 Ryosuke Kurauchi
Ryosuke Kurauchi Hiroi Kasai3
Hiroi Kasai3- 1Department of Human Health Science, School of Medicine, Kyoto University, Kyoto, Japan
- 2School of Medicine, Hokkaido University, Sapporo, Japan
- 3Institute for Advancement of Clinical and Translational Science, Kyoto University Hospital, Kyoto, Japan
Medical products comprising human cells, genes, and tissues have been developed for clinical applications worldwide, and their developmental environment has been established. These products can be imported and exported, but marketing authorization regulations are complicated among regions. This investigation was conducted to identify the characteristics of medical products comprising human cells, genes, and tissues. We used website data, books from survey companies, and reports from public agencies to conduct two investigations. We used website data to conduct a general information survey of 143 cell-therapy and gene-therapy products sold in 24 countries and public assessment reports to individually survey non-clinical and clinical developments of 18 cell-therapy and gene-therapy products developed in Japan and the European Union (EU). The first survey revealed that the numbers of products used in orthopedic surgery and dermatology have increased since 2000, and the numbers of hematological products have increased since 2011. The second investigation revealed that fewer orphaned products were developed in Japan than in the EU. The most appropriate dose was 1.2 × 108 cells per injection per adult. Clinical trials to determine the most appropriate dose were conducted in the EU but not in Japan. No non-clinical immunogenicity tests for autogenous products were conducted in Japan or the EU. Pharmacokinetics tests were not individually performed for sheet-form products. Both in vivo and in vitro pharmacological tests were more likely to be conducted in the EU, while only one or the other was conducted in Japan. Furthermore, in Japan, carcinogenicity tests were performed based on non-clinical technical guidance, while in the EU, these tests were determined according to each product's features. Fewer clinical trials were performed, and fewer subjects per product were used in Japan than in the EU. Many aspects of the clinical and non-clinical development of medical products comprising human cells, genes, and tissues differ between Japan and the EU. Analyzing these differences will enable the safe and rapid distribution of these products to clinical sites.
Introduction
Medical products comprising human cells, genes, and tissues (including products approved as transplants and medicinal devices in the past) have been developed for clinical applications worldwide, and their developmental environment has been established (Kondo et al., 2017). From 2014 to 2017, 45 of 143 product developments (31%) were suspended, and half of those were due to plan reformation (BB-Bridge, Inc, 2017). Although plans are frequently reformed when developing new therapies such as cell-based and gene-based therapies, proper development plans that reduce reformation are important for accelerating medical product development. Hanna et al. and Maciulaitis et al. found that most advanced therapy medicinal products (ATMPs), i.e., medical products comprising human cells, genes, and tissues in the European Union (EU), are developed by non-commercial sponsors and small- and medium-sized enterprises (SMEs) rather than by large pharmaceutical companies (Maciulaitis et al., 2012; Hanna et al., 2016). SMEs and start-up companies with biotechnology methods for engineering tissues and modifying genes strive to make their technologies available for clinical applications and lead novel therapies. Large pharmaceutical companies must reduce the cost of developing ATMPs and regenerative medicine products (RMPs), i.e., medical products comprising human cells, genes, and tissues in Japan, because their product development budgets are limited. Product development may avoid failure risk by using good results from research conducted by SMEs (Maciulaitis et al., 2012; Hanna et al., 2016). The number of imported and exported products is expected to increase, and regulatory bodies have attempted to establish common principles (Petricciani et al., 2017); however, developing methods for product approval differs by region. Each region has an expedited program for promoting clinical development including a priority review process and conditional marketing authorization (Detela and Lodge, 2019). For example, Japan has a conditional, time-limited authorization system and “SAKIGAKE” designation scheme (preferential review). The conditional, time-limited authorization is effective for 5–7 years (Daisaku, 2014). The EU has conditional marketing authorization systems and a Priority Medicines (PRIME) scheme. In the EU, conditional marketing authorization is effective for 1 year and is renewed annually (European Medicines Agency, 2020d). Thus, developmental standards for medical products comprising human cells, genes, and tissues remain to be clearly established worldwide. To date, no report has focused on developmental schemes and comparisons of products between Japan and the EU. This study was conducted to review the regulatory and developmental trends for medical products comprising human cells, genes, and tissues in the EU and Japan over two decades. We identified the characteristics of the clinical and non-clinical development of medical products comprising human cells, genes, and tissues between the EU and Japan.
Materials and Methods
The data sources included information from reports online of survey companies and public agencies. In this study, we conducted two investigations. The first compared the differences in regulations and systems related to medical products comprising human cells, genes, and tissues between the EU and Japan, and we then investigated the number of developed products and their review terms. We used market survey data from BB-Bridge, Inc (2017) and Seed Planning, Inc (2017), and from online information regarding medical products comprising human cells, genes, and tissues (Mitsubishi Chemical Research Corporation, 2009, 2012; Patent Office, 2009; Office of Regulation and System Reform, 2012; Seed Planning, Inc, 2013; Alimchandani, 2017; Arthur D Little, Inc, 2017; Chen et al., 2017; Foundation for Intellectual Property, 2017; Cuende et al., 2018; Shahryari et al., 2019) as of 1 May 2020. We identified 142 cell-based and gene-based therapy products, including products that overlapped or were withdrawn, approved in 24 countries according to disease and approval period. We also surveyed the examination periods of 9 RMPs of the Pharmaceuticals and Medical Devices Agency (PMDA) in Japan (Pharmaceuticals and Medical Devices Agency, 2007; Pharmaceuticals and Medical Devices Agency, 2012a, 2015, 2016a,b, 2019a,b,c,d, 2020a,b; Japan Tissue Engineering Co. Ltd, 2012, 2016, 2019; Terumo Corporation, 2015; JCR Pharmaceuticals Co. Ltd, 2016; AnGes Inc, 2019; Nipro corporation, 2019; Novartis Pharma, 2019) and 19 ATMPs of the European Medicines Agency (EMA) in Europe (European Medicines Agency, 2009, 2011, 2012, 2013, 2014, 2016a,b,c,d, 2017a,b,c, 2018a,b, 2019a,b, 2020a,b), for a total of 28 products, including withdrawn products, as of 1 May 2020. We accessed data sources from public assessment reports of 17 out of 28 products on the EMA or PMDA websites as of 1 May 2020. We also focused on the examination period at each stage of the EMA products using public assessment reports.
In the second investigation, we compared individual product development between Japan and the EU using online product review reports. We examined nonclinical tests and clinical trials for 9 RMPs of the PMDA (Pharmaceuticals and Medical Devices Agency, 2007; Pharmaceuticals and Medical Devices Agency, 2012a, 2015, 2016a,b, 2019a,b,c,d, 2020a,b; Japan Tissue Engineering Co. Ltd, 2012, 2016, 2019; Terumo Corporation, 2015; JCR Pharmaceuticals Co. Ltd, 2016; AnGes Inc, 2019; Nipro corporation, 2019; Novartis Pharma, 2019) and 9 ATMPs of the EMA (European Medicines Agency, 2016a,b,c, 2017a,b,c, 2018a,b, 2019a). Table 1 shows the generic names, brand names, approved countries or regions, developers, and target diseases, as well as whether the drugs were designated as orphan medical products, whether their genes were changed, and whether they were autologous or allogenic as of 1 May 2020. Table 2 provides an overview of the products.
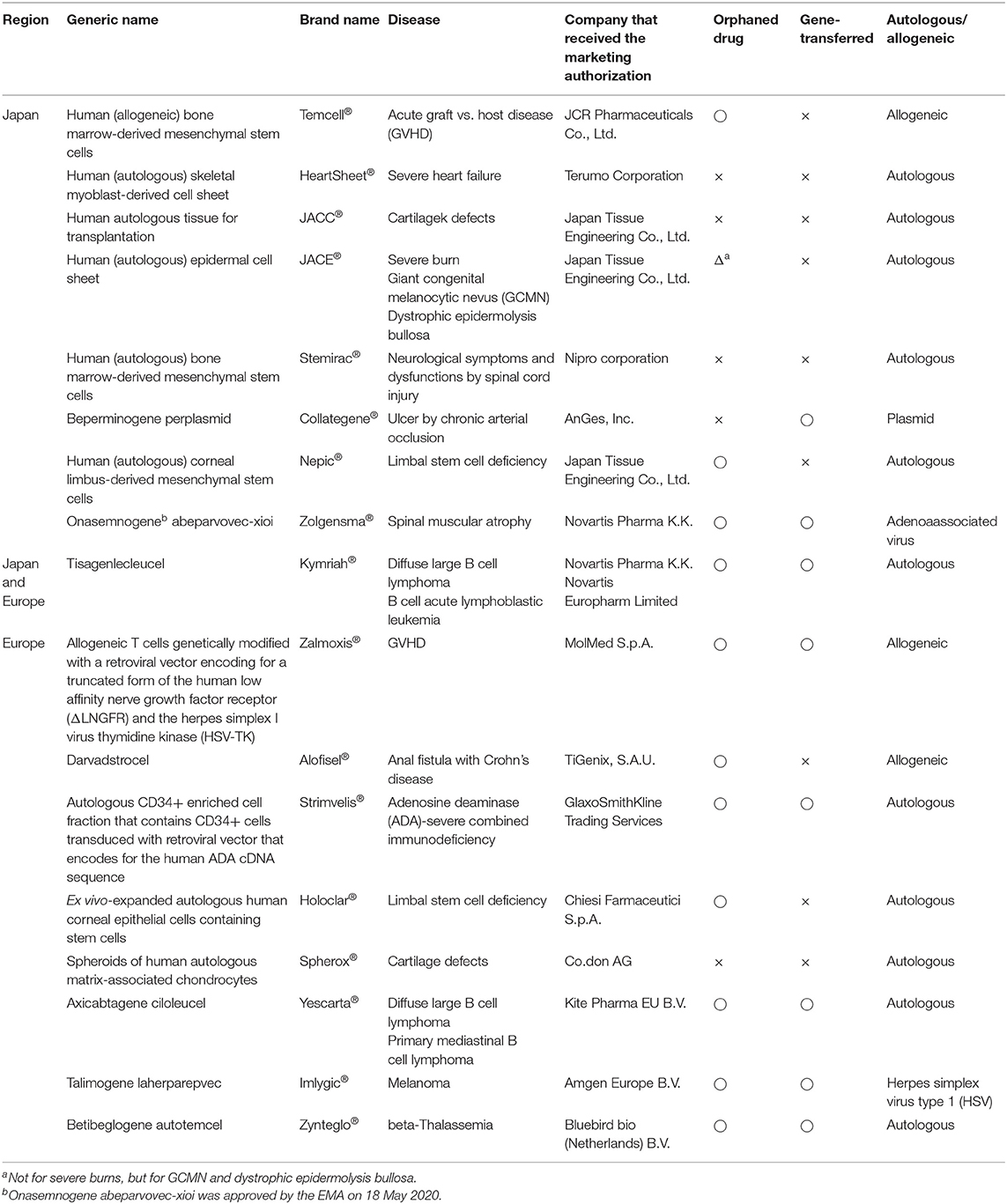
Table 1. Medical products comprising human cells, genes, and tissues in 2009–2019 as of 1 May 2020.
