 Rosa Sagita
Rosa Sagita Wim J. Quax
Wim J. Quax Kristina Haslinger
Kristina Haslinger
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Bioeng. Biotechnol., 15 March 2021
Sec. Bioprocess Engineering
Volume 9 - 2021 | https://doi.org/10.3389/fbioe.2021.649906
This article is part of the Research TopicEndophytes and Their Biotechnological ApplicationsView all 9 articles
The bioprospecting of secondary metabolites from endophytic fungi received great attention in the 1990s and 2000s, when the controversy around taxol production from Taxus spp. endophytes was at its height. Since then, hundreds of reports have described the isolation and characterization of putative secondary metabolites from endophytic fungi. However, only very few studies also report the genetic basis for these phenotypic observations. With low sequencing cost and fast sample turnaround, genetics- and genomics-based approaches have risen to become comprehensive approaches to study natural products from a wide-range of organisms, especially to elucidate underlying biosynthetic pathways. However, in the field of fungal endophyte biology, elucidation of biosynthetic pathways is still a major challenge. As a relatively poorly investigated group of microorganisms, even in the light of recent efforts to sequence more fungal genomes, such as the 1000 Fungal Genomes Project at the Joint Genome Institute (JGI), the basis for bioprospecting of enzymes and pathways from endophytic fungi is still rather slim. In this review we want to discuss the current approaches and tools used to associate phenotype and genotype to elucidate biosynthetic pathways of secondary metabolites in endophytic fungi through the lens of bioprospecting. This review will point out the reported successes and shortcomings, and discuss future directions in sampling, and genetics and genomics of endophytic fungi. Identifying responsible biosynthetic genes for the numerous secondary metabolites isolated from endophytic fungi opens the opportunity to explore the genetic potential of producer strains to discover novel secondary metabolites and enhance secondary metabolite production by metabolic engineering resulting in novel and more affordable medicines and food additives.
Endophyte is an all-encompassing term that refers to organisms which, within a certain period of their life, colonize the interior organs of their plant hosts (Box 1). Among them, endophytic fungi represent one of the largest communities with conservatively estimated at least 1 million species (Rashmi et al., 2019). More than 100 years of research indicate that most, if not all of the billions of living land plants are host for different endophytic fungi in natural ecosystems. This makes this group of microorganisms one of the largest untapped natural resources for the bioprospecting of secondary metabolites and biosynthetic enzymes (Manganyi and Ateba, 2020).
Box 1. Definition of “endophyte.”
The molecular mechanisms underlying endophytism are to this date uncertain and several fungal strains have been observed to be pathogenic in one host plant and neutral or mutualistic in another (Kogel et al., 2006). It has been speculated that endophytic lineages have evolved from plant pathogenic ancestors (Delaye et al., 2013), while other scientists argue for the opposite (Xu X. H. et al., 2014). The “endophytic continuum” model (Schulz and Boyle, 2005) suggests that the outcome of the plant-fungus interaction, which can range from mutualism to parasitism, depends on not only the fungal species, but also the host genetic background and the environment (Kogel et al., 2006). As pointed out by Delaye et al. (2013), fungi can easily switch at the evolutionary or ecological timescale between the symptomless endophytic life style in host tissue and the life as a necrotrophic pathogen that kills its host. Therefore, it is impossible to deduce the lifestyle of a fungus solely from a species database. It appears that currently the most commonly used indicator for endophytism is the healthy appearance of the host plant that the putative endophyte was isolated from. Clearly, stronger experimental evidence would be desirable, such as observations of the fungal behavior on stressed or wounded host plants, or during several developmental stages of the host plant from seed to senescence.
For the purpose of this review we use the term “endophyte” with the broad definition of a fungus isolated from surface sterilized plant material that does not show visual signs of disease. Of course, we rely on the accurate reporting of the scientists who performed the experiments and their appropriate use of experimental controls to avoid contamination from other sources. Hopefully in the future it will be easier to classify fungal isolates as endophytes by yet to be identified shared genomic or metabolomic traits.
Since 1981, almost 40% of FDA approved small molecule drugs were discovered or derived from natural sources (Newman and Cragg, 2020a), including medicinal plants and their endophytes. The discovery of endophytes dates far back to 1898 (Vogl, 1898), but it did not receive much attention until the past two decades, when it became evident that endophytes, especially endophytic fungi, harbor an enormous potential for secondary metabolite production with relevant bioactivities and diverse molecular structures, which are hardly mimicked by synthetic chemistry. Most notably, Stierle and Strobel (1993) pioneered the exploration of an endophytic fungus associated with Taxus spp. for its proposed independent synthesis of taxol (1). Numerous studies have since then reported on the isolation of new and previously known secondary metabolites from endophytic fungi as recently reviewed by Manganyi and Ateba (2020) and Newman and Cragg (2020b).
Endophytic fungi play a profound role for the survival and fitness of plants (Dubey et al., 2020; Gupta et al., 2020). Considering the intricate balance between overcoming host barriers and establishing a mutualistic relationship with the host, endophytes are assumed to adapt to their symbiotic microenvironments by genetic variation, including the uptake of foreign DNA. Such DNA uptake is overall rare in fungi, but has been shown to occur with microbial and plant donors (Richards et al., 2009, 2011; Armijos Jaramillo et al., 2013). This mechanism is often suggested to explain the detection of secondary metabolites that were originally identified as phytochemicals associated with the host plants (Strobel, 2003), however, experimental evidence has not been presented yet. The concept was furthermore refuted by Heinig et al. (2013) as they did not find genetic evidence for production of 1 in several endophytic fungi associated with Taxus spp. At the same time the authors identified several caveats in the experimental approach of studies detecting 1 from endophytic fungi and called for appropriate experimental controls in order to rule out the false-positive detection of metabolites and the carry-over of enzymes or nucleic acids from the host plant during isolation and cultivation of the fungus. The controversy regarding the biosynthesis of putative secondary metabolites by endophytic fungi has progressively increased since and has strikingly impacted the field.
While the controversy was sparked around the high-value phytochemical 1, the same arguments of proper experimental controls and genetic evidence can be made for other secondary metabolites isolated from endophytic fungi. Most bioprospecting studies in the field focus on the isolation and characterization of bioactive compounds from one-time sampling instead of a time-course observation of the fungal culture. Often, the identification of the underlying biosynthetic pathway is not well-presented with an experimental verification to provide appropriate evidence of the proposed secondary metabolite production by endophytic fungi. Several studies reported instability of secondary metabolite production by the native host in axenic culture supposedly due to pathway silencing and inactivation, or enzyme attenuation (El-Hawary et al., 2016; Gupta et al., 2018; El-Sayed et al., 2019). In contrast to the widely claimed promise of endophytic fungi as a highly prolific secondary metabolite producer, secondary metabolite bioprospecting appears far-fetched at this point considering that their underlying biosynthetic pathways are elusive and that so many fungal strains are even unculturable under laboratory conditions (Wu B. et al., 2019).
In the current post-genomic era, these problems should be able to be resolved. With the advanced development of sequencing technologies, sequencing cost and turnaround time are dramatically reduced (Goodwin et al., 2016), making genomics and metagenomics widely accessible. Genetics- and genomics-based strategies have risen as comprehensive approaches to study natural products from a wide-range of organisms (Hover et al., 2018; Schorn et al., 2019; Walker et al., 2020). They allow the elucidation of underlying pathways for secondary metabolites isolated from organisms and facilitate the computational discovery of secondary metabolite biosynthetic pathways. From there, further exploration of the biosynthetic potential of a producer strain is made possible to activate silent pathways and to conduct rational de novo design of novel molecules.
However, given the scarcity of genomic information from endophytic fungi and difficulties in experimental verification of putative Biosynthetic Gene Clusters (BGCs), the majority of secondary metabolite biosynthetic pathways are still undiscovered. In this review, we will provide a brief overview of the steps needed for a successful bioprospecting study that provides both phenotypic and genotypic evidence for secondary metabolite production by endophytic fungi. As depicted in Figure 1, there are two possible starting points, but the overall steps to be taken are the same for both approaches. A study with phenotypic observation starts from the isolation and characterization of secondary metabolites (phenotyping), followed by a generation of hypothesis on the biosynthetic pathway, and genotyping of the responsible biosynthetic genes and/or cluster in the proposed pathway. On the other hand, a study might start from a genotypic observation via genome mining. This is also followed by the hypothesis generation of the biosynthetic pathway, all the way to the detection of secondary metabolite (phenotyping) in an experimental verification. Both approaches offer identification of the responsible biosynthetic genes and associating them with the expressed phenotype through experimental verification that will provide the fundamental evidence of independent secondary metabolite production by an organism.
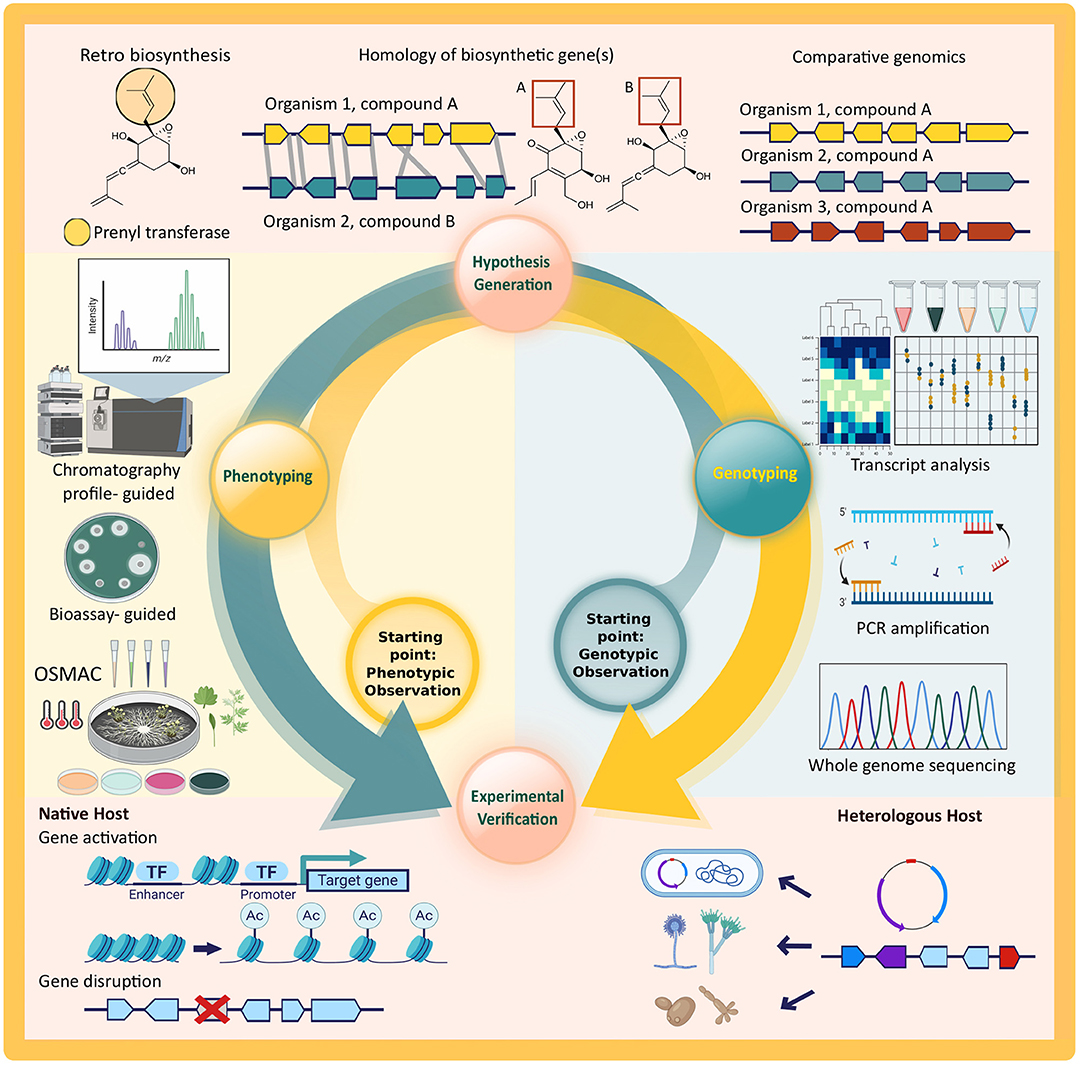
Figure 1. Schematic overview of secondary metabolite discovery and pathway elucidation steps discussed in this review. All steps of the cycle are required to fully connect phenotypic observations to the genotype of an organism and vice versa. Key technologies depicted for each step are: whole genome sequencing (WGS), amplicon sequencing after PCR amplification, and transcriptomics and reverse transcriptase quantitative PCR (RT-qPCR) for genotyping; retro-biosynthesis, homology searches, and comparative genomics to generate hypotheses on the link between genotype and phenotype and vice versa; chromatography- and bioassay-guided metabolite discovery and analysis, possibly coupled to differential culturing of the native host (OSMAC) for phenotyping; gene activation and deletion in the native host as well as recombinant expression of target genes in heterologous hosts for experimental verification. Created with graphical elements from BioRender.com.
In this review we will first provide an overview of the phenotyping efforts of endophytic fungi including the best practices and a few interesting bioactive metabolites identified with each strategy. For a larger survey of bioactive secondary metabolites reported for endophytic fungi, we would like to refer to other recent reviews by Manganyi and Ateba (2020) and Newman and Cragg (2020a). Second, genotyping strategies applied in endophytic fungi will be presented, followed by a brief overview of general strategies for hypothesis generation and experimental verification employed to establish a link between phenotype and genotype. For detailed reviews on general methods we would like to refer to the recent reviews by Hautbergue et al. (2018) and Kjærbølling et al. (2019). Third, we will highlight studies that drive the success in the bioprospecting of endophytic fungi by establishing the link between phenotype and genotype in endophytic fungi to date. Lastly, we will discuss the shortcomings and benefits of the different starting points and discuss the future opportunities in the field. Bioprospecting studies presented in this review focus on isolated fungal endophytes, and for a recent review on bioprospecting from multi-omics datasets from a wide range of organisms we would like to refer to van der Hooft et al. (2020).
A large number of potentially high-value bioactive compounds with pharmaceutical importance were discovered from cultivable endophytic fungi as reviewed by Manganyi and Ateba (2020). In this review we want to focus on the main strategies employed and the best practices to prevent experimental error in phenotyping endophytic fungi. The presented examples are only a miniscule fraction of the published phenotyping studies and were selected to illustrate specific strategies. We do not intend to cast judgement on the overall quality and merit of the omitted studies. The mentioned secondary metabolites are depicted in Figure 2.
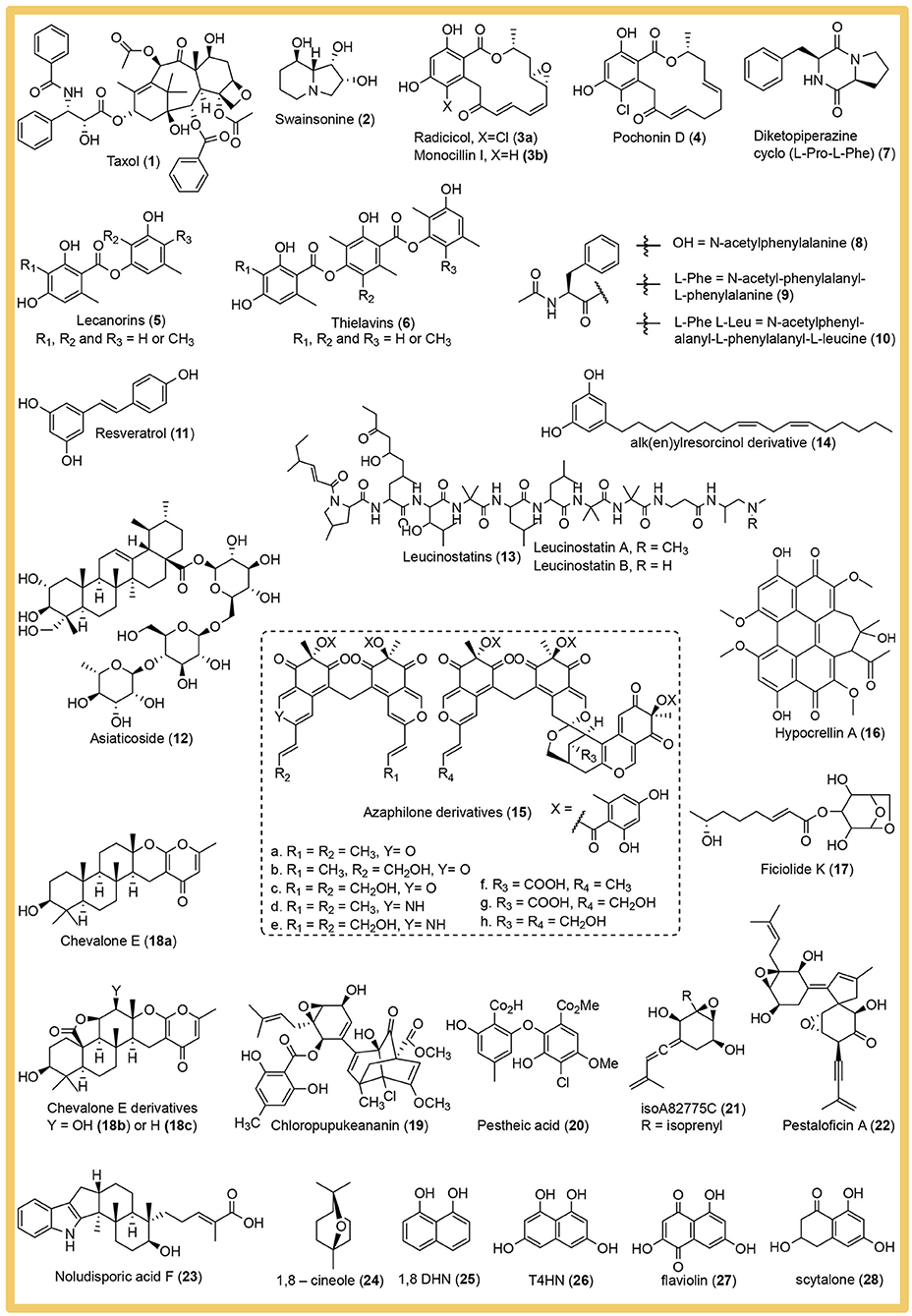
Figure 2. Structures of the bioactive secondary metabolites isolated from endophytic fungi and named in this review.
Metabolic phenotyping is often targeted toward certain chemical entities, or bioactivities (Hautbergue et al., 2018). Therefore, it is mainly based on (1) the detection of target compounds in culture extracts by chromatographic methods, either gas or liquid chromatography (GC or LC, respectively) coupled to mass spectrometry (MS) and nuclear magnetic resonance (NMR) spectroscopy (chromatography profile-guided experiment), or (2) the observation of target biological effects by a bioassay-guided experiment. An example for the first approach is a study targeting the detection of swainsonine (2), a cytotoxic fungal alkaloid, leading to the discovery of an independent producer of 2, an endophytic fungus from Astragalus mollissimus (Braun et al., 2003). Meanwhile as an example for the bioassay-guided phenotyping approach, Turbyville et al. (2006) successfully discovered radicicol (3a) and monocillin I (3b) as Hsp90 inhibitors with potential anticancer activity in fractionated extracts of the endophytic fungus Chaetomium chiversii and Paraphaeosphaeria quadriseptata.
