 Wei Luo
Wei Luo Jinglong Xu1
Jinglong Xu1 Huili Zhang
Huili Zhang Peilong Yang
Peilong Yang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Bioeng. Biotechnol., 23 September 2021
Sec. Industrial Biotechnology
Volume 9 - 2021 | https://doi.org/10.3389/fbioe.2021.747404
Compared with low-yield extraction from plants and environmentally unfriendly chemical synthesis, biocatalysis by asparagine synthetase (AS) for preparation of L-asparagine (L-Asn) has become a potential synthetic method. However, low enzyme activity of AS and high cost of ATP in this reaction restricts the large-scale preparation of L-Asn by biocatalysis. In this study, gene mining strategy was used to search for novel AS with high enzyme activity by expressing them in Escherichia coli BL21 (DE3) or Bacillus subtilis WB600. The obtained LsaAS-A was determined for its enzymatic properties and used for subsequent preparation of L-Asn. In order to reduce the use of ATP, a class III polyphosphate kinase 2 from Deinococcus ficus (DfiPPK2-Ⅲ) was cloned and expressed in E. coli BL21 (DE3), Rosetta (DE3) or RosettagamiB (DE3) for ATP regeneration. A coupling reaction system including whole cells expressing LsaAS-A and DfiPPK2-Ⅲ was constructed to prepare L-Asn from L-aspartic acid (L-Asp). Batch catalytic experiments showed that sodium hexametaphosphate (>60 mmol L−1) and L-Asp (>100 mmol L−1) could inhibit the synthesis of L-Asn. Under fed-batch mode, L-Asn yield reached 90.15% with twice feeding of sodium hexametaphosphate. A final concentration of 218.26 mmol L−1 L-Asn with a yield of 64.19% was obtained when L-Asp and sodium hexametaphosphate were fed simultaneously.
As a common amino acid, L-asparagine (L-Asn) is widely used in medicine, food and other fields. L-Asn can be used as a precursor for some drugs (Bunev et al., 2014), a green catalyst or a precursor of synthetic catalysts (Garg et al., 2020; Safari et al., 2020), and also has antioxidant and other effects that is useful in food manufacturing (Echavarría et al., 2013). L-Asn can be prepared by a variety of synthetic methods, including chemical synthesis, extraction from plants and biosynthesis. Compared with the former two, biosynthesis has the advantages of simple production process, low equipment requirements, high production efficiency, low energy consumption and low pollution.
In plants, biosynthesis of L-Asn is derived from L-aspartic acid (L-Asp) by the catalysis of asparagine synthetase (AS) using NH4+ or glutamine as amide donor. The NH4+-dependent asparagine synthetase (AS-A) encoded by asnA gene only use NH4+ as an amide donor and exists in prokaryotes, while glutamine-dependent asparagine synthetase (AS-B) encoded by asnB gene can use either NH4+ or glutamine as an amide donor and widely exists in all kinds of organisms (Gaufichon et al., 2010).
Although AS has been successfully cloned and expressed (Kasai et al., 2000), research on the preparation of L-Asn by the catalysis of AS is limited. Due to the low activity of AS and the need to add expensive ATP, this reaction is not economically viable. Moreover, accumulation of by-products (AMP or ADP) during the reaction derived from the addition of ATP, which not only increases the difficulty of product separation and purification, but also causes product inhibition (Iwamoto et al., 2007). Therefore, it is necessary to build a cost-effective ATP regeneration system in order to overcome these problems.
The common way of ATP regeneration depend on kinase, such as acetate kinase (Yan et al., 2014), which has been used for the preparation of 3-phosphate glycerol (Li et al., 2001). Polyphosphate kinase (PPK) is another important one catalyzes reversible synthesis of ATP from ADP using polyphosphate as a phosphate donor and such ATP regeneration system has been used in the synthesis of d-allose (Xiao et al., 2019). However, a novel class III polyphosphokinase 2 (PPK2-Ⅲ) can catalyze both AMP and ADP phosphorylation to generate ATP (Motomura et al., 2014). Considering the simple operation (addition of a single enzyme), high availability and low cost of polyphosphate, PPK2-Ⅲ obviously represents as a more feasible candidate for ATP regeneration. Suzuki et al. (2018) co-expressed PPK2-Ⅲ and tyrosine synthase A in Escherichia coli BL21 (DE3) for whole-cell catalysis, in which 6.7 mmol mL−1 L-trp-L-pro was produced in 24 h with the addition of only 10 mmolmL−1AMP.
Here, AS with high enzyme activity was screened by gene mining strategy and LsaAS from Lactobacillus salivarius was isolated. LsaAS coupled with an ATP regeneration system using polyphosphokinase 2 from D. ficus (DfiPPK2-III) was constructed in an one-pot whole-cell reaction for synthesis of L-Asn. The reaction conditions were investigated in order to obtain the optimal production of L-Asn.
The strains used here were purchased from the General Microorganisms Deposit Management Center of Jiangnan University (Supplementary Table S1). The tool plasmids used were previously deposited in our laboratory. The genes and primers were synthesized by Suzhou Jinweizhi Biotechnology Co., Ltd. Kits for genome extraction, plasmid extraction, DNA recovery and DNA purification were purchased from Shanghai Shenggong Co., Ltd. Restriction enzymes and DNA recombinases were obtained from Dalian TAKARA, Co., Ltd.
