 Leijie Li
Leijie Li Yujia Zhang1
Yujia Zhang1 Yongyong Ren
Yongyong Ren Hongyu Zhao
Hongyu Zhao Hui Lu
Hui Lu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Bioeng. Biotechnol. , 13 May 2022
Sec. Cell and Gene Therapy
Volume 10 - 2022 | https://doi.org/10.3389/fbioe.2022.849798
This article is part of the Research Topic Single Cell Intelligence and Tissue Engineering View all 10 articles
Upper gastrointestinal cancer (UGIC) is an aggressive carcinoma with increasing incidence and poor outcomes worldwide. Here, we collected 39,057 cells, and they were annotated into nine cell types. By clustering cancer stem cells (CSCs), we discovered the ubiquitous existence of sub-cluster CSCs in all UGICs, which is named upper gastrointestinal cancer stem cells (UGCSCs). The identification of UGCSC function is coincident with the carcinogen of UGICs. We compared the UGCSC expression profile with 215,291 single cells from six other cancers and discovered that UGCSCs are specific tumor stem cells in UGIC. Exploration of the expression network indicated that inflammatory genes (CXCL8, CXCL3, PIGR, and RNASE1) and Wnt pathway genes (GAST, REG1A, TFF3, and ZG16B) are upregulated in tumor stem cells of UGICs. These results suggest a new mechanism for carcinogenesis in UGIC: mucosa damage and repair caused by poor eating habits lead to chronic inflammation, and the persistent chronic inflammation triggers the Wnt pathway; ultimately, this process induces UGICs. These findings establish the core signal pathway that connects poor eating habits and UGIC. Our system provides deeper insights into UGIC carcinogens and a platform to promote gastrointestinal cancer diagnosis and therapy.
Upper gastrointestinal cancer (UGIC), including head and neck squamous cell carcinoma (HNSCC), esophageal cancer (EC), and gastric cancer (GC), is one of the malignant tumors that seriously threaten the human health (Yamada et al., 2011). Its occurrence is mainly associated with unhealthy eating habits and lifestyle and their consequences, including low intake of fruits and vegetables (Akhtar, 2013), smoking (Gandini et al., 2008), drinking (Goldstein et al., 2010; Zhang et al., 2012; González et al., 2013), and high body mass index (BMI). The global incidence of UGIC has significantly increased in recent years (Bray et al., 2018). Patients with UGIC account for a large proportion of all patients with malignant tumors (Sung et al., 2021). UGIC has a poorer prognosis and lower overall survival rate than other cancers (Sung et al., 2021). GC is the fifth most prevalent cancer and the third leading death cause of patients with cancers on a global scale (Yin et al., 2020). The 5-year survival rate of patients with EC is not more than 20% worldwide (Zhang, 2013). Because of the increasing incidence, the high relapse and metastasis rate, and the low overall survival rate, studies on the molecular mechanism of UGICs or gastrointestinal pan-cancer are imperative.
In recent years, the growing number of patients has prompted many studies on gastrointestinal tumors (Chakravarthy et al., 2018; Yang et al., 2020; Cui et al., 2021). At present, researchers have discovered many biomarkers for the diagnosis and treatment of gastrointestinal cancer, including human epidermal growth factor receptor2 (HER2) (Li Z. et al., 2020), mismatch repair deficiency/microsatellite instability (dMMR/MSI-H) (Dhakras et al., 2020), and programmed death-ligand 1 (PD- L1) (Dai et al., 2021). In addition, there are many new biomarkers under investigation, including neurotrophic-tropomyosin receptor kinase (NTRK) (Westphalen et al., 2019), claudin-18 (CLDN18) (Zhang et al., 2020), Rho GTPase-activating protein 26 (ARHGAP26) (Dhakras et al., 2020), fibroblast growth factor receptor (FGFR) (Babina and Turner, 2017), lymphocyte-activation gene 3 (LAG3) (Saleh et al., 2019), and T-cell immunoglobulin and mucin-domain containing-3 (TIM3) (Wang et al., 2017). However, only few clinical trials on UGIC patients have shown positive curative effects; the underlying mechanisms remain elusive so far. Nearly 50% of patients in good conditions will still suffer from local recurrence or systematic metastasis after aggressive treatment (Dhakras et al., 2020; Sung et al., 2021). It seems that most of the works aim at general tumor cells rather than cancer stem cells (CSCs) in UGIC. It is because CSCs are difficult to isolate due to the limitation of early experimental conditions and heterogeneity of CSCs (Clarke et al., 2006; Sreepadmanabh and Toley, 2018). Considering that the digestive tract organs share a common external environment and perform similar functions in a system, diet-induced mucosal lesions may have similar effects on cancer of the mouth, esophagus, and stomach (Haas et al., 2012). Therefore, it is necessary to take oral cancer, esophageal cancer, and gastric cancer as a whole, that is, UGIC, for integration research.
Some laboratories have conducted pan-cancer research on UGICs. Tran et al.'s pan-cancer study on somatic mutations found that leukocyte antigen-restricted T-cell receptors targeted the KRAS (G12D) hotspot driver mutation found in many human gastrointestinal cancers (Tran et al., 2015). Another study observed that IL-6 is the main communication medium for tumor cells and cancer-related fibroblasts in a murine model (Johnson et al., 2018). IL-6 deletion inhibits the occurrence of gastrointestinal tumors through STAT3 and MEK1/2 signals (Karakasheva et al., 2018). Dana–Farber Cancer Institute discovered the new immune checkpoint biomarker TET1 and PD-1 ligands (CD274 and PDCD1LG2) (Thienpont et al., 2016; Bu et al., 2021; Rahman et al., 2021). But fewer studies focus on CSCs. The problems of poor prognosis and a high recurrence rate still require more intensive studies in UGIC.
In this work, to verify the pathogenesis and therapeutic targets of UGIC, we performed the pan-cancer analysis on UGIC. Our results identified the unique CSCs in UGIG, which are named upper gastrointestinal common cancer stem cells (UGCSCs). The core regulation network of UGCSCs suggested that inflammation-related genes, namely, CXCL8, PIGR, and CXCL3, and Wnt pathway-related genes, namely, GAST, REG1A, TFF3, and ZG16B, are activated. Further analysis indicated that mucosal damage and inflammation caused by poor dietary habits trigger the Wnt pathway and eventually induce UGIC. In addition, GAST and TFF3 activate phosphatidylinositol 3-kinase (PI3k)/Ras to enhance the metastasis and invasion of UGIC. Taken together, these results pave the way for the better diagnosis and treatment of UGIC.
The data were collected from the published literature (Table 1). For different sequencing methods of single-cell data, specific analysis procedures were applied. For Drop-seq single-cell data, Cell Ranger software (Freytag et al., 2018) (3.0.1) was adopted to calculate the cell expression counts. For Smart-seq2 single-cell sequencing data, we operated cell expression matrixes provided in the original article. The expression matrix file was then imported into R 3.6.2 for subsequent analysis.
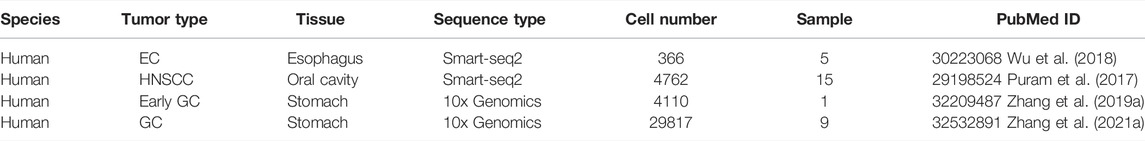
TABLE 1. Single-cell RNA-seq data of UGIC.
First, we used Seurat (Stuart et al., 2019) (3.1.4) to filter the quality of cells and delete all cells with more than 6000 expressed genes or less than 201 genes. A total of 39,057 UGIC cells and 215,291 other cancer cells were obtained. Next, standardized integration processing was performed on the cell level, sample level, and study level.