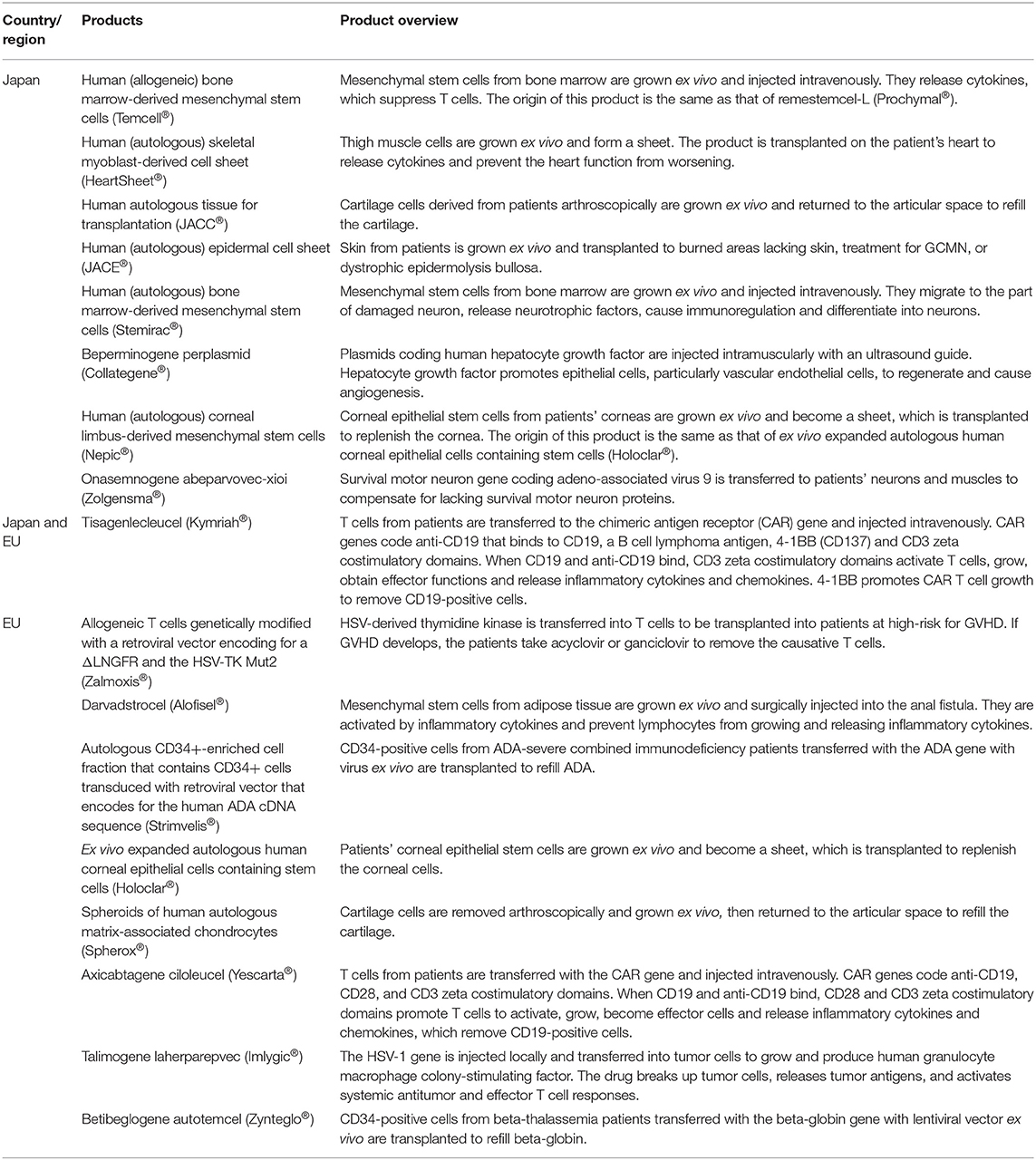
Table 2. Overview of medical products comprising human cells, genes, and tissues.
Results
Regulatory Frameworks of Medical Products Comprising Human Cells, Genes, and Tissues in the EU and Japan
Three regulatory bodies regulate the quality, safety, and efficacy of medical products comprising human cells, genes, and tissues using a risk-based approach. Clinical development must be performed under consistent, common rules (e.g., Good Gene, Cellular, and Tissue-based Products Manufacturing Practice, Good Manufacturing Practice, Good Tissue Practice, Good Laboratory Practice, and Good Clinical Practice). However, new marketing authorization schemes and systems for advanced treatments, i.e., cell-based and gene-based therapy products and unmet-need medicines, have recently been introduced in each region. Table 3 summarizes the regulatory frameworks for the two regions.
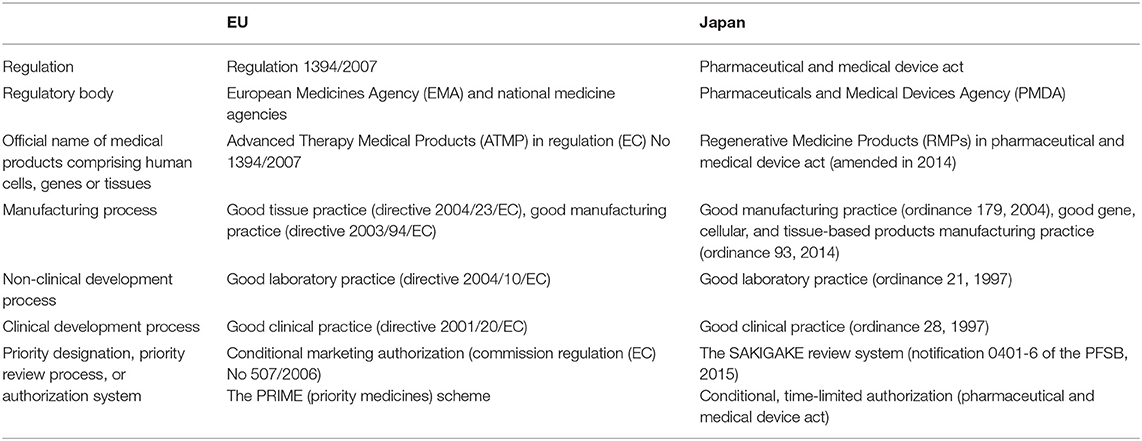
Table 3. Regulatory frameworks in the EU and Japan.
In the EU, ATMPs are new treatments for humans based on gene therapy, somatic-cell therapy, or tissue engineering, and the EMA offers conditional marketing authorization and the PRIME scheme (Table 4) (European Medicines Agency, 2020c,d). In Japan, RMPs are defined as human or animal cells processed and used for restructuring, repairing, or forming body structures or functions or treating or preventing diseases and gene-transferred human cells for gene therapy (Pharmaceutical and Medical Device Act, 2016). Since 2014, two schemes have promoted RMP development: the conditional time-limited authorization system and the SAKIGAKE Designation System (Table 4) (Pharmaceutical and Medical Device Act, 2016; Ministry of Health Labour and Welfare, 2019).

Table 4. Expedited programs in the EU and Japan.
Development and Approval of Medical Products Comprising Human Cells, Genes, and Tissues
Figure 1 indicates the number of approved medical products comprising human cells, genes, and tissues each year from 2000 to 2020. The number of products approved in the fields of orthopedic surgery and dermatology has increased since 2000, and the number of hematology-associated products has increased since 2011. Other medical fields currently have fewer numbers of approved products.
