 Vidyasrilekha Yele
Vidyasrilekha Yele Bharat Kumar Reddy Sanapalli
Bharat Kumar Reddy Sanapalli Ashish D. Wadhwani3
Ashish D. Wadhwani3 Afzal Azam Mohammed
Afzal Azam Mohammed
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Bioeng. Biotechnol., 17 June 2021
Sec. Synthetic Biology
Volume 9 - 2021 | https://doi.org/10.3389/fbioe.2021.669728
This article is part of the Research TopicInnovative Strategies from Synthetic Biology and Bacterial Pathways to Master Biochemical Environmental ChallengesView all 7 articles
Antibacterial resistance (ABR) is a major life-threatening problem worldwide. Rampant dissemination of ABR always exemplified the need for the discovery of novel compounds. However, to circumvent the disease, a molecular target is required, which will lead to the death of the bacteria when acted upon by a compound. One group of enzymes that have proved to be an effective target for druggable candidates is bacterial DNA topoisomerases (DNA gyrase and ParE). In our present work, phenylacetamide and benzohydrazides derivatives were screened for their antibacterial activity against a selected panel of pathogens. The tested compounds displayed significant antibacterial activity with MIC values ranging from 0.64 to 5.65 μg/mL. Amongst 29 title compounds, compounds 5 and 21 exhibited more potent and selective inhibitory activity against Escherichia coli with MIC values at 0.64 and 0.67 μg/mL, respectively, and MBC at onefold MIC. Furthermore, compounds exhibited a post-antibiotic effect of 2 h at 1× MIC in comparison to ciprofloxacin and gentamicin. These compounds also demonstrated the concentration-dependent bactericidal activity against E. coli and synergized with FDA-approved drugs. The compounds are screened for their enzyme inhibitory activity against E. coli ParE, whose IC50 values range from 0.27 to 2.80 μg/mL. Gratifyingly, compounds, namely 8 and 25 belonging to the phenylacetamide series, were found to inhibit ParE enzyme with IC50 values of 0.27 and 0.28 μg/mL, respectively. In addition, compounds were benign to Vero cells and displayed a promising selectivity index (169.0629–951.7240). Moreover, compounds 1, 7, 8, 21, 24, and 25 (IC50: <1 and Selectivity index: >200) exhibited potent activity in reducing the E. coli biofilm in comparison with ciprofloxacin, erythromycin, and ampicillin. These astonishing results suggest the potential utilization of phenylacetamide and benzohydrazides derivatives as promising ParE inhibitors for treating bacterial infections.
Antibacterial resistance (ABR) is one of the significant public health concerns of the twenty-first century that threatens the prevention and treatment of an ever-increasing range of various pathogenic infections (Fischbach and Walsh, 2009; Silver, 2011; Michael et al., 2014). Though, WHO has taken many initiatives to combat ABR, lack of adequate surveillance in many parts leaves large gaps in existing knowledge for dissemination and extent of the ABR (Simpkin et al., 2017; Huttner et al., 2019; Piddock, 2019). Infections caused by resistant strains are speculated to cause millions of deaths in the near future unless suitable actions are taken to overwhelm the risk (Nathan, 2004; Davies and Davies, 2010; Roca et al., 2015). Thus, there is an urgent need to discover newer chemo-types with a novel mode of action to overcome the ABR (Miller and Waldrop, 2010). Among all the validated bacterial targets, DNA topoisomerases are considered efficient for the growth of bacterial cells and possess all the prerequisites of an “Ideal” target (Maxwell, 1997; Maxwell and Lawson, 2003). The ATP-dependent bacterial topoisomerases comprise DNA gyrase (Gyrase A and Gyrase B) and topoisomerase IV (ParC and ParE). These two enzymes play a vital role in DNA replication, leading to bacterial cell proliferation (Drlica and Zhao, 1997). DNA Gyrase belongs to the class II topoisomerase, a heterotetramer, involved in introducing negative supercoils into constrained DNA strands. Topoisomerase IV, primarily responsible for the decatenation of daughter chromatids at the end of replication (Heisig, 2001). Fluoroquinolone class of drugs is the first line of therapeutics to inhibit the catalytic subunits of DNA gyrase and topoisomerase IV. However, their use has been limited because of its side effects, toxicity, and emerging bacterial resistances (Jones, 1989; Booker et al., 2005).
GyrB and Topo IV (ParE), bacterial enzymes possess structural similarities in conserved regions, quinolones and coumarins sensitivity and, subunit organization. However, the most considerable difference between these enzyme structures is the long α-helices of each monomer’s C-terminus (Tourova et al., 2010). In E. coli, it is clear that fluoroquinolones preferentially bind to ParE enzyme than GyrB. Indeed, ParE is an unexplored target and could not establish any type of drug resistance. So, we concentrated on exploring the efficiency of small molecule inhibitors targeting ParE enzyme.
E. coli ParE ATPase catalytic 24-kDa domain (residues 1–217) consists of five α-helices and eight β-sheet strands along with a highly conserved amino acid sequence across the bacterial species. Novobiocin is an ATP-competitive inhibitor of the topoisomerases; however, activity is always greater for ParE than gyrase. This difference in the potency of novobiocin is due to a single residue (M74 in ParE) (Wigley et al., 1991). The ATP substrate binds to the catalytic pocket and is involved in hydrogen bond donating and accepting interactions between ligand and ParE crystal structure (Brino et al., 2000; Azam et al., 2015). These interactions are homologous to the donor/acceptor motif, explored in drug design and development of kinase inhibitors. Indeed, pyrrolopyrimidines, azaindoles, pyridylureas, quinazolidinediones series exploit these donor/acceptor interactions with the ParE protein.
In our earlier work, we reported the synthesis, minimum inhibitory concentration (MIC), minimum bactericidal concentration (MBC), time-kill kinetics of phenylacetamide and benzohydrazides derivatives (1–29) (Supplementary Table 1) (Yele et al., 2020, 2021). In continuation of our work, we report in vitro MIC and MBC against Escherichia coli (NCIM 2065) (E. coli), methicillin-resistant Staphylococcus aureus (MRSA) (NCIM 5021 and ATCC 43300). Whereas, time-kill kinetics, synergy testing, post-antibiotic effect (PAE) and anti-biofilm studies were performed against E. coli. Furthermore, we determined the IC50 values of title compounds (1–29) against E. coli ParE enzyme by malachite green assay.
The panel of bacteria selected for the current study are Escherichia coli (NCIM 2065) (E. coli), methicillin-resistant Staphylococcus aureus (MRSA) (NCIM 5021 and ATCC 43300). These strains were retrieved from the National Collection of Industrial Microorganisms (NCIM), Pune, and American Type Culture Collection repositories. All the strains were cultured using Mueller-Hinton broth before the study.
AST was performed on the compounds (1–29) (Supplementary Table 1) (Yele et al., 2020, 2021) concerning the standard protocol (CLSI, 2017). The lowest concentration of a compound at which inhibition of bacterial growth was observed is termed MIC. The microbroth dilution method in Hi-media, Mueller Hinton Broth (MHB), was used to calculate the MIC. Bacterial cultures were diluted at a ratio of 1:1,000 MHB to achieve turbidity of 0.5 McFarland. DMSO and ciprofloxacin were used as a negative and positive control, respectively. The bacterial inoculum was added into a series of 96-well plates containing various test compounds ranging from 0.625 to 20 μg/mL. Plates were incubated for 20–24 h at 37°C. The absorbance was recorded at 600 nm of wavelength using Tecan-i-control, 1.7.1.12. MIC determinations for each compound were carried out in triplicate using a duplicate sample each time (Eloff, 1998; Agyare et al., 2012).
MBC can be defined as the minimum concentration of a compound required to kill the bacteria over a fixed period under precise conditions. MBC was determined for the synthesized compounds (1–29) (Supplementary Table 1; Yele et al., 2020, 2021) concerning the standard protocol (CLSI, 2017). Three successive concentrations above the MIC were employed, and 20 μL of the aliquot were sub-cultured onto the prepared nutrient agar plates, followed by incubation at 37°C for 24 h. Colonies were counted, and colony forming unit (CFU) per milliliter was calculated. MBC value is the concentration at which the ≥99% reduction (3 log 10) of bacterial growth is observed (Ahmad et al., 2015).
Bactericidal activity was evaluated for the compounds by the time-kill kinetics. The bacterial strain E. coli were diluted up to ∼106 μg/mL and treated with a compound of concentration 1× and 0.5× MIC of compounds 5 and 21 and ciprofloxacin in MHB and incubated at 37°C. 50 μL aliquot of the culture was inoculated at regular time intervals of 0, 2, 4, 8, 16, and 24 h onto the solidified agar plates, followed by incubation at 37°C for 24 h. Ciprofloxacin is used as a reference throughout the study. CFU was calculated by the colonies formed on the agar plate at each time interval. Time-kill curves were plotted using the CFU/mL of surviving bacteria at each time point in the absence or presence of the compound (Tsuji et al., 2008).
The checkerboard method1 was used to determine the synergy between the test sample and the antibiotics (ciprofloxacin, gentamicin, rifampicin and doxycycline) against a panel of bacteria (Pandey et al., 2017). Serial twofold dilutions of each antibiotic to at least double the MIC were freshly prepared before testing. According to CLSI guidelines, the test compounds were serially diluted along the ordinate ranged from 0.625 to 20 μg/mL. In comparison, the antibiotics were serially diluted as shown along the abscissa ranged from 0.625 to 5 μg/mL/ml in 96 well microtiter plate. An inoculum ∼106 CFU/mL of specific bacterial strain was prepared, and 100 μL was inoculated in each microtiter well, and the plates were incubated at 37°C for 24 h under aerobic conditions. The ΣFICs (fractional inhibitory concentrations) were calculated as follows:
Where, A and B refers to the MIC of each antibiotic in combination (in a single well), and MICA and MICB are the MIC of each drug individually. FIC A is the MIC of drug A in the combination/MIC of drug A alone, and FIC B is the MIC of drug B in the combination/MIC of drug B alone. The combination is considered synergistic when the ΣFICs is ≤ 0.5, indifferent when the ΣFIC is > 0.5–4, and antagonistic when the ΣFICs is > 4 (Vogelman and Craig, 1985; Domínguez et al., 2001; Odds, 2003; Odenholt et al., 2003).
