 Yun Tian
Yun Tian Qiong Liu
Qiong Liu Yafang Zhou
Yafang Zhou Xiao-Yu Chen5
Xiao-Yu Chen5 Yongcheng Pan
Yongcheng Pan Hongwei Xu
Hongwei Xu Zhuanyi Yang
Zhuanyi Yang- 1Department of Geriatrics, Xiangya Hospital, Central South University, Changsha, China
- 2National Clinical Research Center for Geriatric Disorders, Xiangya Hospital, Central South University, Changsha, China
- 3Department of Neurology, Xiangya Hospital, Central South University, Changsha, China
- 4Key Laboratory of Hunan Province in Neurodegenerative Disorders, Central South University, Changsha, China
- 5Department of Neurosurgery, Xiangya Hospital, Central South University, Changsha, China
Vanishing white matter disease (VWM) is one of the most common childhood inherited leukoencephalopathies with autosomal recessive inheritance. Mutations in five genes, EIF2B1-5, have been identified as the major cause of VWM. In this study, a targeted gene capture sequencing panel comprising 160 known pathogenic genes associated with leukoencephalopathies was performed in a large Han Chinese family affected by adult-onset VWM, and a novel heterozygous missense mutation (c.1337G > A [p. R446H]) in EIF2B4 (NM_001034116.2) was detected. Further functional studies in HEK 293 cells showed dramatically reduced EIF2Bδ protein levels in the mutated group compared with the wild-type group. This study revealed that a heterozygous missense mutation (c.1337G > A [p. R446H]) in EIF2B4 was potentially associated with the adult-onset mild phenotype of VWM. In contrast to previous reports, autosomal dominant inheritance was also observed in adult-onset VWM.
Introduction
Vanishing white matter disease (VWM), also known as childhood ataxia with central nervous system hypomyelination (CACH), is one of the most common childhood inherited leukoencephalopathies. It is an autosomal recessive disease characterized by progressive ataxia, spasticity, and leukoencephalopathy. The age of the VWM onset varies from before birth to adulthood, with most patients presenting with an onset between the ages of 2 and 6 years (Wu et al., 2009). Mental function is better preserved than motor function. For instance, intellectual problems are relatively rare in VWM, and psychiatric symptoms may occur as an initial manifestation in adult-onset patients. Episodic neurological regression and coma provoked by fever and head trauma usually appear during the progressive course of the disease (Van Der Knaap et al., 1997; Bugiani et al., 2010). Some female patients also experience ovarian failure (Fogli et al., 2003). The clinical phenotypes of VWM vary between studies; notably, the earlier the age of onset, the more severe the disease (Hamilton et al., 2018). Most individuals with VWM display a severe progressive phenotype in early childhood that may end in early death (Van Der Knaap et al., 2003), while a few instances have developed in adulthood and presented much milder progression (Pronk et al., 2006). Compared to early childhood-onset VWM, adult-onset VWM has rarely been reported. The clinical course of adult-onset VWM may be milder and slower, but it is still progressive, and many patients ultimately lose the ability to perform daily living activities (Labauge et al., 2009). Typical brain magnetic resonance imaging (MRI) shows extensive leukoencephalopathy with diffuse fluid-like signals as a strong indicator of VWM (Van Der Knaap et al., 1999).
The eIF2B complex, which consists of five subunits (eIF2Bα, β, γ, δ, and ε) encoded by EIF2B1-5 genes, is critical for the initiation of protein translation (Pavitt, 2005; Lorsch and Dever, 2010; Hinnebusch, 2011). Defects in any of these five subunits impair protein synthesis by reducing eIF2B activity (Pavitt et al., 1997; Kimball et al., 1998; Sonenberg and Hinnebusch, 2009; Elsby et al., 2011). Mutations in the EIF2B1-5 genes have been identified as the major cause of VWM and follow an autosomal recessive inheritance mode (Leegwater et al., 2001; Van Der Knaap et al., 2002). Patients diagnosed with VWM have been reported to have either homozygous or compound heterozygous variants in one of the aforementioned five genes, while very few harbor heterozygous mutations (Wu et al., 2009; Zhang et al., 2015). To date, more than 150 mutations in EIF2B have been reported (Zhang et al., 2015). Most of the reported patients with VWM are Caucasian, while Han Chinese patients with EIF2B gene mutations have rarely been reported. Moreover, none of the Chinese affected individuals had adult-onset (Wu et al., 2009; Zhang et al., 2015).
