 Rong Li
Rong Li Yao Chen
Yao Chen- Department of Biological Engineering, Beijing University of Chemical Technology, Beijing, China
For the multimeric enzymes R-ω-transaminase (RTA), alanine dehydrogenase (AlaDH), and formate dehydrogenase (FDH), peptide bond formation between the hetrosubunits has been achieved by the intein-mediated in vivo subunit splicing. The subunit ligation is triggered by the heterodimerization of an arginine rich leucine zipper motif with a glutamic acid rich leucine zipper motif. The one-by-one ligation of hetrosubunits constructs the pairing enzymes RTA&AlaDH and AlaDH&FDH. The ligation modes were analyzed based on blue native polyacrylamide gel electrophoresis (BN-PAGE). The spectra of circular dichroism (CD), fluorescence, and two-dimensional FTIR provide information on the secondary structures and stability of the pairing enzymes. The enzyme-substrate interaction was analyzed based on microscale thermophoresis analysis. In contrast to the mixed three enzymes RTA + AlaDH + FDH, the ligated enzymes RTA&AlaDH + AlaDH&FDH exhibited a much larger substrate affinity, higher stability, and significantly enhanced activity.
Introduction
Chiral amines are used for the synthesis of pharmaceuticals (Hannes et al., 2014; Simon et al., 2014). The optically pure amines have been used for the synthesis of biologically active compounds (Koszelewski et al., 2009). ω-Transaminases (ω-TAs) are multimeric enzymes and have been investigated for preparation of chiral amines (Han et al., 2019; Rocha et al., 2019; Isupov et al., 2019; Liao et al., 2019). ω-Transaminases are efficient biocatalysts for reductive amination of ketones to corresponding amines (Kelly et al., 2018; Padrosa et al., 2019). Alanine is commonly used as amine donor and the coproduct is pyruvate (Pressnitz et al., 2013). However, equilibrium is unfavored for the reductive amination. For circumventing this issue, ω-TAs have been combined with various enzymes to establish multienzyme networks for removing the coproduct, combining with lactate dehydrogenase (LDH) to convert pyruvate into lactic acid, incorporating pyruvate decarboxylase (PDC) to convert pyruvate to acetaldehyde and CO2 (Shin and Kim, 1999; Hohne et al., 2008). 2-Propanamine was also used as a cheap and achiral amine donor (Cassimjee et al., 2010). Yeast alcohol dehydrogenase was used for removing the coproduct acetone to displace the equilibrium.
Herein, we pay attention to another approach using an alanine dehydrogenase (AlaDH) to cooperate with R-ω-transaminase (RTA). In this approach, alanine is used as amine donor. The coproduct pyruvate is recycled back to alanine by consuming ammonia and formate (Koszelewski et al., 2008) (Figure 1). Formate dehydrogenase is integrated into the network to recycle NADH, which is needed for the reductive amination of pyruvate to alanine. However, we found that for some ketones, like acetophenone and 2-octanone, the conversions are very low under the catalysis of the mixed enzymes RTA + AlaDH + FDH. When these three enzymes are simply mixed together, the orientation and distance between the enzymes cannot be controlled. It is speculated that for efficient conversion of those ketones, the amine donor alanine and coproduct pyruvate as well as NADH and NAD+ must be efficiently transferred between RTA and AlaDH and between AlaDH and FDH, respectively. The distances between RTA and AlaDH and between AlaDH and FDH affect the transfer efficiency. Ideally, the enzymes are in close proximity with a molecular distance. Through gene fusion, two enzymes can be linked together (Lindbladh et al., 1994). However, for these multimeric enzymes, gene fusion does not work (exhibiting no activity). It is possibly because the fusion enzyme cannot fold correctly in the Escherichia coli cell.
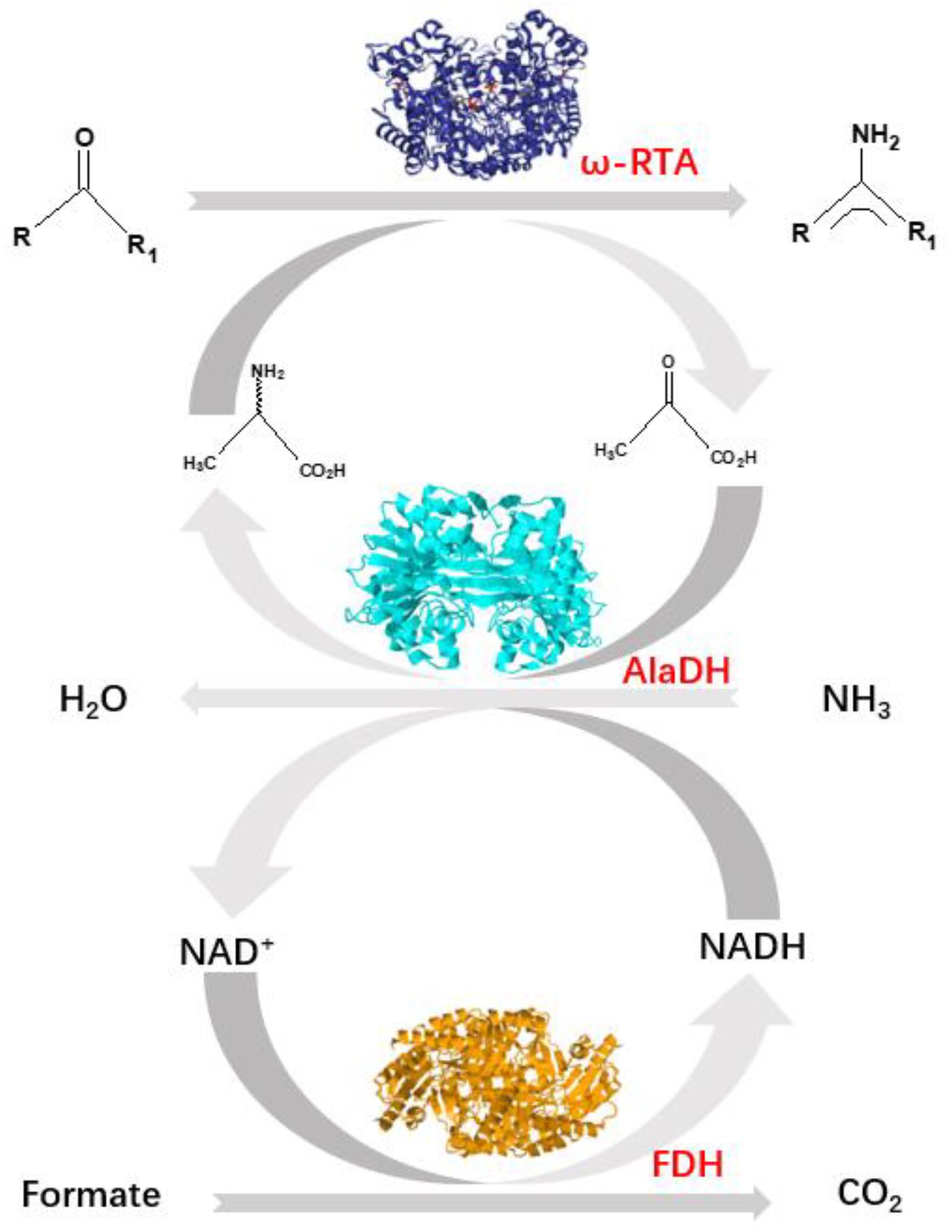
Figure 1. Shifting the equilibrium in RTA catalyzed reactions with alanine dehydrogenase (AlaDH) in combination with formate dehydrogenase (FDH).
Except the above mentioned low catalysis efficiency by the mixed enzymes, there is a significant concern for enzyme stability (Börner et al., 2017; Santos et al., 2018). The enzymes RTA, AlaDH, and FDH are multimeric enzymes. Experimental conditions may weaken the subunit-subunit interactions for multimeric enzymes (Fernandez, 2009). A sufficient operational stability of the enzymes is necessary for high productivity in chiral amine synthesis.
Previously, we have specifically ligated two multimeric enzymes with native peptides through intein-mediated in vivo subunit splicing (Du et al., 2017; Li et al., 2018). Herein, we develop the methodology, by using the coiled-coil association of an arginine-rich leucine zipper motif (ZR) (Moll et al., 2001) and a glutamic acid-rich leucine zipper motif (ZE) (Moll et al., 2001) to trigger the intein-mediated in vivo subunit splicing. RTA is specifically ligated to AlaDH and AlaDH is specifically linked to FDH with peptide bonds. The specific ligations were achieved through in vivo subunit splicing. Thus, the cycle of alanine → pyruvate → alanine between RTA and AlaDH and the cycle of NADH → NAD+→ NADH between AlaDH and FDH are formed. Evaluation of the three-enzyme system has been performed in terms of enzyme secondary structure, enzyme stability, and catalysis efficiency.
Materials and Methods
Materials
DNA ligase, DNA polymerase, and restriction enzymes were purchased from Thermo Fisher Scientific. Oligonucleotide primers were synthesized by BioMed Tech. Escherichia coli strain DH5α used as a host for DNA manipulation and strain BL21 (DE3) as a host for expressing enzymes were both purchased from Zoman Bio. And all other reagents were purchased from Sigma-Aldrich (Shanghai, China).
Construction of Plasmids
The alanine dehydrogenase (AlaDH) gene was synthesized based on the genome sequence of Archaeoglobus fulgidus (EC:1.4.1.1) (Agren et al., 2008) and cloned in pUC57 plasmid by Genewiz (Suzhou, China). The AlaDH gene was purified using a DNA extraction kit (Omega Bio-tek). The purified AlaDH gene was digested with both BamHI and EcoRI restriction endonucleases, which then was ligated into the plasmid pETDuet-1 cleaved with BamHI and EcoRI restriction endonucleases already. The AlaDH gene was amplified by PCR using the paired primers AlaDH-fp and AlaDH-rp (Supplementary Table S1), at the amplification conditions (Supplementary Table S1). The ZE-IntC-link gene was amplified by PCR using the ZE-IntC-fp and ZE-IntC-rp primers (Supplementary Table S1). The purified ZE-IntC-link gene was digested with NcoI and BamHI restriction endonucleases, then was ligated into the vector AlaDH-pETDuet that had been cleaved with NcoI and BamHI restriction endonucleases. The constructed expression plasmid was designated as ZE-IntC-link-AlaDH-pETDuet.
The R-ω-transaminase (RTA) gene used here was the same as that in our previous work (Sun et al., 2018). The RTA gene was amplified by PCR using primers RTA-fp and RTA-rp (Supplementary Table S1). The purified RTA gene was digested with both NdeI and KpnI restriction endonucleases, and then was ligated into the plasmid ZE-IntC-link-AlaDH-pETDuet, which had been cleaved with NdeI and KpnI restriction endonucleases. Therefore, the plasmid IntC-link-AlaDH/RTA-pETDuet in Escherichia coli was constructed. The gene of link-IntN-ZR was amplified by PCR using the IntN-ZR-fp and IntN-ZR-rp primers (Supplementary Table S1). The purified link-IntN-ZR gene was digested with both KpnI and XhoI restriction endonucleases, and then was ligated into the plasmid ZE-IntC-link-AlaDH-pETDuet, which had been cleaved with KpnI and XhoI restriction endonucleases. Then plasmid for coexpressing the precursors RTA-link-IntN-ZR and ZE-IntC-link-AlaDH was finally constructed. It is ZE-IntC-link-AlaDH/RTA-link-IntN-ZR-pETDuet, as illustrated in Supplementary Figure S1a. The FDH gene (Fogal et al., 2015) was amplified. The plasmid for coexpressing the precursors ZE-IntC-link-AlaDH and FDH-link-IntN-ZR was constructed similarly, designated as ZE-IntC-link-AlaDH/FDH-link-IntN-ZR-pETDuet as illustrated in Supplementary Figure S1b.