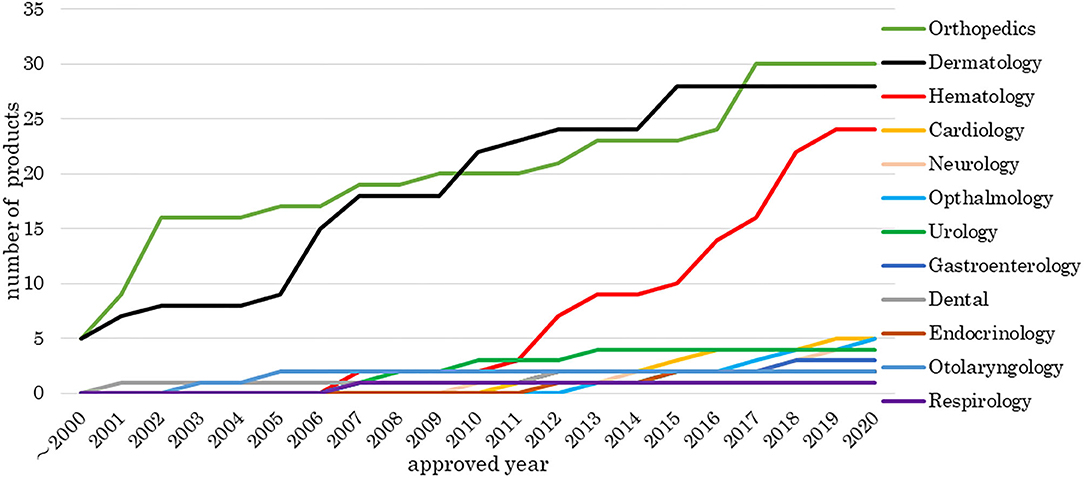
Figure 1. Number of medical products comprising human cells, genes, and tissues by medical field. The data sources included information from online reports of survey companies and public agencies as of 1 May 2020. There were 142 cell-based and gene-based therapy products, including products that overlapped or were withdrawn, approved in 24 countries. The number of products approved in the fields of orthopedic surgery and dermatology has increased since 2000 and the number of hematology-associated products has increased since 2011.
Figure 2 shows the number of approved medical products comprising human cells, genes, or tissues, and Figure 3 shows the review times for the EU and Japan over two decades. Although the number of approved products differs between the EU and Japan, their review periods tended to shorten over two decades.
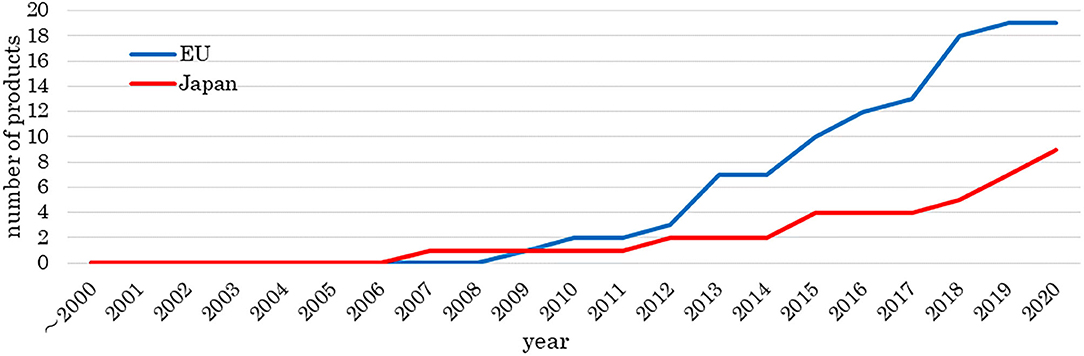
Figure 2. Number of approved medical products comprising human cells, genes, and tissues. The data sources included information from online reports of survey companies and public agencies as of 1 May 2020. The number of approved products is increasing.
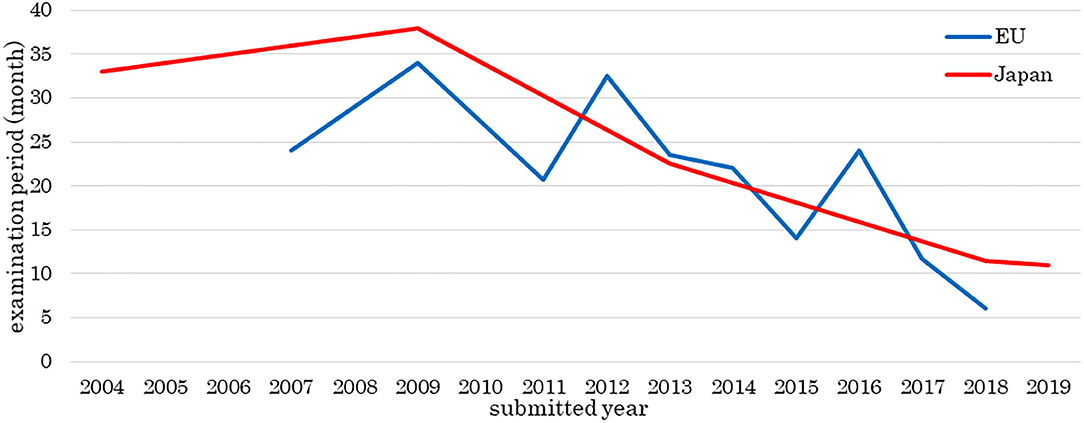
Figure 3. Examination period of medical products comprising human cells, genes, and tissues. The data sources included information from the assessment reports of public agencies as of 1 May 2020. The review periods tended to become shorter over the two decades. The horizontal line shows the year of a products submission, not approval.
The review times in 2009, 2012, and 2016 were longer than those of other years. Alipogene tiparvovec (Glybera®) was assessed three times, and more time was taken to answer the list of questions or the list of outstanding issues for spheroids of human autologous matrix-associated chondrocytes (Spherox®) or darvadstrocel (Alofisel®) than other products.
General Information on RMPs and ATMPs
We focused on approved RMPs and ATMPs in Japan and the EU. The PMDA and EMA designated five of nine products (56%) and eight of nine products (89%) as orphan medical products, respectively. The SAKIGAKE Designation System reviewed human (autologous) bone marrow-derived mesenchymal stem cells (Stemirac®), and the SAKIGAKE Designation System and PRIME scheme reviewed onasemnogene abeparvovec-xioi (Zolgensma®) (January 2017–July 2019). In Japan, three of nine products (33%) were gene transferred; in the EU, six of nine (67%) were gene transferred. In Japan, 6 of 9 products (67%) were autogenic; in the EU, six of nine (67%) were autogenic.
Table 5 summarizes the products' injection doses and shows the number of cells injected by body weight. The most suitable liquid dose was 1.2 × 108 cells per injection per adult, indicating that the most appropriate RMP and ATMP doses were independently decided according to disease type. In Japan, clinical trials for determining the best dose were conducted only for onasemnogene abeparvovec-xioi (Zolgensma®) and tisagenlecleucel (Kymriah®). In the EU, these trials were not conducted for the three ATMPs: an autologous CD34+ enriched cell fraction that contains CD34+ cells transduced with a retroviral vector encoding the human adenosine deaminase (ADA) complementary deoxyribonucleic acid (cDNA) sequence (Strimvelis®), ex vivo expanded autologous human corneal epithelial cells containing stem cells (Holoclar®) and betibeglogene autotemcel (Zynteglo®). In Japan, the dose was determined from non-clinical data; in the EU, the dose was determined from clinical data.
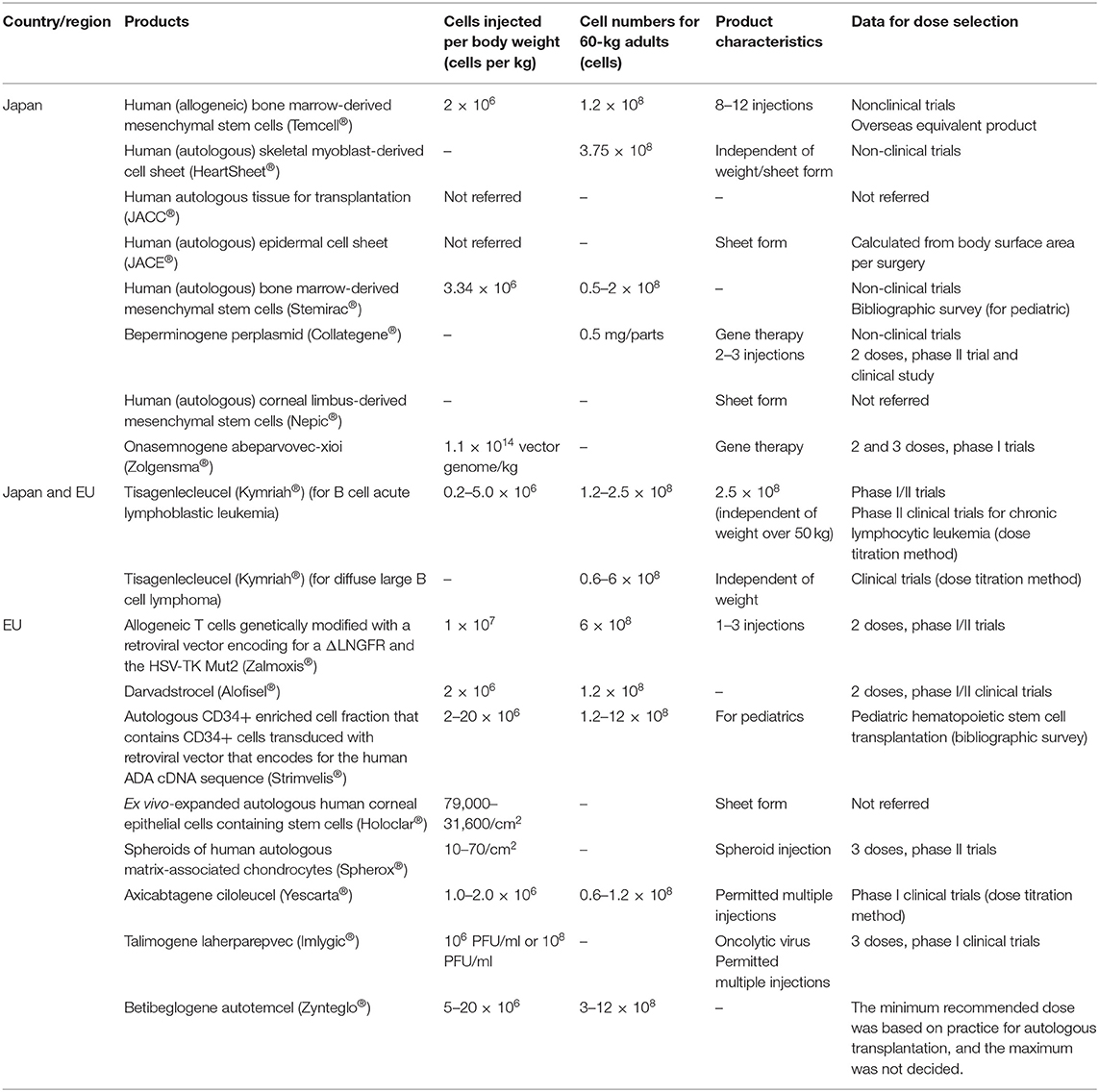
Table 5. Doses of medical products comprising human cells, genes, and tissues.