In this study, we performed targeted gene capture sequencing followed by Sanger sequencing validation in a Chinese family affected by adult-onset VWM and identified a novel heterozygous missense mutation (c.1337G > A [p. R446H]) in EIF2B4, which led to dramatically reduced EIF2B4 protein levels when overexpressed in HEK 293T cells.
Materials and Methods
Subjects
A Han Chinese VWM-affected family and healthy controls were recruited for this study. All study participants were carefully evaluated by at least two trained neurologists. VWM was diagnosed according to the clinical manifestation of motor and/or cognitive dysfunction accompanied by typical leukoencephalopathy with diffuse fluid-like signals on brain MRI. Clinical data and blood samples were collected from all participants.
This study was approved by the Ethics Committee of Xiangya Hospital of Central South University in China. Written informed consent was obtained from all the participants.
Mutation Screening
A QIAGEN kit was used to extract genomic DNA from peripheral blood leukocytes. To screen for mutations in the probands, we used a targeted gene capture sequencing panel composed of 160 genes reported to be associated with leukoencephalopathies, including AARS2, ABAT, ABCD1, ACOX1, ACTA2, ADH1C, AIMP1, ALDH3A2, APBB2, ARSA, ASPA, BCKDHA, BCKDHB, BCS1L, CHM, CLCN2, COA5, COX15, COX6B1, CSF1R, CTC1, CYP27A1, DARS2, DBT, DDC, DLD, DNAJC13, DNAJC6, ECM1, EIF2B1, EIF2B2, EIF2B3, EIF2B4, and EIF2B5 (Supplementary Table S1). Biotinylated RNA probes were used to capture the known DNA sequences from the GRCh37/hg19 human reference sequence. After QC was used for quality control of sequencing data, low-quality reads were filtered out. The sequence reads were then aligned to the GRCh37/hg19 human reference genome using the Burrows–Wheeler Aligner (BWA, v.0.7.15). Raw variant calling was performed using the Genome Analysis Toolkit (GATK, v.3.2). The variants, including single nucleotide variants (SNVs) and indels (insertions and deletions), were annotated using ANNOVAR. Public databases, including 1,000 human genome datasets (1,000 Genomes) and the Genome Aggregation Database dataset (gnomAD, v2.11), were used to screen mutations among previously obtained SNVs and indels. The variants were filtered in accordance with allele frequencies, with minor allele frequencies (MAFs) < 0.001. Damaging missense and splicing mutations were predicted using the database for nonsynonymous SNPs’ functional prediction descriptions (dpNSFP, v.4.0), Sorting Intolerant from Tolerant (SIFT) (Alanazi et al., 2011) and Polymorphism Phenotyping v2 (PolyPhen-2) (Adzhubei et al., 2010). Amino acid conservation around mutated positions was evaluated using T-Coffee (v.11.00) (Di Tommaso et al., 2011), to blast multiple sequences of eIF2Bδ proteins from different species. A 3D structural protein model was predicted using Phyre2 (Kelley et al., 2015). In addition, reported mutations were screened using the Human Gene Mutation Database (HGMD) and the ClinVar database. All variants were classified based on the sequence variant standards and guidelines of the American College of Medical Genetics and Genomics (ACMG) (Li et al., 2017). Further Sanger sequencing validation of family members was also performed.
Expression Plasmids
The wild-type EIF2B4 plasmid was generated by cloning the complementary DNA (cDNA) of EIF2B4 (NM_001034116.2) into mammalian expression vectors (pcDNA3.1) with a FLAG tag at the C-terminus. EIF2B4 variants (p. R374C and p. R446H) were generated using a QuickChange II site-directed mutagenesis kit (Stratagene, 200523). The authenticity of all constructs was confirmed using Sanger sequencing.
Antibodies
The primary antibodies used in this study included FLAG (Sigma, F1804), vinculin (Cell Signaling Technology, 13901), and β-actin (Cell Signaling Technology, 4967L). The secondary antibodies were donkey anti-rabbit and donkey anti-mouse antibodies (Jackson ImmunoResearch).
Cell Culture and Transfection
Human embryonic kidney (HEK) 293 cells were grown at 37°C under 5% CO2 in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum and 100 U/mL of penicillin/streptomycin. Expression plasmids were transfected into cells using Lipofectamine 2000 reagent (Invitrogen), according to the manufacturer’s protocol. The cells were collected for Western blotting 48 hours after transfection.