For the hetrosubunit splicing without using the coiled-coil association of ZE and ZR to promote the splicing, the plasmids were constructed using the same procedures as described above. The plasmid for coexpressing the precursors RTA-link-IntN and IntC-link-AlaDH was constructed, designated as ZE-IntC-link-AlaDH/RTA-link-IntN- ZR-pETDuet, as illustrated in Supplementary Figure S1c. The plasmid for coexpressing the precursors IntC-link-AlaDH and FDH-link-IntN was constructed, designated as IntC-link-AlaDH/FDH-link-IntN-pETDuet as illustrated in Supplementary Figure S1d.
Enzyme Expression and in vivo Subunit Splicing
The genes were transferred into Escherichia coli BL21 (DE3) cells. The cells were cultured in Luria-Bertani medium (50 μg/ml ampicillin) under shaking (220 rpm) at 37°C for 3 h. At this time, the OD600 reached about 0.4, and the cells grew at exponential phase. Then the cells were induced with 0.5 mM isopropyl β-D-1-thiogalactopyranoside (IPTG), and the culture temperature was changed to 25°C. At this temperature the cells grew for 8, 12, and 16 h, respectively. During the co-expression of the precursors ZE-IntC-link-RTA and AlaDH-link-IntN-ZR, the in vivo subunit splicing occurred simultaneously. Both SDS-PAGE and blue native polyacrylamide gel electrophoresis (BN-PAGE) were used to monitor and analyze the crude proteins released from the cells. It was found that 8 h is enough for the cell growth to accomplish the in vivo subunit splicing, determined by observing the bands for the splicing products.
The E. coli cells were collected after 30 min centrifugation at 7000 g at 4°C. The cells were then disrupted under ultrasonication on ice. The cell debris was removed by centrifugation at 7,000 g at 4°C for 30 min. The supernatant was collected into a fresh tube. The enzymes were first purified by passing through a chromatography column (GE Life Sciences) filled with Ni Sepharose beads (GE Life Sciences). Five hundred mM imidazole was used as the elution liquid, which was separated from the purified enzymes by dialysis into a buffer (500 mM NaCl, 50 mM Tris-Cl pH 7.5, 1 mM EDTA). The purified enzymes were further fractionated by ultrafiltration (GE Healthcare, 50000 Da cut off). The splicing products were finally purified using a gel filtration chromatography (AKTA Pure chromatography system, GE Healthcare, Superose 6 column).
Microscale Thermophoresis (MST)
The experiments were performed on an instrument of microscale thermophoresis (MST) (Nano Temper technologies). The splicing product RTA&AlaDH was not labeled by fluorescence. The dialysis liquid consisted of 50 mM Tris-Cl (PH 8.0), 150 mM NaCl, and 10 mM MgCl2. After dialysis, RTA&AlaDH (200 nM) was mixed with the ligand 2-octanone (100 mM), and then incubated at 30°C for an hour. A 16 step dilution series from 10 mM to 0.25 μM were prepared by DMSO. 10 μL of each step was filled into 16 fresh small micro reaction tubes and was well mixed by pipetting up and down. The measurements were performed at 20% IR-laser power and 40% MST power. Using the software MO. Affinity Analysis, the binding coefficient Kd values were obtained by fitting the fraction of bound protein to the quadratic solution of the binding reaction equilibrium. MST benefits from very low sample consumption and short measurement times.
Attenuated Total Reflectance Fourier Transform Infrared (ATR-FTIR)
FTIR- attenuated total reflection (ATR) spectra were recorded on a spectrometer (Bruker Tensor 27) at a nominal resolution of 2 cm–1. Each spectrum is the result of the accumulation and averaging of 512 interferograms. Ultrapure nitrogen was continuously introduced into the sample compartment to minimize the spectral contribution of atmospheric water. The enzyme was dissolved in 10 mM deuterated potassium phosphate buffer (pH 7.4) with a concentration of 15 mg/mL. The enzyme solution was rapidly prepared at 10°C, thus condensation of atmospheric water into the solution can be avoided. D2O was added into the sample of the enzyme solution, and the measurement of FTIR spectra was started. The hydrogen-deuterium exchange experiment was carried out for 90 min. The pure spectrum of enzyme was obtained by subtracting the spectra of D2O.
Enzymatic Activity
For transformation of the amines, the enzymatic catalysis was performed at 30°C under shaking at 200 rpm. The enzyme concentrations were: 0.5 mg/mL RTA&AlaDH + 0.5 mg/mL AlaDH&FDH; 0.5 mg/mL RTA#AlaDH + 0.5 mg/mL AlaDH#FDH. The concentration of substrate 2-octanone was 50 mM. The substrate solution was prepared by dissolving in phosphate buffered solution (mix 100 mM Na2HPO4 and 100 mM NaH2PO4 in proportion to pH 8.0), then alanine (5 mM), pyridoxal-5′-phosphate (1 mM), 1 mM NAD+, and 100 mM ammonium formate were added. The reaction volume was 2 mL. The chromatographic pure methanol and ultra pure water were respectively filtered by the filter membrane, and then the ultrasonic treatment was carried out to exhaust bubbles for half an hour. The concentration of the substrate 2-octanone was monitored by HPLC (Shimadzu LC-10A) with ultraviolet detector at 280 nm using a C18 column (Diamonsil C18 250 × 4.6 mm, 5 μm). Setup column thermostat at 30°C, and the mobile phase was methanol/water (60:40 by vol) at 0.8 mL/min, and the injected sample was 20 μL.
Results and Discussion
In vivo Hetrosubunit Splicing
Herein, the split intein was utilized to specifically ligate multimeric enzymes for potential industrial application. These multimeric enzymes have large molecular weights. The ligations were accomplished through intein-mediated in vivo subunit splicing, triggered by the coiled-coil association of an arginine-rich leucine zipper motif (ZR) and a glutamic acid-rich leucine zipper motif (ZE) (Moll et al., 2001). The developed methodology was used to ligate the enzymes for constructing three-enzyme network, as illustrated in Figure 2.
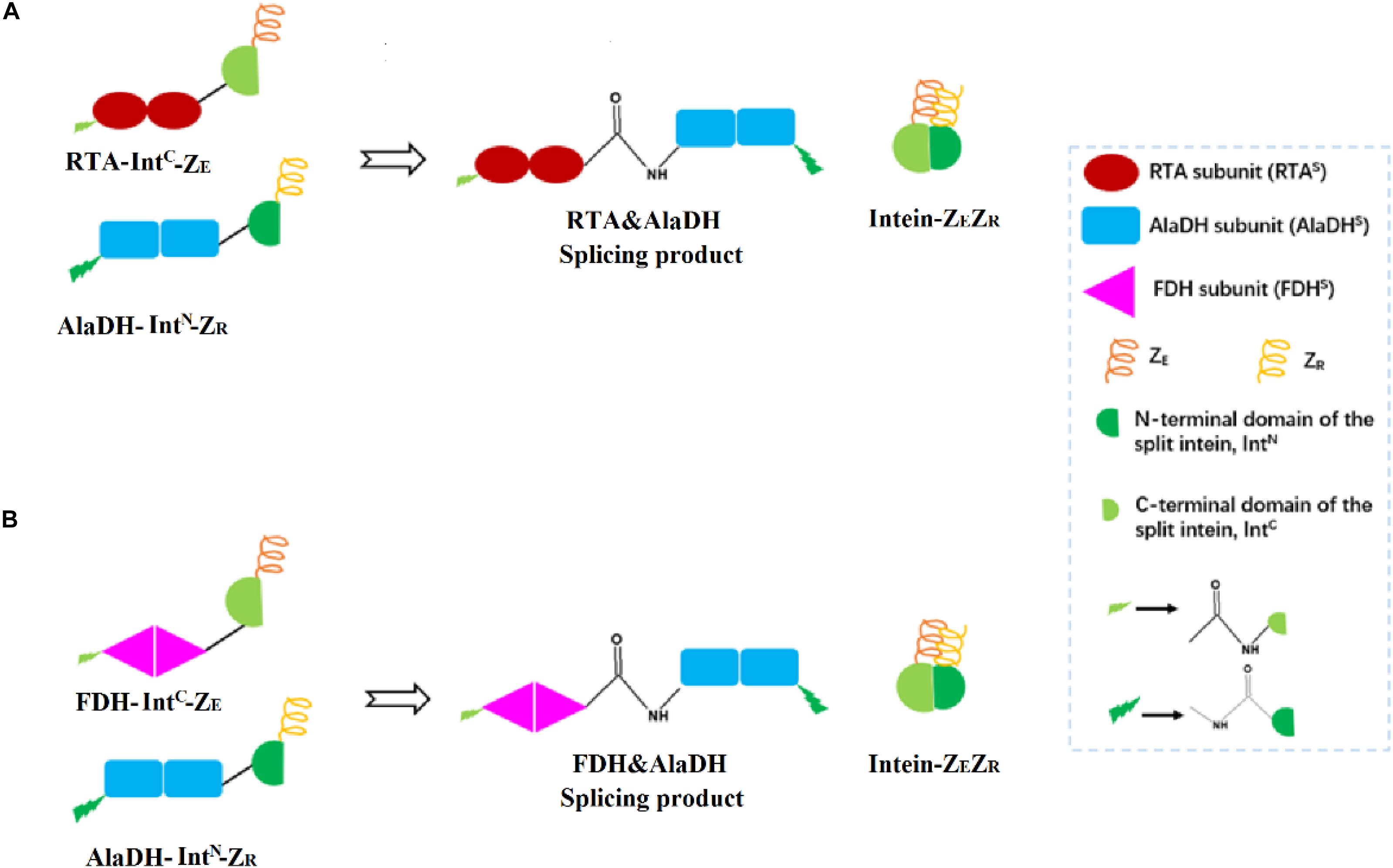
Figure 2. Schematic presentation for the ligations of multienzymes with peptide bonds through hetrosubunit splicing triggered by the dimerization of ZE and ZR. (A) The RTA subunit (RTAS) is ligated to the AlaDH subunit (AlaDHS) forming the splicing product RTA&AlaDH. (B) The FDH subunit (FDHS) is ligated to AlaDHS forming the splicing product FDH&AlaDH. ZR, arginine-rich leucine zipper; ZE, glutamic acid-rich leucine zipper.