For gene mining, AS-A and AS-B from E. coli were selected as probes (Wang et al., 2005), and BLAST procedures were conducted in the NCBI database to search for AS-A and AS-B with different sequence identities. In order to obtain enzymes with different enzymatic properties, representative proteins with different sequence identities were selected for gene cloning, expression and analyses of enzyme activities.
Guided with the primers in Supplementary Table S2, different AS genes (asn) and polyphosphokinase 2 genes were amplified by polymerase chain reaction (PCR) using the genome of each strain as a template. Each asn was connected to pET28a and pMA5 and the recombinant plasmids were transformed into E. coli JM109 for gene cloning. Then the recombinant plasmids were extracted and transformed into E. coli BL21 (DE3) and Bacillus subtilis WB600, respectively. On the other hand, PPK2-Ⅲ genes from Deinococcus ficus and D. phoenicis were linked to pET21a by using DNA recombinase and transformed into E. coli strains.
The recombinant E. coli cells harboring expression plasmids were cultured in the TB liquid media supplementated with kanamycin (final concentration of 50 μg ml−1) until the optical density (OD600) reaching 0.4–0.6. Then isopropyl-β-d-Thiogalactopyranoside (IPTG) was added with a final concentration of 0.1 mmol L−1 and incubated at 16°C for 24 h. The recombinant B. subtilis WB600 cells harboring expression plasmids were cultured in the SB liquid media with kanamycin (final concentration of 50 μg ml−1) at 36°C for 24 h.
The collected cells were washed, resuspended in lysis buffer (20 mM Tris-HCl buffer, pH 8.0). Then cells were crushed by two rounds of ultrasound treatment and centrifuged at 12,000 rpm for 20 min at 4°C. Crude enzyme extract was prepared by filtering supernatant with a 0.22 μm microporous membrane for use. The crude extract was loaded onto a Ni-NTA column pre-equilibrated with buffer. The binding solution and eluent were used as the mobile phase for linear elution (eluent ratio: 0–100%), followed by SDS-PAGE analysis of the collected eluate.
The 5 ml reaction mixture contained 0.1 mol L−1 Tris-HCl (pH 8.0), 0.1–0.5 mol L−1 L-Asp, 0.1–0.8 mol L−1 NH4Cl, 1–10 mmol L−1 ATP, 5–250 mmol L−1 MgCl2, 5–150 mmol L−1 sodium hexametaphosphate and 1% Triton X-100. The reaction began with the addition of recombinant cells of AS and PPK2-Ⅲ. The reaction temperature is 25–45°C and the rotation speed is 180 rpm. Whole-cell catalytic reaction is terminated in a boiling water bath for 5 min.
The activity of AS was measured in 1 ml reaction system containing 100 mmol L−1 Tris-HCl buffer (pH 8.0), 20 mmol L−1 L-Asp, 10 mmol L−1 MgCl2, 10 mmol L−1 NH4Cl (or L-Gln), 10 mmol L−1 ATP and 0.1 ml enzyme solution. After incubation at 37°C for 15 min, the sample was boiled for 5 min to terminate the reaction. Then the sample was centrifuged at 12,000 rpm and 4°C for 5 min, and the supernatant was filtered through a microporous (0.22 μm) membrane and poured into a sample bottle for content determination. The L-Asn content was determined by HPLC (High Performance Liquid Chromatography) with orthophthalaldehyde (OPA) on-line derivatization, and the amount of enzyme required to produce 1 μmol L-Asn from L-Asp per minute was defined as one enzyme activity unit.
The activity of PPK2-Ⅲ was measured in 1 ml reaction system containing 100 mmol L−1 Tris-HCl (pH 8.0), 10 mmol L−1 MgCl2, 50 mmol L−1 polyphosphate and 1 mmol L−1 AMP. The reaction was conducted at 37°C for 10 min, and the sample was heated at 80°C for 10 min to terminate the reaction. After centrifuge at 12,000 rpm for 10 min, the supernatant was collected to determine the ATP content by ATP detection kit. As for the definition of activity of PPK2-Ⅲ, the amount of enzyme required to produce 1 μmol of ATP per minute is defined as a unit of enzyme activity.
Concentrations of L-Asn, L-Asp and glutamine were determined by HPLC on-line o-phthalaldehyde (OPA)-derivation method with an Agilent Zorbax Eclipse AAA column (4.6 × 150 mm, 5 μm). The control conditions were set as follows, column temperature 40°C, detection wavelength 338 nm, flow rate 1.0 ml min−1, sample volume 10 μL. The mobile phase contained A (40 mmol L−1 Na2HPO4, pH 7.8) and B (acetonitrile: methanol: water = 45: 45: 10). The linear elution programme represented as the proportion of mobile phase B was as follows, 5% at 0–2 min, 5%–57% at 2–12 min, 100% at 13–17 min, 5% at 18–24 min.