Western Blot
Transfected cells were lysed in ice-cold RIPA buffer containing a protease inhibitor cocktail and PMSF. The protein samples were separated using 10% SDS-PAGE and transferred onto a PVDF membrane (Millipore). After blocking, the blots were probed overnight with the appropriate antibodies. Blots were developed using an ECL prime chemiluminescence kit (GE Healthcare). Images were obtained using ChemiDoc XRS+ with Image Lab software (Bio-Rad).
Results
Clinical Assessment of the VWM-Affected Family
The pedigree with VWM included three affected individuals (Figure 1A).
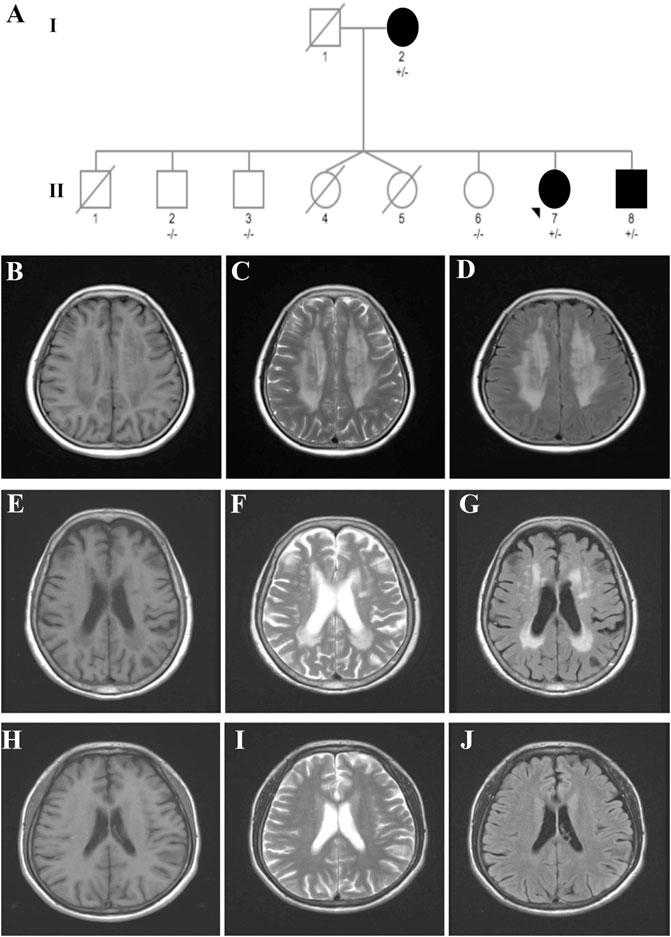
FIGURE 1. Pedigree of a family affected by VWM. (A) Pedigree of the VWM-affected family. (B–D) Brain MRI of the proband (case II-7) indicating typical symmetric and diffuse periventricular leukoencephalopathy and a cerebrospinal fluid-like signal within the area on T1-weighted (B), T2-weighted (C), and flair images (D). (E–G) Brain MRI of the mother (case I-2) showing moderate leukoencephalopathy. (H–J) Brain MRI of the brother (case II-8) revealed no leukoencephalopathy.
The proband case (case II-7) was a 53-year-old woman who presented with progressive cognitive decline for 3 years. At the age of 50, she experienced memory loss and became easily lost in new places. Four days before admission, her symptoms worsened after staying up late. Patients with ovarian failure or other medical history were excluded. The neurological examination revealed memory loss, a decline in calculation ability, language dysfunction, and visuospatial disorders. Her Mini-Mental State Examination (MMSE) score was 14, and the Montreal Cognitive Assessment (MoCA) Scale score was 12. Magnetic resonance imaging (MRI) revealed symmetric and diffuse leukoencephalopathy in the periventricular and cerebrospinal fluid-like signal within the area on T1-weighted, T2-weighted, and flair images (Figures 1B–D).
The mother of the proband (case I-2), an 85-year-old woman, was referred to our hospital with a 20-year history of cognitive dysfunction. Neurological examination revealed mild cognitive impairment. Her MMSE score was 18, with 0-year education. Moderate leukoencephalopathy was observed on her brain MRI, while no fluid-like signal was observed (Figures 1E–G).
The younger brother of the proband (case II-8) was a 50-year-old man who presented with abnormal behavior at the age of 44. He had been diagnosed with schizophrenia 5 years before and had received risperidone for treatment. He had been experiencing memory loss since childhood and was unable to walk until 7 years of age. However, no leukoencephalopathy was observed on his brain MRI (Figures 1H–J).
It is noteworthy that case II:1 died at the age of 3 years for unknown reasons. Coincidently, cases II-4 and 5, a pair of twin sisters, died a few days after birth. It is possible that these three patients may have been affected by VWM and died young. However, further confirmation was difficult because we were unable to obtain their exact medical history and DNA samples.