Instead of targeting compounds with known chemical structures or bioactivity, other studies focus on seeking “hidden gems” in metabolomic profiles of organisms to search for novel compounds, which is often employed in the study of endophytic fungi. Considering that the nature of many pathways in fungi is silent or attenuated under unfavorable experimental conditions (Keller, 2019), the qualitative and quantitative profiles of metabolites in organisms exposed to different growth conditions provide a holistic overview of their biochemical status and allows for an exploitation of their natural biogenetic capability. This approach is known under the acronym OSMAC, One Strain-Many Compounds (Bode et al., 2002), and has led to several interesting secondary metabolite discoveries in a wide range of organisms, including endophytic fungi (Pan et al., 2019). The discovery is mainly guided by (1) uncharacterized features in the analytical profile of fungal extracts, (2) bioassays with fungal extracts, or (3) a combination of both. Using the first strategy, novel lecanorin (5) and thielavin (6) polyketides were discovered from the extract of a guava endophytic fungus Setophoma sp. (De Medeiros et al., 2015). The authors studied the chromatographic profile (absorbance and MS) and discovered unknown features by comparing several different growth media. The compounds were isolated and characterized based on their absorbance, NMR, and MS spectroscopic properties for structure elucidation. The advantage of this approach is that the risk of rediscovery is decreased, and new scaffolds and molecules can be identified efficiently. However, since this approach is guided by only physicochemical traits of the compound, there is no guarantee for identifying relevant bioactivity. Therefore, other studies rely more to the bioassay-guided approach with consecutive fractionations of the fungal extract to the smallest fraction containing a single active substance with the target bioactivity. Based on this strategy, a diketopiperazine cyclo (L-Pro-L-Phe) (7), with antibacterial activity against Salmonella enterica was discovered for the first time in the endophytic fungus Paraphaeosphaeria sporulosa (Carrieri et al., 2020). To avoid rediscovery of known compounds, a combination of chromatography profile- and bioassay-guided isolation is preferred. With the combination strategy, Tawfike et al. (2018) investigated the metabolic profile of the endophyte Culvularia sp. isolated from the leaves of Terminalia laxiflora. First, they performed comparative metabolite profiling using high resolution LC-MS to assess the chemical diversity from different culture conditions and proceeded with interesting metabolites to the bioactivity assay on NF-κB and the myelogenous leukemia cell line K562. From there, mass spectral data of extracts with the desired bioactivity were analyzed by multivariate analysis using principal component analysis to identify the presence of exclusive metabolites. Those metabolites were dereplicated using the Dictionary of Natural Product database to prevent rediscovery and further isolated to elucidate their structures. This led the authors to discover N-acetylphenylalanine (8), N-acetyl-phenylalanyl-L-phenylalanine (9), and N-acetylphenylalanyl-L-phenylalanyl-L-leucine (10), which are predicted to be responsible for the observed bioactivities (Tawfike et al., 2018).
Overall, comparative metabolic profiling provides a more comprehensive and efficient approach to discover bioactive novel secondary metabolites in a high-throughput manner compared to targeting a single secondary metabolite.
Many endophytic fungi were seen to be difficult to grow in axenic condition and/or to lose the production capability for secondary metabolites over repeat passages. Several factors might explain this phenomenon, e.g., the attenuation of biosynthetic gene expression, and/or the lack of pathway precursors or critical transcription factors as stimuli for secondary metabolite production during subculture (Li et al., 1998; Kusari et al., 2009a,b; Shweta et al., 2010; Vasanthakumari et al., 2015; El-Sayed et al., 2019). Due to this problem, numerous studies keep the passaging during strain isolation and cultivation to a minimum. As pointed out by Heinig et al., this may lead to carry-over of other microorganisms, plant metabolites, or enzymes, which leads to a false positive observation of secondary metabolites. It is therefore crucial to include the right experimental controls (Table 1). This could be as simple as repeat metabolite extractions to observe the build-up of metabolites over time, or the use of negative control cultures with the addition of fungicides. According to Heinig et al., the major culprit for carry-over of 1 from the host plant is its accumulation in the endophytic cell wall due to its physicochemical characteristics. To rule out the possibility of carry-over, a study by Shi et al. (2012) showed a time-course experiment on resveratrol (11) production by Alternaria sp. MG1, which showed accumulation of 11 starting at day 1 and coinciding with an increase in cell dry weight. They also showed the modulation of 11 production during optimization of the growth condition that supports the finding of independent production of 11 by the endophytic fungus. Similar observations were made by Gupta et al., who followed asiaticoside (12) production in an endophytic fungus associated with Centella asiatica over time across multiple passages. Although the secondary metabolite production was lost after eight passages, the build-up of 12 within each generation provided strong evidence for the independent production of 12 by this fungus (Gupta et al., 2018).
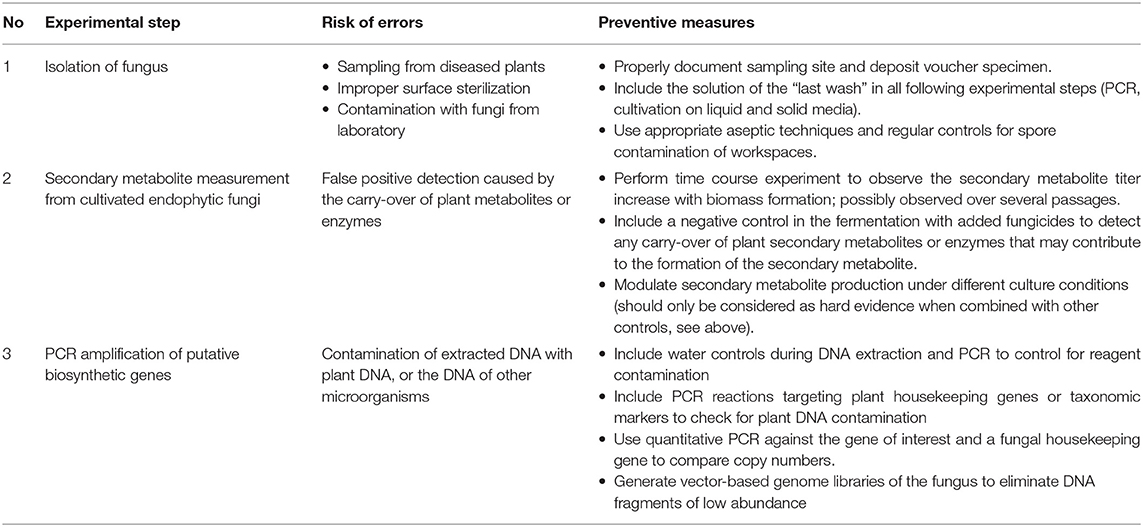
Table 1. Possible sources of error when working with endophytic fungi and commonly used or suggested preventive measures.
Despite the considerable risks of error, phenotypic observation has led to the successful discovery of numerous natural products with biological activities for almost a century since the discovery of penicillin (Aldridge et al., 1999). Alongside with its long-standing history, the analytical tools in chromatography and spectrometry (e.g., high resolution MS), data analysis (e.g., principal component analysis), and experimental strategies (e.g., OSMAC, dereplication) for metabolic phenotyping have emerged to facilitate the laborious isolation work and overcome the redundant analysis of metabolomic studies from natural sources (Covington et al., 2017). However, given the many experimental challenges associated with studying metabolites of endophytic fungi, there are a large number of studies that unfortunately lack the experimental controls and analytical depth to form reliable grounds for rational and efficient bioprospecting in endophytic fungi. But even in the carefully conducted studies reviewed in this section, often the elucidation of the underlying pathways has not been possible. In the next section we will review the opposite strategy for secondary metabolite discovery beginning with a genotypic, rather than a phenotypic observation.
A genotypic observation can also be the starting point for a secondary metabolite discovery project. This observation can either be the presence of signature genes identified by (1) polymerase chain reaction (PCR) amplification, (2) the co-regulation of a set of genes observed on the transcript level, or (3) the computational identification of interesting BGCs in whole genome sequencing (WGS) data. In the first strategy, degenerate PCR primers are designed based on the sequences of known biosynthetic genes in order to screen isolated genomic or environmental DNA for the presence of a certain gene, and obtain its sequence. Depending on the design of the primer binding site, the screening can be highly targeted toward a gene encoding a specialized enzyme, e.g., taxadiene synthase (Staniek et al., 2009; Heinig et al., 2013), or allow for a rather untargeted detection of genes encoding an enzyme family, e.g., polyketide synthases (PKS) (Wang et al., 2008). In the case of endophytic fungi, Heinig et al. clearly demonstrated the risk for contamination with host plant DNA and false-positive detection of DNA fragments. Appropriate controls are needed to rule out these problems. One of them is to use PCR primers targeting plant housekeeping genes or taxonomic markers to check for plant DNA contamination. Another option is to perform quantitative PCR of the gene of interest and compare it to an internal control, such as single-copy housekeeping genes of the endophytic fungus. A different approach for reducing DNA contamination in the PCR amplification strategy is to generate vector-based genome libraries of the fungus, as done for C. chiversii using the E. coli F-plasmid for a fosmid library (Wang et al., 2008). Lambda phage libraries were constructed from genomic DNA from three Taxus spp. endophytic fungi (Heinig et al., 2013). Since the construction of these libraries is a stochastic process, DNA fragments of low abundance, such as contaminations from plant parts or environmental DNA, are more likely to be lost.
The second genotyping strategy uses transcript analysis by reverse transcription quantitative PCR (RT-qPCR) or RNA sequencing technology (RNAseq) in order to identify genes that are transcriptionally co-regulated. As an example, RNAseq of Alternaria sp. MG1 was done to identify putative genes involved in the proposed biosynthetic pathway of 11 (Che et al., 2016). An RNAseq library was constructed, sequenced by Illumina technology, assembled to unigenes, annotated and analyzed for gene and pathway expression. Several candidates for pathway genes were identified, although no experimental verification was presented. This strategy is particularly useful, when combined with experimental strategies to modulate gene expression, such as deletion or overexpression of a regulatory gene, or an OSMAC strategy. As an example, different culture conditions and qRT-PCR analysis were used to determine the BGC boundaries in the elucidation of the leucinostatin (13) biosynthetic pathway in Purpureocillium lilacinum (Wang et al., 2016).
The last genotyping strategy is WGS, which allows for the computational search of biosynthetic genes and gene clusters (genome mining). There are two principal strategies of in silico genome mining (Weber and Kim, 2016). The rule-based strategy is used to identify gene clusters based on the presence of scaffolding enzymes, such as PKSs, non-ribosomal peptide synthetases (NRPS) and terpene synthases (TS), or signatures of ribosomally synthesized and translationally modified peptides (RiPPs). This search for genes with high sequence similarity to reference genes can be done by an automated BGC finder, e.g., antiSMASH (Medema et al., 2011), or manually with the basic local alignment search tool (BLAST) (Altschul et al., 1990). A combination of BLAST, multiple sequence alignments and homology modeling is often used to manually inspect and curate the results of antiSMASH. For example in the study of 5-alk(en)ylresorcinol biosynthesis, the authors searched the genome of the endophytic fungus Shiraia sp Slf14 for putative type III PKS genes (Yan et al., 2018). The authors identified one gene encoding for SsARS and confirmed its PKS activity by heterologous expression in E. coli and yeast, followed by metabolite analysis. The second principal strategy uses a rule-independent, machine learning-based approach for automated phylogenomic analyses and/or prediction of transcriptional co-regulation of genes. It is important to note that these models are only as good as the accuracy and depth of the genomic and biochemical data used for training (Weber and Kim, 2016). When this review was written, the use of the latter approach has not been reported in endophytic fungal research. Please refer to an extensive review by Chavali and Rhee (2018) on tools and platforms for computational mining and to van der Lee and Medema (2016) for detailed computational techniques in genome-based natural product discovery in fungi.
Genome mining can be carried out under various goals, e.g., pathway elucidation of a specific secondary metabolite, homolog search for pathway engineering purposes, discovery of novel bioactive secondary metabolites, and many more (Ziemert et al., 2016). Genome mining allows the exploration of the entire genome and thus enables us to discover novel secondary metabolites. Filamentous fungi are prolific producers of bioactive secondary metabolite, yet most of them are merely expressed under experimental conditions (Keller, 2019), which prohibits their discovery through metabolic phenotyping. This is illustrated by a study in Pestalotopsis fici reporting only 10 out 74 gene clusters to be active under axenic conditions (Wang et al., 2015). In another study, 43 BGCs were found computationally in Penicillium dangeardii, but culture extracts were dominated by rubratoxins. A metabolic shunting strategy by deleting the key gene for rubratoxins biosynthesis was applied and led to the activation of cryptic BGC encoding several novel monomeric, dimeric and trimeric azaphilones (some are depicted in Figure 2, 15) (Wei et al., 2020). Considering the vast amount of silent BGCs in endophytic fungi, genome mining can be seen as a high-throughput strategy for “treasure hunting” of novel secondary metabolites. Furthermore, it will significantly broaden the possibility of synthetic biology efforts, allowing for refactoring and de novo engineering of BGCs, and facilitating the systematization of BGCs to engineer novel biosynthetic pathways. However, in order to achieve these high goals, it is of utmost importance to connect the metabolomic/phenotypic with the computational/genotypic observations, regardless of the starting point of the study. In the next section we will review the steps to complete the circle.
Regardless whether a study started from phenotypic or genotypic observation, a crucial part in conducting successful bioprospecting studies is to connect the two with experimental evidence. Initially, a hypothesis needs to be generated on the link between the observed secondary metabolite (phenotype) and the responsible biosynthetic genes or clusters (genotype). Next, an experimental investigation is required to verify the proposed hypothesis.
The three main strategies for hypothesis generation are (1) retro biosynthesis, (2) homology of biosynthetic gene(s), and (3) comparative genomics, as recently reviewed (Kjærbølling et al., 2019). In brief, the retro biosynthesis approach is based on the prediction of enzymes involved in the biosynthesis of a certain (class of) secondary metabolite, as seen in the study of Hypocrellin A biosynthesis (16), where four enzymatic steps were predicted based on the known biosynthetic route of a structurally similar compound (Zhao et al., 2016). Retro-biosynthesis utilizes general knowledge on biosynthetic enzymes, e.g., substrate scope, reaction mechanisms, conserved domain architectures, etc. and is often combined with the other two techniques to predict a full BGC. Since no prior knowledge from related species is required, this is a commonly used strategy to propose the biosynthetic pathway of a compound with known structure, as seen in Table 2. The second strategy, homology search of gene(s), is based on the fact that enzymes of similar function and substrate scope usually share a certain degree of sequence similarity and structural similarity. Therefore, target enzymes can be identified by searching for homologs of genes of known function from other organisms, as seen in the study of the PKS encoding-gene, pfmaE, in the pfma cluster of P. fici (Zhang et al., 2017). Structural 3D models can be generated based on structural information from homologs, and substrate specificities can be investigated by in silico docking, as done on hyp3 from Hypoxylon sp. (Shaw et al., 2015). The third strategy focuses on a set of genomes, which are subjected to comparative analysis to find shared BGCs across species, or presence and absence of BGCs in secondary metabolite producer and non-producer strains, respectively. This strategy led Cook et al. (2017) to successfully link secondary metabolite 2 to its SWN BGC across the producers. Which strategy to use critically depends on the starting point and the initial knowledge of a study. A combination of several strategies is most often needed to propose a reasonable hypothesis with a strong logic.
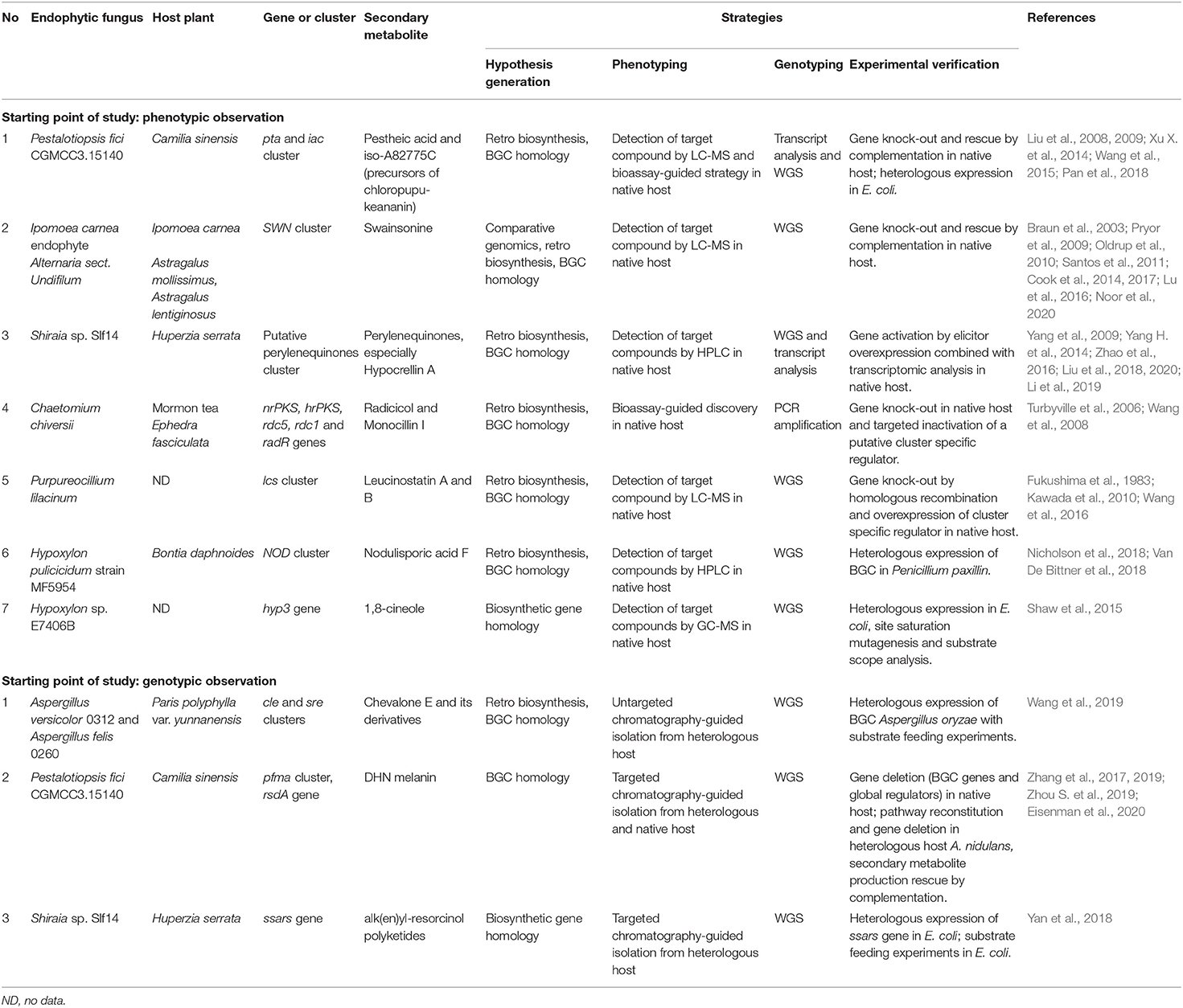
Table 2. Successful studies in linking phenotype and genotype of the related bioactive secondary metabolites from plant endophytic fungi supported by experimental verifications.