The logarithmic percentage of gene expression in cells was adopted as the standardized integration of data between different cells in the sample (Butler et al., 2018). The value of the expression of gene x in a cell was divided by the value of the expression of all genes in this cell and multiplied by the scale amplification factor, which is set to 10000 in this experiment. Then, the logarithm of this value is the normalized value of the expression of gene x in the cell. This process can reduce the deviation of gene expression values caused by different sequencing depths and sequencing methods. The formula (1) is described as follows:
where
We diminished gene features to avert the dimension disaster problem in the single-cell expression matrix. First, the logarithm of gene expression means and variances was calculated. Next, we fitted a line regression model to represent the relationship between the two values using the local polynomial regression. Next, we normalized the gene expression value through the mean value and expected variance of the model. Finally, the top variable 2000 gene features were selected for the subsequent analysis based on the normalized expression value.
We conducted an integrated analysis of multiple samples, by looking for similar sites between cells. First, the dimensionality was reduced by using canonical correlation analysis (CCA) (Andrew et al., 2013). Next, similarity anchor points were constructed, according to the similarity of sample expression matrixes. Finally, the data were integrated between different studies, according to the identified anchor points.
After PCA dimensionality reduction was performed on 39,057 UGIC cells, nine cell sets were obtained by T-distributed stochastic neighbor embedding (t-SNE) clustering. In order to identify the cell types, we calculated highly expressed genes on each cluster through the FindMarkers (Butler et al., 2018) function in Seurat. Then, through the artificial gene annotation on the CellMarker (RRID:SCR_018503) database (Zhang X. et al., 2019), the marker genes and the corresponding cell type were finally annotated. We show the statistical graph of cell types identified by EPCAM in the published articles as an example in the CellMarker (RRID:SCR_018503) database (Supplementary Figure S5). Then, we analyzed the subtypes of cancer stem cells, obtained a total of six subclasses, and calculated the differentially expressed genes (DEGs) of each subclass.
A total of 71 single-cell sequencing data (Supplementary Table S2) from six other cancers were collected. We used the same method to process other tumor single-cell data to ensure the consistency of the analysis process. First, the quality control of single-cell data obtained a total of 215,291 cells. After standardization at the cell level, sample level, and study level, we used PCA and t-SNE visualization to reduce the dimension of those single-cell data and obtained 29 cell collections. We calculated the highly expressed genes of 29 cell collections and used the CellMarker database (Zhang X. et al., 2019) to annotate the cell types. Then, we marked cancer stem cells, which are subtypes 4 and 7. The relevant marker annotations are shown in Supplementary Figure S3.
We gathered bulk RNA-seq data of UGICs in the TCGA database (Aldape et al., 2015). We obtained the expression matrix data using the cBioPortal (Cerami et al., 2012; Gao et al., 2013), including 522 HNSCC samples, 185 EC samples, and 415 GC samples. Three types of UGICs were congregated with the data label “hnsc_tcga,” “esca_tcga,” and “stad_tcga”. DESeq2 (RRID:SCR_000154) (Love et al., 2014) (1.26.0) software was used to measure the DEGs in the cancer sample and the corresponding normal sample.
We annotated the function and pathway information of the significantly different genes in the Gene Ontology (GO) (Ashburner et al., 2000; Ashburner, 2021) database and Kyoto Encyclopedia of Genes and Genomes (KEGG) (RRID:SCR_012773) (Kanehisa et al., 2021) database using the clusterProfiler (RRID:SCR_016884) (Yu et al., 2012) (3.14.3) package in R (3.6.2) software. The top 15 terms are presented in Figure 4.
We adjusted the gene set enrichment score between the specific differential genes and the cancer-related gene sets through the Gene Set Enrichment Analysis (GSEA) (Subramanian et al., 2005) software (4.1.0). “C6: oncogenic signatures” was selected as the existing cancer-related gene set in GSEA software. We filtered several parameters to draw gene enrichment results. The normalized enrichment score (NES) was larger than 1. The normalized significance level (NSL) p-value was lower than 0.05.
We collected all human entries in the String database (RRID:SCR_005223) and deleted low-quality and text mining entries (Szklarczyk et al., 2021). After removing the duplicated edges and self-loops, we constructed a human protein–protein interaction network (PPIN) with 19,267 proteins and 1,689,887 edges by Cytoscape software (RRID:SCR_003032) (Shannon et al., 2003) (3.7.1). Then, 174 genes specifically expressed in UGCSC were mapped to the PPIN. After removing outlier proteins, a regulatory sub-network composed of 144 protein nodes and 545 edges was constructed. Next, we appraised the topological attributes of the network and selected the degree and clustering coefficient (CC) to measure the function of the sub-network (Sporns, 2013). The degree represents the number of connections through a particular node, which measures the importance of the node in the network. CC represents the closeness of connections between a node and the surrounding nodes, which demonstrates the network closeness and function similarity. The formula (2) is described as follows:
where
We manually reviewed the tumor-related literature studies published since 2000 to screen functions and pathways of DEGs. Then, we formulated UGCSC function networks by integrating different genes and the known inflammation and Wnt pathways (Figure 6).
We collected 39,057 tumor single-cell sequencing data from 30 patients including 4762 HNSCC cells, 366 EC cells, and 33,927 GC cells (Figure 1A). It is noteworthy that EC samples applied Smart-seq2 single-cell sequencing technology (Picelli et al., 2014) which is manually sequencing each cell. So the EC group has few cells but higher confidence. After quality filtering (see Methods) and removing the batch effect, more than 70 million transcripts were obtained from 39,057 cells. Subsequently, we classified cells into different clusters by using T-distributed stochastic neighbor embedding (t-SNE) methods in Seurat software (Supplementary Figure S1). Through marker genes, these identified cell clusters could be assigned to known cell lineages: T cells, B cells, epithelial cells, natural killer cells, fibroblasts, plasma cells, cancer stem cells, mast cells, and endothelial cells (Figure 1B). To corroborate these profiles, we showed the high expression gene distribution heatmap of each cell type and the expression abundance of marker genes of each type (Figure 1C; Supplementary Table S1). Each cell type has specific marker genes: CD3D, KRT8, MS4A1, PDGFRA, IGHG3, EPCAM, ECSCR, TPSB2, and CCL3 (Figure 1D). The violin plot of marker genes shows that the expression of most marker genes is specific, which indicates that the classification of cell types is accurate and is very helpful for subsequent analysis (Figure 1E). Taken together, these results indicate that the cell classification was accurate, and most of the cells were classified into the correct cell type. The distribution of samples and cancer types is shown in Supplementary Figures S1, S2. We also counted the number and frequency of all cell types in HNSCC, EC, and GC and provided the results in Supplementary Figure S6.