To determine the PAE, young culture of E. coli was diluted in MHBII ∼106 CFU/mL and exposed to 0.5× and 1× MIC of ciprofloxacin, gentamicin, and compounds 5 and 21 incubated at 37°C for 1 h (Suller and Lloyd, 2002). The culture was centrifuged and washed twice with MHBII to remove the traces of antibiotics used. The cells obtained were resuspended in antibiotic or sample free MHBII and incubated at 37°C. Samples collected for every 1 h were inoculated on the agar plate followed by the enumeration of CFU. PAE can be calculated using the following equation:
T refers to difference in time required for 1 Log 10 increase in CFU vs. CFU observed immediately after removing the test sample or antibiotic. C refers to similarly treated sample free control.
In vitro enzyme, the inhibitory assay was performed for the synthesized compounds 1–29 using a commercial ParE assay kit (ProFoldin, Hudson, United States). The assay was performed as per the standard protocol provided by the ProFoldin. The title compounds were screened at six concentrations ranging from 0.1 to 3.2 μg/mL. ParE hydrolyzes ATP as the source of molecular energy to perform decatenation reactions. Fluoroquinolones and aminocoumarins usually block ATPase activity to hinder the biological activities of topoisomerase II. Thus, the topoisomerase ATPase assay can be used for high-throughput screening of topoisomerase inhibitors. The ATPase activity of the E. coli ParE enzyme was based on the detection of inorganic phosphate produced by the hydrolysis of ATP in the presence of DNA. The phosphate is detected using a microplate reader at an absorbance wavelength of 650 nm. The selected compounds were tested for the inhibition of ATPase activity of ParE enzyme. The total volume of ATPase assay reaction mixture consists of 18 μL of H2O, 3 μL of 2 mM ATP, 3 μL of 10 × Buffer, 3 μL of 10 × DNA, 3 μL of 10 × E. coli ParE and different concentrations of the test sample were dissolved in DMSO. The reaction mixture was incubated for 1 h at 37°C and then quenched with 45 μL of Dye. Incubate for 5 min, and the absorbance was measured at 650 nm. The IC50 values were determined from the absorbance readings using no enzyme and no compound controls. They were calculated using the non-linear regression curve fit method.
Vero cells were centrifuged, and cell count was adjusted to ∼105 cells/mL using Hi-media, Dulbecco’s Modified Eagle Medium (DMEM) containing 10% FBS. 100 μL (approximately 10,000 cells/well) of the diluted cell suspension was added to each well of a 96-well plate and incubated 24 h (Patel et al., 2009). Then the cells were centrifuged, obtained pellets were resuspended with 100 μL of different concentrations of test samples prepared in maintenance media and incubated at 37°C for 48 h in a 5% CO2 atmosphere. Microscopic examination of plates was carried out, and observations were recorded every 24 h. 20 μL of 3-(4,5 dimethyl thiazole-2-yl)- 2,5-diphenyl tetrazolium bromide (MTT, 2 mg/mL) was added, and plates were gently shaken and incubated for 2 h at 37°C in 5% CO2 atmosphere. To solubilize the formed formazan crystals, 100 μL of DMSO was added and gently shaken. The absorbance was measured using a microplate reader at a wavelength of 540 nm. The % of cell viability was calculated, and the concentration of drug or test samples required to inhibit the cell growth by 50% was generated from the dose-response curves.
E. coli (NCIM 2065) were grown overnight in 1% tryptic soy broth (TSB) medium at 37°C, followed by dilution (1:100) in fresh TSB medium. The freshly diluted culture (200 μL) was transferred into 96 well plates, covered with adhesive foil for maintaining low oxygen inside the plate and incubated for 48 h at 37°C in static condition. The media was decanted, and the plate was rinsed gently with phosphate buffer saline (PBS) (pH 7.4) to remove planktonic E. coli. Plates were refilled with different drug concentrations prepared in TSB and incubated at 37°C for 24 h. The media was decanted, washed thrice with PBS, and fixation of biofilm was achieved after incubating the plate at 60°C for 1 h. The formed biofilm is stained using 0.06% crystal violet for 10 min, washed with PBS, and dried at room temperature. Biofilm quantification was carried out by eluting the crystal violet using 30% acetic acid (200 μL) (Kwasny and Opperman, 2010). The absorbance was recorded at 600 nm of wavelength using Tecan-i-control, 1.7.1.12.
The 3D-structures of 29-compounds and X-ray crystallized protein structure were prepared using LigPrep available in Maestro (Schrödinger suite). Further, low energy conformers were selected for docking. The X-ray crystal structure of protein was prepared using Protein preparation wizard available in Maestro (Schrödinger suite) (Sastry et al., 2013). Hydrogens were added and water molecules were deleted in the protein crystal structures using Epik module. Addition of missing side chains and loops followed by generation of tautomeric states and protonation of amino acids using the module Prime (Jacobson et al., 2004). Protein minimization was carried out with a RMSD of 0.30 Å for crystallographic heavy atoms using OPLS3e forcefield (Roos et al., 2019). Furthermore, a grid box was generated at the centroid of active site (co-crystallized ligand) keeping the partial charge cutoff and van der Waals scaling at 0.25 and 1 Å, respectively. The obtained conformers from LigPrep were docked using Glide in extra-precision (XP) mode without smearing any constraints (Friesner et al., 2006). The best-docked poses of ligands were selected based on the Glide emodel, Glide Gscore, and Glide energy.
The BFG of synthesized ligands were computed using molecular mechanics-generalized born surface area (MM-GBSA)/Prime approach with OPLS3e forcefield (Roos et al., 2019). Energy minimization of protein-ligand complexes and simulation were carried out using Prime and VSGB 2.0 energy model (Li et al., 2011), respectively. Optimized implicit solvation model and physics-based corrections included in this energy model for all types of interactions (Hydrogen bonding, self-contact, hydrophobic, π-π stacking, π-cationic interactions).
Mean absorbances and their standard deviations (SDs) were computed for tested strains and controls, determined in triplicate and repeated three times. A two-way analysis of variance was used to determine the statistical significance. Statistical analysis was performed in GraphPad Prism v5.01 software (San Diego, CA, United States). A p-value ≤ 0.01 or lower was considered as statistically significant.
AST of selected compounds was determined using the micro-broth dilution method according to CLSI (2017). The MIC of the selected compounds was determined in the concentration range of 0.625–20 μg/mL against E. coli and MRSA strains. The MIC of compounds against other organisms was already reported (Yele et al., 2020, 2021). The obtained MIC values are compared with standard drug ciprofloxacin, and the results are given in Table 1.
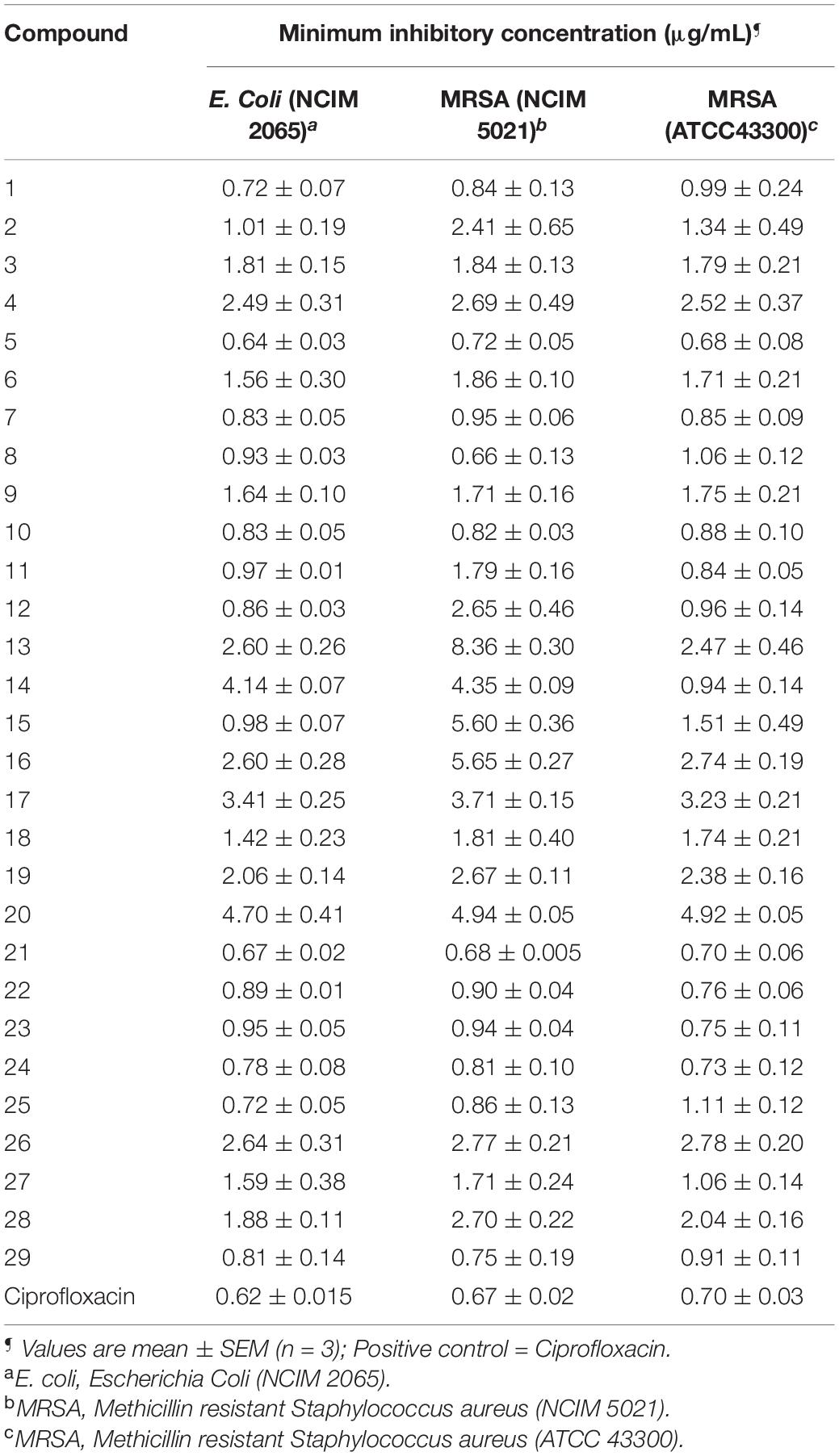
Table 1. Minimum inhibitory concentration (MIC) of synthesized compounds (1–29) against the selected bacterial strains.