Since the number of phenotypic studies on endophytic fungi, which at least proposed underlying biosynthetic pathways for the observed secondary metabolites, is so vast and were recently reviewed by Dubey et al. (2020), we will not give more specific examples here. In section Successful Elucidation of Biosynthetic Pathways by Linking Phenotype and Genotype of Endophytic Fungi of this review, we provide a deeper analysis of the studies that encompass the entire pathway elucidation process and successfully connect the secondary metabolite discovery and BGC identification.
Depending on the starting point of the study, the experimental verification will require the same sets of experiments as described in sections Secondary Metabolite Discovery by Metabolic Phenotyping of Endophytic Fungi and Secondary Metabolite Discovery in Endophytic Fungi Starting From Genotypic Observations. For a study starting from the phenotypic observation, genotyping needs to be performed with targeted or untargeted sequencing strategies, while for studies starting with an in silico analysis and prediction of BGCs, targeted or untargeted metabolomics need to be performed to detect the pathway products and intermediates in the native host or a recombinant host (Figure 1). Individual gene functions need to be verified by targeted overexpression or gene disruption followed by metabolite analysis. Detailed techniques and methods for this step in the workflow were extensively reviewed recently (Hautbergue et al., 2018; Kjærbølling et al., 2019).
The native host strategy is used when a target microorganism is cultivable and amenable to genetic modification. The major strategies are gene deletions and gene/gene cluster activation. Gene disruption, or knock-out by homologous recombination coupled to metabolite analysis is by far the most common method for dissecting the function of genes in BGCs, since the gene disruption should lead to the absence of the target secondary metabolite and/or accumulation of pathway intermediates (Kjærbølling et al., 2019). This strategy has been successfully applied in several studies on endophytic fungi starting from phenotypic observation (see section Studies Starting From Phenotypic Observation), e.g., the elucidation of the pathway of 2 in Metarrhizium robertsii (Cook et al., 2017). In studies starting from genotypic observation, this approach is more challenging, since BGCs are often silent, however, some positive examples are summarized in section Studies Starting From Genotypic Observation. A general challenge in this approach is that the common strategy to delete gene(s) by homologous recombination is inefficient or even impossible often due to repeat regions (Zhang et al., 2019). This has been overcome with the recent innovation of CRISPR/cas9 (Clustered Regularly Interspaced Short Palindromic Repeats/cas9) technology in filamentous fungi, which improves recombination efficiency and allows for targeted and multiplexed gene editing without or with fewer selectable markers as reviewed by Song et al. (2019). For example, even in the presence of “TG” repeats in the promoter region that hampered conventional gene deletion by homologous recombination, CRISPR/cas9 gene editing was performed to disrupt the pfmaF gene in the endophytic fungus P. fici and thereby elucidate its role as a global regulator (Zhang et al., 2019).
Gene activation is another strategy in the native host, since many BGCs of endophytic fungi become silent under artificial experimental conditions. Physicochemical triggers and the use of interspecies crosstalk are reported to successfully activate silent genes in many microbes (Pan et al., 2019). However, the specific requirements to induce expression from such gene clusters are not well-understood, since it is not possible to predict the complex regulatory circuits involved in an endophytic fungal biosynthetic pathway. Thus, other global activation strategies, such as epigenetic re-modeling, are promising avenues toward exploring silent gene clusters. Small molecule inhibitors of DNA methylating and histone acetylating enzymes (Toghueo et al., 2020), and targeted knock-out of the corresponding genes, e.g., the hdaA gene (Yang X. L. et al., 2014; Mao et al., 2015; Bai et al., 2018; Ding et al., 2020) can be employed to stimulate secondary metabolite production. This is reported in the study of the endophytic fungus P. fici, where Ficiolide K (17) was found along with 14 new polyketides upon knock-out of hdaA (Wu et al., 2016). Another global approach was performed by Zheng et al. (2017) to discover five novel Pestaloficins, including Pestaloficin A (22), by deleting the PfcsnE gene. Other chromatin remodeling strategies remain to be explored in fungi and promise an even wider application of this gene cluster activation strategy (Collemare and Seidl, 2019). Overall, the global gene activation strategy is mainly used to discover novel compounds, but it could also be used to pinpoint specific BGCs by studying the effect of the manipulation on transcript level in endophytic fungi. As reported for other cultivable organisms (Kang et al., 2019), we see great potential for connecting phenotype and genotype with the combination of metabolomics and transcriptomics following global gene activation in endophytic fungi.
In contrast to the global gene activation approach, targeted overexpression or knock-out of specific, positive or negative transcriptional regulators, respectively, provides a precise tool to study a certain set of co-regulated genes. This method is widely applicable, as up to 50% of fungal BGCs contain putative cluster-specific regulators (Keller, 2019), and it requires less genetic manipulation compared to promoter replacement strategies targeting individual genes (Scherlach and Hertweck, 2009). As an example, knock-out of the RadR gene encoding the cluster-specific positive regulator was performed to characterize functional genes in the BGC of 3 in C. chiversii (Wang et al., 2008). In another study, Wei et al. (2020) presented experimental evidence for the putative azaphilone BGC by showing that deletion and overexpression of the cluster-specific transcription factor danS in the endophytic fungus Penicillium dangeardii lead to changes in the production levels of 15.
Overall, the native host strategy has been applied successfully many times to investigate the gene function in endophytic fungi. However, native host strategies cannot be used for unculturable organisms or isolates that are not amenable to classic genetic modification. In section Future Directions for Successful Secondary Metabolite Bioprospecting From Endophytic Fungi, we will review some technology advances that might help to overcome this hurdle.
Synthetic biology offers vast opportunity to investigate the function of BGCs, even cryptic ones or those identified in metagenomic assemblies, in a heterologous host. The possibility to generate long synthetic DNA fragments (Eisenstein, 2020) and advanced DNA assembly strategies (Bartley et al., 2020) allow for synthesis of entire clusters including de novo design of BGCs with host promoters and regulatory elements for better heterologous expression. Selection of a host and the design of DNA constructs suitable for the host (presence/absence of introns, codon usage, choice of vectors, and burden of foreign DNA) are the most important success limiting factors in heterologous expression, but they can be overcome with new design tools. Recent advances in heterologous expression systems for fungal BGCs were recently reviewed by Qiao et al. (2019) and Lin et al. (2020).
The most commonly used bacterial hosts for BGC reconstitution are Escherichia coli, Streptomyces, or Bacillus subtilis, and the most popular eukaryotic hosts are Saccharomyces cerevisiae and filamentous fungi, such as Penicillium and Aspergillus. Although there are some impressive examples of complex plant (Nakagawa et al., 2016; Chen et al., 2018; Pramastya et al., 2021) and fungal (Matthes et al., 2012; Zobel et al., 2015) pathways expressed in bacteria, we have only found studies where individual, cytosolic enzymes from endophytic fungi were expressed in bacterial hosts (Shaw et al., 2015; Pan et al., 2018; Yan et al., 2018). For reconstitution of multiple genes, fungal hosts appear to be preferred (Zhang et al., 2017; Van De Bittner et al., 2018; Wang et al., 2019). This can most likely be attributed to the challenge of intron prediction, the large size of fungal BGCs, and the lack of post-translational modifications and compartmentalization/membrane trafficking in prokaryotes. Fungi are also more likely to provide the required supporting enzymes and metabolic precursors, allowing exploration of the cloned genes or clusters even with limited information on upstream and downstream pathway modules. The main difficulty is that most fungi themselves dispose of a myriad of BGCs, which may result in crosstalk with the heterologous pathway. This was observed by Xie et al. (2011) where the recombinant biosynthetic genes of the fungal endophyte triggered the inactivation of a negative regulator in the host Fusarium, leading to the production of the mycotoxin fusaric acid instead of the target mycoepoxydiene. In order to eliminate cross-talk between the native and the recombinant pathway, Zhang et al. (2017) chose to integrate their heterologous genes into the locus of the native pathway genes of the host and confirmed the success of their strategy on transcript level.
Besides crosstalk, the often-complex metabolite profile of filamentous fungi as prolific secondary metabolite producers themselves may obscure the products of the heterologous pathway. The recent engineering of platform strains with low secondary metabolite background, e.g., the Penicillium rubens 4xKO (Pohl et al., 2020) and A. nidulans LO8030 (Chiang et al., 2016), will help overcome this hurdle. Wang et al. (2019) used an A. oryzae mutant with low secondary metabolite background, which enabled them to elucidate the biosynthetic pathway of Chevalone E (18a) and its analogs (18b and 18c) by investigating two heterologous gene clusters from the endophytic fungi A. versicolor 0312 and A. felis 0260. This study also highlights the unique capability of fungal hosts to accept large foreign DNA constructs, here divided over multiple plasmids. Other recently developed strategies, e.g., the HEx platform for Saccharomyces cerevisiae (Harvey et al., 2018) and the construction and delivery of fungal artificial chromosomes (Bok et al., 2015; Clevenger et al., 2017) to fungal hosts are major break-throughs in the field and will facilitate the study of BGCs from endophytic fungi in heterologous hosts.
Despite the previously described challenges in phenotyping and genotyping efforts, several studies were reported to successfully link secondary metabolite phenotype and its genotype supported by meticulous experimental verification, either with phenotypic observation or genotypic observation as the starting point of the study. Here we review these studies as cornerstones of future bioprospecting efforts of endophytic fungi (Table 2).
The first example starting from phenotypic observation is the elucidation of the biosynthetic pathway of chloropupukeananes in P. fici (Figures 3A,B). It started from the isolation of a novel secondary metabolite class with significant antitumor and anti-HIV activity, later named chloropupukeananins (19), by a bioassay-guided fractionation of culture extracts of P. fici, an endophytic fungus of tea plant (Liu et al., 2008). Based on retro-biosynthesis, two main precursors, pestheic acid (20) and isoA82775C (21), were proposed (Liu et al., 2008, 2009) and confirmed with a series of extensive experiments (Xu X. et al., 2014; Pan et al., 2018). In the 2014 study, Xu et al. hypothesized that a non-reducing PKS would be the key enzyme of the BGC of 20, based on its structural similarity with other fungal diphenyl ethers. Homology-based genome mining was employed to find the gene encoding this enzyme from the genome of P. fici. The thus identified pta cluster was examined for the presence of genes encoding for putative tailoring enzymes in the pathway proposed by retro-biosynthesis. Transcriptional analysis by RT-qPCR demonstrated a correlation between increased transcript levels from the pta cluster (ptaA, ptaM, ptaE, ptaH) and an increase of 20 production, verifying the involvement of this BGC in the biosynthesis of 20. Experimental verification was conducted by disrupting several genes including ptaA, ptaE, and ptaM in the native host, which resulted in abolishment of 20 and 19 production, whereas no function could be assigned to ptaK by gene disruption. PtaE was found to be the key phenolic coupling enzyme in the pathway. Heterologous expression of the key chlorinating enzyme PtaM in E. coli was performed to elucidate its biochemical mechanism and identify important intermediates (Xu X. et al., 2014). Since the production of the other precursor, 21, was not affected by the disruption the mentioned key enzymes in the pta BGC, it was confirmed that a separate BGC was responsible for 21 production. In 2018, Pan et al. identified the iac BGC in the genome of P. fici based on gene homology and connected it to the biosynthesis of 21 by disrupting eight genes in the cluster, resulting in the loss of 21 and 19 production. This phenotype was successfully rescued with complementation of the key gene iacE, thus providing strong evidence for the involvement of the iac cluster in biosynthesis of 21. Moreover, heterologous expression of the prenyl-transferase enzyme IacE in E. coli was also performed for enzymatic and kinetic study. Unexpectedly, gene deletion of iacE led the authors to the discovery of four new chloropestolides from P. fici (Pan et al., 2018). Genome sequencing, comparative genome analysis, and transcriptional analysis of BGCs from this organism were further reported by Wang et al. (2015), revealing the potential of P. fici as a prolific secondary metabolite producer.
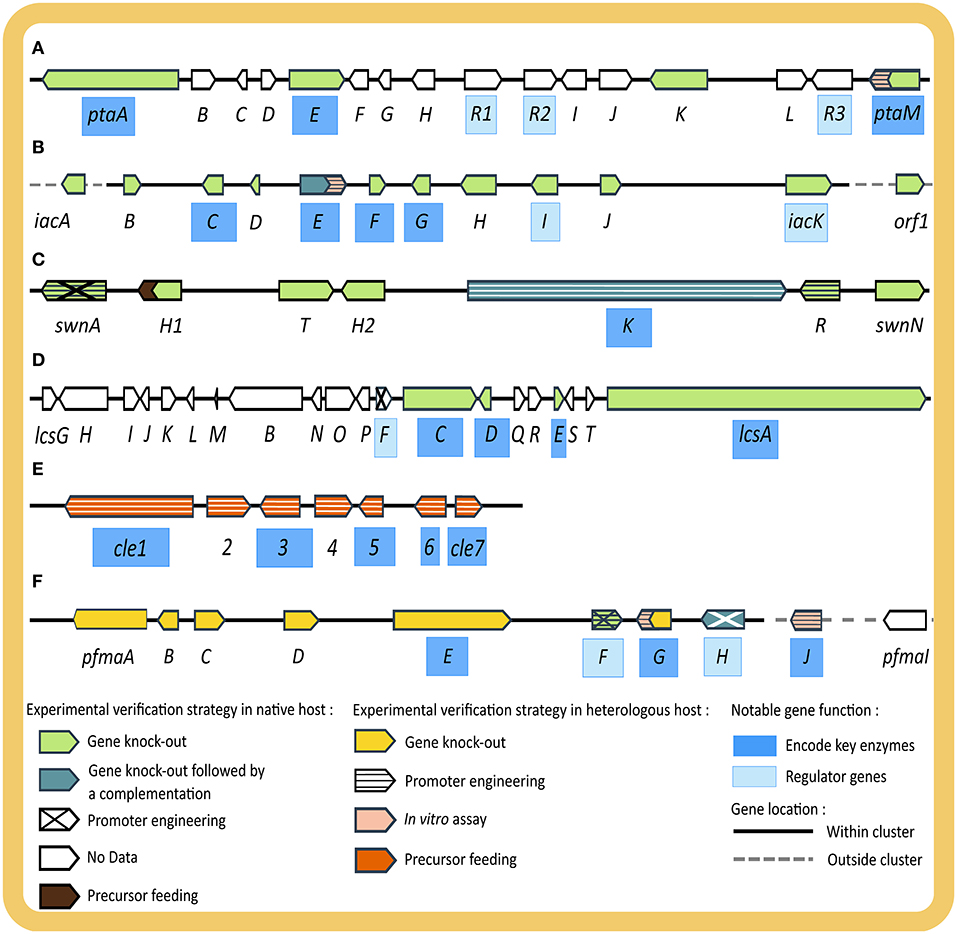
Figure 3. Illustration of four BGCs discovered in studies starting from phenotypic observation (A–D) and two BGCs from genotypic observation (E,F) including the experimental strategies used to verify and characterize their functions. (A) pta giving rise to pestheic acid (20), (B) iac encoding enzymes for isoA82775C (21), (C) SWN shown to be essential for swainsonine (2) production, (D) lcs giving rise to leucinostatin (13), (E) cle involved in the biosynthesis of chevalone E (18a) and its derivatives (18b, 18c), and (F) pfma essential for the production of 1,8 DHN (25) melanin. Genes are indicated as arrowheads, with names of genes encoding key enzymes highlighted in dark blue, (putative) regulatory genes in light blue. Fill and pattern of arrowheads depict experimental evidence (native host: light green—gene knock-out, dark green—gene knock-out followed by rescue via gene complementation, diagonal cross—promoter engineering, no pattern with white fill—no experimental data, brown—precursor feeding experiment; heterologous host: yellow—gene knock-out, horizontal stripe—promoter engineering, pink—in vitro assay, orange—precursor feeding experiment). Genes located within BGC are connected with a solid black line while genes outside the BGC with dotted gray line.
The second example is the elucidation of the swainsonine BGC (SWN) (Figure 3C) across several studies in multiple endophytic fungi (Lu et al., 2016; Cook et al., 2017; Noor et al., 2020). Swainsonine (2) is an indolizidine alkaloid, originally observed in plants due to its toxicity to livestock, with potential applications in cancer therapy (Santos et al., 2011). It was originally isolated from several plant species but later found to be a fungal rather than a plant secondary metabolite (Cook et al., 2013). The first step toward pathway elucidation of swainsonine was taken in 2016, when Lu et al. sequenced the whole genome of Alternaria sect. Undifilum and proposed genes involved in the biosynthesis of 2 based on retro-biosynthesis and gene homology. In 2017, Cook et al. performed comparative genomics on the known producers of 2, Metarhizium robertsii, Slafractonia leguminicola and Alternaria sect. Undifilum, and uncovered the SWN BGC (Cook et al., 2017). The authors searched for orthologous BGCs with a PKS gene that also carries an adenylation domain to load the pipecolic acid starter unit. The thus identified PKS gene was named swnK and its function was verified by gene disruption in M. robertsii, which resulted in abolishment of 2 production. In addition, the authors demonstrated a rescue of 2 production by homologous recombination using a complementary swnK gene. The amino acid sequence of the ketoacyl synthase domain of SwnK was later found to be essentially identical among all 2 producing Alternaria species (Noor et al., 2020). The genes adjacent to swnK in the genome were also analyzed by functional in silico evaluation to identify putative genes involved in 2 biosynthesis based on the retro-biosynthetic strategy (Cook et al., 2017). With this approach the authors predicted the full SWN cluster, including swnN and swnR (an Nmr-A like, and a nicotinamide dinucleotide-binding Rossman-fold reductase gene), swnH1 (2-oxoglutarate-dependent oxidase gene), swnH2 [Fe (II)-dependent dioxidase gene], swnA (aminotransferase gene), and swnT (transmembrane choline transporter gene). Extended comparative genomics were performed on published fungal genomes and identified the SWN in five different orders of filamentous Ascomycota from different ecological niches. Cultivation and secondary metabolite analysis of representatives of these orders, showed the presence of 2, which indicates that swainsonine production is not a specialized trait of plant-associated fungi (Cook et al., 2017). In a recent study, the essential functions of swnH1 and swnH2 were confirmed by gene deletion in M. robertsii, whereas the swnN, swnR and swnT knock-out mutants still produced 2 (Luo F. et al., 2020). The role of SwnA in producing the pipecolic acid precursor for SwnK was confirmed by gene deletion and overexpression, however, it appears that pipecolic acid can also be produced by other (unconfirmed) pathways in M. robertsii, involving genes outside SWN BGC. Even after meticulous experiments, the exact role of SwnN, SwnR, and SwnT remain elusive. However, important pathway intermediates were identified, which allowed the conclusion that SwnH1 catalyzes the final step in 2 biosynthesis.