Through sequence alignment, the enzyme molecules exhibiting 20–100% identity with the amino acid sequence of the probe were selected in order to obtain a novel AS with different enzymatic property from the probe. Thereafter, eight genes of asnB and four genes of asnA were selected for clone and heterologous expression (Supplementary Table S1). Heterologous expression of these enzyme proteins in E. coli and B. subtilis is indicated by SDS-PAGE (Supplementary Figure S1 and Supplementary Figure S2). Theoretically, the molecular weights of AS-B and AS-A are ∼60 and ∼40 kDa, respectively. EcoAS-B, SplAS-B, HalAS-B and EcoAS-A were achieved high soluble protein expression and consistent with the theoretical value, while VruAS-B, SmaAS-B had less soluble protein expression but consistent with the theoretical value in E. coli. B. subtilis is considered to be a good host of protein soluble expression and was selected as host for these enzyme proteins expression. SDS-PAGE shows that HalAS-A, LsaAS-A, EcoAS-B, SplAS-B, HalAS-B, VruAS-B and SmaAS-B are expressed in soluble state in B. subtilis WB600 (Supplementary Figure S2). Enzyme activity assys in Table 1 shows B. subtilis is more suitable as a host for heterologous expression of these enzyme proteins, except for EcoAS-A. LsaAS-A expressed in B. subtilis produced highest enzyme activity (1356.7 mU·mL−1) and this value increased to as high as 3850.0 mU·mL−1 under optimal culture condition in triangular flask.

TABLE 1. Activities of recombinant AS expressed in E. coli BL21 (DE3) and B. subtilis WB600.
Figure 1A shows that the activity of LsaAS-A was temperature-dependent with an optimal temperature around 40°C. When temperature exceeded 50°C, the enzyme activity decreased rapidly. In order to obtain the half-life data of LsaAS-A, the first-order exponential decay formula was used to determine the inactivation rate constant by plot of the decay formula (Figure 1B). Through calculation by inactivation rate constant, the half-lives of LsaAS-A at 30, 40 and 50°C were 857.9, 94.6 and 51.0 min, respectively. In previous reports, the AS-A derived from E. coli had a half-life of less than 1 min at 45°C and was completely inactivated after being stored at 50°C for 5 min (Humbert and Simoni, 1980), suggesting that LsaAS-A was significantly more stable than E. coli-derivived AS-A. Moreover, effect of pH on LsaAS-A activity was also investigated by incubating the reaction solution under buffers with different pH. Figure 2A shows that LsaAS-A is pH-dependent with an optimal pH of 8.0. The suitable reaction buffer seems to be acetate buffer and Tris-HCl buffer, rather than boric acid buffer. In addition, pH stability of LsaAS-A was also investigated by storing LsaAS-A at 4°C for 24 h under different pH conditions and its residual activity was measured. LsaAS-A was stable in the acetate buffer at pH 5, while storage of LsaAS-A in buffer with pH 4 or 6–9 resulted in partial loss of enzyme activity. When storage pH was up to 10, only 29.8% of the residual enzyme activity was retained (Figure 2B). On the other hand, metal ions were added to the reaction system to investigate the effect of them on enzyme activity and Table 2 shows that Mg2+, Mn2+, Fe2+, and Zn2+ have an activation effect on the enzyme activity. Among them, Fe2+ has the highest activation performance (136%), while Zn2+ gave a relatively low activation effect (23.61%). The lost of LsaAS-A activity without addition of metal indicates that LsaAS-A is a metal-dependent enzyme. The main reason is that metal ion including Mg2+ binds to the C-terminal active site of AS and participates in the reaction (Richards and Schuster, 1998).
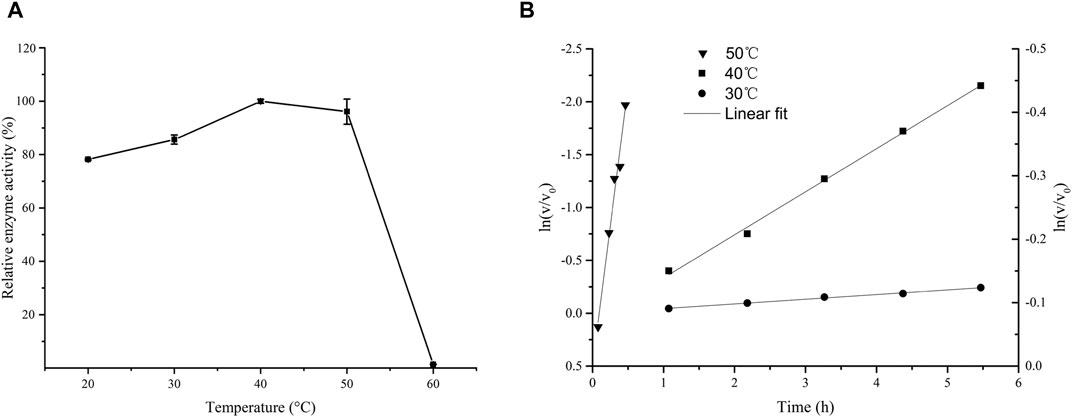
FIGURE 1. Effect of temperature on the activity of LsaAS-A. (A) Optimal temperature of LsaAS-A. (B) Plot of the first-order exponential decay formula to show the thermal stability of LsaAS-A at 30, 40 and 50°C. The calculated values of the ln at 30°C and 40°C were calibrated by the left ordinate, while the counterpart at 50°C was calibrate by the right ordinate.