Identification of Novel Heterozygous Mutations in EIF2B4
Mutation screening was performed in the proband by using a targeted gene capture sequencing panel composed of 160 genes associated with leukoencephalopathies. A novel heterozygous missense mutation (c.1337G > A [p. R446H]) in EIF2B4 (NM_001034116.2) was identified, but no other potential variants in reported genes associated with leukoencephalopathies were detected. Sanger sequencing validation showed that none of the cases had other pathogenic or likely pathogenic mutations in this gene (Figure 2A).
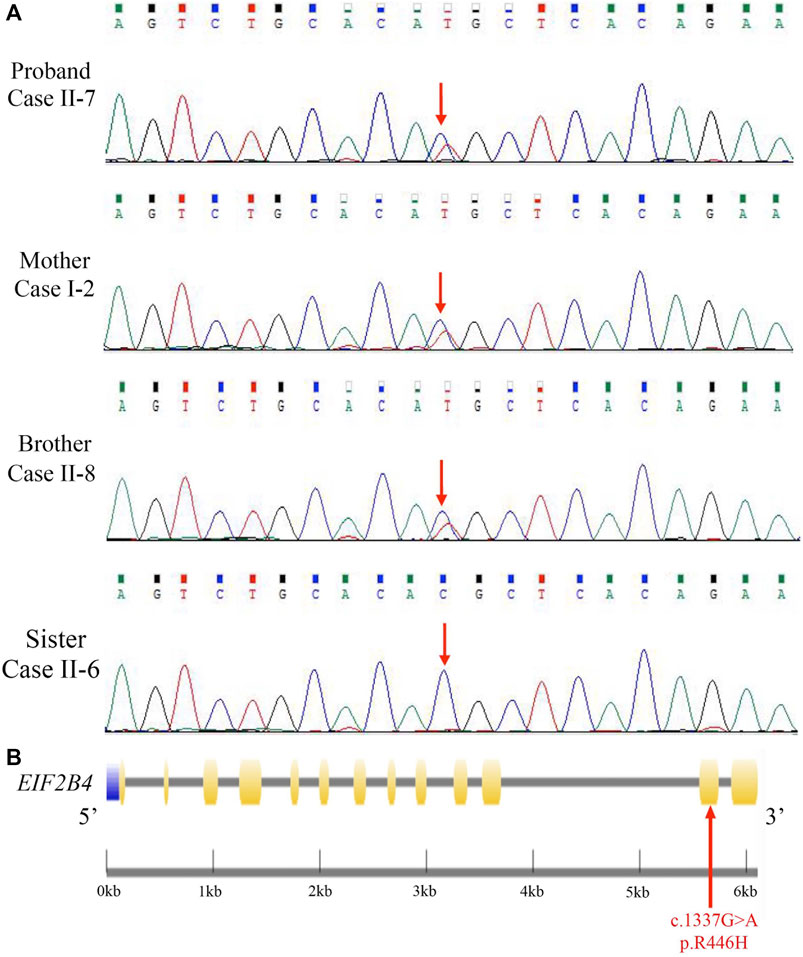
FIGURE 2. Identification of a novel heterozygous EIF2B4 mutation. (A) Sanger sequencing validated a missense mutation (c.1337G > A [p. R446H]) in the EIF2B4 gene in family members. (B) Schematic presentation of the EIF2B4 gene with an R446H substitution located in exon 12. Purple: upstream; yellow: exons; black line: introns.
The EIF2B4 gene is located on chromosome 2p23.2, encoding a 67-kD protein. This mutation lies in exon 11 (Figure 2B), resulting in an amino acid substitution of arginine to histidine (R446H) at the C-terminus of the eIF2Bδ protein. The majority of mutations reported to contribute to VWM or ovarioleukodystrophy (OLD) are missense mutations in the EIF2B4 gene (Fogli et al., 2004; Scali et al., 2006), but this mutation has not been reported in the HGMD and ClinVar databases. We reanalyzed the sequencing data from 12 previously unrelated patients with non-vascular leukoencephalopathies, and no other patients were found to carry the R446H mutation. We further checked whole-exome sequencing data from 1,017 patients with neurological disorders, and none of them carried this mutation. The p. R446H substitution is absent from the human population database 1,000 Genomes, and the frequency of the p.R446H variant is 0.00006 in the gnomAD.