A third example is the pathway investigation and enhancement of the biosynthesis of perylenequinones, including Hypocrellin A (16), in the endophytic fungus S. sp. Slf14 from Huperzia serrata. These studies were started from the phenotypic observation of 16 with high pharmaceutical importance from an endophytic fungus S. bambusicola (Yang et al., 2009). Using de novo transcriptome assembly and retro-biosynthesis based on structural similarity of 16 with cercosporin, perylenequinones biosynthesis was suggested to involve the a type I PKS, an O-methyltransferase/FAD-dependent monooxygenase, an hydroxylase and another methyltransferase in S. bambusicola (Zhao et al., 2016). In order to further study the regulatory mechanisms of the PKS, the methyltransferase and hydroxylase genes, Liu et al. performed RT-qPCR of S. sp. Slf14 cultured under different Ca2+ concentrations, and in the presence of Ca2+ signaling antagonists (Liu et al., 2018). Under high Ca2+ conditions, perylenequinones production was enhanced, and the transcription dynamics of the putative pathway genes were similar to those of the known Ca2+ sensors cam, cna, and crz1. Later on, the same pathway genes were pinpointed by transcript analysis in S. bambusicola S4201, and genes encoding a putative O-methyltransferase/FAD-dependent monooxygenase, an Flavin-dependent oxidoreductase, a multicopper oxidase and a Zink-finger transcription factor (Li et al., 2019). Recently, comparative transcriptomics were used to study the transcription of the genes in the proposed BGC in S. sp. Slf14 grown with different carbon sources (Liu et al., 2020). The highest perylenequinones production was observed with fructose and the transcription of the putative perylenequinones biosynthetic genes including the Zink-finger transcription factor, and important enzymes in precursor supply were upregulated, whereas competing pathways, such as fatty acid synthesis were downregulated. This could be attributed to the activity of global regulators, e.g., Cre1, PaC (upregulated in presence of fructose) and LaeA (downregulated) (Liu et al., 2020). To summarize, even though the whole genome sequence of S. sp. Slf14 was obtained (Yang H. et al., 2014), no successful gene deletion of the putative BGC has been reported to date that would unambiguously confirm the importance of the BGC for 16 production. Nevertheless, many years of extensive work have provided convincing evidence for the function of this BGC and remarkable insight into the regulation of perylenequinones biosynthesis.
The fourth example is reported by Wang et al. (2008) in the re-isolation of radicicol (3a) from the endophytic fungus C. chiversii and the elucidation of its biosynthetic pathway. 3a is a known fungal polyketide with prospective anticancer activity, and was originally isolated by a bioassay-guided fractionation based on its inhibitory activity against Hsp90 (Turbyville et al., 2006). Based on the similarity of its scaffold with fungal Resorcylic Acid Lactones (RALs), retro-biosynthesis led to the hypothesis that a highly reducing PKS (hrPKS) and a non-reducing PKS (nrPKS) would form the core structure. A fosmid library of the genome was constructed and screened for the putative biosynthetic genes using degenerate PCR primers designed with the sequence of a close homolog. In the sequence of the fosmid hits, a putative BGC encoding the expected PKS enzymes and a number of tailoring enzymes, were identified. Next, targeted disruptions of the core biosynthetic genes, namely the ccRads1 (hrPKS), ccRads2 (nrPKS), and radR (a gene encoding a putative positive transcriptional regulator) in the native host led to a loss of 3a production. The disruption of one of the putative tailoring enzymes, RadP, led to the accumulation of Pochonin D (4), now shown to be a pathway intermediate in 3a and 3b biosynthesis (Wang et al., 2008).
In the fifth example, Purpureocillium lilacinum, an endophyte with a well-known biocontrol use against various plant pathogens in agriculture, was shown to be a prolific producer of leucinostatins (12) (Wang et al., 2016), which are peptaibiotics with a wide range of activities including, antibiotic (Fukushima et al., 1983) and antitumor effects (Kawada et al., 2010). Based on BGC homology and a retro-biosynthesis approach, Wang et al. hypothesized that the lcs gene cluster involving 20 genes would be responsible for biosynthesis of 12. From there, gene knock-outs on lcsC, lcsD, lcsE, and lcsA were performed by homologous recombination in the native host to verify their function in the pathway (Figure 3D). In addition, the OSMAC strategy coupled with RT-qPCR and RNAseq analysis was employed to determine the boundaries of the BGC. Lastly, the overexpression of a cluster specific regulator, LcsF, rounded up the study by confirming the co-regulation of the genes in the newly identified BGC.
In the sixth example, BGC homology and retro-biosynthesis were applied to propose a biosynthetic pathway for nodulisporic acids, bioactive indole diterpenes produced in the endophytic fungus Hypoxylon pulicicidum strain MF5954, from Bontia daphnoides (Nicholson et al., 2018). To confirm the involvement of the genes in the proposed gene cluster (NOD), Van De Bittner et al. (2018) used a multigene assembly strategy to reconstitute parts of the biosynthetic pathway in the heterologous host Penicillium paxillin. Thereby, they characterized the function of four genes involved in the biosynthesis of the noludisporic acid core compound, noludisporic acid F (23).
Although the above-mentioned elucidation efforts of several genes in a BGC offer comprehensive insight into the biosynthesis of secondary metabolites in endophytic fungi, studying a single yet specific biosynthetic enzyme can be promising for bioprospecting as well. In this regard, a success story was reported for Hypoxylon sp., which was discovered to produce a series of volatile organic compounds, including 1,8-cineole (24), a monoterpene with high commercial and pharmaceutical value (Tomsheck et al., 2010). Since up to that point, no fungal monoterpene synthases were known, Shaw et al. selected eight putative terpene synthase genes from the whole genome sequence of the fungus based on sequence homology to plant terpene synthase genes. They expressed these genes heterologously in E. coli and analyzed the product range (Shaw et al., 2015). In this way, they identified the first fungal monoterpene synthase Hyp3 and established efficient production of 24. Kinetic studies, homology modeling and site directed mutagenesis identified the crucial active site residues for catalysis and substrate selectivity of Hyp3. Taken together, this extensive study was an important step in the bioprospecting of an endophytic fungal enzyme with potential for industrial application.
In the first example starting from genotypic observation, Wang et al. (2019) successfully identified the plausible biosynthetic pathways of Chevalone E (18a) and its novel analogs by studying the genome of the endophytic fungus A. versicolor 0312 (Figure 3E). First, WGS, assembly, and annotation were conducted. Since hybrid polyketide-diterpenoid (PK-DT) scaffolds are often part of molecules with a wide range of bioactivities and only few BGCs giving rise to this scaffold were known at the time, this study was aimed to accelerate the discovery of interesting novel molecules from this scaffold. The authors performed rule-based genome mining based on BGC homology. Initially, a BLAST search was performed to find a genomic region that encodes both a PKS and a geranylgeranyl pyrophosphate synthase, and the genome neighborhood was analyzed for putative tailoring enzymes, resulting to the discovery of a proposed cle BGC. Given that the cle cluster was significantly different from known PK-DT hybrid clusters, it was deemed an interesting target. PCR amplification was used to clone the target genes for expression in a heterologous host, A. oryzae, for experimental verification. Several gene combinations were tested to study the function and order of all enzymes in the pathway, as well as the intermediates produced. Lastly, the authors designed a combinatorial pathway by co-expressing parts of the cle cluster with the terpene synthase of the homologous sre cluster from A. felis 0260 to obtain novel molecules (18b and 18c). These were seen to enhance the efficacy of doxorubicin against breast cancer cells. More importantly, these molecules possess a characteristic five-membered lactone ring, which is very rare in meroterpenoids and has never been seen in fungal meroterpenoids (Wang et al., 2019).
The second example started from the discovery of a cryptic BGC, encoding for enzymes later found to produce 1,8-dihydroxynaphthalene (DHN) (25) melanin in P. fici (Figure 3F) (Zhang et al., 2017, 2019). DHN melanin plays significant roles in secondary metabolite production, UV protection, oxidative stress and pathogenesis of fungi (Eisenman et al., 2020). The study aimed at identifying fungal pigments and their physiological roles and the authors searched for PKS genes, typically involved in melanin biosynthesis (Zhang et al., 2017). Based on sequence homology the authors identified the putative PKS gene pfmaE in the genome of P. fici. Deletion of this gene showed a complete abolishment of melanin biosynthesis, as seen by decreased pigmentation and incomplete spore development, which were later rescued by feeding of pathway intermediates. However, since phenotyping efforts with different extraction methods and analyses by HPLC and LC-MS analysis could not distinguish differences in the chemical profile of wild type and pfmaE mutant due to the low concentration or cryptic nature of melanin biosynthesis, the authors performed further experimental verification by heterologous expression of the predicted pfma cluster (pfmaA-D, G, H+E) in A. nidulans. Metabolite profiles of several recombinant strains were compared to assign enzyme functions and identify intermediates. Prominent intermediates of the melanin pathway, T4HN (26), which is unstable and easily oxidized to form flaviolin (27), and scytalone (28) were observed in the strains expressing pfmaE, while they were absent in a strain only expressing the other pfma genes, thus suggesting the contribution of PfmaE in the pathway. To further study the function of each gene in the cluster, gene deletions in the pfma expressing heterologous host were performed and only PfmaE and G were found to be essential for the formation of the pathway intermediates. In conclusion, the authors suggested that PfmaE produces 26 which is reduced by PfmaG to form 28. Based on homology search, the authors suggested PFICI_02498 as syctalone dehydratase or PfmaD as vermelone dehydratase to be responsible in the final steps of melanin biosynthesis. However, experimental verification was not done on those genes. Co-expression of a putative regulatory gene, pfmaF, was performed which resulted in a significant increase of 26–28 in the heterologous host, indicating an involvement in the melanin biosynthetic pathway. Since the function of two genes encoding transcription factors in the pfma cluster, pfmaF and pfmaH were not clearly characterized in the first study, a follow up experimental verification using gene deletion in the native host was conducted (Zhang et al., 2019). With transcriptomic analysis and chemical studies, the authors showed the cluster-specific role of PfmaH in the melanin biosynthetic pathway. Another transcription factor, PfmaF, was verified to be a global regulator of secondary metabolite biosynthesis in P. fici, including its role in assisting PfmaH in promoting melanin synthesis. The function of other genes was also verified in this study by in vitro characterization, namely PfmaJ previously named PFICI_02498 as scytalone dehydratase and PfmaG or PFICI_05460 as T4HN reductase. In another study, a second global regulator, RsdA, was also reported to be involved in melanin biosynthesis of P. fici. Experimental verification by gene deletion showed a remarkable increase in melanin production, which was suggested to be achieved by its modulation of PfmaE PKS expression (Zhou S. et al., 2019).
As studying the entire genes in a pathway might be difficult considering their complexity and the big size of BGCs, some studies focus on ascertaining the presence of key genes encoding the enzymes which could serve as markers in the pathway. As an example, a the ssars gene encoding 5-alk(en)ylresorcinol synthase was identified in the genome of S. sp. Slf14, an endophytic fungus from Huperzia serrata based on sequence homology to known Type 3 PKSs (Yan et al., 2018). Yan et al. studied the gene in two heterologous host, E. coli and S. cerevisiae by cloning the intron free DNA of ssars through reverse transcriptase PCR. Substrate feeding with fatty acids showed the potential of the enzyme to generate a wide range of fatty acid primed resorcinol derivatives, including 14, using the exogenously supplied precursors in the heterologous hosts (Yan et al., 2018). With this strategy, bioprospecting efforts can be efficiently directed to the use of specific enzymes in the production of secondary metabolites with biopharmaceutical importance.
Despite the great potential of starting a study from genotypic observation, successful stories from this approach are much less prevalent than when starting from phenotypic observation. In the next section of this review, we will provide some insight into the major hurdles in the field.
Compared to other microorganisms, such as bacteria, the genotyping effort of endophytic fungi appears to be lagging behind. This may be attributed to the fact that genomics of fungi in general is still in its infancy and that the currently available BGCs in the databases might be of low predictive value for fungal BGCs (van der Lee and Medema, 2016).
Even though technologies of next generation sequencing (NGS) have changed dataflow dramatically as the amount of sequencing data is growing at a significant pace (Goodwin et al., 2016), only a miniscule fraction of endophytic fungi have their whole genomes sequenced and published. From a thorough data base search (NCBI whole genome sequences and pubmed), we have compiled a list of publicly accessible WGS projects of proven endophytic fungi (Table 3). Based on a search of the JGI project database and the Mycocosm (JGI, 2019), we believe that many more genomes will be published in the near future, but certainly not as many as one would hope for. This is most likely due to the uncultivability of many endophytic fungi outside their habitat, which impedes their isolation process (Rashmi et al., 2019). Thus, in the future directions section we will discuss several advances in environmental genomics that we think will have a positive impact on the field of endophyte biology in general, and in particular fungal genomics.
Table 3. List of endophytic fungi subject to whole genome sequencing projects referenced in the literature.
In terms of genome mining and BGC discovery, several hurdles remain to be overcome when studying endophytic fungi. The first problem is the low predictive value of known BGCs for the annotation of fungal BGCs in bioinformatic tools with a conventional rule-based approach. A global analysis of BGCs in the genomes of 24 Penicillium species found that only 16% of the PKS- and NRPS-derived BGCs can be connected to a pathway and the respective secondary metabolites, while the rest have no predictive value for secondary metabolite (Nielsen et al., 2017). The prediction of fungal BGCs is problematic since most of the bioinformatic tools were developed for bacterial genomes, which have a different architecture: fungal BGCs are more often split over multiple loci and/or have a bidirectional orientation in which the cis-regulatory region is shared between a set of genes (van der Lee and Medema, 2016). Beyond that, fungi possess a large number of BGCs lacking even the homologs of scaffolding enzymes, so-called exotic BGCs, which are even more difficult to annotate (van der Lee and Medema, 2016). In order to solve such annotation problems, transcriptomic studies are often employed in other organisms, e.g., bacteria and plants, however, since many of these genes are also transcriptionally silent under experimental conditions this technique is often not applicable, which makes them frequently misannotated (van der Hooft et al., 2020). Similar to bacterial BGCs, plant biosynthetic genes are often also unsuitable for comparative analysis to predict endophytic fungal BGCs. Many studies propose that the biosynthetic pathways in endophytic fungi are most likely different from those in their host plants (Kusari et al., 2009b; Yang et al., 2014; Kang et al., 2019; Qiao et al., 2020) even though lateral gene transfer from plant to endophytic fungi was originally hypothesized. Two prominent examples for molecules present in fungi and plants yet formed by different pathways are gibberellins and indole acetic acid (Salazar-Cerezo et al., 2018). Taken together, the prediction of fungal BGCs is at high risk of being inaccurate with the conventional rule-based approach.
Even in case of successful functional annotation of BGC boundaries and all the contained genes, it remains challenging to predict the structure of the secondary metabolites produced. Unlike studies starting from phenotypic observations with the elucidated chemical structure in hand, judging the biological or biotechnological relevance of a predicted secondary metabolite purely based on the genes encoded in a BGC is nearly impossible to date (Weber and Kim, 2016; van der Hooft et al., 2020). Platforms like SANDPUMA (Chevrette et al., 2017), or a combination of individual module-level prediction tools like antiSMASH (Medema et al., 2011) and PRISM (Skinnider et al., 2017) offer various algorithms to predict full chemical structures, yet often lead to a massive amount of combinatorial possibilities with error-prone results, especially for unusual and rare modifications from less studied organisms, like endophytic fungi. In addition, an effective method to sort the prediction results based on their match rate with the actual structures has not been reported (van der Hooft et al., 2020).
Despite the mentioned challenges, genome mining allows to explore cryptic and novel BGCs. Current drawbacks are mainly due to the early stages of genetics and genomics compared to the phenotyping methods. As reviewed in the next section, we expect that some of these problems will be overcome with the availability of more functional genomics studies in fungi and especially endophytic fungi.
Despite the current shortcomings and limitations in the field, we observe some emerging technologies and tools that might advance the bioprospecting effort of endophytic fungi in the future. In this last section we will review these developments within three major categories: (1) the cultivation and isolation process, (2) culture-independent techniques, and (3) genome editing techniques.
Studying endophytes in axenic experimental conditions is highly challenging considering the absence of metabolites and stimuli provided by the host plant and other members of the microbial community. It appears that the lack of essential, life-supporting factors leads to the majority of endophytic fungi being uncultivable under experimental conditions and hampering their isolation process. Currently we have no reliable information on the ratio of cultivable to uncultivable fungal endophytes, but environmental DNA surveys suggest that the diversity of species is far greater than the diversity of cultivated isolates (Wu B. et al., 2019). A study on the endophytic fungal community of Dysphania ambrosioides, found that almost 10 times more species can be identified by amplicon sequencing than can be isolated by cultivation (Parmar et al., 2018). In order to increase the number of cultivable species and study their biosynthetic potential, it is of utmost importance to improve cultivation and isolation procedures. Next to the technological advances discussed in the following sections, closer collaboration of bioprospectors with and training from experienced mycologists would also help solve the cultivability problem.
Recently, Libor et al. (2019) developed a Fungal one-step IsolatioN Device (FIND) which allows the isolation of rare fungi from terrestrial and marine samples. This technology is an adaptation of the Isolation chip (iChip), originally developed to grow bacteria from environmental samples using the naturally occurring nutrients and growth factors from their own habitat (Berdy et al., 2017). Both technologies allow for high throughput isolation of microbes by utilizing several hundred miniature diffusion chambers in a growth device, each inoculated with a single cell (environmental isolates or engineered variants of a single strain). This growth device is brought back into its natural environment (e.g., submerged in sea water or buried in damp soil) to allow for diffusion of nutrients and signaling molecules from the environment and between the microbes in the individual chambers. This setup facilitates the cultivation of novel species without the need to optimize artificial growth conditions and allows for capturing broader biological diversity. The use of this technology has not been reported for plant-associated fungi, but a so-called “rhizochip” was developed to study bacteria in the rhizosphere of the canola plant (Gurusinghe et al., 2019). For the study of epiphytes, living on the surface of a plant, we could imagine that techniques to attach a FIND to a plant could be developed. However, developing a similar system for endophytes, where an incision on a leaf, the stem or bark would be required, appears challenging. An easier strategy could be to use the FIND submerged in a plant liquid culture or in media spiked with plant exudates. Clearly, there is still a lot of room for innovation in this field of research, but we think that facilitating the isolation and cultivation process of endophytic fungi would really accelerate the possibilities to mine for secondary metabolites and biosynthetic genes.