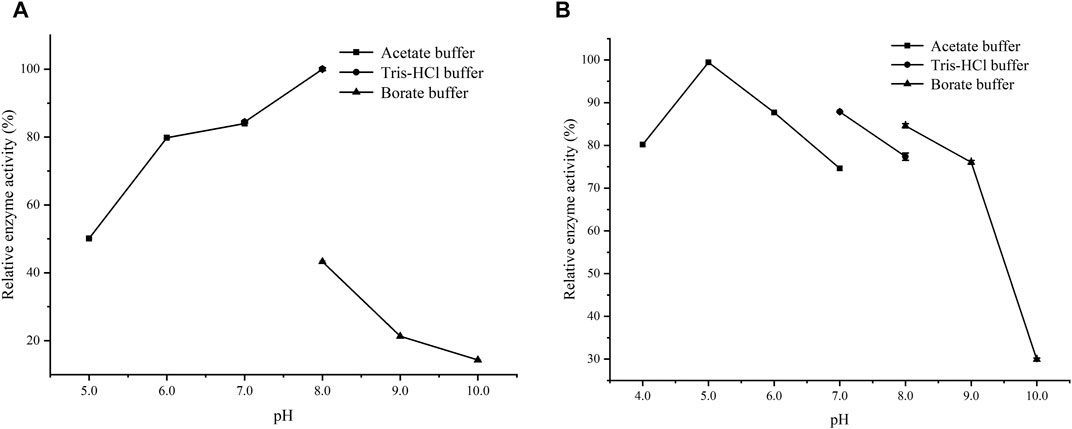
FIGURE 2. Effect of pH on the activity of LsaAS-A. (A) Optimal pH of LsaAS-A. (B) Stability of LsaAS-A at different pH buffer solution.
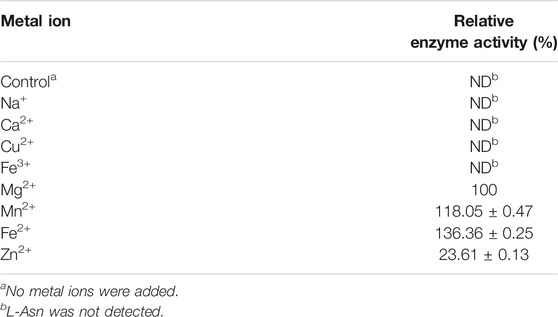
TABLE 2. Effect of metal ions on the activity of LsaAS-A.
In order to obtain Km and Vmax, double reciprocal plot was used to determine these kinetic parameters and the results were shown in Table 3. The Km of LsaAS-A for Asp is 0.78 mmol L−1, which is similar to AS from Klebsiella aerogenes (Richards and Schuster, 1998), indicating that LsaAS-A has a high affinity for Asp. The Km of LsaAS-A for NH4+ is 6.5 mmol L−1, which is similar to AS from Streptococcus bovis (Burchall et al., 1964). The Km of LsaAS-A for ATP is 2.24 mmol L−1, indicating that the affinity to ATP is high.

TABLE 3. Kinetic parameters of LsaAS-A.
PPK2-III from Deinococcus ficus and D. phoenicis was cloned and expressed in RosettagamiB (DE3), Rosetta (DE3) and BL21 (DE3) of E. coli, respectively. Electrophoretic analysis shows that they were expressed effectively (Supplementary Figure S3) and their enzyme activities were shown in Table 4. Each host can express ppk2-III, while E. coli Rosetta (DE3) exhibits the best expression performance among three hosts. The reason may be that E. coli Rosetta (DE3) can enhance the soluble expression of genes containing rare codons when compared with E. coli BL21 (DE3). Although E. coli RosettagamiB (DE3) also has this function, its slow growth restricts the improvement of enzyme activity. Considering that the polymerization degree has a great influence on the activity of most PPK (Ishige et al., 2002; Motomura et al., 2014; Meng et al., 2016), polyphosphates with different polymerization degrees were used to evaluate the activity of DfiPPK2-Ⅲ. Among the measured polyphosphates, sodium hexametaphosphate and sodium polyphosphate could be used as substrates for DfiPPK2-Ⅲ to synthesize ATP, while sodium pyrophosphate and sodium tripolyphosphates was not suitable as phosphate donors (Table 5).
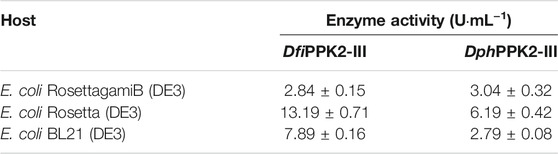
TABLE 4. Enzyme activity of PPK2-III in different hosts.
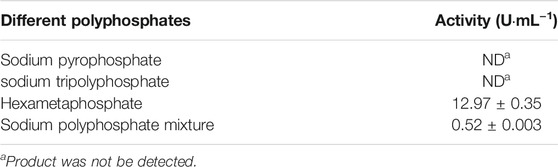
TABLE 5. Effect of polyphosphate with different degrees of polymerization on the enzyme activity of DfiPPK2-III.