All compounds exhibited good antibacterial activity with MIC values ranging from 0.64 to 5.65 μg/mL. However, it should be noticed that the sensitivity of bacterial strains toward title compounds was in general different. The compounds 5 and 21 appeared to be most active against E. coli with an MIC of 0.64 and 0.67 μg/mL followed by compound 1 (MIC, 0.72 μg/mL), 24 (0.78 μg/mL) and 25 (0.72 μg/mL).
In addition, compounds 8 (0.66 μg/mL) and 21 (0.68 μg/mL) showed significant antibacterial activity against MRSA (NCIM 5021). While MRSA (ATCC 43300) appeared to be sensitive against compound 5 with MIC at 0.68 μg/mL.
MBC is the minimum concentration of a compound required to kill 99.9% of bacteria. The MBC/MIC ratio of ≤2 demonstrates that the compound has bactericidal and tolerance development if the ratio is ≥4. The MBC of the selected compounds was tested against E. coli and MRSA strains. The MBC of compounds against other organisms was already reported (Yele et al., 2020, 2021). All the compounds exhibited significant bactericidal activity against E. coli and MRSA strains (Table 2).
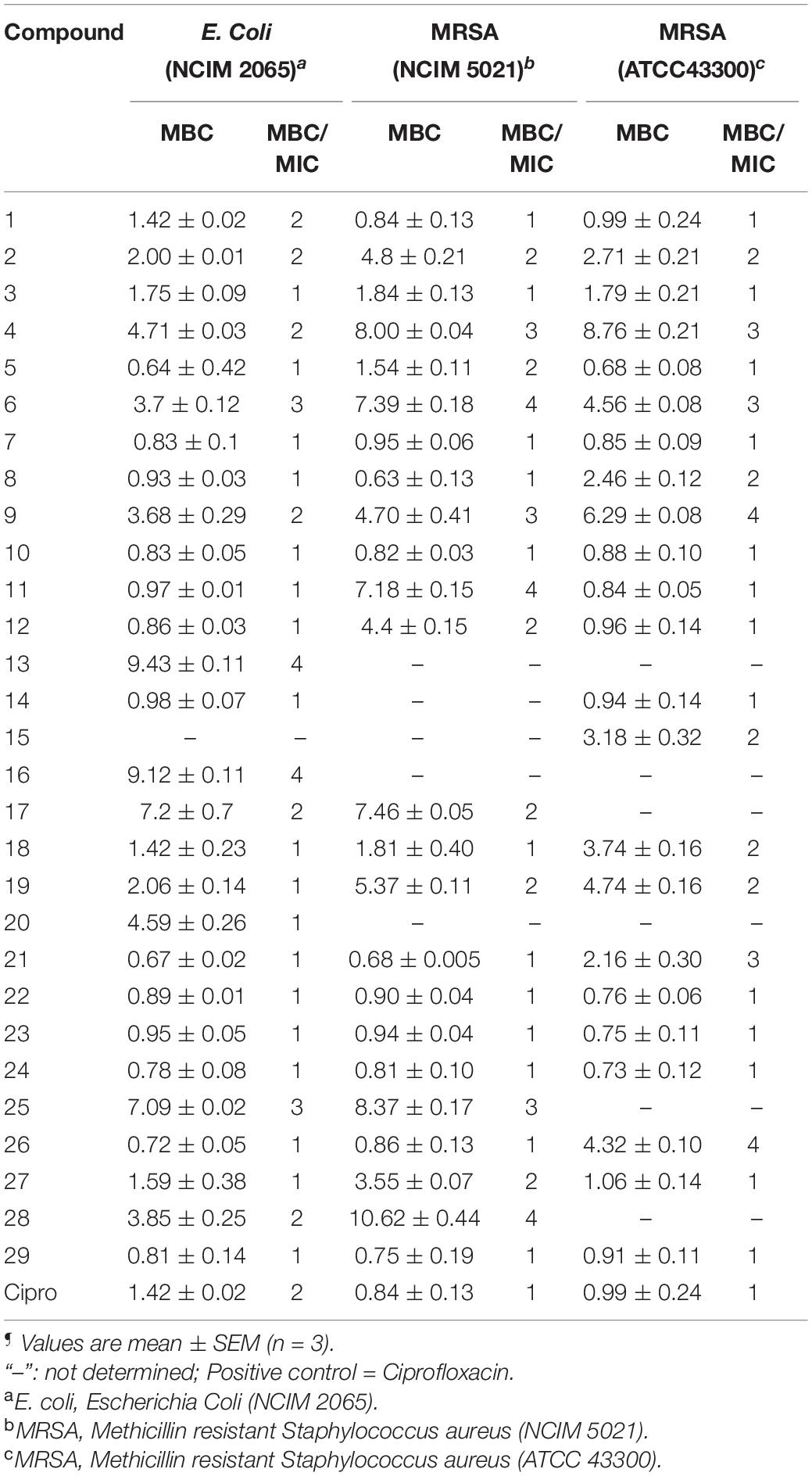
Table 2. Minimum bactericidal concentration (MBC) (μg/mL)¶ and MBC/MIC ratio of benzohydrazides and phenylacetamide derivatives (1–29) against a panel of organisms.
Compounds 5 and 21 exhibited significant MIC (0.64 and 0.67 μg/mL), and MBC values at onefold MIC against E. coli are considered for further evaluation. Time-kill kinetics demonstrated the bactericidal property of compounds 5 (Figure 1A) and 21 (Figure 1B) against E. coli. The E. coli bacterial culture was serially diluted to attain ∼105 CFU/mL concentration and treated with test samples and ciprofloxacin at concentrations of 0.5× and 1.0× MIC. The 50 μL was collected and sub-cultured on an agar plate at regular time intervals of 0, 2, 4, 8, 16, and 24 h, respectively. The cultured agar plates were incubated at 37°C. CFU was calculated from the colonies formed on the agar plate at each time interval. Time-kill curves were plotted using the CFU/mL of surviving bacteria at each time point in the compound’s absence or presence. Tested samples exhibited significant bactericidal property against E. coli compared to the untreated E. coli and culture with ciprofloxacin.
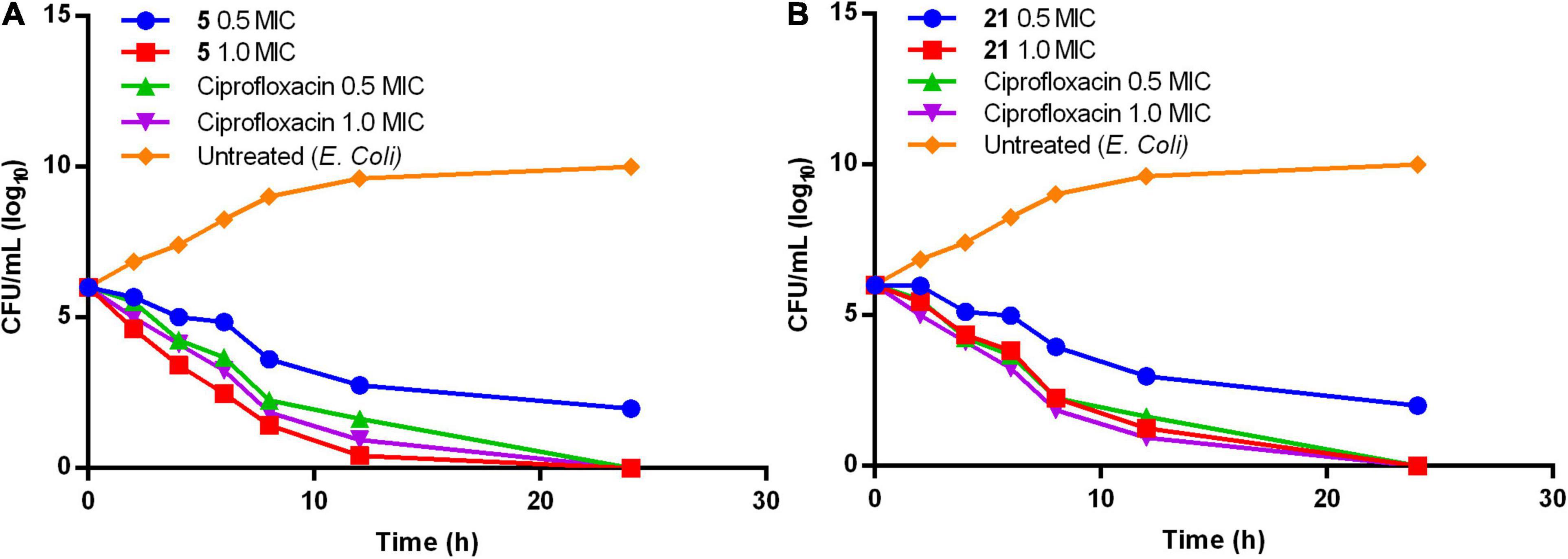
Figure 1. Time-kill kinetics of compound (A) 5 and (B) 21.
The checkerboard method was employed to determine the synergy between compounds 5, 21, and antibiotics ciprofloxacin, gentamicin, rifampicin and doxycycline. The results have shown that compounds 5 (Table 3) and 21 (Table 4) synergized with the FDA-approved antibiotics, demonstrating significant potential to be a part of a multi-drug regimen.
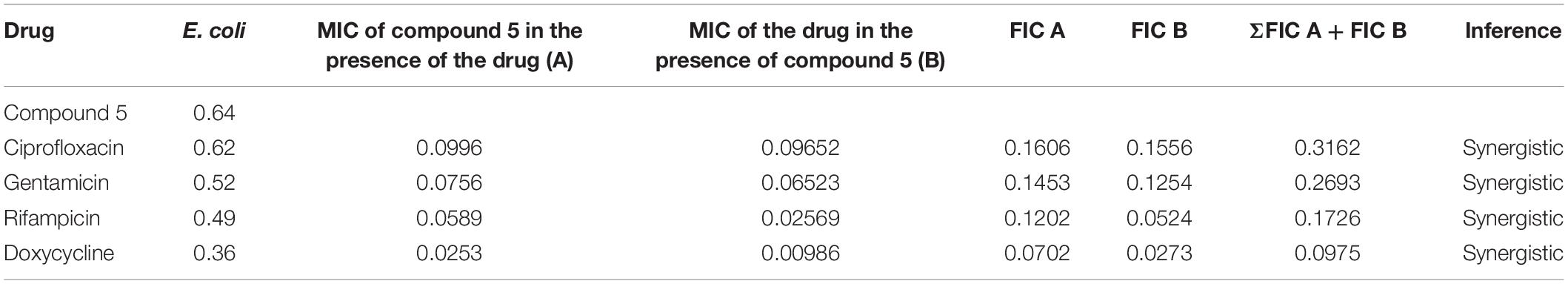
Table 3. Determination of synergy of compound 5 with approved antibiotics.