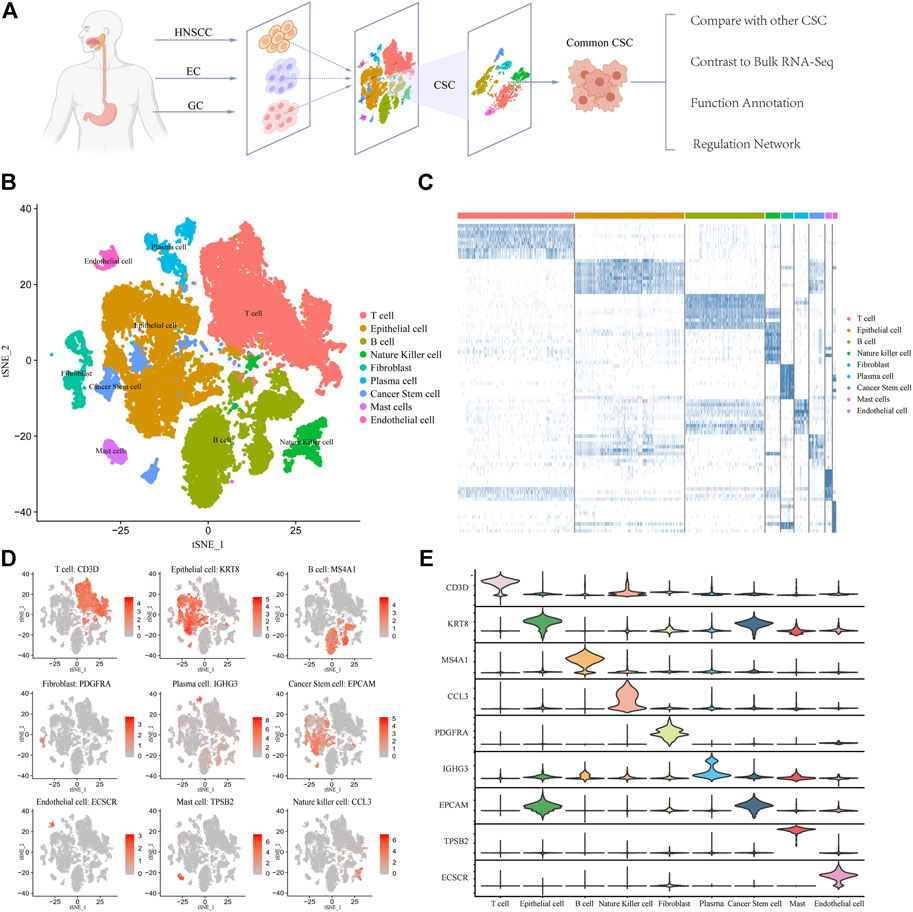
FIGURE 1. Expression profiling of 39,057 single cells in UGIC. (A) Workflow of sample processing, cell type annotation, and functional analysis for 30 samples in UGIC. (B) t-SNE of 39,057 cells profiled here, with each cell color-coded for the associated cell type. (C) Heatmap of the expression pattern in each cell type. (D) Expression of marker genes for the cell types defined above each panel. (E) Expression trend of marker genes for each cell type in the violin chart.
We focused on cancer stem cell types in order to reveal the pathogenesis and distant metastasis mechanism of UGIC. We collected a total of 1,586 CSCs (Figures 2A,B) including 136 HNSCC cells, 23 EC cells, and 1427 GC cells. Due to the heterogeneity of CSCs, there are differences in the same type of cancer while similarities exist in different types of cancers, coincident with the characteristics of the remote metastasis and recurrence of the cancers. Therefore, we performed a cluster analysis of CSCs, and a total of six sub-clusters were found. After annotating and analyzing all sub-clusters, sub-cluster 0 is ubiquitous in UGICs, including 19 EC stem cells, 356 GC stem cells, and 114 HNSCC stem cells, which proves that sub-cluster 0 preliminarily meets the characteristics of common CSCs (Figures 2C,D). Therefore, we concentrated on sub-cluster 0 in the follow-up analysis.
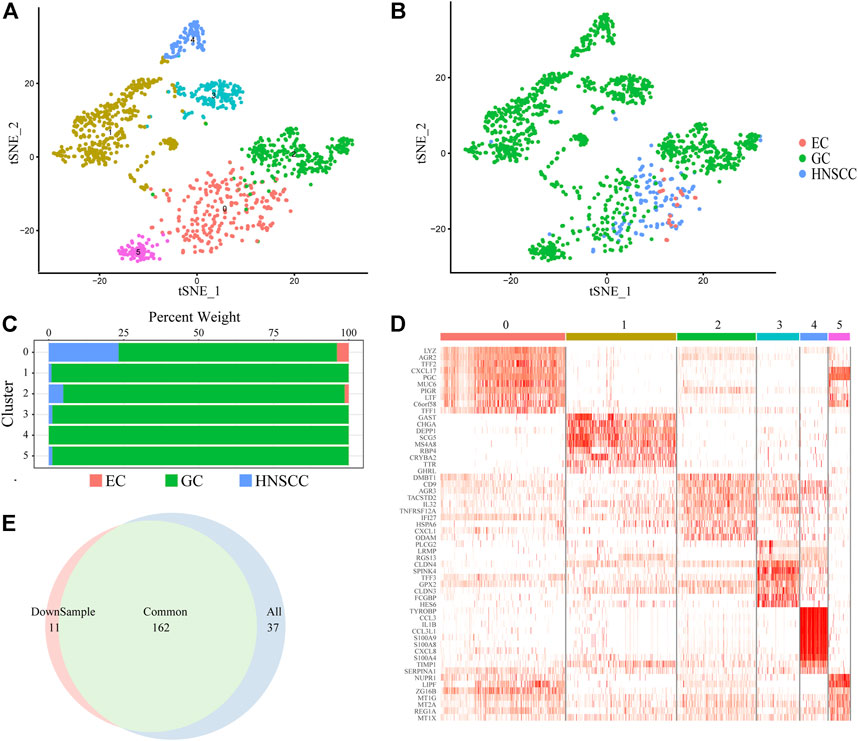
FIGURE 2. Cancer stem cell clusters. (A) t-SNE plot of 1,586 CSC cells color-coded by sub-clusters. (B) t-SNE plot color-coded for cancer type of origin. (C) Histogram of cell numbers in each CSC cancer type. (D) Heatmap of the expression pattern of six sub-clusters in CSC. (E) Venn diagram of DEGs in downsample CSCs and All CSCs.
To verify whether sub-cluster 0 reflects the characteristics of UGIC rather than only GC, we performed a down-sampling process in sub-cluster 0 since there are more than 70% GC stem cells in sub-cluster 0. We randomly selected the same number of GC cells as the HNSCC cells and named new sub-cluster 0. Subsequently, we compared the differential genes between sub-cluster 0 and the new sub-cluster 0 in CSCs. The merge ratio is 77.14% (Figure 2E), which means these two sub-clusters share the same differential gene set. These results indicate that the differentially expressed genes (DEGs) of sub-cluster 0 represent the features of UGICs.
To validate the specificity of UGCSCs, we compared UGCSCs with other tumor cells. We collected 71 samples (215,291 cells) from six types of cancers including glioma (GLM), melanoma (MELA), osteosarcoma (OSTC), breast cancer (BC), ovarian cancer (OVC), and stellate cell cancer (SCC) (Supplementary Table S2). After normalizing the cells and removing the batch effect (see Method), all the cells were gathered into 29 sub-clusters (Figures 3A,C). After annotating all cancer cells in the CellMarker database, we noticed that there are plenty of cell types due to the complexity of tissue types involved. Therefore, we only annotated CSCs by using marker genes. The tumor stem cells were obviously aggregated with CXCR4 markers (Figure 3B; Supplementary Figure S3), which are sub-clusters 4 and 7 and contain 21323 cells, as circled in Figure 3A. We compared CSCs of other cancers with CSCs of UGICs. We re-clustered and obtained 31 sub-clusters in all CSCs, which reveals the differences between CSCs of different tumor types (Figure 3D). But at the same time, the cluster distribution of CSCs from different tissues is uniform, which indicates that there are similarities between different tissues in CSCs (Figures 3E,F). This phenomenon is also coincident with the heterogeneity of tumors. The cluster annotation of cancer types shows that the UGCSC is self-clustering and far away from other tumor CSCs (Figure 3E). Therefore, the UGCSC is the specific cancer stem cell in UGIC while UGCSC does not exist in other cancers.