A coupling reaction system for L-Asn preparation was constructed using LsaAS-A and DfiPPK2-Ⅲ ATP regeneration system (Supplementary Figure S4) either including free enzymes or whole cells. From the point of view of application, free enzyme and whole cell have their own advantages and disadvantages. Table 6 shows that the catalysis efficiency of whole cell as a catalyst is higher than that of crude enzyme solution whether ATP or AMP was used as energy donor. The reason may be that endogenous PPase in living cell can eliminate the inhibitory effect of pyrophosphate (Suzuki et al., 2018; Lelièvre et al., 2020) and LsaAS-A and DfiPPK2-III remained in orininal cells were more stable than the free enzymes. Table 5 also indicates that the addition of surfactants to the whole-cell catalytic system can improve L-Asn titer, which may be ascribed to the fact that surfactant could enhance the permeability of the cell membrane and thereby improve the effect of mass transfer.

TABLE 6. Effect of different catalyst forms and energy donors on the production of L-Asn.
Considering that polyphosphate is the phosphate donor for regeneration of ATP via the catalysis of PPK2-Ⅲ, the amount of polyphosphate added in the reaction system were investigated. Figure 3A shows that L-Asn production is hexametaphosphate-dependent. When the concentration of sodium hexametaphosphate added in the reaction solution was lower than 60 mmol L−1, the production of L-Asn increases with the enhancement of sodium hexametaphosphate concentration. While the concentration of sodium hexametaphosphate is greater than 60 mmol L−1, the production of L-Asn gave an opposite trend. The main reason may be that high concentration of sodium hexametaphosphate will chelate with Mg2+ to reduce enzyme activity (Iwamoto et al., 2007). Furthermore, sodium hexametaphosphate is loaded with high negative charge and is able to combine with enzyme, thereby reducing enzyme activity (Kameda et al., 2001).
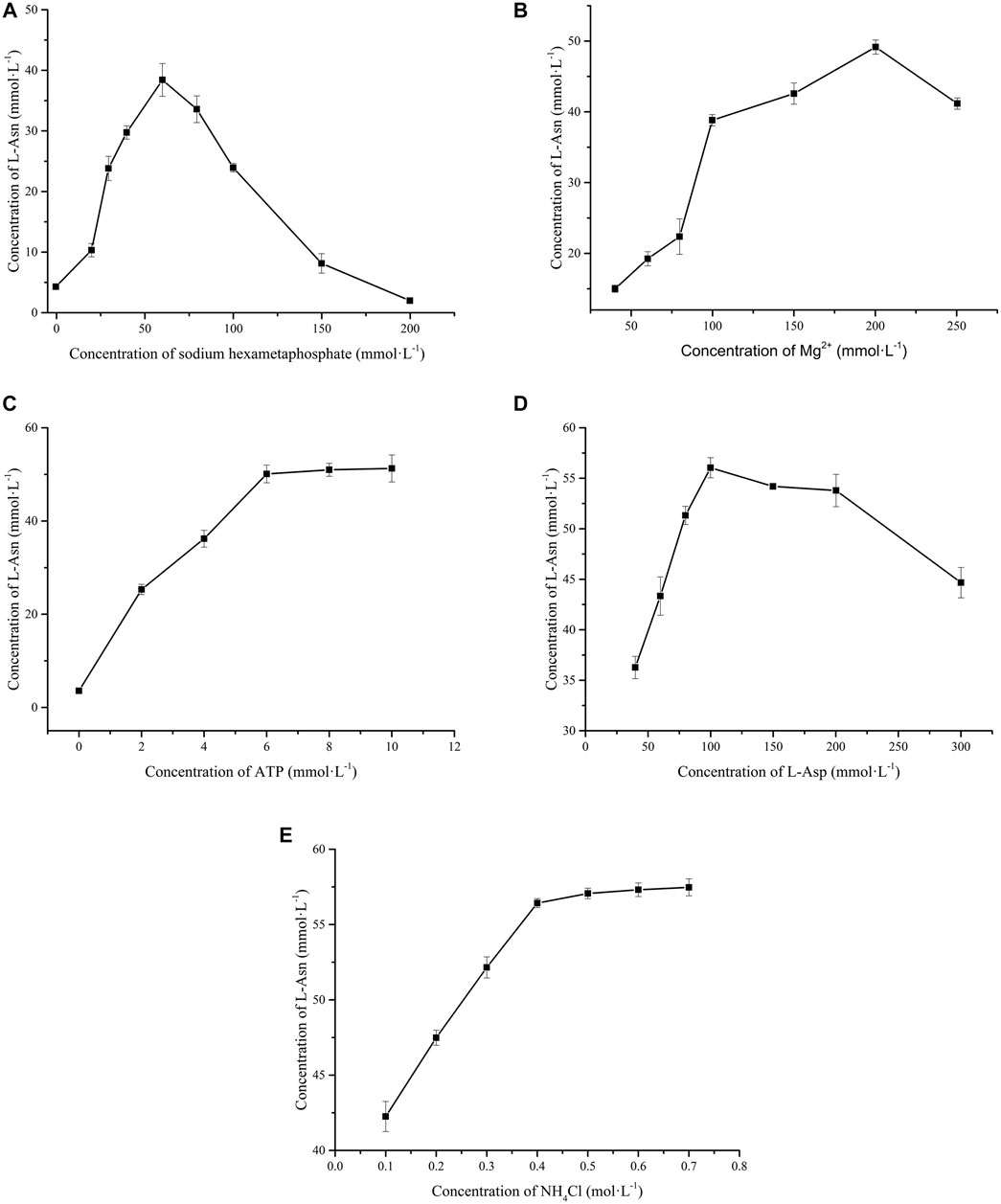
FIGURE 3. Effects of substrates and co-factor on whole cell catalytic preparation of L-Asn. (A) Sodium hexametaphosphate. (B) Mg2+. (C) ATP. (D) L-Asp. (E) NH4+.