The p.R446H substitution was predicted to affect protein function by SIFT and was expected to be probably damaged by PolyPhen-2. The mutation lies in the IF-2B domain of the eIF2Bδ protein (Figure 3A). IF-2B resembles a domain that belongs to the initiation factor 2 subunit family, which includes IF-2Bα, β, and δ subunits from eukaryotes and some other proteins of unknown function from archaebacteria and prokaryotes (Kyrpides and Woese, 1998). The amino acid sequence alignments remained highly conserved across vertebrates within this region (Figure 3B), indicating that these residues play a critical role in eI2Bδ function. The 3D structural protein model revealed that residue R446 is located close to the predicted pocket region (Figure 3C), which might be functionally important.
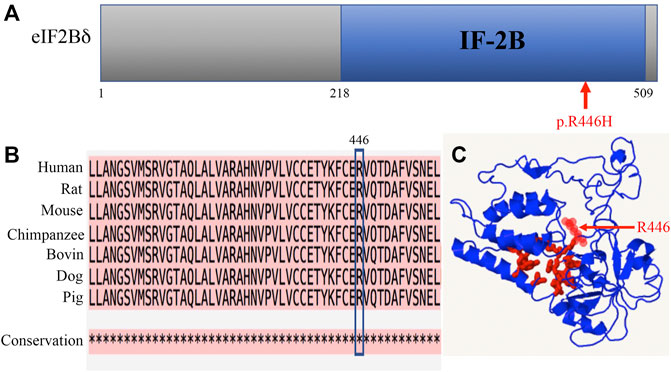
FIGURE 3. Amino acid R446 is highly conserved among mammals. (A) Location of the R446H mutation in the eIF2Bδ protein. (B) Amino acid sequence alignments across species indicate high conservation around residue R446. * fully conserved. (C) 3D structural prediction of the eIF2Bδ protein. Red: pocket; red arrow: residue R446.
Furthermore, the EIF2B4 (c.1337G > A [p. R446H]) mutation was found in all affected individuals but not in unaffected family members of the pedigree (Figure 2A and Supplementary Figure S1). According to the ACMG guidelines, the novel R446H substitution is classified as a variant of uncertain significance (VUS), indicating that a heterozygous (c.1337G > A [p. R446H]) mutation in the EIF2B4 gene may be associated with VWM.
The p. R446H Substitution Significantly Reduces eIF2Bδ Protein Levels
To evaluate the pathogenicity of the R446H substitution, vectors containing wild-type and mutant EIF2B4 (R374C and R446H, respectively) with a C-terminal flag tag were transfected into HEK 293 cells for functional in vitro studies. The R374C variant is also a missense mutation that has been previously reported to be related to VWM before (Van Der Knaap et al., 2002). The expression levels of R374C and R446H were measured using an anti-flag antibody and were found to significantly decrease compared to wild type (Figure 4). These results suggest that both p.R446H and p.R374C substitutions lead to haploinsufficiency of eIF2B, δ, which may contribute to VWM.

FIGURE 4. Expression of wild-type and mutant eIF2Bδ in transfected HEK 293 cells. The levels of eIF2Bδ−R374C and R446H were significantly reduced compared with the wild-type protein.
Discussion
To date, at least 250 patients with VWM and 150 mutations in EIF2B genes have been reported (Zhang et al., 2015). Most EIF2B gene mutations have been reported in Caucasian populations, and the only study that was conducted in a Chinese population reported 34 patients with VWM and 37 EIF2B gene mutations (Zhang et al., 2015). Among all reported EIF2B mutations, EIF2B4 gene mutations were observed in both Caucasian and Chinese patients with frequencies of 17% and 22%, respectively (Scali et al., 2006; Zhang et al., 2015). Rare mutations in the EIF2B4 gene have been reported to account for 10–15% of Caucasian patients with VWM (Fogli et al., 2004; Pronk et al., 2006) and 18% of Han Chinese patients with VWM (Zhang et al., 2015). Most of the reported pathogenic mutations are missense mutations, which are mainly distributed in the highly conserved C-terminal region of eIF2B (Pavitt, 2005).
Patients with VWM with EIF2B4 gene mutations are either homozygous or compound heterozygous, and the majority develop symptoms before adulthood. Consistent with patients with VWM with other EIF2B gene mutations, EIF2B4 gene mutations are usually associated with motor dysfunction, rather than cognitive decline, as cognitive function is preserved relatively well. Some patients present motor development delays, speech delays, or school difficulties before the disease onset (Fogli et al., 2004; Ohlenbusch et al., 2005; Zhang et al., 2015). Only two adult-onset patients with EIF2B4 gene mutations have been previously reported, both of which were compound heterozygous variants. Furthermore, both patients experienced cognitive impairment and motor symptoms, with mild progression (Kanbayashi et al., 2015).