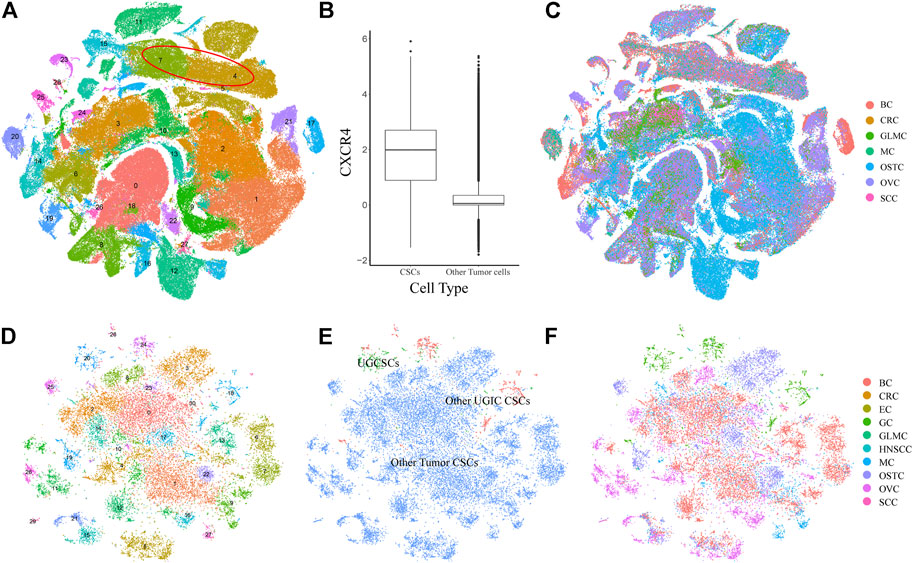
FIGURE 3. Expression profiling of 215,291 single cells in six cancer types. (A) t-SNE plot of 215,291 single cells in six cancer types, color-coded by 29 clusters. Clusters 4 and 7 are cancer stem cells, which are marked by a red circle. (B) Box plot shows the expression of the CSC marker gene CXCR4. The x-axis represents the cell type. The y-axis represents the log value of the normalized CXCR4 expression. (C) t-SNE plot of other cancer cells, color-coded by the cancer type. (D) t-SNE plot of all CSCs, color-coded by clusters. (E) t-SNE plot of all CSCs, color-coded by the CSC type. (F) t-SNE plot of all CSCs, color-coded by cancer types.
We comprehensively analyzed the distribution and function of UGCSCs. The cell sources of UGCSC cancers were analyzed and counted (Figure 4A). As shown in Figure 4, UGCSCs are averagely expressed in UGIC patients, including 10 GC patients, four EC patients, and nine HNSCC patients. In summary, the UGCSCs are distributed uniformly, which proves that the UGCSC is common in upper gastrointestinal patients.
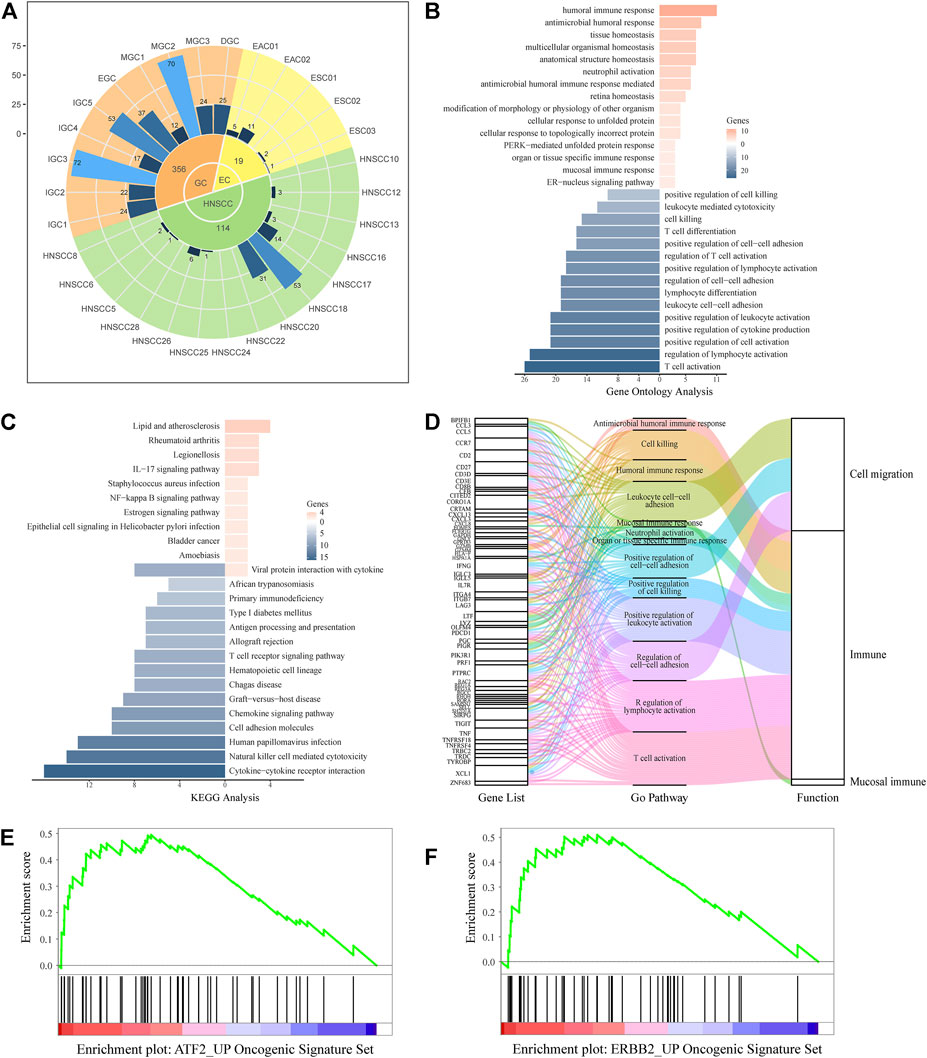
FIGURE 4. UGCSC function annotation. (A) Sample source of UGCSCs. Green, yellow, and orange represent HNSCC, EC, and GC, respectively. The inner circle is for three types of cancer, and the middle circle is the number of cells in each cancer. The outer circle represents the number of cells in each sample. (B) Gene ontology function annotation of UGCSC DEGs. (C) Pathway analysis of UGCSC differentially expressed genes. (D) Gene function integration in UGCSCs. (E) UGCSC differentially expressed gene set enrichment analysis in the ATF2_UP tumor set. (F) UGCSC differentially expressed gene set enrichment analysis in the ERBB2_UP tumor set.
We analyzed the expression network of UGCSCs. First, we compared the expression profiles of UGCSCs and all other tumor stem cells and obtained 174 genes with significant differences, including 33 upregulated genes and 141 downregulated genes (Supplementary Data S1). We uncovered that the gene information function reflects the characteristics of UGCSCs as a digestive system and as cancer stem cells by analyzing the function annotation (Ashburner et al., 2000) of DEGs (Figures 4B,D). The upregulated genes are related to antibacterial response, such as “antibacterial humoral response,” “antibacterial humoral immune response-mediated,” and “mucosal immune response,” which are consistent with the function of the digestive tract in the human body. In addition, the functions of downregulated genes mainly focus on reducing the activity of T cells and lymphocytes and downregulating cell killing which could reduce the body’s immune response and enhance the survival rate of tumor cells, and these are also the characteristics of cancer stem cells. The downregulated genes also play a role in the regulation of cell–cell adhesion to facilitate the distant metastasis of tumors, which is in line with the feature of metastasis. Through the analysis of the KEGG pathway (Kanehisa et al., 2021), we observed that the significantly differentially expressed genes are enriched in inflammation-related mucosal infections such as “Staphylococcus aureus infection,” “epithelial cell signaling in Helicobacter pylori infection,” “IL-17 signaling pathway,” and “chemokine signal pathway” (Figure 4C). These results uncovered a potential carcinogenic factor of UGIC, that is, mucosal damage induced activation and mutation in inflammatory pathways. Through gene set enrichment analysis (GSEA), we found that the significantly differentially expressed genes were significantly enriched in ATF2- and ERBB2-related cancer genes (Figures 4E,F). To further confirm the reliability of these genes, we calculated the DEGs between UGCSCs and 30,267 cells of normal tissues in the upper alimentary tract (Cillo et al., 2020; Zhang X. et al., 2021) (Supplementary Figure S7A). The DEGs of UGCSCs and tumor cells are few and share fewer genes with the DEGs of UGCSCs and normal cells (Supplementary Figure S7B). Also, the functional analysis of DEGs of UGCSCs and normal cells indicates that the functional pathways are related to cell development, which is a common feature of tumors (Supplementary Figure S7C). In summary, these results indicate that the DEGs of UGCSCs and tumor cells are oncogenes related to the function of the digestive tract.