R-ω-Transaminase (RTA) and alanine dehydrogenase (AlaDH) are dimeric enzymes, each consisting of two subunits. ZE was fused to the C-terminal domain of the split NpuDnaE intein (IntC), the ZE-IntC segment was fused to the N-terminus of the RTA subunit (RTAS), constructing ZE-IntC-RTAS. ZR was fused to the N-terminal domain of the split NpuDnaE intein (IntN), the ZR-IntN segment was fused to the C-terminus of the AlaDH subunit (AlaDHS), constructing ZR-IntN-AlaDHS (Shah et al., 2011). In between ZR-IntN and AlaDHS and in between ZE-IntC and RTAS, rigid linkers (EPPPPLPPPPLPPPPEPPPP) are required, in order to reduce the steric hindrance for the subunit splicing. Dimerization of ZE and ZR can naturally occur due to the coiled-coil association between ZE and ZR (Shah et al., 2011). Herein, the dimerization of ZE and ZR was utilized to promote the self-excision of IntC and IntN and concomitant ligation of the subunits of RTAS and AlaDHS. The splicing product is designated as RTA&AlaDH (Figure 2A). Another splicing product FDH&AlaDH is obtained similarly, as shown in Figure 2B. Thus, the cycle of alanine → pyruvate → alanine between RTA and AlaDH and the cycle of NADH → NAD+→ NADH between AlaDH and FDH are formed (Figure 1).
The splicing products RTA&AlaDH and AlaDH&FDH were purified sequentially by Ni-Sepharose beads, ultrafiltration fractionation, and gel filtration chromatography. They were analyzed through SDS-PAGE (Figure 3). The band indicated by the black arrow in Figure 3A is ascribed to the ligation of the RTA subunit (RTAS) with the AlaDH subunit (AlaDHS), the ligated subunit of RTAS-AlaDHS. The molecular weight of RTAS-AlaDHS is 75.9 kDa. The band of RTAS-AlaDHS is below the position 97.2 kDa of a mark protein. The band indicated by the black arrow in Figure 3B is attributed to the ligation of AlaDHS with the FDH subunit (FDHS). The band of the ligated subunit AlaDHS-FDHS has a molecular weight of 83.1 kD. Appearance of the bands of RTAS-AlaDHS and AlaDHS-FDHS confirmed the ligations of RTAS with AlaDHS and AlaDHS with FDHS, through the in vivo posttranslational hetrosubunit splicing triggered by the dimerization of ZE and ZR.
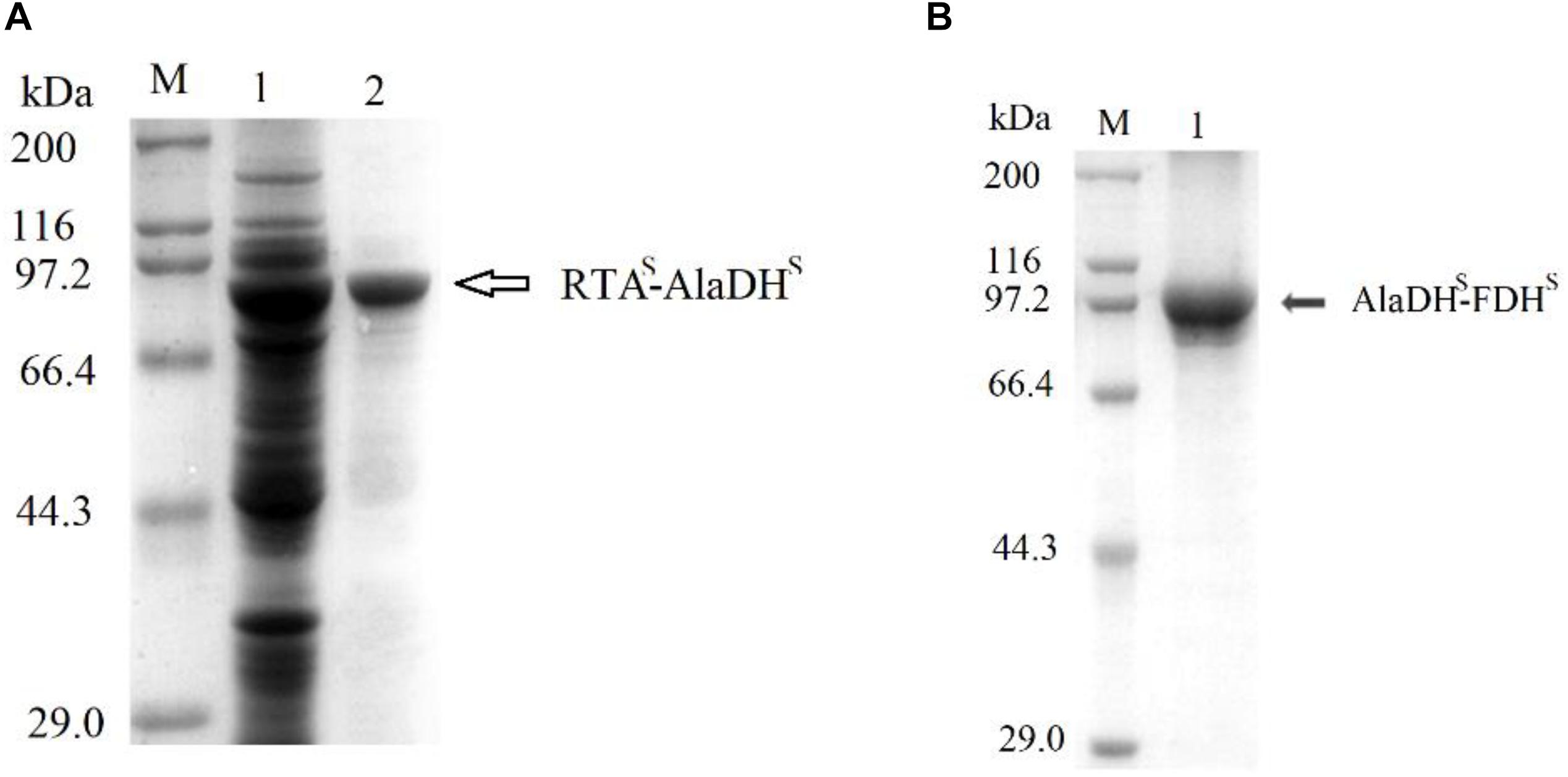
Figure 3. SDS-PAGEs for the splicing products. (A) Lane 1 for total proteins, lane 2 for RTA&AlaDH. (B) Lane 1 for AlaDH&FDH. M, marker protein.
Ligation Mode Based on BN-PAGE Analysis
We have tried to use matrix-assisted laser desorption and ionization time-of-flight mass spectrometry (MALDI-TOF-MS) to identify the fractions of the splicing products RTA&AlaDH and AlaDH&FDH. However, the measured data are not useful, due to the lack of reliability of the data. This is ascribed to the large molecular weights of the fractions of RTA&AlaDH and AlaDH&FDH. In an alternative approach, blue native polyacrylamide gel electrophoresis (BN-PAGE) was used to analyze RTA&AlaDH and AlaDH&FDH. BN-PAGE has been proved to be effective for identifying proteins from multiprotein complexes (Wiedemann et al., 2003; Wittig et al., 2016). The BN-PAGE of RTA&AlaDH is shown in Figure 4A. Eight bands appeared. It is meant that RTA&AlaDH comprises eight fractions. Marker proteins are shown on Lane M. Based on the bands on lane M, the electrophoretic mobility of the marker proteins is plotted against the molecular weight (Supplementary Figure S2), and an equation was obtained. The apparent molecular weights (AMWs) corresponding to the eight bands were estimated, based on the comparison between the electrophoretic mobility of marker proteins and that of the eight bands. The AMWs of the bands I, II, III, IV, V, VI, VII, and VIII were determined to be 254.8, 311.9, 388.0, 422.4, 480.7, 564.9, 674.2, and 741.3 kDa, respectively (Supplementary Table S2). Based on the band intensity, the relative abundances of the eight bands were determined to be 26.5, 2.3, 5.8, 20.4, 11.8, 6.8, 7.6, 19.1% for bands I, II, III, IV, V, VI, VII, and VIII, respectively. For each fraction of RTA&AlaDH, the mode of hetrosubunit ligation was searched. The criterion is that the theoretical molecular weight (TMW) predicated on the basis of the ligation mode is in accordance with the AMW. Based on the molecular weights of intein, ZR-IntN-RTAS, and ZE-IntC-AlaDHS, the TMW of the possible ligation mode were predicated. Supplementary Table S2 lists those ligation modes with TMWs being in accordance with the AMWs of the eight bands. The ligation modes were finally determined to be, RTA∼AlaDH∼RTA (band I), RTA∼AlaDH∼RTA∼AlaDH (band II), AlaDH∼RTA∼ AlaDH∼RTA∼AlaDH (band III), RTA∼AlaDH∼RTA∼AlaDH∼RTA∼AlaDH (band IV), RTA∼ AlaDH∼RTA∼AlaDH∼RTA∼AlaDH∼RTA (band V), AlaDH∼ RTA∼ AlaDH∼RTA∼AlaDH∼RTA∼AlaDH (band VI), RTA∼ AlaDH∼RTA∼AlaDH∼RTA∼ AlaDH∼RTA∼AlaDH (band VII), RTA∼AlaDH∼RTA∼AlaDH∼RTA∼AlaDH∼RTA ∼Ala DH∼RTA∼AlaDH (band VIII).