Microfluidic platforms have been used as a powerful approach to evaluate the responses of plant cells to external perturbations, such as nutrients, temperature, light and other abiotic or even biotic factors like a pathogen (Meier et al., 2010; Busch et al., 2012; Jiang et al., 2014). It has a higher throughput compared to conventional cultivation techniques and a more precisely controlled environment. However, to date only a few studies use this technology to study biotic interactions of plants with their symbionts. A recent breakthrough using this technology was reported by researchers at the U.S. Department of Energy's Argonne National Laboratory. They successfully developed a Root Microbiome Interaction (RMI) chip to study the interaction between Populus tremuloides (aspen tree) and its bacterial root microbes (Noirot-Gros et al., 2020). The microfluidic setup in the RMI-chip is coupled with advanced live imaging microscopy to allow for direct visualization of the dynamic interactions at cellular resolution at real-time. However, it only enables the observation of the root, which is relatively easy since it can be immersed and continuously perfused to monitor the biochemical interactions. Studying other plant parts is not as easy since the system needs to facilitate gas exchange and light exposure.
Since bacterial colonization is significantly different from fungal, optimization of these microfluidic devices is necessary to study fungal endophytes. Advances in microfluidic devices for fungi were recently reviewed (Zhou W. et al., 2019) but we'd like to highlight one chip design recently published by Millet et al. in 2019: a ready-to-use microfluidic device with chambers and boundary channels forming the letters “ORNL” was established to study binary interactions of various organisms, e.g., a plant-growth promoting bacterium and the fungus Mortierella elongata NVP64+, and Nicotiana attenuata seedlings and the fungus. In these binary interactions it was observed that the microbes did not influence each other strongly, although a small part of the bacterial population showed altered and previously unknown cell morphologies. With the plant-fungus co-culture, however, clear colonization of the plant roots by the endophyte was observed (Millet et al., 2019). The ease of use of these ready-to-use ORNL devices might make them widely applicable to study endophytic fungi and their BGCs in the presence of their host plants and allow for the tracking of their dynamics at high spatial and temporal resolution. Given that plant development from a single protoplast has been studied in microfluidic devices before (Sakai et al., 2019), we could even imagine using a similar setup to study the reciprocal influence of microbiota and plants with such devices. This would be particularly interesting for aerial parts of plants rather than roots and might allow for downstream metabolomics analysis and secondary metabolite discovery.
Bioprospecting of natural products is evolving from studying the biosynthetic potential of isolated organisms to a more comprehensive approach of studying microbial communities as a whole (meta-omics) or individual organism without prior isolation by cultivation (single-cell -omics). The meta-omics strategy has been reviewed in detail recently (van der Hooft et al., 2020) but we will highlight some developments specific to fungi. Single-cell analysis of fungi is still in its infancy and we will give a broader perspective on where we see the field going.
With decreasing cost of sequencing and advanced computational analysis pipelines, shotgun metagenomics has become an important technique to study microbial communities (Pérez-Cobas et al., 2020), particularly for uncultivable species. With sufficient sequencing depth, shotgun metagenomics has been shown to provide high quality assemblies of BGCs (Kim et al., 2012; Bengtsson-Palme et al., 2014; Donia et al., 2014; Sugimoto et al., 2019; Youngblut et al., 2020), entire chromosomes or even whole genomes from a microbial community (Chen et al., 2020). Unfortunately, to date many shotgun metagenomics projects are focused on bacteria and exclude fungal DNA in the sample preparation stage, thereby limiting the number of useful datasets for researchers interested in fungal BGCs. Donovan et al. have, however, developed a computational pipeline to identify fungal genes/contigs in metagenomic datasets with their FindFungi pipeline and identified several fungi in animal-associated microbiomes (Donovan et al., 2018). Several other tools that are not specific to fungi but can be applied to identify bacterial and eukaryotic contigs from metagenomic samples have been developed and recently compared in terms of performance (Ye et al., 2019). The most recent one is worth mentioning since it is an extension of the highly performant MMSeqs2 platform (Mirdita et al., 2020) that has gained significant traction in the environmental genomics field over the last few years (Steinegger and Söding, 2017; Santos et al., 2020; Segawa et al., 2020). As discussed by Ye et al. (2019), all of the tools heavily rely on reference genomes in public data bases and are therefore error-prone in undersampled branches of the tree of life. MMSeqs2 taxonomy appears to fare much better than its competitors in this regard, since it cross-validates its hits and thereby finds the last common ancestor of the query sequence (Mirdita et al., 2020). Another common problem in metagenomics analysis is the contamination of reference genomes with contigs that were incorrectly assigned to the respective species. Also here the developers of MMSeqs2 have recently developed a tool to “clean-up” these contaminations and therefore improve the quality and reliability of the database entries (Steinegger and Salzberg, 2020). With these improved reference databases and with the most recent tools, the analysis of existing and new metagenomic datasets will be dramatically improved and hopefully yield many interesting fungal contigs for exploration.
Another promising strategy is the use of long-read sequencing technologies, i.e., Pacific Bioscience or Oxford Nanopore sequencing on metagenomic libraries. Despite the relatively high error rates observed in these technologies, in particular Oxford Nanopore has been shown to provide excellent recovery of functional genomic units, entire chromosomes, and whole genomes of bacteria (Tyler et al., 2018; Arumugam et al., 2019; Moss et al., 2020) and fungi (Dutreux et al., 2018; Liu et al., 2019; McKenzie et al., 2020). Recent developments in real-time selective sequencing also allow for the depletion or enrichment of specific sequences during the sequencing run (Loose et al., 2016; Edwards et al., 2019; Masutani and Morishita, 2019; Kovaka et al., 2020; Payne et al., 2020). One could imagine that this strategy could be applied to suppress the sequencing of highly abundant species in a microbiome and thereby enhance the recovery of species with lower abundance.
In addition to these culture-independent genomics strategies, meta-transcriptomics and meta-metabolomics can be employed to give a more comprehensive view of a microbial community, since they allow for the analysis of gene expression levels and biochemical profiles thus revealing the details of the metabolic status of a community within a specific time frame. Please refer to van der Hooft et al. (2020) for a review on the emerging strategies and tools used to integrate metagenomics and metabolomic datasets. One of the biggest challenges of multi-omics strategies is the large quantity of data produced, which makes data integration a daunting task. Refinement and standardization of data structures might be a solution to allow the programmatic access by the different tools, but the application is yet to be seen in the field. Despite its great opportunities, there is to our knowledge no study applying meta-omics to study endophytic fungi. There is one very recent study integrating holobiont metabolomics with amplicon sequencing of the endophytic fungi of separated plant parts of Ephedra sinica to correlate the microbiome composition with the metabolite profiles (Zhang et al., 2020). Since culture-independent techniques are inherently high-throughput strategies and allow for the study of uncultivable organisms, we expect to see many more studies soon that lead to the discovery of novel BGCs and secondary metabolites.
Single-cell -omics aims to gather information from a single cell of an organism, or an individual microbial cell from a community. In the latter case, this allows for the study of uncultivable organisms, and/or community members of low abundance. Herein, single-cell genomics is the most advanced of the -omics techniques and has even yielded near-complete genome assemblies of environmental microbes (Seeleuthner et al., 2018; Pachiadaki et al., 2019; Dam et al., 2020). In this technology, sorting of cells by flow cytometry is used for single cell isolation, then multiple displacement amplification is conducted followed by sequencing. Although this technology rarely yields full genome coverage, it offers a bright future to facilitate genomic exploration of unculturable organisms (Woyke et al., 2017). A recent study on bacterial environmental samples even suggests that the recovery of genomes with the single-cell approach is higher than that of a shotgun metagenomics approach (Dam et al., 2020). The major difficulty in using this technology on endophytic fungi is probably the preparation of single cells. The two studies applying single-cell genomics to fungi used spores that can easily be harvested from cultivable fungi (Ahrendt et al., 2018; Montoliu-Nerin et al., 2020). However, for uncultivable organisms this is not possible and other strategies need to be developed. Looking at the recent advances in plant single-cell genomics (Luo C. et al., 2020), the most promising strategies, will most likely be the preparation of protoplasts by cell wall digestion (host plant and fungal cells need to be digested), and/or the isolation of nuclei (Montoliu-Nerin et al., 2020). Once a suspension of cells or nuclei is prepared, they need to be separated by manual sorting or by Fluorescence-activated cell sorting (FACS). Depending on the presence of suitable markers for staining needed for FACS analysis and the overall numbers of harvested cells or nuclei, one strategy might be better than the other. It appears that in the plant community manual sorting is more common at this stage than FACS (Luo C. et al., 2020) and this might also turn out to be the better strategy for endophytic fungi. However, since flow cytometry of filamentous fungi has been demonstrated (Bleichrodt and Read, 2019), it is possible that such a high-throughput approach might become applicable for the sorting of fungal endophytes.
Recent advances in animal single-cell technologies have also allowed for multimodal single-cell -omics as reviewed by several groups (Stuart and Satija, 2019; Lee et al., 2020; Ma et al., 2020). These workflows aim to gather genomic, transcriptomic and proteomic information from each isolated cell and are expected to even be combined with other physiologic observations prior to these destructive analyses. We envision that similar developments are also on the horizon for the study of environmental microbial isolates, such as endophytic fungi.
Experimental verification of BGC identity in the native host is a major challenge in filamentous fungi. Several strategies for genome editing were recently reviewed in detail (Mei et al., 2019). Here, we'd like to emphasize only recent developments in the field of CRISPR/cas9 genome engineering of filamentous fungi that promise to also be applicable to organisms without an elaborate engineering toolbox at hand. As reviewed by Mei et al. (2019), Schuster and Kahmann (2019), Song et al. (2019), Ouedraogo and Tsang (2020), and Ullah et al. (2020), the CRISPR/cas9 strategy most commonly used in a variety of organisms including filamentous fungi relied mostly on the delivery of the Cas9 gene and guideDNA in expression cassettes for in vivo generation of the active cas9/guideRNA complex. This requires the availability of a suitable DNA shuttle, selective markers, and promoter and terminator sequences functional in the native host, which is often not available for new isolates. Recently, in vitro preparation of the Cas9-gRNA ribonucleoprotein complex and transformation into protoplasts of filamentous fungi has become available (Pohl et al., 2016; Al Abdallah et al., 2017; Wang et al., 2018). This system is reported to have a higher efficiency, fewer mistargets, and allows for rapid disruption using direct protoplast transformation. Although it has not been applied in endophytic fungi, this is a promising strategy to advance the bioprospecting effort of endophytic fungi with limited molecular tools. Not only in gene editing, CRISPR/Cas9 also advances the development of gene expression for biotechnological purposes using the recently developed CRISPR interference and CRISPR activation systems which offer novel strategies to regulate gene expression as reviewed by Zheng et al. (2019). Taken together, the use of CRISPR/Cas9 promises easier experimental verification of BGCs under investigation, and expedited construction of suitable expression systems for biotechnological processes involving endophytic fungi.
Fungi are prolific producers of secondary metabolites and have been exploited for industrial production of pharmaceuticals and nutraceuticals for almost a century. Endophytic fungi, living in plant tissues are a very species-rich, yet somewhat elusive group of fungi that have been chased after by bioprospectors over the last three decades. Despite prevalent difficulties in cultivation and stability of metabolite production of these organisms, phenotypic studies describing the observation of novel metabolites are numerous. The real challenge appears to be the discovery of the underlying biosynthetic pathways that would allow the rational engineering, or refactoring of these pathways for industrial purposes. In our detailed literature survey presented in this review, we were only able to find nine examples of endophyte secondary metabolites, whose biosynthetic gene clusters were conclusively elucidated. The overall small number of whole genome sequences of endophytic fungi, and the often-low predictive value of known BGCs for fungal BGCs, make genome mining and BGC discovery challenging. We expect that some of these problems will be overcome with the availability of more functional genomics studies in fungi and especially endophytic fungi. For this endeavor, rule-independent approaches for BGC prediction will be instrumental, as well as advanced workflows for cultivation and experimental verification. Moreover, the increasing number of biochemical studies will allow for better training data sets for current and future machine learning-based platforms. Continuous improvements in the development of genetics and genomics tools have significantly supported the bioprospecting of natural products and will continue to do so.
RS and KH developed a concept for the review and collected the literature. RS wrote the manuscript with input from WQ and KH. All authors contributed to the article and approved the submitted version.
This project has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No 893122. RS was an awardee of the Indonesia Endowment Fund for Education (LPDP) scholarship.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors were grateful to Sadaf Deravi and Kahraman Gercek, who compiled the list of publicly accessible genomes of endophytic fungi as part of their B.Sc. theses.
BGC, biosynthetic gene cluster; BLAST, basic local alignment search tool; CRISPR/cas9, clustered regularly interspaced short palindromic repeats/cas9; DHN, dihydroxynaphthalene; DNA, deoxyribonucleic acid; FACS, fluorescence-activated cell sorting; FAD, flavin adenine dinucleotide; FDA, United States Food and Drug Administration; FIND, Fungal one-step IsolatioN Device; GC, gas chromatography; HIV, human immunodeficiency virus; HPLC, high pressure liquid chromatography; IChip, isolation chip; JGI, Joint Genome Institute of the U.S. Department of Energy; LC, liquid chromatography; MS, mass spectrometry; NCBI, National Center for Biotechnology Information of the U.S. National Institutes of Health; ND, no data; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; NGS, Next Generation Sequencing; NMR, Nuclear Magnetic Resonance; NRPS, non-ribosomal peptide synthetase; OSMAC, one strain many compounds; PCR, polymerase chain reaction; PKS, polyketide synthase (hr—highly reducing, nr—non-reducing); PK-DT, polyketide-diterpenoidqRT—quantitative PCR; RAL, resorcylic acid lactones; RMI, root microbiome interaction; RNA, ribonucleic acid; RNAseq, RNA sequencing technology; RT-qPCR, reverse transcriptase qPCR; U.S., United States of America; REEIS, Research, Education and Economics Information System of the US Department of Agriculture; WGS, whole genome sequencing.