In order to study the pathogenic mechanisms that may exist in UGCSCs, we mapped 174 proteins into the human protein–protein interaction network (PPIN). We constructed PPIN and deleted low-quality text mining terms in the String database (Szklarczyk et al., 2021). After mapping 174 DEGs in UGCSCs into PPIN, an interaction network consisting of 144 proteins and 545 edges was obtained (Figure 5A). Through the analysis of the topological properties of the network, we found that the degree of DEGs in PPIN is 362.174, which is significantly higher than 175.418 in the human total network (Figure 5B). This result indicates that the shortest path through different genes is significantly higher than the average value (Figure 5B), which implied that these genes are hub genes in the UGCSC network. Furthermore, another topological property, the clustering coefficient (CC), is significantly higher than the background network, which points out that the 144 genes are closely linked compared with the random gene set in the network. The close interaction means a similar or synergistic function in cells. Through the comprehensive analysis of degree and CC, we inferred that the 144 genes are tightly connected hub genes in PPIN, which means that they play an important function in UGCSCs as a co-operative hub gene set.

FIGURE 5. Network analysis of UGCSCs. (A) Protein regulation networks of DEGs in UGCSCs. (B) Topological attribute comparison of UGCSC sub-networks and whole PPIN.
We have performed functional annotations on the possible functions of these genes and inferred regulatory pathways with the aim to explore the possible pathogenic mechanisms and potential therapeutic targets in UGIC. We analyzed the regulation pathway of those genes through published articles and proved that the upregulated genes are basically related to cancer (Supplementary Table S3). Here are some exciting discoveries. Some genes are related to inflammatory pathways, such as CXCL8 (Ha et al., 2017), BPIFB1 (Li J. et al., 2020), PIGR (Kakiuchi et al., 2020), CXCL3, and RNASE1 (Wang et al., 2006), and some genes are related to specific functions of the digestive tract, such as GAST (Giraud et al., 2016), REG1A (Sha et al., 2019), and TFF3 (Braga Emidio et al., 2020). These results illustrated that there may have similar pathogenic mechanisms and common regulatory pathways in some UGICs. We speculated that mucosal damage is induced by long-term unhealthy eating habits, which include smoking, drinking, and hot food breed inflammation. Persistent inflammation leads to carcinogenic mutations and early gastrointestinal tumors. These conjectures have been confirmed in the specific regulatory network of UGCSCs. Based on the detected differentially expressed genes and the mining of relevant research literature studies, we speculated the pathogenesis of the disease, as shown in the Figure 6. Inflammation-associated interleukin (CXCL8 and CXCL3) and inflammation defense-related BPIFB1, PIGR, and RNASE1 are activated in UGCSCs. Combined with the epidemiological investigation of gastrointestinal cancer, there is a hypothesis that chronic inflammation is incited by mucosal damage due to long-term bad eating habits. We present that the cancerous chronic inflammation is activated by GAST, REG1A, TFF3, and ZG16B in the Wnt signaling pathway. Upregulated hPG80 and TFF3 induce PI3K/Ras and lead to tumor cell growth and invasion, which may be one reason for the poor prognosis of UGIC. Hence, this resource provides a novel view for the occurrence and development of UGIC and the advancement of gastrointestinal cancer diagnosis and therapy.
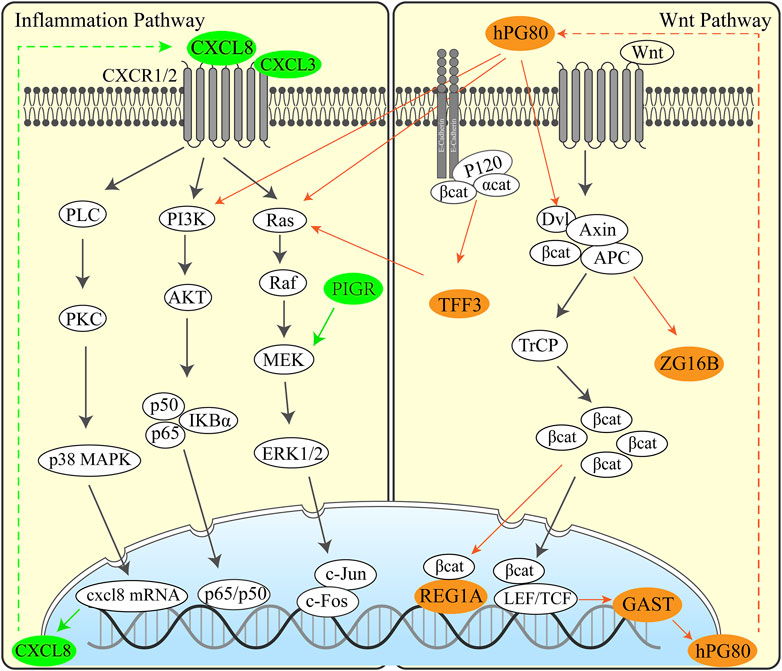
FIGURE 6. Regulation pathway in UGCSCs. Inflammatory pathway (left) and Wnt pathway (right) activated in UGCSC. The green genes represent inflammatory factors that are highly expressed in UGCSCs. The brown–yellow genes represent Wnt-related factors that are highly expressed in UGCSCs.
In this article, we collectively analyzed single-cell sequencing data of HNSCC, EC, and GC and identified a specific cancer stem cell type in UGIC: UGCSCs. Then, we presented the unique expression pattern and hub gene set in UGCSCs by comparing it with other tumors’ single-cell RNA-seq data. We declared the common carcinogens of UGICs that the mucosa damage of the digestive tract induces chronic inflammation due to unhealthy eating habits. The hub gene set provides promising entry points for the design of novel therapies including CXCL8, CXCL3, GAST, TFF3, PIGR, and RNASE1.
Here, we provided a comprehensive catalog of human UGICs at single-cell resolution. In the integrative analysis of UGICs, we confirmed that there are specific cancer stem cells in UGIC, which are named UGCSCs. This discovery provides a new perspective for scientific analysis of the poor prognosis and easy recurrence of UGIC. By comparing the tumor stem cells of six cancers, we extracted the core gene set that plays an important role in UGCSCs and explored the possible pathogenic pathway of UGIC and core genes including GAST, CXCL8, CXCL3, PIGR, REG1A, and TFF3. With further in-depth research, these genes can also be used as diagnostic markers or possible therapeutic targets for gastrointestinal cancers.
However, all cell types and subtypes cannot possibly be described here; some key results emerge. On one hand, the distribution of all cell types in UGIC is shown in the cell clustering figure (Figure 1B). On the other hand, through the comparative analysis with bulk RNA-seq sequencing data, the DEGs between single-cell data and bulk RNA-seq data varied significantly. Therefore, we performed further research only on cancer stem cells. Intriguing questions remain as to whether there are specific immune cells in UGIC and whether the immune cell counts would have an impact on the prognosis of UGIC.
The single-cell data of UGIC and the six cancer types are composed of cells from different patients. Some sub-clusters of cell types have different abundances due to sample differences, according to the results of cell clustering. We removed batch effects and deleted outlier cells from the clustering result. In this way, the impact of samples from different patient sources is reduced.