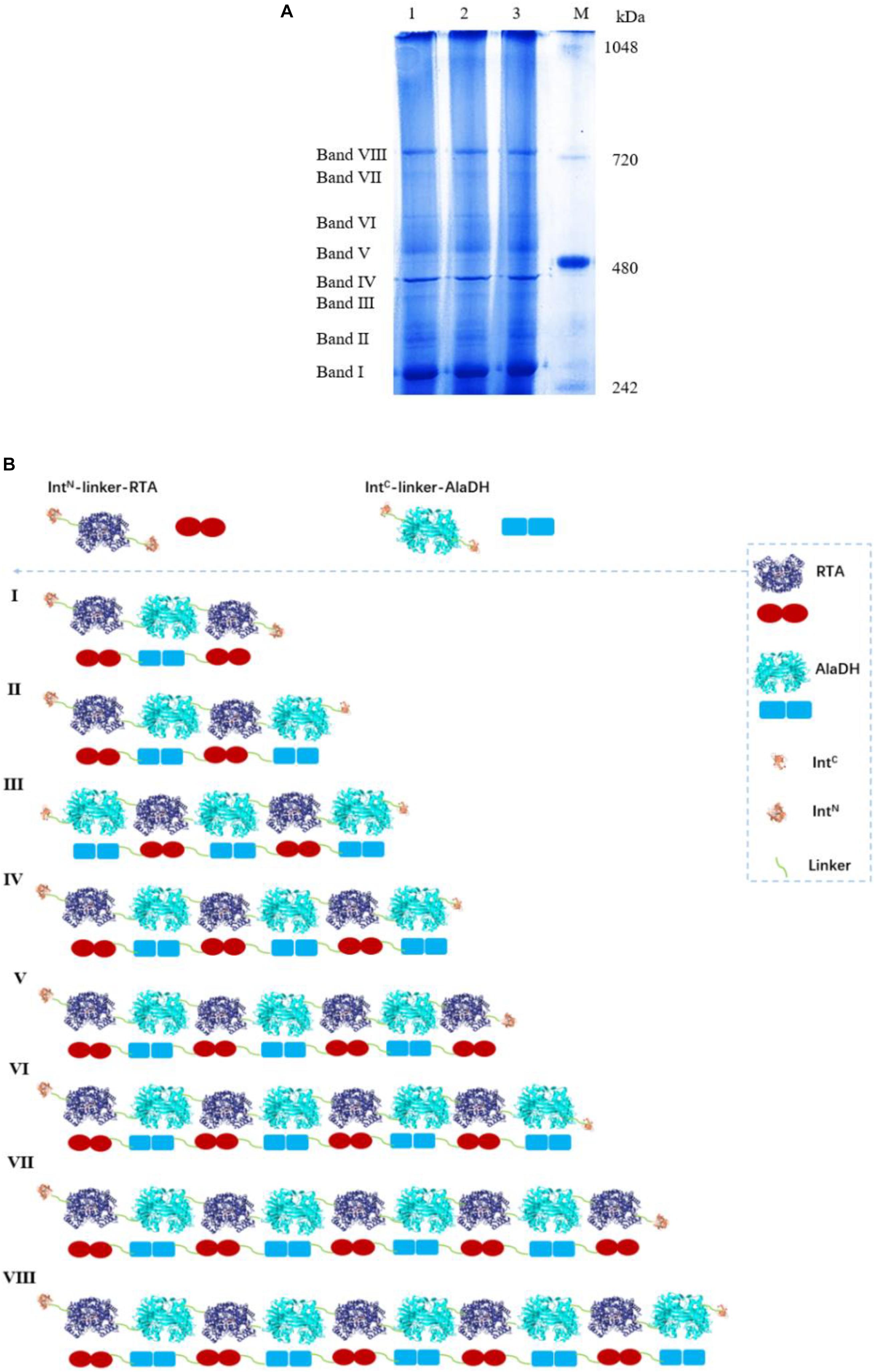
Figure 4. (A) Analysis of the splicing product RTA&AlaDH by BN-PAGE showing eight bands. Lane M: marker. Lanes 1, 2, 3 are for three samples. (B) Schematic diagram representing the RTA&AlaDH fractions derived from the ligation modes.
Figure 4B schematically illustrates the ligation modes corresponding to the eight bands. The paired subunit RTAS-AlaDHS (as illustrated by in Figure 4B) is formed by the hetrosubunit ligation with a peptide bond through the subunit splicing, and it is the key subunit in the RTA&AlaDH fractions. Based on the relative abundances and eight ligation modes, the amount of paired subunit RTAS-AlaDHS is determined to be 5.4 mol per mole of RTA&AlaDH.
For comparison, the subunit splicing was carried out without using the promotion by the heterodimerization of ZR and ZE. The obtained SDS-PAGE (Supplementary Figure S3a) is similar to that in Figure 3, confirming the ligation of RTA with AlaDH through the in vivo subunit splicing. The splicing product is designated as RTA#AlaDH. The BN-PAGE analysis is shown in Supplementary Figure S3b. Four bands appeared, with apparent molecular weights being 271.2, 346.7, 435.1, and 761.7 kDa, corresponding to bands I, II, III, and IV, respectively, and the relative abundances of the four bands are 25.4, 43.9, 10.1, 20.6%, respectively. The ligation modes are shown in Supplementary Figure S3c. The amount of paired subunit RTAS-AlaDHS is determined to be 4.2 mol per mole of RTA#AlaDH. The amount of paired subunit RTAS-AlaDHS in RTA&AlaDH is 28.5% higher than that in RTA#AlaDH.
The BN-PAGE for AlaDH&FDH shown in Figure 5A. The seven bands correspond to seven fractions. Based on Supplementary Figure S4, the AMWs of the bands I, II, III, IV, V, VI, and VII were determined to be 210.6, 278.7, 327.0, 399.4, 469.7, 533.8, and 665.1 kDa, respectively. The relative abundances of the seven bands are 27.4, 20.4, 14.8, 24.3, 3.4, 3.8, and 5.9%, respectively. The ligation modes were, FDH∼AlaDH (band I), FDH∼AlaDH∼FDH (band II), FDH∼AlaDH∼FDH∼AlaDH (band III), AlaDH∼FDH∼AlaDH∼FDH∼AlaDH (band IV), AlaDH∼FDH∼AlaDH∼FDH∼AlaDH∼FDH (band V), AlaDH∼FDH∼AlaDH∼ FDH∼AlaDH∼FDH∼AlaDH (band VI), AlaDH∼FDH∼AlaDH∼FDH∼AlaDH∼FDH∼ AlaDH∼FDH (band VII). Figure 5B schematically illustrates the ligation modes corresponding to the seven bands. The paired subunit AlaDHS-FDHS is indicated by in Figure 5B. Based on the relative abundances and seven ligation modes, the amount of paired subunit AlaDHS-FDHS is determined to be 2.9 mol per mole of AlaDH&FDH. The splicing product AlaDH#FDH was obtained without using the promotion by the heterodimerization of ZR and ZE. The SDS-PAGE (Supplementary Figure S5a) confirmed the ligation of AlaDH with FDH through the in vivo subunit splicing. The BN-PAGE for AlaDH#FDH is shown in Supplementary Figure S5b. Five bands appeared, with apparent molecular weights being 205.8, 280.9, 331.6, 403.8, and 679.3 kDa, corresponding to bands I, II, III, IV, and V, respectively, and the relative abundances of the five bands are 24.4, 22.7, 20.7, 27.4, and 4.8%, respectively. The ligation modes are shown in Supplementary Figure S5c. The amount of paired subunit AlaDHS-FDHS is 2.8 mol per mole of AlaDH#FDH. The amount of paired subunit RTAS-AlaDHS in AlaDH&FDH is comparable to that in AlaDH#FDH.
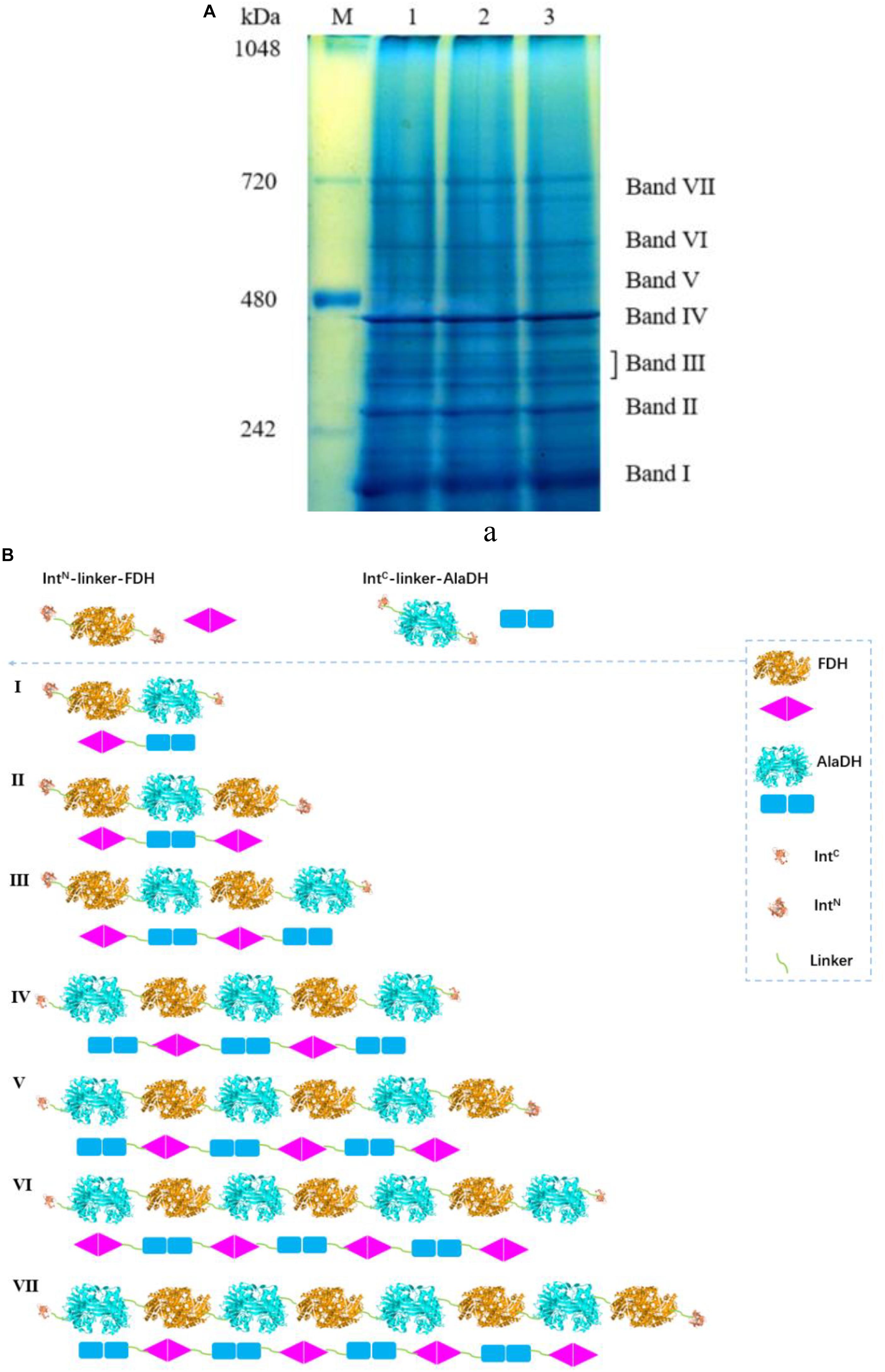
Figure 5. (A) Analysis of the splicing product AlaDH&FDH by BN-PAGE showing seven bands. Lane M: marker. Lanes 1, 2, 3 are for three samples. (B) Schematic diagram representing the AlaDH&FDH fractions derived from the ligation modes.
Alpha-Helical Content Analysis by Circular Dichroism (CD) Spectra
Here we presented two splicing products. RTA&AlaDH was obtained through the in vivo hetrosubunit splicing triggered by the coiled-coil association of ZE and ZR. For comparison, RTA#AlaDH was obtained through the in vivo hetrosubunit splicing without using the coiled-coil association of ZE and ZR. CD spectra were measured for the splicing products RTA&AlaDH and RTA#AlaDH (Figure 6), with the CD spectrum of the mixed enzymes RTA + AlaDH as control. The CD absorption at 222 nm is sensitive to the change of alpha-helical content of proteins (Fasshauer et al., 1997). In comparison to RTA + AlaDH, both RTA&AlaDH and RTA#AlaDH exhibited a more negative ellipticity at 222 nm (Figure 6). The CD spectra demonstrated that the splicing products RTA&AlaDH and RTA#AlaDH contain more alpha-helical structure than the mixed enzymes RTA+AlaDH. This is possibly ascribed to the formation of paired subunit RTAS-AlaDHS in the splicing products. The CD spectra also show that RTA&AlaDH contains more alpha-helical structure than RTA#AlaDH. Supplementary Figure S6 shows the CD spectra for the splicing products AlaDH&FDH and AlaDH#FDH and the mixed enzymes AlaDH + FDH. The difference between the spectra is not significant.