In our study, we identified a novel heterozygous missense mutation (c.1337G > A [p. R446H]) in the EIF2B4 gene in a family with a milder form of adult-onset VWM with cognitive decline or abnormal behavior as their chief complaint. The younger brother of the proband (case II-8) had motor developmental delays in early childhood. Unlike previously reported adult-onset cases of EIF2B4 mutations, no motor symptoms were observed in this study.
The R446H variant identified here is located in a highly conserved region within the C-terminus of eIF2Bδ. The eIF2Bδ protein is ubiquitously expressed in human organs and is one of the five subunits of the eIF2B complex. The eIF2B complex is involved in translation initiation, where its guanine nucleotide exchange (GEF activity) turns inactive GDP-eIF2 to an active GTP-bound form (Leng et al., 2011). Furthermore, the eIF2B complex is composed of two α, β, and δ subunits in the central region, with one γε heterodimer on each side (Kashiwagi et al., 2019). The catalytic center of the γε subcomplex interacts with eIF2 and exhibits GEF activity (Pavitt, 2005), while the α2β2δ2 subcomplex recognizes phosphorylated eIF2α and inhibits GEF activity (Kashiwagi et al., 2019). The C-terminus of the δ subunit shares a similar sequence with the α and β subunits, annotated as the initiation factor 2 subunit family, which suggests that these three subunits may interact with each other to regulate the eIF2B complex (Kyrpides and Woese, 1998; Pavitt, 2005).
Our functional study revealed that overexpression of the R446H variant resulted in a significant reduction in eIF2Bδ levels, which may reduce the activity of the eIF2B complex and impair its physiological function (Bugiani et al., 2010). It is reasonable to postulate that the R446H variant may cause VWM, even in a heterozygous status, by reducing the activity of eIF2B in patients.
In conclusion, our work indicated that a heterozygous missense mutation (c.1337G > A [p. R446H]) in the EIF2B4 gene might be associated with an adult-onset milder form of VWM, and the genetic and clinical spectrum of VWM was extended. Further research on how the heterozygous R446H variant contributes to VWM is required in the future.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material; further inquiries can be directed to the corresponding author.
Ethics Statement
This study involving human participants was reviewed and approved by the Ethics Committee of Xiangya Hospital of Central South University in China. Written informed consent was obtained from all participants.
Author Contributions
YT carried out most of the clinical assessment, data analysis, and manuscript writing. QL carried out the in vitro experiments and participated in manuscript writing. YZ contributed to clinical assessment and data analysis. XC performed the data analysis. YP, HX, and LS helped in the experimental design. ZY contributed to experimental design and manuscript revising.
Funding
This work was funded by the Natural Science Foundation of Hunan Province (Grant 2020JJ5923) and Scientific Research Projects of Health Commission of Hunan Province (Grant 202104090550).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We would like to thank the participants for permitting us to publish this information in this study.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fbioe.2022.901452/full#supplementary-material
References
Adzhubei, I. A., Schmidt, S., Peshkin, L., Ramensky, V. E., Gerasimova, A., Bork, P., et al. (2010). A Method and Server for Predicting Damaging Missense Mutations. Nat. Methods 7, 248–249. doi:10.1038/nmeth0410-248
Alanazi, M., Abduljaleel, Z., Khan, W., Warsy, A. S., Elrobh, M., KhanFAU - Al Amri, Z. A., et al. (2011). In Silico Analysis of Single Nucleotide Polymorphism (SNPs) in Human β-Globin Gene. PloS One 6 (10), e25876. doi:10.1371/journal.pone.0025876