Comparison of Non-clinical Trials Between Japan and EU
Tables 6, 7 summarize the results of non-clinical trials and carcinogenicity tests, respectively. Neither Japan nor the EU tested the immunogenicity of autogenous products, but they both conducted allogeneic testing. For talimogene laherparepvec (Imlygic®), a genetically modified herpes simplex virus (HSV) for tumor lysis, beperminogene perplasmid (Collategene®), a plasmid-coding hepatocyte growth factor, and onasemnogene abeparvovec-xioi (Zolgensma®), a genetically modified adeno-associated virus 9, no studies have investigated whether host cells immunologically recognize these drugs as foreign substances. Thirteen of 18 products (72%) underwent non-clinical trials with animal disease models. Regarding non-clinical pharmacology tests, no in vivo tests were conducted in Japan if no appropriate animal model was available. Four of nine products (57%) in Japan and eight of nine products (88%) in the EU underwent non-clinical in vitro trials for efficacy. Thus, both in vivo and in vitro tests were likely to be conducted in the EU, whereas only one or the other was likely to be conducted in Japan. Pharmacokinetics tests were not individually conducted for sheet-form products, except for human (autologous) skeletal myoblast-derived cell sheets (Heartsheet®) and human (autologous) corneal limbus-derived mesenchymal stem cells (Nepic®), but these tests were individually conducted for all liquid-form products except human autologous tissue for transplantation (JACC®) and autologous CD34+ enriched cell fractions that contain CD34+ cells transduced with a retroviral vector encoding the human ADA cDNA sequence (Strimvelis®). For nonclinical carcinogenicity tests, six of nine products were investigated by karyotyping, soft-agar colony testing, and in vivo tumor-formation testing following technical guidance in Japan (Pharmaceuticals and Medical Devices Agency, 2016c). In the EU, karyotyping and soft-agar colony tests were performed on only two products, in vivo tumor-formation testing was performed on six products, and carcinogenicity was tested using various methods such as testing for gene expression involving carcinogenicity. Thus, in Japan, some tests were performed based on non-clinical technical guidance, whereas in the EU, trial contents were considered according to purpose, administration form, and the location of each administered product.
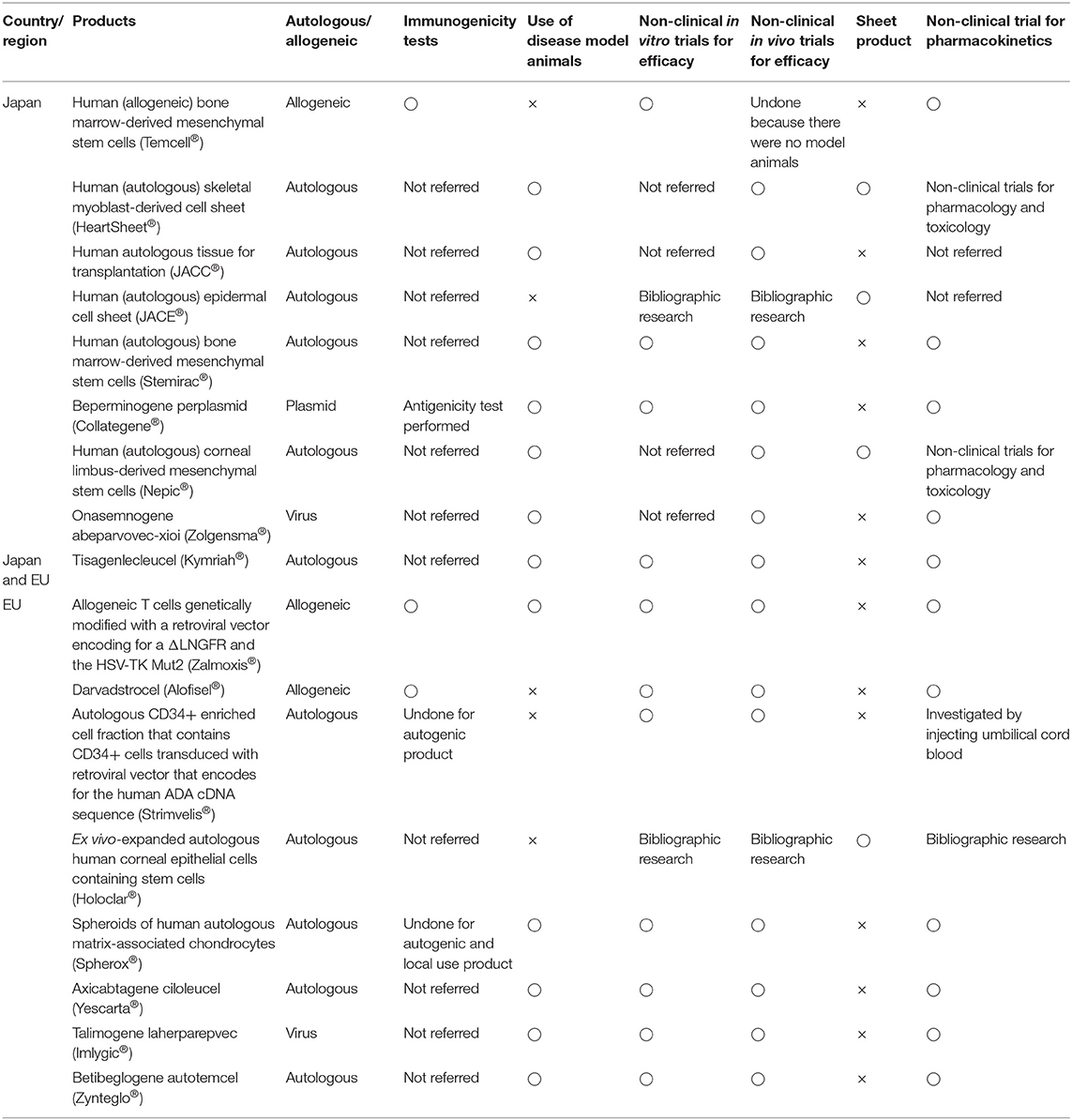
Table 6. Implementation status of non-clinical trials.
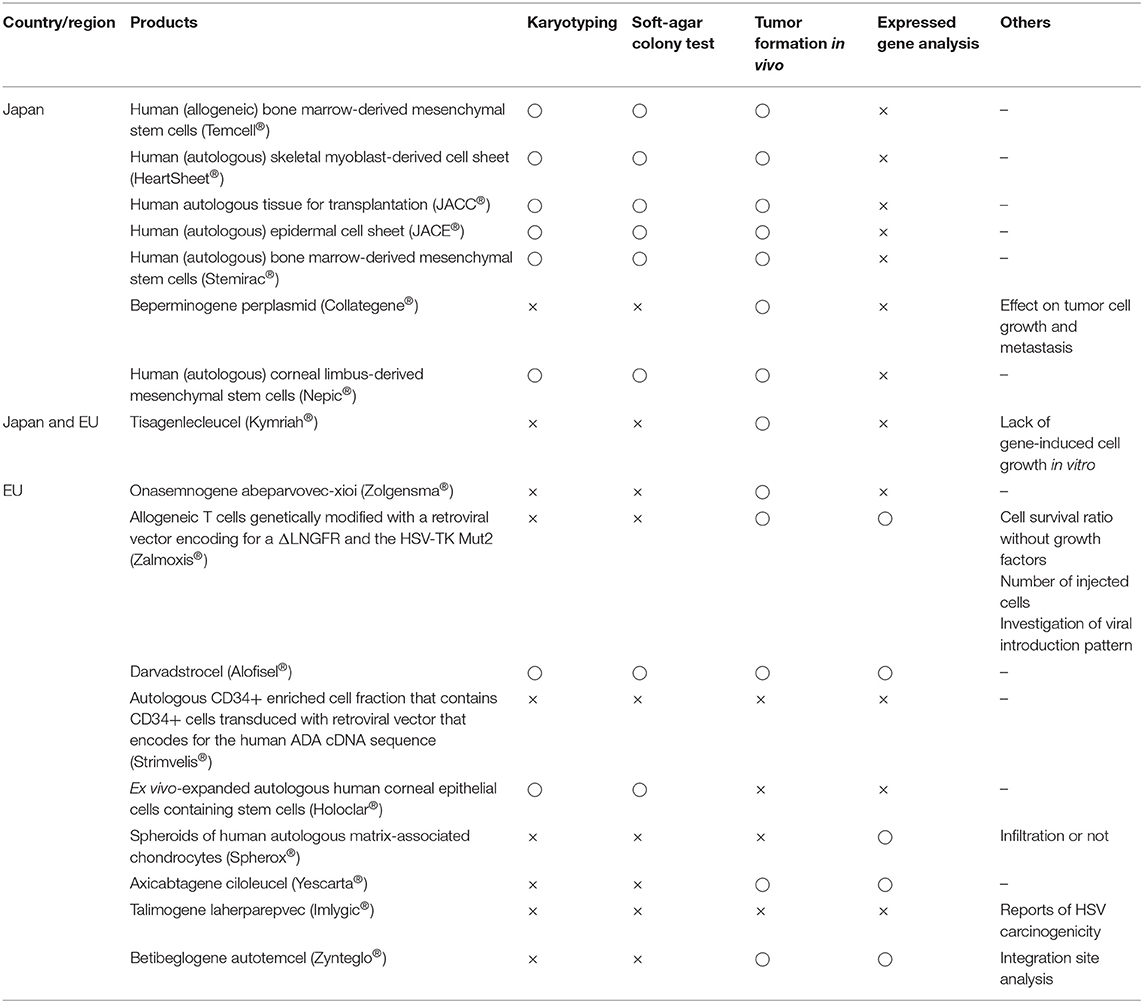
Table 7. Implementation status of carcinogenicity in non-clinical trials.