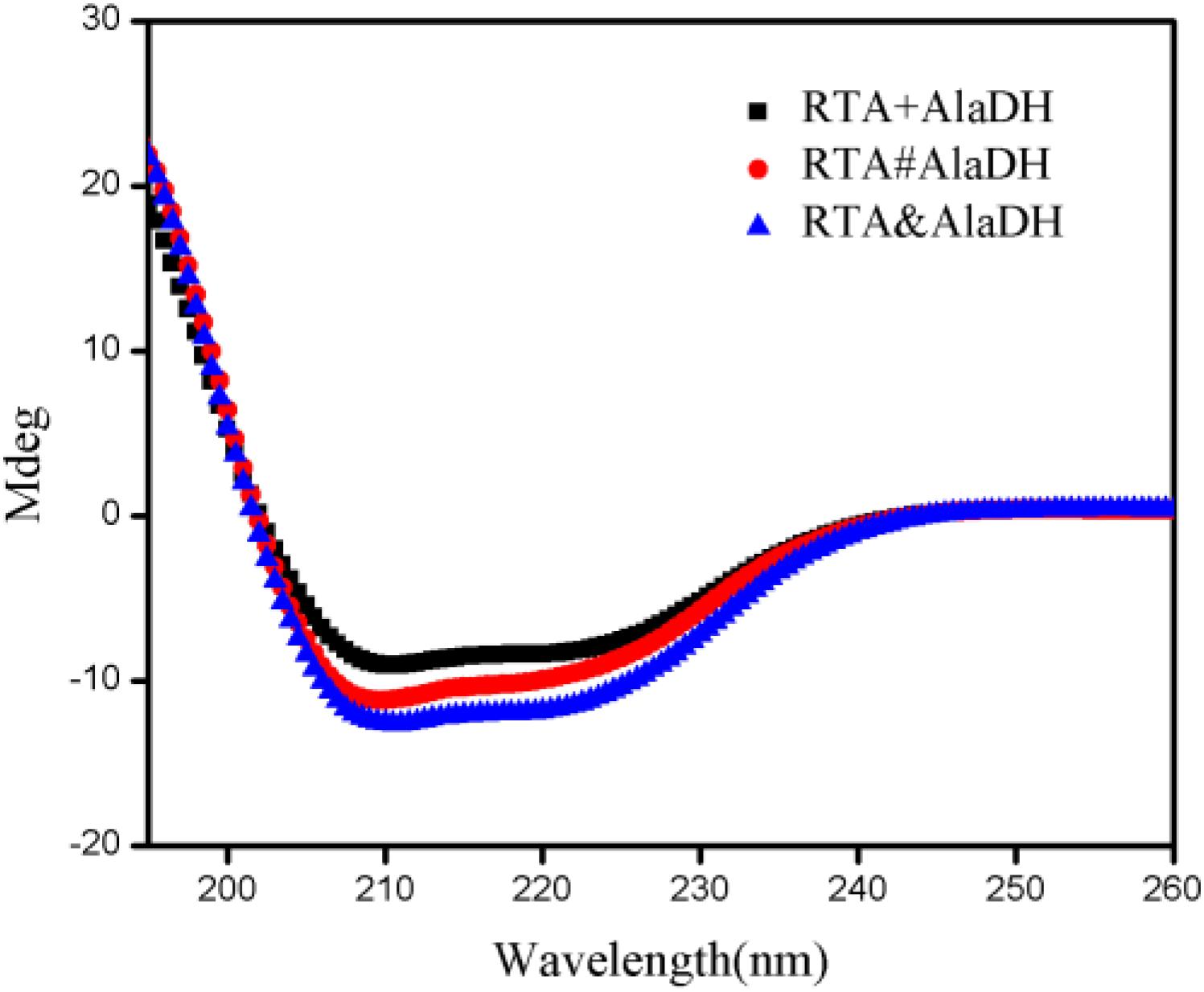
Figure 6. CD spectra for the splicing products RTA&AlaDH and RTA#AlaDH and the mixed enzymes RTA + AlaDH.
Analysis of Enzyme Structures by Two-Dimensional FTIR Spectra
The CD spectra (Figure 6) showed that the splicing products RTA&AlaDH and RTA#AlaDH comprise more alpha-helical structure then the mixed enzymes RTA+AlaDH. Herein, to further get insight into the structures of the splicing products, RTA&AlaDH and RTA#AlaDH as well as RTA + AlaDH were subjected to the measurement of two-dimensional FTIR spectra, as described in the experimental section. Through two-dimensional correlation spectroscopy, individual bands with a high resolution can be identified. Upon external perturbation, the sequential order of spectral intensity changes of the enzyme structures (unordered, α-helix, and β-sheet) can be obtained. By combining the sign of the peaks in the synchronous and asynchronous maps, the sequence of the spectral variations can be determined (Noda, 1989).
Infrared spectra as a function of the deuteration time were measured as shown in Supplementary Figure S7. Based on Supplementary Figure S7, two-dimensional spectra are obtained for RTA&AlaDH (Figure 7A), RTA#AlaDH (Figure 7B), and RTA + AlaDH (Figure 7C). During the measurements, D2O was added, hence hydrogen-deuterium (H-D) exchange was the external perturbation. The main correlation peaks observed in the synchronous and asynchronous maps of Figure 7 are summarized in Supplementary Table S3. Published literature on infrared spectroscopy of proteins provides the basis for the band assignment (Richard et al., 2005; Mecozzi et al., 2011). The sequential order of H-D exchange is determined based on Noda’s rules (Noda, 1989). For RTA&AlaDH (Figure 7A), the cross-peaks (1650, 1450) and (1610, 1450) can be identified from the synchronous and asynchronous maps. The Noda’s rules confirm that the unordered structure (1450 cm–1) exhibited a faster H-D exchange than the α-helix (1650 cm–1) and β-sheet (1610 cm–1) structures. For RTA#AlaDH, the cross-peaks (1639, 1430) and (1665, 1437) can be identified from the synchronous and asynchronous maps are shown Figure 7B. The unordered structure (1430 cm–1) exhibited a faster H-D exchange than the α-helix (1639 cm–1), and the unordered structure represented by 1437 cm–1 exchanged before the β-sheet structure (1665 cm–1). For RTA + AlaDH (Figure 7C), the cross-peaks (1645, 1425) and (1680, 1427) can be identified. The unordered structure (1425 cm–1) exhibited a faster H-D exchange than the α-helix (1645 cm–1), and the unordered structure represented by 1427 cm–1 exchanged before the β-sheet structure (1680 cm–1).
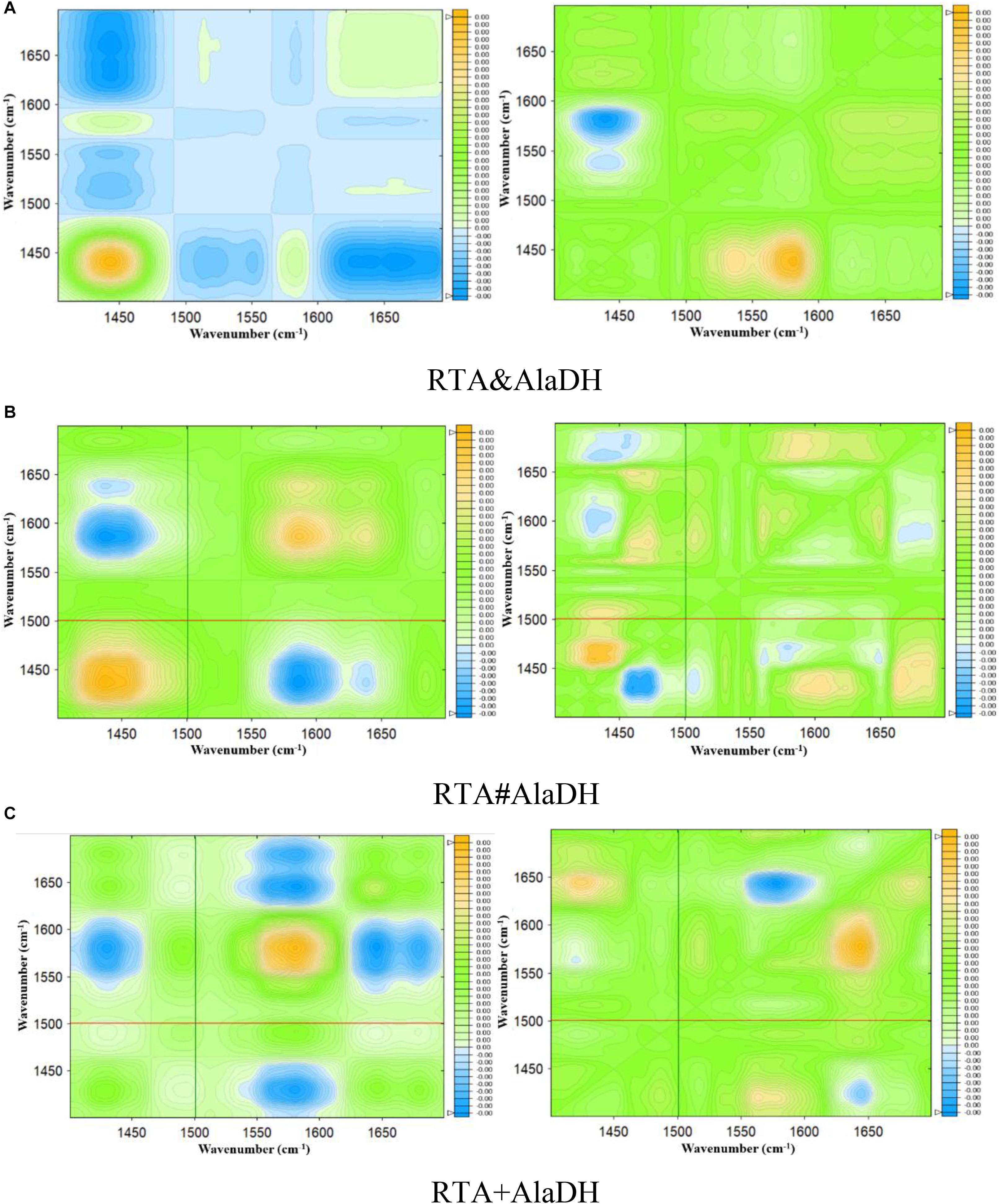
Figure 7. Synchronous (left) and asynchronous (right) 2D correlation spectra of the enzymes obtained for 90 min of deuteration. (A) Splicing product RTA&AlaDH; (B) splicing product RTA#AlaDH; (C) mixed enzyme RTA + AlaDH.
