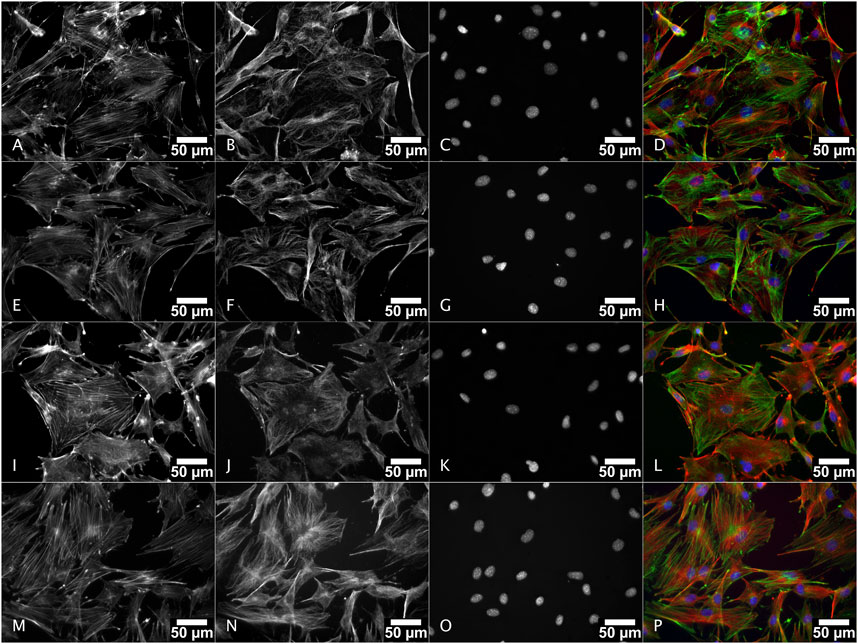
FIGURE 10. Morphology of MC3T3-E1 cells cultured in presence of M18H5, M18H5/MePS_1 and M18H5/MePS_2 powder extracts after staining of actin cytoskeleton and ɑ-tubulin. M18H5 (A–D), M18H5/MePN_1 (E–H), M18H5/MePN_2 (I–L) and complete culture medium (M–P). A, E, I and M: actin cytoskeleton; B, F, J and N: ɑ-tubulin; C, G, K, and O: nuclei; D, H, L and P: merge. Scale bar: 50 μm. n = 3 independent experiments.
The present study demonstrated that biphasic calcium phosphates can be synthesized by the solid-state chemical reaction method from a natural calcified material from the recycling of oyster shells, providing an easy and economical approach for mass producing BCP powders with MePS.
Ball milling for 18 h effectively produces calcite powders with a suitable particle size, and heat treatment at 1,050°C can transform oyster shell powders into BCP powders with a high content of β-TCP. Additionally, anodization of silicon wafers can be utilized to synthesize mesoporous silicon with pore lengths ranging from 5 to 50 nm. Furthermore, calcium phosphate mixtures with mesoporous silicon particles are biocompatible.
It would be interesting to further investigate the effects of diluted extracts prepared from powders calcium phosphates and mesoporous silicon on osteoblastic cells based on their specific surface area. Although some positive effects of silicon on bone cells have been described in the literature, it is challenging to compare these findings due to differences in methodology, silicon source, and chemical state. As such, it would be valuable to study the impact of low doses of silicon on osteoblastic cells using these specific materials.
The original contributions presented in the study are included in the article/supplementary materials, further inquiries can be directed to the corresponding author.
MA-A, HR, and PM, three students, designed the synthesis of the powders and analyzed their results in the frame of their internships. AM carried out the biocompatibility assessment with the supervision of EC, GG, and TD guided the three students for the manufacturing process of mesoporous silicon particles. CR formulated the idea, supervised the three students for the calcium phosphates particles production and wrote the paper. CA-L carried out XRD analysis and Rietvelt Refinement. NP carried out FT-IR analysis and TGA. Funding acquisition: CR All authors have read and agreed to the submitted version of the manuscript.
This research was funded by laboratory own funds and MATCCAP-BIO project (Grant No. 2017-0047222), project of Centre Val de Loire Region, France. The publication of this work was supported by MATV2L, CNRS Research Federation.