Clinical Trials for RMPs and ATMPs in Japan and EU
Table 8 lists subject numbers and clinical trials. The median numbers of subjects in Japan and the EU were 32 and 182, respectively. The median numbers of clinical trials in Japan and the EU were three and five, respectively. Conditional, time-limited authorization or conditional marketing authorization enables applicants to obtain marketing authorization without confirmatory trials, as with human (autologous) skeletal myoblast-derived cell sheets (Heartsheet®), human (autologous) bone marrow-derived mesenchymal stem cells (Stemirac®), beperminogene perplasmid (Collategene®), allogeneic T cells genetically modified with a retroviral vector encoding a truncated form of the human low affinity nerve growth factor receptor (ΔLNGFR), herpes simplex I virus thymidine kinase (HSV-TK Mut2) (Zalmoxis®), ex vivo expanded autologous human corneal epithelial cells containing stem cells (Holoclar®), and betibeglogene autotemcel (Zynteglo®).

Table 8. Features of clinical trials.
We also analyzed clinical trial results for the rates of effectiveness, stable disease (SD), ineffectiveness, and number of dropout subjects for human (allogeneic) bone marrow-derived mesenchymal stem cells (Temcell®) and human (autologous) skeletal myoblast-derived cell sheets (Heartsheet®) with conventional treatments including steroid (Murata et al., 2013) and cardiac resynchronization therapy (Birnie and Tang, 2006) (Figures 4, 5). Interestingly, in the clinical trials for human (allogeneic) bone marrow-derived mesenchymal stem cells (Temcell®), the effective rate decreased as the trial phase progressed (Figure 4), which is unusual in medical product development. In the phase II/III trial in Japan, the applicant did not set a control group, but an appropriated control group was used in other countries. After the phase II/III trial, the applicant found that the severity of the inclusion criteria in these clinical trials differed between Japan and the USA. Thus, they compared the active drug group in Japan with the reset control group, whose degree of patient severity was consistent between Japan and the USA. Therefore, the effective rate in the phase II/III clinical trial was higher than that in the reset control group.
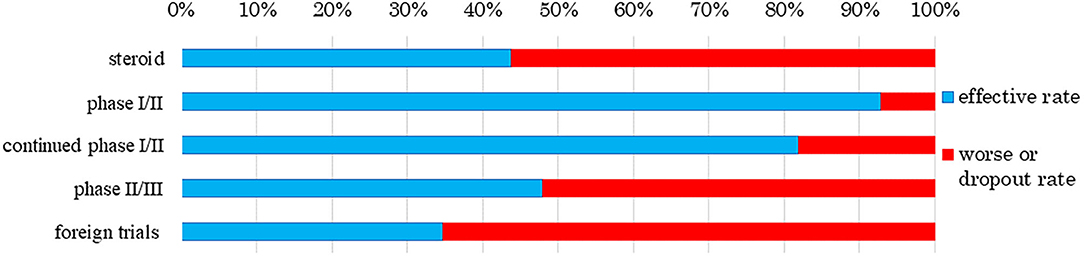
Figure 4. Response rate to human (allogeneic) bone marrow-derived mesenchymal stem cells (Temcell®). The data sources were assessment reports and application materials for human (allogeneic) bone marrow-derived mesenchymal stem cells (Temcell®), which are used for corticosteroid-refractory acute GVHD following hematopoietic stem cell transplantation. The foreign trials were performed using an equivalent product, remestemcel-L (Prochymal®) in the USA, Canada, Australia, Germany, Spain, and the UK. The effective rate decreased as the trial phase progressed. The rate of steroid use was 43.8% as reported by Murata et al. (2013). While 13 subjects showed effective treatment in a phase I/II trial of human (allogeneic) bone marrow-derived mesenchymal stem cells (Temcell®), one subject worsened or dropped out. In the continued phase I/II trial, 12 subjects showed effective treatment and two worsened or dropped out. Twelve subjects showed effective treatment in the phase II/III trial and 13 worsened or dropped out. In a trial of remestemcel-L (Prochymal®) in foreign countries, 60 subjects showed effective treatment whereas 113 had worsened disease.
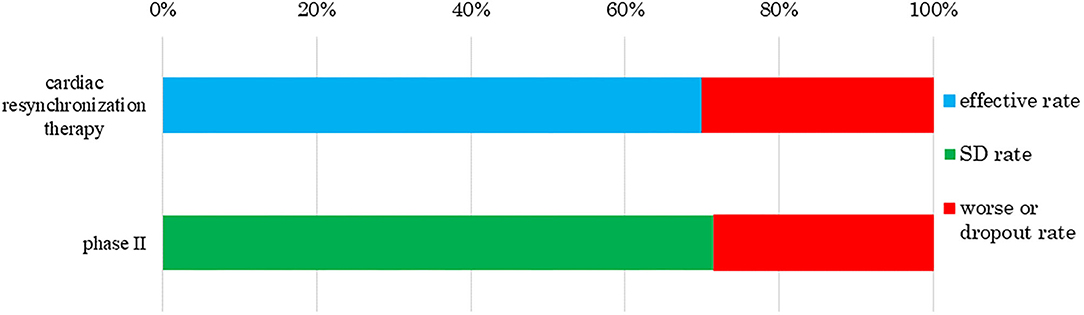
Figure 5. Response rate to human (autologous) skeletal myoblast-derived cell sheets (HeartSheet®). The data sources were assessment reports and application materials for human (autologous) skeletal myoblast-derived cell sheets (Heartsheet®). Cardiac resynchronization therapy was used for patients with severe heart failure who did not respond to standard drug therapies. Human (autologous) skeletal myoblast-derived cell sheets (Heartsheet®) were used for patients with severe heart failure unresponsive to standard treatments including drug and invasive therapies. This treatment mediated its effect by stabilizing cardiovascular function, i.e., stable disease (SD). The rate of cardiac resynchronization therapy was 70%, as reported by Birnie and Tang (2006). No subject showed effective treatment in a phase II trial of human (autologous) skeletal myoblast-derived cell sheets (Heartsheet®), but five achieved SD and two worsened or dropped out.
Human (autologous) skeletal myoblast-derived cell sheets (Heartsheet®) were assessed by their ability to stabilize cardiovascular function (Figure 5). The trial subjects had severe heart failure, which can only be effectively treated by heart transplantation. A PMDA officer wrote, as a personal opinion, “through the assessment, human (autologous) skeletal myoblast-derived cell sheets (Heartsheet®) have a certain effect on typical drug-resistant severe heart failure (European Medicines Agency, 2008; Maruyama, 2015).” Therefore, human (autologous) skeletal myoblast-derived cell sheets (Heartsheet®) secured conditional time-limited approval, although it did not improve patients' heart function. As with human (autologous) skeletal myoblast-derived cell sheets (Heartsheet®), clinical issues must be clarified, and effectiveness standards must be defined when developing medical products comprising human cells, genes, and tissues.
Discussion
We investigated the development and approval of medical products comprising human cells, genes, and tissues worldwide. Numbers of medical products comprising human cells, genes, and tissues in orthopedics and dermatology were increased at the beginning of the 2000's and in hematology in 2011. Most orthopedic products aim to refill cartilage, which tends to be damaged through traumatic or sports-related injury. Injected medical products comprising human cells, genes, and tissues remain in the joint cavity, which has no blood vessels. These products are unlikely to spread into the systemic circulation even if undifferentiated cells are present; therefore, the risk of tumorigenesis is low. Dermatological products are locally administered as sheets, which can easily and comparatively cure patients, for example, by reducing inflammation. Thus, orthopedic and dermatological products were the primary products being developed. Seven hematological products were approved from 2011 to 2013, of which five were for hematopoietic stem cell transplantation. At that time, it was simple to obtain approval, and this made it easier to develop medical products comprising human cells, genes, and tissues. Since then, the number of products in hematology has increased. Furthermore, the numbers of medical products comprising human cells, genes, and tissues approved by the EU and Japan have increased. Among the two agencies, the review times have decreased, while the median approval time for new active substances has not significantly changed (Centre for Innovation in Regulatory Science, 2019), indicating these trends are similar between the two agencies. In the EMA, more time was taken in 2009, 2012, and 2016 than in other years. “Steps taken for the assessment of the product” stated in the public assessment report shows which review stages took longer. Alipogene tiparvovec (Glybera®), the first gene therapy product in the EU, was submitted by UniQure Biopharma B.V. in 2009. Alipogene tiparvovec (Glybera®) was assessed three times. The first assessment started on 23 December 2009, and the Committee for Human Medicinal Products issued a negative opinion on 23 June 2011. It was then reexamined from 23 June to 20 October 2011. The third assessment started on 23 January 2012, and the Committee for Human Medicinal Products finally issued a positive opinion on 19 July 2012 (European Medicines Agency, 2012). Thus, the review term was 2.5 years. For spheroids of human autologous matrix-associated chondrocytes (Spherox®), submitted by Co.don AG in 2012, the list of questions took longer to answer than those of other products in 2013 (European Medicines Agency, 2017b) because the biopharmaceutical company had to collect clinical trial data. It also took longer to answer the list of outstanding issues for darvadstrocel (Alofisel®), submitted by Tigenix, S.A.U. in 2016, than for other products in 2016 (European Medicines Agency, 2017a). The public assessment report showed that the EMA found no difference in the quality of life between the darvadstrocel (Alofisel®) and control groups and that all patients in the clinical trials received the same dose regardless of the number of fistulas.