As mentioned in enzymatic properties of LsaAS-A, metal ions are necessary for LsaAS synthesis of L-Asn, so DfiPPK2S also requires Mn2+ or Mg2+ as cofactors (Sato et al., 2007; Motomura et al., 2014). However, the present study showed that Mn2+, Fe2+ and Zn2+ had no stimulating effect on L-Asn production (data not shown). The reason may be that their strong binding force with sodium hexametaphosphate leads to the formation of precipitation, so these ions can not bind to LsaAS to play the role of cofactor (Larsen et al., 1999). As for Mg2+, Figure 3B shows that the situation is different. When the concentration of Mg2+ is no more than 200 mmol L−1, the production of L-Asn is stimulated by the enhancement of the concentration of Mg2+. However, the titer of L-Asn would be reduced if Mg2+ exceeds 200 mmol L−1 since chelation derived from high concentration Mg2+ is easy to genarate precipitation (Kameda et al., 2001).
In order to reduce costs, effect of initial ATP concentration (0–10 mmol L−1) on whole-cell catalytic synthesis of L-Asn was investigated. Without the addition of ATP, only 3.56 mmol L−1 of L-Asn was produced. When 6 mmol L−1 of ATP was added, the L-Asn output reached 50.10 mmol L−1 (Figure 3C). Theoretically, 1 mol ATP is consumed for every 1 mol L-Asn produced, so the ATP regeneration system has recycled ATP for 7.35 times. The final concentraion of L-Asn was not restricted by ATP when its intial concentraion was no less than 6 mmol L−1, so addition amount of 6 mmol L−1 is a suitable value in this ATP regeneration system.
Under the aforementioned optimal conditions, effects of L-Asp concentration (40–300 mmol L−1) and NH4+ concentration (0.1–0.7 mol L−1) on the whole-cell catalytic synthesis of L-Asn were investigated. When the concentration of L-Asp is 100 mmol L−1, the yield of L-Asn reaches the maximum (55.87 mmol L−1). L-Asp concentration high than 100 mmol L−1 would lead to reduction in L-Asn titer, which may be ascribed to substrate inhibition (Figure 3D). As for another substrate NH4+, Figure 3E shows that enhanced concentration of NH4+ improves the production of L-Asn, but 0.4 mol L−1 may be an optimal value since more addition of NH4+ is not able to bring about more production of L-Asn.
Above results indicate that addition of high concentration of sodium hexametaphosphate will cause inhibition in L-Asn production due to its chelation of Mg2+ and inhibition of enzyme activity. In order to release the inhibitory effect of sodium hexametaphosphate, a fed-batch strategy for addition of sodium hexametaphosphate was used. After 12 h, the conversion rate reached 90.15% (Figure 4A), which was nearly 80% higher than that of adding 180 mmol L−1 sodium hexametaphosphate directly, indicating that feeding sodium hexametaphosphate in batches can suppress its negative effect (Kameda et al., 2001). Furthermore, the strategy of feeding L-Asp and sodium hexametaphosphate simultaneously was adopted in order to obtain higher yield of L-Asn. At 4, 8, 12, 16, and 20 h after the start of the reaction, L-Asp and sodium hexametaphosphate were fed at a final concentration of 60 mmol L−1. At the end of the catalytic reaction, 218.26 mmol L−1 L-Asn was produced with a yield of 64.19% (Figure 4B).
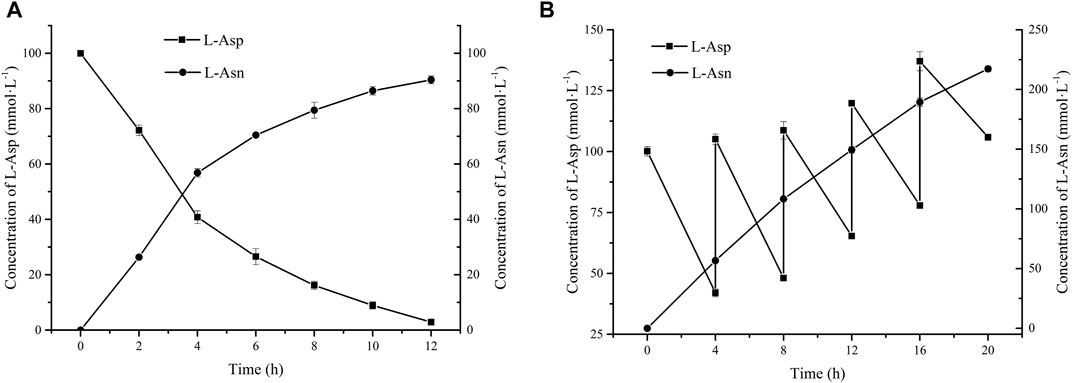
FIGURE 4. Effects of feeding substrates on whole cell catalytic preparation of L-Asn. (A) Sodium hexametaphosphate (60 mmol L−1) was added at 4 and 8 h, respectively. (B) Sodium hexametaphosphate (60 mmol L−1) and L-Asp (60 mmol L−1) were added at 4, 8, 12, 16, and 20 h, respectively.