Bugiani, M., Boor, I., Powers, J. M., Scheper, G. C., and Van Der Knaap, M. S. (2010). Leukoencephalopathy with Vanishing White Matter: A Review. J. Neuropathol. Exp. Neurol. 69, 987–996. doi:10.1097/nen.0b013e3181f2eafa
Di Tommaso, P., Moretti, S., Xenarios, I., OROBITG, M., Montanyola, A., Chang, J.-M., et al. (2011). T-coffee: a Web Server for the Multiple Sequence Alignment of Protein and RNA Sequences Using Structural Information and Homology Extension. Nucleic Acids Res. 39, W13–W17. doi:10.1093/nar/gkr245
Elsby, R., Heiber, J. F., Reid, P., Kimball, S. R., Pavitt, G. D., and Barber, G. N. (2011). The Alpha Subunit of Eukaryotic Initiation Factor 2B (eIF2B) Is Required for eIF2-Mediated Translational Suppression of Vesicular Stomatitis Virus. J. Virol. 85, 9716–9725. doi:10.1128/jvi.05146-11
Fogli, A., Rodriguez, D., Eymard-Pierre, E., Bouhour, F., Labauge, P., Meaney, B. F., et al. (2003). Ovarian Failure Related to Eukaryotic Initiation Factor 2B Mutations. Am. J. Hum. Genet. 72, 1544–1550. doi:10.1086/375404
Fogli, A., Schiffmann, R., Bertini, E., Ughetto, S., Combes, P., Eymard-Pierre, E., et al. (2004). The Effect of Genotype on the Natural History of eIF2B-Related Leukodystrophies. Neurology 62, 1509–1517. doi:10.1212/01.wnl.0000123259.67815.db
Hamilton, E. M. C., Van Der Lei, H. D. W., Vermeulen, G., Gerver, J. A. M., Lourenço, C. M., Naidu, S., et al. (2018). Natural History of Vanishing White Matter. Ann. Neurol. 84, 274–288. doi:10.1002/ana.25287
Hinnebusch, A. G. (2011). Molecular Mechanism of Scanning and Start Codon Selection in Eukaryotes. Microbiol. Mol. Biol. Rev. 75, 434–467. doi:10.1128/mmbr.00008-11
Kanbayashi, T., Saito, F., Matsukawa, T., Oba, H., Hokkoku, K., Hatanaka, Y., et al. (2015). Adult-onset Vanishing White Matter Disease with Novel Missense Mutations in a Subunit of Translational regulator,EIF2B4. Clin. Genet. 88, 401–403. doi:10.1111/cge.12554
Kashiwagi, K., Yokoyama, T., Nishimoto, M., Takahashi, M., Sakamoto, A., Yonemochi, M., et al. (2019). Structural Basis for eIF2B Inhibition in Integrated Stress Response. Science 364, 495–499. doi:10.1126/science.aaw4104
Kelley, L. A., Mezulis, S., Yates, C. M., Wass, M. N., and Sternberg, M. J. E. (2015). The Phyre2 Web Portal for Protein Modeling, Prediction and Analysis. Nat. Protoc. 10, 845–858. doi:10.1038/nprot.2015.053
Kimball, S. R., Fabian, J. R., Pavitt, G. D., Hinnebusch, A. G., and Jefferson, L. S. (1998). Regulation of Guanine Nucleotide Exchange through Phosphorylation of Eukaryotic Initiation Factor eIF2α. J. Biol. Chem. 273, 12841–12845. doi:10.1074/jbc.273.21.12841
Kyrpides, N. C., and Woese, C. R. (1998). Archaeal Translation Initiation Revisited: The Initiation Factor 2 and Eukaryotic Initiation Factor 2B α-β-δ Subunit Families. Proc. Natl. Acad. Sci. U.S.A. 95, 3726–3730. doi:10.1073/pnas.95.7.3726
Labauge, P., Horzinski, L., Ayrignac, X., Blanc, P., Vukusic, S., Rodriguez, D., et al. (2009). Natural History of Adult-Onset eIF2B-Related Disorders: a Multi-Centric Survey of 16 Cases. Brain 132, 2161–2169. doi:10.1093/brain/awp171
Leegwater, P. A. J., Vermeulen, G., Könst, A. A. M., Naidu, S., Mulders, J., Visser, A., et al. (2001). Subunits of the Translation Initiation Factor eIF2B Are Mutant in Leukoencephalopathy with Vanishing White Matter. Nat. Genet. 29, 383–388. doi:10.1038/ng764
Leng, X., Wu, Y., Wang, X., Pan, Y., Wang, J., Li, J., et al. (2011). Functional Analysis of Recently Identified Mutations in Eukaryotic Translation Initiation Factor 2Bɛ (eIF2Bɛ) Identified in Chinese Patients with Vanishing White Matter Disease. J. Hum. Genet. 56, 300–305. doi:10.1038/jhg.2011.9