Clinical trials to determine suitable cell doses are seldom conducted in Japan but are required in the EU. The PMDA requires an investigation of the most appropriate dose and usage, which seems to be effective throughout drug development in terms of efficacy, while developers must pay attention to dose-dependent risks and the risk of immune responses caused by repeat injections (Pharmaceuticals and Medical Devices Agency, 2016c). According to the EMA guidelines, different doses should be applied, and the dose-response relationship should be tested if different doses cannot be applied (European Medicines Agency, 2008, 2018c). The EMA is likely to require a stricter investigation of a dose than the PMDA. Furthermore, Japan and the EU might consider non-clinical immunogenicity tests for autogenous products unnecessary. Guidance in Japan suggests that although non-clinical immunogenicity tests are required for allogenic and autogenic products, they are not always conducted for self-recognizing autogenic products (Pharmaceuticals and Medical Devices Agency, 2000; Pharmaceuticals and Medical Devices Agency, 2008a,b,c,d, 2012b,c). EMA guidelines do not describe autogenic product immunogenicity tests. This allows developers to save costs related to immunogenicity tests. As a result, autogenic products are cheaper than allogenic products (Shukla et al., 2019). In the guidance and guidelines, conventional stand-alone tests for pharmacokinetics are unnecessary in Japan and the EU (Pharmaceuticals and Medical Devices Agency, 2016c; European Medicines Agency, 2017d). However, when products are used directly or target specific organs, developers in Japan must clarify the locality and the impact on efficacy and safety through pharmacokinetic tests (Pharmaceuticals and Medical Devices Agency, 2000; Pharmaceuticals and Medical Devices Agency, 2008a,b,c,d, 2012b,c), and in the EU, biodistribution data may not be required if the cellular distribution potential is thought to be limited (European Medicines Agency, 2018c). Thus, in Japan, but not the EU, biodistribution data for sheet-form products must always be investigated.
Only in vitro or in vivo non-clinical pharmacological tests are conducted in Japan, while both are conducted in the EU. In Japan, applicants can submit the results of either in vitro or in vivo tests to suggest product mechanisms. A proof of concept must be shown by using an experimental animal model or suitable cells as per the PMDA guidance (Pharmaceuticals and Medical Devices Agency, 2008a,b,c,d, 2012b,c). Similarly, if relevant animal models cannot be developed, in vitro studies may replace animal studies as per the EMA guidelines (European Medicines Agency, 2017d). To date, no products have been approved without non-clinical in vivo tests in the EU except for ex vivo expanded autologous human corneal epithelial cells containing stem cells (Holoclar®). Regarding non-clinical pharmacological tests, the EMA seems to undertake a stricter review process compared with the PMDA.
Other regulations are related to carcinogenicity. The PMDA provides simple integrated guidance, which is easily understood for conducting testing (Pharmaceuticals and Medical Devices Agency, 2016c), whereas the EU guidelines state that tests must be performed along with written product characteristics (European Medicines Agency, 2008, 2018c). Clinical trials in Japan used fewer subjects than those in the EU. The number of subjects can be compensated for in two ways: using the results of clinical studies from other countries and postmarketing clinical trials for conditional, time-limiting approval or conditional marketing authorization. The former was the case for human (allogeneic) bone marrow-derived mesenchymal stem cells (Temcell®), which are equivalent to remestemcel-L (Prochymal®), whose clinical trials were conducted in the USA for rare diseases. Using many subjects from other countries improved the limited results obtained using fewer subjects in Japan. Postmarketing clinical trials for conditional, time-limiting approval were imposed on human (autologous) skeletal myoblast-derived cell sheets (Heartsheet®), beperminogene perplasmid (Collategene®), and human (autologous) bone marrow-derived mesenchymal stem cells (Stemirac®), which were only developed in Japan. Their safety was assessed via the results of a phase II trial, which secured a conditional, time-limiting approval. The confirmatory efficacy was investigated in a postmarketing clinical trial. In the EU, ex vivo expanded autologous human corneal epithelial cells containing stem cells (Holoclar®), allogeneic T cells genetically modified with a retroviral vector encoding ΔLNGFR and HSV-TK Mut2 (Zalmoxis®) and betibeglogene autotemcel (Zynteglo®) received conditional marketing authorization because ex vivo expanded autologous human corneal epithelial cells containing stem cells (Holoclar®) were approved with retrospective data, and allogeneic T cells genetically modified with a retroviral vector encoding ΔLNGFR and HSV-TK Mut2 (Zalmoxis®) and betibeglogene autotemcel (Zynteglo®) were approved with limited clinical data (Detela and Lodge, 2019). Furthermore, it is difficult to define the effectiveness of medical products comprising human cells, genes, and tissues in clinical trials, i.e., when maintaining heart function in the human (autologous) skeletal myoblast-derived cell sheet (Heartsheet®) clinical trials and when determining similar criteria between domestic and overseas trials for human (allogeneic) bone marrow-derived mesenchymal stem cells (Temcell®). Tisagenlecleucel (Kymriah®) was developed in Japan and the EU. The nonclinical tests and clinical data reviewed by the two regulatory bodies were similar although there are some different points in the guidelines between Japan and the EU. This utilization of the non-clinical and clinical data can save developmental costs. However, the review term by the PMDA in Japan was longer than the target review period in the SAKIGAKE scheme, which caused a delay in approval in Japan. If the developers apply EU data to a review by the PMDA, they should recognize the differences between guidelines and consult with the PMDA from an early stage of product development (Pharmaceuticals and Medical Devices Agency, 2019a).
Overall, EMA guidelines undergo stricter investigation than those of the PMDA and many products have been developed in compliance with these guidelines; however, some products in the EU and Japan have been developed without conducting their required tests. The developers must consult with regulatory agencies and judged to have effectively conducted the relevant examinations. The developers also utilize the SAKIGAKE scheme or PRIME scheme for fast track applications. Especially, the PRIME scheme is a supportive system for SMEs and academia. Products designated for the PRIME scheme can apply for accelerated assessment (European Medicines Agency, 2020c) but are automatically designated for the SAKIGAKE scheme (Ministry of Health Labour and Welfare, 2019). This difference may affect the development schedule of medical products comprising human cells, genes, and tissues. In addition, the review term of the SAKIGAKE scheme was shorter than that of the PRIME scheme (median time = 5.1 and 8 months, respectively) (Office of Pharmaceutical Industry Research, 2020). These points are the disadvantages of the PRIME scheme compared with the SAKIGAKE scheme. Furthermore, the SAKIGAKE scheme has other advantages including accompaniment consultation and the potential for conditional approval. In terms of conditional approval, developers in the EU must renew it annually, whereas conditional and time-limited approval in Japan is effective for 5–7 years. In addition, national insurance reimbursement is paid for products even under conditional approval in Japan. Therefore, developers can use their funds for further development in Japan compared with the EU. However, a disadvantage of the SAKIGAKE scheme is that it is designated only for products that are developed first in Japan. This makes it difficult for companies outside Japan, which do not have a Japanese subsidiary, to apply through the SAKIGAKE scheme (Nagai, 2019).
We focused on the characteristics of medical products comprising human cells, genes, and tissues developed over two decades. Our study had some limitations. First, the number of products was small. The numbers of cell-therapy medicinal products and in vivo gene therapy products were only 15 and 3, respectively. To increase accuracy, we will investigate products approved by the Food and Drug Administration and the Therapeutic Goods Administration (the Australian regulatory agency) and products that were once approved by the EMA but have since been withdrawn. Second, we did not study products developed worldwide, just those in the EU and Japan. The first gene therapy product was developed in the USA followed by China, Russia, and the Philippines (Seed Planning, Inc, 2017; Shahryari et al., 2019). To date, many products have been developed in the USA and South Korea. Third, we conducted this research with limited information obtained from development data and review results online and from documents. Based on this research, we suggest that the development pattern of medical products comprising human cells, genes, and tissues is extremely complex for each product, and researchers are perhaps rushing to summarize the findings from their experience and knowledge; however, a more measured approach might be required to establish their development patterns.
Conclusion
EMA guidelines require stricter investigations than those of the PMDA. Although there are similar fast track systems in Japan and the EU, the details of each system, for example, the designation conditions, are different. The results of this study combined with further investigations will enable the rapid and safe clinical application of medical products comprising human cells, genes, and tissues.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary materials, further inquiries can be directed to the corresponding author/s.
Author Contributions
RK conceived this research, collected and analyzed the data for the EU and Japan, and drafted the manuscript. HK and TI analyzed and coordinated this research. All authors read and approved the final manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Professor Yasutomo Fujii, Department of Human Health Science, School of Medicine, Kyoto University, for helping with and supporting this paper. We thank Traci Raley, MS, ELS, and J. Ludovic Croxford, PhD from Edanz Group (https://en-author-services.edanzgroup.com/ac) for editing a draft of this manuscript.
References
Alimchandani, M. (2017). Epicel HDE# BH990200 Pediatric Advisory Committee (PAC) Meeting. Available online at: https://www.fda.gov/media/103668/download (accessed June 8, 2018).
AnGes, Inc. (2019). Application Materials of Collategene. Available online at: https://www.pmda.go.jp/regenerative_medicines/2019/20190419001/index.html (accessed September 11, 2019).
Arthur D Little, Inc. (2017). Investigation on Proper Aid for “Radical Treatment”. Available online at: https://www.meti.go.jp/meti_lib/report/H29FY/000111.pdf (accessed Febuary 6, 2019).
BB-Bridge, Inc. (2017). 2017 Edition Overview of Currents State and Future Prospects of Cell Medicine Development in the World. Tokyo: BB-Bridge, Inc.