Through gene mining, a novel asparagine synthetase LsaAS-A with good catalytic performance was isolated. The optimal temperature and pH of LsaAS-A was 41°C and 8.0, respectively; the half-life in the buffer solution at pH 8 and 30°C was 857.85 min. The enzyme was a metal-dependent enzyme and has a high affinity for L-Asp. In order to solve the problem of ATP supply in the reaction process, we expressed PPK2-III from Deinococcus ficus in E. coli Rosetta (DE3), which can use polyphosphate to synthesize ATP. A coupling catalytic reaction system including LsaAS-A, DfiPPK2-Ⅲ and substrate was then constructed to prepare L-Asn from L-Asp. Biotransformation experiments showed that the whole-cell reaction system could obtain higher product yield than free enzyme catalysis, so preparation of L-Asn from L-Asp by whole cell catalysis was optimized under batch modes. In order to reduce substrate inhibition, fed-batch strategy was used to control substrate concentration and 218.26 mmol L−1 L-Asn was obtained with a yield of 64.19%.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
WL, JX, PY, and XY conceived and planned the experiments. WL, HC, and PY performed the conceptualization, methodology, experiments, data analyses, and writing. WL, JX, and HZ took part in the design of experiments, reviewed, and edited the draft. All authors approved the final manuscript.
This work was supported by the Key Technology Research Plan Project of the Inner Mongolia Autonomous Region (No. 2019GG302) and the Program of the Key Laboratory of Carbohydrate Chemistry and Biotechnology, Ministry of Education, China (No. KLCCB-KF201405), the Open Project Program of Key Laboratory of Feed Biotechnology, Ministry of Agriculture and Rural Affairs (No. KLFB-FRI-202001).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fbioe.2021.747404/full#supplementary-material
Bunev, A. S., Rudakova, Y. I., Peregudov, A. S., and Ostapenko, G. I. (2014). Synthesis of Thiazole-Containing Amino Acids Based on Asparagine. Russ. Chem. Bull. 63, 1232–1234. doi:10.1007/s11172-014-0579-6
Burchall, J. J., Reichelt, E. C., and Wolin, M. J. (1964). Purification and Properties of the Asparagine Synthetase of Streptococcus Bovis. J. Biol. Chem. 239, 1794–1798. doi:10.1016/s0021-9258(18)91260-3
Echavarría, A. P., Pagán, J., and Ibarz, A. (2013). Antioxidant Activity of the Melanoidin Fractions Formed from DGlucose and D-Fructose with L-Asparagine in the Maillard Reaction. Sci. Agropecu 4, 45–54. doi:10.17268/sci.agropecu.2013.01.05
Garg, N., Bera, S., Rastogi, L., Ballal, A., and Balaramakrishna, M. V. (2020). Synthesis and Characterization of L-Asparagine Stabilised Gold Nanoparticles: Catalyst for Degradation of Organic Dyes. Spectrochimica Acta A: Mol. Biomol. Spectrosc. 232, 118126. doi:10.1016/j.saa.2020.118126
Gaufichon, L., Reisdorf-Cren, M., Rothstein, S. J., Chardon, F., and Suzuki, A. (2010). Biological Functions of Asparagine Synthetase in Plants. Plant Sci. 179, 141–153. doi:10.1016/j.plantsci.2010.04.010
Humbert, R., and Simoni, R. D. (1980). Genetic and Biomedical Studies Demonstrating a Second Gene Coding for Asparagine Synthetase in Escherichia coli. J. Bacteriol. 142, 212–220. doi:10.1128/jb.142.1.212-220.1980
Ishige, K., Zhang, H., and Kornberg, A. (2002). Polyphosphate Kinase (PPK2), a Potent, Polyphosphate-Driven Generator of GTP. Proc. Natl. Acad. Sci. 99, 16684–16688. doi:10.1073/pnas.262655299
Iwamoto, S., Motomura, K., Shinoda, Y., Urata, M., Kato, J., Takiguchi, N., et al. (2007). Use of an Escherichia coli Recombinant Producing Thermostable Polyphosphate Kinase as an ATP Regenerator to Produce Fructose 1,6-Diphosphate. Appl. Environ. Microbiol. 73, 5676–5678. doi:10.1128/aem.00278-07