The authors acknowledge the Centre d'Études et de Recherche Technologiques en Microéléctronique (Certem) for providing all the necessary facilities, including SEM and XRD, for the characterization of the powders. Additionally, the authors express their gratitude to Professor G. Manivasagam from the Vellore Institute of Technology (VIT) in India for facilitating the exchange of master students and supporting some physical-chemical characterizations. Special recognition is also given to Emeline Renaudie from the University of Limoges for her technical assistance in the biological evaluation of the powders.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Astrova, E., and Tolmachev, V. (2000). Effective refractive index and composition of oxidized porous silicon films. Mat. Sci. Eng. B 69–70, 142–148. doi:10.1016/S0921-5107(99)00236-6
Bobbio, A. (1972). The first endosseous alloplastic implant in the history of man. Bull. Hist. Dent. 20, 1–6. PMID: 4505221.
Bose, S., Fielding, G., Tarafder, S., and Bandyopadhyay, A. (2013). Understanding of dopant-induced osteogenesis and angiogenesis in calcium phosphate ceramics. Trends Biotechnol. 31, 594–605. doi:10.1016/j.tibtech.2013.06.005
Bowditch, A., Waters, K., Gale, H., Rice, P., Scott, E., Canham, L., et al. (1998). In-vivo assessment of tissue compatibility and calcification of bulk and porous silicon. MRS Proc. 536, 149. doi:10.1557/PROC-536-149
Brundavanam, R-K., Fawcett, D., and Jai Poinern, G-E. (2017). Synthesis of a bone like composite material derived from waste pearl oyster shells for potential bone tissue bioengineering applications. Int. J. Res. Med. Sci. 5 (6), 2454–2461. doi:10.18203/2320-6012.ijrms20172428
Camaioni, A., Cacciotti, I., Campagnolo, L., and Bianco, A. (2015). “Silicon-substituted hydroxyapatite for biomedical applications,” in Hydroxyapatite (HAp) for biomedical applications. Editor M. Mucalo (Elsevier: Elsevier), 343–373. Woodhead Publishing Series in Biomaterials: Number 95, Chap. 15. doi:10.1016/B978-1-78242-033-0.00015-8
Chadwick, G., Clarkin, O. M., and Tanner, D. A. (2010). Hydroxyapatite formation on metallurgical grade nanoporous silicon particles. J. Mat. Sci. 45 (23), 6562–6568. doi:10.1007/s10853-010-4745-4
Chen, T-Y., Huang, H-C., Cao, J-L., Xin, Y-J., Luo, W-F., and Ao, N-J. (2016). Preparation and characterization of alginate/HACC/oyster shell powder biocomposite scaffolds for potential bone tissue engineering applications. RSC Adv. 6, 35577–35588. doi:10.1039/C5RA26805B
Choi, U-K., Lee, O-H., and Kim, Y-C. (2011). Effect of calcinated oyster shell powder on growth, yield, spawn run, and primordial formation of king oyster mushroom (pleurotus eryngii). Molecules 16, 2313–2322. doi:10.3390/molecules16032313
Cuadrado Rica, H. (2017). Étude du comportement de bétons de coproduits coquilliers pour une utilisation en récifs artificiels. France: Phd-Report, University of Caen-Normandie.
Dalilottojari, A., Yin Tong, W., McInnes, S. J. P., and Voelcker, N. H. (2015). “Biocompatibility and bioactivity of porous silicon,” in 17, section III: Biomedical applications in porous silicon – from formation to application – biomedical and sensor applications- volume II, sci. Editor G. Korotcenkov (Boca Raton, Florida: CRC Press).
Destainville, A., Champion, E., Bernache-Assollant, D., and Laborde, E. (2003). Synthesis, characterization and thermal behavior of apatitic tricalcium phosphate. Mater. Chem. Phys. 80, 269–277. doi:10.1016/s0254-0584(02)00466-2
Dorozhkin, S. V. (2009). Calcium orthophosphates in nature, biology and medicine. Materials 2, 399–498. doi:10.3390/ma2020399
Elliott, J. C. (1994). Structure and chemistry of the apatites and other calcium orthophosphates. Amsterdam: Elsevier Science.