Birnie, D. H., and Tang, A. S. (2006). The problem of non-response to cardiac resynchronization therapy. Curr. Opin. Cardiol. 21, 20–26. doi: 10.1097/01.hco.0000198983.93755.99
Centre for Innovation in Regulatory Science. (2019). New Drug Approvals in Six Major Authorities 2009-2018. Available online at: https://www.cirsci.org/wp-content/uploads/2020/02/CIRS-RD-Briefing-70-New-approvals-in-six-regulatory-authorities-2009-18.pdf (accessed April 14, 2020).
Chen, Y. C., Cheng, H. F., and Yeh, M. K. (2017). Cell therapy regulation in Taiwan. Cell Transplant. 26, 483–492. doi: 10.3727/096368916X693293
Cuende, N., Rasko, J. E. J., Koh, M. B. C., Dominici, M., and Ikonomou, L. (2018). Cell, tissue and gene products with marketing authorization in 2018 worldwide. Cytotherapy 20, 1401–1413. doi: 10.1016/j.jcyt.2018.09.010
Daisaku, S. (2014). Regenerative Medicines Products: Conditional, Time-Limited Authorization -for Fulfilling Conditions. Available online at: https://www.pmda.go.jp/files/000199506.pdf (accessed April 4, 2020).
Detela, G., and Lodge, A. (2019). EU regulatory pathways for ATMPs: standard, accelerated and adaptive pathways to marketing authorisation. Mol. Ther. Methods Clin. Dev. 13, 205–232. doi: 10.1016/j.omtm.2019.01.010
European Medicines Agency. (2008). Guideline on Human Cell-Based Medical Products. Available online at: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-human-cell-based-medicinal-products_en.pdf (accessed April 2, 2020).
European Medicines Agency. (2009). ChondroCelect: EPR - Public Assessment Report. Available online at: https://www.ema.europa.eu/documents/assessment-report/chondrocelect-epar-public-assessment-report_en.pdf (accessed April 29, 2020).
European Medicines Agency. (2011). Withdrawal Assessment Report for Cerepro. Available online at: https://www.ema.europa.eu/documents/withdrawal-report/withdrawal-assessment-report-cerepro_en-0.pdf (accessed April 29, 2020).
European Medicines Agency. (2012). Glybera: EPR - Public Assessment Report. Available online at: https://www.ema.europa.eu/documents/assessment-report/glybera-epar-public-assessment-report_en.pdf (accessed April 29, 2020).
European Medicines Agency. (2013). Provenge: EPR - Public Assessment Report. Available online at: https://www.ema.europa.eu/documents/assessment-report/provenge-epar-public-assessment-report_en.pdf (accessed April 29, 2020).
European Medicines Agency. (2014). Maci: EPR - Public Assessment Report. Available online at: https://www.ema.europa.eu/documents/assessment-report/maci-epar-public-assessment-report_en.pdf (accessed April 29, 2020).
European Medicines Agency. (2016a). Holoclar : EPR - Public Assessment Report. Available online at: https://www.ema.europa.eu/documents/assessment-report/holoclar-epar-public-assessment-report_en.pdf (accessed May 11, 2018).
European Medicines Agency. (2016b). Imlygic : EPR - Public Assessment Report. Available online at: https://www.ema.europa.eu/documents/assessment-report/imlygic-epar-public-assessment-report_en.pdf (accessed June 21, 2016).
European Medicines Agency. (2016c). Zalmoxis : EPR - Public Assessment Report. Available online at: https://www.ema.europa.eu/documents/assessment-report/zalmoxis-epar-public-assessment-report_en.pdf (accessed March 30, 2018).
European Medicines Agency. (2016d). Heparesc: EPR - Public Assessment Report. Available online at: https://www.ema.europa.eu/documents/assessment-report/heparesc-epar-public-assessment-report_en.pdf (accessed April 29, 2020).
European Medicines Agency. (2017a). Alofisel: EPR - Public Assessment Report. Available online at: https://www.ema.europa.eu/documents/assessment-report/alofisel-epar-public-assessment-report_en.pdf (accessed May 26, 2018).
European Medicines Agency. (2017b). Spherox: EPR - Public Assessment Report. Available online at: https://www.ema.europa.eu/documents/assessment-report/spherox-epar-public-assessment-report_en.pdf (accessed October 2, 2018).
European Medicines Agency. (2017c). Strimvelis: EPR - Public Assessment Report. Available online at: https://www.ema.europa.eu/documents/assessment-report/strimvelis-epar-public-assessment-report_en.pdf (accessed April 17, 2017).
European Medicines Agency. (2017d). Development of Non-Substantially Manipulated Cell-Based ATMPs: Flexibility Introduced Via the Application of the Risk-Based Approach. Available online at: https://www.ema.europa.eu/en/documents/regulatory-procedural-guideline/development-non-substantially-manipulated-cell-based-advanced-therapy-medicinal-products-flexibility_en.pdf (accessed April 2, 2020).
European Medicines Agency. (2018a). Kymriah : EPR - Public Assessment Report. Available online at: https://www.ema.europa.eu/documents/assessment-report/kymriah-epar-public-assessment-report_en.pdf (accessed October 3, 2018).
European Medicines Agency. (2018b). Yescarta : EPR - Public Assessment Report. Available online at: https://www.ema.europa.eu/documents/assessment-report/yescarta-epar-public-assessment-report_en.pdf (accessed October 3, 2018).
European Medicines Agency. (2018c). Guideline on the uality, Non-Clinical and Clinical Aspects of Gene Therapy Medicinal Products. Available online at: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-quality-non-clinical-clinical-aspects-gene-therapy-medicinal-products_en.pdf (accessed April 2, 2020).
European Medicines Agency. (2019a). Zynteglo: EPR - Public Assessment Report. Available online at: https://www.ema.europa.eu/documents/assessment-report/zynteglo-epar-public-assessment-report_en.pdf (accessed April 29, 2020).
European Medicines Agency. (2019b). Luxturna: EPR - Public Assessment Report. Available online at: https://www.ema.europa.eu/documents/assessment-report/luxturna-epar-public-assessment-report_en.pdf (accessed April 29, 2020).
European Medicines Agency. (2020a). Hyalograft C autograft: Withdrawal of the Marketing Authorisation Application. Available online at: https://www.ema.europa.eu/en/medicines/human/withdrawn-applications/hyalograft-c-autograft (accessed April 29, 2020).
European Medicines Agency. (2020b). OraNera: Withdrawal of the Marketing Authorisation Application. Available online at: https://www.ema.europa.eu/en/medicines/human/withdrawn-applications/oranera (accessed April 29, 2020).
European Medicines Agency. (2020c). PRIME: Priority Medicines. Available online at: https://www.ema.europa.eu/en/human-regulatory/research-development/prime-priority-medicines (accessed April 4, 2020).
European Medicines Agency. (2020d). Conditional Marketing Authorisation. Available online at: https://www.ema.europa.eu/en/human-regulatory/marketing-authorisation/conditional-marketing-authorisation (accessed April 2, 2020).
Foundation for Intellectual Property. (2017). Investigative Research Report Relating to Current Status and Problems about Patent Protection of Regenerative Medicines, Drugs for in vitro Diagnostic and so on Which are Under Patent Act Article 2 Paragraph 2. Available online at: https://www.jpo.go.jp/shiryou/toushin/chousa/pdf/zaisanken/2016_06.pdf (accessed May 13, 2018).
Hanna, E., Rémuzat, C., Auquier, P., and Toumi, M. (2016). Advanced therapy medicinal products: current and future perspectives. J. Mark Access Health Policy. 4, 417–429. doi: 10.3402/jmahp.v4.31036
Japan Tissue Engineering Co. Ltd. (2012). Application Materials of JACC. Available online at: http://www.pmda.go.jp/medical_devices/2012/M201200024/index.html (accessed October 13, 2017).
Japan Tissue Engineering Co. Ltd. (2016). Application Materials of JACE. Available online at: http://www.pmda.go.jp/regenerative_medicines/2016/R20161005001/index.html (accessed 17 Oct 2017).
Japan Tissue Engineering Co. Ltd. (2019). Application Materials of JACE. Available online at: https://www.pmda.go.jp/regenerative_medicines/2019/R20190109001/index.html (accessed January 15, 2019).
JCR Pharmaceuticals Co. Ltd. (2016). Application Materials of Temcell HS Injection. Available online at: http://www.pmda.go.jp/regenerative_medicines/2015/R20151009001/index.html (accessed April 25, 2017).
Kondo, H., Hata, T., Ito, K., Koike, H., and Kono, N. (2017). The current status of SAKIGAKE designation in Japan, PRIME in the European Union, and breakthrough therapy designation in the United States. Ther. Innov. Regul. Sci. 51, 51–54. doi: 10.1177/2168479016662682
Maciulaitis, R., D'Apote, L., Buchanan, A., Pioppo, L., and Schneider, C. K. (2012). Clinical development of advanced therapy medicinal products in Europe: evidence that regulators must be proactive. Mol. Ther. 20, 479–482. doi: 10.1038/mt.2012.13
Maruyama, R. (2015). Overview of Approval Examination of Two New Products. Available online at: https://www.pmda.go.jp/files/000210025.pdf (accessed January 16, 2018).
Ministry of Health Labour Welfare. (2019). Overview about SAKIGAKE Review System. Available online at: https://www.mhlw.go.jp/stf/seisakunitsuite/bunya/kenkou_iryou/iyakuhin/tp150514-01_00001.html (accessed April 4, 2020).
Mitsubishi Chemical Research Corporation. (2009). 2008 Edition Report of Investigation for Assisting Small and Medium-Sized Enterprises Smaller Businesses (including Investigation about Tasks which Prevent Regenerative Medicine Business from Developing). Available online at: https://www.meti.go.jp/policy/mono_info_service/mono/bio/H20chousa.pdf (accessed May 10, 2018).