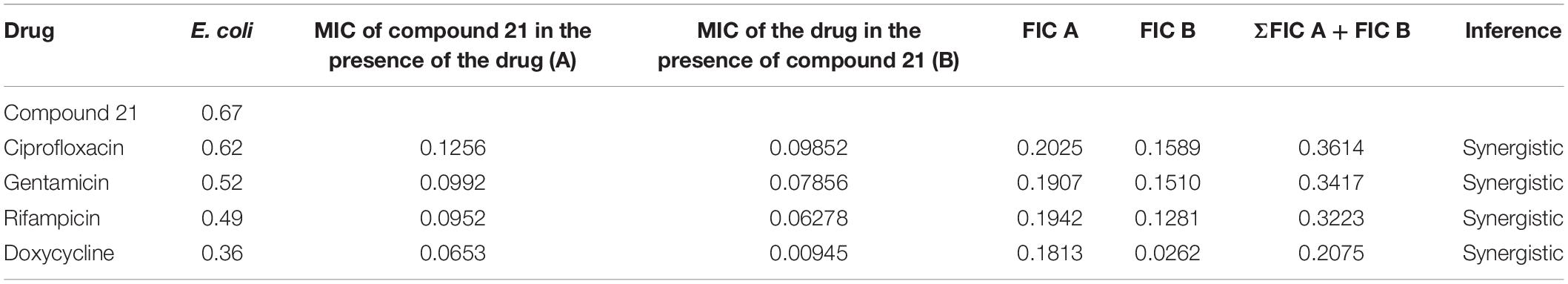
Table 4. Determination of synergy of compound 21 with approved antibiotics.
PAE of compounds 5 and 21 were evaluated with ciprofloxacin and gentamicin as controls. The results demonstrated that compounds 5 and 21 exhibited a PAE of ∼2 h at 10× MIC compared to ciprofloxacin and gentamicin (Table 5).

Table 5. Post-antibiotic effect of compound 5 and 21.
E. coli ParE enzyme inhibition assay was performed for the synthesized compounds 1–29 using commercial ParE assay kit (ProFoldin, Hudson, United States). The compounds displayed IC50 values (Figure 2) in the range of 0.27–2.80 μg/mL. Among, the assayed molecules, the two compounds namely 8 and 25 belonging to the phenylacetamide series were found to inhibit ParE enzyme with IC50 values of 0.27 and 0.28 μg/mL, respectively (Figure 3). The other compounds 1 (0.99 μg/mL), 3 (0.95 μg/mL), 4 (0.95 μg/mL), 5 (0.35 μg/mL), 7 (0.95 μg/mL), 14 (0.36 μg/mL) 17 (0.86 μg/mL), 21 (0.50 μg/mL), and 24 (0.48 μg/mL) belonging to both phenylacetamide and benzohydrazides series displayed significant inhibitory activity against ParE enzyme with IC50 values less than <1 μg/mL. The compounds 6 (1.19 μg/mL), 10 (1.84 μg/mL) 11 (1.81 μg/mL), 15 (1.33 μg/mL), 18 (1.81 μg/mL), 19 (1.80 μg/mL), 22 (1.01 μg/mL), 23 (1.76 μg/mL), 26 (1.54 μg/mL), 27 (1.30 μg/mL), and 28 (1.93 μg/mL) exhibited variable activity against E. coliParE enzyme with IC50 values ranging from 1 to 2 μg/mL. Only few compounds 2 (2.33 μg/mL), 9 (2.80 μg/mL), 12 (2.34 μg/mL), 13 (2.73 μg/mL), 16 (2.62 μg/mL), 20 (2.78 μg/mL), and 29 (2.77 μg/mL) displayed partial significant inhibition against ParE enzyme with IC50 values in the range of 2–2.80 μg/mL.
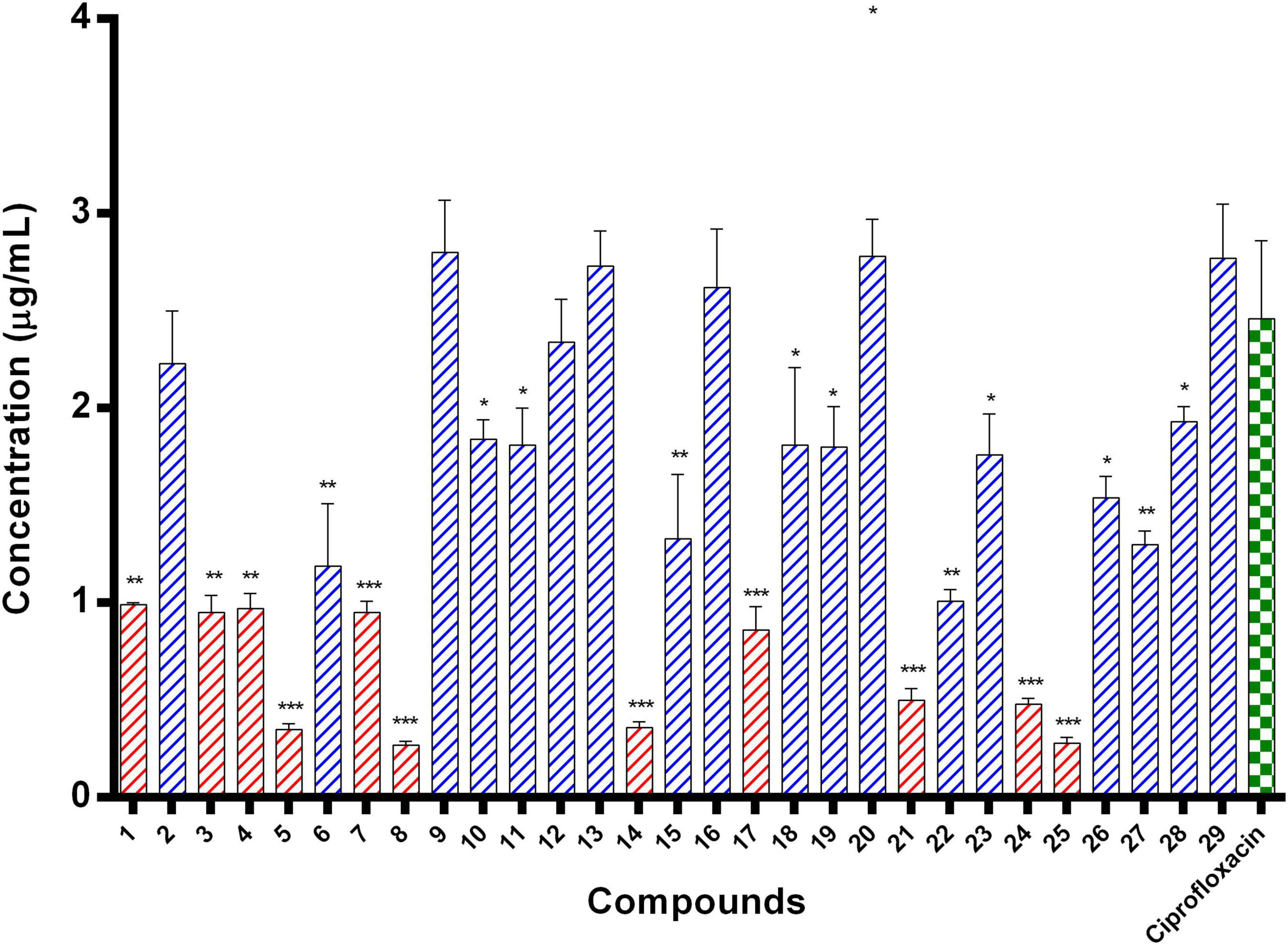
Figure 2. IC50 values of 29 compounds against E. coli ParE enzyme. Data are expressed as mean ± SD (n = 3). *p ≤ 0.05, **p ≤ 0.005, ***p ≤ 0.001.
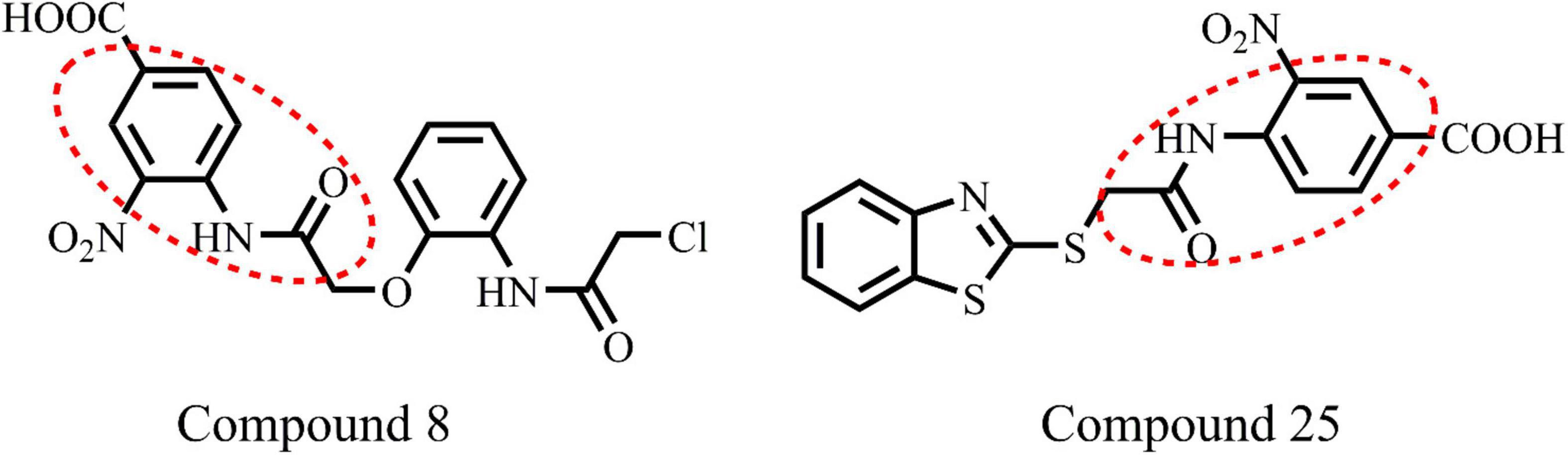
Figure 3. Chemical structure of highly active compounds 8 and 25 in phenylacetamide series.