Ahrendt, S. R., Quandt, C. A., Ciobanu, D., Clum, A., Salamov, A., Andreopoulos, B., et al. (2018). Leveraging single-cell genomics to expand the fungal tree of life. Nat. Microbiol. 3, 1417–1428. doi: 10.1038/s41564-018-0261-0
Akone, S. H., Mándi, A., Kurtán, T., Hartmann, R., Lin, W., Daletos, G., et al. (2016). Inducing secondary metabolite production by the endophytic fungus Chaetomium sp. through fungal–bacterial co-culture and epigenetic modification. Tetrahedron 72, 6340–6347. doi: 10.1016/j.tet.2016.08.022
Al Abdallah, Q., Ge, W., and Fortwendel, J. R. (2017). A simple and universal system for gene manipulation in Aspergillus fumigatus: in vitro-assembled Cas9-guide RNA ribonucleoproteins C. mSphere 2:e00446-17. doi: 10.1128/msphere.00446-17
Aldridge, S., Parascandola, J., and Sturchio, J. L. (1999). “The discovery and development of penicillin 1928–1945,” in An International Historic Chemical Landmark, 1st Edn (The Alexander Fleming Laboratory Museum; American Chemical Society and the Royal Society of Chemistry). Available online at: http://www.acs.org/content/acs/en/education/whatischemistry/landmarks/flemingpenicillin.html (accessed February 24, 2021).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Armijos Jaramillo, V. D., Vargas, W. A., Sukno, S. A., and Thon, M. R. (2013). Horizontal transfer of a subtilisin gene from plants into an ancestor of the plant pathogenic fungal genus Colletotrichume. PLoS ONE 8:e59078. doi: 10.1371/journal.pone.0059078
Arumugam, K., Bagci, C., Bessarab, I., Beier, S., Buchfink, B., Gorska, A., et al. (2019). Annotated bacterial chromosomes from frame-shift-corrected long read metagenomic data. Microbiome 7:61. doi: 10.1101/511683
Bai, J., Mu, R., Dou, M., Yan, D., Liu, B., Wei, Q., et al. (2018). Epigenetic modification in histone deacetylase deletion strain of Calcarisporium arbuscula leads to diverse diterpenoids. Acta Pharma. Sin. B 8, 687–697. doi: 10.1016/j.apsb.2017.12.012
Bartley, B. A., Beal, J., Karr, J. R., and Strychalski, E. A. (2020). Organizing genome engineering for the gigabase scale. Nat. Commun. 11:689. doi: 10.1038/s41467-020-14314-z
Bengtsson-Palme, J., Boulund, F., Fick, J., Kristiansson, E., and Joakim Larsson, D. G. (2014). Shotgun metagenomics reveals a wide array of antibiotic resistance genes and mobile elements in a polluted lake in India. Front. Microbiol. 5:648. doi: 10.3389/fmicb.2014.00648
Berdy, B., Spoering, A. L., Ling, L. L., and Epstein, S. S. (2017). In situ cultivation of previously uncultivable microorganisms using the ichip. Nat. Protoc. 12, 2232–2242. doi: 10.1038/nprot.2017.074
Bhargavi, S. D., Praveen, V. K., Anil Kumar, M., and Savitha, J. (2018). Comparative study on whole genome sequences of Aspergillus terreus (soil fungus) and Diaporthe ampelina (endophytic fungus) with reference to lovastatin production. Curr. Microbiol. 75, 84–91. doi: 10.1007/s00284-017-1353-4
Bleichrodt, R. J. J., and Read, N. D. (2019). Flow cytometry and FACS applied to filamentous fungi. Fungal Biol. Rev. 33, 1–15. doi: 10.1016/j.fbr.2018.06.001
Bode, H. B., Bethe, B., Höfs, R., and Zeeck, A. (2002). Big effects from small changes: possible ways to explore nature's chemical diversity. ChemBioChem 3, 619–627. doi: 10.1002/1439-7633(20020703)3:7<619::AID-CBIC619>3.0.CO;2-9
Bok, J. W., Ye, R., Clevenger, K. D., Mead, D., Wagner, M., Krerowicz, A., et al. (2015). Fungal artificial chromosomes for mining of the fungal secondary metabolome. BMC Genomics 16:343. doi: 10.1186/s12864-015-1561-x
Braun, K., Romero, J., Liddell, C., and Creamer, R. (2003). Production of swainsonine by fungal endophytes of locoweed. Mycol. Res. 107, 980–988. doi: 10.1017/S095375620300813X
Busch, W., Moore, B. T., Martsberger, B., MacE, D. L., Twigg, R. W., Jung, J., et al. (2012). A microfluidic device and computational platform for high-throughput live imaging of gene expression. Nat. Methods 9, 1101–1106. doi: 10.1038/nmeth.2185
Cain, J. W., Miller, K. I., Kalaitzis, J. A., Chau, R., and Neilan, B. A. (2020). Genome mining of a fungal endophyte of Taxus yunnanensis (Chinese yew) leads to the discovery of a novel azaphilone polyketide, lijiquinone. Microb. Biotechnol. 13, 1415–1427. doi: 10.1111/1751-7915.13568
Carrieri, R., Borriello, G., Piccirillo, G., Lahoz, E., Sorrentino, R., Cermola, M., et al. (2020). Antibiotic activity of a Paraphaeosphaeria sporulosa-produced diketopiperazine against Salmonella enterica. J. Fungi 6:83. doi: 10.3390/jof6020083
Chavali, A. K., and Rhee, S. Y. (2018). Bioinformatics tools for the identification of gene clusters that biosynthesize specialized metabolites. Brief. Bioinformatics 19, 1022–1034. doi: 10.1093/bib/bbx020
Che, J., Shi, J., Gao, Z., and Zhang, Y. (2016). Transcriptome analysis reveals the genetic basis of the resveratrol biosynthesis pathway in an endophytic fungus (Alternaria sp. MG1) isolated from Vitis vinifera. Front. Microbiol. 7:1257. doi: 10.3389/fmicb.2016.01257
Chen, G. E., Canniffe, D. P., Barnett, S. F. H., Hollingshead, S., Brindley, A. A., Vasilev, C., et al. (2018). Complete enzyme set for chlorophyll biosynthesis in Escherichia coli. Sci. Adv. 4:eaaq1407. doi: 10.1126/sciadv.aaq1407
Chen, L. X., Anantharaman, K., Shaiber, A., Murat Eren, A., and Banfield, J. F. (2020). Accurate and complete genomes from metagenomes. Genome. earch 30, 315–333. doi: 10.1101/gr.258640.119
Chevrette, M. G., Aicheler, F., Kohlbacher, O., Currie, C. R., and Medema, M. H. (2017). SANDPUMA: ensemble predictions of nonribosomal peptide chemistry reveal biosynthetic diversity across Actinobacteria. Bioinformatics 33, 3202–3210. doi: 10.1093/bioinformatics/btx400
Chiang, Y. M., Ahuja, M., Oakley, C. E., Entwistle, R., Asokan, A., Zutz, C., et al. (2016). Development of genetic dereplication strains in Aspergillus nidulans results in the discovery of aspercryptin. Angew. Chem. Int. Ed. 55, 1662–1665. doi: 10.1002/anie.201507097
Clevenger, K. D., Bok, J. W., Ye, R., Miley, G. P., Verdan, M. H., Velk, T., et al. (2017). A scalable platform to identify fungal secondary metabolites and their gene clusters. Nat. Chem. Biol. 13, 895–901. doi: 10.1038/nchembio.2408
Collemare, J., and Seidl, M. F. (2019). Chromatin-dependent regulation of secondary metabolite biosynthesis in fungi: is the picture complete? FEMS Microbiol. Rev. 43, 591–607. doi: 10.1093/femsre/fuz018
Cook, D., Beaulieu, W. T., Mott, I. W., Riet-Correa, F., Gardner, D. R., Grum, D., et al. (2013). Production of the alkaloid swainsonine by a fungal endosymbiont of the ascomycete order Chaetothyriales in the host Ipomoea carnea. J. Agric. Food Chem. 61, 3797–3803. doi: 10.1021/np400274n
Cook, D., Donzelli, B. G. G., Creamer, R., Baucom, D. L., Gardner, D. R., Pan, J., et al. (2017). Swainsonine biosynthesis genes in diverse symbiotic and pathogenic fungi. G3 Genes Genomes Genetics 7, 1791–1797. doi: 10.1534/g3.117.041384
Cook, D., Gardner, D. R., and Pfister, J. A. (2014). Swainsonine-containing plants and their relationship to endophytic fungi. J. Agric. Food Chem. 62, 7326–7334. doi: 10.1021/jf501674r
Covington, B. C., McLean, J. A., and Bachmann, B. O. (2017). Comparative mass spectrometry-based metabolomics strategies for the investigation of microbial secondary metabolites. Nat. Prod. Rep. 34, 6–24. doi: 10.1039/c6np00048g
Dam, H. T., Vollmers, J., Sobol, M. S., Cabezas, A., and Kaster, A. K. (2020). Targeted cell sorting combined with single cell genomics captures low abundant microbial dark matter with higher sensitivity than metagenomics. Front. Microbiol. 11:1377. doi: 10.3389/fmicb.2020.01377
De Medeiros, L. S., Abreu, L. M., Nielsen, A., Ingmer, H., Larsen, T. O., Nielsen, K. F., et al. (2015). Dereplication-guided isolation of depsides thielavins S-T and lecanorins D-F from the endophytic fungus Setophoma sp. Phytochemistry 111, 154–162. doi: 10.1016/j.phytochem.2014.12.020
Delaye, L., García-Guzmán, G., and Heil, M. (2013). Endophytes versus biotrophic and necrotrophic pathogens-are fungal lifestyles evolutionarily stable traits? Fungal Divers. 60, 125–135. doi: 10.1007/s13225-013-0240-y
Ding, Z., Zhou, H., Wang, X., Huang, H., Wang, H., Zhang, R., et al. (2020). Deletion of the histone deacetylase HdaA in endophytic fungus Penicillium chrysogenum Fes1701 induces the complex response of multiple bioactive secondary metabolite production and relevant gene cluster expression Zhuang. Molecules 25:3657. doi: 10.3390/molecules25163657
Donia, M. S., Cimermancic, P., Schulze, C. J., Wieland Brown, L. C., Martin, J., Mitreva, M., et al. (2014). A systematic analysis of biosynthetic gene clusters in the human microbiome reveals a common family of antibiotics. Cell 158, 1402–1414. doi: 10.1016/j.cell.2014.08.032
Donovan, P. D., Gonzalez, G., Higgins, D. G., Butler, G., and Ito, K. (2018). Identification of fungi in shotgun metagenomics datasets. PLoS ONE 13:e0192898. doi: 10.1371/journal.pone.0192898
Dubey, A., Malla, M. A., Kumar, A., Dayanandan, S., and Khan, M. L. (2020). Plants endophytes: unveiling hidden agenda for bioprospecting toward sustainable agriculture. Crit. Rev. Biotechnol. 40, 1210–1231. doi: 10.1080/07388551.2020.1808584
Dutreux, F., Da Silva, C., D'agata, L., Couloux, A., Gay, E. J., Istace, B., et al. (2018). Data descriptor: de novo assembly and annotation of three Leptosphaeria genomes using oxford nanopore minion sequencing. Sci. Data 5:180235. doi: 10.1038/sdata.2018.235
Edwards, H. S., Krishnakumar, R., Sinha, A., Bird, S. W., Patel, K. D., and Bartsch, M. S. (2019). Real-time selective sequencing with RUBRIC: read until with basecall and reference-informed criteria. Sci. Rep. 9:11475. doi: 10.1038/s41598-019-47857-3
Eisenman, H. C., Greer, E. M., and McGrail, C. W. (2020). The role of melanins in melanotic fungi for pathogenesis and environmental survival. Appl. Microbiol. Biotechnol. 104, 4247–4257. doi: 10.1007/s00253-020-10532-z
Eisenstein, M. (2020). Enzymatic DNA synthesis enters new phase. Nat. Biotechnol. 38, 1107–1109. doi: 10.1038/s41587-020-0693-y
El-Hawary, S. S., Mohammed, R., Abouzid, S. F., Bakeer, W., Ebel, R., Sayed, A. M., et al. (2016). Solamargine production by a fungal endophyte of Solanum nigrum. J. Appl. Microbiol. 120, 900–911. doi: 10.1111/jam.13077
El-Sayed, A. S. A., Mohamed, N. Z., Safan, S., Yassin, M. A., Shaban, L., Shindia, A. A., et al. (2019). Restoring the taxol biosynthetic machinery of Aspergillus terreus by Podocarpus gracilior pilger microbiome, with retrieving the ribosome biogenesis proteins of WD40 superfamily. Sci. Rep. 9:11534. doi: 10.1038/s41598-019-47816-y
Fill, T. P., Baretta, J. F., de Almeida, L. G. P., Malavazi, I., Cerdeira, L. T., Samborskyy, M., et al. (2018). Draft genome sequence of the fungus Penicillium brasilianum (Strain LaBioMMi 136), a plant endophyte from Melia azedarach. Microbiol. Resour. Announc 7:e01235-18. doi: 10.1128/mra.01235-18
Firrincieli, A., Otillar, R., Salamov, A., Schmutz, J., Khan, Z., Redman, R. S., et al. (2015). Genome sequence of the plant growth promoting endophytic yeast Rhodotorula graminis WP1. Front. Microbiol. 6:978. doi: 10.3389/fmicb.2015.00978
Fukushima, K., Arai, T., Mori, Y., Tsuboi, M., and Suzuki, M. (1983). Studies on peptide antibiotics, leucinostatins I. separation, physicochemical properties and biological activities of leucinostatins A and B. J. Antibiot. 36, 1606–1612. doi: 10.7164/antibiotics.36.1606
Gao, Q., Jin, K., Ying, S. H., Zhang, Y., Xiao, G., Shang, Y., et al. (2011). Genome sequencing and comparative transcriptomics of the model entomopathogenic fungi Metarhizium anisopliae and M. acridum. PLoS Genet. 7:1001264. doi: 10.1371/journal.pgen.1001264
Gazis, R., Kuo, A., Riley, R., LaButti, K., Lipzen, A., Lin, J., et al. (2016). The genome of Xylona heveae provides a window into fungal endophytism. Fungal Biol. 120, 26–42. doi: 10.1016/j.funbio.2015.10.002
Gianoulis, T. A., Griffin, M. A., Spakowicz, D. J., Dunican, B. F., Alpha, C. J., Sboner, A., et al. (2012). Genomic analysis of the hydrocarbon-producing, cellulolytic, endophytic fungus Ascocoryne sarcoides. PLoS Genet. 8:e1002558. doi: 10.1371/journal.pgen.1002558
Goodwin, S., McPherson, J. D., and McCombie, W. R. (2016). Coming of age: ten years of next-generation sequencing technologies. Nat. Rev. Genet. 17, 333–351. doi: 10.1038/nrg.2016.49
Gramaje, D., Berlanas, C., Martínez-Diz, M. D. P., Diaz-Losada, E., Antonielli, L., Beier, S., et al. (2020). Comparative genomic analysis of Dactylonectria torresensis strains from grapevine, soil and weed highlights potential mechanisms in pathogenicity and endophytic lifestyle. J. Fungi 6:255. doi: 10.3390/jof6040255
Guarnaccia, V., Gehrmann, T., Silva-Junior, G. J., Fourie, P. H., Haridas, S., Vu, D., et al. (2019). Phyllosticta citricarpa and sister species of global importance to Citrus. Mol. Plant Pathol. 20, 1619–1635. doi: 10.1111/mpp.12861
Gupta, S., Bhatt, P., and Chaturvedi, P. (2018). Determination and quantification of asiaticoside in endophytic fungus from Centella asiatica (L.) Urban. World J. Microbiol. Biotechnol. 34:111. doi: 10.1007/s11274-018-2493-9
Gupta, S., Chaturvedi, P., Kulkarni, M. G., and Van Staden, J. (2020). A critical review on exploiting the pharmaceutical potential of plant endophytic fungi. Biotechnol. Adv. 39, 107462. doi: 10.1016/j.biotechadv.2019.107462
Gurusinghe, S., Brooks, T. L., Barrow, R. A., Zhu, X., Thotagamuwa, A., Dennis, P. G., et al. (2019). Technologies for the selection, culture and metabolic profiling of unique rhizosphere microorganisms for natural product discovery. Molecules 24:1955. doi: 10.3390/molecules24101955
Harvey, C. J. B., Tang, M., Schlecht, U., Horecka, J., Fischer, C. R., Lin, H., ching, Li, J., et al. (2018). HEx: a heterologous expression platform for the discovery of fungal natural products. Sci. Adv. 4:eaar5459. doi: 10.1126/sciadv.aar5459
Hautbergue, T., Jamin, E. L., Debrauwer, L., Puel, O., and Oswald, I. P. (2018). From genomics to metabolomics, moving toward an integrated strategy for the discovery of fungal secondary metabolites. Nat. Prod. Rep. 35, 147–173. doi: 10.1039/c7np00032d
Heinig, U., Scholz, S., and Jennewein, S. (2013). Getting to the bottom of Taxol biosynthesis by fungi. Fungal Divers. 60, 161–170. doi: 10.1007/s13225-013-0228-7
Hover, B. M., Kim, S. H., Katz, M., Charlop-Powers, Z., Owen, J. G., Ternei, M. A., et al. (2018). Culture-independent discovery of the malacidins as calcium-dependent antibiotics with activity against multidrug-resistant Gram-positive pathogens. Nat. Microbiol. 3, 415–422. doi: 10.1038/s41564-018-0110-1
Jeon, J., Park, S. Y., Kim, J. A., Yu, N. H., Park, A. R., Kim, J. C., et al. (2019). Draft genome sequence of Amphirosellinia nigrospora JS-1675, an endophytic fungus from Pteris cretica. Microbiol. Resour. Announc. 8:e00069-19. doi: 10.1128/MRA.00069-19
JGI (2019). JGI Genome Portal–Home. University of California. Available online at: https://genome.jgi.doe.gov/portal/
Jiang, H., Xu, Z., Aluru, M. R., and Dong, L. (2014). Plant chip for high-throughput phenotyping of Arabidopsis. Lab Chip 14, 1281–1293. doi: 10.1039/c3lc51326b
Kang, X., Liu, C., Shen, P., Hu, L., Lin, R., Ling, J., et al. (2019). Genomic characterization provides new insights into the biosynthesis of the secondary metabolite Huperzine A in the endophyte Colletotrichum gloeosporioides Cg01. Front. Microbiol. 9:3237. doi: 10.3389/fmicb.2018.03237
Kawada, M., Inoue, H., Ohba, S. I., Masuda, T., Momose, I., and Ikeda, D. (2010). Leucinostatin A inhibits prostate cancer growth through reduction of insulin-like growth factor-I expression in prostate stromal cells. Int. J. Cancer 126, 810–818. doi: 10.1002/ijc.24915
Keller, N. P. (2019). Fungal secondary metabolism: regulation, function and drug discovery. Nat. Rev. Microbiol. 17, 167–180. doi: 10.1038/s41579-018-0121-1
Kim, H., Han, H., Ahn, J., Lee, J., Cho, N., Jang, H., et al. (2012). “Shotgun DNA synthesis” for the high-throughput construction of large DNA molecules. Nucleic Acids Res. 40:e140. doi: 10.1093/nar/gks546
Kim, J. A., Jeon, J., Kim, K. T., Choi, G., Park, S. Y., Lee, H. J., et al. (2017a). Draft genome sequence of an endophytic fungus, Gaeumannomyces sp. strain JS-464, isolated from a reed plant, Phragmites communis. Genome Announc. 5:e00734-17. doi: 10.1128/genomeA.00734-17
Kim, J. A., Jeon, J., Park, S., Jeon, J., Yeo, J., and Lee, Y. (2020). Draft genome sequence of Daldinia childiae JS-1345, an endophytic fungus isolated from stem tissue of Korean fir. Microbiol. Resour. Announc. 9:e01284-19. doi: 10.1128/MRA.01284-19
Kim, J. A., Jeon, J., Park, S., Kim, K., Choi, G., Lee, H., et al. (2017b). Genome sequence of an endophytic fungus, Fusarium solani JS-169, which has antifungal activity. Genome Announc. 5:e01071-17. doi: 10.1128/genomeA.01071-17
Kjærbølling, I., Mortensen, U. H., Vesth, T., and Andersen, M. R. (2019). Strategies to establish the link between biosynthetic gene clusters and secondary metabolites. Fungal Genet. Biol. 130, 107–121. doi: 10.1016/j.fgb.2019.06.001
Knapp, D. G., Németh, J. B., Barry, K., Hainaut, M., Henrissat, B., Johnson, J., et al. (2018). Comparative genomics provides insights into the lifestyle and reveals functional heterogeneity of dark septate endophytic fungi. Sci. Rep. 8:6321. doi: 10.1038/s41598-018-24686-4
Kogel, K. H., Franken, P., and Hückelhoven, R. (2006). Endophyte or parasite–what decides? Curr. Opin. Plant Biol. 9, 358–363. doi: 10.1016/j.pbi.2006.05.001
Kovaka, S., Fan, Y., Ni, B., Timp, W., and Schatz, M. C. (2020). Targeted nanopore sequencing by real-time mapping of raw electrical signal with UNCALLED. Nat. Biotechnol. doi: 10.1038/s41587-020-0731-9. [Epub ahead of print].