We performed the same analysis on bulk RNA sequencing data; however, due to the varieties of cell types and the low proportion of CSCs in cancer tissues, the pathways and therapeutic targets were not discovered. We collected 1,122 patients and 1,966 normal samples of bulk RNA-seq data in TCGA database. The differentially expressed genes of the three cancers were compared with those of UGCSCs. The result suggests that the merge ratio is only 0.79%. Moreover, the function of differentially expressed genes is mostly about cell cycle-related pathways in bulk RNA-seq data (Supplementary Figure S4). We inferred that plenty of cell types in UGIC generates noises in UGIC expression profile information and makes some core pathways and genes undetectable, while single-cell RNA-seq can filter noise signals by extracting specific cell types.
Last, we constructed a regulatory network of UGCSCs under the framework of the existing experimental knowledge atlas. More and other types of data such as downstream genes and mutation information of the core regulatory network need to be further studied. However, we proposed UGCSCs and their regulatory networks based on the analysis of single-cell data from more than 100 patients and more than 25,000 cells, which has strong robustness. These data build a framework for a deeper understanding of the molecular mechanisms of UGCSCs and the regulation network of hub genes and might be applied to screen for molecular target drugs to improve the efficacy and outcomes for UGIC patients.
The UGIC single-cell sequencing data are available through SRR6133148 (Wu et al., 2018), GSE103322 (Puram et al., 2017), and GSE134520 (Zhang et al., 2019a) in the Gene Expression Omnibus (GEO) and HRA000051 (Zhang et al., 2021a) in Genome Sequence Archive (GSA). The normal single cell data of upper alimentary tract are available through GSE139324 (Cillo et al., 2020) and GSE160269 (Zhang et al., 2021b) in the GEO database and HRA000051 (Zhang et al., 2021a) in GSA database. The other cancer single-cell sequencing data are available through GEO database: GSE152048 (Zhou et al., 2020), GSE89567 (Venteicher et al., 2017), GSE72056 (Tirosh et al., 2016), GSE75688 (Chung et al., 2017), GSE84465 (Darmanis et al., 2017), and GSE102130 (Filbin et al., 2018) in GEO database and https://lambrechtslab.sites.vib.be/ (Qian et al., 2020) website. Gene interactions networks were identified using the STRING database (https://string-db.org) (Szklarczyk et al., 2021). GO term analysis was performed by using the GENEONTOLOGY database (http://geneontology.org/) (Ashburner et al., 2000). Tumor gene sets enrichment analysis was performed using the MSigDB database (www.gseamsigdb.org) (Subramanian et al., 2005). All other data are included in the article and its Supplementary Information files or available from the corresponding authors upon reasonable request.
Study concept and design: LL and HL; acquisition of data: LL and YjZ; analysis and interpretation of data: LL, YjZ, YR, ZC, YnZ, XW, HZ, and HL; drafting of the manuscript: LL, HZ, and HL. All authors reviewed and approved the final draft of the manuscript.
This work was supported by the National Key R&D Program of China (2018YFC0910500), the SJTU-Yale Collaborative Research Seed Fund, and the Neil Shen’s SJTU Medical Research.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fbioe.2022.849798/full#supplementary-material
Akhtar, S. (2013). Areca Nut Chewing and Esophageal Squamous-Cell Carcinoma Risk in Asians: A Meta-Analysis of Case-Control Studies. Cancer Causes Control 24, 257–265. doi:10.1007/s10552-012-0113-9
Aldape, K., Zadeh, G., Mansouri, S., Reifenberger, G., and von Deimling, A. (2015). Glioblastoma: Pathology, Molecular Mechanisms and Markers. Acta Neuropathol. 129, 829–848. doi:10.1007/s00401-015-1432-1
Andrew, G., Arora, R., Bilmes, J., and Livescu, K. (2013). “Deep Canonical Correlation Analysis,” in International Conference on Machine Learning, PMLR, Atlanta, GA, USA, Jun 16-21, 2013, 1247–1255.
Ashburner, M., Ball, C. A., Blake, J. A., Botstein, D., Butler, H., Cherry, J. M., et al. (2000). Gene Ontology: Tool for the Unification of Biology. Nat. Genet. 25, 25–29. doi:10.1038/75556
Ashburner, (2021). The Gene Ontology Resource: Enriching a GOld Mine. J. Nucl. Acids Res. 49 (D1), D325–D334.
Babina, I. S., and Turner, N. C. (2017). Advances and Challenges in Targeting FGFR Signalling in Cancer. Nat. Rev. Cancer 17, 318–332. doi:10.1038/nrc.2017.8
Braga Emidio, N., Brierley, S. M., Schroeder, C. I., and Muttenthaler, M. (2020). Structure, Function, and Therapeutic Potential of the Trefoil Factor Family in the Gastrointestinal Tract. ACS Pharmacol. Transl. Sci. 3, 583–597. doi:10.1021/acsptsci.0c00023
Bray, F., Ferlay, J., Soerjomataram, I., Siegel, R. L., Torre, L. A., and Jemal, A. (2018). Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 68, 394–424. doi:10.3322/caac.21492
Bu, X., Juneja, V. R., Reynolds, C. G., Mahoney, K. M., Bu, M. T., McGuire, K. A., et al. (2021). Monitoring PD-1 Phosphorylation to Evaluate PD-1 Signaling during Antitumor Immune Responses. Cancer Immunol. Res. 9, 1465–1475. doi:10.1158/2326-6066.CIR-21-0493
Butler, A., Hoffman, P., Smibert, P., Papalexi, E., and Satija, R. (2018). Integrating Single-Cell Transcriptomic Data across Different Conditions, Technologies, and Species. Nat. Biotechnol. 36, 411–420. doi:10.1038/nbt.4096
Cerami, E., Gao, J., Dogrusoz, U., Gross, B. E., Sumer, S. O., Aksoy, B. A., et al. (2012). The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2, 401–404. doi:10.1158/2159-8290.CD-12-0095
Chakravarthy, A., Furness, A., Joshi, K., Ghorani, E., Ford, K., Ward, M. J., et al. (2018). Pan-cancer Deconvolution of Tumour Composition Using DNA Methylation. Nat. Commun. 9, 1–13. doi:10.1038/s41467-018-05570-1
Chung, W., Eum, H. H., Lee, H.-O., Lee, K.-M., Lee, H.-B., Kim, K.-T., et al. (2017). Single-cell RNA-Seq Enables Comprehensive Tumour and Immune Cell Profiling in Primary Breast Cancer. Nat. Commun. 8, 1–12. doi:10.1038/ncomms15081
Cillo, A. R., Kürten, C. H. L., Tabib, T., Qi, Z., Onkar, S., Wang, T., et al. (2020). Immune Landscape of Viral- and Carcinogen-Driven Head and Neck Cancer. Immunity 52, 183–199. doi:10.1016/j.immuni.2019.11.014