The compounds whose IC50 is <1 μg/mL were tested for cytotoxicity profile against Vero cells using MTT assay. CC50 is referred to as the lowest concentration of the compound, leads to a 50% cell viability reduction with doxorubicin as a positive control. In addition, the selectivity index (SI) was also calculated by dividing CC50 with antibacterial activity. Table 6 explains that compounds are non-toxic to Vero cells (61.7583–256.9655) and exhibited a selectivity index (169.0629–951.7240).
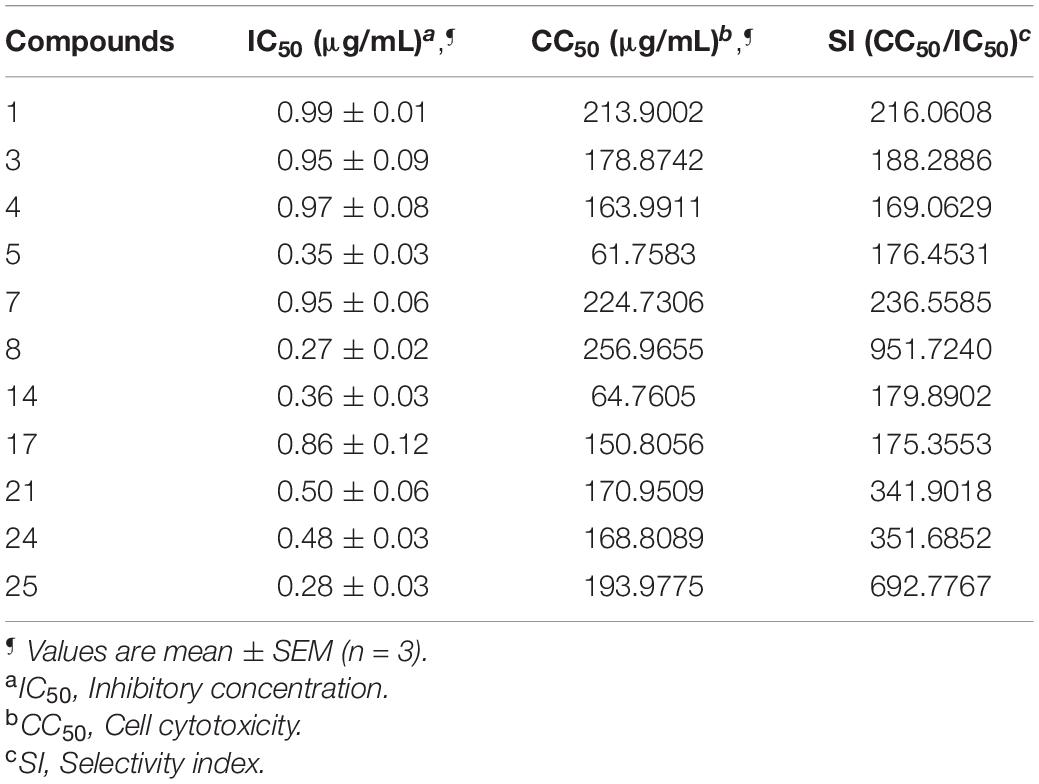
Table 6. Cytotoxicity profile against Vero cells and Selectivity index (SI) of selected compounds against E. coli.
Upon analysis of results obtained through enzyme inhibition assay and selectivity index, we selected compounds 1, 7, 8, 21, 24, and 25 (IC50: <1 and Selectivity index: >200) for E. coli antibiofilm activity. The compound 1, 7, 8, 24, and 25 at 10× MIC displayed potent anti-biofilm activity in reducing biofilm by 76, 81, 76, 71, 62, and 43%, respectively. On the other hand, a similar reduction in biofilm was achieved at 10 × MIC of ciprofloxacin (86% reduction) and erythromycin (71% reduction), while ampicillin reduced only 63%. Thus, compounds 1, 7, 8, and 21 are more potent in reducing biofilm than ampicillin (Figure 4).
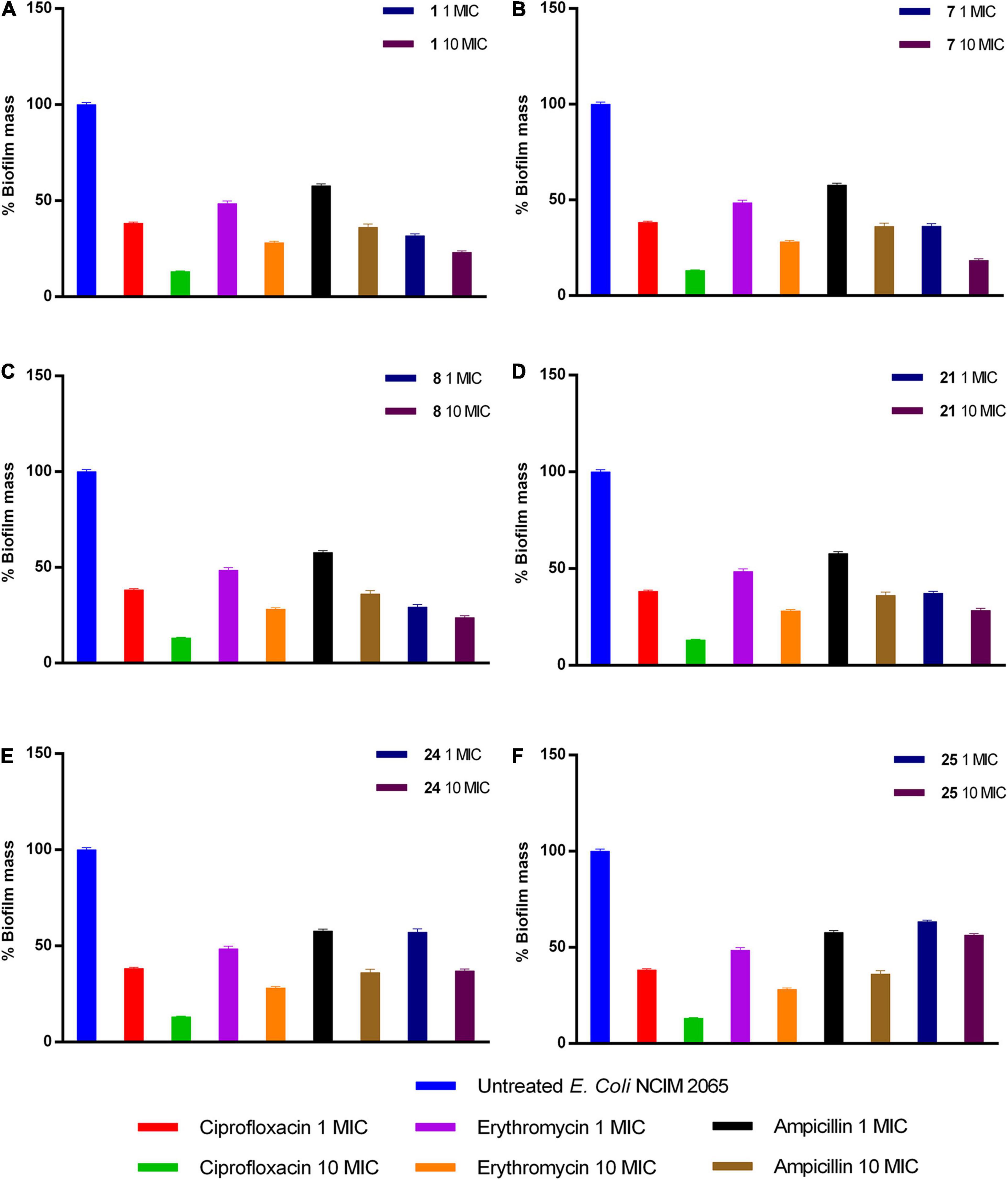
Figure 4. Activity of (A) 1, (B) 7, (C) 8, (D) 21, (E) 24, and (F) 25 compounds against preformed E. coli biofilm.
The ligand binds to the protein crystal structure with high binding affinity (high negative glide Gscore). The results of XP-molecular docking were provided in Table 7. The Gscore of all the compounds were found in the range of –2.822 to –5.375 kcal/mol. XP-docked poses of the compounds exhibited significant interactions like hydrogen bonding, self-contact, hydrophobic, π-π stacking, π-cationic interactions with conserved N-terminal domain residues Asp45, Asp69, Arg72, Gly73, His79, Ala86, Ile90, Ile116, and Ser117 (Supplementary Figures 1–29). The results were compared by redocking of the co-crystal into the binding pocket of E. coli ParE. The Gscore of co-crystal was found to be –7.603 kcal/mol. Further, the high active compounds 8 (Figure 4A) and 25 (Figure 4B) exhibited significant inhibitory activity as evident by their Gscore –5.412 and –5.375 kcal/mol.