The analysis of the two-dimensional FTIR spectra (Figure 7) demonstrated that the three two-enzyme systems, RTA&AlaDH, RTA#AlaDH, and RTA + AlaDH, exhibited the same sequence of the spectral variations upon the external perturbation of D2O. That is the unordered structures exhibited a faster H-D exchange than the structures of α- helices and β-sheets. Ascribing to the presence of strong hydrogen bonds in α-helices and β-sheets, the protons of α-helix and β-sheet are more difficult to exchange than those of unordered structures (Richard et al., 2005; Mecozzi et al., 2011). There is no regular hydrogen bonds in the unordered structures, the amide protons can be exchanged more easily than those of α-helices and β-sheets. The sequential order of H-D exchange demonstrated that the in vivo hetrosubunit splicing had little effect on the hydrogen bonding interactions presented, that maintain the secondary structures of α-helices and β-sheets of the enzymes. These secondary structures play an important role in the function of the enzymes.
Anti-denaturation Through Hetrosubunit Splicing
Multienzymes comprise of subunits that are associated through non-covalent interactions, which play an important role in maintaining the quaternary structures. However, the non-covalent interactions can be weakened or partially destroyed under certain experimental conditions. The dissociation of the subunits leads to the inactivation of multimeric enzymes. Multimeric enzymes face the concern of stability, especially when applied at industrial scale (Fernandez, 2009). For the two-enzyme systems, RTA&AlaDH, RTA#AlaDH, and RTA+AlaDH, fluorescence spectra were measured to investigate enzyme stability. Urea (2 M) was used as the denaturing agent. Acrylamide was used to quench the intrinsic fluorescence of the enzymes. The spectra of fluorescence intensity changing with the acrylamide concentration are shown in Supplementary Figure S8. Stern–Volmer equation F0/F = 1+ KSV[A] was used to evaluate the concentration-dependence quenching (Georlette et al., 2004). In the equation, F and F0 are the fluorescence intensities in the presence and absence of acrylamide, respectively. [A] is the acrylamide concentration. The quenching constant KSV reflects the conformational change of enzymes (Georlette et al., 2004). The ratio of F0/F is plotted against acrylamide concentration as shown in Figure 8. A larger KSV corresponds to a larger conformational change of the protein. The KSV value of RTA&AlaDH is 2.57, which is smaller than 3.48 for RTA#AlaDH and much smaller than 6.63 for RTA + AlaDH. In comparison to RTA&AlaDH and RTA#AlaDH, RTA + AlaDH is more inclined to expose the tryptophan residues upon interfering by urea. The splicing products are stable than the mixed two enzymes RTA+AlaDH. The splicing products resulted from the covalent ligation of the RTA subunit with the AlaDH subunit, making the splicing products become more rigid than mixed enzymes. It is indicated that RTA&AlaDH is stable than RTA#AlaDH. Based on the BN-PAGE analysis (Figure 4 and Supplementary Figure S3b), the amount of the paired subunit RTAS-AlaDHS in RTA&AlaDH is 28.5% higher than that in RTA#AlaDH. This results in a lower subunit mobility in RTA&AlaDH, leading to a more rigid structure. The stabilities of AlaDH&FDH, AlaDH#FDH, and AlaDH+FDH have also been analyzed based on the fluorescence intensity (in the legend of Supplementary Figure S9).
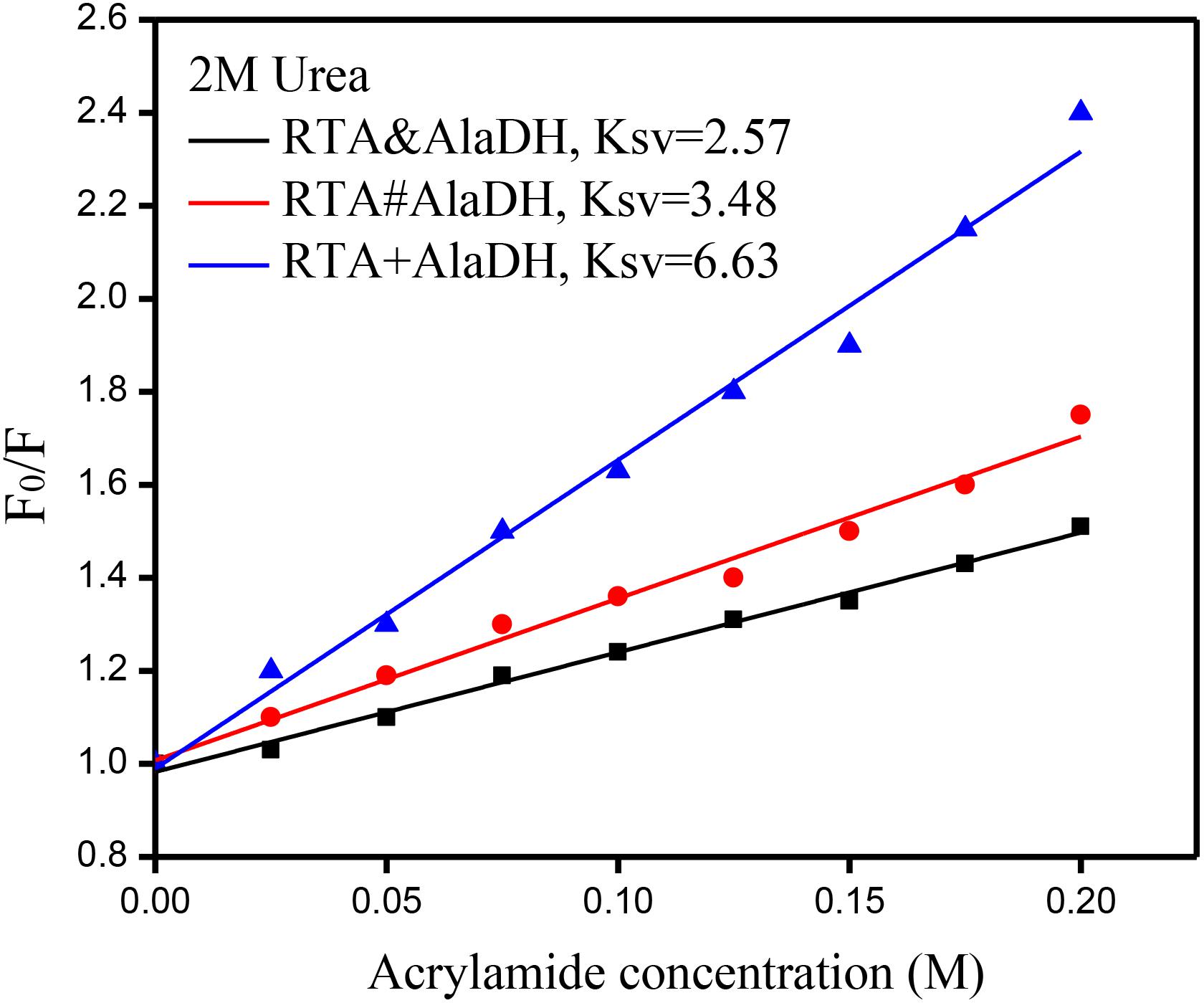
Figure 8. Stern–Volmer plots for the quenching of protein fluorescence by acrylamide. F0 and F are the fluorescence intensities in the absence and presence of acrylamide, respectively.
Enzyme-Substrate Interaction Based on Microscale Thermophoresis Analysis
The analysis of enzyme binding to substrate can provide the information of enzyme affinity toward the substrate. Microscale thermophoresis (MST) is a relatively recent technique based on the unique physical principle of thermophoresis. It can be used to quantify the interactions of proteins and small molecules (Seidel et al., 2013). The two-enzyme systems, RTA&AlaDH, RTA#AlaDH, and RTA+AlaDH, were subjected to MST measurements to quantify binding coefficients (Kd). Figure 9 shows the fraction bound versus ligand concentration. The substrate 2-octanone was the ligand. RTA&AlaDH exhibited a Kd value of 0.68 ± 0.15 mM, which is smaller than the Kd value of 2.33 ± 3.22 mM by RTA#AlaDH, and much smaller than the Kd value of 14.91 ± 10.13 mM by RTA+AlaDH. It is indicated that RTA&AlaDH exhibited the affinity to 2-octanone 3.4 times stronger than that by RTA#AlaDH, and 6.5 times stronger than that by RTA+AlaDH.
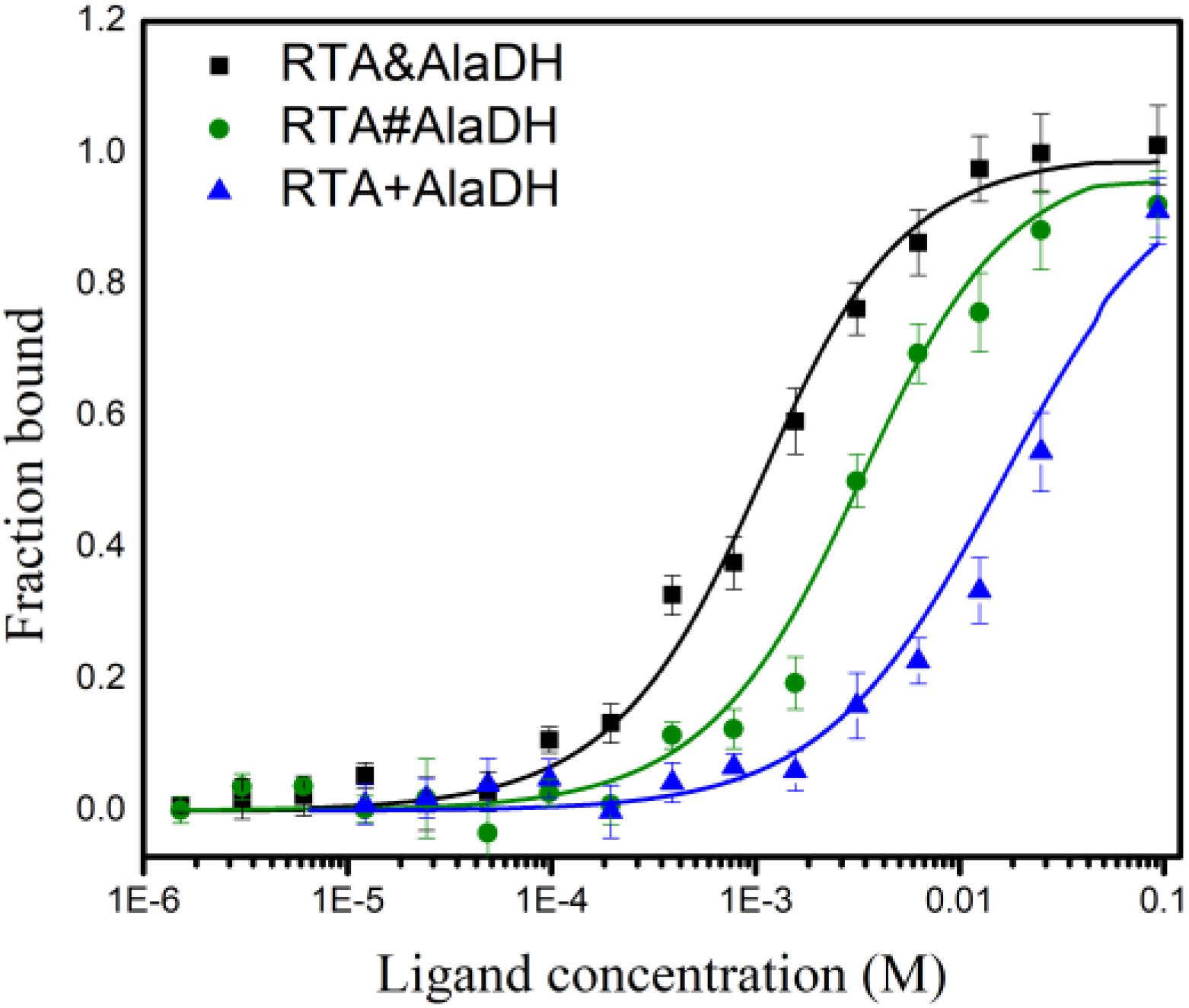
Figure 9. Binding of the two-enzyme systems to the substrate 2-octanone measured by MST. The curves are obtained from the specific change in the thermophoretic mobility upon 2-octanone (ligand) titration to the enzymes. The error bars represent the standard deviations of triplicate samples.