Clarke, M. F., Dick, J. E., Dirks, P. B., Eaves, C. J., Jamieson, C. H. M., Jones, D. L., et al. (2006). Cancer Stem Cells-Perspectives on Current Status and Future Directions: AACR Workshop on Cancer Stem Cells. Cancer Res. 66, 9339–9344. doi:10.1158/0008-5472.CAN-06-3126
Cui, Y., Guo, W., Li, Y., Shi, J., Ma, S., and Guan, F. (2021). Pan-cancer Analysis Identifies ESM1 as a Novel Oncogene for Esophageal Cancer. Esophagus 18, 326–338. doi:10.1007/s10388-020-00796-9
Dai, L., Huang, Z., and Li, W. (2021). Analysis of the PD-1 Ligands Among Gastrointestinal Cancer Patients: Focus on Cancer Immunity. Front. Oncol. 11, 525. doi:10.3389/fonc.2021.637015
Darmanis, S., Sloan, S. A., Croote, D., Mignardi, M., Chernikova, S., Samghababi, P., et al. (2017). Single-cell RNA-Seq Analysis of Infiltrating Neoplastic Cells at the Migrating Front of Human Glioblastoma. Cel Rep. 21, 1399–1410. doi:10.1016/j.celrep.2017.10.030
Dhakras, P., Uboha, N., Horner, V., Reinig, E., and Matkowskyj, K. A. (2020). Gastrointestinal Cancers: Current Biomarkers in Esophageal and Gastric Adenocarcinoma. Transl. Gastroenterol. Hepatol. 5, 55. doi:10.21037/tgh.2020.01.08
Filbin, M. G., Tirosh, I., Hovestadt, V., Shaw, M. L., Escalante, L. E., Mathewson, N. D., et al. (2018). Developmental and Oncogenic Programs in H3K27M Gliomas Dissected by Single-Cell RNA-Seq. Science 360, 331–335. doi:10.1126/science.aao4750
Freytag, S., Tian, L., Lönnstedt, I., Ng, M., and Bahlo, M. (2018). Comparison of Clustering Tools in R for Medium-Sized 10x Genomics Single-Cell RNA-Sequencing Data. F1000Res 7, 1297. doi:10.12688/f1000research.15809.2
Gandini, S., Botteri, E., Iodice, S., Boniol, M., Lowenfels, A. B., Maisonneuve, P., et al. (2008). Tobacco Smoking and Cancer: A Meta-Analysis. Int. J. Cancer 122, 155–164. doi:10.1002/ijc.23033
Gao, J., Aksoy, B. A., Dogrusoz, U., Dresdner, G., Gross, B., Sumer, S. O., et al. (2013). Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal. 6, pl1. doi:10.1126/scisignal.2004088
Giraud, J., Failla, L. M., Pascussi, J.-M., Lagerqvist, E. L., Ollier, J., Finetti, P., et al. (2016). Autocrine Secretion of Progastrin Promotes the Survival and Self-Renewal of Colon Cancer Stem-like Cells. Cancer Res. 76, 3618–3628. doi:10.1158/0008-5472.CAN-15-1497
Goldstein, B. Y., Chang, S.-C., Hashibe, M., La Vecchia, C., and Zhang, Z.-F. (2010). Alcohol Consumption and Cancers of the Oral Cavity and Pharynx from 1988 to 2009: an Update. Eur. J. Cancer Prev. 19, 431–465. doi:10.1097/CEJ.0b013e32833d936d
González, C. A., Sala, N., and Rokkas, T. (2013). Gastric Cancer: Epidemiologic Aspects. Helicobacter 18, 34–38. doi:10.1111/hel.12082
Ha, H., Debnath, B., and Neamati, N. (2017). Role of the CXCL8-CXCR1/2 axis in Cancer and Inflammatory Diseases. Theranostics 7, 1543–1588. doi:10.7150/thno.15625
Haas, S. L., Ye, W., and Löhr, J.-M. (2012). Alcohol Consumption and Digestive Tract Cancer. Curr. Opin. Clin. Nutr. Metab. Care 15, 457–467. doi:10.1097/MCO.0b013e3283566699
Johnson, D. E., O'Keefe, R. A., and Grandis, J. R. (2018). Targeting the IL-6/JAK/STAT3 Signalling axis in Cancer. Nat. Rev. Clin. Oncol. 15, 234–248. doi:10.1038/nrclinonc.2018.8
Kakiuchi, N., Yoshida, K., Uchino, M., Kihara, T., Akaki, K., Inoue, Y., et al. (2020). Frequent Mutations that Converge on the NFKBIZ Pathway in Ulcerative Colitis. Nature 577, 260–265. doi:10.1038/s41586-019-1856-1
Kanehisa, M., Sato, Y., and Kawashima, M. (2021). KEGG Mapping Tools for Uncovering Hidden Features in Biological Data. Protein Sci. 31, 47–53. doi:10.1002/pro.4172
Karakasheva, T. A., Lin, E. W., Tang, Q., Qiao, E., Waldron, T. J., Soni, M., et al. (2018). IL-6 Mediates Cross-Talk between Tumor Cells and Activated Fibroblasts in the Tumor Microenvironment. Cancer Res. 78, 4957–4970. doi:10.1158/0008-5472.CAN-17-2268
Li, J., Xu, P., Wang, L., Feng, M., Chen, D., Yu, X., et al. (2020a). Molecular Biology of BPIFB1 and its Advances in Disease. Ann. Transl. Med. 8, 651. doi:10.21037/atm-20-3462
Li, Z., Chen, S., Feng, W., Luo, Y., Lai, H., Li, Q., et al. (2020b). A Pan-Cancer Analysis of HER2 index Revealed Transcriptional Pattern for Precise Selection of HER2-Targeted Therapy. EBioMedicine 62, 103074. doi:10.1016/j.ebiom.2020.103074
Love, M. I., Huber, W., and Anders, S. (2014). Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 15, 1–21. doi:10.1186/s13059-014-0550-8
Picelli, S., Faridani, O. R., Björklund, Å. K., Winberg, G., Sagasser, S., and Sandberg, R. (2014). Full-length RNA-Seq from Single Cells Using Smart-Seq2. Nat. Protoc. 9, 171–181. doi:10.1038/nprot.2014.006
Puram, S. V., Tirosh, I., Parikh, A. S., Patel, A. P., Yizhak, K., Gillespie, S., et al. (2017). Single-cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell 171, 1611–1624. doi:10.1016/j.cell.2017.10.044
Qian, J., Olbrecht, S., Boeckx, B., Vos, H., Laoui, D., Etlioglu, E., et al. (2020). A Pan-Cancer Blueprint of the Heterogeneous Tumor Microenvironment Revealed by Single-Cell Profiling. Cell Res. 30, 745–762. doi:10.1038/s41422-020-0355-0
Rahman, S. A., Yagnik, B., Bally, A. P., Morrow, K. N., Wang, S., Vanderford, T. H., et al. (2021). PD-1 Blockade and Vaccination Provide Therapeutic Benefit against SIV by Inducing Broad and Functional CD8 + T Cells in Lymphoid Tissue. Sci. Immunol. 6, eabh3034. doi:10.1126/sciimmunol.abh3034
Saleh, R. R., Peinado, P., Fuentes-Antrás, J., Pérez-Segura, P., Pandiella, A., Amir, E., et al. (2019). Prognostic Value of Lymphocyte-Activation Gene 3 (LAG3) in Cancer: a Meta-Analysis. Front. Oncol. 9, 1040. doi:10.3389/fonc.2019.01040
Sha, Y.-L., Liu, S., Yan, W.-W., and Dong, B. (2019). Wnt/β-catenin Signaling as a Useful Therapeutic Target in Hepatoblastoma. Biosci. Rep. 39, BSR20192466. doi:10.1042/BSR20192466
Shannon, P., Markiel, A., Ozier, O., Baliga, N. S., Wang, J. T., Ramage, D., et al. (2003). Cytoscape: a Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 13, 2498–2504. doi:10.1101/gr.1239303
Sporns, O. (2013). Network Attributes for Segregation and Integration in the Human Brain. Curr. Opin. Neurobiol. 23, 162–171. doi:10.1016/j.conb.2012.11.015