Faiz Mohd Yusoff, M., Hayaty Abu Kasim, N., Harun Himratul-Aznit, W., Saidin, S., Genasan, K., Kamarul, T., et al. (2021). Physicochemical, antibacterial and biocompatibility assessments of silver incorporated nano-hydroxyapatite synthesized using a novel microwave-assisted wet precipitation technique. Mater. Charact. 178, 111169. doi:10.1016/j.matchar.2021.111169
Gomes, S., Renaudin, G., Mesbah, A., Jallot, E., Bonhomme, C., Babonneau, F., et al. (2010). Thorough analysis of silicon substitution in biphasic calcium phosphate bioceramics: A multi-technique study. Acta Biomater. 6, 3264–3274. doi:10.1016/j.actbio.2010.02.034
Gunasekaran, S., Anbalagan, G., and Pandi, S. (2006). Raman and infrared spectra of carbonates of calcite structure. J. Raman Spectrosc. 37, 892–899. doi:10.1002/jrs.1518
Habraken, W., Habibovic, P., Epple, M., and Bohner, M. (2016). Calcium phosphates in biomedical applications: Materials for the future? Mater. Today 19 (2), 69–87. doi:10.1016/j.mattod.2015.10.008
Hasan Mahmud, Md., Abdus Salam, K., Gafur, M. A., Alam Rana, A., Rakibul Qadir, M., Masum, S- Md., et al. (2015). Chemical characteristics of hydroxyapatite from oyster shell by thermo-chemical process. IJRSET 4 (7), 5039–5047. doi:10.15680/IJIRSET.2015.0407002
Herino, R., Bomchil, G., Barla, K., Bertrand, C., and Ginoux, J. L. (1987). Porosity and pore size distributions of porous silicon layers. J. Electrochem. Soc. 134 (8), 1994–2000. doi:10.1149/1.2100805
Hernández-Montelongo, J., Muñoz-Noval, A., Torres-Costa, V., Martín-Palma, R. J., and Manso-Silván, M. (2015). Nanostructured porous silicon: The winding road from photonics to cell scaffolds - a review. Front. Bioeng. Biotechnol. 3, 60. doi:10.3389/fbioe.2015.00060
Ho, W-F., Hsu, H-C., Hsu, S-K., Hung, C-W., and Wu, S-C. (2013). Calcium phosphate bioceramics synthesized from eggshell powders through a solid state reaction. Ceram. Int. 39 (6), 6467–6473. doi:10.1016/j.ceramint.2013.01.076
Ho, W-F., Lee, M-H., Thomas, J-L., Li, J-A., Wu, S-C., Hsu, H-C., et al. (2021). Porous biphasic calcium phosphate granules from OysterShell promote the differentiation of induced pluripotent stem cells. Int. J. Mol. Sci. 22, 9444. doi:10.3390/ijms22179444
Jordana, F., Le Visage, C., and Weiss, P. (2017). Matériaux pour la médecine de demain. Substituts osseux, Med. Sci. Paris. 33, 60–65. doi:10.1051/medsci/20173301010
Karbassian, F. (2018). in Porous silicon. Porosity -process, technologies and applications. Editor T. H. Ghrib (London: IntechOpen).
Lehmann, V., Stengl, R., and Luigart, A. (2000). On the morphology and the electrochemical formation mechanism of mesoporous silicon. Mater. Sci. Eng. B 69, 11–22. doi:10.1016/S0921-5107(99)00286-X
Low, S. P., and Voelcker, N. H. (2014). Handbook of porous silicon. Switzerland: Springer International Publishing Switzerland.