Kusari, S., Zühlke, S., Košuth, J., Cellárová, E., and Spiteller, M. (2009b). Light-independent metabolomics of endophytic Thielavia subthermophila provides insight into microbial hypericin biosynthesis. J. Nat. Prod. 72, 1825–1835. doi: 10.1021/np9002977
Kusari, S., Zühlke, S., and Spiteller, M. (2009a). An endophytic fungus from Camptotheca acuminata that produces camptothecin and analogues. J. Nat. Prod. 72, 2–7. doi: 10.1021/np800455b
Lai, Y., Liu, K., Zhang, X., Zhang, X., Li, K., Wang, N., et al. (2014). Comparative genomics and transcriptomics analyses reveal divergent lifestyle features of nematode endoparasitic fungus Hirsutella minnesotensis. Genome Biol. Evol. 6, 3077–3093. doi: 10.1093/gbe/evu241
Larriba, E., Jaime, M. D. L. A., Carbonell-Caballero, J., Conesa, A., Dopazo, J., Nislow, C., et al. (2014). Sequencing and functional analysis of the genome of a nematode egg-parasitic fungus, Pochonia chlamydosporia. Fungal Genet. Biol. 65, 69–80. doi: 10.1016/j.fgb.2014.02.002
Lebreton, A., Corre, E., Jany, J. L., Brillet-Guéguen, L., Pèrez-Arques, C., Garre, V., et al. (2020). Comparative genomics applied to Mucor species with different lifestyles. BMC Genomics 21:135. doi: 10.1186/s12864-019-6256-2
Lee, J., Hyeon, D. Y., and Hwang, D. (2020). Single-cell multiomics: technologies and data analysis methods. Exp. Mol. Med. 52, 1428–1442. doi: 10.1038/s12276-020-0420-2
Li, D., Zhao, N., Guo, B. J., Lin, X., Chen, S. L., and Yan, S. Z. (2019). Genetic overexpression increases production of hypocrellin A in Shiraia bambusicola S4201. J. Microbiol. 57, 154–162. doi: 10.1007/s12275-019-8259-8
Li, J. Y., Sidhu, R. S., Ford, E. J., Long, D. M., Hess, W. M., and Strobel, G. A. (1998). The induction of taxol production in the endophytic fungus–Periconia sp from Torreya grandifolia. J. Ind. Microbiol. Biotechnol. 20, 259–264. doi: 10.1038/sj.jim.2900521
Libor, B., Harms, H., Kehraus, S., Egereva, E., Crüsemann, M., and König, G. M. (2019). Isolation of fungi using the diffusion chamber device FIND technology. Beilstein J. Org. Chem. 15, 2191–2203. doi: 10.3762/bjoc.15.216
Lin, Z., Nielsen, J., and Liu, Z. (2020). Bioprospecting through cloning of whole natural product biosynthetic gene clusters. Front. Bioeng. Biotechnol. 8:526. doi: 10.3389/fbioe.2020.00526
Liu, B., Bao, J., Zhang, Z., Yan, R., Wang, Y., Yang, H., et al. (2018). Enhanced production of perylenequinones in the endophytic fungus Shiraia sp. Slf14 by calcium/calmodulin signal transduction. Appl. Microbiol. Biotechnol. 102, 153–163. doi: 10.1007/s00253-017-8602-0
Liu, F., Chen, S., Ferreira, M. A., Chang, R., Sayari, M., Kanzi, A. M., et al. (2019). Draft genome sequences of five Calonectria species from Eucalyptus plantations in China, Celoporthe dispersa, Sporothrix phasma and Alectoria sarmentosa. IMA Fungus 10:22. doi: 10.1186/s43008-019-0023-5
Liu, L., Li, Y., Liu, S., Zheng, Z., Chen, X., Zhang, H., et al. (2009). Chloropestolide A, an antitumor metabolite with an unprecedented spiroketal skeleton from Pestalotiopsis fici. Org. Lett. 11, 2836–2839. doi: 10.1021/ol901039m
Liu, L., Liu, S., Jiang, L., Chen, X., Guo, L., and Che, Y. (2008). Chloropupukeananin, the first chlorinated pupukeanane derivative, and its precursors from Pestalotiopsis fici. Org. Lett. 10, 1397–1400. doi: 10.1021/ol800136t
Liu, Z., Bao, J., Yang, H., Zhang, Z., Yan, R., and Zhu, D. (2020). Transcriptome analysis on fructose as the sole carbon source enhancing perylenequinones production of endophytic fungus Shiraia sp. Slf14. 3 Biotech. 10:190. doi: 10.1007/s13205-020-02181-w
Loose, M., Malla, S., and Stout, M. (2016). Real-time selective sequencing using nanopore technology. Nature Methods 13, 751–754. doi: 10.1038/nmeth.3930
Lu, H., Quan, H., Ren, Z., Wang, S., Xue, R., and Zhao, B. (2016). The Genome of Undifilum oxytropis provides insights into swainsonine biosynthesis and locoism. Sci. Rep. 6:30760. doi: 10.1038/srep30760
Lu, Y., Ye, C., Che, J., Xu, X., Shao, D., Jiang, C., et al. (2019). Genomic sequencing, genome-scale metabolic network reconstruction, and in silico flux analysis of the grape endophytic fungus Alternaria sp. MG1. Microb. Cell Factor. 18:13. doi: 10.1186/s12934-019-1063-7
Luo, C., Fernie, A. R., and Yan, J. (2020). Single-cell genomics and epigenomics: technologies and applications in plants. Trends Plant Sci. 25, 1030–1040. doi: 10.1016/j.tplants.2020.04.016
Luo, F., Hong, S., Chen, B., Yin, Y., Tang, G., Hu, F., et al. (2020). Unveiling of swainsonine biosynthesis via a multibranched pathway in fungi. ACS Chem. Biol. 15, 2476–2484. doi: 10.1021/acschembio.0c00466
Ma, A., McDermaid, A., Xu, J., Chang, Y., and Ma, Q. (2020). Integrative methods and practical challenges for single-cell multi-omics. Trends Biotechnol. 38, 1007–1022. doi: 10.1016/j.tibtech.2020.02.013
Manganyi, M. C., and Ateba, C. N. (2020). Untapped potentials of endophytic fungi: a review of novel bioactive compounds with biological applications. Microorganisms 8:1934. doi: 10.3390/microorganisms8121934
Mao, X. M., Xu, W., Li, D., Yin, W. B., Chooi, Y. H., Li, Y. Q., et al. (2015). Epigenetic genome mining of an endophytic fungus leads to the pleiotropic biosynthesis of natural products. Angew. Chem. Int. Ed. 54, 7592–7596. doi: 10.1002/anie.201502452
Masutani, B., and Morishita, S. (2019). A framework and an algorithm to detect low-abundance DNA by a handy sequencer and a palm-sized computer. Bioinformatics 35, 584–592. doi: 10.1093/bioinformatics/bty663
Matthes, D., Richter, L., Müller, J., Denisiuk, A., Feifel, S. C., Xu, Y., et al. (2012). In vitro chemoenzymatic and in vivo biocatalytic syntheses of new beauvericin analogues. Chem. Commun. 48, 5674–5676. doi: 10.1039/c2cc31669b
McKenzie, S. K., Walston, R. F., and Allen, J. L. (2020). Complete, high-quality genomes from long-read metagenomic sequencing of two wolf lichen thalli reveals enigmatic genome architecture. Genomics 112, 3150–3156. doi: 10.1016/j.ygeno.2020.06.006
Medema, M. H., Blin, K., Cimermancic, P., De Jager, V., Zakrzewski, P., Fischbach, M. A., et al. (2011). AntiSMASH: rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res. 39, W330–W346. doi: 10.1093/nar/gkr466
Meena, N., Vasundhara, M., Reddy, M. S., Suravajhala, P., Raghavender, U. S., and Medicherla, K. M. (2018). Draft genome sequence of a fungus (Fusarium tricinctum) cultured from a monoisolate native to the Himalayas. Genome Announc. 6:e00365-18. doi: 10.1128/genomeA.00365-18
Mefteh, F. Ben, D. A., Bouket, A. C., Thissera, B., Kadri, Y., Cherif-Silini, H., Eshelli, M., et al. (2018). Date palm trees root-derived endophytes as fungal cell factories for diverse bioactive metabolites. Int. J. Mol. Sci. 19:1986. doi: 10.3390/ijms19071986
Mei, Y. Z., Zhu, Y. L., Huang, P. W., Yang, Q., and Dai, C. C. (2019). Strategies for gene disruption and expression in filamentous fungi. Appl. Microbiol. Biotechnol. 103, 6041–6059. doi: 10.1007/s00253-019-09953-2
Meier, M., Lucchetta, E. M., and Ismagilov, R. F. (2010). Chemical stimulation of the Arabidopsis thaliana root using multi-laminar flow on a microfluidic chip. Lab Chip 10, 2147–2153. doi: 10.1039/c004629a
Meng, L., Sun, P., Tang, H., Li, L., Draeger, S., Schulz, B., et al. (2011). Endophytic fungus Penicillium chrysogenum, a new source of hypocrellins. Biochem. Syst. Ecol. 39, 163–165. doi: 10.1016/j.bse.2011.02.003
Millet, L. J., Aufrecht, J., Labbé, J., Uehling, J., Vilgalys, R., Estes, M. L., et al. (2019). Increasing access to microfluidics for studying fungi and other branched biological structures. Fungal Biol. Biotechnol. 6:8. doi: 10.1186/s40694-019-0071-z
Mirdita, M., Steinegger, M., Breitwieser, F., Söding, J., and Levy Karin, E. (2020). Fast and sensitive taxonomic assignment to metagenomic contigs. bioRxiv. doi: 10.1101/2020.11.27.401018
Montoliu-Nerin, M., Sánchez-García, M., Bergin, C., Grabherr, M., Ellis, B., Kutschera, V. E., et al. (2020). Building de novo reference genome assemblies of complex eukaryotic microorganisms from single nuclei. Sci. Rep. 10:1303. doi: 10.1038/s41598-020-58025-3
Moss, E. L., Maghini, D. G., and Bhatt, A. S. (2020). Complete, closed bacterial genomes from microbiomes using nanopore sequencing. Nat. Biotechnol. 38, 701–707. doi: 10.1038/s41587-020-0422-6
Nakagawa, A., Matsumura, E., Koyanagi, T., Katayama, T., Kawano, N., Yoshimatsu, K., et al. (2016). Total biosynthesis of opiates by stepwise fermentation using engineered Escherichia coli. Nat. Commun. 7:10390. doi: 10.1038/ncomms10390
Newman, D. J., and Cragg, G. M. (2020a). Natural products as sources of new drugs over the nearly four decades from 01/1981 to 09/2019. J. Nat. Prod. 83, 770–803. doi: 10.1021/acs.jnatprod.9b01285
Newman, D. J., and Cragg, G. M. (2020b). Plant endophytes and epiphytes: burgeoning sources of known and unknown cytotoxic and antibiotic agents? Planta Med. 86, 891–905. doi: 10.1055/a-1095-1111
Nguyen, H. T., Kim, S., Yu, N. H., Park, A. R., Yoon, H., Bae, C. H., et al. (2019). Antimicrobial activities of an oxygenated cyclohexanone derivative isolated from Amphirosellinia nigrospora JS-1675 against various plant pathogenic bacteria and fungi. J. Appl. Microbiol. 126, 894–904. doi: 10.1111/jam.14138
Nicholson, M. J., Van de Bittner, K. C., Ram, A., Bustamante, L. Y., Scott, B., and Parker, E. J. (2018). Draft genome sequence of the filamentous fungus Hypoxylon pulicicidum ATCC 74245. Genome Announc. 6:e01380-17. doi: 10.1128/genomeA.01380-17
Nielsen, J. C., Grijseels, S., Prigent, S., Ji, B., Dainat, J., Nielsen, K. F., et al. (2017). Global analysis of biosynthetic gene clusters reveals vast potential of secondary metabolite production in Penicillium species. Nat. Microbiol. 2:17044. doi: 10.1038/nmicrobiol.2017.44
Noirot-Gros, M. F., Shinde, S. V., Akins, C., Johnson, J. L., Zerbs, S., Wilton, R., et al. (2020). Functional imaging of microbial interactions with tree roots using a microfluidics setup. Front. Plant Sci. 11:408. doi: 10.3389/fpls.2020.00408
Noor, A. I., Neyaz, M., Cook, D., and Creamer, R. (2020). Molecular characterization of a fungal ketide synthase gene among swainsonine-producing Alternaria species in the USA. Curr. Microbiol. 77, 2554–2563. doi: 10.1007/s00284-020-02111-2
Oldrup, E., Mclain-Romero, J., Padilla, A., Moya, A., Gardner, D., and Creamer, R. (2010). Localization of endophytic Undifilum fungi in locoweed seed and influence of environmental parameters on a locoweed in vitro culture system. Botany 88, 512–521. doi: 10.1139/B10-026
Ouedraogo, J. P., and Tsang, A. (2020). CRISPR_Cas systems for fungal research. Fungal Biol. Rev. 34, 189–201. doi: 10.1016/j.fbr.2020.10.002
Pachiadaki, M. G., Brown, J. M., Brown, J., Bezuidt, O., Berube, P. M., Biller, S. J., et al. (2019). Charting the complexity of the marine microbiome through single-cell genomics. Cell 179, 1623–1635. doi: 10.1016/j.cell.2019.11.017
Pan, R., Bai, X., Chen, J., Zhang, H., and Wang, H. (2019). Exploring structural diversity of microbe secondary metabolites using OSMAC strategy: a literature review. Front. Microbiol. 10:294. doi: 10.3389/fmicb.2019.00294
Pan, Y., Liu, L., Guan, F., Li, E., Jin, J., Li, J., et al. (2018). Characterization of a prenyltransferase for Iso-A82775C biosynthesis and generation of new congeners of chloropestolides. ACS Chem. Biol. 13, 703–711. doi: 10.1021/acschembio.7b01059
Parmar, S., Li, Q., Wu, Y., Li, X., Yan, J., Sharma, V. K., et al. (2018). Endophytic fungal community of Dysphania ambrosioides from two heavy metal-contaminated sites: evaluated by culture-dependent and culture-independent approaches. Microb. Biotechnol. 11, 1170–1183. doi: 10.1111/1751-7915.13308
Payne, A., Holmes, N., Clarke, T., Munro, R., Debebe, B. J., and Loose, M. (2020). Readfish enables targeted nanopore sequencing of gigabase-sized genomes. Nat. Biotechnol. doi: 10.1038/s41587-020-00746-x. [Epub ahead of print].
Pérez-Cobas, A. E., Gomez-Valero, L., and Buchrieser, C. (2020). Metagenomic approaches in microbial ecology: an update on whole-genome and marker gene sequencing analyses. Microb. Genomics 6, 1–22. doi: 10.1099/mgen.0.000409
Pohl, C., Kiel, J. A. K. W., Driessen, A. J. M., Bovenberg, R. A. L., and Nygård, Y. (2016). CRISPR/Cas9 based genome editing of Penicillium chrysogenum. ACS Synth. Biol. 5, 754–764. doi: 10.1021/acssynbio.6b00082
Pohl, C., Polli, F., Schütze, T., Viggiano, A., Mózsik, L., Jung, S., et al. (2020). A Penicillium rubens platform strain for secondary metabolite production. Sci. Rep. 10:7630. doi: 10.1038/s41598-020-64893-6
Pramastya, H., Xue, D., Abdallah, I. I., Setroikromo, R., and Quax, W. J. (2021). High level production of amorphadiene using Bacillus subtilis as an optimized terpenoid cell factory. New Biotechnol. 60, 159–167. doi: 10.1016/j.nbt.2020.10.007
Pryor, B. M., Creamer, R., Shoemaker, R. A., McLain-Romero, J., and Hambleton, S. (2009). Undifilum, a new genus for endophytic Embellisia oxytropis and parasitic Helminthosporium bornmuelleri on legumes. Botany 87, 178–194. doi: 10.1139/B08-130
Qiao, W., Tang, T., and Ling, F. (2020). Comparative transcriptome analysis of a taxol-producing endophytic fungus, Aspergillus aculeatinus Tax-6, and its mutant strain. Sci. Rep. 10:10558. doi: 10.1038/s41598-020-67614-1
Qiao, Y. M., Yu, R. L., and Zhu, P. (2019). Advances in targeting and heterologous expression of genes involved in the synthesis of fungal secondary metabolites. RSC Adv. 9, 35124–35134. doi: 10.1039/c9ra06908a
Rashmi, M., Kushveer, J. S., and Sarma, V. V. (2019). A worldwide list of endophytic fungi with notes on ecology and diversity. Mycosphere 10, 798–1079. doi: 10.5943/mycosphere/10/1/19
Richards, T. A., Leonard, G., Soanes, D. M., and Talbot, N. J. (2011). Gene transfer into the fungi. Fungal Biol. Rev. 25, 98–110. doi: 10.1016/j.fbr.2011.04.003
Richards, T. A., Soanes, D. M., Foster, P. G., Leonard, G., Thornton, C. R., and Talbot, N. J. (2009). Phylogenomic analysis demonstrates a pattern of rare and ancient horizontal gene transfer between plants and fungi. Plant Cell 21, 1897–1911. doi: 10.1105/tpc.109.065805
Rodrigues, C. M., Takita, M. A., Silva, N. V., Ribeiro-Alves, M., and MacHado, M. A. (2019). Comparative genome analysis of Phyllosticta citricarpa and Phyllosticta capitalensis, two fungi species that share the same host. BMC Genomics 20:554. doi: 10.1186/s12864-019-5911-y
Rogério, F., Boufleur, T. R., Ciampi-Guillardi, M., Sukno, S. A., Thon, M. R., Massola Júnior, N. S., et al. (2020). Genome sequence resources of Colletotrichum truncatum, C. plurivorum, C. musicola, and C. sojae: four species pathogenic to soybean (Glycine max). Phytopathology 110, 1497–1499. doi: 10.1094/PHYTO-03-20-0102-A
Sakai, K., Charlot, F., Saux, T., Le, B. S., Nogué, F., Palauqui, J. C., et al. (2019). Design of a comprehensive microfluidic and microscopic toolbox for the ultra-wide spatio-temporal study of plant protoplasts development and physiology. Plant Methods 15:79. doi: 10.1186/s13007-019-0459-z
Salazar-Cerezo, S., Martínez-Montiel, N., García-Sánchez, J., Pérez-y-Terrón, R., and Martínez-Contreras, R. D. (2018). Gibberellin biosynthesis and metabolism: a convergent route for plants, fungi and bacteria. Microbiol. Res. 208, 85–98. doi: 10.1016/j.micres.2018.01.010
Santos, C. D., Sarmento, H., de Miranda, F. P., Henrique-Silva, F., and Logares, R. (2020). Uncovering the gene machinery of the Amazon River microbiome to degrade rainforest organic matter. Microbiome 8:151. doi: 10.1101/585562
Santos, F. M., Latorre, A. O., Hueza, I. M., Sanches, D. S., Lippi, L. L., Gardner, D. R., et al. (2011). Increased antitumor efficacy by the combined administration of swainsonine and cisplatin in vivo. Phytomedicine 18, 1096–1101. doi: 10.1016/j.phymed.2011.06.005
Scherlach, K., and Hertweck, C. (2009). Triggering cryptic natural product biosynthesis in microorganisms. Org. Biomol. Chem. 7, 1753–1760. doi: 10.1039/b821578b
Schmidt-Heydt, M., Stoll, D., and Geisen, R. (2019). Whole-genome sequencing of the fungus Penicillium citrinum reveals the biosynthesis gene cluster for the mycotoxin citrinin. Microbiol. Resour. Announc. 8, 1419–1437. doi: 10.1128/mra.01419-18
Schorn, M. A., Jordan, P. A., Podell, S., Blanton, J. M., Agarwal, V., Biggs, J. S., et al. (2019). Comparative genomics of cyanobacterial symbionts reveals distinct, specialized metabolism in tropical Dysideidae sponges. mBio 10:e00821-19. doi: 10.1128/mBio.00821-19
Schulz, B., and Boyle, C. (2005). The endophytic continuum. Mycol. Res. 109, 661–686. doi: 10.1017/S095375620500273X