Li, M. M., Datto, M., Duncavage, E. J., Kulkarni, S., Lindeman, N. I., Roy, S., et al. (2017). Standards and Guidelines for the Interpretation and Reporting of Sequence Variants in Cancer. J. Mol. Diagnostics 19, 4–23. doi:10.1016/j.jmoldx.2016.10.002
Lorsch, J. R., and Dever, T. E. (2010). Molecular View of 43 S Complex Formation and Start Site Selection in Eukaryotic Translation Initiation. J. Biol. Chem. 285, 21203–21207. doi:10.1074/jbc.r110.119743
Ohlenbusch, A., Henneke, M., Brockmann, K., Goerg, M., Hanefeld, F., Kohlschütter, A., et al. (2005). Identification of Ten Novel Mutations in Patients with eIF2B-Related Disorders. Hum. Mutat. 25, 411. doi:10.1002/humu.9325
Pavitt, G. D. (2005). eIF2B, a Mediator of General and Gene-specific Translational Control. Biochem. Soc. Trans. 33 (Pt 6), 1487–1492. doi:10.1042/BST20051487
Pavitt, G. D., Yang, W., and Hinnebusch, A. G. (1997). Homologous Segments in Three Subunits of the Guanine Nucleotide Exchange Factor eIF2B Mediate Translational Regulation by Phosphorylation of eIF2. Mol. Cell Biol. 17, 1298–1313. doi:10.1128/mcb.17.3.1298
Pronk, J. C., van Kollenburg, B., Scheper, G. C., and Van Der Knaap, M. S. (2006). Vanishing White Matter Disease: a Review with Focus on its Genetics. Ment. Retard. Dev. Disabil. Res. Rev. 12 (2), 123–128. doi:10.1002/mrdd.20104
Scali, O., Di Perri, C., and Federico, A. (2006). The Spectrum of Mutations for the Diagnosis of Vanishing White Matter Disease. Neurol. Sci. 27, 271–277. doi:10.1007/s10072-006-0683-y
Sonenberg, N., and Hinnebusch, A. G. (2009). Regulation of Translation Initiation in Eukaryotes: Mechanisms and Biological Targets. Cell 136, 731–745. doi:10.1016/j.cell.2009.01.042
Van Der Knaap, M. S., Barth, P. G., Gabreëls, F. J., Franzoni, E., Begeer, J. H., Stroink, H., et al. (1997). A New Leukoencephalopathy with Vanishing White Matter. Neurology 48 (4), 845–855. doi:10.1212/wnl.48.4.845
Van Der Knaap, M. S., Breiter, S. N., Naidu, S., Hart, A. A. M., and Valk, J. (1999). Defining and Categorizing Leukoencephalopathies of Unknown Origin: MR Imaging Approach. Radiology 213, 121–133. doi:10.1148/radiology.213.1.r99se01121
Van Der Knaap, M. S., Leegwater, P. A. J., Könst, A. A. M., Visser, A., Naidu, S., Oudejans, C. B. M., et al. (2002). Mutations in Each of the Five Subunits of Translation Initiation Factor eIF2B Can Cause Leukoencephalopathy with Vanishing White Matter. Ann. Neurol. 51, 264–270. doi:10.1002/ana.10112
Van Der Knaap, M. S., van Berkel, C. G. M., Herms, J., van Coster, R., Baethmann, M., Naidu, S., et al. (2003). eIF2B-Related Disorders: Antenatal Onset and Involvement of Multiple Organs. Am. J. Hum. Genet. 73, 1199–1207. doi:10.1086/379524
Wu, Y., Pan, Y., Du, L., WangFAU - Gu, J. Q., GuFAU - Gao, Q. Z., GaoFAU - Li, Z. J., et al. (2009). Identification of Novel EIF2B Mutations in Chinese Patients with Vanishing White Matter Disease. J. Hum. Genet. 54 (2), 74–77. doi:10.1038/jhg.2008.10
Keywords: vanishing white matter disease, EIF2B4, heterozygous missense mutation, loss of function, targeted gene capture sequencing panel
Citation: Tian Y, Liu Q, Zhou Y, Chen X-Y, Pan Y, Xu H and Yang Z (2022) Identification of a Novel Heterozygous Mutation in the EIF2B4 Gene Associated With Vanishing White Matter Disease. Front. Bioeng. Biotechnol. 10:901452. doi: 10.3389/fbioe.2022.901452
Received: 22 March 2022; Accepted: 23 May 2022;
Published: 04 July 2022.
Edited by:
Youmei Bao, Yale University, United StatesReviewed by:
Rachita Yadav, Massachusetts General Hospital and Harvard Medical School, United StatesNguyen Quoc Khanh Le, Taipei Medical University, Taiwan
Copyright © 2022 Tian, Liu, Zhou, Chen, Pan, Xu and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhuanyi Yang, bmV1cm95enlAY3N1LmVkdS5jbg==
†These authors have contributed equally to this work