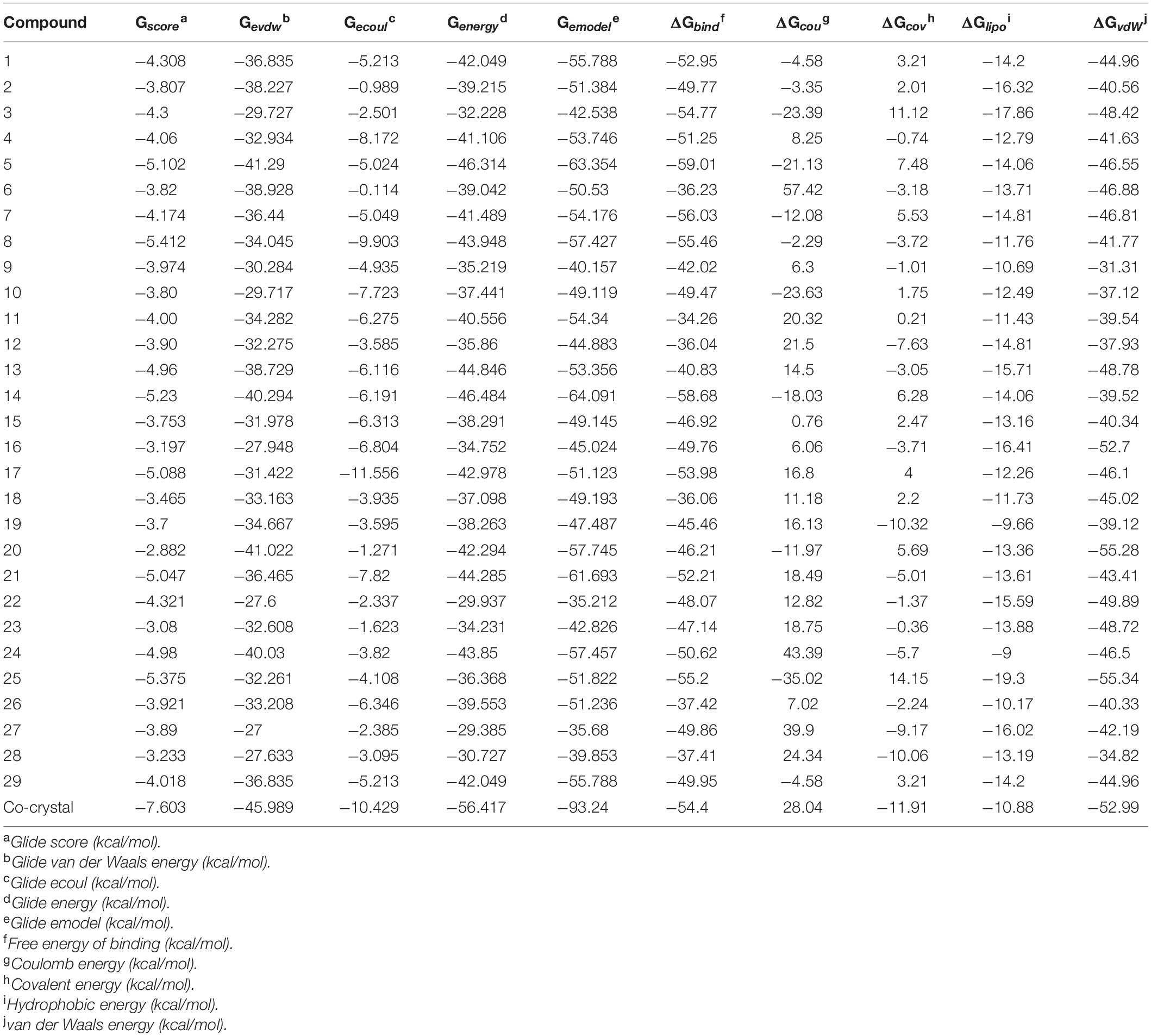
Table 7. XP-docking and binding free energy (kcal/moL) results of synthesized compounds (1–29) against E. coli ParE enzyme (Pdb.3FV5).
The BFG of compounds with E. coli ParE was computed using MM-GBSA/Prime approach. The ΔGbind values were observed in the range of –34.26 to –55.20 kcal/mol. All the energy terms of Prime calculations were represented in the Table 7. Amongst, all the prime energy terms, van der Waals energy was found to be in the range of –31.31 to –55.28 kcal/mol.
In this article, the synthesized phenylacetamide and benzohydrazides derivatives were screened for their ParE inhibitory activity. Further, the compounds displayed potent antibacterial activity against E. coli and MRSA strains.
Both phenylacetamide and benzohydrazides series exhibited optimal activity against selected strains (Table 1). Gratifyingly, most of the compounds shown potential inhibition of E. coli than MRSA strains. The compounds which exhibited potency against E. coli were observed to inhibit MRSA (ATCC43300) at MIC <1 μg/mL. The activity in the phenylacetamide series may be attributed to amide linkage (29) or the presence of nitro (1, 7, 8) and carboxyl (5, 8) groups on the phenyl ring. In addition, compounds (23, 24, 25) have shown promising inhibition of bacteria because of benzothiazole ring flanked by substituted phenyl ring via an amide linkage. Moreover, the compounds in the benzohydrazides series also shown prominent inhibitory activity that may be ascribed to the presence of formylformohydrazide (O = C-NH-NH-C = O) (12, 15) or benzothiazole ring, which is linked to the substituted phenyl rings through the formylformohydrazide (21, 22) group.
Both phenylacetamide and benzohydrazides series displayed potent bactericidal activity against selected pathogens. Besides, compounds 5 (0.64 μg/mL) and 21 (0.64 μg/mL) exhibited significant bactericidal property against E. coli at onefold MIC. However, these two compounds also exhibited significant bactericidal properties against MRSA strains. The activity of compound 5 may be attributed to substitutions, such as carboxyl and bromine groups on the phenyl ring and amide moiety flanked between the two aromatic rings. On the other hand, compound 21 belonging to the benzohydrazides series also exhibited potential bactericidal activity may be due to the presence of formylformohydrazide connecting benzothiazole ring and nitro substituted phenyl ring. Furthermore, time-kill kinetics of compounds 5 and 21 (Figure 1) revealed the potential bactericidal property against E. coli compared to the ciprofloxacin and untreated E. coli.
After analyzing the activity of these compounds, we performed synergy testing using a checkerboard assay with different approved drugs like ciprofloxacin, gentamicin, rifampicin, and doxycycline. The results obtained were astounding, exemplifying that the selected compounds 5 and 21 were considered the best candidates in multi-drug regimens (Tables 3, 4).
In addition, compounds exhibited significant PAE at 10× MIC in 2 h compared to ciprofloxacin. In contrast, both compounds exhibited more activity at 10× MIC in comparison with gentamicin (Table 5).
All the compounds were screened for E. coli ParE inhibitory activity using an assay kit. The results revealed that all the tested compounds displayed significant inhibitory activity against ParE with the IC50 values in the range of 0.27 and 0.28 μg/mL (Figure 2). Almost 50% of the compounds exhibited significant activity against E. coli ParE with IC50 <1 μg/mL. While 20% exhibited very potent activity with IC50 values <0.5 μg/mL. The promising inhibition of these compounds may be attributed to the electron-withdrawing groups on the phenyl ring at positions two and four. In the phenylacetamide series, the high active compounds, namely 8 and 25, exhibited potential inhibition of E. coli ParE at IC50 of 0.27 ± 0.02 μg/mL and 0.28 ± 0.03 μg/mL, respectively. Fortunately, both compounds share common functional groups, such as carboxyl at position four and nitro at position two on the phenyl ring. However, the two compounds are dissimilar at the other side of molecular structure, substituted phenoxy moiety (8) and 2-mercaptobenzothiazole group (25) (Figure 3). Further, compounds 5 and 24 displayed promising activity with IC50 values of 0.35 ± 0.03 and 0.48 ± 0.06 μg/mL, which may be attributed to the presence of electron-withdrawing groups, such as carboxyl and nitro at position four of the phenyl ring. The variation among the activities of both the compounds maybe because of the substituted phenoxy moiety present in compound 5. However, the replacement of electron-withdrawing groups with chlorine atoms shown detrimental to the inhibitory activity (10, 11).
In the benzohydrazides series, the compounds 14 and 21 shown significant inhibitory activity against E. coli ParE at IC50 of 0.36 ± 0.02 and 0.50 ± 0.06 μg/mL, respectively. The potent activity of these compounds may be because of the presence of electron-withdrawing group NO2 at position four of the phenyl ring. However, on the other side, the molecules possess different chemical moieties (substituted phenoxy moiety; 14 and 2-mercaptobenzothiazole group; 21) responsible for the variation in the activity. The compounds possessing electron-releasing groups (12, 13, 19, 20) have exhibited partial inhibitory activity against E. coli ParE.
Furthermore, the compounds whose IC50 is <1 μg/mL were tested for cytotoxicity profile against Vero cells using MTT assay using doxorubicin as the reference standard. A perusal of cytotoxicity data reveals that all compounds are non-toxic to Vero cells in the range of 61.7583–256.9655 μg/mL and displayed potential SI [>20; (169.0629 to 951.7240)]. The higher SI value indicates that the drug was more effective and safer during in vivo treatment (Table 6).
Besides, compounds, which displayed promising SI (>200), were selected for antibiofilm activity. Bacteria under different stresses make biofilm protect themselves, leading to prolonged therapeutic intervention and increased drug-resistance. All the approved antibiotics possess minimal activity against biofilm because of the altered physiological state; thus, it is essential to determine the antibiofilm activity of compounds under development. Results explained that compounds 1, 7, 8, 21, 24, and 25 exhibited potent activity in biofilm reduction of E. coli at 10× MIC. Except compounds 24 and 25, the remaining compounds exhibited greater potency in antibiofilm activity than ampicillin (Figure 4).
Computational studies, such as molecular docking and binding free energy revealed better binding affinity of the compounds within the catalytic pocket of E. coli ParE (Table 7). From the Figure 5A that the active 8 (IC50 of 0.27 μg/mL) is placed well within the active site and occupied N-terminal domain of the pocket. It exhibited five hydrogen bonding interactions with Asn42, Gly73, Ile90, Ile116, Thr163 with carbonyl groups of the amide linker and carboxyl group. Further, benzene ring of compound formed a salt-bridge and π-cationic interactions with the protonated Arg72. Besides compound was stabilized by forming a π-π stacking interaction between the electron clouds of aromatic ring (compound 8) and imidazoline ring (His79). Like compound 8, another high active inhibitor 25 (0.28 μg/mL) (Figure 5B) also occupied N-terminal domain of the receptor and showed three hydrogen bonding interactions between the carbonyl groups of amide fragment and carboxylic acid with Asn42, Asp69, and Ile116. A salt bridge was also observed between the nitro group and protonated Arg72. Moreover, the compound stabilized by the π-π stacking interaction with His79. Similar interactions were observed from the docking pose of co-crystallized ligand (Supplementary Figure 30). The binding strength of the compounds 8 (57.427 kcal/mol) and 25 (57.822) indicated higher binding of the ligands in the catalytic pocket of enzyme. The non-polar solvation energy (ΔGsolv: –9.00 to 16.41 kcal/mol) term was moderately favorable while coulombs (ΔGcou: 43.39 to –23.63 kcal/mol) and covalent (ΔGcov: 11.12 to –17.86 kcal/mol) energy terms were unfavorable for the inhibitory activity. Further, it should be noted that van der Waals energy (ΔGvdW: –31.31 to –55.28 kcal/mol) term was found to be favorable and is the key driving forced for the ligand binding. This is in agreement with the Glide Emodel (–29.937 to –46.484 kcal/mol) having significant weighting of the force field.