Kameda, A., Shiba, T., Kawazoe, Y., Satoh, Y., Ihara, Y., Munekata, M., et al. (2001). A Novel ATP Regeneration System Using Polyphosphate-AMP Phosphotransferase and Polyphosphate Kinase. J. Biosci. Bioeng. 91, 557–563. doi:10.1263/jbb.91.557
Kasai, K., Fujie, M., Nakanishi, Y., Murooka, Y., Usami, S., and Yamada, T. (2000). Molecular Cloning and Characterization of Two cDNAs Encoding Asparagine Synthetase from Astragalus sinicus Nodules. J. Biosci. Bioeng. 89, 559–563. doi:10.1016/s1389-1723(00)80057-2
Larsen, T. M., Boehlein, S. K., Schuster, S. M., Richards, N. G. J., Thoden, J. B., Holden, H. M., et al. (1999). Three-Dimensional Structure ofEscherichia coliAsparagine Synthetase B: A Short Journey from Substrate to Product†,‡. Biochemistry 38, 16146–16157. doi:10.1021/bi9915768
Lelièvre, C. M., Balandras, M., Petit, J. L., Vergne‐Vaxelaire, C., and Zaparucha, A. (2020). ATP Regeneration System in Chemoenzymatic Amide Bond Formation with Thermophilic CoA Ligase. ChemCatChem 12, 1184–1189. doi:10.1002/cctc.201901870
Li, T., Yeh, H., Kim, J.-H., and Ryu, D. D. Y. (2001). Performance of Batch Membrane Reactor: Glycerol-3-Phosphate Synthesis Coupled with Adenosine Triphosphate Regeneration. Biotechnol. Bioeng. 74, 326–334. doi:10.1002/bit.1123
Meng, Q., Zhang, Y., Ju, X., Ma, C., Ma, H., Chen, J., et al. (2016). Production of 5-Aminolevulinic Acid by Cell Free Multi-Enzyme Catalysis. J. Biotechnol. 226, 8–13. doi:10.1016/j.jbiotec.2016.03.024
Motomura, K., Hirota, R., Okada, M., Ikeda, T., Ishida, T., and Kuroda, A. (2014). A New Subfamily of Polyphosphate Kinase 2 (Class III PPK2) Catalyzes Both Nucleoside Monophosphate Phosphorylation and Nucleoside Diphosphate Phosphorylation. Appl. Environ. Microbiol. 80, 2602–2608. doi:10.1128/aem.03971-13
Richards, N. G., and Schuster, S. M. (1998). Mechanistic Issues in Asparagine Synthetase Catalysis. Adv. Enzymol. Relat. Areas Mol. Biol. 72, 145–198. doi:10.1002/9780470123188.ch5
Safari, J., Shokrani, Z., and Zarnegar, Z. (2020). Asparagine as a Green Organocatalyst for the Synthesis of 2-Aminothiazoles. Polycyclic Aromatic Compd. 40, 1105–1111. doi:10.1080/10406638.2018.1528287
Sato, M., Masuda, Y., Kirimura, K., and Kino, K. (2007). Thermostable ATP Regeneration System Using Polyphosphate Kinase from Thermosynechococcus Elongatus BP-1 for D-Amino Acid Dipeptide Synthesis. J. Biosci. Bioeng. 103, 179–184. doi:10.1263/jbb.103.179
Suzuki, S., Hara, R., and Kino, K. (2018). Production of Aminoacyl Prolines Using the Adenylation Domain of Nonribosomal Peptide Synthetase with Class III Polyphosphate Kinase 2-mediated ATP Regeneration. J. Biosci. Bioeng. 125, 644–648. doi:10.1016/j.jbiosc.2017.12.023
Wang, H., Liu, D., Sun, J., and Zhang, A. (2005). Asparagine Synthetase Gene TaASN1 from Wheat Is Up-Regulated by Salt Stress, Osmotic Stress and ABA. J. Plant Physiol. 162, 81–89. doi:10.1016/j.jplph.2004.07.006
Xiao, Q., Niu, J., Liu, H., Liu, Y., and Zhou, X. (2019). High Conversion of D-Fructose into D-Allulose by Enzymes Coupling with an ATP Regeneration System. Mol. Biotechnol. 61, 432–441. doi:10.1007/s12033-019-00174-6
Keywords: l-Asparagine, asparagine synthase, class III polyphosphate kinase, ATP regeneration system, biocatalysis and biotransformation
Citation: Luo W, Xu J, Chen H, Zhang H, Yang P and Yu X (2021) Synthesis of L-asparagine Catalyzed by a Novel Asparagine Synthase Coupled With an ATP Regeneration System. Front. Bioeng. Biotechnol. 9:747404. doi: 10.3389/fbioe.2021.747404
Received: 26 July 2021; Accepted: 09 August 2021;
Published: 23 September 2021.
Edited by:
Xiao-Jun Ji, Nanjing Tech University, ChinaReviewed by:
Huabao Zheng, Zhejiang Agriculture and Forestry University, ChinaCopyright © 2021 Luo, Xu, Chen, Zhang, Yang and Yu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wei Luo, d2x1b0BqaWFuZ25hbi5lZHUuY24=; Peilong Yang, eWFuZ3BlaWxvbmdAY2Fhcy5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.