Sreepadmanabh, M., and Toley, B. J. (2018). Investigations into the Cancer Stem Cell Niche Using Iin-Vvitro 3-D Tumor Models and Microfluidics. Biotechnol. Adv. 36, 1094–1110. doi:10.1016/j.biotechadv.2018.03.009
Stuart, T., Butler, A., Hoffman, P., Hafemeister, C., Papalexi, E., Mauck, W. M., et al. (2019). Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902. doi:10.1016/j.cell.2019.05.031
Subramanian, A., Tamayo, P., Mootha, V. K., Mukherjee, S., Ebert, B. L., Gillette, M. A., et al. (2005). Gene Set Enrichment Analysis: a Knowledge-Based Approach for Interpreting Genome-wide Expression Profiles. Proc. Natl. Acad. Sci. U.S.A. 102, 15545–15550. doi:10.1073/pnas.0506580102
Sung, H., Ferlay, J., Siegel, R. L., Laversanne, M., Soerjomataram, I., Jemal, A., et al. (2021). Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 71, 209–249. doi:10.3322/caac.21660
Szklarczyk, D., Gable, A. L., Nastou, K. C., Lyon, D., Kirsch, R., Pyysalo, S., et al. (2021). The STRING Database in 2021: Customizable Protein-Protein Networks, and Functional Characterization of User-Uploaded Gene/measurement Sets. Nucleic Acids Res. 49, D605–D612. doi:10.1093/nar/gkaa1074
Thienpont, B., Steinbacher, J., Zhao, H., D’Anna, F., Kuchnio, A., Ploumakis, A., et al. (2016). Tumour Hypoxia Causes DNA Hypermethylation by Reducing TET Activity. Nature 537, 63–68. doi:10.1038/nature19081
Tirosh, I., Izar, B., Prakadan, S. M., Wadsworth, M. H., Treacy, D., Trombetta, J. J., et al. (2016). Dissecting the Multicellular Ecosystem of Metastatic Melanoma by Single-Cell RNA-Seq. Science 352, 189–196. doi:10.1126/science.aad0501
Tran, E., Ahmadzadeh, M., Lu, Y.-C., Gros, A., Turcotte, S., Robbins, P. F., et al. (2015). Immunogenicity of Somatic Mutations in Human Gastrointestinal Cancers. Science 350, 1387–1390. doi:10.1126/science.aad1253
Venteicher, A. S., Tirosh, I., Hebert, C., Yizhak, K., Neftel, C., Filbin, M. G., et al. (2017). Decoupling Genetics, Lineages, and Microenvironment in IDH-Mutant Gliomas by Single-Cell RNA-Seq. Science 355, eaai8478. doi:10.1126/science.aai8478
Wang, L., Zhu, J.-S., Song, M.-Q., Chen, G.-Q., and Chen, J.-L. (2006). Comparison of Gene Expression Profiles between Primary Tumor and Metastatic Lesions in Gastric Cancer Patients Using Laser Microdissection and cDNA Microarray. World J. Gastroenterol. 12, 6949. doi:10.3748/wjg.v12.i43.6949
Wang, Z., Yin, N., Zhang, Z., Zhang, Y., Zhang, G., and Chen, W. (2017). Upregulation of T-Cell Immunoglobulin and Mucin-Domain Containing-3 (Tim-3) in Monocytes/macrophages Associates with Gastric Cancer Progression. Immunol. Invest. 46, 134–148. doi:10.1080/08820139.2016.1229790
Westphalen, C. B., Preinfalk, A., Kruger, S., Haas, M., Renz, B. W., Riener, M.-O., et al. (2019). Neurotrophic Tropomyosin Receptor Kinase (NTRK) and Nerve Growth Factor (NGF) Are Not Expressed in Caucasian Patients with Biliary Tract Cancers: Pooled Data from Three Independent Cohorts. Clin. Transl. Oncol. 21, 1108–1111. doi:10.1007/s12094-018-02030-6
Wu, H., Yu, J., Li, Y., Hou, Q., Zhou, R., Zhang, N., et al. (2018). Single-cell RNA Sequencing Reveals Diverse Intratumoral Heterogeneities and Gene Signatures of Two Types of Esophageal Cancers. Cancer Lett. 438, 133–143. doi:10.1016/j.canlet.2018.09.017
Yamada, T., Alpers, D. H., Kalloo, A. N., Kaplowitz, N., Owyang, C., and Powell, D. W. (2011). Textbook of Gastroenterology. Seattle, Washington: John Wiley & Sons.
Yang, Y., Zhang, J., Chen, Y., Xu, R., Zhao, Q., and Guo, W. (2020). MUC4, MUC16, and TTN Genes Mutation Correlated with Prognosis, and Predicted Tumor Mutation burden and Immunotherapy Efficacy in Gastric Cancer and pan‐cancer. Clin. Transl. Med. 10, e155. doi:10.1002/ctm2.155
Yin, J., Wu, X., Li, S., Li, C., and Guo, Z. (2020). Impact of Environmental Factors on Gastric Cancer: A Review of the Scientific Evidence, Human Prevention and Adaptation. J. Environ. Sci. 89, 65–79. doi:10.1016/j.jes.2019.09.025
Yu, G., Wang, L.-G., Han, Y., and He, Q.-Y. (2012). clusterProfiler: An R Package for Comparing Biological Themes Among Gene Clusters. OMICS 16, 284–287. doi:10.1089/omi.2011.0118
Zhang, H.-Z., Jin, G.-F., and Shen, H.-B. (2012). Epidemiologic Differences in Esophageal Cancer between Asian and Western Populations. Chin. J. Cancer 31, 281–286. doi:10.5732/cjc.011.10390
Zhang, P., Yang, M., Zhang, Y., Xiao, S., Lai, X., Tan, A., et al. (2019a). Dissecting the Single-Cell Transcriptome Network Underlying Gastric Premalignant Lesions and Early Gastric Cancer. Cel Rep. 27, 1934e1935–1947. doi:10.1016/j.celrep.2019.04.052
Zhang, X., Lan, Y., Xu, J., Quan, F., Zhao, E., Deng, C., et al. (2019b). CellMarker: a Manually Curated Resource of Cell Markers in Human and Mouse. Nucleic Acids Res. 47, D721–D728. doi:10.1093/nar/gky900
Zhang, J., Dong, R., Dong, R., and Shen, L. (2020). Evaluation and Reflection on Claudin 18.2 Targeting Therapy in Advanced Gastric Cancer. Chin. J. Cancer Res. 32, 263–270. doi:10.21147/j.issn.1000-9604.2020.02.13
Zhang, M., Hu, S., Min, M., Ni, Y., Lu, Z., Sun, X., et al. (2021a). Dissecting Transcriptional Heterogeneity in Primary Gastric Adenocarcinoma by Single Cell RNA Sequencing. Gut 70, 464–475. doi:10.1136/gutjnl-2019-320368
Zhang, X., Peng, L., Luo, Y., Zhang, S., Pu, Y., Chen, Y., et al. (2021b). Dissecting Esophageal Squamous-Cell Carcinoma Ecosystem by Single-Cell Transcriptomic Analysis. Nat. Commun. 12, 1–17. doi:10.1038/s41467-021-25539-x
Zhang, Y. (2013). Epidemiology of Esophageal Cancer. World J. Gastroenterol. 19, 5598. doi:10.3748/wjg.v19.i34.5598
Keywords: upper gastrointestinal cancers, cancer stem cell, single-cell sequence data, pan-cancer analysis, oncogene
Citation: Li L, Zhang Y, Ren Y, Cheng Z, Zhang Y, Wang X, Zhao H and Lu H (2022) Pan-Cancer Single-Cell Analysis Reveals the Core Factors and Pathway in Specific Cancer Stem Cells of Upper Gastrointestinal Cancer. Front. Bioeng. Biotechnol. 10:849798. doi: 10.3389/fbioe.2022.849798
Received: 06 January 2022; Accepted: 07 April 2022;
Published: 13 May 2022.
Edited by:
Zhaoyuan Fang, Zhejiang University, ChinaReviewed by:
Binhua Tang, Hohai University, ChinaCopyright © 2022 Li, Zhang, Ren, Cheng, Zhang, Wang, Zhao and Lu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hui Lu, aHVpbHVAc2p0dS5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.