Melhem, A., De Sousa Meneses, D., Andreazza-Vignolle, C., Defforge, T., Gautier, G., Sauldubois, A., et al. (2017). Structural, optical, and thermophysical properties of mesoporous silicon layers: Influence of substrate characteristics. J. Phys. Chem. C 121 (14), 7821–7828. doi:10.1021/acs.jpcc.6b13101
Ramakrishna, C., Thenepalli, T., Nam, S-Y., Kim, C., and Ahn, J-W. (2018). Oyster Shell waste is alternative sources for Calcium carbonate (CaCO3) instead of Natural limestone. J. Energy Eng. 27 (1), 59. doi:10.5855/ENERGY.2018.27.1.059
Raynaud, S., Champion, E., Bernache-Assollant, D., and Laval, J.-P. (2001). Determination of calcium/phosphorus atomic ratio ofCalcium phosphate apatites using X-ray diffractometry. Am. Ceram. Soc. 84 (2), 359–366. doi:10.1111/j.1151-2916.2001.tb00663.x
Rhee, S-H. (2002). Synthesis of hydroxyapatite via mechanochemical treatment. Biomaterials 23, 1147–1152. doi:10.1016/s0142-9612(01)00229-0
Rujitanapanich, S., Kumpapan, P., and Wanjanoi, P. (2014). Synthesis of hydroxyapatite from oyster shell via precipitation. Energy Procedia 56, 112–117. doi:10.1016/j.egypro.2014.07.138
Salonen, J., and Lehto, V.-P. (2008). Fabrication and chemical surface modification of mesoporous silicon for biomedical applications. Chem. Eng. J. 137, 162–172. doi:10.1016/j.cej.2007.09.001
Silva, T. H., Mesquita-Guimarães, J., Henriques, B., Silva, F. S., and Fredel, M. C. (2019). The potential use of oyster shell waste in new value-added by-product. Resources 8 (13), 13–15. doi:10.3390/resources8010013
Szurkowska, K., and Kolmas, J. (2017). Hydroxyapatites enriched in silicon – bioceramic materials for biomedical and pharmaceutical applications. Prog. Nat. Sci. Mater. Int. 27, 401–409. doi:10.1016/j.pnsc.2017.08.009
Uhlir, A. (1956). Electrolytic shaping of germanium and silicon. Bell Syst. Tech. J. 35 (2), 333–347. doi:10.1002/j.1538-7305.1956.tb02385.x
Wiens, M., Wang, X., Schröder, H. C., Kolb, U., Schloßmacher, U., Ushijima, H., et al. (2010). The role of biosilica in the osteoprotegerin/RANKL ratio in human osteoblast-like cells. Biomaterials 31, 7716–7725. doi:10.1016/j.biomaterials.2010.07.002
Wu, S-C., Hsu, H-C., Wu, Y-N., and Ho, W-F. (2011a). Hydroxyapatite synthesized from oyster shell powders by ball milling and heat treatment. Mater. Charact. 62, 1180–1187. doi:10.1016/j.matchar.2011.09.009
Wu, S-C., Hsu, H-C., Hsu, S-K., Wang, W-H., and Ho, W-F. (2011b). Preparation and characterization of four different compositions of calcium phosphate scaffolds for bone tissue engineering. Mater. Charact. 62 (5), 526–534. doi:10.1016/j.matchar.2011.03.014
Yoon, G-L., Kim, B-T., Kim, B-O., and Han, S-H. (2003). Chemical–mechanical characteristics of crushed oyster-shell. Waste Manag. 23, 825–834. doi:10.1016/S0956-053X(02)00159-9
Zaman, S. U., Irfan, M., Irfan, M., Muhammad, N., Zaman, M. K. U., Rahim, A., et al. (2020). Overview of hydroxyapatite; composition, structure, synthesis methods and its biomedical uses. Biomed. Lett. 6 (1), 84–99. ISSN 2410-955X.
Zhang, X. G. (2004). Morphology and formation mechanisms of porous silicon. J. Electrochem. Soc. 151 (1), C69–C80. doi:10.1149/1.1632477
Zhou, X., Moussa, F. M., Mankoci, S., Ustriyana, P., Zhang, N., Abdelmagid, S., et al. (2016). Orthosilicic acid, Si(OH)4, stimulates osteoblast differentiation in vitro by upregulating miR-146a to antagonize NF-κB activation. Acta Biomater. 39, 192–202. doi:10.1016/j.actbio.2016.05.007
Keywords: hydroxyapatite (HAP), tricalcium phosphate (TCP), biocompatibility, ball milling method, mesoporous silicon
Citation: Richard C, Alfred-Arulrasa M, Ramadas H, Mahagamage PT, Defforge T, Gaultier G, Autret-Lambert C, Poirot N, Champion E and Magnaudeix A (2023) Synthesis by solid route and physicochemical characterizations of blends of calcium orthophosphate powders and mesoporous silicon particles. Front. Bioeng. Biotechnol. 11:1101513. doi: 10.3389/fbioe.2023.1101513
Received: 17 November 2022; Accepted: 09 March 2023;
Published: 20 March 2023.
Edited by:
Geetha Manivasagam, VIT University, IndiaReviewed by:
Nenad L. Ignjatovic, Institute of Technical Sciences (SASA), SerbiaCopyright © 2023 Richard, Alfred-Arulrasa, Ramadas, Mahagamage, Defforge, Gaultier, Autret-Lambert, Poirot, Champion and Magnaudeix. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Caroline Richard, Y2Fyb2xpbmUucmljaGFyZEB1bml2LXRvdXJzLmZy
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.