Enzymatic Activity
In the conversion of the ketones to amines under the catalysis of RTA, alanine was used as amine donor, and pyruvate being the co-product. Maintaining alanine with a low concentration facilitates subsequent purification of the product. In addition, recycling of pyruvate back to alanine can reduce the inhibitory effect of by-product. Alanine with a small concentration (5 mM) was used, which is tenth of that of 2-octanone (50 mM). The conversion was catalyzed under the three-enzyme systems RTA&AlaDH + AlaDH&FDH, RTA#AlaDH + AlaDH#FDH, and RTA + AlaDH + FDH. Figure 10 shows the 2-octanone conversion versus reaction time. RTA + AlaDH + FDH (control) achieved a conversion of 32% after 2 h reaction, and 79.2% of 2-octanone was converted by RTA#AlaDH + AlaDH#FDH. In contrast, RTA&AlaDH + AlaDH&FDH achieved a 2-octanone conversion of 98.8% after 2 h reaction. For the conversion of acetophenone (Supplementary Figure S10), after 6 h reaction, 65, 45, and 18% were achieved by RTA&AlaDH + AlaDH&FDH, RTA#AlaDH + AlaDH#FDH, and RTA + AlaDH + FDH, respectively. RTA&AlaDH + AlaDH&FDH exhibited a higher catalysis efficiency than RTA#AlaDH + AlaDH#FDH, and much higher efficiency than RTA + AlaDH + FDH.
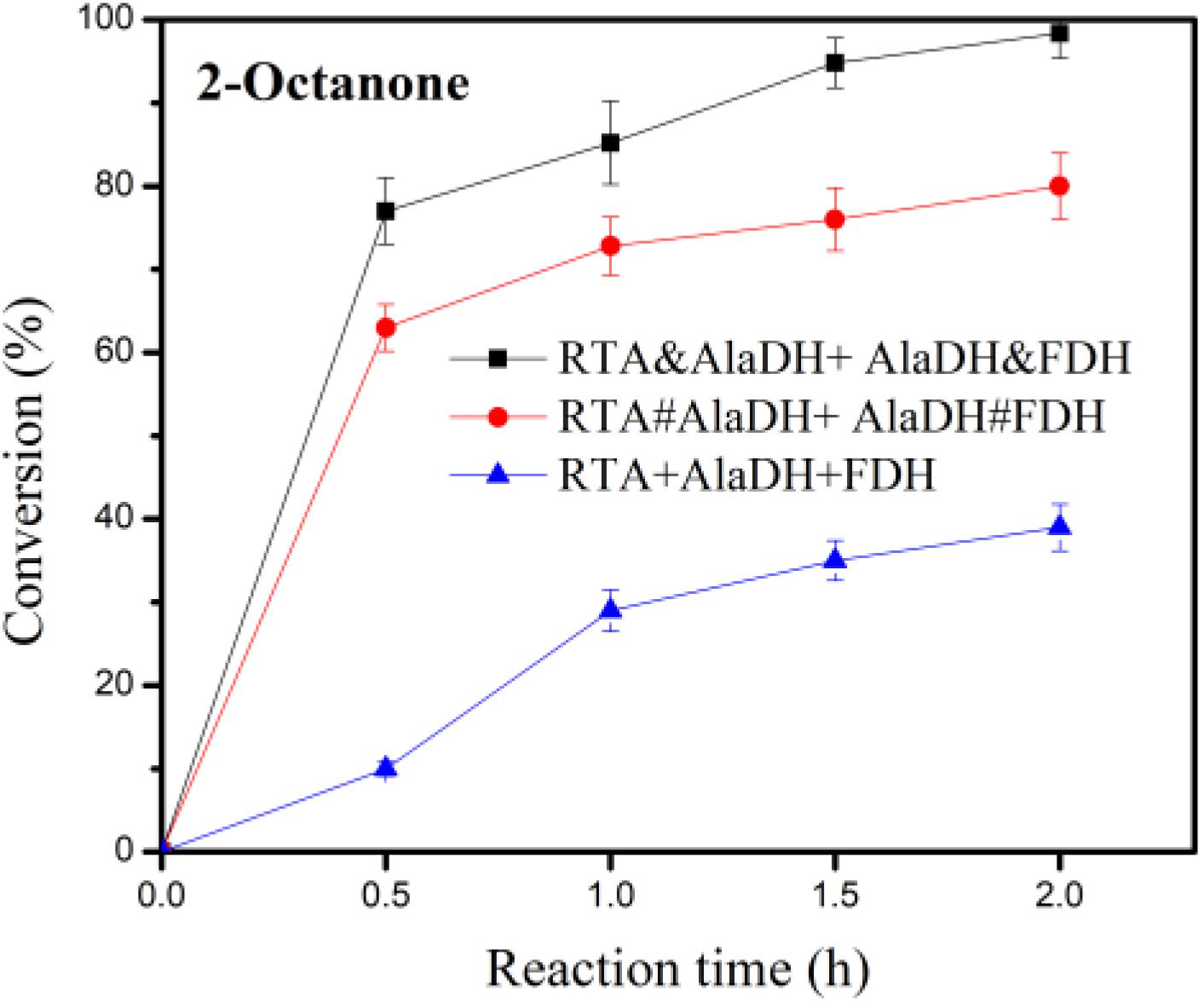
Figure 10. Conversion of 2-octanone under the catalysis of RTA&AlaDH + AlaDH&FDH, RTA#AlaDH + AlaDH#FDH, and RTA + AlaDH + FDH. The error bars represent the standard deviations of triplicate samples.
Supplementary Table S4 lists the published results using ω-transaminases combining AlaDH and FDH for the conversion of 2-octanone (50 mM). In the catalysis, alanine was used as amine donor. The amounts of alanine used are 250 mM by Mutti et al. (2011, 2012) and 280 mM by Koszelewski et al. (2010). In contrast, this work used alanine with a small concentration (5 mM), which is tenth of that of 2-octanone (50 mM). The conversion of 2-octanone achieved by this work is higher than the conversions by the three-enzyme systems of the published articles (Supplementary Table S4). Through the hetrosubunit splicing, the hetrosubunits are paired. This paves a short pathway for alanine recycling. The amount of alanine used is much less than that published, demonstrating that the transferring and recycling of alanine are more efficient than the three-enzyme systems (Supplementary Table S4). In addition, the reaction time of this work is 2 h in comparison to 24 h required by the catalysis in the published articles.
For the mixed enzymes RTA + AlaDH + FDH, the interactions between the enzymes are not specific interactions. Under shaking condition during the reaction, occasionally pairing of RTA with AlaDH and AlaDH with FDH occurred. However, the instantaneous pairing of the enzymes cannot be controlled and maintained for the whole reaction time, attributed to the non-specific interaction between the enzymes. This results in that, the transfers of alanine and pyruvate between RTA and AlaDH and NADH and NAD+ between AlaDH and FDH cannot be directional. In contrast, RTA&AlaDH + AlaDH&FDH and RTA#AlaDH + AlaDH#FDH achieved the paired hetrosubunits, RTAS-AlaDHS and AlaDHS-FDHS, as illustrated in Figure 2. Based on the software (Discovery Studio), the linker length is 3.3 nm. Hence the largest distance between the pairing hetrosubunits is 6.6 nm. Thus, the hetrosubunit pairing paves short transferring-recycling pathways for transferring pyruvate and NAD+ and recycling alanine and NADH. Thus the transferring and recycling are directional and oriented. The hetrosubunit pairing through peptide bonds makes the splicing products become stable in comparison to the mixed enzymes. In addition, the splicing products have exhibited much higher affinity toward the model substrate 2-octanone. All these contribute to the splicing products exhibiting much higher catalysis efficiency than the mixed enzymes.
Based on the BN-PAGE analysis for RTA&AlaDH, RTA#AlaDH, AlaDH&FDH, and AlaDH#FDH, the linking mode corresponding to each band has been determined, and the fraction of each band has also been obtained. Based on the information, the number of the pairing hetersubunits RTAS-AlaDHS in RTA&AlaDH is 28.5% higher than that in RTA#AlaDH, and the number of AlaDHS-FDHS in AlaDH&FDH is comparable to that in AlaDH#FDH. Possibly, this is the reason that RTA&AlaDH contains more alpha-helical structure than RTA#AlaDH. It is implied that, RTA&AlaDH has more alanine→pyruvate→alanine cycles than RTA#AlaDH. Due to having more pairing hetrosubunits, RTA&AlaDH + AlaDH&FDH is more stable than RTA#AlaDH + AlaDH#FDH. In addition, RTA&AlaDH + AlaDH&FDH exhibited much higher affinity toward the substrate 2-octanone than RTA#AlaDH + Ala DH#FDH. As a result, RTA&AlaDH + AlaDH&FDH exhibited a higher catalysis efficiency than RTA#AlaDH + AlaDH#FDH.
Conclusion
The heterodimerization of leucine zipper motifs promoted the in vivo hetrosubunit splicing, and the pairing enzymes RTA&AlaDH and AlaDH&FDH have been constructed. The hetrosubunit splicing has little effect on the intro-hydrogen bonding interactions, and can confer RTA&AlaDH having more alpha-helical structure and a higher stability. Ascribing to the hetrosubunit pairing through peptide bonds, the transferring pyruvate and NAD+ and recycling alanine and NADH are directional and oriented. In contrast to the mixed three enzymes RTA+AlaDH+FDH, the ligated enzymes RTA&AlaDH+AlaDH&FDH exhibited a much larger substrate affinity, higher stability, and significantly enhanced activity.
Data Availability Statement
All datasets generated for this study are included in the article/Supplementary Material.
Author Contributions
RL and WF designed the experiment study and prepared the manuscript. RL contributed the expression plasmids ZE-IntC-link-AlaDH/RTA-link-IntN-ZR-pETDuet and ZE-IntC-link-AlaDH/FDH-link-IntN-ZR-pETDuet, performed the experimental setup and procedure. RL, YC, and KD carried out the analysis and evaluation of the results. All the authors approved the final manuscript.