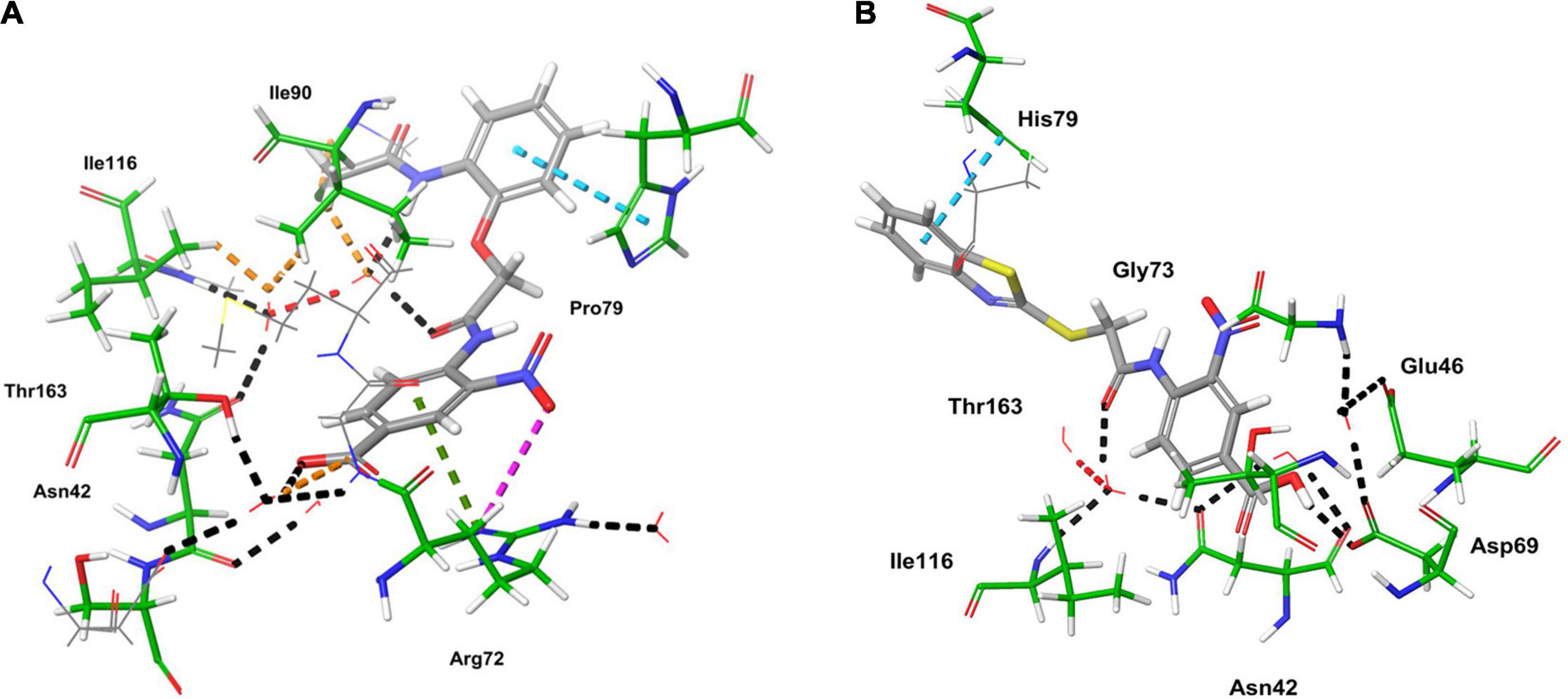
Figure 5. 3D-interaction diagrams of (A) compound 8 and (B) compound 25 exposing key interactions with the catalytic residues of E. coli ParE enzyme.
The broad structural activity relationship was summarized in Figure 6. Although, compounds have shown significant potential inhibition of E. coli ParE, the variation in their inhibitory concentrations might be because of the chemical substitutions at different positions on the aromatic rings.
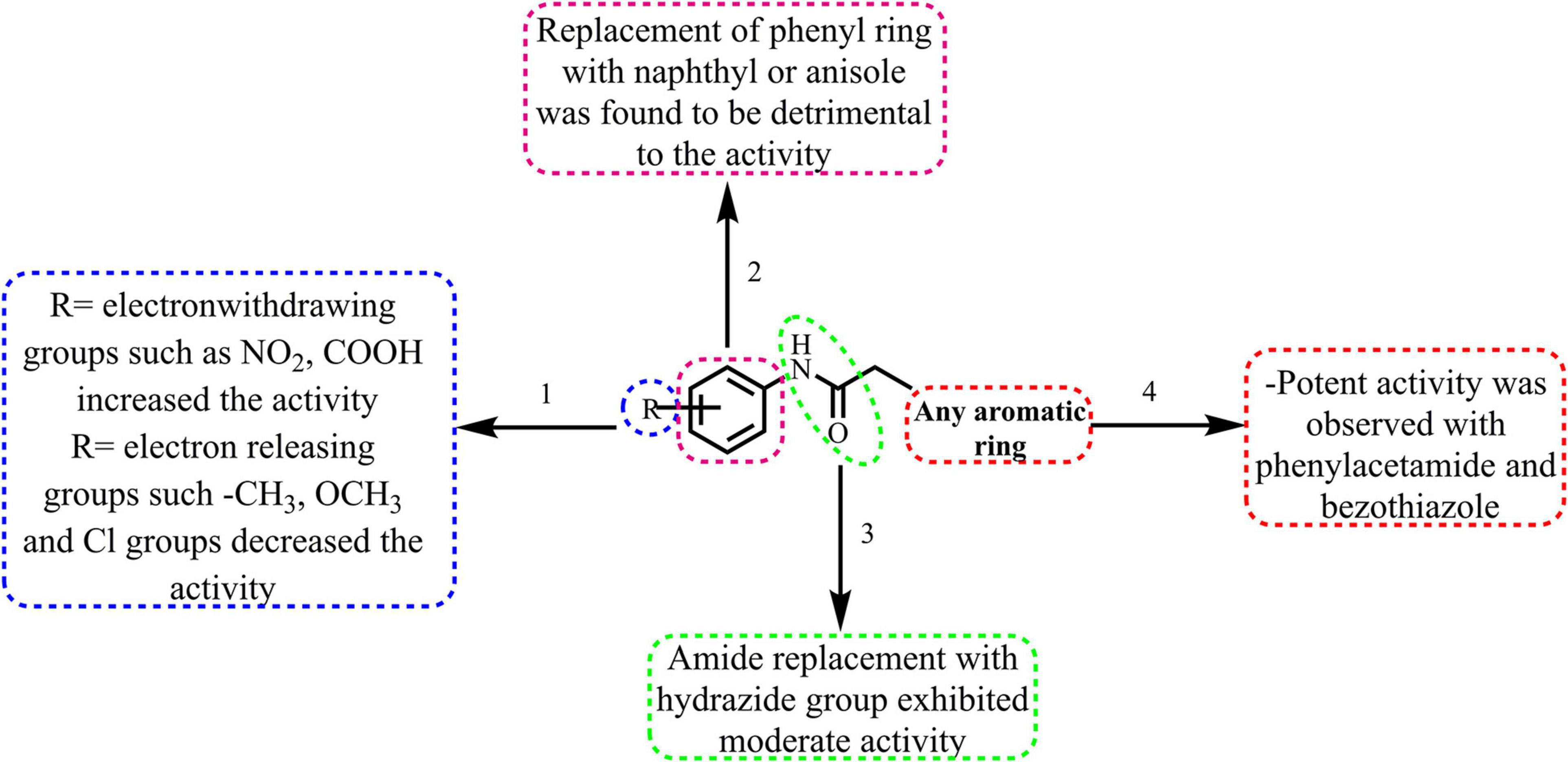
Figure 6. Broad structure-activity relationship of the phenylacetamide and benzohydrazides.
(1) From the results, it is clear that compounds possessing electron-withdrawing groups, such as nitro and carboxyl moieties (8 and 25) increased the activity toward the E. coli ParE. However, electron releasing groups like methyl, methoxy groups decreased the inhibitory activity (12, 13, and 20). The halogen substitution on the aromatic rings possesses substantial activity of these compounds against E. coli ParE (11, 15, 28, and 29).
(2) Further, the replacement of the phenyl ring with fused aromatic system or with anisole markedly decrease the inhibitory activity (28 and 29).
(3) Both amide and hydrazide linker were found to be necessary for the activity as evident by their IC50s and docking results.
(4) Potent activity was observed with both the phenylacetamide and benzothiazole ring systems (8 and 25).
We have previously synthesized and tested a series of phenylacetamide and benzohydrazides, which displayed interesting antibacterial activity against selected bacteria strains. In the current paper, we have successfully performed antibacterial screening for E. coli and MRSA strains. Amongst the title compounds, compounds 5 and 21 exhibited significant MIC and MBC. Further, these compounds showed PAE at 2 h compared to ciprofloxacin and gentamicin and established concentration-dependent bactericidal property. The compounds exhibited synergistic activity with FDA-approved drugs indicating the better potential for the multi-drug regimen. Besides, the molecular structures of the compounds 8 and 25 were found to be favorable for E. coli ParE inhibition as evident by their IC50 values IC50 of 0.27 ± 0.02 μg/mL and 0.28 ± 0.03 μg/mL, respectively. The significant values of CC50 and selectivity index indicated that drugs were more effective and safer during in vivo treatment.
Furthermore, compounds 8 and 25 also exhibited significant antibiofilm activity, as evidenced by their biofilm mass reduction. In summary, the phenylacetamide series (compounds 8 and 25) represent one of the most exciting chemical scaffolds in the bacterial armamentarium. As a novel chemical that addressing ABR by targeting unexplored targets like ParE, the representative compounds 8 and 25 can further be investigated to develop potent antibacterial agents for use against nosocomial pathogens.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
VY synthesized the literature, drafted the manuscript, and revised the manuscript. BS and AW were involved in the in vitro studies. AM provided the conceptual inputs. All authors read and approved the final manuscript.