Mitsubishi Chemical Research Corporation. (2012). Edition Report of Investigation for Assisting Small and Medium-Sized Enterprises (Including Comparison of Systems in other Countries about Practical Realization for Industrialization of Regenerative Medicine). Available online at: https://www.meti.go.jp/policy/mono_info_service/mono/bio/H24kaigaiseido.pdf (accessed March 14, 2020).
Murata, M., Nakasone, H., Kanda, J., Nakane, T., Furukawa, T., Fukuda, T., et al. (2013). Clinical factors predicting the response of acute graft-versus-host disease to corticosteroid therapy: an analysis from the GVHD Working Group of the Japan society for hematopoietic cell transplantation. Biol. Blood Marrow Transplant. 19, 1183–1189. doi: 10.1016/j.bbmt.2013.05.003
Nagai, S. (2019). Flexible and expedited regulatory review processes for innovative medicines and regenerative medical products in the US, the EU, and Japan. Int. J. Mol. Sci. 20:3801. doi: 10.3390/ijms20153801
Nipro corporation. (2019). Application Materials of Stemirac. Available online at: https://www.pmda.go.jp/regenerative_medicines/2019/R20190125001/index.html (accessed Febuary 26, 2019).
Novartis Pharma, K. K. (2019). Application Materials of Kymriah. Available online at: https://www.pmda.go.jp/regenerative_medicines/2019/R20190423001/index.html (accessed September 11, 2019).
Office of Regulation System Reform. (2012). Government Revitalization Unit Regulatory and Institutional Reforms Committee “Intensive discussion”. Available online at: https://www.cao.go.jp/sasshin/kisei-seido/meeting/2012/togi/life/121129/item0.pdf (accessed March 10, 2018).
Office of Pharmaceutical Industry Research. (2020). Comparison of New Drug Approval and Examination Period in Japan, the US and Europe on 2018. Available online at: http://www.jpma.or.jp/opir/news/058/no058_10.html (accessed October 19, 2020).
Patent Office. (2009). 2008 Edition Report of Investigation about Trend of Techniques which are Applied for Patent: Regenerative Medicine. Available online at: https://www.jpo.go.jp/shiryou/pdf/gidou-houkoku/20saisei_iryou.pdf (accessed November 18, 2020).
Petricciani, J., Hayakawa, T., Stacey, G., Trouvin, J. H., and Knezevic, I. (2017). Scientific considerations for the regulatory evaluation of cell therapy products. Biologicals 50, 20–26. doi: 10.1016/j.biologicals.2017.08.011
Pharmaceutical Medical Device Act. (2016). Available online at: https://elaws.e-gov.go.jp/search/elawsSearch/elaws_search/lsg0500/detail?lawId=335AC0000000145 (accessed April 4, 2020).
Pharmaceuticals Medical Devices Agency. (2000). Technological Guidance for Securing Quality and Safety of Medicines made of Components of Human or Animals. Available online at: https://www.pmda.go.jp/files/000205395.pdf (accessed April 2, 2020).
Pharmaceuticals Medical Devices Agency. (2007). Review Reports of JACE. Available online at: https://www.pmda.go.jp/files/000203222.pdf (accessed October 17, 2017).
Pharmaceuticals Medical Devices Agency. (2008a). Guidance for Securing Quality and Safety of Medicines and Medical Devices made of Human (Autogenic) Somatic Stem Cell. Available online at: https://www.pmda.go.jp/files/000205400.pdf (accessed April 2, 2020).
Pharmaceuticals Medical Devices Agency. (2008b). Questions and Answers about Technological Guidance for Securing Quality and Safety of Medicines and Medical Devices made of Human (Autogenic) Somatic Stem Cell. Available online at: https://www.pmda.go.jp/files/000205397.pdf (accessed April 2, 2020).
Pharmaceuticals Medical Devices Agency. (2008c). Technological Guidance for Securing Quality and Safety of Medicines and Medical Devices made of Human (Allogenic) Somatic Stem Cell. Available online at: https://www.pmda.go.jp/files/000205401.pdf (accessed April 2, 2020).
Pharmaceuticals Medical Devices Agency. (2008d). Questions and Answers about Technological Guidance for Securing Quality and Safety of Medicines and Medical Devices made of Human (Allogenic) Somatic Stem Cell. Available online at: https://www.pmda.go.jp/files/000205399.pdf (accessed April 2, 2020).
Pharmaceuticals Medical Devices Agency. (2012a). Review Reports of JACC. Available online at: https://www.pmda.go.jp/files/000203221.pdf (accessed October 13, 2017).
Pharmaceuticals Medical Devices Agency. (2012b). Technological Guidance for Securing Quality and Safety of Medicines and Medical Devices made of Cells or Tissues of Human (Autogenic). Available online at: https://www.pmda.go.jp/files/000205396.pdf (accessed April 2, 2020).
Pharmaceuticals Medical Devices Agency. (2012c). Technological Guidance for Securing Quality and Safety of Medicines and Medical Devices made of Cells or Tissues of Human (Allogenic). Available online at: https://www.pmda.go.jp/files/000205398.pdf (accessed April 2, 2020).
Pharmaceuticals Medical Devices Agency. (2015). Review Reports of HeartSheet. Available online at: http://www.pmda.go.jp/regenerative_medicines/2015/R20151008001/470034000_22700FZX00002_A100_2.pdf (accessed June 4, 2017).
Pharmaceuticals Medical Devices Agency. (2016a). Review Reports of JACE. Available online at: http://www.pmda.go.jp/regenerative_medicines/2016/R20161005001/340938000_21900FZX00039001_A100_1.pdf (accessed October 17, 2017).
Pharmaceuticals Medical Devices Agency. (2016b). Review Reports of Temcell HS Inj. Available online at: http://www.pmda.go.jp/regenerative_medicines/2015/R20151009001/530210000_22700FZX00001_A100_4.pdf (accessed April 25, 2017).
Pharmaceuticals Medical Devices Agency. (2016c). Technical Guidance about Conduction of Quality Control, Non-Clinical and Clinical Trial of Regenerative Medicine Products (Processed Human Cell Products). Available online at: https://www.pmda.go.jp/files/000212850.pdf (accessed July 6, 2017).
Pharmaceuticals Medical Devices Agency. (2019a). Review Reports of Kymriah. Available online at: https://www.pmda.go.jp/regenerative_medicines/2019/R20190423001/300242000_23100FZX00002000_A100_1.pdf (accessed April 28, 2019).
Pharmaceuticals Medical Devices Agency. (2019b). Review Reports of Collategene. Available online at: https://www.pmda.go.jp/regenerative_medicines/2019/20190419001/111298000_23100FZX00001000_A100_1.pdf (accessed April 25, 2019).
Pharmaceuticals Medical Devices Agency. (2019c). Review Reports of JACE. Available online at: https://www.pmda.go.jp/regenerative_medicines/2019/R20190109001/340938000_21900FZX00039001_A100_1.pdf (accessed April 24, 2019).
Pharmaceuticals Medical Devices Agency. (2019d). Review Reports of Stemirac. Available online at: https://www.pmda.go.jp/regenerative_medicines/2019/R20190125001/530100000_23000FZX00001_A100_1.pdf (accessed April 24, 2019).
Pharmaceuticals Medical Devices Agency. (2020a). Review Reports of Zolgensma. Available online at: https://www.pmda.go.jp/regenerative_medicines/2020/R20200407001/300242000_30200FZX00001000_A100_2.pdf (accessed April 23, 2020).
Pharmaceuticals Medical Devices Agency. (2020b). Review Reports of Nepic. Available online at: https://www.pmda.go.jp/regenerative_medicines/2020/R20200413001/340938000_30200FZX00002_A100_1.pdf (accessed April 23, 2020).
Seed Planning Inc. (2013). 2012 Edition Report of Investigation for Assisting Small and Medium-Sized Enterprises (Including Investigation About Practical Realization and Industrialization of Regenerative Medicine). Available online at: https://www.meti.go.jp/policy/mono_info_service/mono/bio/H24chousazimu.pdf (accessed May 11, 2018).
Seed Planning Inc. (2017). 2017 Edition Future Prospect of Gene Therapy Products. Tokyo: Seed Planning, Inc.
Shahryari, A., Saghaeian Jazi, M., Mohammadi, S., Razavi Nikoo, H., Nazari, Z., Hosseini, E. S., et al. (2019). Development and clinical translation of approved gene therapy products for genetic disorders. Front Genet. 10:868. doi: 10.3389/fgene.2019.00868
Shukla, V., Seoane-Vazquez, E., Fawaz, S., Brown, L., and Rodriguez-Monguio, R. (2019). The landscape of cellular and gene therapy products: authorization, discontinuations, and cost. Hum. Gene Ther. Clin. Dev. 30, 102–113. doi: 10.1089/humc.2018.201
Terumo Corporation. (2015). Application Materials of HeartSheet. Available online at: http://www.pmda.go.jp/regenerative_medicines/2015/R20151008001/index.html (accessed July 4, 2017).
Keywords: advanced therapy medicinal products (ATMP), cell therapy, gene therapy, marketing access, regenerative medicine, regenerative medicine products, public assessment reports
Citation: Kurauchi R, Kasai H and Ito T (2020) Characteristics of Medical Products Comprising Human Cells, Genes, or Tissues Developed in Japan and the European Union Compared via Public Assessment Reports. Front. Bioeng. Biotechnol. 8:606606. doi: 10.3389/fbioe.2020.606606
Received: 15 September 2020; Accepted: 13 November 2020;
Published: 23 December 2020.
Edited by:
Abhay Pandit, National University of Ireland Galway, IrelandReviewed by:
Pavel Makarevich, Lomonosov Moscow State University, RussiaNatividad Cuende, Andalusian Health Service, Spain
Copyright © 2020 Kurauchi, Kasai and Ito. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ryosuke Kurauchi, kurauchi_ryosuke_46m@eis.hokudai.ac.jp