Schuster, M., and Kahmann, R. (2019). CRISPR-Cas9 genome editing approaches in filamentous fungi and oomycetes. Fungal Genet. Biol. 130, 43–53. doi: 10.1016/j.fgb.2019.04.016
Seeleuthner, Y., Mondy, S., Lombard, V., Carradec, Q., Pelletier, E., Wessner, M., et al. (2018). Single-cell genomics of multiple uncultured stramenopiles reveals underestimated functional diversity across oceans. Nat. Commun. 9:310. doi: 10.1038/s41467-017-02235-3
Segawa, T., Takeuchi, N., Mori, H., Rathnayake, R. M. L. D., Li, Z., Akiyoshi, A., et al. (2020). Redox stratification within cryoconite granules influences the nitrogen cycle on glaciers. FEMS Microbiol. Ecol. 96:fiaa199. doi: 10.1093/femsec/fiaa199
Shaw, J. J., Berbasova, T., Sasaki, T., Jefferson-George, K., Spakowicz, D. J., Dunican, B. F., et al. (2015). Identification of a fungal 1,8-cineole synthase from Hypoxylon sp. with specificity determinants in common with the plant synthases. J. Biol. Chem. 290, 8511–8526. doi: 10.1074/jbc.M114.636159
Shi, J., Zeng, Q., Liu, Y., and Pan, Z. (2012). Alternaria sp. MG1, a resveratrol-producing fungus: isolation, identification, and optimal cultivation conditions for resveratrol production. Appl. Microbiol. Biotechnol. 95, 369–379. doi: 10.1007/s00253-012-4045-9
Shweta, S., Zuehlke, S., Ramesha, B. T., Priti, V., Mohana Kumar, P., Ravikanth, G., et al. (2010). Endophytic fungal strains of Fusarium solani, from Apodytes dimidiata E. Mey. ex Arn (Icacinaceae) produce camptothecin, 10-hydroxycamptothecin and 9-methoxycamptothecin. Phytochemistry 71, 117–122. doi: 10.1016/j.phytochem.2009.09.030
Skinnider, M. A., Merwin, N. J., Johnston, C. W., and Magarvey, N. A. (2017). PRISM 3: Expanded prediction of natural product chemical structures from microbial genomes. Nucleic Acids Res. 45, W49–W54. doi: 10.1093/nar/gkx320
Sobreira, A. C. M., Pinto, F. das C. L., Florêncio, K. G. D., Wilke, D. V., Staats, C. C., Streit, R. de, A. S., et al. (2018). Endophytic fungus Pseudofusicoccum stromaticum produces cyclopeptides and plant-related bioactive rotenoids. RSC Adv. 8, 35575–35586. doi: 10.1039/C8RA06824K
Song, R., Zhai, Q., Sun, L., Huang, E., Zhang, Y., Zhu, Y., et al. (2019). CRISPR/Cas9 genome editing technology in filamentous fungi: progress and perspective. Appl. Microbiol. Biotechnol. 103, 6919–6932. doi: 10.1007/s00253-019-10007-w
Staniek, A., Woerdenbag, H. J., and Kayser, O. (2009). Taxomyces andreanae: a presumed paclitaxel producer demystified? Planta Med. 75, 1561–1566. doi: 10.1055/s-0029-1186181
Steinegger, M., and Salzberg, S. L. (2020). Terminating contamination: large-scale search identifies more than 2,000,000 contaminated entries in GenBank. Genome Biol. 21:115. doi: 10.1186/s13059-020-02023-1
Steinegger, M., and Söding, J. (2017). MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 35, 1026–1028. doi: 10.1038/nbt.3988
Stierle, A., and Strobel, G. S. D. (1993). Taxol and taxane production by Taxomyces andreanae, an endophytic fungus of Pacific yew. Science 260, 214–216. doi: 10.1007/s40011-017-0924-2
Strobel, G. A. (2003). Endophytes as sources of bioactive products. Microbes Infect. 5, 535–544. doi: 10.1016/S1286-4579(03)00073-X
Stuart, T., and Satija, R. (2019). Integrative single-cell analysis. Nat. Rev. Genet. 20, 257–272. doi: 10.1038/s41576-019-0093-7
Sugimoto, Y., Camacho, F. R., Wang, S., Chankhamjon, P., Odabas, A., Biswas, A., et al. (2019). A metagenomic strategy for harnessing the chemical repertoire of the human microbiome. Science 366:eaax9176. doi: 10.1126/science.aax9176
Sun, X., Wang, N., Li, P., Jiang, Z., Liu, X., Wang, M., et al. (2020). Endophytic fungus Falciphora oryzae promotes lateral root growth by producing indole derivatives after sensing plant signals. Plant Cell Environ. 43, 358–373. doi: 10.1111/pce.13667
Tawfike, A. F., Abbott, G., Young, L., and Edrada-Ebel, R. (2018). Metabolomic-guided isolation of bioactive natural products from Curvularia sp, an endophytic fungus of Terminalia laxiflora. Planta Med. 84, 182–190. doi: 10.1055/s-0043-118807
Toghueo, R. M. K., Sahal, D., and Boyom, F. F. (2020). Recent advances in inducing endophytic fungal specialized metabolites using small molecule elicitors including epigenetic modifiers. Phytochemistry 174:112338. doi: 10.1016/j.phytochem.2020.112338
Tomsheck, A. R., Strobel, G. A., Booth, E., Geary, B., Spakowicz, D., Knighton, B., et al. (2010). Hypoxylon sp., an endophyte of Persea indica, producing 1,8-cineole and other bioactive volatiles with fuel potential. Microb. Ecol. 60, 903–914. doi: 10.1007/s00248-010-9759-6
Tulsook, K., Isarangkul, D., Sriubolmas, N., Kittakoop, P., and Wiyakrutta, S. (2020). Draft genome sequence of Diaporthe sp. strain HANT25, an endophytic fungus producing mycoepoxydiene. Microbiol. Resour. Announc. 9:e00805-20. doi: 10.1128/mra.00805-20
Turbyville, T. J., Wijeratne, E. M. K., Liu, M. X., Burns, A. M., Seliga, C. J., Luevano, L. A., et al. (2006). Search for Hsp90 inhibitors with potential anticancer activity: isolation and SAR studies of radicicol and monocillin I from two plant-associated fungi of the Sonoran desert. J. Nat. Prod. 69, 178–184. doi: 10.1021/np058095b
Tyler, A. D., Mataseje, L., Urfano, C. J., Schmidt, L., Antonation, K. S., Mulvey, M. R., et al. (2018). Evaluation of Oxford Nanopore's MinION sequencing device for microbial whole genome sequencing applications. Sci. Rep. 8:10931. doi: 10.1038/s41598-018-29334-5
Ullah, M., Xia, L., Xie, S., and Sun, S. (2020). CRISPR/Cas9-based genome engineering: a new breakthrough in the genetic manipulation of filamentous fungi. Biotechnol. Appl. Biochem. 67, 835–851. doi: 10.1002/bab.2077
Van De Bittner, K. C., Nicholson, M. J., Bustamante, L. Y., Kessans, S. A., Ram, A., Van Dolleweerd, C. J., et al. (2018). Heterologous biosynthesis of nodulisporic acid F. J. Am. Chem. Soc. 140, 582–585. doi: 10.1021/jacs.7b10909
van der Hooft, J. J. J., Mohimani, H., Bauermeister, A., Dorrestein, P. C., Duncan, K. R., and Medema, M. H. (2020). Linking genomics and metabolomics to chart specialized metabolic diversity. Chem. Soc. Rev. 49, 3297–3314. doi: 10.1039/d0cs00162g
van der Lee, T. A. J., and Medema, M. H. (2016). Computational strategies for genome-based natural product discovery and engineering in fungi. Fungal Genet. Biol. 89, 29–36. doi: 10.1016/j.fgb.2016.01.006
Vasanthakumari, M. M., Jadhav, S. S., Sachin, N., Vinod, G., Shweta, S., Manjunatha, B. L., et al. (2015). Restoration of camptothecine production in attenuated endophytic fungus on re-inoculation into host plant and treatment with DNA methyltransferase inhibitor. World J. Microbiol. Biotechnol. 31, 1629–1639. doi: 10.1007/s11274-015-1916-0
Vogl, A. (1898). Mehl Und Die Anderen Mehlprodukte Der Cerealien Und Leguminosen. Z Nahrungsmittel Untersuchung Hyg Warenkunde 12, 25–29.
Walker, A. K., Frasz, S. L., Seifert, K. A., Miller, J. D., Mondo, S. J., LaButti, K., et al. (2016). Full genome of Phialocephala scopiformis DAOMC 229536, a fungal endophyte of spruce producing the potent anti-insectan compound rugulosin. Genome Announc. 4:e01768-15. doi: 10.1128/genomeA.00510-16
Walker, M. C., Eslami, S. M., Hetrick, K. J., Ackenhusen, S. E., Mitchell, D. A., and Van Der Donk, W. A. (2020). Precursor peptide-targeted mining of more than one hundred thousand genomes expands the lanthipeptide natural product family. BMC Genomics 21:387. doi: 10.1186/s12864-020-06785-7
Wang, G., Liu, Z., Lin, R., Li, E., Mao, Z., Ling, J., et al. (2016). Biosynthesis of antibiotic leucinostatins in bio-control fungus Purpureocillium lilacinum and their inhibition on Phytophthora revealed by genome mining. PLoS Pathog. 12:e1005685. doi: 10.1371/journal.ppat.1005685
Wang, Q., Cobine, P. A., and Coleman, J. J. (2018). Efficient genome editing in Fusarium oxysporum based on CRISPR/Cas9 ribonucleoprotein complexes. Fungal Genet. Biol. 117, 21–29. doi: 10.1016/j.fgb.2018.05.003
Wang, S., Xu, Y., Maine, E. A., Wijeratne, E. M. K., Espinosa-Artiles, P., Gunatilaka, A. A. L., et al. (2008). Functional characterization of the biosynthesis of radicicol, an Hsp90 inhibitor resorcylic acid lactone from Chaetomium chiversii. Chem. Biol. 15, 1328–1338. doi: 10.1016/j.chembiol.2008.10.006
Wang, W. G., Du, L. Q., Sheng, S. L., Li, A., Li, Y. P., Cheng, G. G., et al. (2019). Genome mining for fungal polyketide-diterpenoid hybrids: discovery of key terpene cyclases and multifunctional P450s for structural diversification. Org. Chem. Front. 6, 571–578. doi: 10.1039/c8qo01124a
Wang, X., Zhang, X., Liu, L., Xiang, M., Wang, W., Sun, X., et al. (2015). Genomic and transcriptomic analysis of the endophytic fungus Pestalotiopsis fici reveals its lifestyle and high potential for synthesis of natural products. BMC Genomics 16:28. doi: 10.1186/s12864-014-1190-9
Weber, T., and Kim, H. U. (2016). The secondary metabolite bioinformatics portal: computational tools to facilitate synthetic biology of secondary metabolite production. Synth. Syst. Biotechnol. 1, 69–79. doi: 10.1016/j.synbio.2015.12.002
Wei, Q., Bai, J., Yan, D., Bao, X., Li, W., Liu, B., et al. (2020). Genome mining combined metabolic shunting and OSMAC strategy of an endophytic fungus leads to the production of diverse natural products. Acta Pharma. Sin. B 11, 572–587. doi: 10.1016/j.apsb.2020.07.020
Woyke, T., Doud, D. F. R., and Schulz, F. (2017). The trajectory of microbial single-cell sequencing. Nat. Methods 14, 1045–1054. doi: 10.1038/nmeth.4469
Wu, B., Hussain, M., Zhang, W., Stadler, M., Liu, X., and Xiang, M. (2019). Current insights into fungal species diversity and perspective on naming the environmental DNA sequences of fungi. Mycology 10, 127–140. doi: 10.1080/21501203.2019.1614106
Wu, G., Zhou, H., Zhang, P., Wang, X., Li, W., Zhang, W., et al. (2016). Polyketide production of pestaloficiols and macrodiolide ficiolides revealed by manipulations of epigenetic regulators in an endophytic fungus. Org. Lett. 18, 1832–1835. doi: 10.1021/acs.orglett.6b00562
Wu, J. C., Hou, Y., Xu, Q., Jin, X. J., Chen, Y., Fang, J., et al. (2019). (±)-Alternamgin, a pair of enantiomeric polyketides, from the endophytic fungi Alternaria sp. MG1. Org. Lett. 21, 1551–1554. doi: 10.1021/acs.orglett.9b00475
Wu, W., Davis, R. W., Tran-Gyamfi, M. B., Kuo, A., LaButti, K., Mihaltcheva, S., et al. (2017). Characterization of four endophytic fungi as potential consolidated bioprocessing hosts for conversion of lignocellulose into advanced biofuels. Appl. Microbiol. Biotechnol. 101, 2603–2618. doi: 10.1007/s00253-017-8091-1
Xie, Y., Zhang, W., Li, Y., Wang, M., Cerny, R. L., Shen, Y., et al. (2011). Transformation of Fusarium verticillioides with a polyketide gene cluster isolated from a fungal endophyte activates the biosynthesis of fusaric acid. Mycology 2, 24–29. doi: 10.1080/21501203.2011.554907
Xu, X., Liu, L., Zhang, F., Wang, W., Li, J., Guo, L., et al. (2014). Identification of the first diphenyl ether gene cluster for pestheic acid biosynthesis in plant endophyte Pestalotiopsis fici. ChemBioChem 15, 284–292. doi: 10.1002/cbic.201300626
Xu, X. H., Su, Z. Z., Wang, C., Kubicek, C. P., Feng, X. X., Mao, L. J., et al. (2014). The rice endophyte Harpophora oryzae genome reveals evolution from a pathogen to a mutualistic endophyte. Sci. Rep. 4:5783. doi: 10.1038/srep05783
Yan, H., Sun, L., Huang, J., Qiu, Y., Xu, F., Yan, R., et al. (2018). Identification and heterologous reconstitution of a 5-alk(en)ylresorcinol synthase from endophytic fungus Shiraia sp. Slf14. J. Microbiol. 56, 805–812. doi: 10.1007/s12275-018-8278-x
Yang, H., Wang, Y., Zhang, Z., Yan, R., and Zhu, D. (2014). Whole-genome shotgun assembly and analysis of the genome of Shiraia sp. strain Slf14, a novel endophytic fungus producing huperzine A and hypocrellin A. Genome Announc. 2:e00011-14. doi: 10.1128/genomeA.e00011-14
Yang, H., Xiao, C., Ma, W., and He, G. (2009). The production of hypocrellin colorants by submerged cultivation of the medicinal fungus Shiraia bambusicola. Dyes Pigments 82, 142–146. doi: 10.1016/j.dyepig.2008.12.012
Yang, X. L., Huang, L., and Ruan, X. L. (2014). Epigenetic modifiers alter the secondary metabolite composition of a plant endophytic fungus, Pestalotiopsis crassiuscula obtained from the leaves of Fragaria chiloensis. J. Asian Nat. Prod. Res. 16, 412–417. doi: 10.1080/10286020.2014.881356
Yang, Y., Liu, X., Cai, J., Chen, Y., Li, B., Guo, Z., et al. (2019). Genomic characteristics and comparative genomics analysis of the endophytic fungus Sarocladium brachiariae. BMC Genomics 20:782. doi: 10.1186/s12864-019-6095-1
Yang, Y., Zhao, H., Barrero, R. A., Zhang, B., Sun, G., Wilson, I. W., et al. (2014). Genome sequencing and analysis of the paclitaxel-producing endophytic fungus Penicillium aurantiogriseum NRRL 62431. BMC Genomics 15:69. doi: 10.1186/1471-2164-15-69
Ye, S. H., Siddle, K. J., Park, D. J., and Sabeti, P. C. (2019). Benchmarking metagenomics tools for taxonomic classification. Cell 178, 779–794. doi: 10.1016/j.cell.2019.07.010
Youngblut, N. D., de la Cuesta-Zuluaga, J., Reischer, G. H., Dauser, S., Schuster, N., Walzer, C., et al. (2020). Large-scale metagenome assembly reveals novel animal-associated microbial genomes, biosynthetic gene clusters, and other genetic diversity. mSystems 5:e01045-20. doi: 10.1128/msystems.01045-20
Zhang, P., Wang, X., Fan, A., Zheng, Y., Liu, X., Wang, S., et al. (2017). A cryptic pigment biosynthetic pathway uncovered by heterologous expression is essential for conidial development in Pestalotiopsis fici. Mol. Microbiol. 105, 469–483. doi: 10.1111/mmi.13711
Zhang, P., Zhou, S., Wang, G., An, Z., Liu, X., Li, K., et al. (2019). Two transcription factors cooperatively regulate DHN melanin biosynthesis and development in Pestalotiopsis fici. Mol. Microbiol. 112, 649–666. doi: 10.1111/mmi.14281
Zhang, Q., Xue, X. Z., Miao, S. M., Cui, J. L., and Qin, X. M. (2020). Differential relationship of fungal endophytic communities and metabolic profiling in the stems and roots of Ephedra sinica based on metagenomics and metabolomics. Symbiosis 81, 115–125. doi: 10.1007/s13199-020-00685-w
Zhao, N., Lin, X., Qi, S. S., Luo, Z. M., Chen, S. L., and Yan, S. Z. (2016). De novo transcriptome assembly in Shiraia bambusicola to investigate putative genes involved in the biosynthesis of hypocrellin A. Int. J. Mol. Sci. 17:311. doi: 10.3390/ijms17030311
Zheng, Y., Ma, K., Lyu, H., Huang, Y., Liu, H., Liu, L., et al. (2017). Genetic manipulation of the COP9 signalosome subunit PfCsnE leads to the discovery of pestaloficins in Pestalotiopsis fici. Org. Lett. 19, 4700–4703. doi: 10.1021/acs.orglett.7b02346
Zheng, Yi, Su, T., and Qi, Q. (2019). Microbial CRISPRi and CRISPRa systems for metabolic engineering. Biotechnol. Bioprocess Eng. 24, 579–591. doi: 10.1007/s12257-019-0107-5
Zhou, S., Zhang, P., Zhou, H., Liu, X., Li, S. M., Guo, L., et al. (2019). A new regulator RsdA mediating fungal secondary metabolism has a detrimental impact on asexual development in Pestalotiopsis fici. Environ. Microbiol. 21, 416–426. doi: 10.1111/1462-2920.14473
Zhou, W., Le, J., Chen, Y., Cai, Y., Hong, Z., and Chai, Y. (2019). Recent advances in microfluidic devices for bacteria and fungus research. Trends Anal. Chem. 112, 175–195. doi: 10.1016/j.trac.2018.12.024
Ziemert, N., Alanjary, M., and Weber, T. (2016). The evolution of genome mining in microbes-a review. Nat. Prod. Rep. 33, 988–1005. doi: 10.1039/c6np00025h
Zobel, S., Kumpfmüller, J., Süssmuth, R. D., and Schweder, T. (2015). Bacillus subtilis as heterologous host for the secretory production of the non-ribosomal cyclodepsipeptide enniatin. Appl. Microbiol. Biotechnol. 99, 681–691. doi: 10.1007/s00253-014-6199-0
Keywords: genome mining, biosynthetic gene cluster, biosynthetic pathway elucidation, culture-dependent, culture-independent, secondary metabolite discovery
Citation: Sagita R, Quax WJ and Haslinger K (2021) Current State and Future Directions of Genetics and Genomics of Endophytic Fungi for Bioprospecting Efforts. Front. Bioeng. Biotechnol. 9:649906. doi: 10.3389/fbioe.2021.649906
Received: 05 January 2021; Accepted: 16 February 2021;
Published: 15 March 2021.
Edited by:
Adeline Su Yien Ting, Monash University Malaysia, MalaysiaReviewed by:
Nicholas Oberlies, University of North Carolina at Greensboro, United StatesCopyright © 2021 Sagita, Quax and Haslinger. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kristina Haslinger, ay5oYXNsaW5nZXJAcnVnLm5s
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.