The authors acknowledge the All India Council of Technical Education, National Doctoral Fellowship (NDF)—56149, for providing the funding support.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fbioe.2021.669728/full#supplementary-material
Agyare, C., Koffuor, G. A., Boamah, V. E., Adu, F., Mensah, K. B., and Adu-Amoah, L. (2012). Antimicrobial and anti-inflammatory activities of Pterygota macrocarpa and Cola gigantea (Sterculiaceae). Evid. Based Complement. Alternat. Med. 2012:902394.
Ahmad, N., Shinwari, Z. K., Hussain, J., Ahmad, I., Perveen, R., and Rehman, S. (2015). Phytochemical screening and biological activities of different parts of Centaurea montana. Int. J. Plant Sci. Ecol. 1, 237–240.
Azam, M. A., Thathan, J., and Jubie, S. (2015). Dual targeting DNA gyrase B (GyrB) and topoisomerse IV (ParE) inhibitors: a review. Bioorgan. Chem. 62, 41–63. doi: 10.1016/j.bioorg.2015.07.004
Booker, B. M., Smith, P. F., Forrest, A., Bullock, J., Kelchlin, P., Bhavnani, S. M., et al. (2005). Application of an in vitro infection model and simulation for reevaluation of fluoroquinolone breakpoints for Salmonella enterica serotype Typhi. Antimicrob. Agents Chemother. 49, 1775–1781. doi: 10.1128/aac.49.5.1775-1781.2005
Brino, L., Urzhumtsev, A., Mousli, M., Bronner, C., Mitschler, A., Oudet, P., et al. (2000). Dimerization of Escherichia coli DNA-gyrase B provides a structural mechanism for activating the ATPase catalytic center. J. Biol. Chem. 275, 9468–9475. doi: 10.1074/jbc.275.13.9468
CLSI (2017). M100. Performance Standards for Antimicrobial Susceptibility Testing, 27th Edn. Wayne, PA: Clinical and Laboratory Standards Institute (CLSI).
Davies, J., and Davies, D. (2010). Origins and evolution of antibiotic resistance. Microbiol. Mol. Biol. Rev. 74, 417–433.
Domínguez, M. C., de la Rosa, M., and Borobio, M. V. (2001). Application of a spectrophotometric method for the determination of post-antibiotic effect and comparison with viable counts in agar. J. Antimicrob. Chemother. 47, 391–398. doi: 10.1093/jac/47.4.391
Drlica, K., and Zhao, X. (1997). DNA gyrase, topoisomerase IV, and the 4-quinolones. Microbiol. Mol. Biol. Rev. 61, 377–392. doi: 10.1128/.61.3.377-392.1997
Eloff, J. N. (1998). A sensitive and quick microplate method to determine the minimal inhibitory concentration of plant extracts for bacteria. Planta Med. 64, 711–713. doi: 10.1055/s-2006-957563
Fischbach, M. A., and Walsh, C. T. (2009). Antibiotics for emerging pathogens. Science 325, 1089–1093. doi: 10.1126/science.1176667
Friesner, R. A., Murphy, R. B., Repasky, M. P., Frye, L. L., Greenwood, J. R., Halgren, T. A., et al. (2006). Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein- ligand complexes. J. Med. Chem. 49, 6177–6196. doi: 10.1021/jm051256o
Heisig, P. (2001). Inhibitors of bacterial topoisomerases: mechanisms of action and resistance and clinical Aspects1. Planta Med. 67, 3–12. doi: 10.1055/s-2001-10635
Huttner, B., Saam, M., Moja, L., Mah, K., Sprenger, M., Harbarth, S., et al. (2019). How to improve antibiotic awareness campaigns: findings of a WHO global survey. BMJ Global Health 4:e001239. doi: 10.1136/bmjgh-2018-001239
Jacobson, M. P., Pincus, D. L., Rapp, C. S., Day, T. J., Honig, B., Shaw, D. E., et al. (2004). A hierarchical approach to all-atom protein loop prediction. Proteins Struct. Funct. Bioinform. 55, 351–367. doi: 10.1002/prot.10613
Jones, R. N. (1989). Should novobiocin be clinically re-evaluated? Diagn. Microbiol. Infect. Dis. 12, 363–365. doi: 10.1016/0732-8893(89)90105-3
Kwasny, S. M., and Opperman, T. J. (2010). Static biofilm cultures of Gram-positive pathogens grown in a microtiter format used for anti-biofilm drug discovery. Curr. Protoc. Pharmacol. 50, 13A. 18.11–13A. 18.23.
Li, J., Abel, R., Zhu, K., Cao, Y., Zhao, S., and Friesner, R. A. (2011). The VSGB 2.0 model: a next generation energy model for high resolution protein structure modeling. Proteins Struct. Funct. Bioinform. 79, 2794–2812.
Maxwell, A. (1997). DNA gyrase as a drug target. Trends Microbiol. 5, 102–109. doi: 10.1016/s0966-842x(96)10085-8
Maxwell, A., and Lawson, D. M. (2003). The ATP-binding site of type II topoisomerases as a target for antibacterial drugs. Curr. Top. Med. Chem. 3, 283–303. doi: 10.2174/1568026033452500
Michael, C. A., Dominey-Howes, D., and Labbate, M. (2014). The antimicrobial resistance crisis: causes, consequences, and management. Front. Public Health 2:145. doi: 10.3389/fpubh.2014.00145
Miller, J. R., and Waldrop, G. L. (2010). Discovery of novel antibacterials. Expert Opin. Drug Discov. 5, 145–154. doi: 10.1517/17460440903493449
Odds, F. C. (2003). Synergy, antagonism, and what the chequerboard puts between them. J. Antimicrob. Chemother. 52:1. doi: 10.1093/jac/dkg301
Odenholt, I., Löwdin, E., and Cars, O. (2003). Postantibiotic, postantibiotic sub-MIC, and subinhibitory effects of PGE-9509924, ciprofloxacin, and levofloxacin. Antimicrob. Agents Chemother. 47, 3352–3356. doi: 10.1128/aac.47.10.3352-3356.2003
Pandey, M., Singh, A. K., Thakare, R., Talwar, S., Karaulia, P., Dasgupta, A., et al. (2017). Diphenyleneiodonium chloride (DPIC) displays broad-spectrum bactericidal activity. Sci. Rep. 7:11521.
Patel, S., Gheewala, N., Suthar, A., and Shah, A. (2009). In-vitro cytotoxicity activity of Solanum nigrum extract against Hela cell line and Vero cell line. Int. J. Pharm. Pharm. Sci. 1, 38–46.
Piddock, L. J. (2019). The global antibiotic research and development partnership (GARDP): researching and developing new antibiotics to meet global public health needs. MedChemComm 10, 1227–1230. doi: 10.1039/c9md90010a
Roca, I., Akova, M., Baquero, F., Carlet, J., Cavaleri, M., Coenen, S., et al. (2015). The global threat of antimicrobial resistance: science for intervention. New Microbes New Infect. 6, 22–29.
Roos, K., Wu, C., Damm, W., Reboul, M., Stevenson, J. M., Lu, C., et al. (2019). OPLS3e: extending force field coverage for drug-like small molecules. J. Chem. Theory Comput. 15, 1863–1874. doi: 10.1021/acs.jctc.8b01026
Sastry, G. M., Adzhigirey, M., Day, T., Annabhimoju, R., and Sherman, W. (2013). Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Design 27, 221–234. doi: 10.1007/s10822-013-9644-8
Simpkin, V. L., Renwick, M. J., Kelly, R., and Mossialos, E. (2017). Incentivising innovation in antibiotic drug discovery and development: progress, challenges and next steps. J. Antibiot. 70, 1087–1096. doi: 10.1038/ja.2017.124
Suller, M., and Lloyd, D. (2002). The antibacterial activity of vancomycin towards Staphylococcus aureus under aerobic and anaerobic conditions. J. Appl. Microbiol. 92, 866–872. doi: 10.1046/j.1365-2672.2002.01594.x
Tourova, T., Korshunova, A., Mikhailova, E., Sokolova, D. S., Poltaraus, A., and Nazina, T. (2010). Application of gyrB and parE sequence similarity analyses for differentiation of species within the genus Geobacillus. Microbiology 79, 356–369. doi: 10.1134/s0026261710030124
Tsuji, B. T., Yang, J. C., Forrest, A., Kelchlin, P. A., and Smith, P. F. (2008). In vitro pharmacodynamics of novel rifamycin ABI-0043 against Staphylococcus aureus. J. Antimicrob. Chemother. 62, 156–160. doi: 10.1093/jac/dkn133
Vogelman, B. S., and Craig, W. A. (1985). Postantibiotic effects. J. Antimicrob. Chemother. 15 (suppl. A), 37–46. doi: 10.1093/jac/15.suppl_a.37
Wigley, D. B., Davies, G. J., Dodson, E. J., Maxwell, A., and Dodson, G. (1991). Crystal structure of an N-terminal fragment of the DNA gyrase B protein. Nature 351, 624–629. doi: 10.1038/351624a0
Yele, V., Azam, M. A., and Wadhwani, A. D. (2021). Synthesis, molecular docking and biological evaluation of 2-Aryloxy-N-Phenylacetamide and N’-(2-Aryloxyoxyacetyl) benzohydrazide derivatives as potential antibacterial agents. Chem. Biodiv. 18:e2000907.
Keywords: benzohydrazide derivatives, phenylacetamide derivatives, ParE, antibacterial activity, antibiofilm activity
Citation: Yele V, Sanapalli BKR, Wadhwani AD and Mohammed AA (2021) Benzohydrazide and Phenylacetamide Scaffolds: New Putative ParE Inhibitors. Front. Bioeng. Biotechnol. 9:669728. doi: 10.3389/fbioe.2021.669728
Received: 22 March 2021; Accepted: 24 May 2021;
Published: 17 June 2021.
Edited by:
Thomas Dandekar, Julius Maximilian University of Würzburg, GermanyReviewed by:
Harichandra D. Tagad, National Institutes of Health (NIH), United StatesCopyright © 2021 Yele, Sanapalli, Wadhwani and Mohammed. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Afzal Azam Mohammed, YWZ6YWw5YXphbUBob3RtYWlsLmNvbQ==; YWZ6YWxAanNzdW5pLmVkdS5pbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.