Funding
This work was supported by National Natural Science Foundation of China (21776011 and 21978018).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fbioe.2020.00686/full#supplementary-material
References
Agren, D., Stehr, M., Berthold, C. L., Kapoor, S., Oehlmann, W., Singh, M., et al. (2008). Three-dimensional structures of apo- and holo-L-alanine dehydrogenase from mycobacterium tuberculosis reveal conformational changes upon coenzyme binding. J. Mol. Biol. 377, 1161–1173. doi: 10.1016/j.jmb.2008.01.091
Börner, T., Rämisch, S., Reddem, E. R., Bartsch, S., Vogel, A., and Thunnissen, A. M. W. H. (2017). Explaining operational instability of amine transaminases: substrate-induced inactivation mechanism and influence of quaternary structure on enzyme–cofactor intermediate stability. ACS Catal. 7, 1259–1269. doi: 10.1021/acscatal.6b02100
Cassimjee, K. E., Branneby, C., Abedi, V., Wells, A., and Berglund, P. (2010). Transaminations with isopropyl amine: equilibrium displacement with yeast alcohol dehydrogenase coupled to in situ cofactor regeneration. Chem. Commun. 46, 5569–5571. doi: 10.1039/c0cc00050g
Du, K., Zhao, J., Sun, J., and Feng, W. (2017). Specific ligation of two multimeric enzymes with native peptides and immobilization with controlled molar ratio. Bioconjugate Chem. 28, 1166–1175. doi: 10.1021/acs.bioconjchem.7b00043
Fasshauer, D., Otto, H., Eliason, W. K., Jahn, R., and Brünger, A. T. (1997). Structural changes are associated with soluble N-ethylmaleimide-sensitive fusion protein attachment protein receptor complex formation. J. Biol. Chem. 272, 28036–28041. doi: 10.1074/jbc.272.44.28036
Fernandez, L. R. (2009). Stabilization of multimeric enzymes: strategies to prevent subunit dissociation. Enzym. Microb. Technol. 45, 405–418. doi: 10.1016/j.enzmictec.2009.08.009
Fogal, S., Beneventi, E., Cendron, L., and Bergantino, E. (2015). Structural basis for double cofactor specificity in a new formate dehydrogenase from the acidobacterium Granulicella mallensis MP5ACTX8. Appl. Microbiol. Biotechnol. 99, 9541–9554. doi: 10.1007/s00253-015-6695-x
Georlette, D., Blaise, V., Bouillenne, F., Damien, B., Thorbjarnardóttir, S. H., Depiereux, E., et al. (2004). Adenylation-dependent conformation and unfolding pathways of the NAD+-dependent DNA ligase from the thermophile Thermus scotoductus. Biophys. J. 86, 1089–1104. doi: 10.1016/s0006-3495(04)74184-3
Han, S. W., Jang, Y., and Shin, J. S. (2019). In vitro and in vivo one-pot deracemization of chiral amines by reaction pathway control of enantiocomplementary ω-Transaminases. ACS Catal. 9, 6945–6954. doi: 10.1021/acscatal.9b01546
Hannes, K., Fabian, S. M., and Matthias, H. (2014). Recent achievements in developing the biocatalytic toolbox for chiral amine synthesis. Curr. Opin. Chem. Biol. 19, 180–192. doi: 10.1016/j.cbpa.2014.02.021
Hohne, M., Kuhl, S., Robins, K., and Bornscheuer, U. T. (2008). Efficient asymmetric synthesis of chiral amines by combining transaminase and pyruvate decarboxylase. Chem. Bio. Chem. 9, 363–365. doi: 10.1002/cbic.200700601
Isupov, N. M., Boyko, M. K., Sutter, J. M., James, P., Sayer, C., Schmidt, M., et al. (2019). Thermostable branched-chain amino acid transaminases from the archaea Geoglobus acetivorans and Archaeoglobus fulgidus: biochemical and structural characterization. Front. Bioeng. Biotechnol. 7:7. doi: 10.3389/fbioe.2019.00007
Kelly, S. A., Pohle, S., Wharry, S., Mix, S., Allen, C. R. C., Moody, T. S., et al. (2018). Application of ω-transaminases in the pharmaceutical industry. Chem. Rev. 118, 349–367. doi: 10.1021/acs.chemrev.7b00437
Koszelewski, D., Clay, D., Rozzell, D., and Kroutil, W. (2009). Deracemisation of α-chiral primary amines by a one-pot, two-step cascade reaction catalysed by ω-transaminases. Eur. J. Org. Chem. 14, 2289–2292. doi: 10.1002/ejoc.200801265
Koszelewski, D., Gçritzer, M., Clay, D., Seisser, B., and Kroutil, W. (2010). Synthesis of optically active amines employing recombinant ω-transaminases in E. coli cells. ChemCatChem 2, 73–77. doi: 10.1002/cctc.200900220
Koszelewski, D., Lavandera, I., Clay, D., Guebitz, G. M., Rozzell, D., and Kroutil, W. (2008). Formal asymmetric biocatalytic reductive amination. Angew. Chem. Int. Ed. 47, 9337–9340. doi: 10.1002/anie.200803763
Li, R., Zhou, X., Liu, D., and Feng, W. (2018). Enhancing the activity and stability of Mn-superoxide dismutase by one-by-one ligation to catalase. Free Radic. Biol. Med. 129, 138–145. doi: 10.1016/j.freeradbiomed.2018.09.018
Liao, L. T., Meng, Y. L., Wang, R. M., Jia, B. L., and Li, P. W. (2019). Coupling and regulation of porous carriers using plasma and amination to improve the catalytic performance of glucose oxidase and catalase. Front. Bioeng. Biotechnol. 7:426. doi: 10.3389/fbioe.2019.00426
Lindbladh, C., Rault, M., Hagglund, C., Small, W. C., Mosbach, K., Bülow, L., et al. (1994). Preparation and kinetic characterization of a fusion protein of yeast mitochondrial citrate synthase and malate dehydrogenase. Biochemistry 33, 11692–11698. doi: 10.1021/bi00205a004
Mecozzi, M., Pietroletti, M., and TornambeÌ, A. (2011). Molecular and structural characteristics in toxic algae cultures of Ostreopsis ovata and Ostreopsis spp. evidenced by FTIR and FTNIR spectroscopy. Spectrochim. Acta Part A 78, 1572–1580. doi: 10.1016/j.saa.2011.02.002
Moll, J. R., Ruvinov, S. B., Pastan, I., and Vinson, C. (2001). Designed heterodimerizing leucine zippers with a range of pIs and stabilities up to 10-15 M. Protein Sci. 10, 649–655. doi: 10.1110/ps.39401
Mutti, G. F., Fuchs, S. C., Pressnitz, D., Sattler, H. J., and Kroutil, W. (2011). Stereoselectivity of four (R)-selective transaminases for the asymmetric amination of ketones. Adv. Synth. Catal. 353, 3227–3233. doi: 10.1002/adsc.201100558
Mutti, G. F., Fuchs, S. C., Pressnitz, D., Turrini, G. N., Sattler, H. J., Lerchner, A., et al. (2012). Amination of ketones by employing two new (S)-selective ω-transaminases and the His-tagged ω-TA from vibrio fluvialis. Eur. J. Org. Chem. 2012, 1003–1007. doi: 10.1002/ejoc.201101476
Noda, I. (1989). Two-dimensional infrared spectroscopy. J. Am. Chem. Soc. 111, 8116–8118. doi: 10.1021/ja00203a008
Padrosa, D. R., Alaux, R., Smith, P., Dreveny, I., López-Gallego, F., and Paradisi, F. (2019). Enhancing PLP-binding capacity of class-III ω-transaminase by single residue substitution. Front. Bioeng. Biotechnol. 7:282. doi: 10.3389/fbioe.2019.00282
Pressnitz, D., Fuchs, C. S., Sattler, J. H., Knaus, T., Macheroux, P., Mutti, F. G., et al. (2013). Asymmetric amination of tetralone and chromanone derivatives employing ω-transaminases. ACS Catal. 3, 555–559. doi: 10.1021/cs400002d
Richard, J. A., Kelly, I., Marion, D., Auger, M., and Pezolet, M. (2005). Structure of β-purothionin in membranes: a two-dimensional infrared correlation spectroscopy study. Biochemistry 44, 52–61. doi: 10.1021/bi048443t
Rocha, J. F., Pina, A. F., Sousa, S. F., and Cerqueira, N. M. F. S. A. (2019). PLP-dependent enzymes as important biocatalysts for the pharmaceutical, chemical and food industries: a structural and mechanistic perspective. Catal. Sci. Technol. 9, 4864–4876. doi: 10.1039/c9cy01210a
Santos, D. R. D., Souza, R. O., Dias, L. B., Ribas, T. B., de Oliveira, L. C. F., Sumida, D. H., et al. (2018). The effects of storage time and temperature on the stability of salivary phosphatases, transaminases and dehydrogenase. Arch. Oral Biol. 85, 160–165. doi: 10.1016/j.archoralbio.2017.10.016
Seidel, S. A. I., Dijkman, P. M., Lea, W. A., van den Bogaart, G., Jerabek-Willemsen, M., Lazic, A., et al. (2013). Microscale thermophoresis quantifies biomolecular interactions under previously challenging conditions. Methods 59, 301–315. doi: 10.1016/j.ymeth.2012.12.005
Shah, N. H., Vila-Perelló, M., and Muir, T. W. (2011). Kinetic control of one pot trans-splicing reactions by using a wild type and designed split intein. Angew. Chem. Int. Ed. 50, 6511–6515. doi: 10.1002/anie.201102909
Shin, J. S., and Kim, B. G. (1999). Asymmetric synthesis of chiral amines with ω-transaminase. Biotechnol. Bioeng. 65, 206–211. doi: 10.1016/j.cbpa.2017.12.007
Simon, R. C., Richter, N., Busto, E., and Kroutil, W. (2014). Recent developments of cascade reactions involving ω-transaminases. ACS. Catal. 4, 129–143. doi: 10.1021/cs400930v
Sun, J., Fu, Y. Q., Li, R., and Feng, W. (2018). Multifunctional hollow-shell microspheres derived from cross-linking of MnO2 nanoneedles by zirconium-based coordination polymer: enzyme mimicking, micromotors, and protein immobilization. Chem. Mater. 30, 1625–1634. doi: 10.1021/acs.chemmater.7b04945
Wiedemann, N., Kozjak, V., Chacinska, A., Schönfisch, B., Rospert, S., Ryan, M. T., et al. (2003). Machinery for protein sorting and assembly in the mitochondrial outer membrane. Nature 424, 565–571. doi: 10.1038/nature01753
Keywords: R-ω-transaminase, alanine dehydrogenase, formate dehydrogenase, hetrosubunit splicing, leucine zipper motifs
Citation: Li R, Chen Y, Du K and Feng W (2020) Peptide Bond Formation Between the Hetrosubunits of ω-Transaminase, Alanine Dehydrogenase, and Formate Dehydrogenase Through Subunit Splicing Promoted by Heterodimerization of Leucine Zipper Motifs. Front. Bioeng. Biotechnol. 8:686. doi: 10.3389/fbioe.2020.00686
Received: 19 March 2020; Accepted: 02 June 2020;
Published: 30 June 2020.
Edited by:
Ken’ichiro Matsumoto, Hokkaido University, JapanCopyright © 2020 Li, Chen, Du and Feng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wei Feng, ZmVuZ3dlaUBtYWlsLmJ1Y3QuZWR1LmNu